
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of ring-type structures in nonconventional luminescent polyurethane derivatives†
Chang-Yi
Zhu
a,
Ya-Jie
Meng
b,
Yan-Hong
Xu
 a,
Chun-Qiu
Xia
a,
Nan
Jiang
*a,
Jia-Wei
Xu
*b and
Martin R.
Bryce
*c
a,
Chun-Qiu
Xia
a,
Nan
Jiang
*a,
Jia-Wei
Xu
*b and
Martin R.
Bryce
*c
aKey Laboratory of Preparation and Applications of Environmental Friendly Materials, Key Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Jilin Normal University, Changchun 130103, China. E-mail: jiangn270@jlnu.edu.cn
bMinistry-of-Education Key Laboratory of Numerical Simulation of Large-Scale Complex System (NSLSCS) and School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China. E-mail: jwxu_njnu@sina.com
cDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 28th March 2025
Abstract
Nonconventional luminescent materials have been extensively studied in recent years. However, the ambiguity of their luminescence mechanism seriously hinders their development and the establishment of detailed structure–activity relationships. Based on this background, polyurethane derivatives (PUs) with different photophysical behaviors were synthesized by introducing into the backbone either a spirobicyclic structure with an orthogonal conformation of two acetal rings (PU1) or a monocyclic acetal structure giving a stretched linear conformation (PU2 and PU3). Detailed experimental and theoretical calculations show that the spirocyclic acetal in PU1 imparts an orthogonal conformation, reducing the intermolecular contacts, making molecules more inclined to form intramolecular interactions, which are detrimental to the intermolecular interactions and the aggregation behavior. The acetal ring in PU2/PU3 gives stretched conformations, enabling more intermolecular interactions. Meanwhile, high-temperature synthesis facilitates molecular aggregation, which indirectly red-shifts the emission. The proof-of-concept applications in cells and as luminescent inks are reported. Overall, this work contributes new understanding of the significance of ring-type structures without π electrons and gives a new perspective for the design of nonconjugated luminescent polymers.
 Nan Jiang | Nan Jiang is currently working in the Key Laboratory of Preparation and Applications of Environmental Friendly Materials, Jilin Normal University. She received her PhD degree in analytical chemistry from Northeast Normal University in 2022, under the supervision of Prof. Dong-Xia Zhu. In 2020–2021, she was a joint PhD student at Durham University (UK) under the supervision of Prof. Martin R. Bryce. Since 2025, she is a visiting scholar at Zhejiang University. Her dreams are scientific research, world travel and music. Her research is mainly on the development of functional polymeric photoelectric materials and intelligent response-type gel materials. |
Introduction
Recently, nonconventional luminescent materials without typical chromophores such as polycyclic aromatic hydrocarbons or extended π-conjugated structures, instead containing only electron-rich units (such as N, O, P, S, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C
C, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, etc.) have attracted wide attention because of their importance in theoretical research and potential applications.1–6 However, due to their unique structures and the presence of different luminescent species, the origin and the mechanism of luminescence is still not unified and is unclear.7–10 Most studies have focused on the through-space conjugation (TSC) formed by overlapping electron clouds with isolated lone pair electrons.11 However, only a few reports have considered how the photophysical properties of nonconventional chromophores can be affected by regulating the spatial configuration and conformation, and by balancing the rigidity/flexibility of the molecules, or changing the electronic properties. Studies should also include the internal electron distribution, intermolecular interactions, and the interaction between molecules and the environment.12–15
C, etc.) have attracted wide attention because of their importance in theoretical research and potential applications.1–6 However, due to their unique structures and the presence of different luminescent species, the origin and the mechanism of luminescence is still not unified and is unclear.7–10 Most studies have focused on the through-space conjugation (TSC) formed by overlapping electron clouds with isolated lone pair electrons.11 However, only a few reports have considered how the photophysical properties of nonconventional chromophores can be affected by regulating the spatial configuration and conformation, and by balancing the rigidity/flexibility of the molecules, or changing the electronic properties. Studies should also include the internal electron distribution, intermolecular interactions, and the interaction between molecules and the environment.12–15
The importance of incorporating saturated ring structures (including folded or twisted rings) into the backbone is often overlooked in nonconventional luminescent materials without π-conjugated groups. However, unlike benzene or polycyclic aromatic rings with a large planar conformation, saturated non-planar ring structures will avoid π–π stacking interactions which are often not conducive to luminescence, and can retain their rigidity and unique three-dimensional structures, to assist in the regulation of the properties of nonconjugated chromophores.16–19 Saturated ring-type structures have been widely studied to regulate the physicochemical properties of traditional conjugated molecules. For example, Wang et al. used norbornene spacer units to restrict conjugation and tune the emission color in polymers with through-space charge transfer.13 Jannasch et al. incorporated vanillin with a rigid spiroacetal structure into an amorphous polyester, which enhanced the Tg and thermal stability of the polymer and improved the oxygen barrier performance of its cast film.20 Hong et al. incorporated spirobicromane units into thermotropic polyesters, influencing the chain arrangement in the mesomorphic state.21 Ren et al. reported high-efficiency thermally activated delayed fluorescence polymers with a spatially confined conjugated backbone.22 However, this ring strategy is rarely discussed in nonconjugated luminescent molecules, where most mechanistic studies focus only on TSC,11 without exploring in detail how such ring-type structures affect their luminescence behavior. Our group incorporated estradiol as a linear ring structure into a polyurethane (PU) derivative, but there was no comparison in that work with an orthogonally structured analog.23
