 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synchrotron X-ray nanoprobe and correlative electron microscopy reveal the role of surface chemistry of self-assembling peptides in calcium phosphate nucleation†
Reham
Gonnah
 ab,
Julia E.
Parker
a,
Robert P. W.
Davies
b and
Maisoon
Al-Jawad
*b
ab,
Julia E.
Parker
a,
Robert P. W.
Davies
b and
Maisoon
Al-Jawad
*b
aDiamond Light Source, Diamond House, Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK
bDivision of Oral Biology, School of Dentistry, University of Leeds, Leeds, UK. E-mail: m.al-jawad@leeds.ac.uk
First published on 19th March 2025
Abstract
A biomimetic peptide (P11-4), which is predominantly negatively-charged, facilitates the nucleation of hydroxyapatite (HAp). P11-4 self-assembles into fibrils via β-sheet formation, creating a 3D-gel-network. Here, X-ray nanoimaging and correlative scanning electron microscopy (SEM) investigated P11-4's surface chemistry and its ability to nucleate HAp in the absence of the 3D-gel-network. P11-4 was deposited on silicon nitride (SiN) windows, which were immersed in a mineralising solution (MS) and then mapped using nano-X-ray fluorescence (n-XRF) and differential phase contrast imaging at the hard X-ray nanoprobe beamline (I14) at Diamond Light Source. Elemental calcium and phosphorus maps were extracted using n-XRF, and compared with and without P11-4. The windows were subsequently mapped using SEM and Energy Dispersive Spectroscopy (EDS) to confirm the morphology and elemental compositions of the formed structures. The calcium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :phosphorus ratios were calculated to identify the phases formed. P11-4 increased the calcium and phosphorus signals with time in MS compared to the control (without P11-4). After 12 hours in MS, calcium ions accumulated on the deposited β-sheets, attracting phosphorus ions at later time points. From the morphology in the images and EDS analysis, the spherical calcium phosphate (CaP) structures appeared to be amorphous, indicating the formation of precursors, likely amorphous CaP, at early time points. In the presence of P11-4, these structures grew and fused into larger CaP formations over time, unlike in the control. Nano-imaging techniques highlighted that P11-4's surface chemistry accelerates the kinetics and controls the initial CaP crystallisation process, resulting in an amorphous CaP phase.
:phosphorus ratios were calculated to identify the phases formed. P11-4 increased the calcium and phosphorus signals with time in MS compared to the control (without P11-4). After 12 hours in MS, calcium ions accumulated on the deposited β-sheets, attracting phosphorus ions at later time points. From the morphology in the images and EDS analysis, the spherical calcium phosphate (CaP) structures appeared to be amorphous, indicating the formation of precursors, likely amorphous CaP, at early time points. In the presence of P11-4, these structures grew and fused into larger CaP formations over time, unlike in the control. Nano-imaging techniques highlighted that P11-4's surface chemistry accelerates the kinetics and controls the initial CaP crystallisation process, resulting in an amorphous CaP phase.
1. Introduction
Calcium phosphate mineralisation is a critical process that is relevant to biological systems, particularly in the development of hard tissues like dental enamel and bone. During the natural biomineralisation process of hard tissues, the organic matrix proteins play a critical role in guiding and controlling the mineralisation process, reportedly through the formation of calcium phosphate precursor phases, following a non-classical nucleation route. For example, during dental enamel formation (amelogenesis), enamel matrix proteins (e.g. amelogenins) are responsible for guiding the hydroxyapatite (HAp) mineralisation process, via the formation of amorphous calcium phosphate (ACP), to give rise to enamel's highly organised hierarchical structure with exceptional mechanical properties.1–5 For instance, the charged C-terminal functional domain of amelogenin, which contains negatively charged amino acids such as glutamic acid (Glu) and aspartic acid (Asp), has been reported to play a role in the organisation of crystals.6 It has been suggested that the C-terminal region may be involved in the self-assembly of amelogenin through the interaction of the Glu and Asp residues with the calcium ions.6,7 However, these matrix proteins degrade as the tissue matures and do not provide a reparative mechanism throughout the tissue's lifetime. Consequently, there is a critical need for function-mimicking templates to promote biomimetic mineralisation, enabling the restoration of the tissue's structural and mechanical properties following damage.In the recent years, self-assembling peptide P11-4 has gained enormous attention due to its reported ability to nucleate HAp for dental enamel remineralisation when demineralised (e.g. due to early caries), making the peptide a promising candidate for promoting biomimetic mineralisation. P11-4 was reported to self-assemble to form a 3D gel-like scaffold, made up of assembled fibrils, as a function of concentration to support nucleation.8,9 To achieve this gel state, there are distinct concentration dependent hierarchical phases. The simplest assembly manifests as a result of the peptide adopting a β-sheet conformation in a single dimension through hydrogen bonding.8 The β-sheets then continue to self-assemble into nanotapes that have a helical structure, which then stack to form nanoribbons composed of two tapes. As the nanoribbons continue to stack, they form nanofibrils, which, if stable, develop into fibres that create the scaffold.10 The modulation of the hierarchical fibrils, although intrinsically concentration dependant, can be controlled by the ionic state of the amino acids of the primary sequence. In the case of P11-4, at a high pH (≥8), the P11-4 stays in a “monomeric state” (not self-assembled).11 Acidic pH (less than 7.4) and the “presence of cations” activate the self-assembling process of P11-4.12
It has been suggested that P11-4 forms HAp directly by chelating calcium ions (Ca2+) on negatively-charged glutamic acid residues, spaced with the same geometry as the Ca2+ ions found in HAp.13 However, this suggestion was based on in silico modelling and lacks experimental data for validation. P11-4's structure was reported to result in de novo HAp nucleation, however the peptide's precise mechanism, its nucleation route (i.e. classical or non-classical nucleation including precursor phase formation) and early kinetics of action remain unclear. Specifically, the role of surface chemistry in the hydroxyapatite nucleation process is poorly understood. Studying and understanding the process of calcium phosphate nucleation in the presence of promising biomimetic materials are critical for their development for tissue engineering applications to treat various pathological conditions that affect hard tissues in the human body.
