
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethods to achieve tissue-mimetic physicochemical properties in hydrogels for regenerative medicine and tissue engineering
Rabia
Fatima
and
Bethany
Almeida
 *
*
Department of Chemical and Biomolecular Engineering, Clarkson University, Potsdam, NY 13699, USA. E-mail: balmeida@clarkson.edu
First published on 16th August 2024
Abstract
Hydrogels are water-swollen polymeric matrices with properties that are remarkably similar in function to the extracellular matrix. For example, the polymer matrix provides structural support and adhesion sites for cells in much of the same way as the fibers of the extracellular matrix. In addition, depending on the polymer used, bioactive sites on the polymer may provide signals to initiate certain cell behavior. However, despite their potential as biomaterials for tissue engineering and regenerative medicine applications, fabricating hydrogels that truly mimic the physicochemical properties of the extracellular matrix to physiologically-relevant values is a challenge. Recent efforts in the field have sought to improve the physicochemical properties of hydrogels using advanced materials science and engineering methods. In this review, we highlight some of the most promising methods, including crosslinking strategies and manufacturing approaches such as 3D bioprinting and granular hydrogels. We also provide a brief perspective on the future outlook of this field and how these methods may lead to the clinical translation of hydrogel biomaterials for tissue engineering and regenerative medicine applications.
 Rabia Fatima | Rabia Fatima is currently a PhD Student in Chemical Engineering conducting research on advanced hydrogels for cartilage tissue engineering in the Almeida Lab. Prior to attending Clarkson University, Rabia completed her M.S. in Industrial Biotechnology in 2022 from the National University of Science & Technology and her B.S. in Biochemistry in 2020 from the Kinnaird College for Women University. |
 Bethany Almeida | Dr Bethany Almeida is an Assistant Professor at Clarkson University. The Almeida Lab's research focuses on engineering advanced, functional biomaterials to modulate mesenchymal stem cell behavior with applications in tissue engineering and regenerative medicine. Dr Almeida is the recipient of several awards, including a 2020 BMES Career Development Award and a 2020 Rising Star in Engineering in Health. In addition to her B.S. and PhD in Biomedical Engineering, Dr Almeida also has a B.S. in Professional Writing and is passionate about science communication and K-12 outreach. |
1. Introduction
Tissue engineering and regenerative medicine are among the fastest growing biomedical fields, seeking to repair, reconstruct, or regenerate human tissues using a combination of cells, biomaterials, and bioactive physicochemical factors. Hydrogels in particular have received significant attention in recent years as a powerful biomaterial for tissue engineering and regenerative medicine applications. Hydrogels are capable of providing structural support to cells, serving as a scaffold onto which the cells may adhere and grow.1–4 In addition, the shear-thinning properties of many hydrogels protect cells during injection to wound sites, maintaining cellular viability and preventing clearance of the cells.3,5 In addition, depending on the materials and manufacturing approaches used, the hydrogel may provide bioactive signals to the cells, resulting in the activation of different signaling pathways.1,4–6Since the 1993 Langer and Vacanti review paper introducing the concept of tissue engineering,7 the primary goal of tissue engineering and regenerative medicine applications has been to develop new tissues or regenerate tissues/heal wounds such that the new tissue is functionally mature with essentially no phenotypic or genotypic differences from native tissue. In seeking to understand how cells form mature tissues, various historical studies were conducted to evaluate cellular response to physicochemical cues. For example, in the mesenchymal stem cell (MSC) field, Pittenger was the first to demonstrate the multilineage potential of MSCs by evaluating their responses to different chemical signals.8 Engler later demonstrated that physical cues could also control MSC lineage specificity, demonstrating that MSCs cultured on elastic substrates with soft moduli underwent neurogenesis, cells cultured on medium stiffness substrates underwent myogenesis, and cells on stiff substrates underwent osteogenesis.9 Kilian took this a step further, showing that the geometric shape of individual cells could also control MSC differentiation, and that the effects of this physical cue were independent of chemical signaling from differentiation culture media.10
Stemming from these and other seminal works, recent trends over the past couple of decades have been to use biomaterials that are capable of mimicking the physicochemical properties of the native extracellular matrix (ECM) in order to achieve this goal. The ECM is a three-dimensional (3D) matrix of proteins and polysaccharides surrounding cells in vivo.11 In the human body, the ECM provides structural and adhesive support for cells, as well as bioactive physicochemical signaling to modulate cellular behavior.11–16 Since the ECM is the natural way in which our bodies control cellular behavior, and hydrogels share many similarities to ECM, it is perhaps obvious why hydrogels have garnered such significant interest as the biomaterial of choice for tissue engineering and regenerative medicine.
Physicochemical properties, such as mechanical strength, porosity, swelling behavior, biodegradability, bioactivity, and stimuli-responsiveness play a critical role in determining the functionality and suitability of hydrogels for various tissue engineering and regenerative medicine applications. These properties may be tuned in hydrogels by altering how the hydrogel is made, such as crosslinking strategies and manufacturing methods, as well as through incorporation of bioactive molecules and nanomaterials. However, fabricating hydrogels that truly mimic the physicochemical properties of ECM to physiologically-relevant values is easier said than done. It is also important to consider that the requirements for different tissues and organs are different. For example, bone is a stiff tissue with a compact region that is highly organized and predominantly calcified to withstand high loads. On the other hand, skin has a similar layered structure, yet is significantly softer and more viscoelastic to allow for stretching and bending.
When considering how to make hydrogels that mimic human tissues, the first thought that many researchers have is to change the material used to make the hydrogel.17 Hydrogels are fabricated from polymers, commonly proteins,18,19 polysaccharides,19,20 or synthetic polymeric molecules.1,3 Naturally derived proteins and polysaccharides form hydrogels with the required bioactive signaling, yet they are mechanically weak, with moduli significantly lower than native tissues.2,5,21 While synthetic polymers are capable of achieving the physical properties of native tissues, they lack integrin binding sites and bioactive signaling.2,5 One recent trend is to form composite hydrogels from both naturally-derived and synthetic polymers in order to obtain the beneficial properties of both.22,23 However, as naturally derived polymers and synthetic polymers typically form hydrogels using different gelation mechanisms, combining these requires advanced methodologies.
Thus, current research is focused on the design of hydrogels using advanced materials science and engineering approaches to achieve tissue-mimetic physicochemical properties24 (Fig. 1). For example, the formation of stimuli-responsive hydrogels can make hydrogels that release bioactive molecules in the presence of specific triggers, resulting in hydrogels with biochemical properties mimetic of the ECM. In addition, bioconjugation strategies can also covalently adhere bioactive molecules to the polymeric backbone. Similarly, the formation of nanocomposites, which are hydrogels laden with nanomaterials, allows for the sustained, localized delivery of bioactive molecules within the hydrogel, as well as improvement of physical properties such as pore size and matrix stiffness and viscosity depending on the nanomaterial type and incorporation method.
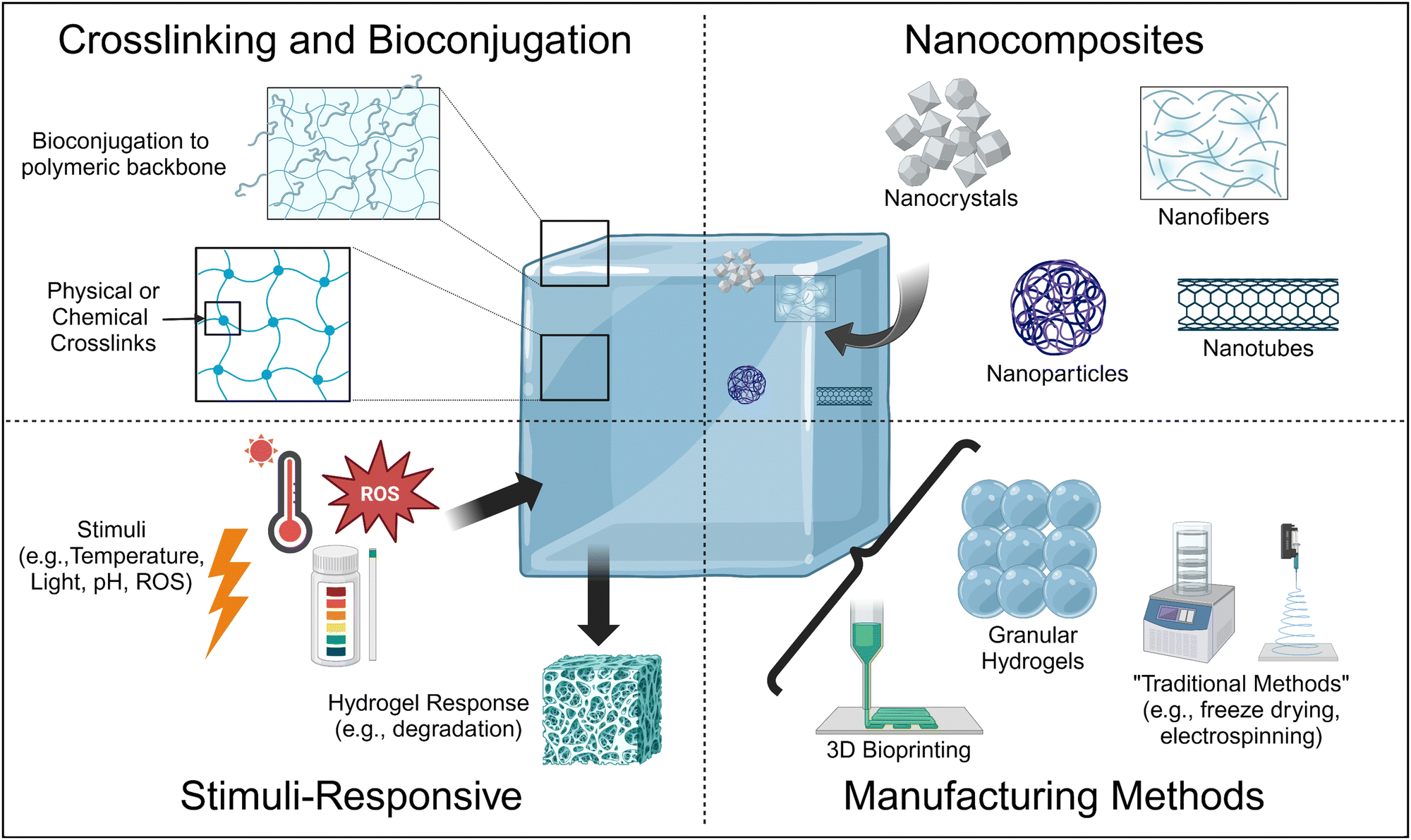 | ||
| Fig. 1 Schematic depicting the various advanced materials science and engineering methods currently used in the field to enhance the physicochemical properties of hydrogels to tissue-mimetic values for tissue engineering and regenerative medicine applications. Created using BioRender. | ||
In this review, we will discuss some of the most promising, upcoming methods by which researchers are fabricating hydrogels with physiologically-relevant properties, with an emphasis on improving the physical properties of hydrogels, for tissue engineering and regenerative medicine applications, with an emphasis on literature from the last five years. We focus on these upcoming methods as we are primarily interested in understanding what engineering advances are needed in order to achieve physiologically relevant properties and tunability such that these physiologically relevant physicochemical properties can be obtained independent of the tissue system mimic. For the studies detailed, we will relate their methods and results to understanding how the method utilized in the study improved a specific physicochemical property toward physiologically-relevant values for the desired target tissue/organ.
2. Crosslinking
In this section, we discuss the effects of crosslinking the polymeric matrix on enhancing the physical and chemical properties of hydrogels towards tissue-relevant values. There are known relationships between crosslinking of a polymeric network and changes to hydrogel matrix stiffness (compressive modulus), viscoelasticity (rheology), and pore size. Here, we define crosslinking as the connection between two or more polymer strands to form a 3D polymeric matrix that forms the backbone of the hydrogel; a hydrogel is a water-swollen, crosslinked polymeric network, wherein the crosslinks provide structure to the polymeric backbone.25A key aspect of tissue engineering and regenerative medicine is the synthesis of hydrogels that possess low viscosity prior to injection and quickly solidify within the physiological tissue environment where they are needed. This process is primarily achieved through crosslinking, which facilitates the sol–gel transition and gel formation.26 Injectable hydrogels can be crosslinked either in vitro during preparation or in situ after injection. In addition, these crosslinks can be reversible or irreversible, which affects their biodegradability, as well as their mechanical properties.25 This section briefly explores current advances in physical and chemical crosslinking strategies before exploring the rapidly growing field of dual-network crosslinking. Physical, thermal, and ionic crosslinking are forms of reversible (non-covalent) crosslinking mechanisms, and chemical crosslinking is irreversible (covalent). A dual network crosslinked network is one that uses one or more types of crosslinking to form the hydrogel.
