
N-(3-Aminoalkyl)proline derivatives with potent antigycation activity†
Abstract
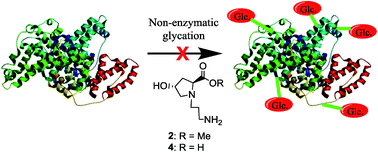
The importance of amino acids in the therapy of conditions such as renal failure, neurological disorders and congenital defects has been documented. Some amino acids such as lysine and glycine have also been reported to have antiglycating activity. Herein we report the synthesis of a new series of N-(3-aminoalkyl)proline derivatives which are non-natural in nature. The compounds were unambiguously characterized by NMR, mass and IR spectroscopy. Their in vitro antiglycation activity was studied by circular dichroism and fluorescence spectrometry. The mechanism of action was also studied and found to take place by inhibition of Amadori product formation. The inhibition of AGE formation was further confirmed by western blot and LC-MS/MS analyses and the IC50 values of the potent compounds were determined. Compounds containing hydroxyl substituents at C4 were found to have superior antiglycation properties than those containing azide substituents at the same position. The compounds were additionally found to possess good anti-oxidant properties, which could lead to further reduction in AGE formation. Moreover, the title compounds were found to have low cytotoxicity in mammalian cells, another important attribute. Thus, the title compounds represent a novel promising class of antiglycating agents.
 Please wait while we load your content...
Please wait while we load your content...