Characterising biomaterials to understand how they affect calcium phosphate mineralisation to elucidate their role in biomimetic mineralisation requires the use of cutting-edge tools to achieve nanoscale resolutions. Formed mineral structures at early time points can be too small to be detected using the conventional characterisation methods in the lab, necessitating the application of high-resolution imaging techniques, such as synchrotron-based X-ray nanoprobe imaging to better understand these processes. To complement the X-ray nanoprobe imaging, lab-based electron microscopy techniques can be used to study mineralisation at different length scales to build a full picture on how biomaterials interact with minerals to enhance their development. At the time of writing, no reported studies have made use of X-ray nanoprobe techniques to understand how biomimetic materials influence the nucleation of calcium phosphate phases.
The secondary conformation and the resulting surface chemistry of the peptide can affect the nucleation pathways. Therefore, this study aims to apply synchrotron X-ray nanoprobe techniques on the I14 beamline at Diamond Light Source (DLS),14 along with correlative scanning electron microscopy, to investigate these effects in the absence of 3D confinement on the hydroxyapatite nucleation process by depositing the peptide on a substrate. This is to enhance our understanding of crystal growth, in the presence of P11-4, at the nano- and microscale levels and to elucidate the peptide's role in biomimetic mineralisation by addressing the question: does the peptide form hydroxyapatite through a similar pathway to natural enamel biomineralisation? These techniques can be applied to characterise biomimetic peptides and proteins of biological significance. In this study, P11-4 is employed as a model system for characterisation.
2. Experimental
For all the experiments, a 10 mg mL−1 P11-4 (Credentis AG, Switzerland) solution was prepared and adjusted to pH 7 using 1% ammonia. At pH 7, the solution appeared as a pre-gel, viscous liquid containing fibrillar assemblies.112.1 Optimising P11-4 β-sheet deposition on silicon nitride windows
For X-ray nanoprobe imaging, the P11-4 solution was deposited on 5 × 5 mm silicon nitride (SiN) windows before immersion in mineralising solution (MS) for different durations. Prior to nano-X-ray fluorescence (n-XRF) and differential phase contrast (DPC) imaging,15 an assessment by Fourier Transform Infrared Spectroscopy (FTIR) was conducted to establish the avidity of the fibrils to the SiN windows after immersion in MS and establish a deposition protocol that results in the most prominent signal at the 1620 cm−1 wave number, indicating the presence of β-sheets, to investigate how they affect nucleation of mineral.(1) Method 1 – Drop-coating: one 6 μL drop of P11-4 solution was placed onto the center of a SiN window and allowed to dry.
(2) Method 2 – Blotting: one 6 μL drop of P11-4 solution was placed onto the center of a silicon nitride window, excess peptide solution was then blotted with tissue paper. It was then left to dry.
(3) Method 3 – Immersion: SiN windows were dipped in P11-4 solution, removed and re-dipped, and then left to dry.
After the windows were dry, they were assessed by FTIR (Nicolet iS50 FT-IR) to determine which method resulted in the highest proportion of β-sheets by assessing the intensity of the peak at 1620 cm−1 wave number. A bespoke 3D-printed sample holder was designed to hold the sample securely in place during conformational analysis. The following parameters were set for the FTIR scans: number of scans = 32, resolution = 16, final format was absorbance and no correction was specified. From the collected spectra, the amide I region (1720 and 1580 cm−1) was evaluated to indicate the presence of P11-4 on the window.