2.1 Physical, thermal, and ionic crosslinking
Physical crosslinking is triggered by physical stimuli including temperature27 and pH.28 This approach offers several advantages, such as simplicity and the absence of exogenous crosslinking agents that can result in cytotoxicity. Hydrogels crosslinked due to a response to changes in temperature and pH have attracted significant interest in the literature due to their soft physical properties, biocompatibility, bioactive potential, and biodegradability.29 These properties have been widely studied in tissue engineering using predominantly natural polymers. However, these methods are susceptible to potential variations in the encapsulation and release of bioactive small molecules, variability in water content management, and variability in temperature and pH sensitivity. Another method used to enhance the physicochemical properties of hydrogels for tissue engineering and regenerative medicine is through the use of ionic crosslinking agents, which is known to improve the mechanical performance of hydrogels compared to temperature and pH-crosslinked hydrogels.30 However, the use of ionic crosslinking is affected by complex synthesis procedures, primarily if the polymers are not inherently ionic, and it can become difficult to carefully control the swelling ratio and other physicochemical properties as a result of ion leaching. Additional limitations to physically-crosslinked hydrogels are the overall poor mechanical strength that comes with the predominant use of naturally derived polymers, as well as fast biodegradation.Despite their limitations, there has been some success in this area. For example, Pan et al. conducted a study to enhance the physical properties of chitosan (CS) hydrogels by adjusting the water content.31 The hydrogel was fabricated by temperature-induced crosslinking of the polymeric backbone and water-swollen to approximately 94%. Subsequent dehydration produced a xerogel (XG) with about 3% water, whereas rehydration resulted in a water content of about 56%. The authors found that these modifications significantly affected the hydrogen bonding and molecular structure of the hydrogel, leading to a wide range of compressive strengths (0.312–139 MPa) and moduli (0.2408–1094 MPa). In addition, the authors noted that MC3T3-E1 cells cultured in the hydrogel and xerogel exhibited excellent cell adhesion, viability, and proliferation, with viability rates reaching 90.7% for the hydrogel and 99.3% for the xerogel by day five. Therefore, by simply combining the temperature-sensitive, physical crosslinking approach with the dehydration/rehydration steps, the authors were able to demonstrate significant tunability in their system and achieve compressive moduli (matrix stiffness) that covers a wide array of tissue-relevant values.
However, despite this, significant variability in hydrogel properties and complex synthesis processes in order to attempt to alleviate the limitations of physical crosslinking yield hydrogels that are unlikely to be clinically effective. For these reasons, chemical crosslinking remains the gold standard.
2.2 Chemical crosslinking
Despite being the ‘gold standard’ for hydrogel synthesis in tissue engineering and regenerative medicine applications, traditional chemically crosslinked hydrogel approaches may have lower biocompatibility because of the potential toxicity of residual polymerization initiators and organic solvents.32 This requires investigation in order to determine the optimal crosslinking moieties that yield favorable physicochemical properties without significant toxicity. To address this limitation, researchers have investigated several chemical crosslinking approaches that have been found to be more cytocompatible, including photo-polymerization,33 enzyme-induced crosslinking,34 and bioorthogonal click chemistry,35 such as the Diels–Alder reaction,36 Schiff base formation,37 and Michael type-addition.38Photo-crosslinking is of interest because it initiates chemical reactions in polymer chains by subjecting them to light with or without the use of photoinitiators.39 Photoinitiators can absorb photons and subsequently transform light energy into chemical energy,40 and the generated free radicals are then covalently bound to intra- or extra-molecular groups. Among the various photoresponsive moieties employed in biomaterials; tyrosine,41 tyramine,42 methacryloyl,43 cinnamoyl,44 benzophenone,45 norbornene,46 and aryl azide,47 are most utilized. Other researchers have sought to improve chemical crosslinking via the use of enzymatic crosslinking, which has demonstrated significant potential in the fabrication of injectable hydrogels. A variety of enzymes, including tyrosinase, transglutaminase, phosphates, thermolysin, oxidases, and peroxidases, are used as catalysts in this process.48 Tyramine is particularly of use due to its simplicity in functionalizing it through the amidation of various polymers, both synthetic and natural, including collagen,49 polyethylene glycol (PEG),50 and hyaluronic acid (HA).51Table 1 presents some examples of recent literature using photo-crosslinking and enzymatic crosslinking approaches, listing their key findings (i.e., which physicochemical property was affected the most and its relation to the target tissue/organ), the role of the crosslinking, and limitations with the method.
| Crosslinking Mechanism | Description | Targeted organ/tissue | Key findings | Role of crosslinking | Limitations | Ref. |
|---|---|---|---|---|---|---|
| NHS, CHO, methacrylate | Col type I and HA composites | Cartilage | – 2.5–4 min gelation time | – Methacrylate crosslinking enhanced mechanical properties of hydrogels | – Causes toxicity due to residual polymerization initiator/organic solvent | 52 |
| – Storage modulus of methacrylate ∼7 kPa, NHS & CHO 5 kPa at 2 Hz | – Improved chondrogenesis, cell viability | |||||
| Irgacure 2959, LAP along UV light | Gelatin methacrylate hydrogels | Vascular construct | – Cell viability decreased with high photoinitiator concentration (0.7–0.9% w/v) | – Fast crosslinking for structural stability | – Higher photo-initiator concentrations affect the viability of cells | 53 |
| – Good cell viability maintained by low concentrations (0.3–0.5% w/v) | – Enhanced mechanical properties | – Irgacure 2959 caused large pore size (up to 50 um), resulting in fast degradation | ||||
| – LAP maintained high cell viability (at 0.7% and 0.9% w/v) at higher concentrations | Provides tunable physical properties | – optimization is required for crosslinking and cell viability | ||||
| Enzymatic crosslinking by HRP | Col/HA-Tyr hydrogels crosslinked using HRP | Skin, cartilage, intervertebral disc | – Higher HA-Tyr & HRP content formed microfibrillar network along synergistic mechanical properties | – Provides tunable compositions and gelation conditions | – Requires precise control of HRP concentration | 49 |
| – High resistance against enzymatic degradation and high cell viability | – Improved physical properties and thermal stability | – High HA content is required to maintain hydration | ||||
| Enzymatic (horseradish peroxidase + H2O2 crosslinking | HA-TA using HRP and H2O2, 8-arm PEG hydrogels- | Cartilage, soft tissue | – Gelation time within secs under physiological conditions | – Provides fast gelation | – Precise control of enzyme and polymer concentration is required | 51 |
| – Higher gel content >95% | – Improved biocompatibility | – Potential variability in degradation time based on composition | ||||
| – Storage moduli up to 22.4 kPa | – Supports cell viability | – Potential cytotoxicity of crosslinking byproducts | ||||
| – Degradation time increased due to PEG up to several weeks | ||||||
| – Encapsulated human chondrocytes produced more than 10-fold type II collagen than control hydrogels | ||||||
| Diels–Alder reaction; HA/PEG hydrogels | Injectable hydrogel for intraarticular delivery of MSC-sEVs | Osteoarthritis (Joints) | – Sustained release of MSC-derived small extracellular vesicles by degradation control | – Provides gelation within 1800 s per injectability | – Precise control of crosslinking reaction conditions | 54 |
| – Preserves therapeutic function of MSCs-sEVs | – Regulates biodegradability and release kinetics | |||||
| – Enhanced bioavailability | ||||||
| Schiff base reaction; adipic dihydrazide (ADH)-modified poly(L-glutamic acid) (PLGA-ADH) and benzaldehyde-terminated poly(ethylene glycol) (PEG-CHO) | PLGA/PEG hydrogels | Cartilage | – Swelling ratio decreased four-fold from 5–11% mass fraction and increased two-fold with 0.2–0.8 molar ratio | – Regulates biocompatibility and biodegradability | – Immune response and integration with host tissue need thorough evaluation in long-term studies to ensure no adverse effects | 55 |
| – 5% mass fraction resulted in complete degradation in 2 weeks while 7–11% mass fraction caused <20% degradation after 5 weeks | – Promoted chondrogenesis | |||||
| – Type II Col expression increased | ||||||
| Schiff base reaction; Dialdehyde-modified HA (AHA), Cystamine dihydrochloride 9Cys) | Pure HA hydrogels | Tissue engineering, drug delivery, bioprinting | – Gelation between dialdehyde groups on AHA and amino groups on Cys | – Fast gelation due to Schiff base reaction | – Potential susceptibility to mechanical disruption under extreme conditions | 56 |
| – Mechanical properties improved due to an increase in Cys content | – Provides dynamic and reversible bonds for self healing | |||||
| Michael addition reaction; Bisphosphonate-functionalized PAA (PAA-BP) | Hydroxyethyl methacrylate (HEMA) hydrogel | Bone tissue engineering | – Strong hydrogel with an elastic modulus of 83 kPa | – Adds excellent mineralization ability | – Achieving consistent mechanical properties is challenging due to variability in mineralization | 57 |
| – pH responsiveness | – Facilitates cell adhesion after mineralization | – pH sensitivity may pose a challenge for applications where pH conditions are variable/hard to control | ||||
| Michael type reaction; 4-arm acrylate-PEG, HA-SH, GSE4-SH | HA/PEG hydrogel | Lung cells | – Promotes differentiation of BMSCs into alveolar epithelial type 2 cells (AEC2) with a differentiation rate of 58.6% | Facilitates in situ crosslinking under physiological conditions at pH 7.8 | – Depends on specific pH and reaction condition for effective crosslinking | 58 |
| – Forms 3D porous network structure | ||||||
Perhaps the most rapidly growing chemical crosslinking approach for hydrogels with applications in tissue engineering and regenerative medicine, however, is bioorthogonal click chemistry,35 recent examples of which are also detailed in Table 1. Bioorthogonal click chemistry reactions have several benefits that make them of particular interest to these applications, including substantial yields at moderate, cell culture- and physiologically-relevant conditions, low to no cytotoxic byproduct formation, and great selectivity and specificity.
For example, Li et al., developed an injectable hydrogel for cartilage tissue engineering using a Schiff base reaction between adipic dihydrazide-modified poly (L-glutamic acid) and benzaldehyde-terminated PEG (Fig. 2A).55 Utilization of the Schiff base reaction yielded a biocompatible, self-healing hydrogel with tunable mechanical properties. The authors noted that high mass fractions lead to a an approximately four-fold decreased swelling ratio from a mass fraction of 5% to a mass fraction of 11%, while increasing the Molar ratio between the crosslinking moieties from 0.2 to 0.8 enhanced the swelling ratio approximately two-fold (Fig. 2B). Swelling is an inherently important consideration in cartilage tissue engineering. Swelling can reduce compressive strength of the hydrogel, but can also facilitate the delivery of nutrients via diffusion. Cartilage, particularly articular cartilage, requires high compressive strengths, so significant swelling may not be favorable. However, as an avascular tissue, the increased diffusion of nutrients would support cell survival.
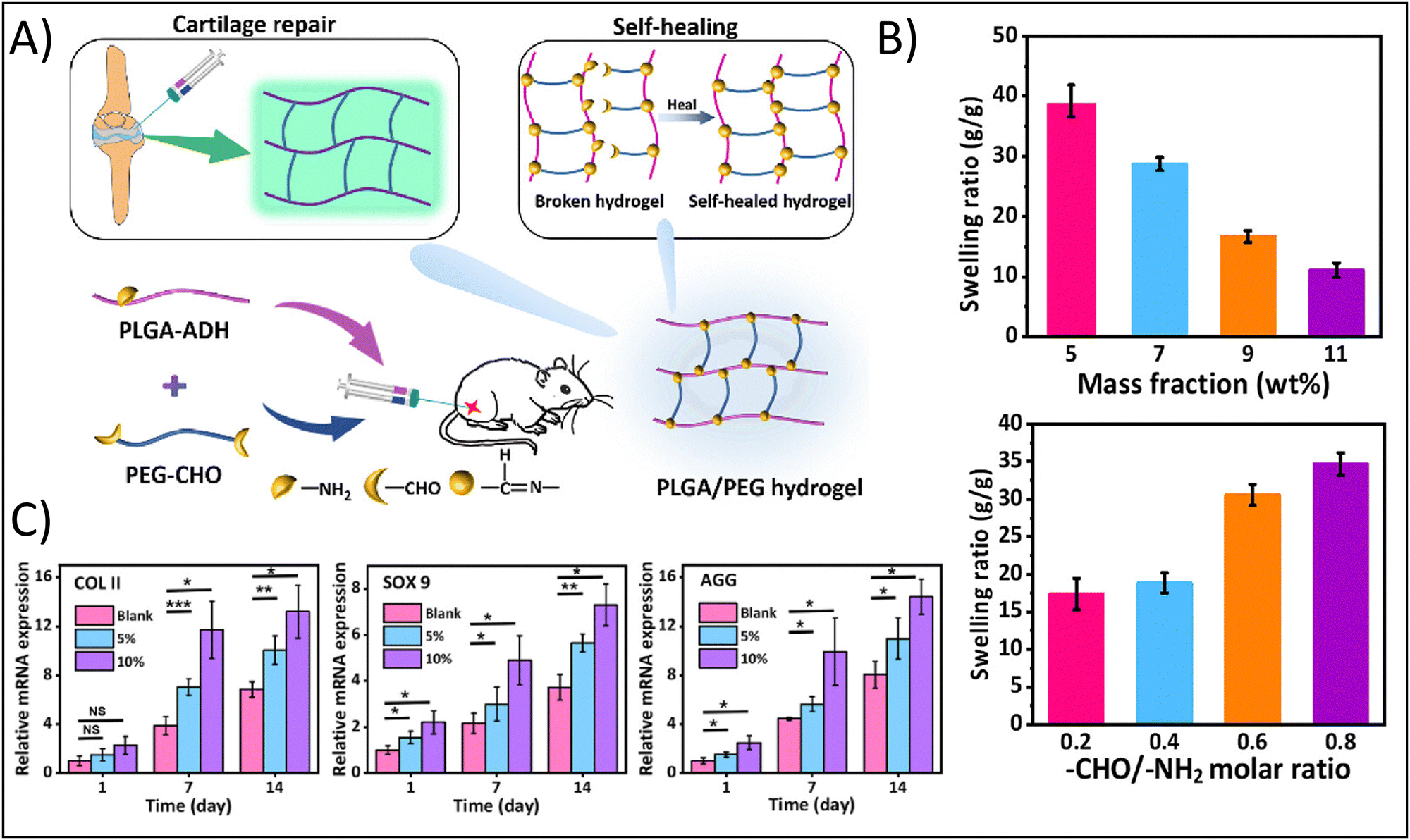 | ||
| Fig. 2 Enhancing physicochemical properties of hydrogels through chemical crosslinking approaches. (A) Fabrication of the self-healing poly(L-glutamic acid) (PLGA)-based hydrogel demonstrating functionalization of PLGA with aldehyde (ADH) and functionalization of poly(ethylene glycol) (PEG) with dialedehyde (CHO). (B) Swelling ratio as a function of mass fraction and Molar ratio between functional groups. (C) Relative gene expression of the chondrogenic factors, type II collagen (COLII), SOX9, and aggrecan (AGG). Adapted with permission from ref. 55 copyright 2023 American Chemical Society. | ||
The authors further demonstrated how mass fraction affected hydrogel degradation: increasing mass fraction from 5% to 7–11% resulting in a change from complete degradation in 2 weeks to less than 20% degradation after 5 weeks, showing that degradation can be tuned to align with typical cellular remodeling timelines. In addition, hydrogels with a 5% and 10% gel concentration were found to support MSC proliferation and the expression of cartilage-specific genes. The authors noted a nearly two-fold increased expression after 14 days for type II collagen in 10% gels compared to blanks and a nearly 1.5-fold increase in type II collagen expression for 5% gels compared to blanks; similar results were obtained for SOX9 and aggrecan expression (Fig. 2C).