Due to the limitations of the theoretical calculation of electronic structure, the structure–activity relationships of nonconventional luminescent materials are mainly limited to small molecular systems.24–27 However, compared with small molecules, the segmental structure and richer conformational changes of macromolecules give them greater potential for interesting photophysical properties.28–38 More in-depth experimental and computational characterization of macromolecular systems could lead to more practical and commercial applications. Based on the above background, polyurethane derivatives PU1, PU2 and PU3 with different luminescent properties were synthesized by introducing into the backbone either a spirocyclic bis-acetal structure with an orthogonal conformation of the two acetal rings (PU1) or a monocyclic acetal structure with a stretched linear conformation, with parallel chains (PU2 and PU3). The aim of this study is to clarify how the ring structure affects the photoluminescence properties at the molecular level and to deepen the understanding of the internal working mechanisms of non-traditional chromophores. PU derivatives are attractive candidates for testing these aspects because they are readily available polymers with high global demand and varied applications in everyday life and in industry.39–41
Results and discussion
Synthesis
The PUs were synthesized by reacting trimethylhexa-1,6-diyl diisocyanate, polyethylene glycol monomethyl ether, and diol monomers (3,9-bis(1,1-dimethyl-2-hydroxyethyl)-2,4,8,10-tetraoxaspiro[5.5]undecane) for PU1 or 1,4-dioxane-2,5-diol for PU2 and PU3 in one-pot reactions at different temperatures (75 °C for PU1/PU2, and 130 °C for PU3). The detailed synthetic routes and structural characterization are shown in Fig. S1–S7 and Table S1 (ESI†). The structures of the PUs (Fig. 1a) were validated by nuclear magnetic resonance (NMR) spectroscopy, and the hydrogen atoms in different environments are clearly assigned (Fig. S3–S6, ESI†). In the Fourier-transform infrared (FT-IR) spectra, the appearance of characteristic peaks of CO, C–H and C–O–C further proves the successful synthesis of the PU derivatives (Fig. S7a, ESI†). To exclude the effect of hydrogen bonding in the aggregates, IR spectra of dilute solutions of PU2 and PU3 (0.1 mg mL−1 in DMSO) were also obtained. Fig. S7b (ESI†) shows that that PU2 and PU3 possess the same characteristic functional groups. Fig. S7c–f (ESI†) show that all the PUs display a broad amorphous peak centered near 20° in their X-ray diffraction patterns, consistent with the disordered hard-segment domains of polyurethanes.42Mw values of the PUs were within the range 1877–1963 g mol−1 (Table S1, ESI†).
 | ||
| Fig. 1 (a) Chemical structures of PU1, PU2, PU3 and the spirobicyclic acetal (blue) and acetal (orange) starting reagents. The individual polymer structures are shown in Fig. S1 and S2 in ESI.† (b) Emission spectra of PU1, PU2, PU3, and their photographs under 365 nm UV illumination. (c) Schematics of the spirocyclic acetal with orthogonal conformation (left) or the monocyclic acetal with linear conformation (right) in the main chain of polyurethanes. | ||
Photophysical and structural properties
Ultraviolet-visible (UV-vis) absorption spectra showed that PU1, PU2 and PU3 have a broad absorption in the visible region (Fig. S8, ESI†). Fig. S9 and S10 (ESI†) show the microstructural morphology of the diol monomers and the PUs measured by scanning electron microscopy (SEM). The diol monomers have irregular block-shaped structures. The product PUs, however, present nanoaggregate structures of varying sizes, which are more conducive to the spatial electron overlap and the consequent cluster-induced photoluminescence properties.3,24,27,43 This could be why the PUs possess high light utilization properties. Although heating can promote molecular aggregation and redshift the emission, it is detrimental to the lifetime and photoluminescence quantum yield QY in the solid state (Table S2, ESI†). PU1 and PUX synthesized from the same diol monomer at 75 °C and 130 °C, respectively, both showed very similar blue fluorescence (Fig. S11, ESI†). Therefore, PUX was not studied further. However, by simply regulating the reaction temperature, PU2 and PU3 with the same chemical structure showed yellow and red fluorescence, respectively, under a 365 nm UV lamp (Fig. 1b and Table S2, ESI†).The conformation influences the luminescence of these PUs. Their powders exhibited wide multi- or single-emission peaks, which is typical for nonconjugated polymers (Fig. 1b), due to the presence of various emissive species with different extents of through-space conjugation (TSC).44–47 To investigate this difference among the PUs, multi-excitation emission spectra were collected. Fig. 2a and b show that solid powders of PU1 and PU2 exhibit excitation-dependent characteristics, consistent with luminescent clusters with diverse energy levels.48 However, in the excitation wavelength range of 325 nm to 605 nm, the excitation-dependence of PU3 powder is significantly weakened (Fig. 2c and Fig. S12, ESI†). We suspect that this may be due to closer interactions in the PU3 aggregates, resulting in a more uniform TSC. Emission spectra of PUs in different solvents were also collected. As shown in Fig. S13a–c (ESI†) the emission peaks of the PUs all showed a blue shift trend with the increase of solvent polarity; it is shown that they all exhibit locally excited (LE) state excitation characteristics, and PU3 has the most obvious LE state emission. The relevant discussion is included in the theoretical calculation section below.