2.2 Mapping of calcium phosphate phases – hard X-ray nanoprobe
2.3 Complementary scanning electron microscopy (SEM) and energy dispersive spectrometry (EDS)
The samples mapped on the beamline were subsequently mapped using SEM (JEOL JSM-6610LV) and EDS using Aztec (Oxford Instruments) for quantitative analysis. The secondary electron images (SEI) mode was selected, and the voltage was set to 20 kV. The working distance (WD) was 11 mm and the spot size was set to 60 nm. The images were taken at various magnifications. The calcium–phosphorus ratio was calculated from the atomic compositions.3. Results
3.1 Optimising P11-4 β-sheet deposition on silicon nitride windows
Three deposition methods, drop-coating, immersion, and blotting, were tested for depositing P11-4 onto SiN windows to determine which method yielded the highest deposition of β-sheets to subsequently carry out n-XRF and DPC mapping of mineralisation events on the I14 beamline. The amide I region (1720–1580 cm−1) was analysed to confirm P11-4's presence, with a peak at 1620 cm−1 indicating β-sheets. While all methods facilitated some β-sheet deposition, blotting yielded the least amount as shown in Fig. 1 (full spectra in Fig. D in the ESI†). The drop-coating method resulted in the highest mean absorbance signal, and most consistent result, indicating substantial and reproducible β-sheet formation compared to the two other methods (Fig. 1). Consequently, drop-coating was chosen for further sample preparation. To evaluate the stability of the P11-4 adsorbed-layer on SiN windows, P11-4 was deposited using the drop-coating method and immersed in 700 μL of MS for 4–5 hours. The FTIR spectra (see Fig. E in the ESI†) after 4–5 hours in MS exhibited a persistent peak at 1620 cm−1, with a calculated mean absorbance signal of 0.128 ± 0.004 arbitrary units (n = 3). | ||
| Fig. 1 Mean absorbance signal at 1620 cm−1 (wave number), indicative of the presence of the β-sheet conformation, assessed by Fourier Transform Infrared Spectroscopy (FTIR) using three different deposition methods: blotting, drop-coating, and immersion. The error bars represent the standard deviation (SD). | ||
3.2 Mapping of calcium phosphate phases – hard X-ray nanoprobe
In the control sample, fewer structures were observed in the mapped areas using DPC compared to the SiN windows with deposited P11-4 as shown in Fig. 2. These structures were confirmed to be calcium phosphate, as evidenced by Fig. 3, which presents the elemental calcium and phosphorus maps from the same mapped areas as in Fig. 2 for the equivalent time points. | ||
| Fig. 2 Typical Differential Phase Contrast (DPC) images of SiN windows with deposited P11-4 and without (control) mapped on the I14 beamline at different time points, depicting the morphology of the crystals formed after immersion in mineralising solution. The area shown is 15 μm × 15 μm, with a step size of 50 nm. | ||
 | ||
| Fig. 3 Typical calcium and phosphorus elemental maps at different time points (6 hours, 12 hours, 16 hours, 24 hours and 2 weeks) comparing the control to the mapped SiN windows with deposited P11-4, extracted using n-XRF mapping on the I14 beamline. These maps depict the morphology of the formed calcium phosphate structures. The area shown is 15 μm × 15 μm, with a step size of 50 nm. | ||
Additionally, Fig. 2 shows that P11-4 induces the formation of spherical structures, which increase in number with time, and appear to migrate and fuse together over time, forming the biggest calcium phosphate deposits at the longest time point (2 weeks of mineralisation) (Fig. 2). The spherical structures observed in Fig. 2 (in the presence of P11-4 and in the control after 2 weeks of mineralisation) can be observed in the calcium and phosphorus maps in Fig. 3 at the early time points (12, 16 and 24 hours) in the presence of P11-4, confirming that these structures are a calcium phosphate phase. These spherical structures were absent in the control SiN windows imaged using DPC during the early time points as shown in Fig. 2. In addition, the control elemental maps (Fig. 3) do not manifest similar spherical structures as observed in the presence of P11-4. Moreover, the calcium phosphate structures formed in the absence of P11-4 (i.e. the control) are smaller, and do not appear to form an interconnected network with neighbouring calcium phosphate structures as manifested in the presence of P11-4 at the early time points. However, the morphology of the mineralised structures formed after 2 weeks in the absence of P11-4 (control sample) is similar to that in the presence of P11-4 as shown in Fig. 2. At 2 weeks, larger calcium phosphate structures form in the absence of P11-4 (control), and appear to be networking with neighbouring structures. From our results, the needle-like nanostructural morphology characteristic of HAp could not be observed, even after 2 weeks in mineralising solution.
In Fig. 3, calcium phosphate can be observed at 6 hours in the presence of P11-4, indicating the presence of small calcium phosphate structures that are approximately 50 nm in diameter. With time in mineralising solution, the calcium phosphate structures grow in the presence of P11-4 compared to the control.