2.3 Dual network crosslinking
To exploit the benefits of both chemical and physical crosslinking methods, hybrid hydrogels that include two or more precursors are made using a dual stage crosslinking approach. By combining different physical and chemical crosslinking strategies together, researchers are able to tune the specific properties that make the most sense for their target tissue/organ. In the examples presented below, the studies target bone tissue engineering; given the high mechanical strength of bone, it is very difficult to mimic the properties of bone using traditional hydrogel crosslinking strategies. However, all of the studies demonstrate strong potential of their hydrogel for bone tissue engineering, showing the potential of dual crosslinking strategies. Min et al. fabricated a composite hydrogel consisting of glycol CS with amino-functionalized bioactive glass nanoparticles, which were crosslinked dually with genipin and PEG diglycidyl ether towards applications in bone tissue engineering.59 The bioactive glass nanoparticles sought to mimic the biochemical properties of bone, while also seeking to provide additional compressive strength from the ceramic material and the dual crosslinking approach. The authors showed that the dual crosslinked hydrogels had a storage modulus of 6.9 kPa and yielding strains of over 60%, in comparison to around 3 kPa for the counterpart single crosslinked hydrogel, demonstrating the increased durability and elasticity that results from the dual crosslinking approach. This result is particularly important when considering that the tissue of interest is bone, which is highly elastic and has very high yielding strains and compressive moduli. Additionally, these hydrogels supported the growth of MC3T3-E1 cells, resulting in an approximately 1.3-fold increase in type I collagen (predominant in bone ECM) production after 14 days in comparison to counterpart single-crosslinked hydrogels.In a different study, Ghorbani et al. developed a dual crosslinked hydrogel composed of alginate (Alg), oxidized alginate (OAlg), and silk fibroin (SF) towards bone tissue engineering applications with the goal of overcoming the known limitations of low mechanical strength and rapid degradation associated with traditional Alg-based hydrogels.60 Briefly, the authors utilized a combination of ionic gelation through the addition of CaCO3-GDL for physical crosslinking of the polymeric matrix and a Schiff-base reaction for covalent chemical crosslinking of the polymeric matrix (Fig. 3A). Similar to the use of the bioactive glass in the previous example, the calcium carbonate would provide biochemical mimicry, while the covalent Schiff-base reaction would improve the biophysical mimicry. Using this approach, the authors demonstrated that the hydrogels had a significant increase in compressive modulus from 28 kPa for Alg alone to 67 kPa for Alg/OAlg/SF (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:8 ratio); the authors attributed this increase in compressive modulus to the covalent imine bond formed using the Schiff base reaction (Fig. 3B). Although 67 kPa is still quite low compared to native bone, the 2.3-fold increase in compressive modulus was able to yield biological results. The dual crosslinked hydrogels had an approximately 10-fold enhanced alkaline phosphatase expression, an approximately 1.75-fold increased OCN expression, an approximately 1.75-fold increased RUNX expression, and an approximately 2.5-fold increased OXN expression compared to control hydrogels after 14 days, demonstrating the osteogenic capabilities of these hydrogels (Fig. 3C).
:1:8 ratio); the authors attributed this increase in compressive modulus to the covalent imine bond formed using the Schiff base reaction (Fig. 3B). Although 67 kPa is still quite low compared to native bone, the 2.3-fold increase in compressive modulus was able to yield biological results. The dual crosslinked hydrogels had an approximately 10-fold enhanced alkaline phosphatase expression, an approximately 1.75-fold increased OCN expression, an approximately 1.75-fold increased RUNX expression, and an approximately 2.5-fold increased OXN expression compared to control hydrogels after 14 days, demonstrating the osteogenic capabilities of these hydrogels (Fig. 3C).
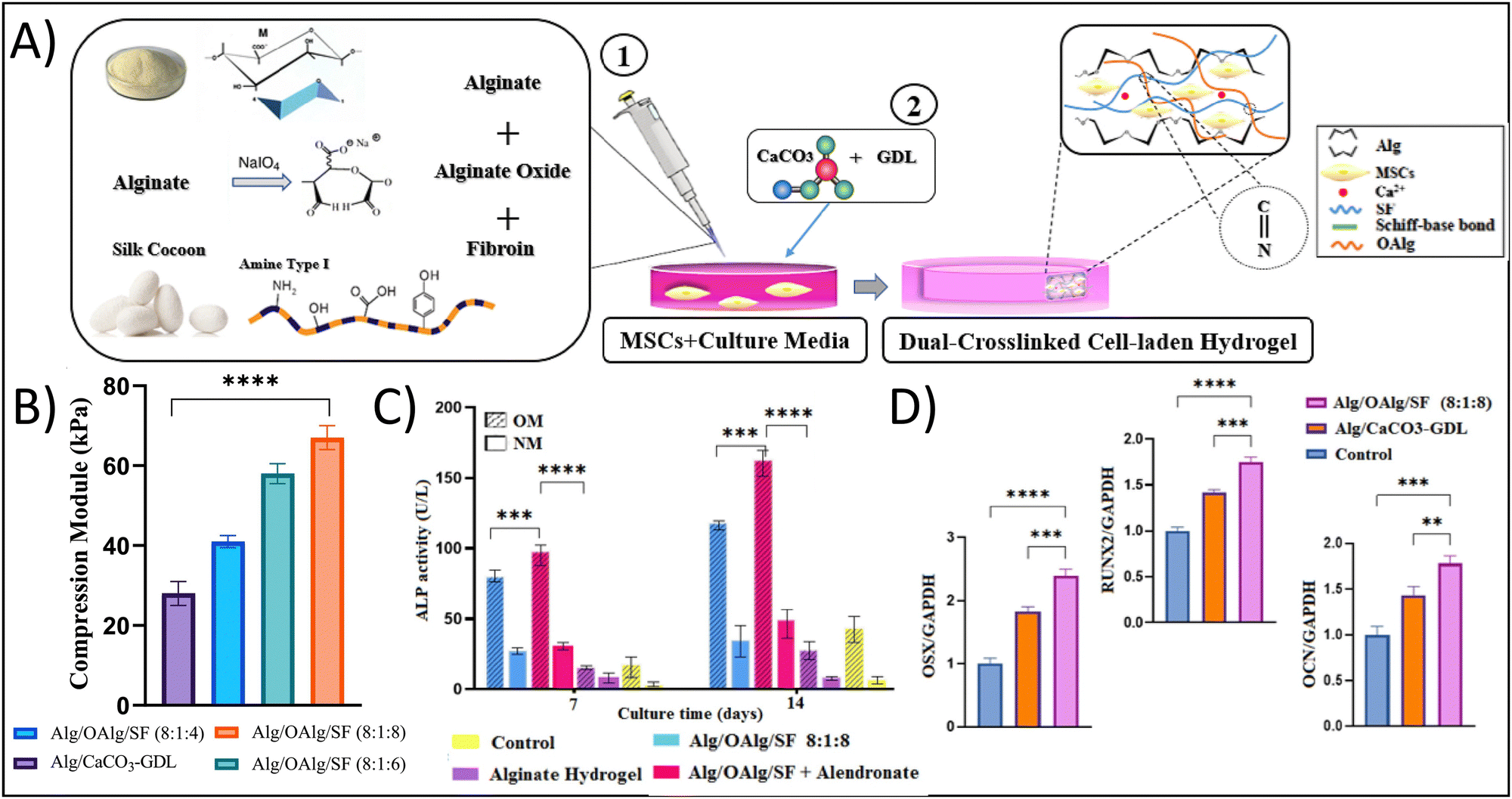 | ||
| Fig. 3 Enhancing physicochemical properties of hydrogels through dual network crosslinking approaches – using physical, ionic crosslinking combined with biorthogonal click, Schiff-base reactions. (A) Schematic depicting the fabrication of the dual network crosslinked hydrogel showing a Schiff-base reaction between oxidized alginate and fibroin. Sodium alginate and calcium carbonate are added for ionic crosslinking. (B) Compressive modulus of the different hydrogel formulations; abbreviations: alginate (alg), oxidized alginate (OAlg), silk fibroin (SF). (C) Alkaline phosphatase (ALP) activity and (D) osteogenic gene expression of cells cultured in the various hydrogels. Adapted with permission from ref. 60 from Elsevier. | ||
Finally, He et al. approached bone tissue engineering using dual crosslinking by fabricating a hydrogel composed of sulfated methacrylated HA, alendronate, and Ca2+ and Mg2+ ions.61 Briefly, a sequential crosslinking mechanism was utilized; first, physical, ionic crosslinking was initiated through the presence of the ion-induced nanoclusters. The soft, relatively amorphous properties of the hydrogel due to the physical crosslinking allowed for a hydrogel that could fit to irregular bone defects. Following this, the hydrogel was photocrosslinked using UV light to increase the mechanical properties of the hydrogel towards bone-relevant values. The authors noted an elastic modulus of between approximately 27 kPa and 96 kPa depending on the concentration of ions present, with increasing ion concentration increasing moduli. These moduli values were found to be sufficient for craniofacial applications, where the compressive strength of bone is not quite as high. Indeed, the dual crosslinked hydrogel demonstrated increased bone regeneration in a rat skull defect model due to the added effects of the ions, which resulted in approximately two-fold enhanced angiogenesis, as a function of vascular endothelial factor (VEGF) production, for hydrogels with the greatest ion concentration compared to control hydrogels without ions.
3. Manufacturing approaches
The method by which the hydrogel is manufactured can have significant effects on tuning the physical and chemical properties of a hydrogel. In this section, we describe the use of “traditional” manufacturing approaches as well as “advanced” manufacturing approaches in the fabrication of hydrogels with tissue-mimetic physicochemical properties.3.1 “Traditional” methods
Many current studies in the literature still employ “traditional” hydrogel manufacturing methods. Among the most common are freeze-drying,62 salt leaching,63,64 electrospinning,65 and gas foaming.66 Freeze-drying is a widely utilized technique for fabricating porous, biocompatible hydrogels for tissue engineering applications.1 Briefly, freeze-drying entails the formation of ice crystals within the hydrogel precursor solution followed by the sublimation of ice into vapor, which results in the formation of pores in a dried polymeric scaffold. The dried scaffold can then be rehydrated to achieve a hydrogel wherein the size of the pores is directly proportional to cell attachment, viability, and migration. However, freeze-drying is incapable of mimicking the unique micro-nano fibrous characteristics of ECM and the applications can be highly restricted.Salt leaching is particularly useful for the introduction of microporosity.64 Briefly, hydrogel precursor solutions are mixed with salt particles of different sizes, which act as porogens during gelation. After gelation, the salt can be dissolved away, leaving behind pores that are influenced by the size of the salt particles. However, it is difficult to obtain consistent pore sizes, and salt leaching (or insufficient salt leaching), as well as the inconsistency of the pore sizes, can result in changes to the physical properties of the hydrogel. Salt leaching is also not capable of mimicking the unique micro-nano fibrous characteristics of ECM.