 | ||
| Fig. 2 Emission spectra of (a) PU1, (b) PU2 and (c) PU3 powders with different excitation wavelengths at room temperature. Concentration-dependent PL spectra of (d) PU1, (e) PU2, and (f) PU3 in DMSO; plots of wavelength and emission intensity versus concentration of (g) PU1, (h) PU2, and (i) PU3 in DMSO under 365 nm excitation. | ||
Concentration-dependent absorption and emission spectra were also obtained. Fig. S13d–f (ESI†) shows that within a certain concentration range, the PUs display concentration-dependent absorption enhancement. However, the concentration-dependent emission behavior is different within the series (Fig. 2d–f). For PU1, with the concentration rising from 0.1 mg mL−1 to 20 mg mL−1, the emission peak wavelength remained almost unchanged, while the emission intensity gradually increased (Fig. 2g). However, For PU2 and PU3, in a certain concentration range, their emission intensity at 365 nm decreased rapidly after a short rise, and the emission peak gradually redshifted (Fig. 2h and i).
To visualize the luminescence behavior of aggregated states the microscopic aggregation patterns of the PUs were studied at different concentrations in DMSO solution by scanning electron microscopy (SEM). Fig. 3a–d and Fig. S14 (ESI†) show that at low concentrations (0.1 mg mL−1) sparse nano-spherical aggregates are the primary morphology of PU1. With increasing concentration PU1 gradually transforms into non-nanoscale sheet structures. Fig. 3e–h show that for PU3 as the solution concentration increased from 0.1 mg mL−1 to 20 mg mL−1, the morphology gradually changed from spherical nanostructures to compact cluster structures. Similar morphological changes to PU3 are also observed for PU2 (Fig. S15, ESI†). However, the aggregation structure of PU2 at high concentrations is intermediate between a sheet structure and a spherical structure, and the packing pattern is irregular.
 | ||
| Fig. 3 (a) SEM images of PU1 (a) 0.1 mg mL−1, (b) 5 mg mL−1, (c) 10 mg mL−1, and (d) 20 mg mL−1 in DMSO solution. SEM images of PU3 (e) 0.1 mg mL−1, (f) 5 mg mL−1, (g) 10 mg mL−1, and (h) 20 mg mL−1 in DMSO solution. | ||
The comparison of SEM results suggests that the difference in the photophysical behavior of PU1 and PU2/PU3 is closely related to the evolution of their microscopic aggregation behavior with an increase in concentration. If the polymer chains show a highly dense and uniform cluster nanostructure like PU3, this is more conducive to long-wavelength luminescence. The flocculent aggregate structure of PU3 helps to uniform TSC, which in turn stimulate wavelength independence, while the flake/block aggregate structure of PU1/PU2 helps to stimulate wavelength dependence (Fig. 2, 3 and Fig. S15, ESI†). Dynamic light scattering (DLS) results show that the size of the aggregates of the PUs increases with the increase of solution concentration, and PU3 solution is more likely to produce large-size aggregates, which is consistent with its long-wavelength emission and the largest aggregates observed in SEM (Fig. S16, ESI†).
However, as mentioned above, the final formation of this aggregation structure is complex, and future experimental work may unravel the internal mechanism of this process. This would be of great significance for the establishment of a universal non-traditional photophysical theory for nonconventional luminescence. Establishment of a universal non-traditional photophysical theory for nonconventional luminescence.
Theoretical calculations
To understand the relationship between the molecular structure of the PUs and the microscopic aggregation structure and luminescence behavior at the molecular level, molecular dynamics (MD) simulations were performed based on two repeating units of PUs using the GROMACS software suite. Hydrogen bonds are crucial interactions in polyurethane derivatives49–51 and in related polymers incorporating –NH–C(O)-units.52,53 Thus, the molecular conformation and intra- or inter-molecular interactions in the prepared polymers were investigated using hydrogen bond interactions as the carrier. Aggregate systems including 50 molecules of PU1 or PU2/PU3 were constructed and MD simulations were then performed, including three stages: (1) MD1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50.0 ns initial equilibrium at 298.15 K; (2) MD2:50.0 ns heating at the synthesis temperature; and (3) MD3:50.0 ns annealing at 298.15 K. NEVPT2/MM calculations based on snapshots from the MD3 trajectory show that the PUs’ light response originates from the amide moieties. Leading conformations adopted by PU1 and PU2/PU3 show a significant difference, which acts as a dominant factor influencing the number of intermolecular hydrogen bond interactions. As shown by a representative snapshot in Fig. 4a, the spirocyclic acetal rings in PU1 impart an orthogonal conformation (marked as yellow lines in Fig. 4a), which favors intramolecular agglomeration of the molecules. Therefore, hydrogen bond interactions between molecules are reduced, which is not conducive to forming large aggregates. The radius of gyration (Rg) during the MD3 trajectory shows that due to the difference in molecular conformation, PU1 still has a smaller averaged Rg (0.83 nm) compared with PU2 (1.02 nm) and PU3 (0.98 nm), even with a longer molecular length. The acetal ring in PU2/PU3 gives stretched conformations (Fig. 4b), enabling more inter-molecular hydrogen bond interactions. Therefore, it is easier for PU2/PU3 to form large aggregates with strong electron delocalization and conjugation.