Looking at the distribution of structures over a relatively larger area (400 μm × 400 μm), fewer calcium phosphate structures were observed on the control SiN windows (Fig. 4 and 5) at the early time points compared to the windows with P11-4. For the control, the distribution of calcium phosphate appears to be more random compared to the SiN windows with deposited P11-4 as shown in Fig. 4 and 5. In Fig. 4, at 12 hours, the calcium signal, indicated by the red colour, appears to be higher than the phosphorus signal in the presence of P11-4. After 12 hours of mineralisation, the phosphorus signal increases to indicate phosphorus ion attraction at later time points compared to the calcium ions.
 | ||
| Fig. 4 Combined calcium and phosphorus elemental maps to detect the calcium phosphate distribution across the mapped areas (using n-XRF on the I14 beamline) at different time points under 24 hours, comparing the control to the mapped SiN windows with deposited P11-4. Red = calcium signal, blue = phosphorus signal and pink = both. The area shown is 400 μm × 400 μm with a step size of 1000 nm. | ||
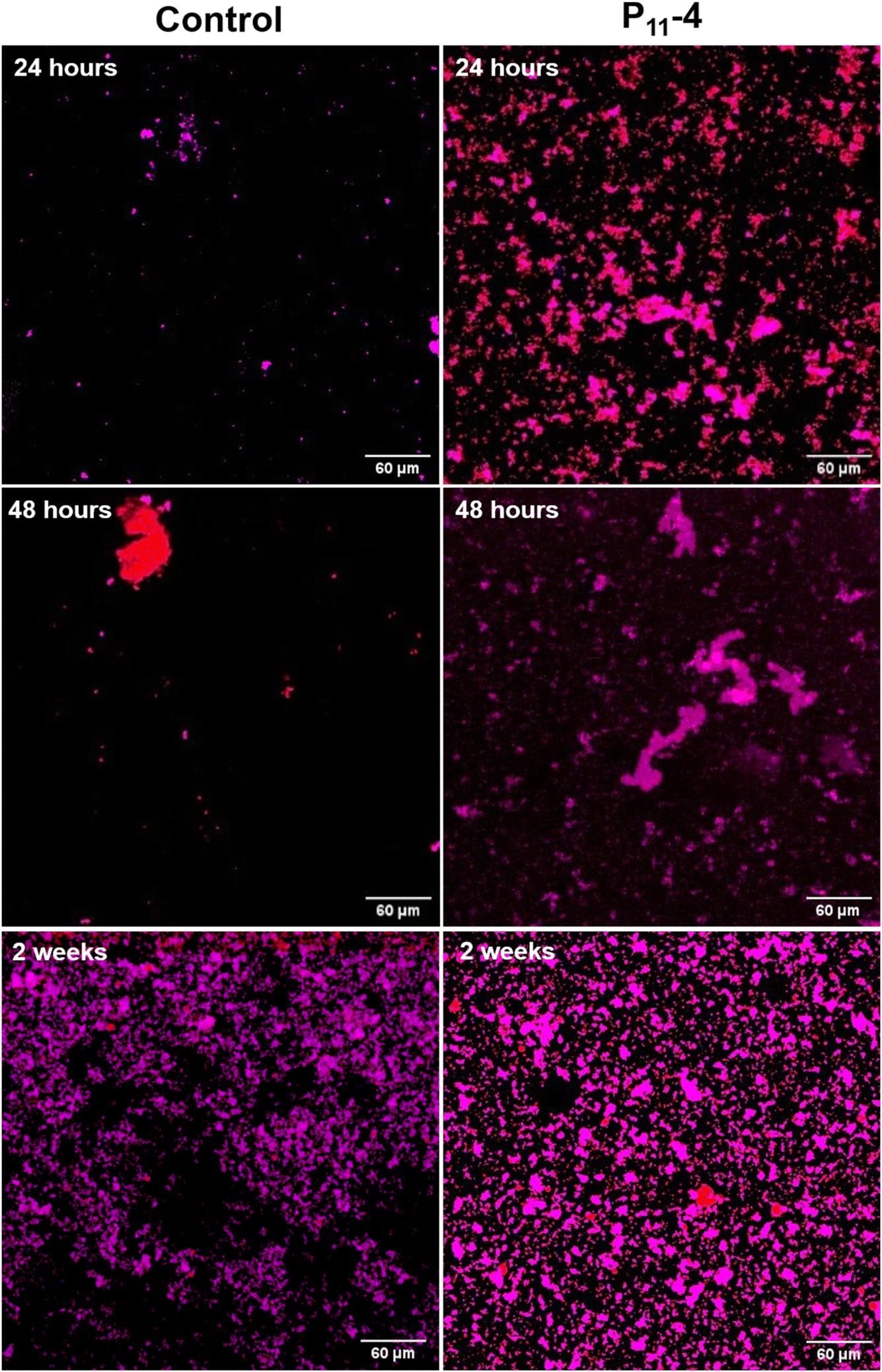 | ||
| Fig. 5 Combined calcium and phosphorus elemental maps to detect the calcium phosphate distribution across the mapped areas (using n-XRF on the I14 beamline) at different time points over 24 hours, comparing the control to the mapped SiN windows with deposited P11-4. Red = calcium signal, blue = phosphorus signal and pink = both. The area shown is 400 μm × 400 μm with a step size of 1000 nm. | ||
As shown in Fig. 6, the mean calcium and phosphorus intensities generally increased with time in mineralising solution in the presence of P11-4. The mean calcium intensity was higher in the presence of P11-4 compared to the control for the early time points, except for the time point at 12 hours. However, after 2 weeks in mineralising solution, the calcium signal was significantly higher in the control, which was also the case for the phosphorus signal. The phosphorus signal was higher in the control compared to the SiN window with deposited P11-4 at 6 hours and 12 hours. At 16 hours, the signal dropped below the level of the SiN treated with P11-4. By 24 hours, the signals, in the presence and absence of P11-4 (control), were nearly equal, however at 48 hours, it was lower again in the control compared to the SiN window with deposited P11-4.