To address the limitations of not mimicking the micro-nano fibrous characteristics of ECM, another common, traditional manufacturing approach utilized is electrospinning.65 A variety of naturally-derived and synthetic polymers are conducive to electrospinning, such as collagen, gelatin, SF, polycaprolactone (PCL), poly(lactic-co-glycolic acid), and polyurethane, with a broad range of applications, including the regeneration of skin, skeletal, and cardiac tissues. However, electrospinning is only capable of moderate elasticity, making it difficult to use in applications like bone tissue engineering.
A final commonly used traditional manufacturing method is gas foaming.66 Briefly, gas foaming is a process by which polymeric foam solutions are saturated with inert gas at high pressure. As the gas dissolves, pores are formed. However, limitations including long-term cell viability and proliferation due to the potential cytotoxicity of the gas, exist.
Unfortunately, many of the “traditional” methods for hydrogel manufacturing rely on a “top-down” approach, wherein the hydrogel scaffold is made in its entirety without cells, then cells are seeded on top of the hydrogel and cultured to hopefully migrate through the pores of the hydrogel. However, rather than a homogenous distribution of cells or a specifically controlled localization of cells throughout the hydrogel, this often leads to a high concentration of cells on the hydrogel surface with minimal and heterogeneous migration and distribution. Therefore, “bottom-up” approaches such as 3D bioprinting and granular hydrogels are making their way as the preferred methods for hydrogel manufacturing.
3.2 3D bioprinting
3D bioprinting has the potential to mimic a wide array of physicochemical properties of native ECM. For example, 3D bioprinting can mimic biochemical cues by including bioactive molecules in the bioink or using inherently bioactive, naturally-derived polymers, it can mimic the compressive and tensile moduli, as well as the viscoelasticity, of native tissues by tuning polymer concentration and crosslinking density, and crosslinking density can also affect porosity, which affects the migration of cells and delivery of encapsulated molecules. 3D bioprinting achieves this through the precise placement of biomaterials, bioactive molecules, and living cells to create complex biological structures mimicking the structure of human tissues and organs. It is particularly important to highlight the ability of 3D bioprinting to mimic highly complex tissue structure as this is perhaps what makes 3D bioprinting stand apart from traditional methods; as a result, it has been hailed in the field as having the potential to revolutionize regenerative medicine and tissue engineering.1,67Table 2 highlights the different 3D bioprinting techniques that may be used in the fabrication of 3D bioprinted hydrogels.| Bioprinting techniques | Descriptions | Organ/Tissue | Printability | Challenges | Ref. |
|---|---|---|---|---|---|
| Inkjet based | Structures are created by depositing bioink droplets via the use of methods i.e. Continuous inkjet (CI), Drop-on-demand (DOD) | Skin, cartilage, bone | – <30 μm high resolution | – Risk of nozzle clogging | 68–70 |
| – Higher printing speed | – Precise droplet positioning | – Heat may cause an impact on cell viability | |||
| – Less expensive | |||||
| Extrusion based | Employs pressure to dispense bioink from a syringe, considered an ideal for highly viscous material | Bone, cartilage, skin | – Suitable for high-viscosity bio-ink | – High pressure can reduce cell viability | 71 |
| – Simple, most used technique | – Frequent nozzle clogging | ||||
| Laser-assisted | Laser-induced forward transfer is utilized for higher precision of cells/biomaterial printing | Blood vessels, nerves | – High resolution | – Can cause potential cell damage from laser interaction | 69,70 |
| – Handle different viscosities | – High cost | ||||
| Vat photopolymerization | Photo initiators are utilized for crosslinking to create high resolution tissue construct | Various tissue i.e. skin, cartilage | – Enhanced printing resolution for detailed structures | – Limited selection of biocompatible materials | 72 |
| – Single bio-resin usage | |||||
In the fabrication of 3D bioprinted hydrogels, bioinks are the essential precursor solutions that serve as the building blocks; these may be formulated using a variety of polymeric solutions mixed with or without the bioactive molecules and cells or cell spheroids, and the selection of materials for bioinks involves several factors including swellability, stability, printability, and biocompatibility.73,74 Therefore, based on the components of the bioink, 3D bioprinted hydrogels may have bioactive properties as well as mimic the physical cues of native tissue through the complex, 3D architecture and enhanced mechanical strength. For tissue engineering and regenerative medicine applications, bioinks may be fabricated from a variety of naturally-derived or synthetic polymers. Common polymers are Alg,75,76 gelatin,77,78 agarose,79 CS,80 collagen,81 PEG,82 PEGDMA,83 and polyurethane (PU).84 To achieve effective biomimicry of the tissue/organ of interest, Table 3 provides a comprehensive summary of 3D bioprinting applications for tissue engineering and regenerative medicine, detailing the specific bioink used, the printing techniques employed, and the outcomes achieved for different tissues and organs.
| Tissue/organ | Bioinks | Printing techniques | Printability | Challenges | Ref. |
|---|---|---|---|---|---|
| Articular cartilage | MeHA Col-I, Col-I/Col-II, Col-II | 3D printing with gelatin slurry | – Methacrylation enhances the mechanical properties of materials, | – Poor physiochemical properties of HA and Col II | 85 |
| – Col I hydrogels exhibit stability during 3DP, | – Difficult to incorporate into injectable 3DP hydrogels | ||||
| – Col II enhances MSCs chondrogenesis | |||||
| Nanocellulose fibrils (NFCs), nanocellulose crystals (NCCs), and nanocellulose blend (NCBs) with Alg (75:25 v/v) |
3D bioprinting | – NFCs: superior print resolution | – The structural differences in nano celluloses affecting the printability, chondrogenesis, and biocompatibility | 86 | |
| – NCBs: best port printing shape fidelity | |||||
| – NCCs: highest chondrogenicity | |||||
| Bone | Chitosan, nano-hydroxyapatite, sodium alendronate, hydroxyethyl-cellulose | 3D bioprinting | – Hydroxyethyl cellulose enhanced printability; a sustained drug release for 50 days | – Ensuring scaffold biocompatibility and efficient drug delivery | 87 |
| – Crosslinking with KOH/citric acid; coatings of Col & gelatin enhanced biocompatibility | |||||
| 7% GelMA, 3% AlgMA, articular cartilage progenitor cells (ACPCs), BMSCs | Dual channel extrusion bioprinting | – Superior printability at room temperature; maintained high cell viability | Simultaneous cartilage and bone regeneration to achieve | 88 | |
| – Differential spatial regulation for chondro-osteogenesis | |||||
| Skin | GelMA, chitosan NPs, curcumin-loaded NPs | Extrusion based 3D bioprinting | – High swelling ratio (1200%), efficient drug release for 5 days | – Controlled drug release | 89 |
| – Enhanced cell proliferation and mimics ECM | – Optimization of NP concentration for cell behavior | ||||
| – To ensure proper wound healing and tissue regeneration, stability, and functionality in vivo | |||||
| Heart | AlgGel with cardiac spheroids, freely suspended cardiac cells, acellular | Extrusion based 3D bioprinting | – Synthesis of hydrogel-patches preserved cardiac spheroids | – To understand immune response and gene expression mechanisms | 90 |
| – Enhanced cardiac function and maintained cell viability | – Optimization of patch composition for efficient therapy | ||||
| – To ensure proper integration and efficacy in vivo | |||||
| Spinal cord | Dual network hydrogel (HA derivatives, N-cadherin modified sodium Alg), gelatin/cellulose nanofiber hydrogel | Coaxial 3D bioprinting | – Efficient encapsulation, controlled release of bioactive cues | – To ensure scaffold stability | 91 |
| – The inner layer supported endogenous neural stem cell migration/differentiation | – Optimization of mechanical support and biochemical cues | ||||
| – The outer layer protected endogenous neural stem cells via oxidative stress inhibition | |||||
| – Enhanced motor function recovery in spinal cord injury rats | |||||
Perhaps among the most common polymer used in bioinks is gelatin77 due to its biocompatibility, affordable cost,92 and ability to be combined with other polymers including SF,93 methylacrylate,94 CS,95 and Alg.96 For example, Schneider et al. fabricated a 3D bioprinted hydrogel for soft tissue engineering applications composed of SF, tyramine-substituted gelatin, and human placenta chorion ECM.97 The authors demonstrated that the bioink yielded hydrogels with good mechanical properties mimetic of soft tissues, with compressive moduli at day 7 ranging from 0.5 kPa for SF hydrogels only to 2.5 kPa for hydrogels with equal concentrations of SF and gelatin to 1.8 kPa for hydrogels with 2% SF, 1.5% gelatin, and 0.5% placenta. Increasing concentration of the placenta ECM to 1% and decreasing gelatin to 1% decreased the compressive modulus to 1.3 kPa. The compressive moduli of the hydrogels was also found to remain consistent up to 28 days of culture, demonstrating that culture conditions did not affect the hydrogels and that the effects of mimicking the mechanical properties of soft tissues would remain constant. In addition, the 3D bioprinted hydrogel supported cell growth, with the hydrogel containing 2% SF, 1% gelatin, and 1% placenta ECM having a 10-fold increase in metabolic activity compared to SF hydrogels only at day 7.
As noted previously, the combination of naturally-derived polymers such as gelatin with synthetic polymers including polyvinyl alcohol (PVA),98 PCL,99 and polylactic acid (PLA)100 has the potential to fabricate hydrogels exhibiting the bioactive properties of the naturally-derived polymer, as well as the mechanical properties of the synthetic polymer. This is the strategy employed by Jian et al., who utilized 3D bioprinting to fabricate a hydrogel composed of PCL and gelatin methacrylate (GelMA) encapsulated with meniscal fibrocartilage chondrocytes (Fig. 4A) towards cartilage tissue engineering.99 To form the bioink, the authors used decellularized meniscus tissue, which gave the bioink the necessary biochemical mimicry. The use of PCL, on the other hand, provided the mechanical integrity and mimicry that the authors would not have achieved with a naturally-derived polymer alone. In addition, the authors printed the meniscal scaffold by modeling the native structure of meniscus, further providing structurally signaling to the cells. As a result of these efforts, the authors showed that GelMA with PCL upregulated Col I and Col II expression 1.25-fold and two-fold, respectively, compared to GelMA alone, but SOX9 expression was unchanged, indicating the chondrogenic potential of the 3D bioprinted hydrogel (Fig. 4B). Further, cells encapsulated in the composite hydrogel were visually elongated, with dendritic shapes and mesh-like connections, mimicking native meniscal cells.
