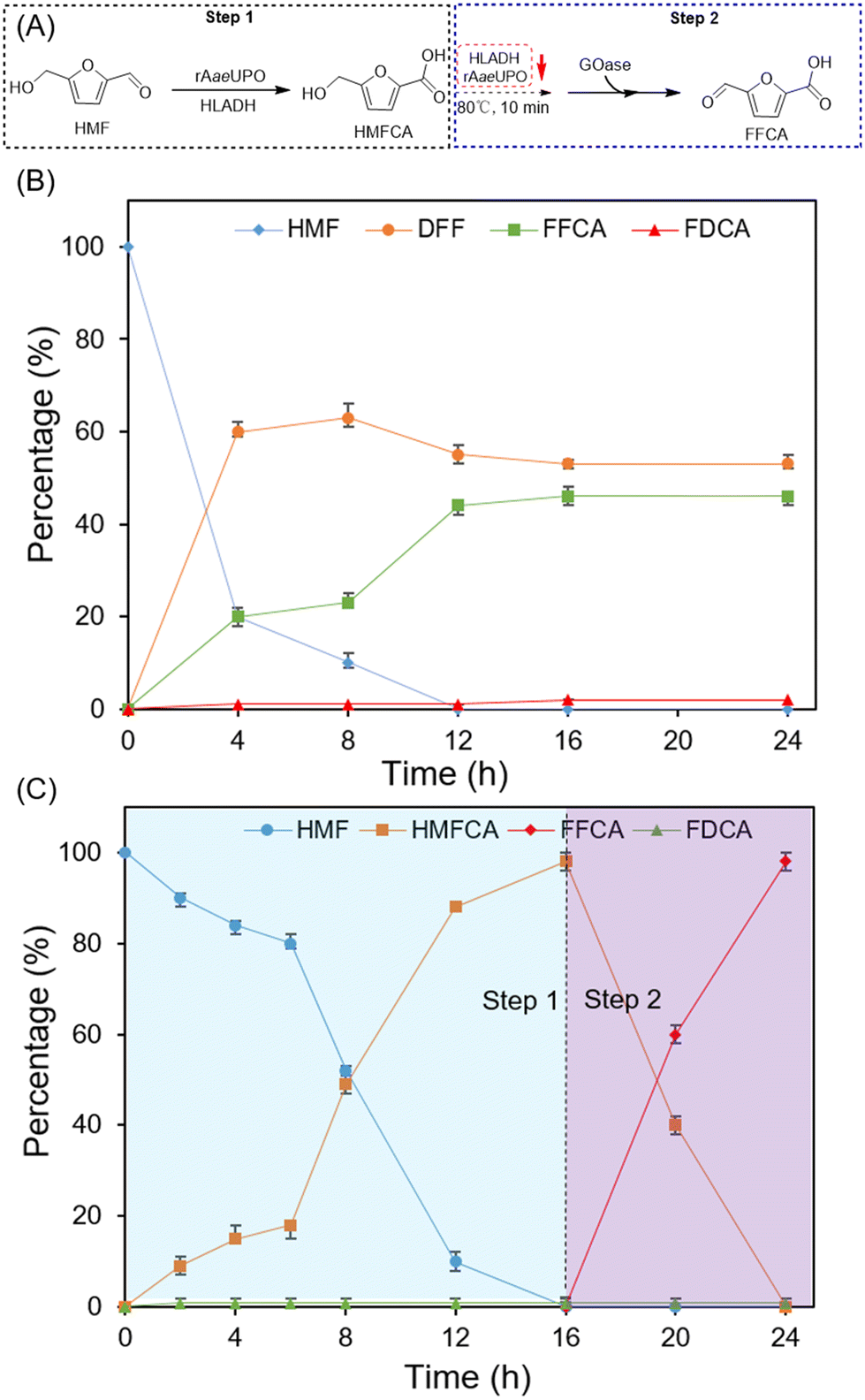
DOI:
10.1039/D4GC04735D
(Paper)
Green Chem., 2025,
27, 642-649
Constructing a chemoenzymatic strategy for enhancing the efficiency of selectively transforming 5-hydroxymethylfurfural into furan carboxylic acids†
Received
21st September 2024
, Accepted 5th December 2024
First published on 6th December 2024
Abstract
Furan carboxylic acids including 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 5-formyl-2-furancarboxylic acid (FFCA) and 2,5-furandicarboxylic acid (FDCA) are important bio-based chemicals and widely applied in the fields of pharmaceuticals and materials. However, selectively obtaining these value-added compounds from biomass-derived 5-hydroxymethylfurfural (HMF) is difficult due to the readily reactive alcohol/aldehyde groups of the generated intermediates during the oxidative process. Biotransformation using enzyme catalysis is a sustainable and promising approach, but there are remaining challenges including low efficiency and selectivity due to the chaotic and complex biocatalytic process, loss of enzyme activity and inevitable increase in reaction volume caused by exogenous addition of H2O2. In these contexts, a natural flavin cofactor mimic (NFCM)-mediated chemoenzymatic system consisting of H2O2-dependent peroxygenase, NAD+-dependent alcohol dehydrogenase and galactose oxidase was constructed for the first time for selectively converting HMF to each furan carboxylic acid. The bifunctional catalyst NFCM enabled the in situ generation of H2O2 and regeneration of NAD+, which skillfully alleviated the problems caused by exogenous addition of H2O2. Each furan carboxylic acid was achieved with excellent yield (>99%) and selectivity (>99%). Furthermore, an immobilization technique based on a hydrogel material was employed for the first time, which greatly promoted the stability and reusability of each biocatalyst.