 | ||
| Fig. 6 Mean total signal intensity of (a) calcium and (b) phosphorus with time (6 hours, 12 hours, 16 hours, 24 hours, 48 hours and 2 weeks) comparing the control to the SiN windows with deposited P11-4. The 400 μm × 400 μm map was split into eight regions, from which the calcium and phosphorus signals intensity was calculated to calculate the mean total signal intensity value of calcium and phosphorus. The error bars indicate the standard deviation of values. | ||
Fig. 7 shows no obvious trend in the control, with the ratio increasing from 6 to 12 hours, decreasing until the 48-hour time point, and then increasing drastically at 2 weeks. On the other hand, the calcium-to-phosphorus ratio with time is shown to have a trend in the presence of P11-4 (where the ratio increases until 16 hours, decreases slightly at the 24-hour time point, and then plateaus) compared to the control. The ratio of the mean intensity values increases to 33.7 after 2 weeks for the control, while the ratio of the mean intensity values after 2 weeks is 28.3 in the presence of P11-4, which is within the range of ratio values after 12–48 hours in the presence of P11-4.
 | ||
| Fig. 7 Mean calcium-to-phosphorus signal intensity ratio against time, comparing the control to the SiN windows with deposited P11-4. The 400 μm × 400 μm map was split into eight regions, from which the calcium and phosphorus signals intensity was calculated. The mean signal intensity value of calcium and phosphorus was calculated from the eight regions, and the ratio of the values was plotted on the y-axis against time on the x-axis. The dark grey line indicates the values calculated for the control while the red line indicates the values in the presence of P11-4. | ||
3.3 Complementary scanning electron microscopy (SEM) and energy dispersive spectrometry (EDS)
The samples imaged at the hard X-ray nanoprobe were subsequently imaged using lab-based SEM for complementary analysis of the SiN windows. SEM imaging enabled imaging the whole SiN window for each sample, as shown in Fig. 8. EDS was also conducted on the windows to confirm the elements within certain regions on the SiN windows to confirm and complement the previous findings (Fig. A–C in the ESI†). | ||
| Fig. 8 Scanning electron microscopy (SEM) images of the SiN windows that were imaged on the I14 beamline to show the whole window (magnification = ×50) at the different time points, comparing the control to the mapped SiN windows with deposited P11-4. The secondary electron detector was used at an energy of 20 kV, a working distance of 11 mm and a spot size of 60 nm. | ||
Fig. 8 showed reduced amount of calcium phosphate structures on the control SiN windows when compared to the SiN windows with P11-4 for the early time points. EDS spectra of the control SiN windows detected no calcium or phosphorus signals for the early-time-point samples (6–48 hours). However, Fig. 8 shows that mineralisation has occurred in the control after 2 weeks. EDS analysis of the control after 2 weeks in MS showed that the formed structures were a mixture of calcium phosphate and sodium chloride. On the SiN window with deposited peptide, no sodium or chlorine signals were detected.
As shown in Fig. 8, SEM imaging confirmed the gradual formation of calcium phosphate in the presence of P11-4. Comparing the peptide-treated SiN windows to the control, the peptide is shown to induce the formation of calcium phosphate structures, as confirmed by n-XRF imaging in the previous section, with time for the early time points (6–48 hours). For the 6-hour time point, little crystallisation can be observed on the whole SiN window and the EDS analysis of the window showed no calcium or phosphorus peaks in the spectrum to indicate very little or no calcium or phosphorus signals.