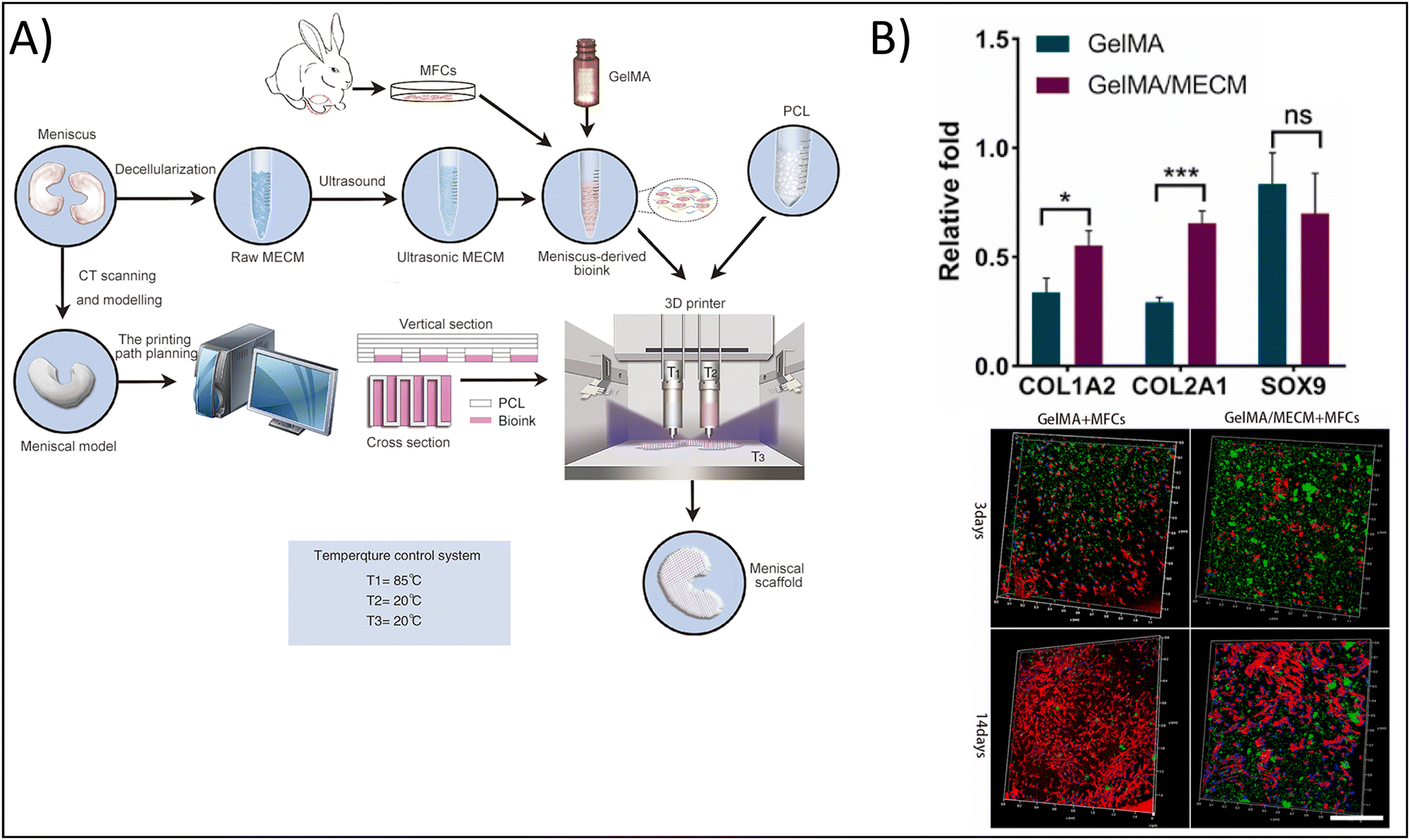 | ||
| Fig. 4 Enhancing physicochemical properties of hydrogels via 3D bioprinting. (A) 3D bioprinted gelatin methacrylate (GelMA) and meniscus-derived extracellular matrix (ECM) hydrogel. (B) Chondrogenic gene expression for control GelMA hydrogels vs. composite hydrogels demonstrating enhanced chondrogenic potential in the composite hydrogel (top). Type I collagen staining (green), nuclei (blue), and actin (red) for cells encapsulated in control and composite hydrogels after 3 and 14 days, scale = 500 μm (bottom). Adapted from ref. 99 with permission by the authors. | ||
In another study, Kamaraj et al. fabricated a 3D bioprinted sodium Alg and PEG hydrogel modified with acrylate and methacrylate moieties to support chemical crosslinking of the polymeric backbone.101 This crosslinking approach combined with 3D bioprinting yielded hydrogels with enhanced mechanical properties towards mimicry of bone tissue. They demonstrated that PEG diacrylate and sodium Alg hydrogels had elastic moduli of approximately 0.45 MPa compared to 0.2 MPa for PEG methacrylate alone, thereby increasing the elastic modulus closer to that of bone. The authors do note that the slight increase in elastic modulus, which still remains lower than that of native bone, was not sufficient alone to have a biological effect, showing that PEG methacrylate with sodium Alg hydrogels had slightly increased alkaline phosphatase in comparison to PEG diacrylate with sodium Alg hydrogels when cultured in the absence of osteogenic induction medium. However, when combined with biochemical mimicry through the addition of osteogenic induction medium, this result was reversed, with PEG diacrylate and sodium Alg hydrogels having nearly double the alkaline phosphatase expression compared to PEG methacrylate and sodium Alg hydrogels in osteogenic induction medium. In addition, PEG methacrylate with sodium Alg hydrogels had increased alkaline phosphatase gene expression, decreased type I collagen gene expression and increased RUNX2 gene expression compared with PEG diacrylate with sodium Alg hydrogels, a trend that remained consistent regardless of culture with or without osteogenic induction media. It is notable that the authors were able to demonstrate the osteogenic capability of the hydrogels even in the absence of osteoinduction media, demonstrating that the mechanical and inherent bioactive properties of the hydrogel were sufficiently mimetic of bone tissue and that this hydrogel has significant potential for bone tissue engineering applications.
3.3 Granular hydrogels
While 3D bioprinting has been an active area of research for some time now, granular hydrogels are an exciting, new avenue of exploration. Granular hydrogels are hydrogels formed by the dense packing, or jamming, of microgels.102,103 The microgels form the individual units, or building blocks, of the macroscale hydrogel, each with their own intrinsic properties that can provide the overall bulk properties to the granular hydrogel.104,105 These microgels are then packed together in a scaffold, where they stick together typically through the formation of an interstitial crosslinked network surrounding the microgels.102,106 Typically, microfluidic processes are used to fabricate the microgel,102 and the macroscale hydrogel is formed either by casting the microgels in a mold107 or through extrusion bioprinting.102,108,109 This method yields hydrogels with increased porosity and capacity for mass transfer, and it can be applied to a broad range of hydrogel fabrication materials. For example, Mahdieh et al. sought to improve the use of Matrigel for 2- and 3D cell and tissue culture by forming the Matrigel as a granular hydrogel rather than a bulk hydrogel, allowing the Matrigel to not only mimic biochemical ECM cues, but also mimic structural cues.110As a result of this highly innovative method, granular hydrogels have immense potential for achieving tissue-mimetic physicochemical properties as they can be easily tuned to mimic the complex 3D architecture of tissues.103,109 Specifically, it is possible to: (1) tune the mechanical and biochemical properties of the microgel itself by controlling the type, composition, and amount of polymer and bioactive molecules; (2) mix multiple types of microgels together (including those with and without encapsulated cells) in highly specific organizations to impart complex structure; (3) and/or modify the physicochemical properties of the interstitial network separately from the microgels through controlling crosslinking properties. Also, cells can both be encapsulated within the microgels or seeded “top-down”, where they can differentially migrate through the interstitial network and the pores of the microgels.
In addition, the properties of the individual microgel building blocks can be tuned to release bioactive molecules (thereby mimicking chemical cues).111,112 For example, the Segura group developed a new method of nucleic acid delivery based on the post-injection annealing of microparticles into a granular hydrogel containing non-aggregated, hypohilized nucleic acids that increases nucleic acid loading and subsequent delivery.112 By increasing the loading and delivery of the nucleic acids, the biochemical mimicry and bioactive effects of the nucleic acids are improved. In addition, Xu et al. developed a granular HA hydrogel releasing fibromodulin for tendon regeneration,111 and Tigner et al. fabricated PEG granular hydrogels based on bioorthogonal click chemistry for the delivery of neural progenitor cells (as the bioactive ‘molecule’) to spinal cord injuries.113
Granular hydrogels have found significant potential use in musculoskeletal tissue engineering and regenerative medicine applications. For example, chondrogenesis, or chondrogenic differentiation of stem cells, requires spheroidal culture in order to have the physical, cell-to-cell cues necessary to initiate the chondrogenic signaling pathways, and Zhang et al. demonstrated the potential of granular hydrogels to form chondroid spheroids that could easily be adapted for 3D bioprinting applications.108 Keeping in line with cartilage regeneration, Zhu et al. fabricated a photoannealed granular hydrogel comprised of HA, PEG, and gelatin in order to promote cell volume expansion.114 While cell-to-cell contacts is one of the physical cues necessary for chondrogenesis that necessitates spheroidal culture, the other reason is cell volume, allowing the cells to deposit the necessary ECM proteins. Briefly, a microgel precursor solution was fabricated from the mixing of methyl furan-modified HA, methacrylated gelatin, and maleimide-bis-functionalized PEG, which cross-linked into microgels via a Diels–Alder click chemistry water-in-oil emulsion approach; these microgels were then photoannealed into granular hydrogels by mixing with methacrylate HA and irradiated with UV light at 365 nm, forming the crosslinked interstitial matrix. Cells encapsulated in the granular hydrogel had a 2-fold increased area compared with non-granular hydrogels, which is consistent with chondrocyte maturation, where chondrocytes are known to increase in area as they become more chondrogenic, and 0.5- to 3-fold increased mRNA expression of chondrogenic genes, demonstrating their potential for cartilage tissue regeneration. On the osteogenic side of the osteochondral interface, Rao et al. fabricated granular PEG hydrogels to control postmenopausal osteoporosis by mediating osteoporotic MSC clustering. Briefly, the authors fabricated microgels from dibenzocyclooctyne (DBCO)-modified and azide-modified PEG containing RGD peptide. Mixing of the microgels resulted in a granular hydrogel in which the interstitial matrix was crosslinked by DBCO-azide click chemistry of neighboring microgels. The authors found that the size of the microgels affected the macroscale porosity of the granular hydrogel, which subsequently effected the clustering of encapsulated MSCs. Larger diameter microgels yielded larger clusters of cells. MSCs in larger clusters, mimetic of osteoporotic MSC clustering, released a greater number of osteoporotic cytokines. Therefore, the authors conclude that MSC clustering via granular hydrogels could be a useful strategy for the development of MSC-biomaterials to promote or inhibit cellular secretory profiles. In other words, by controlling the size of the microgels, the pores between microgels can be increased in size or decreased in size. In this case, decreasing the size of the pores would reduce MSC clustering, preventing osteoporosis and maintaining an osteogenic phenotype.
Another common application of granular hydrogels due to their increased porosity is neural tissue engineering. The increased number of pores allows for the formation of neurites that can follow the tortuosity of the pores and form synaptic connections with both neighboring neurons and those farther away. Hsu et al. formed granular hydrogels using commercially available HyStem® by fabricating bulk hydrogels using the kit, introducing granularity by hydrogel extrusion through a nylon mesh, and adding additional PEG diacrylate crosslinker to form the crosslinked interstitial space.115 The granular hydrogel demonstrated approximately 25% increased cellular viability due to the enhanced mass transfer capacity, as well as more than double the number of neurite-bearing cells and nearly triple the neurite length at day 7 in comparison to the bulk hydrogels. In addition, Yang et al. tested the effects of an HA granular hydrogel on neuronal regeneration in vivo, demonstrating axon regeneration, regeneration of the myelin sheath, and motor function recovery in rats.116 Therefore, the granular hydrogel system is effective at providing the tortuosity and porosity necessary to mimic neurite formation in mature brain and spinal cord tissue.
However, despite their promise and demonstrated potential in certain tissue engineering and regenerative medicine applications, granular hydrogels remain a nascent field. Therefore, significant research is still being conducted in order to optimize how the granular hydrogels interface with cells and how their properties can be tuned to mimic physicochemical properties. Some of this ongoing research has been conducted in order to support cellular interactions with these granular hydrogels, such as cell adhesion, growth, and migration. In addition, although they have the benefit of increased porosity to increase mass transfer and cellular migration, as well as the benefit of mimicking complex 3D architecture, granular hydrogels often remain mechanically weak in comparison to native tissue. Therefore, an additional primary focus of recent literature has been on enhancing the mechanical properties of granular hydrogels, either through experimental testing104,105,117–125 or through the development of data-driven modeling.126
To highlight the recent efforts in improving cellular interactions with granular hydrogels:
Mendes et al. developed an injectable HA and platelet lysate-derived granular hydrogel with the goal of enhancing cellular adhesion.102 They used bioorthogonal, thiol-norbornene click chemistry to crosslink the covalently-modified HA with the platelet lysate mixed in. The precursor hydrogel solution was passed through a microfluidic device to form the microgels, and jamming of the microgels using 3D printing was conducted by mixing the microgels with a fluorescent thiolated peptide. The thiolated peptide would crosslink with the norbornene-modified HA on the surface of the microgels to form the cross-linked interstitial fluid and provide additional integrin-binding sites for cells. The authors found that the incorporation of the platelet lysate and the peptide promoted MSC adhesion and spreading approximately 3-fold more than HA hydrogels functionalized with RGD peptide and 6-fold more than HA hydrogels alone, demonstrating how the use of bioorthogonal click chemistry can optimize interaction of granular hydrogels with adhesive cells. In another study, Gehlen et al. sought to improve the overall growth of cells in 3D on granular hydrogels fabricated using cellulose nanofibrils.107 The authors specifically chose cellulose nanofibrils due to this material being a naturally-derived polymer with known proclivity for cellular adhesion. In addition, the nanofibril structure mimics the nanofibers of ECM. Briefly, the cellulose nanofibrils were mixed with calcium chloride to pre-crosslink the hydrogel. This precursor hydrogel was then passed through a nylon mesh via syringe to form the granularity, which was subsequently mixed with fibroblasts, cast into a well plate, and crosslinked using RPMI+ cell culture media. The authors demonstrate that increased granularity resulted in a more homogenous distribution of cells within the hydrogel, likely due to the increased void space and increased surface area for cell adhesion and migration. The increased homogeneity supported a more even distribution and attachment of cells, which has the added effect of increased cellular viability. An additionally interesting note is that the opposite can also be true. In tissues where cell distribution is particularly heterogeneous, the overall granularity of the hydrogel can be decreased. In addition, Widener et al. demonstrated the use of guest–host interlinked PEG granular hydrogels to improve cell migration.106 As PEG is a synthetic polymer without cellular binding sites, bulk PEG hydrogels significantly limit cellular infiltration and migration, which further decreases their bioactive mimicry. Therefore, the authors sought to improve cellular infiltration and migration in PEG hydrogels using granularity. Briefly, the authors fabricated microgels of maleimide-functionalized PEG, some of which were further functionalized with β-cyclodextrin, and some of which were further functionalized with adamantane. The β-cyclodextrin and adamantane were then capable of reversible guest–host interactions to form the crosslinked interstitial network. Using a transwell invasion assay, the authors confirmed that the granular hydrogels had greater infiltration of THP-1 monocytes, with a more homogenous distribution throughout the hydrogel, as well as a 1000-fold increased cell density compared to a bulk PEG hydrogel. Therefore, the incorporation of the β-cyclodextrin and adamantane successfully mimicked the integrin binding sites present on native ECM.