:50.0 ns initial equilibrium at 298.15 K; (2) MD2:50.0 ns heating at the synthesis temperature; and (3) MD3:50.0 ns annealing at 298.15 K. NEVPT2/MM calculations based on snapshots from the MD3 trajectory show that the PUs’ light response originates from the amide moieties. Leading conformations adopted by PU1 and PU2/PU3 show a significant difference, which acts as a dominant factor influencing the number of intermolecular hydrogen bond interactions. As shown by a representative snapshot in Fig. 4a, the spirocyclic acetal rings in PU1 impart an orthogonal conformation (marked as yellow lines in Fig. 4a), which favors intramolecular agglomeration of the molecules. Therefore, hydrogen bond interactions between molecules are reduced, which is not conducive to forming large aggregates. The radius of gyration (Rg) during the MD3 trajectory shows that due to the difference in molecular conformation, PU1 still has a smaller averaged Rg (0.83 nm) compared with PU2 (1.02 nm) and PU3 (0.98 nm), even with a longer molecular length. The acetal ring in PU2/PU3 gives stretched conformations (Fig. 4b), enabling more inter-molecular hydrogen bond interactions. Therefore, it is easier for PU2/PU3 to form large aggregates with strong electron delocalization and conjugation.
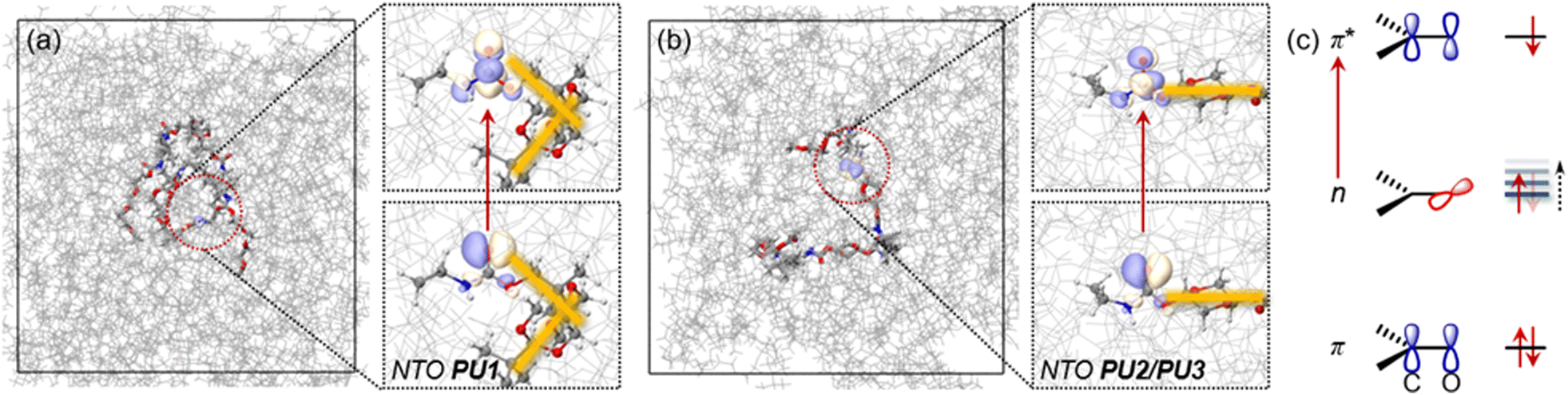 | ||
| Fig. 4 Snapshots taken from the MD3 simulation trajectory along with natural transition orbitals (NTOs) for (a) PU1 and (b) PU2/PU3; (c) n → π* transition feature where the energy level of the n orbital can be adjusted by hydrogen bonding. | ||
Further analysis by comparing the statistical number of hydrogen bonds during the MD simulations is shown in Fig. S17 (ESI†). With the same synthesis temperature, the counts of hydrogen bonds in PU1 (121/112/121 for each stage) are much lower than in PU2 (158/148/157) and PU3 (158, 131, 161). However, PU1 possesses more oxygen atoms in the bis-acetal moieties. This points out that intermolecular hydrogen bonding in these systems is mainly controlled by different conformations adopted by the acetal moieties, not by the number of hydrogen bond donors. As is revealed by the NTOs in Fig. 4, single electron excitation from a lone pair orbital (n) of oxygen to a π* orbital of CO is the major transition feature, which is summarized as Fig. 4c. Forming a hydrogen bond may decrease the electron density occupied by an n orbital and therefore, the n orbital energy level is increased to give a narrower n–π* gap (Fig. 4c). This decreases the energy difference between ground states and excited states finally contributing to the red shift in the emission wavelength of PU2/PU3 relative to PU1. To probe the general influence brought by hydrogen bond interactions on the excitation energy of the n → π* state, considering that hydrogen bonds are determined by electrostatic interactions, we applied an external electrostatic field (EEF) on model systems. Taking the direction of the CO bond as the z-axis, and the carbonyl group in the yz-plane, the excited state energy shift controlled by the EEF strength on three directions was revealed by Fig. S18 (ESI†). The EEF on the x-direction does not have notable influence on the excitation energy, compared with the other directions. This indicates that a π* orbital towards the x-direction contributes little to the excited state energy shift in PU1/PU2/PsU3 systems, which is consistent with the NTO analysis. However, the excited state energy is highly dependent on the strength of EEF on the y- or z-directions (Fig. S18b, ESI†), which corresponds to the n orbitals (lone pair orbitals of the oxygen atom) being shifted as the number of hydrogen bond interactions increases, thereby decreasing the S1 excitation energy and red shifting the emission. Moreover, with a higher synthesis temperature, slightly more hydrogen bond interactions are observed in PU3 (161 after annealing) compared with PU2 (157). Possibly due to the higher synthesis temperature promoting a more compact conformation, PU3 has a slightly smaller radius of rotation than PU2. However, the thermal action of the polymer chains increases aggregation. Therefore, PU3 has a denser and more uniform cluster-like microscopic aggregation structure, which is more conducive to the delocalization of electrons, and ultimately leads to the longest wavelength emission for PU3 in this series.