At higher magnification, Fig. 9b shows that after 12 hours in mineralising solution, spherical calcium phosphate structures, as confirmed by the EDS analysis of the region as shown in Fig. A in the ESI,† start forming in the presence of P11-4. From Fig. 9, it appears that the calcium phosphate structures begin to grow further at 16 hours and even more at 24 hours, migrating towards each other to form larger structures to support the results from the synchrotron experiment. These structures appear to be fusing together with time in mineralising solution. These larger structures start to connect with neighbouring structures to form a scaffold-like network as described earlier. In addition, Fig. B in the ESI† confirmed that the formed structures after 16 hours and 24 hours are made of calcium phosphate as the spatial maps showed calcium, phosphorus and oxygen signals.
 | ||
| Fig. 9 Typical scanning electron microscopy (SEM) images of the SiN windows with deposited P11-4 that were imaged on the I14 beamline (magnification = ×4000). (a) 6 hours, (b) 12 hours, (c) 16 hours, (d) 24 hours, (e) 48 hours and (f) 2 weeks. The secondary electron detector was used at an energy of 20 kV, a working distance of 11 mm and a spot size of 60 nm. | ||
After 48 hours in mineralising solution, the peptide appears to be inducing the formation of bigger calcium structures that form a larger network as shown in Fig. 9e. In the background (behind the large network of calcium phosphate structures), the spherical rounded structures can be observed, in which they appear to be migrating towards each other. The formed network of structures is confirmed to be a calcium phosphate phase as shown in Fig. C in the ESI.† After 2 weeks in mineralising solution, even bigger calcium phosphate structures start forming (Fig. 9f).
The calcium-to-phosphorus atomic ratio is similar at 12 and 16 hours (see Fig. 10), at around 1.4–1.5. The ratio decreases after 24 hours (to ∼1.35) in mineralising solution, and is similar at 48 hours. The ratio increases after 2 weeks to ∼1.5–1.6. As no signal was detected for the control samples at earlier time points (12–48 hours), the ratio could not be calculated.
 | ||
| Fig. 10 Calcium/phosphorus atomic ratios, calculated from the atomic compositions, from the Energy Dispersive Spectrometry (EDS) analysis of the structures formed at 12 hours, 16 hours, 24 hours, 48 hours and 2 weeks. No calcium or phosphorus signals were detected for the control at the 12-, 16-, 24-, and 48-hour time points. | ||
4. Discussion
This study aims to investigate the effect of self-assembling peptide P11-4's fibrillar surface chemistry on calcium phosphate mineralisation in the absence of the 3D-gel network, which has been reported to nucleate hydroxyapatite (HAp).8The results presented in this showed that more calcium phosphate structures were formed on the silicon nitride (SiN) windows at the early time points in the presence of P11-4 compared to the control (in the absence of P11-4). The mean calcium intensity was generally higher in the presence of P11-4 compared to the control for the early time points (except for the 12-hour time point, which appeared to be due to random calcium deposition as revealed by n-XRF elemental mapping of the SiN window). For the control, the distribution of calcium phosphate appeared to be more random compared to the SiN windows with deposited P11-4. This was confirmed by n-XRF imaging and complementary SEM imaging of the SiN windows. EDS spectra of the control SiN windows showed no detectable calcium or phosphorus signals in the early time-point samples, preventing the calculation of the calcium to phosphorus atomic ratios to reveal the phase formed. DPC and n-XRF imaging at high resolution (50 nm step size), in addition to SEM imaging, showed that P11-4 induced the formation of spherical calcium phosphate structures that increased with time, and migrated to fuse together for all the time points, to form bigger calcium phosphate deposits that were observed after 2 weeks. These spherical structures were absent in the control SiN windows during the early time points. Additionally, the calcium phosphate structures formed in the control are smaller and do not appear to form an interconnected network with neighboring calcium phosphate structures, unlike those formed in the presence of P11-4 at early time points. However, the morphology of the mineralised structures formed after 2 weeks in the control was similar to that in the presence of P11-4. The spherical morphology of the formed structures indicate the possible formation of amorphous calcium phosphate (ACP), a known precursor of HAp in biomineralisation, as the morphology aligns with the well-known spherical characteristic of ACP, formed by tightly packed “Posner's clusters” to give rise to “larger spherical particles”.18 This behaviour could be visualised in the presence of P11-4. The interconnections of the structures after 12 hours are likely aggregates of Posner's clusters that give rise to ACP particles (with spherical morphology). In addition, the calcium to phosphorus atomic ratios calculated using EDS for all time points fell within the range reported for ACP.19 However, it is important to note that the range of ratios reported for ACP is broad and includes ratios reported for crystalline phases. The atomic ratios calculated for the 24-hour and 48-hour time points were also close to the ratio reported for octacalcium phosphate (1.33).19 However, taking into account the morphology observed using the imaging techniques in this study, the formed calcium phosphate structures are more likely amorphous. Although some internal crystallinity cannot be ruled out, the results obtained from our chosen methods did not indicate any crystallinity. These results suggest that the presence of P11-4 as a template induces, accelerates and directs the mineralisation process, particularly at early time points, possibly by reducing the activation energy required for initial mineral formation.20 Moreover, they highlight the role of P11-4 in guiding mineral formation.