To highlight the recent efforts in improving the mechanical properties of granular hydrogels:
Yuan et al. developed granular hydrogels with enhanced adhesion, multiple bioactive functionalities, and ultrastrechability for the treatment of chronic wounds.123 They developed this granular hydrogel based on the in situ fusion of zinc-functionalized, hydroxyapatite (HaP) nanoparticle-loaded, acrylic acid nanocomposite microgels by combining hydration-induced physical interactions, as well as carbodiimide (NHS/EDC) covalent bonding to crosslink the interstitial space. In this study, the authors sought to utilize the hydrogel as a bioactive wound dressing; therefore, it is imperative that the granular hydrogel can adhere to and integrate with the existing tissue to cover the wound and share bioactive signaling. The authors report shear strengths of 50–120 kPa for different human tissues, demonstrating significant adhesion of the granular hydrogel to these tissues. In addition, the granular hydrogels showed failure strains of 1000–1800%, demonstrating their ultrastrechability and viscoelastic-mimicry of the native tissues. In a rat full-thickness diabetic wound model, the granular hydrogel had 96% wound closure 14 days post-treatment in comparison to the clinical standard, 3 M Tegaderm, which had approximately 70% wound closure at 14 days. Therefore, mimicking the binding, viscoelasticity, and bioactivity of the native tissue promoted wound healing. In a different study, Hirsch et al. reported on the fabrication of ultra-strong granular hydrogels fabricated from a double network crosslinking approach.120 Briefly, poly 2-acrylamido-2-methylpropane sulfonic acid (PAMPS) microgels were fabricated using a water-in-oil emulsion process. Granular hydrogels were fabricated by casting a bioink made from these microgels into Teflon molds and crosslinking for five minutes under UV light (365 nm), followed by immersing the granular hydrogel overnight in an additional polymeric crosslinking solution then exposing to UV light a second time. The tensile strength of the granular hydrogels increased up to 0.6 MPa with increasing polymer concentration, which is significantly greater than many of the commonly used hydrogels in tissue engineering and regenerative medicine applications. This has particularly beneficial impact for cartilage and tendon tissue engineering, where tissue tears are prominent and tensile strength of the tissue is on the MPa order. Finally, Muir et al. investigated the roles of the microgel composition and interstitial matrix composition on the overall properties of the granular hydrogel.118 Briefly, the authors fabricated HA microgels with compressive moduli ranging from 10–70 kPa and crosslinked the interstitial matrix, at a composition of 0–30 volume%, using photocrosslinking to achieve interstitial matrix compressive moduli ranging from 2–120 kPa (Fig. 5A). They found that the modulus of the microgels affected the flow behavior of bioinks made from these microgels, with increasing microgel moduli yielding increased storage moduli of the granular hydrogel, and the yield strain decreasing with increasing microgel moduli (Fig. 5B). On the other hand, increasing the modulus of the interstitial matrix increased the compressive moduli, failure strains, and failure stresses of the granular hydrogel, yet the previous effects of the microgel moduli were not overwritten by the effects of the interstitial matrix moduli (Fig. 5C). These findings are particularly helpful in understanding how tuning certain microgel and granular hydrogel properties affect the overall mechanical properties. Therefore, the correct interstitial matrix modulus and microgel modulus can be selected in order to get the desired elasticity and viscoelasticity to mimic the tissue or organ of interest.
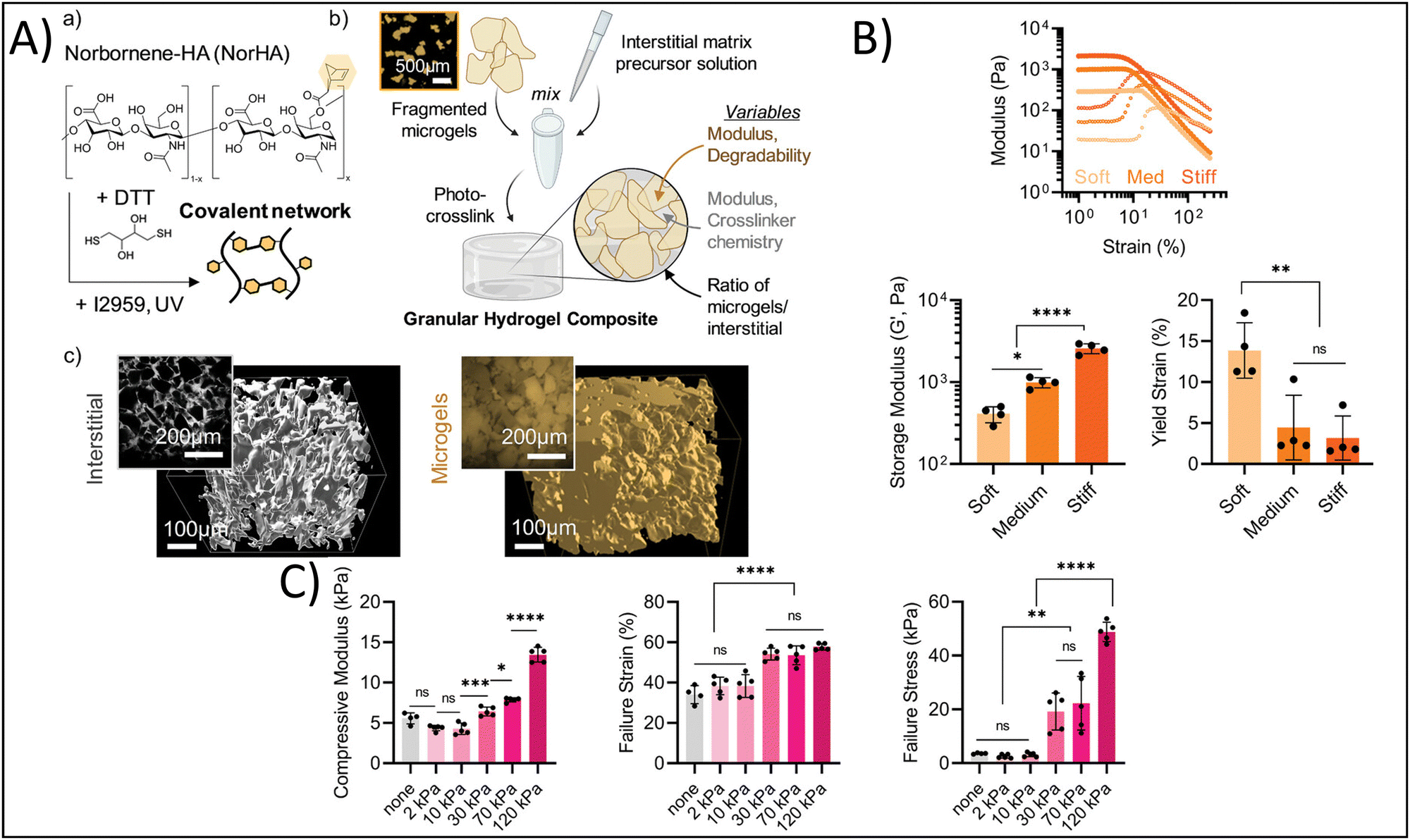 | ||
| Fig. 5 Enhancing physicochemical properties via granular hydrogel fabrication. (A) Schematic depicting granular hydrogel fabrication. Hydrogel crosslinking via norbornene-functionalized HA (NorHA) and photocrosslinked with dithiothreitol (DTT) and Irgacure 2959 to fabricate a bulk hydrogel (a) is followed by fragmentation of the bulk hydrogel (b) and jamming to form granular hydrogels with varying interstitial matrices (c). (B) Rheological characterization of hydrogels with varying formulations. (C) Compressive strength characterization of hydrogels with different formulations. Adapted from ref. 118 with permission by the authors. | ||
4. Conclusions and future perspectives
At the time of writing, there are over 550 results for recruiting, active, or completed studies involving hydrogels on clinicaltrials.gov, many of which are for various tissue engineering and regenerative medicine applications. However, there are only a handful of FDA-approved hydrogels for tissue engineering and regenerative medicine applications on the market today in the USA, such as EUFLEXXA®, INFUSE®, AND OP-1® for spinal fusion and treatment of knee osteoarthritis, as well as Algisyl-LVR® for advanced heart failure.127 The limitations of “traditional” approaches as detailed, such as potential cytotoxicity, weak mechanical properties, incomplete recapitulation of the complexity of native tissues, and a lack of bioactive signaling, to name a few, have limited the clinical use of hydrogels in tissue engineering and regenerative medicine. In this review, we detailed some up and coming methodologies that improve the physicochemical properties of hydrogels towards tissue-mimetic values. For example, dual-network crosslinking approaches allow researchers to obtain the bioactive potential of using naturally-derived polymers, common in physical crosslinking approaches, with the mechanical stability offered by chemical crosslinking. In addition, advances in cytocompatible chemical crosslinking methods, such as bioorthogonal click chemistry, reduce toxicity concerns. Finally, advanced hydrogel manufacturing approaches such as 3D bioprinting and granular hydrogels will enable the fabrication of structurally-complex hydrogels mimicking native tissue.In the coming years, we expect that with continuing advances in the methodologies described herein, more and more hydrogels for tissue engineering and regenerative medicine applications will see clinical effectiveness and come to market. In particular, we expect to see significant advances in the areas of nanocomposite hydrogels with multifunctionalities. For example, these may be nanoparticle-embedded, stimuli-responsive hydrogels for spatiotemporal delivery of drugs and small molecules, or this may be spatially complex, 3D bioprinted hydrogel-nanocomposites. In addition, metal ion cross-linked hydrogels have begun to see recent utilization.128 We also expect to see continued progress in hydrogels fabricated from advanced manufacturing processes, such as self-adhesive hydrogels,129 or hydrogels fabricated using 3D bioprinting with advanced chemical crosslinking or dual network crosslinking approaches or the continued combination of granular hydrogels and 3D bioprinting. An additional method that is rapidly garnering interest in the development of hydrogels with tissue-mimetic physicochemical properties is supramolecular crosslinking.130–132 For example, Li et al. developed a temperature-, ion-, and pH-sensitive biphenyl tripeptide hydrogel formed using supramolecular crosslinking, showing the potential of supramolecular crosslinking to form hydrogels with multifunctional capabilities.132 Additionally, Yu et al. fabricated an injectable, self-healing hydrogel doped with alendronate-Ca2+/Mg2+ using supramolecular chemistry to overcome the limitations of conventional hydrogels, which often exhibit poor mechanical properties and insufficient osteogenic activity.133 The authors demonstrated enhanced bone marrow-derived MSC proliferation, migration, and osteogenic differentiation, as well as enhanced bone regeneration in a rat cranial lesion model. It is these types of advanced, multifunctional hydrogels that we expect to continue to progress to clinical trials and FDA approval.
Additionally, machine learning (ML) has also become an influential tool in tissue engineering and regenerative medicine, driving significant advancements in 3D bioprinting. ML algorithms, including supervised learning, unsupervised learning, and reinforcement learning, can model and predict complex interactions between bioprinting parameters and outcomes, enhancing printability, cell viability, and mechanical properties of the constructs. These algorithms help develop predictive models to optimize bio ink formulations, printing parameters, and scaffold designs, reducing the need for traditional trial-and-error methods. The integration of ML into bioprinting increases efficiency while enabling real-time monitoring and quality control. In the future, ML is expected to lead to the production of more complex, functional, and tailored tissue constructs, thereby accelerating their transition from research to clinical applications.134
However, it is important to note that despite the progress that has been made in the field towards tissue-mimetic hydrogels, several obstacles must be overcome in order to scale up the production of hydrogels and ensure their biocompatibility. One of the most significant challenges is the synthesis of multifunctional biomaterials with the same functional properties and reproducibility outside of laboratory settings. It is essential to ensure that these multifunctional hydrogels possess long-term mechanical stability, biocompatibility, controllable biodegradability, and minimal toxicity or immunogenicity. In addition, the upcoming methods that can be used to fabricate these multifunctional hydrogels must do so in a cost-effective and high-throughput method. As such, it remains essential that the field continue to pursue innovations in multifunctional hydrogel fabrication in order to unlock the full potential of hydrogels and the future of healthcare.
Abbreviations
| 3D | Three-dimensional |
| Alg | Alginate |
| CaCO3/GDL | Calcium carbonate/gluconolactone |
| CHO | Dialdehyde |
| CS | Chitosan |
| DBCO | Dibenzocyclooctyne |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| ECM | Extracellular matrix |
| GDL | Glucono-delta-lactone |
| GelMA | Gelatin methacrylate |
| GSE4 | Glutamate-specific endopeptidase |
| Hap | Hydroxyapatite |
| HA | Hyaluronic acid |
| HA-Tyr | Tyramine-modified Hyaluronic acid |
| LAP | Lithium phenyl-2,4,6-trimethylbenzoylphosphinate |
| MSC | Mesenchymal stem cell |
| NHS | N-Hydroxysulfosuccinimide |
| OAlg | Oxidized alginate |
| PAMPS | Poly 2-acrylamido-2-methylpropane sulfonic acid |
| PCL | Polycaprolactone |
| PEG | Polyethylene glycol |
| PEGDMA | Polyethylene dimethacrylate |
| PLA | Polylactic acid |
| PVA | Polyvinyl alcohol |
| PU | Polyurethane |
| RGD | Arginine–glycine–aspartic acid |
| SF | Silk fibroin |
| UV | Ultraviolet |
| VEGF | Vascular endothelial growth factor |
| XG | Xerogel |
Author contributions
Rabia Fatima: formal analysis; investigation; data curation; writing – original draft; writing – review & editing; visualization. Bethany Almeida: conceptualization; validation; formal analysis; investigation; resources; data curation; writing – original draft; writing – review & editing; visualization; supervision; project administration; funding acquisition.Data availability
There are no data to share.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge funding from the National Science Foundation (Award #2138587).References
- S. Chaudhary and E. Chakraborty, Beni-Suef Univ. J. Basic Appl. Sci., 2022, 11, 3 CrossRef PubMed.