Green Foundation
1. This work demonstrated a green and efficient chemoenzymatic cascade system, which could selectively transform HMF into each value-added furan carboxylic acid.
2. The whole reaction system was carried out in the aqueous phase, and no by-product was accumulated, which facilitated the subsequent separation and purification of products. Furthermore, the E-factor and atom efficiency of this system were calculated, which showed apparent advantages among the reported systems.
3. Further work about enhancing the enzyme tolerance to the substrate concentration through dehydrogenase evolution, which could increase the space–time yields of each furan carboxylic acid, is underway in our lab.
|
1. Introduction
With the growth in the demand and diminishing of the reserves of fossil fuels, it is urgent to develop sustainable sources for fuels and chemicals.1 Lignocellulosic biomass is the most attractive alternative due to its abundance, low cost, and sustainable properties.2 5-Hydroxymethylfurfural (HMF) is one of the top-value platform chemicals derived from biomass.3,4 Based on the active alcohol hydroxyl and aldehyde groups, HMF can be easily oxidized into other value-added furan carboxylic acids which have great application potential in fuels, fine chemicals, polymers, medical intermediates, etc.5 For example, 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) is an important precursor of pharmaceutical intermediates.6,7 5-Formyl-2-furancarboxylic acid (FFCA) and 2,5-furandicarboxylic acid (FDCA) are promising building blocks for the manufacture of polymers.8,9 Generally, the conversion of HMF to these valuable furan carboxylic acids can be divided into two different oxidation pathways: the aldehyde oxidation pathway (route 1) and the alcohol oxidation pathway (route 2) (Scheme 1A). However, due to the readily oxidizing reactive aldehyde/alcohol groups of each compound during the oxidation process, it is difficult to selectively transform HMF into each furan carboxylic acid. Although various valuable chemical methods have been reported for oxidizing HMF, these approaches rely on metals and are carried out under harsh conditions with low selectivity.10–13 Recently, biological oxidation of HMF has emerged as a favorable and promising alternative due to its mild reaction conditions and environmental friendliness.14,15 Several isolated enzyme catalysis approaches using O2-dependent oxidases, H2O2-dependent peroxygenases, and NAD(P)+-dependent dehydrogenases for the oxidation of HMF have been reported.16–19 However, these enzymes usually tend to competitively catalyze the aldehyde/alcohol substrate (intermediate) during the bio-oxidation of HMF.20–23 The resulting chaotic and complex biocatalytic process restricts the selectivity and efficiency of bio-oxidation of HMF to each furan carboxylic acid.20–22 Thus, constructing a simple, orderly, and efficient system that can selectively transform HMF into each furan carboxylic acid is highly desirable.
 |
| | Scheme 1 The previous and current strategies for the selective synthesis of HMFCA, FFCA and FDCA. (A) Previous study. (B) This work. | |
Moreover, H2O2 has been employed as an oxidative alternative to O2 in the biotransformation of HMF, which can alleviate the challenge of oxygen dissolution and mass transfer problem.22,23 However, the enzymes used in the bio-oxidation of HMF are often H2O2 concentration sensitive.22 Exogenous addition of excess amount of H2O2 not only leads to irreversible enzyme deactivation but also inevitably results in the increase of the reaction volume,24–26 which impairs the reaction efficiency. Recently, an in situ H2O2 generation and utilization multi-enzyme catalytic system including dehydrogenase, horseradish peroxidase (HRP), and oxidase was developed to selectively bio-oxidize HMF to furan carboxylic acid (Scheme 1A).23 However, there are still some drawbacks as follows: (1) for the synthesis of HMFCA, exogenous addition of H2O2 was still required; (2) the expensive electron transporter scopoletin27 was required to enable NAD+ regeneration for dehydrogenase during the catalytic process, which generated free radicals and resulted in the loss of enzyme activity; and (3) low turnover numbers (TONs). In these contexts, it is highly attractive to develop a novel and effective in situ H2O2 generation and utilization system that does not impair the enzyme activity to promote the selective biotransformation of HMF into each furan carboxylic acid.
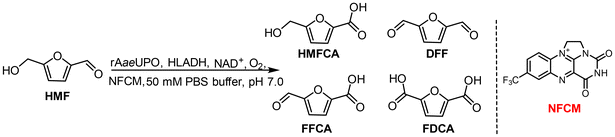
Recently, H2O2-dependent unspecific peroxygenase (UPO) has been demonstrated as an efficient and robust biocatalyst for HMF biotransformation, but the exogenous addition of H2O2 is still required to drive the reaction and the chemoselectivity of UPO was unsatisfactory. Our previous studies have shown that NFCM can oxidize reduced nicotinamide coenzyme NADH to regenerate NAD+ for the ADH-catalyzed oxidation reaction, which could produce H2O2 with the consumption of molecular oxygen.28,29 We wonder if UPO could be coupled with a HLADH–NFCM system, thereby constructing a novel chemoenzymatic system to boost the selective biotransformation of HMF. During the catalytic process, UPO-catalyzed oxidation of HMF or FFCA was enhanced by coupling with NAD+-dependent alcohol dehydrogenase from horse (HLADH). NFCM was responsible for regenerating NAD+ and in situ generating H2O2, which are required for ADH and UPO catalysis, respectively (Scheme 1B). Furthermore, galactose oxidase (GOase) was employed to selectively oxidize HMFCA to FFCA (Scheme 1B).23 Herein, employing UPO, HLADH, and GOase, we proposed a novel one-pot NFCM-mediated chemoenzymatic system that could selectively convert HMF to each value-added furan carboxylic acid with high efficiency (Scheme 1B). The immobilization strategy was further explored to enable the recycling of the biocatalysts, facilitating the further scale-up of HMF derivative production.
2. Results and discussion
2.1 The selective synthesis of HMFCA from HMF
Firstly, we investigated the possibility of UPO-catalyzed selective conversion of HMF to HMFCA. The recombinant evolved unspecific peroxygenase derived from Agrocybe aegerita (rAaeUPO) was chosen for the oxidation reactions.30 In the initial experiments, one equivalent amount of H2O2 was added continuously to drive the rAaeUPO-catalyzed oxidation of HMF. As shown in Table 1 (entry 1), a yield of only 29% HMFCA was obtained. The by-products DFF, FFCA, and FDCA were also observed with yields of 16%, 8%, and 1%, respectively. In addition, the continuous addition of H2O2 led to a 10% increase in the reaction volume and a 30% decrease in the catalytic activity of rAaeUPO (Fig. S1†).22 To alleviate this dilemma, our reported in situ H2O2 generation system using NFCM as the catalyst and NAD(P)H as a hydrogen donor was employed to couple with rAaeUPO for the transformation of HMF.29 The result showed that the conversion and yield of the desired product HMFCA were both increased when compared to that with the continuous addition of H2O2 (Table 1, entry 2), but the chemoselectivity of rAaeUPO remained unsatisfactory. We attributed this to the chaotic oxidative reactivity of rAaeUPO towards alcohol/aldehyde groups. Hence, we speculated that the selectivity and efficiency of this reaction could be enhanced by coupling with NAD+-dependent HLADH, which also can oxidize HMF. Moreover, further considering the high cost of the chemical equivalent NAD(P)H, we also intended to employ HLADH to regenerate this hydrogen donor in the case of HMF oxidation.27 Thus, a NFCM-mediated rAaeUPO–HLADH chemoenzymatic system was proposed to catalyze HMF to HMFCA (Table 1, entry 3). It could be found that the selectivity of this chemoenzymatic system (Fig. 1A, purple line) was significantly enhanced compared with that of the single UPO-catalyzed biotransformation of HMF (Fig. 1A, red line). 90% of HMF was effectively transformed in 12 h (Fig. 1B), and the yield of HMFCA increased to 88%, with only 2% FDCA obtained as a by-product (Table 1, entry 3). We attributed this significant improvement to the effective recycling of H2O2 that alleviated the inhibition of the enzymes and promoted the catalytic efficiency of this reaction.
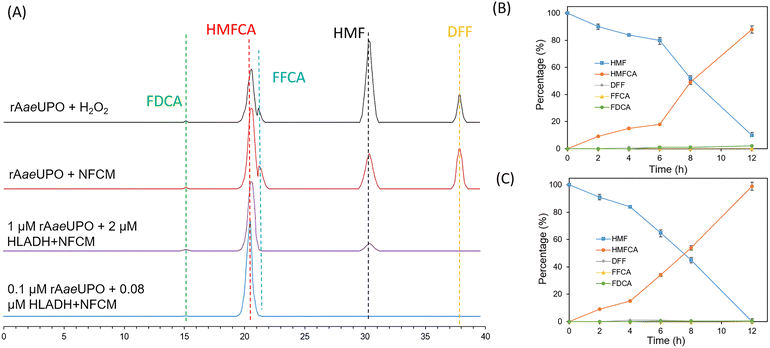 |
| | Fig. 1 The chemoenzymatic selective synthesis of HMFCA from HMF. (A) The final state of the chemoenzymatic reaction under different conditions. (B) Time course for the synthesis of HFMCA catalyzed by the initial rAaeUPO–HLADH system. (C) Time course for the synthesis of HFMCA under optimal conditions. Reaction conditions: 0.1 mM NAD+, 0.02 mM NFCM, 10 mM HMF, 0.08 μM HLADH and 0.1 μM rAaeUPO were added into 1 mL of phosphate buffer (50 mM, pH 7.0) in turn and shaken at 30 °C. Percentage (%) is the ratio of the concentration of each component and calculated according to the formula given in section 1.8 in the ESI†. The detection methods of each compound were the same as those in Table 1. | |
Table 1 The oxidation of HMF to HMFCA catalyzed by rAaeUPO
|
|
| Entry |
rAaeUPO [μM] |
HLADH [μM] |
NFCM [mM] |
H2O2 [mM] |
Conversion [%] |
Yield [%] |
| HMFCA |
DFF |
FFCA |
FDCA |
|
Reaction conditions: 10 mM HMF and 1 μM rAaeUPO were added into 1 mL of phosphate buffer (50 mM, pH 7.0), and 10 mM H2O2 was added continuously over 12 h.
Reaction conditions: 10 mM HMF, 1 μM rAaeUPO, and 10 mM NADH were added into 1 mL of phosphate buffer (50 mM, pH 7.0).
Reaction conditions: 10 mM HMF, 2 μM HLADH, 0.02 mM NFCM, and 0.1 mM NAD+ were added into 1 mL of phosphate buffer (50 mM, pH 7.0).
Reaction conditions: 10 mM HMF, 1 μM rAaeUPO, 2 μM HLADH, 0.02 mM NFCM, and 0.1 mM NAD+ were added into 1 mL of phosphate buffer (50 mM, pH 7.0).
Reaction conditions: 10 mM HMF, 0.1 μM rAaeUPO, 0.08 μM HLADH, 0.02 mM NFCM, and 0.1 mM NAD+ were added into 1 mL of phosphate buffer (50 mM, pH 7.0). The concentration of each product was quantified by HPLC, which was equipped with an Aminex HPX-87H column (300 mm × 7.8 μm), and the methods are listed in section 1.8 of the ESI.†
|
| 1a |
1 |
— |
— |
10 |
54 |
29 |
16 |
8 |
1 |
| 2b |
1 |
— |
0.02 |
— |
80 |
43 |
23 |
13 |
1 |
| 3c |
— |
2 |
0.02 |
— |
16 |
14 |
1 |
1 |
— |
| 4d |
1 |
2 |
0.02 |
— |
90 |
88 |
— |
— |
2 |
| 5e |
0.1 |
0.08 |
0.02 |
— |
100 |
>99 |
— |
— |
— |
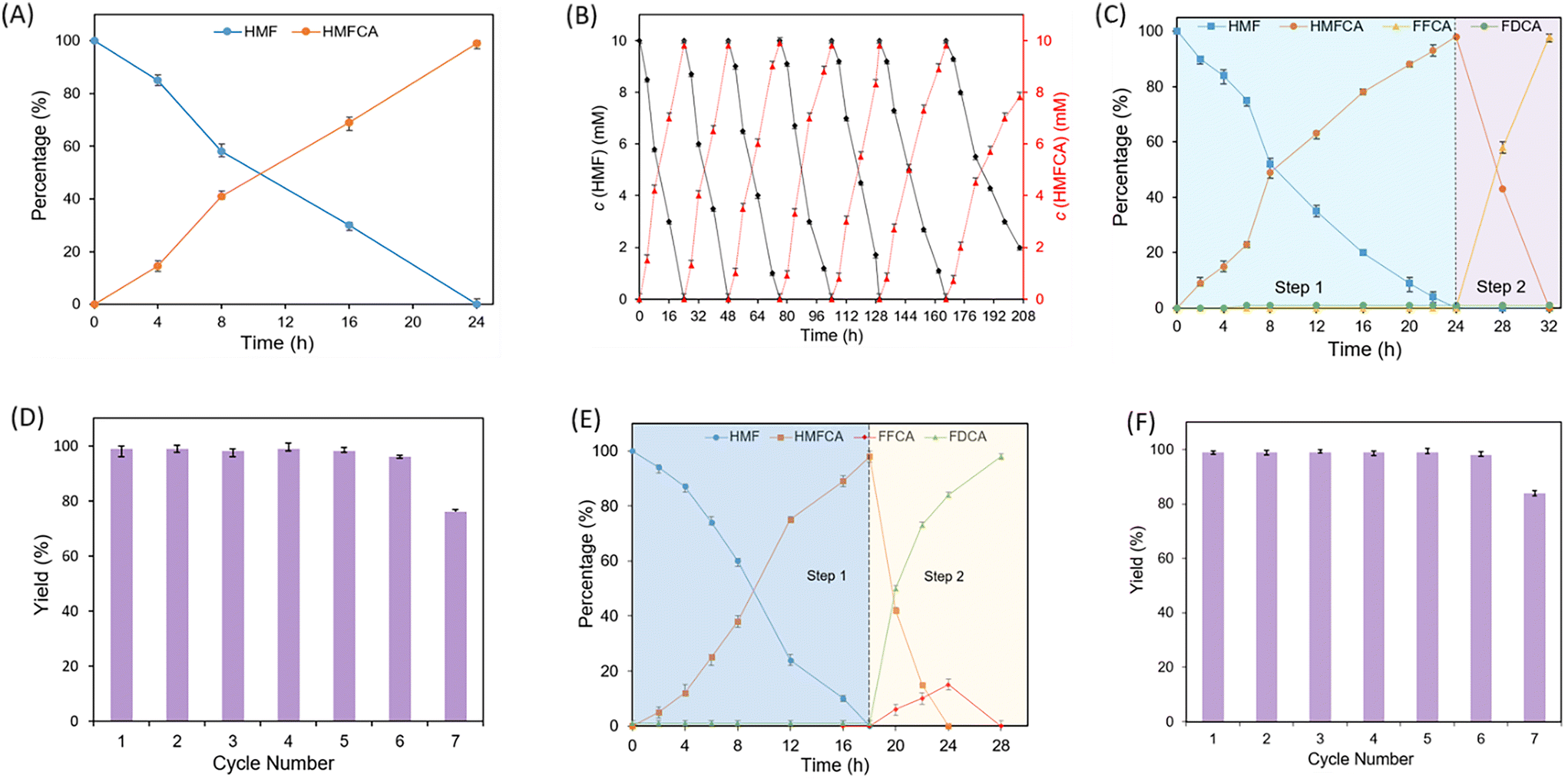
Considering the remaining unconverted HMF and by-product FDCA, further experiments were conducted to promote the efficiency of the reaction. Initially, we hypothesized that different pH values could affect the dual-enzymatic system, and a significant decrease of the conversion and selectivity was observed under acidic or alkaline conditions compared with that under pH 7.0 (Fig. S2†). Then, we surmised that the kinetic balance of the HLADH and rAaeUPO-catalyzed oxidation of HMF was a key factor that affected the reaction efficiency. Hence, we explored if adjusting the quantity of these two enzymes could promote the efficiency of the reaction. As shown in Fig. S3,† increasing the amount of rAaeUPO did not have any impact on the reaction. However, the conversion of HMF gradually decreased with the gradually reduced dosage of rAaeUPO, while the yield of HMFCA was maintained at a high level (98%). This may be because the enzyme activity was impaired by the generated H2O2 that was not consumed by rAaeUPO in time. Based on this result, the impact of the HLADH concentration on the reaction efficiency was further explored. As shown in Fig. S4,† the conversion and selectivity decreased sharply when increasing the dosage of HLADH from 2 to 4 μM. This could also be attributed to the over-generated H2O2, which in turn affected the enzyme activity. In addition, adding a large amount of enzyme can not only increase the reaction cost but also result in low turnover numbers (TONs), which are obviously infeasible. Surprisingly, on reducing the dosage of HLADH from 2 to 0.08 μM, a remarkable enhancement of conversion (100% vs. 90%) and selectivity (>99% vs. 98%) was obtained. We can conclude that the kinetics of HLADH and rAaeUPO were well balanced under these conditions. Notably, the TON of the enzymes (HLADH and rAaeUPO) reached 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, which was nearly 10 times higher than that in the reported literature (Table S3†). Other metrics of our system including space–time yield, E-factor and atom efficiency were also calculated and compared with the reported systems (Table S3†). In the aspects of atom efficiency and E-factor, our system is the best among the reported methods.
000, which was nearly 10 times higher than that in the reported literature (Table S3†). Other metrics of our system including space–time yield, E-factor and atom efficiency were also calculated and compared with the reported systems (Table S3†). In the aspects of atom efficiency and E-factor, our system is the best among the reported methods.
2.2 The chemoenzymatic selective synthesis of FFCA from HMF by a sequential and stepwise process
Based on the above positive results, the selective conversion of HMF to FFCA was investigated. GOase that could transform HMFCA into FFCA23 was further employed to couple with the NFCM-mediated rAaeUPO–HLADH system for selectively oxidizing HMF to FFCA (Fig. 2A). Firstly, a one-pot concurrent approach that simultaneously added GOase and the NFCM-mediated rAaeUPO–HLADH system was proposed. As shown in Fig. 2B, HMF was completely transformed in the first 12 h, but the reaction barely proceeded in the following 12 h with only 47% FFCA detected at the end of the reaction. A large amount of by-product DFF (52%) was detected, which was because GOase could also rapidly convert HMF to DFF.23 Next, a sequential and stepwise process was proposed to enhance the selectivity of the reaction (Fig. 2A). The NFCM-mediated rAaeUPO–HLADH system was firstly added for the transformation of HMF into HMFCA (step 1), and then deactivated by high temperature. Next, GOase was added to the reaction for the following catalysis of HMFCA to FFCA. As expected, HMF was completely transformed in the first 16 h, and then the generated HMFCA was fully oxidized to FFCA by GOase within an additional 8 h (Fig. 2C). The selectivity and yield of the sequential approach were significantly enhanced compared with those of the concurrent approach (99% vs. 46%). Notably, the TON of the enzymes reached 8250, which is higher than that of the previous reported system (Table S4†). The atom efficiency and E-factor of our system are at moderate levels compared to the previously reported system (Table S4†).