After 12 hours of mineralisation, the calcium signal appeared to be higher than the phosphorus signal in the presence of P11-4, indicating calcium ion accumulation on the deposited β-sheets. This supports the idea that P11-4 acts as a template to promote mineralisation by providing nucleation sites, mimicking the role of organic matrix proteins (e.g. amelogenin) in facilitating nucleation and supporting mineral deposition through the presence of functional domains within their structure.21 The results suggest that in the first 12 hours, the calcium ions are attracted to the deposited P11-4 fibrillar assemblies as suggested by in silico modelling.13 It was postulated that P11-4's mechanism involves calcium chelation by the peptide's central glutamic acids, followed by phosphate dehydration and HAp formation.13 However, from our results, the needle-like morphology of HAp could not be observed, even after 2 weeks in mineralising solution. Using in silico modelling, the formation of HAp was postulated to form directly from ions; however, our findings suggest that the mineralisation process likely occurs via a precursor calcium phosphate phase, typical of ACP.
For the control, our results showed that the mean calcium and phosphorus intensities were very low at the early time points (6–48 hours), increasing dramatically after 2 weeks of immersion in mineralising solution. This behaviour is characteristic of ‘homogeneous nucleation’. On the other hand, the mean calcium and phosphorus intensities generally increased with time in mineralising solution in the presence of P11-4, demonstrating a ‘heterogeneous nucleation’ behaviour. The imaging techniques used in this study confirmed the gradual formation of mineral structures in the presence of P11-4. Additionally, in the presence of P11-4, the ratio of the mean intensity values appeared to have a trend, where the ratio increased until 16 hours, decreased slightly at the 24-hour time point, and then plateaued. In contrast, the control showed no obvious trend. This implies that P11-4 controls the formation of calcium phosphate. The trend, in the presence of P11-4, suggests an initial burst of kinetic deposition of a calcium phosphate phase, followed by a shift towards equilibrium. However, this equilibrium is assumed and would require additional time points for confirmation. The results from n-XRF imaging suggest that the calcium ions were attracted to the deposited fibrillar assemblies with time during the initial time points (6–16 hours), after which the ratio of calcium to phosphorus remained relatively stable, demonstrating the stability of the amorphous phase formed.
In the study by Kirkham et al.,8 HAp was reported to form in the presence of a self-assembled, scaffold-like gel. The findings of this study suggest that P11-4's surface chemistry results in the formation of what appears to be ACP rather than hydroxyapatite (HAp). The formation of ACP highlights P11-4's role in biomimetic mineralisation as ACP is a known precursor of HAp during the biomineralisation process of dental enamel.22 During maturation, ACP is thought to transform to HAp in permanent mature enamel and control the crystal formation and growth process during amelogenesis. The transient phase, ACP, was proposed to be stabilised by enamel matrix proteins, like amelogenin, to control the mineralisation process.4 This transformation of ACP to HAp might be dependent on the presence of the assembled P11-4 gel network. Longer studies are required to make better conclusions as the time scales for complete biomineralisation could be longer in vivo. The SiN windows can serve as substrates for characterising similar materials using the hard X-ray nanoprobe beamline (I14) at DLS.
5. Conclusions
An easy and robust method to deposit P11-4 β-sheets to characterise the biomaterial at the nanoscale level, has been established. In comparison to the control (no P11-4), P11-4 induces and promotes the formation of amorphous calcium phosphate structures at early time points by providing sites for nucleation (calcium binding sites). After 12 hours in MS, the calcium signal was high on the SiN window showing that the calcium ions were attracted to the fibrils to form spherical, amorphous calcium phosphate structures, resembling the structure and composition of ACP, as confirmed by n-XRF and EDS. The amorphous ACP-like structures appear to migrate towards each other, forming bigger amorphous calcium phosphate structures that continue to grow to form a network of a calcium phosphate phase that has a scaffold-like morphology. The network of structures is likely composed of calcium phosphate clusters known as Posner's clusters to give rise to the bigger spherical ACP particles. After 2 weeks in MS, more calcium phosphate was deposited on the windows in the absence of P11-4, indicating that the peptide controls the initial crystallisation process, mimicking the role of the enamel matrix proteins. The needle-like morphology of HAp was not observed, even after 2 weeks in the presence of P11-4. Additionally, EDS analysis revealed the calcium phosphate ratios are within the range of ratios reported for amorphous calcium phosphate structures (1.0–2.2),19 suggesting that confinement might be important to complement the effect of P11-4's surface chemistry in HAp formation. The time scale might have been shorter than required. Therefore, longer studies are needed to confirm whether more time was needed for the transformation process to occur. The use of these high-resolution techniques to bridge length scales offers a method for characterising biomaterials for biomineralisation applications, providing complementary structural and chemical insights. As a next step, the nanoimaging techniques at the hard X-ray nanoprobe beamline, which enables in situ measurements, can be applied to obtain dynamic data for the real-time characterisation of biomimetic materials.Data availability
The data for this study was collected as part of a PhD studentship in collaboration with Diamond Light Source (DLS) and is currently archived at DLS under experiment number mg32152. Following the completion of the PhD project, the data will be deposited in a repository. In the interim, the data is available upon request.Author contributions
Reham Gonnah contributed to conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing the original draft, reviewing and editing. Julia Parker contributed to conceptualization, formal analysis, funding acquisition, investigation, methodology, resources, supervision, validation, visualization, writing the original draft, reviewing and editing. Robert Davies contributed to conceptualization, formal analysis, funding acquisition, investigation, methodology, resources, supervision, validation, writing the original draft, reviewing and editing. Maisoon Al-Jawad contributed to conceptualization, funding acquisition, methodology, resources, supervision, validation, writing the original draft, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge Diamond Light Source for the provision of beamtime at the I14 beamline under proposal number mg32152. The authors wish to acknowledge the funders of this project. This work was supported by Diamond Light Source and the University of Leeds in the form of a joint PhD studentship for Reham Gonnah [STU0413].References
- J. P. Simmer and A. G. Fincham, Crit. Rev. Oral Biol. Med., 1995, 6, 84–108 CrossRef CAS PubMed.
- C. Robinson, S. J. Brookes, R. C. Shore and J. Kirkham, Eur. J. Oral Sci., 1998, 106, 282–291 CrossRef CAS PubMed.
- J. P. Simmer, A. S. Richardson, Y. Y. Hu, C. E. Smith and J. Ching-Chun Hu, Int. J. Oral Sci., 2012, 4, 129–134 CrossRef CAS PubMed.
- J. Moradian-Oldak, Front. Biosci., 2012, 17, 1996 CrossRef PubMed.
- Z. Shi, MATEC Web Conf., 2022, 363, 01032 CrossRef CAS.
- E. Beniash, J. P. Simmer and H. C. Margolis, J. Struct. Biol., 2005, 149, 182–190 CrossRef CAS PubMed.
- J. Moradian-Oldak, M. L. Paine, Y. P. Lei, A. G. Fincham and M. L. Snead, J. Struct. Biol., 2000, 131, 27–37 CrossRef CAS PubMed.
- J. Kirkham, A. Firth, D. Vernals, N. Boden, C. Robinson, R. C. Shore, S. J. Brookes and A. Aggeli, J. Dent. Res., 2007, 86, 426–430 CrossRef CAS PubMed.
- L. Kind, S. Stevanovic, S. Wuttig, S. Wimberger, J. Hofer, B. Müller and U. Pieles, J. Dent. Res., 2017, 96, 790–797 CrossRef CAS PubMed.
- R. A. Gulzar, P. Ajitha and H. Subbaiyan, J. Pharm. Res. Int., 2020, 32, 83–89 CrossRef.
- L. M. Carrick, A. Aggeli, N. Boden, J. Fisher, E. Ingham and T. A. Waigh, Tetrahedron, 2007, 63, 7457–7467 CrossRef CAS.
- P. A. Brunton, R. P. W. Davies, J. L. Burke, A. Smith, A. Aggeli, S. J. Brookes and J. Kirkham, Br. Dent. J., 2013, 215, E6 CrossRef CAS PubMed.
- S. Saha, X. B. Yang, N. Wijayathunga, S. Harris, G. A. Feichtinger, R. P. W. Davies and J. Kirkham, Bone, 2019, 127, 602–611 CrossRef CAS PubMed.
- P. D. Quinn, L. Alianelli, M. Gomez-Gonzalez, D. Mahoney, F. Cacho-Nerin, A. Peach and J. E. Parker, J. Synchrotron Radiat., 2021, 28, 1006–1013 CrossRef CAS PubMed.
- P. D. Quinn, F. Cacho-Nerin, M. A. Gomez-Gonzalez, J. E. Parker, T. Poon and J. M. Walker, J. Synchrotron Radiat., 2023, 30, 200–207 CrossRef PubMed.
- C. Whitehouse, J. Fang, A. Aggeli, M. Bell, R. Brydson, C. W. Fishwick, J. R. Henderson, C. M. Knobler, R. W. Owens, N. H. Thomson and D. A. Smith, Angew. Chem., Int. Ed., 2005, 44, 1965–1968 CrossRef CAS PubMed.
- V. A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, Spectrochim. Acta, Part B, 2007, 62, 63–68 CrossRef.
- L. Wang and G. H. Nancollas, Chem. Rev., 2008, 108, 4628–4669 CrossRef CAS PubMed.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 4457–4475 CrossRef CAS PubMed.
- A. L. Boskey and E. Villarreal-Ramirez, Matrix Biol., 2016, 52, 43–59 CrossRef PubMed.
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, 1989 Search PubMed.
- E. Beniash, R. A. Metzler, R. S. Lam and P. U. P.A. Gilbert, J. Struct. Biol., 2009, 166, 133–143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5fd00017c |
| This journal is © The Royal Society of Chemistry 2025 |