- A. Revete, A. Aparicio, B. A. Cisterna, J. Revete, L. Luis, E. Ibarra, E. A. Segura González, J. Molino and D. Reginensi, Int. J. Biomater., 2022, 2022, 1–16 CrossRef PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- D.-M. Radulescu, I. A. Neacsu, A.-M. Grumezescu and E. Andronescu, Polymers, 2022, 14, 799 CrossRef CAS PubMed.
- J. A. Hunt, R. Chen, T. van Veen and N. Bryan, J. Mater. Chem. B, 2014, 2, 5319–5338 RSC.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12, 3323 CrossRef CAS PubMed.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CrossRef CAS PubMed.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- K. A. Kilian, B. Bugarija, B. T. Lahn and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4872–4877 CrossRef CAS PubMed.
- N. K. Karamanos, A. D. Theocharis, Z. Piperigkou, D. Manou, A. Passi, S. S. Skandalis, D. H. Vynios, V. Orian-Rousseau, S. Ricard-Blum, C. E. H. Schmelzer, L. Duca, M. Durbeej, N. A. Afratis, L. Troeberg, M. Franchi, V. Masola and M. Onisto, FEBS J., 2021, 288, 6850–6912 CrossRef CAS PubMed.
- M. Deng, J. Lin, S. Nowsheen, T. Liu, Y. Zhao, P. W. Villalta, D. Sicard, D. J. Tschumperlin, S. Lee, J. Kim and Z. Lou, Sci. Adv., 2020, 6(37), eabb2630 CrossRef CAS PubMed.
- R. B. Diller and A. J. Tabor, Biomimetics, 2022, 7, 87 CrossRef CAS PubMed.
- X. Liao, X. Li and R. Liu, Rev. Endocr. Metab. Disord., 2023, 24, 207–220 CrossRef CAS PubMed.
- A. D. Doyle, S. S. Nazari and K. M. Yamada, Phys. Biol., 2022, 19, 021002 CrossRef PubMed.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 RSC.
- N. Davari, N. Bakhtiary, M. Khajehmohammadi, S. Sarkari, H. Tolabi, F. Ghorbani and B. Ghalandari, Polymers, 2022, 14, 986 CrossRef CAS PubMed.
- M. Gomez-Florit, A. Pardo, R. M. A. Domingues, A. L. Graça, P. S. Babo, R. L. Reis and M. E. Gomes, Molecules, 2020, 25, 5858 CrossRef CAS PubMed.
- Q. Yang, J. Peng, H. Xiao, X. Xu and Z. Qian, Carbohydr. Polym., 2022, 278, 118952 CrossRef CAS PubMed.
- S. Ullah and X. Chen, Appl. Mater. Today, 2020, 20, 100656 CrossRef.
- M. S. B. Reddy, D. Ponnamma, R. Choudhary and K. K. Sadasivuni, Polymers, 2021, 13, 1105 CrossRef CAS PubMed.
- R. Keshavarz, S. Olsen and B. Almeida, Bioeng. Transl. Med., 2024, 9(1), e10598 CrossRef CAS PubMed.
- F. Xu, C. Dawson, M. Lamb, E. Mueller, E. Stefanek, M. Akbari and T. Hoare, Front. Bioeng. Biotechnol., 2022, 10, 849831 CrossRef PubMed.
- H. Pan, B. Zheng, H. Shen, M. Qi, Y. Shang, C. Wu, R. Zhu, L. Cheng and Q. Wang, Chem. Commun., 2020, 56, 3457–3460 RSC.
- K. Lavanya, S. V. Chandran, K. Balagangadharan and N. Selvamurugan, Mater. Sci. Eng., C, 2020, 111, 110862 CrossRef CAS PubMed.
- D. Dehghan-Baniani, B. Mehrjou, D. Wang, R. Bagheri, A. Solouk, P. K. Chu and H. Wu, Int. J. Biol. Macromol., 2022, 205, 638–650 CrossRef CAS PubMed.
- M. Bustamante-Torres, D. Romero-Fierro, B. Arcentales-Vera, K. Palomino, H. Magaña and E. Bucio, Gels, 2021, 7, 182 CrossRef CAS PubMed.
- X. Li and X. Su, J. Mater. Chem. B, 2018, 6, 4714–4730 RSC.
- J. E. Lee, S. W. Heo, C. H. Kim, S. J. Park, S.-H. Park and T. H. Kim, Biomater. Adv., 2023, 147, 213322 CrossRef CAS PubMed.
- P. Pan, J. Wang, X. Wang, Y. Kang, X. Yu, T. Chen, Y. Hao and W. Liu, Int. J. Biol. Macromol., 2024, 257, 128682 CrossRef CAS PubMed.
- S. Zafar, M. Hanif, M. Azeem, K. Mahmood and S. A. Gondal, Polym. Bull., 2022, 79, 9199–9219 CrossRef CAS.
- A. Papaioannou, E. Vasilaki, K. Loukelis, D. Papadogianni, M. Chatzinikolaidou and M. Vamvakaki, Biomater. Adv., 2024, 157, 213737 CrossRef CAS PubMed.
- X. Wang and Q. Wang, Acc. Chem. Res., 2021, 54, 1274–1287 CrossRef CAS PubMed.
- A. Battigelli, B. Almeida and A. Shukla, Bioconjug. Chem., 2022, 33, 263–271 CrossRef CAS PubMed.
- N. H. Dimmitt and C.-C. Lin, Macromolecules, 2024, 57, 1556–1568 CrossRef CAS PubMed.
- X. Li and Y. Xiong, ACS Omega, 2022, 7, 36918–36928 CrossRef CAS PubMed.
- A. Laezza, A. Pepe, N. Solimando, F. Armiento, F. Oszust, L. Duca and B. Bochicchio, ChemPlusChem, 2024, 89, e202300662 CrossRef CAS PubMed.
- S. H. Moon, H. J. Hwang, H. R. Jeon, S. J. Park, I. S. Bae and Y. J. Yang, Front. Bioeng. Biotechnol., 2023, 11, 1127757 CrossRef PubMed.
- S. Maiz-Fernández, L. Pérez-Álvarez, U. Silván, J. L. Vilas-Vilela and S. Lanceros-Mendez, Int. J. Biol. Macromol., 2022, 216, 291–302 CrossRef PubMed.
- Y. Huang, G. Sun, L. Lyu, Y. Li, D. Li, Q. Fan, J. Yao and J. Shao, Soft Matter, 2022, 18, 3705–3712 RSC.
- K. S. Lim, F. Abinzano, P. N. Bernal, A. Albillos Sanchez, P. Atienza-Roca, I. A. Otto, Q. C. Peiffer, M. Matsusaki, T. B. F. Woodfield, J. Malda and R. Levato, Adv. Healthcare Mater., 2020, 9(15), e1901792 CrossRef PubMed.
- S. H. Kim, H. Hong, O. Ajiteru, Md. T. Sultan, Y. J. Lee, J. S. Lee, O. J. Lee, H. Lee, H. S. Park, K. Y. Choi, J. S. Lee, H. W. Ju, I.-S. Hong and C. H. Park, Nat. Protoc., 2021, 16, 5484–5532 CrossRef CAS PubMed.
- A. A. H. Bukhari, N. H. Elsayed and M. Monier, Carbohydr. Polym., 2021, 260, 117771 CrossRef CAS PubMed.
- S. Liu, D. Brunel, K. Sun, Y. Zhang, H. Chen, P. Xiao, F. Dumur and J. Lalevée, Macromol. Rapid Commun., 2020, 41, 2000460 CrossRef CAS PubMed.
- E. M. Plaster, M. K. Eiken and C. Loebel, Carbohydr. Polym. Technol. Appl., 2023, 6, 100360 CAS.
- S. Nam and D. Mooney, Chem. Rev., 2021, 121, 11336–11384 CrossRef CAS PubMed.
- Y. Zhang, Y. Cao, H. Zhao, L. Zhang, T. Ni, Y. Liu, Z. An, M. Liu and R. Pei, J. Mater. Chem. B, 2020, 8, 4237–4244 RSC.
- A. Frayssinet, D. Petta, C. Illoul, B. Haye, A. Markitantova, D. Eglin, G. Mosser, M. D’Este and C. Hélary, Carbohydr. Polym., 2020, 236, 116042 CrossRef CAS PubMed.
- J. Simińska-Stanny, L. Nicolas, A. Chafai, H. Jafari, M. Hajiabbas, G. Dodi, I. Gardikiotis, C. Delporte, L. Nie, D. Podstawczyk and A. Shavandi, Bioact. Mater., 2024, 36, 168–184 Search PubMed.
- R. Wang, X. Huang, B. Zoetebier, P. J. Dijkstra and M. Karperien, Bioact. Mater., 2023, 20, 53–63 CAS.
- W. Kong, Y. Gao, Q. Liu, L. Dong, L. Guo, H. Fan, Y. Fan and X. Zhang, Int. J. Biol. Macromol., 2020, 160, 1201–1211 CrossRef CAS PubMed.
- H. Xu, J. Casillas, S. Krishnamoorthy and C. Xu, Biomed. Mater., 2020, 15, 055021 CrossRef CAS PubMed.
- Y. Yang, Z. Zhu, R. Gao, J. Yuan, J. Zhang, H. Li, Z. Xie and Y. Wang, Acta Biomater., 2021, 128, 163–174 CrossRef CAS PubMed.
- S. Li, D. Niu, T. Shi, W. Yun, S. Yan, G. Xu and J. Yin, ACS Biomater. Sci. Eng., 2023, 9, 2625–2635 CrossRef CAS PubMed.
- S. Li, M. Pei, T. Wan, H. Yang, S. Gu, Y. Tao, X. Liu, Y. Zhou, W. Xu and P. Xiao, Carbohydr. Polym., 2020, 250, 116922 CrossRef CAS PubMed.
- M. N. Guven, B. Balaban, G. Demirci, H. Yagci Acar, O. Okay and D. Avci, Eur. Polym. J., 2021, 159, 110732 CrossRef CAS.
- X. Wang, L. Cui, J. Hong, Z. Wang, J. Li, Z. Liu, Z. Zhu, Y. Hao, G. Cheng and J. Jiang, Polymer, 2023, 272, 125861 CrossRef CAS.
- Q. Min, C. Wang, Y. Zhang, D. Tian, Y. Wan and J. Wu, Nanomaterials, 2022, 12, 1874 CrossRef CAS PubMed.
- M. Ghorbani, E. Vasheghani-Farahani, N. Azarpira, S. Hashemi-Najafabadi and A. Ghasemi, Biomater. Adv., 2023, 153, 213565 CrossRef CAS PubMed.
- W. He, K. Chen, W. Gao, R. Duan, Z. Li, B. Li, J. Xia, Y. Zhao, W. Liu, H. Zhou, X. Xiao, Q. Feng and Y. Yang, Mater. Des., 2024, 237, 112563 CrossRef CAS.
- R. Govindan, F. L. Gu, S. Karthi and E. K. Girija, Mater. Today Commun., 2020, 22, 100765 CrossRef CAS.
- K. R. Coogan, P. T. Stone, N. D. Sempertegui and S. S. Rao, Eur. Polym. J., 2020, 135, 109870 CrossRef CAS.
- K. Yari, G. Gharati and I. Akbari, Int. J. Biol. Macromol., 2023, 227, 1282–1292 CrossRef CAS PubMed.
- B. Yan, Y. Zhang, Z. Li, P. Zhou and Y. Mao, SN Appl. Sci., 2022, 4, 172 CrossRef CAS PubMed.
- Y. Tang, S. Lin, S. Yin, F. Jiang, M. Zhou, G. Yang, N. Sun, W. Zhang and X. Jiang, Biomaterials, 2020, 232, 119727 CrossRef CAS PubMed.
- A. Kjar, B. McFarland, K. Mecham, N. Harward and Y. Huang, Bioact. Mater., 2021, 6, 460–471 CAS.
- S. Ji and M. Guvendiren, APL Bioeng., 2021, 5, 011508 CrossRef PubMed.
- I. Matai, G. Kaur, A. Seyedsalehi, A. McClinton and C. T. Laurencin, Biomaterials, 2020, 226, 119536 CrossRef CAS PubMed.
- Y. He, F. Yang, H. Zhao, Q. Gao, B. Xia and J. Fu, Sci. Rep., 2016, 6, 29977 CrossRef CAS PubMed.
- A. García-García, L. Pérez-Álvarez, L. Ruiz-Rubio, A. Larrea-Sebal, C. Martin and J. L. Vilas-Vilela, Gels, 2024, 10, 126 CrossRef PubMed.
- W. L. Ng, J. M. Lee, M. Zhou, Y.-W. Chen, K.-X. A. Lee, W. Y. Yeong and Y.-F. Shen, Biofabrication, 2020, 12, 022001 CrossRef CAS PubMed.