 |
| | Fig. 2 The chemoenzymatic cascade system for selectively catalyzing the oxidation of HMF to FFCA. (A and C) One-pot two-step method. Reaction conditions: 0.1 mM NAD+, 0.02 mM NFCM, 10 mM HMF, 0.08 μM HLADH, 0.1 μM rAaeUPO and 1 μM GOase were added into 1 mL of phosphate buffer (50 mM, pH 7.0) in turn and shaken at 30 °C (300 rpm). (B) One-pot one-step method. The detection methods of each compound were the same as those in Table 1. Percentage (%) is the ratio of the concentration of each component in the mixture and calculated according to the formula given in section 1.8 in the ESI.† | |
2.3 The chemoenzymatic selective synthesis of FDCA from HMF by a one-pot two-step method
Since the results in Table 1 indicated that rAaeUPO was also a promising biocatalyst for the oxidation of FFCA to FDCA, we investigated if NFCM-mediated rAaeUPO–HLADH could also be employed for the selective transformation of HMF into FDCA. The initial experiment was conducted to explore the possibility of using the NFCM-mediated rAaeUPO–HLADH system to catalyze the transformation of FFCA into FDCA. As shown in Fig. 3B, the yield of FDCA was gradually increased to >99% on adding an in situ H2O2 generation system (NADH and NFCM) and the NAD+ recycling system (ADH, NFCM, and NAD+). This result was similar to that of the above selective transformation of HMF into HMFCA (Table 1). Based on the favorable results, a one-pot, two-step sequential approach using the NFCM-mediated rAaeUPO–HLADH system and GOase for the chemoenzymatic transformation of HMF into FDCA was proposed (Fig. 3A). Notably, unlike the reaction for synthesizing FFCA, HLADH and rAaeUPO should not be inactivated (step 2). The results indicated that HMF could be completely converted in 28 h with excellent selectivity and yield of FDCA (>99%) (Fig. 3C). Moreover, we found that no additional HLADH and rAaeUPO were required during the reaction. After calculation, the TON of the enzymes in the overall catalytic process was about 8250, which was approximately ∼3-fold higher than the reported highest value (Table S5†). Moreover, our constructed chemoenzymatic system also showed apparent advantages in the aspects of E-factor and atom efficiency over other systems (Table S5†).
 |
| | Fig. 3 The selective synthesis of FDCA from HMF. (A and C) One-pot two-step method. Reaction conditions: 0.1 mM NAD+, 0.02 mM NFCM, 10 mM HMF, 0.08 μM HLADH, 0.1 μM rAaeUPO and 1 μM GOase were added into 1 mL of phosphate buffer (50 mM, pH 7.0) in turn and shaken at 30 °C (300 rpm). (B) The oxidation of FFCA to FDCA catalyzed by different chemoenzymatic systems. Reaction conditions and detection methods were the same as those in Table 1. Percentage (%) is the ratio of the concentration of each component in the mixture and calculated according to the formula given in section 1.8 in the ESI.† | |
2.4 The selective synthesis of furan carboxylic acids from HMF catalyzed by immobilized enzymes
Subsequently, we attempted to explore an efficient immobilization method to increase the stability and recyclability of enzymes. Various valuable immobilization strategies including embedding, adsorption, crosslinking and covalent bonding have been proposed.31,32 Hydrogels, as one of the representative materials employed in covalent binding immobilization technology, have been widely used in the medical field due to their excellent biocompatibility, easy availability, safety, and biodegradability.33 Recently, we have investigated a novel sodium alginate hydrogel and applied it to cell immobilization and 3D printing.34 Thus, we wonder if this material could be applied for the immobilization of the above enzymes. As shown in Table 2 (entries 1, 2 and 4) and Fig. S6,† HLADH, rAaeUPO, and GOase can be separately and effectively immobilized to a hydrogel material with a high efficiency ranging from 60% to 78%. Moreover, HLADH and rAaeUPO could also be co-immobilized to this material with an efficiency of 65% (E3). Validation experiments have also shown that immobilized enzymes rarely detach from the hydrogel (Fig. S8†). After confirming the feasibility of the immobilization approach, we first used the co-immobilized E3 for catalyzing HMF to HMFCA. As shown in Fig. 4A, complete conversion was observed at 10 mM HMF in 24 h, proving the viability of the co-immobilization approach. Next, we determined the recyclability of the co-immobilized enzymes. The results in Fig. 4B show that E3 could be reused at least 7 times, while maintaining an above 80% yield. These results clearly demonstrated a significant enhancement in the stability of immobilized enzymes compared with that of free enzymes. Subsequently, we further verified the feasibility of coupling E3 with E4 for the selective transformation of HMF into FFCA. As expected, the reaction proceeded smoothly in the first cycle, yielding 98% FFCA in 32 h (Fig. 4C). Notably, the operation of heat deactivation of the free enzyme-catalysis (Fig. 2) was replaced by the simple operation of taking out of the immobilized E3, which enabled the reuse of E3. With this modified procedure, E3 and E4 could be recycled 6 times. The yield of FFCA of each cycle was maintained above 98% (Fig. 4D). Then, the immobilized enzymes E3 and E4 were applied to catalyze the selective oxidation of HMF to FDCA by a one-pot two-step sequential approach. As shown in Fig. 4E, 99% yield of FDCA was obtained after 28 h. Based on the results, we explored the recyclability of E3 and E4 for converting HMF to FDCA. As expected, the system could also be recycled up to 6 times without any decrease in the yield (98%) (Fig. 4F). Finally, for isolating the product, we changed the analytical scale conditions to a semi-preparative scale (1.26 g). Under these conditions, HMFCA, FFCA, and FDCA were all obtained with excellent analytical yield (>98%) and selectivity (>99%), respectively (Table S6†). Each product was obtained in pure form with 79%–85% isolated yield (Table S6†).
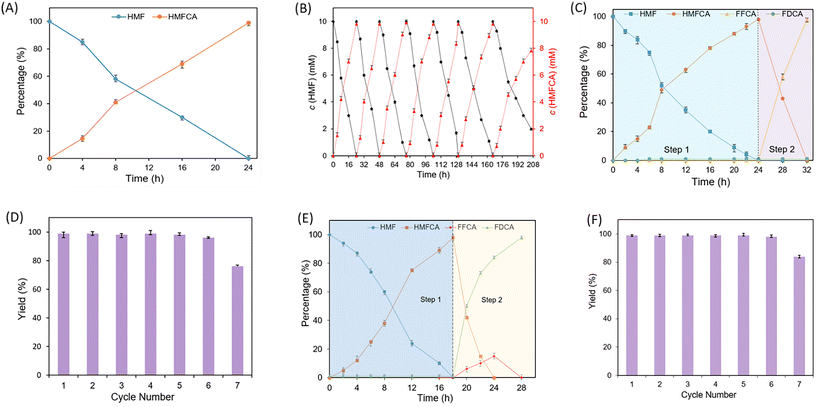 |
| | Fig. 4 The immobilized enzymes selectively oxidized HMF to produce HMFCA, FFCA and FDCA. (A and B) For HMFCA. Reaction conditions: 0.1 mM NAD+, 0.02 mM NFCM, 10 mM HMF, 0.08 or 0.16 μM HLADH and 0.1 or 0.2 μM rAaeUPO were added into 2 mL of phosphate buffer (50 mM, pH 7.0) in turn. (C and D) For FFCA. After HMF was completely transformed, HLADH and rAaeUPO were taken out and 2 μM GOase was added, and the mixture was shaken at 30 °C (300 rpm). (E and F) For FDCA. Reaction conditions: 0.24 μM HLADH and 0.3 μM rAaeUPO were used. Percentage (%) is the ratio of the concentration of each component in the mixture and calculated according to the formula given in section 1.8 in the ESI.† | |
Table 2 The comparison of efficiency before and after enzyme immobilization
| Entry |
Sample |
Activitya [U mg−1] |
Activityb [U mg−1] |
Immobilization efficiencyc [%] |
|
Activity of free enzymes.
Activity of co-immobilized enzymes or immobilized enzymes.
The immobilization efficiency was calculated according to the formula given in section 1.7 in the ESI†.
|
| 1 |
E1 (HLADH) |
0.1 |
0.042 |
70 |
| 2 |
E2 (rAaeUPO) |
257 |
121 |
60 |
| 3 |
E3 (HLADH and rAaeUPO) |
0.1 |
0.037 |
65 |
| 257 |
118 |
| 4 |
E4 (GOase) |
75 |
30 |
78 |
3. Conclusions
In summary, we have developed an orderly and efficient NFCM-mediated chemoenzymatic cascade system, which could selectively transform HMF into each value-added furan carboxylic acid. The organocatalyst NFCM acts as a bifunctional catalyst, which enables in situ generation of H2O2 and regeneration of NAD+ for ADH and UPO-catalyzed oxidation of HMF, thereby promoting efficiency and selectivity of each oxidation reaction. HMFCA, FFCA, and FDCA were all obtained with excellent yield (>99%) and selectivity (>99%). Furthermore, the stability and reusability of each biocatalyst were greatly improved by a sodium alginate hydrogel-based immobilizing strategy. This first example of chemoenzymatic strategy not only offers a promising approach for the selective synthesis of furan carboxylic acids from HMF, but also presents a flexible and mutually beneficial approach that couplesperoxygenase with dehydrogenase for enhancing the efficiency of other selective biocatalytic oxidations. Further work on expanding the application scope of this remarkable system is under investigation in our lab.
Author contributions
Xiaowang Zhang: investigation and writing of the original draft. Zhiyu Zhang: investigation and writing of the original draft. Zhuotao Tan and Ziyi Yu: methodology and resources. Siyu Qi: methodology and investigation. Qi Na: supervision and data curation. Kaichen Zhao: methodology and validation. Hanjie Ying: methodology, supervision, and validation. Chenjie Zhu: supervision, conceptualization, reviewing, and editing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China (grant no. 2022YFC2105900) and the National Natural Science Foundation of China (grant no. 22378195; 22178170; 22208156).
References
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- R. A. Sheldon, Curr. Opin. Green Sustainable Chem., 2018, 14, 89–95 CrossRef.
- X. Wu, M. V. Galkin, T. Stern, Z. Sun and K. Barta, Nat. Commun., 2022, 13, 3376–3387 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- K. Gu, D. Wang, C. Xie, T. Wang, G. Huang, Y. Liu, Y. Zou, L. Tao and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 20253–20258 CrossRef CAS.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS; A. C. Braisted, J. D. Oslob, W. L. Delano, J. Hyde, R. S. McDowell, N. Waal, C. Yu, M. R. Arkin and B. C. Raimundo, J. Am. Chem. Soc., 2003, 125, 3714–3715 CrossRef.
- K. T. Hopkins, W. D. Wilson, B. C. Bender, D. R. McCurdy, J. E. Hall, R. R. Tidwell, A. Kumar, M. Bajic and D. W. Boykin, J. Med. Chem., 1998, 41, 3872–3878 CrossRef CAS PubMed.
- D. Zhang and M.-J. Dumont, J. Polym. Sci., Part A:Polym. Chem., 2017, 55, 1478–1492 CrossRef CAS.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelhob and A. J. D. Silvestrea, Polym. Chem., 2015, 6, 5961–5983 RSC.
- D. Liu, Y. Li, C. Wang, H. Yang, R. Wang, S. Li and X. Yang, Appl. Catal., A, 2024, 669, 119497–119506 CrossRef CAS.
- Y. Tao, S. Fan, X. Li, J. Yang, J. Wang and G. Chen, J. Colloid Interface Sci., 2024, 654, 731–739 CrossRef CAS PubMed.
- S. Xu, P. Zhou, Z. Zhang, C. Yang, B. Zhang, K. Deng, S. Bottle and H. Zhu, J. Am. Chem. Soc., 2017, 139, 14775–14782 CrossRef CAS.
- M. E. Fergani, N. Candu, P. Granger, S. M. Coman and V. I. Parvulescu, Catal. Today, 2022, 405, 267–276 CrossRef.
- Y. Qin, Y. Li, M. Zong, H. Wu and N. Li, Green Chem., 2015, 17, 3718–3723 RSC.
- J. T. Cunha, A. Romaní and L. Domingues, Catalysts, 2022, 12, 202–216 CrossRef CAS.
- S. Shi, X. Zhang, M. Zong, C. Wang and N. Li, Mol. Catal., 2019, 469, 68–74 CrossRef CAS.
- X. Pan, S. Wu, D. Yao, L. Liu, L. Zhang, Z. Yao, Y. Pan, S. Chang and B. Li, React. Chem. Eng., 2020, 8, 1397–1404 RSC.
- A. Araya, N. Guajardo and M. E. Lienqueo, Biochem. Eng. J., 2024, 202, 109157–109162 CrossRef CAS.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., 2014, 126, 6633–6636 CrossRef.
- J. Carro, P. Ferreira, L. Rodríguez, A. Prieto, A. Serrano, B. Balcells, A. Ardá, J. Jiménez-Barbero, A. Gutiérrez, R. Ullrich and M. Hofrichter, FEBS J., 2015, 282, 3218–3229 CrossRef CAS.
- H. Jia, M. Zong, H. Yu and N. Li, ChemSusChem, 2017, 10, 3524–3528 CrossRef CAS PubMed.
- A. Swoboda, S. Zwölfer, Z. Duhović, M. Bürgler, K. Ebner, A. Glieder and W. Kroutil, ChemSusChem, 2024, 17, e202400156 CrossRef CAS PubMed.
- H. Jia, M. Zong, G. Zheng and N. Li, ChemSusChem, 2019, 12, 4764–4768 CrossRef CAS PubMed.
- A. Karich, S. B. Kleeberg, R. Ullrich and M. Hofrichter, Microorganisms, 2018, 6, 1–12 CrossRef.
- D. S. Choi, Y. Ni, E. Fernández-Fueyo, M. Lee, F. Hollmann and C. B. Park, ACS Catal., 2017, 7, 1563–1567 CrossRef CAS.
- W. Zhang, B. O. Burek, E. Fernández-Fueyo, M. Alcalde, J. Z. Bloh and F. Hollmann, Angew. Chem., Int. Ed., 2017, 56, 15451–15455 CrossRef CAS.
- J. H. Jang, J. E. Park and J. S. Han, Eur. J. Pharmacol., 2018, 284, 152–156 CrossRef.
- X. Zhang, Z. Tan, C. Li, S. Qi, M. Xu, M. Li, W. Xiong, W. Zhuang, D. Liu, C. Zhu and H. Ying, Bioresour. Bioprocess., 2021, 8, 1–9 CrossRef.
- Z. Tan, X. Zhang, M. Xu, Y. Fu, W. Zhuang, M. Li, X. Wu, H. Ying, P. Ouyang and C. Zhu, Sci. Adv., 2022, 8, 1912–1921 CrossRef PubMed.
- X. Zhang, Z. Tan, M. Xu, W. Zhuang, H. Ying, Z. Chu and C. Zhu, Green Synth. Catal., 2023, 5, 153–158 CrossRef.
- S. M. McKenna, P. Mines, P. Law, K. Kovacs-Schreiner, W. R. Birmingham, N. J. Turner, S. Leimkühlerd and A. J. Carnell, Green Chem., 2017, 19, 4660–4665 RSC.
- Z. Wu, L. Shi, X. Yu, S. Zhang and G. Chen, Molecules, 2019, 24, 3648–3664 CrossRef CAS PubMed.
- D. del-Bosque, J. Vila-Crespo, V. Ruipérez, E. Fernández-Fernández and J. M. Rodríguez-Nogales, Gels, 2023, 9, 320–336 CrossRef CAS PubMed.
- W. Cheng, J. Zhang, J. Liu and Z. Yu, VIEW, 2020, 1, 20200060–20200067 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Chenjie
Zhu
a and
Chenjie
Zhu