Applications
PUs have excellent solution processability and promising multifunctional fluorescent applications, enhanced by recent encouraging biodegradability studies.54 Since there are no extended π-conjugated units in the PUs’ structure that could lead to cytotoxicity, their future biomedical applications appear promising. PU3 was selected and water-soluble nanoparticles (PU3 NPs) were prepared by a solvent exchange method as described in the ESI.† As shown in Fig. S19 (ESI†), no significant change in the viability of mouse hippocampal neuronal (HT22) cells was observed after treatment with 100 μg mL−1PU3 NPs for 24 h, indicating their low cytotoxicity. Hydrogen peroxide (H2O2) is an abundant reactive oxygen species (ROS) that can freely spread to the interstitial region around cells. ROS are closely related to a variety of pathophysiological processes, including cancer, cardiovascular diseases, inflammation, degenerative diseases, etc. Excessive H2O2 accumulation can lead to oxidative stress in cells and inhibit cell viability, which in turn affects the therapeutic outcome on various diseases.55–57 Therefore, the development of biocompatible agents which can scavenge H2O2 will have practical implications. As shown in Fig. 5a, when treated with H2O2, HT22 cells exhibited dose-dependent mortality, with approximately 91% cell death observed at a concentration of 200 μM. Subsequently, to evaluate the effect of PU3 NPs on the cell destructive ability of H2O2in vitro HT22 cells were incubated with different concentrations of PU3 NPs and H2O2 (0–200 μM) for 24 hours. The viability of H2O2 (200 μM)-treated HT22 cells is low. However, when PU3 NPs were added to the system, the cell viability gradually increased with the increasing amount of PU3 NPs (Fig. 5b). At a concentration of 100 μg mL−1, the survival rate increased from 16.8% to 28.9%, compared to pure H2O2-treated cells, implying that PU3 NPs have some ability to combat excess H2O2. | ||
| Fig. 5 (a) Cell viability was measured after HT22 cells were treated with different concentrations of H2O2 for 24 h. (b) Cell viability was measured after HT22 cells were treated with different concentrations of PU3 NPs (0–100 μg mL−1) for 24 h then incubated with H2O2 (200 μM) for another 24 h. (c) PU3 NPs decreased ROS and O2˙− activity induced by 200 μM H2O2-treated HT22 cells in vitro; CPU3NPs = 100 μg mL−1; scale bars = 50 μm. (d) Photographs of PU1, PU2 and PU3 in daylight and ultraviolet light, and the pixel painting under daylight or 365 nm UV-lamp, respectively. (e) Painting model of the PUs under daylight and 365 nm UV-lamp. Module 1: photos of PU1, PU2 and PU3 pigments and brushes; module 2: fireworks painted by PU2 and PU3 on aluminium plate; module 3: sunrise pattern drawn on the glass by PU1 and PU3; module 4: the Eiffel Tower painted on a wooden board by PU2 and PU3. | ||
For a clearer view of PU3 NPs’ ability to remove H2O2, fluorescent probes 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and dihydroethidium (DHE) were used to monitor changes in ˙OH and O2˙−, respectively, generated in situ from H2O2 (Fig. 5c). Compared to the control group, in the presence of PU3 NPs, the intracellular concentrations of ˙OH and O2˙− decreased significantly, showing that PU3 NPs have the ability to remove H2O2. Although the mechanism by which PU3 NPs act is not clear at present, this work provides a promising direction in biomedical applications of nonconjugated/nonconventional luminescent polymers.
As shown as Fig. 5d and Fig. S20 (ESI†), the PUs serve as assembly modules to convey different images in different situations, due to their distinct colors under daylight or a 365 nm UV-lamp. Under ambient light, the painting of a puppy has an orange background, a flesh-colored body and black ears. However, when illuminated with a 365 nm UV-lamp, the image changed strikingly to a dark-yellow background, blue body and red ears. Besides, due to their ideal viscosity and excellent luminescence properties can also be used as fluorescent painting pigments on a variety of substrates, such as paper, metal, glass, cloth and wood. With the help of molds (Fig. S21, ESI†), the PUs display different patterns like fireworks, sunrise, world architecture, etc. on organic/inorganic substrates (Fig. 5e and Fig. S22, ESI†). The PUs exhibited effective and enduring adhesion on these substrates. These proof-of-concept results show the excellent multifunctional prospect of these PUs in the important contemporary topic of luminescent nanomaterials for security inks with anti-counterfeiting applications.58–60
Conclusion
In summary, introducing into a PU backbone either spiro-biacetal rings with an orthogonal conformation, or a single acetal ring with a linear conformation, has added a new perspective on how nonconjugated structures affect the luminescence of nonconventional macromolecules. Detailed photophysical characterization and theoretical calculations show that the primary source of photoluminescence is the amide moieties. Single electron excitation from a lone pair orbital (n) of oxygen to a π* orbital of CO is the prominent transition feature. Meanwhile, the ring structures are indirectly involved in the regulation of the luminescence by controlling the chain conformation with a balance of rigidity and flexibility. The orthogonal conformation of spiro-biacetal PU1 reduces the number and strength of intermolecular contacts, especially H-bonds, making the molecules more inclined to intramolecular agglomeration, which is detrimental to the bulk aggregation behavior. In contrast, the stretched linear conformation in PU2/PU3 enables more intermolecular H-bonding interactions which decrease the electron density of occupied n orbitals, and increase their energy levels, and consequently the n–π* gap is narrowed. This reduces the energy difference between ground states and excited states and finally contributes to the redshifted emission of PU2/PU3, compared with PU1. Micromorphology monitoring has aided the visualization of the aggregation and luminescence process. Proof-of-concept applications of the PUs in cells and as luminescent inks are reported. This work provides a new strategy for the development of colorful nonconjugated luminescent macromolecules and deepens the understanding of structure–activity relationships in these materials.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Chang-Yi Zhu: investigation, data curation, formal analysis, visualization, writing – original draft; Ya-Jie Meng and Jia-Wei Xu: investigation, software; Yan-Hong Xu: project administration, resources, supervision; Chun-Qiu Xia: formal analysis, methodology; Nan Jiang: conceptualization, funding acquisition, project administration, resources, writing – review & editing; Martin R. Bryce: conceptualization, funding acquisition, supervision, writing – review & editing.Data availability
The data associated with this article are available in the manuscript and ESI.† Additional data can be obtained upon request to the authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work was funded by the Science and Technology Development Program of Jilin Province (YDZJ202301ZYTS305); J.-W. Xu thanks Ministry-of-Education Key Laboratory of Numerical Simulation of Large-Scale Complex System (NSLSCS) for their financial and technical support. M. R. B. thanks EPSRC Grant EP/L02621X/1 for funding. We thank the open-source design community (OpenClipart, C0 1.0) for inspiring the painting ideas.Notes and references
- Z. Wang, H. K. Zhang, S. Q. Li, D. Y. Lei, B. Z. Tang and R. Q. Ye, Top. Curr. Chem., 2021, 379, 14–36 CrossRef CAS.
- N. Jiang, D. X. Zhu, Z. M. Su and M. R. Bryce, Mater. Chem. Front., 2021, 5, 60–75 RSC.
- T. V. Sakhno, Yu. E. Sakhno and S. Ya. Kuchmiy, Theor. Exp. Chem., 2023, 59, 75–106 CAS.
- N. Jiang, C. Y. Zhu, K. X. Li, Y. H. Xu and M. R. Bryce, Macromolecules, 2024, 57, 5561–5577 CrossRef CAS PubMed.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Q. Ye, S. J. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. P. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275–292 CAS.
- K. Bauri, B. Saha, A. Banerjee and P. De, Polym. Chem., 2020, 11, 7293–7315 CAS.
- R. Wang, W. Yuan and X. Zhu, Chin. J. Polym. Sci., 2015, 33, 680–687 CAS.
- Y. Wang, X. Bin, X. Chen, S. Zheng, Y. Zhang and W. Z. Yuan, Macromol. Rapid Commun., 2018, 39, 1800528 Search PubMed.
- H. Wang, Q. Li, P. Alam, H. Bai, V. Bhalla, M. R. Bryce, M. Cao, C. Chen, S. Chen, X. Chen, Y. Chen, Z. Chen, D. Dang, D. Ding, S. Ding, Y. Duo, M. Gao, W. He, X. He, X. Hong, Y. Hong, J. J. Hu, R. Hu, X. Huang, T. D. James, X. Jiang, G. Konishi, R. T. K. Kwok, J. W. Y. Lam, C. Li, H. Li, K. Li, N. Li, W. J. Li, Y. Li, X. J. Liang, Y. Liang, B. Liu, G. Liu, X. Liu, X. Lou, X. Y. Lou, L. Luo, P. R. McGonigal, Z. W. Mao, G. Niu, T. C. Owyong, A. Pucci, J. Qian, A. Qin, Z. Qiu, A. L. Rogach, B. Situ, K. Tanaka, Y. Tang, B. Wang, D. Wang, J. Wang, W. Wang, W. X. Wang, W. J. Wang, X. Wang, Y. F. Wang, S. Wu, Y. Wu, Y. Xiong, R. Xu, C. Yan, S. Yan, H. B. Yang, L. L. Yang, M. Yang, Y. W. Yang, J. Yoon, S. Q. Zang, J. Zhang, P. Zhang, T. Zhang, X. Zhang, X. Zhang, N. Zhao, Z. Zhao, J. Zheng, L. Zheng, Z. Zheng, M. Q. Zhu, W. H. Zhu, H. Zou and B. Z. Tang, ACS Nano, 2023, 17, 14347–14405 CrossRef CAS.
- X. Zhang, Y. Bai, J. Deng, P. Zhuang and H. Wang, Aggregate, 2024, 5, e517–e530 CrossRef CAS.
- Y. Wang, Y. Guo, Y. Liang, Y. Pan, Y. Shi, Y. Wang, S. Zhang, B. Jin and G. Zhao, J. Lumin., 2021, 231, 117783 CrossRef CAS.
- Q. Li, J. Hu, J. Lv, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2020, 59, 20174–20182 CrossRef CAS PubMed.
- T. Y. Hwang, Y. Choi, Y. Song, N. S. A. Eom, S. Kim, H. B. Cho, N. V. Myung and Y. H. A. Choa, J. Mater. Chem. C, 2018, 6, 972–979 RSC.
- J. G. Yang, X. F. Song, G. Cheng, S. Wu, X. Feng, G. Cui, W. P. To, X. Chang, Y. Chen, C. M. Che, C. Yang and K. Li, ACS Appl. Mater. Interfaces, 2022, 14, 13539–13549 CrossRef CAS PubMed.
- R. Ushimaru, L. Cha, S. Shimo, X. Li, J. C. Paris, T. Mori, K. Miyamoto, L. Coffer, M. Uchiyama, Y. Guo, W. Chang and I. Abe, J. Am. Chem. Soc., 2023, 145, 24210–24217 CrossRef CAS.
- S. Ma, D. Mandalapu, S. Wang and Q. Zhang, Nat. Prod. Rep., 2022, 39, 926–945 RSC.
- S. Paul, D. Adelfinsky, C. Salome, T. Fessard and M. K. Brown, Chem. Sci., 2023, 14, 8070–8075 RSC.
- Z. Ran, R. Wang, J. Fu, M. Yang, M. Li, J. Hu, J. He and Q. Li, Adv. Mater., 2023, 35, 2303849–2303858 CAS.
- S. V. Mankar, J. Wahlberg, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 5135–5146 CAS.
- L. L. Lin and J. L. Hong, Polymer, 2000, 41, 4501–4512 CAS.
- Y. Liu, Y. Xie, L. Hua, S. Li, X. Tong, S. Ying, S. Yan and Z. Ren, Adv. Opt. Mater., 2024, 12, 2301811–2301820 CAS.
- N. Jiang, K. X. Li, W. Xie, S. R. Zhang, X. Li, Y. Hu, Y. H. Xu, X. M. Liu and M. R. Bryce, Macromolecules, 2023, 56, 7721–7728 CAS.
- S. X. Tang, T. J. Tang, Z. H. Zhao, T. W. Zhu, Q. Zhang, W. B. W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 CAS.
- T. Rožić, M. Hochlaf, B. R. Said and N. A. Došlić, J. Phys. Chem. A, 2022, 126, 1094–1102 Search PubMed.
- P. Liao, J. Huang, Y. Yan and B. Z. Tang, Mater. Chem. Front., 2021, 5, 6693–6717 CAS.
- H. Zhang and B. Z. Tang, JACS Au., 2021, 1, 1805–1814 CrossRef CAS PubMed.
- Y. Xie, D. Liu, H. Zhang, D. Wang, Z. Zhao and B. Z. Tang, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2022-vznv4.
- W. F. Lai, Mater. Today Chem., 2022, 23, 100712 CrossRef CAS.
- R. Hu, A. Qin and B. Z. Tang, Science, 2020, 100, 101176 CAS.
- C. Shang, N. Wei, H. M. Zhou, Y. M. Shao, Q. Zhang, Z. X. Zhang and H. L. Wang, J. Mater. Chem. C, 2017, 5, 8082–8090 RSC.
- Z. Zhang, W. Yan, D. Dang, H. Zhang, J. Z. Sun and B. Z. Tang, Cell Rep. Phys. Sci., 2022, 3, 100716 CrossRef CAS.
- B. Liu, Y. L. Wang, W. Bai, J. T. Xu, Z. K. Xu, K. Yang, Y. Z. Yang, X. H. Zhang and B. Y. Du, J. Mater. Chem. C, 2017, 5, 4892–4898 RSC.
- Y. Feng, T. Bai, H. Yan, F. Ding, L. Bai and W. Feng, Macromolecules, 2019, 52, 3075–3082 CrossRef CAS.
- H. Lu, L. Feng, S. Li, J. Zhang, H. Lu and S. Feng, Macromolecules, 2015, 48, 476–482 CrossRef CAS.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586–6592 CrossRef CAS PubMed.
- X. Ji, W. G. Tian, K. F. Jin, H. L. Diao, X. Huang, G. J. Song and J. Zhang, Nat. Commun., 2022, 13, 3717–3729 CrossRef CAS PubMed.
- Z. Zhang, Z. Xiong, B. Chu, Z. Zhang, Y. Xie, L. Wang, J. Z. Sun, H. Zhang, X. H. Zhang and B. Z. Tang, Aggregate, 2022, 3, e278–e286 CrossRef CAS.
- T. A. P. Hai, M. Tessman, N. Neelakantan, A. A. Samoylov, Y. Ito, B. S. Rajput, N. Pourahmady and M. D. Burkart, Biomacromolecules, 2021, 22, 1770–1794 CrossRef.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- P. Minei, G. Iasilli, G. Ruggeri, V. Mattoli and A. Pucci, Chemosensors, 2021, 9, 3 CrossRef CAS.
- Y. L. Feng, N. Jiang, D. X. Zhu, Z. M. Su and M. R. Bryce, J. Mater. Chem. C, 2020, 8, 11540–11545 RSC.
- L. H. Bai, H. X. Yan, L. L. Guo, M. M. He, T. Bai and P. F. Yang, Macromol. Chem. Phys., 2021, 222, 2100283 CrossRef CAS.
- B. Chu, H. Zhang, L. Hu, B. Liu, C. Zhang, X. Zhang and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202114117 CrossRef CAS.
- N. Jiang, G. F. Li, B. H. Zhang, D. X. Zhu, Z. M. Su and M. R. Bryce, Macromolecules, 2018, 51, 4178–4184 CrossRef CAS.
- T. Han, J. Xie, F. Chen, P. W. Sze, X. Su, D. Wang and B. Z. Tang, Macromolecules, 2023, 56, 10016–10027 CrossRef CAS.
- K. Chen, Y. Wang, B. Chu, Z. Yan, H. Li, H. Zhang, S. Hu, Y. Yang, B. Liu and X. H. Zhang, J. Mater. Chem. C, 2022, 10, 16420–16429 RSC.
- Y. Y. Wang, Z. A. Liu, J. M. Huang, H. L. Wei, C. J. Jiang, L. Z. Wei, B. L. Jiang, L. M. Zou, H. H. Xie and Y. Y. Gong, Small, 2025, 2411123 CrossRef CAS PubMed.
- X. Li, K. Li, Z. Chen and X. J. Yang, Appl. Polym. Sci., 2022, 139, e53206 CrossRef CAS.
- S. Salimi, Y. Wu, M. I. E. Barreiros, A. A. Natfji, S. Khaled, R. Wildman, L. R. Hart, F. Greco, E. A. Clark, C. J. Roberts and W. Hayes, Polym. Chem., 2020, 11, 3453–3464 RSC.
- A. Z. Tareq, M. Hyder, D. H. Merino, A. M. Chippindale, A. Kaur, J. A. Cooper and W. Hayes, Polymer, 2024, 302, 127052 CrossRef CAS.
- Y. Deng, Q. Zang and D. H. Qu, ACS Mater. Lett., 2023, 5, 480–490 CrossRef CAS.
- S. Efstathiou, G. Nurumbetov, A. Ross, Y. Li and D. M. Haddleton, Mater. Adv., 2024, 5, 3396–3410 RSC.
- P. Bhavsar, M. Bhave and H. K. Webb, World J. Microbiol. Biotechnol., 2023, 39, 122 CrossRef CAS.
- X. Y. Liang, H. Y. Li, X. L. Li, X. X. Tian, A. Zhang, Q. Z. Luo, J. W. Duan, Y. L. Chen, L. Y. Pang, C. Li, X. J. Liang, Y. Zeng and J. Yang, Acta Pharm. Sin. B, 2023, 13, 372–389 CrossRef CAS.
- J. Li, F. Han, J. Ma, H. Wang, J. Pan, G. Yang, H. Zhao, J. Zhao, J. Liu, Z. Liu and B. Li, Adv. Funct. Mater., 2022, 32, 2111208 CrossRef CAS.
- M. Y. Shao, Y. F. Wang, H. Y. Dong, L. Wang, X. Q. Zhang, X. Han, X. A. Sang, Y. N. Bao, M. Y. Peng and G. Cao, Bioact. Mater., 2023, 23, 187–205 CAS.
- P. Kumar, S. Singh and B. K. Gupta, Nanoscale, 2016, 8, 14297–14340 RSC.
- D. Li, Y. Yang, J. Yang, M. Fang, B. Z. Tang and Z. Li, Nat. Commun., 2022, 13, 347 CrossRef CAS.
- R. Martin, M. Echeverri, F. Gonzalez, A. D. Andres, M. C. R. Delgado, A. Concellon, J. L. Serrano and B. Gomez-Lor, Adv. Opt. Mater., 2024, 2400965 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00577a |
| This journal is © The Royal Society of Chemistry 2025 |