- J. M. Unagolla and A. C. Jayasuriya, Appl. Mater. Today, 2020, 18, 100479 CrossRef PubMed.
- D. Chimene, R. Kaunas and A. K. Gaharwar, Adv. Mater., 2020, 32(1), 1902026 CrossRef CAS PubMed.
- K. Ioannidis, R. I. Danalatos, S. Champeris Tsaniras, K. Kaplani, G. Lokka, A. Kanellou, D. J. Papachristou, G. Bokias, Z. Lygerou and S. Taraviras, Front. Bioeng. Biotechnol., 2020, 8, 580889 CrossRef PubMed.
- M. H. Kim and S. Y. Nam, Bioprinting, 2020, 20, e00092 CrossRef.
- R. Schwartz, M. Malpica, G. L. Thompson and A. K. Miri, J. Mech. Behav. Biomed. Mater., 2020, 103, 103524 CrossRef CAS PubMed.
- T. Kreller, T. Distler, S. Heid, S. Gerth, R. Detsch and A. R. Boccaccini, Mater. Des., 2021, 208, 109877 CrossRef CAS.
- H. Budharaju, H. Chandrababu, A. Zennifer, D. Chellappan, S. Sethuraman and D. Sundaramurthi, Int. J. Biol. Macromol., 2024, 260, 129443 CrossRef CAS PubMed.
- Y. He, F. Wang, X. Wang, J. Zhang, D. Wang and X. Huang, Mater. Des., 2021, 202, 109588 CrossRef CAS.
- J. Kim, H. Lee, G. Lee, D. Ryu and G. Kim, Bioact. Mater., 2024, 36, 14–29 CAS.
- K. Zhou, R. Ding, X. Tao, Y. Cui, J. Yang, H. Mao and Z. Gu, Acta Biomater., 2023, 169, 243–255 CrossRef CAS PubMed.
- S.-Y. Chang, T. Ching and M. Hashimoto, Mater. Today Proc., 2022, 70, 179–183 CrossRef CAS.
- K.-C. Cheng, Y.-M. Sun and S. Hsu, J. Mater. Chem. B, 2023, 11, 3592–3606 RSC.
- D. G. O’Shea, T. Hodgkinson, C. M. Curtin and F. J. O’Brien, Biofabrication, 2024, 16, 015007 CrossRef PubMed.
- T. H. Jovic, T. Nicholson, H. Arora, K. Nelson, S. H. Doak and I. S. Whitaker, Carbohydr. Polym., 2023, 321, 121261 CrossRef CAS PubMed.
- S. Afra, M. Koch, J. Żur-Pińska, M. Dolatshahi, A. R. Bahrami, J. E. Sayed, A. Moradi, M. M. Matin and M. K. Włodarczyk-Biegun, Carbohydr. Polym. Technol. Appl., 2024, 7, 100418 CAS.
- Y. Zhang, D. Li, Y. Liu, L. Peng, D. Lu, P. Wang, D. Ke, H. Yang, X. Zhu and C. Ruan, Innovation, 2024, 5, 100542 CAS.
- K. Khoshmaram, F. Yazdian, Z. Pazhouhnia and N. Lotfibakhshaiesh, Biomater. Adv., 2024, 156, 213677 CrossRef CAS PubMed.
- C. D. Roche, H. Lin, Y. Huang, C. E. de Bock, D. Beck, M. Xue and C. Gentile, Bioprinting, 2023, 30, e00263 CrossRef.
- Y. Li, S. Cheng, H. Wen, L. Xiao, Z. Deng, J. Huang and Z. Zhang, Acta Biomater., 2023, 168, 400–415 CrossRef CAS PubMed.
- M. A. Nosenko, A. M. Moysenovich, A. Y. Arkhipova, K.-S. N. Atretkhany, S. A. Nedospasov, M. S. Drutskaya and M. M. Moisenovich, Bioact. Mater., 2021, 6, 3449–3460 CAS.
- Q. Li, S. Xu, Q. Feng, Q. Dai, L. Yao, Y. Zhang, H. Gao, H. Dong, D. Chen and X. Cao, Bioact. Mater., 2021, 6, 3396–3410 CAS.
- Y. Wang, S. Yang, H. Cai, H. Hu, K. Hu, Z. Sun, R. Liu, Y. Wei and L. Han, Sci. Rep., 2024, 14, 4118 CrossRef CAS PubMed.
- J. Xu, H. Fang, Y. Su, Y. Kang, D. Xu, Y. Y. Cheng, Y. Nie, H. Wang, T. Liu and K. Song, Int. J. Biol. Macromol., 2022, 220, 1253–1266 CrossRef CAS PubMed.
- D. Kang, Z. Liu, C. Qian, J. Huang, Y. Zhou, X. Mao, Q. Qu, B. Liu, J. Wang, Z. Hu and Y. Miao, Acta Biomater., 2023, 165, 19–30 CrossRef CAS PubMed.
- K. H. Schneider, B. J. Goldberg, O. Hasturk, X. Mu, M. Dötzlhofer, G. Eder, S. Theodossiou, L. Pichelkastner, P. Riess, S. Rohringer, H. Kiss, A. H. Teuschl-Woller, V. Fitzpatrick, M. Enayati, B. K. Podesser, H. Bergmeister and D. L. Kaplan, Biomater. Res., 2023, 27, 117 CrossRef CAS PubMed.
- A. Jafari, S. Vahid Niknezhad, M. Kaviani, W. Saleh, N. Wong, P. P. Van Vliet, C. Moraes, A. Ajji, L. Kadem, N. Azarpira, G. Andelfinger and H. Savoji, Adv. Funct. Mater., 2024, 34, 2305188 CrossRef CAS.
- Z. Jian, T. Zhuang, T. Qinyu, P. Liqing, L. Kun, L. Xujiang, W. Diaodiao, Y. Zhen, J. Shuangpeng, S. Xiang, H. Jingxiang, L. Shuyun, H. Libo, T. Peifu, Y. Qi and G. Quanyi, Bioact. Mater., 2021, 6, 1711–1726 CAS.
- M. Bahraminasab, N. Doostmohammadi, A. Talebi, S. Arab, A. Alizadeh, A. Ghanbari and A. Salati, Biomed. Eng. Online, 2022, 21, 86 CrossRef PubMed.
- M. Kamaraj, G. Sreevani, G. Prabusankar and S. N. Rath, Mater. Sci. Eng., C, 2021, 131, 112478 CrossRef CAS PubMed.
- B. B. Mendes, A. C. Daly, R. L. Reis, R. M. A. Domingues, M. E. Gomes and J. A. Burdick, Acta Biomater., 2021, 119, 101–113 CrossRef CAS PubMed.
- V. R. Feig, S. Santhanam, K. W. McConnell, K. Liu, M. Azadian, L. G. Brunel, Z. Huang, H. Tran, P. M. George and Z. Bao, Adv. Mater. Technol., 2021, 6, 2100162 CrossRef CAS PubMed.
- V. G. Muir, T. H. Qazi, J. Shan, J. Groll and J. A. Burdick, ACS Biomater. Sci. Eng., 2021, 7, 4269–4281 CrossRef CAS PubMed.
- D. B. Emiroglu, A. Bekcic, D. Dranseike, X. Zhang, T. Zambelli, A. J. deMello and M. W. Tibbitt, Sci. Adv., 2022, 8, eadd8570 CrossRef CAS PubMed.
- A. E. Widener, M. Bhatta, T. E. Angelini and E. A. Phelps, Biomater. Sci., 2021, 9, 2480–2493 RSC.
- D. B. Gehlen, N. Jürgens, A. Omidinia-Anarkoli, T. Haraszti, J. George, A. Walther, H. Ye and L. De Laporte, Macromol. Rapid Commun., 2020, 41(18), 2000191 CrossRef CAS PubMed.
- L. Zhang, H. Tang, Z. Xiahou, J. Zhang, Y. She, K. Zhang, X. Hu, J. Yin and C. Chen, Biofabrication, 2022, 14, 035003 CrossRef CAS PubMed.
- J. Zhang, W. Xin, Y. Qin, Y. Hong, Z. Xiahou, K. Zhang, P. Fu and J. Yin, Chem. Eng. J., 2022, 430, 132713 CrossRef CAS.
- Z. Mahdieh, M. D. Cherne, J. P. Fredrikson, B. Sidar, H. S. Sanchez, C. B. Chang, D. Bimczok and J. N. Wilking, Biomed. Mater., 2022, 17, 045020 CrossRef CAS PubMed.
- X. Xu, Y. Zhang, P. Ha, Y. Chen, C. Li, E. Yen, Y. Bai, R. Chen, B. M. Wu, A. Da Lio, K. Ting, C. Soo and Z. Zheng, Bioeng. Transl. Med., 2023, 8(1), e10355 CrossRef CAS PubMed.
- E. Kurt and T. Segura, Adv. Healthcare. Mater., 2022, 11, 2101867 CrossRef CAS PubMed.
- T. J. Tigner, G. Dampf, A. Tucker, Y. Huang, V. Jagrit, A. J. Clevenger, A. Mohapatra, S. A. Raghavan, J. N. Dulin and D. L. Alge, Adv. Healthcare. Mater., 2024, e2303912 CrossRef PubMed.
- Y. Zhu, Y. Sun, B. Rui, J. Lin, J. Shen, H. Xiao, X. Liu, Y. Chai, J. Xu and Y. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 40674–40687 CrossRef CAS PubMed.
- C.-C. Hsu, J. H. George, S. Waller, C. Besnard, D. A. Nagel, E. J. Hill, M. D. Coleman, A. M. Korsunsky, Z. Cui and H. Ye, Bioact. Mater., 2022, 9, 358–372 CAS.
- J. Yang, C.-C. Hsu, T.-T. Cao, H. Ye, J. Chen and Y.-Q. Li, Neural Regen Res., 2023, 18, 657 CrossRef CAS PubMed.
- T. H. Qazi, J. Wu, V. G. Muir, S. Weintraub, S. E. Gullbrand, D. Lee, D. Issadore and J. A. Burdick, Adv. Mater., 2022, 34(12), 2109194 CrossRef CAS PubMed.
- V. G. Muir, S. Weintraub, A. P. Dhand, H. Fallahi, L. Han and J. A. Burdick, Adv. Sci., 2023, 10, 2206117 CrossRef CAS PubMed.
- V. G. Muir, T. H. Qazi, S. Weintraub, B. O. Torres Maldonado, P. E. Arratia and J. A. Burdick, Small, 2022, 18(36), 2201115 CrossRef CAS PubMed.
- M. Hirsch, A. Charlet and E. Amstad, Adv. Funct. Mater., 2021, 31(5), 2005929 CrossRef CAS.
- Z. Ataie, S. Kheirabadi, J. W. Zhang, A. Kedzierski, C. Petrosky, R. Jiang, C. Vollberg and A. Sheikhi, Small, 2022, 18(37), 2202390 CrossRef CAS PubMed.
- W. Wang, X. Chen, T. Meng and L. Liu, J. Biomater. Appl., 2022, 36, 1852–1862 CrossRef CAS PubMed.
- Z. Yuan, Z. Wan, Z. Tian, Y. Han, X. Huang, Y. Feng, W. Xie, X. Duan, S. Huang, X. Liu and J. Huang, Chem. Eng. J., 2022, 450, 138076 CrossRef CAS.
- C. S. Wyss, P. Karami, P.-E. Bourban and D. P. Pioletti, Soft Matter, 2020, 16, 3769–3778 RSC.
- C. S. Wyss, P. Karami, A. Demongeot, P.-E. Bourban and D. P. Pioletti, Soft Matter, 2021, 17, 7038–7046 RSC.
- C. A. Verheyen, S. G. M. Uzel, A. Kurum, E. T. Roche and J. A. Lewis, Matter, 2023, 6, 1015–1036 CrossRef CAS.
- A. Mandal, J. R. Clegg, A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2020, 5(2), e10158 CrossRef CAS PubMed.
- G. Janarthanan and I. Noh, J. Mater. Sci. Technol., 2021, 63, 35–53 CrossRef CAS.
- Y. Yi, C. Xie, J. Liu, Y. Zheng, J. Wang and X. Lu, J. Mater. Chem. B, 2021, 9, 8739–8767 RSC.
- Y. Zhao, S. Song, X. Ren, J. Zhang, Q. Lin and Y. Zhao, Chem. Rev., 2022, 122, 5604–5640 CrossRef CAS PubMed.
- Y. Lyu and H. S. Azevedo, Molecules, 2021, 26, 873 CrossRef CAS PubMed.
- X. Li, S. Bian, M. Zhao, X. Han, J. Liang, K. Wang, Q. Jiang, Y. Sun, Y. Fan and X. Zhang, Acta Biomater., 2021, 131, 128–137 CrossRef CAS PubMed.
- T. Yu, Y. Hu, W. He, Y. Xu, A. Zhan, K. Chen, M. Liu, X. Xiao, X. Xu, Q. Feng and L. Jiang, Mater Today Bio, 2023, 19, 100558 CrossRef CAS PubMed.
- D. V. Krishna and M. R. Sankar, Ann. 3D Printed Med., 2023, 12, 100132 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |