
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Navigating the landscape of enzyme design: from molecular simulations to machine learning
Jiahui
Zhou
and
Meilan
Huang
 *
*
School of Chemistry and Chemical Engineering, Queen's University, David Keir Building, Stranmillis Road, Belfast BT9 5AG, Northern Ireland, UK. E-mail: m.huang@qub.ac.uk
First published on 11th July 2024
Abstract
Global environmental issues and sustainable development call for new technologies for fine chemical synthesis and waste valorization. Biocatalysis has attracted great attention as the alternative to the traditional organic synthesis. However, it is challenging to navigate the vast sequence space to identify those proteins with admirable biocatalytic functions. The recent development of deep-learning based structure prediction methods such as AlphaFold2 reinforced by different computational simulations or multiscale calculations has largely expanded the 3D structure databases and enabled structure-based design. While structure-based approaches shed light on site-specific enzyme engineering, they are not suitable for large-scale screening of potential biocatalysts. Effective utilization of big data using machine learning techniques opens up a new era for accelerated predictions. Here, we review the approaches and applications of structure-based and machine-learning guided enzyme design. We also provide our view on the challenges and perspectives on effectively employing enzyme design approaches integrating traditional molecular simulations and machine learning, and the importance of database construction and algorithm development in attaining predictive ML models to explore the sequence fitness landscape for the design of admirable biocatalysts.
 Jiahui Zhou | Jiahui Zhou is a PhD student at Queen's University Belfast, working under the supervision of Dr Meilan Huang. He received his BS degree in Pharmaceutical Engineering from Beijing Institute of Technology in 2020. His research focuses on using molecular modeling and simulation methods to unravel complex phenomena in enzyme catalysis. |
 Meilan Huang | Meilan Huang obtained her PhD from Zhejiang University in physical chemistry in 2003. She worked as a research assistant on organic synthesis of anticancer drugs in Prof Fengling Qing's lab in the Key Laboratory of Organoflorine Chemistry at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in 1998. She was a postdoc in the Department of Chemistry at the University of Calgary, Canada in 2003, working with Prof Arvi Rauk on the theoretical study of the oxidation mechanism related to Alzihemer's diseases. Awarded with Welcome Trust International Fellowship on computer-aided drug design, she spent two years in the Laboratory of Physical and Theoretical Chemistry at the University of Oxford, working with Prof W. Graham Richards during 2004–2006 and then moved to Department of Medicine at the University of British Columbia, Canada in 2006, working with Prof Artem Cherkasov on the rational design of infectious disease therapies. She joined Queen's University Belfast as a lecturer in 2007. The research in the Huang group is focused on molecular modelling and theoretical catalysis at the interface of chemistry and biology. |
1. Introduction
Over the past decade, enzyme biocatalysis has become a promising alternative to traditional chemical transformations for the sustainable production of valuable chemicals such as biofuels and pharmaceuticals1–3 and hence has attracted increasing attention from both academia and industries. In order to meet the requirements of large-scale industrial production, new biotechnologies have been developed to discover novel enzymes or optimize existing enzyme biocatalysts to improve their catalytic activities, substrate specificity, selectivity, stability, etc.4–7 The success of structure-based enzyme design strategies has been exemplified in numerous cases in rational design, semi-rational design and de novo design. However, it remains challenging to design novel biocatalysts for specific reactions by navigating the vast protein fitness landscape. Recently, machine learning has emerged as an efficient strategy to harness the available data, accelerating the discovery of enzyme biocatalysts and enabling the accurate prediction of mutation sites to achieve biocatalysts with desirable properties.8–101.1 Structure-based enzyme design
The semi-rational enzyme design approach is based on the prior knowledge of enzyme structure and function to navigate the vast theoretical sequence space by screening a small sequence library generated from random mutagenesis or targeted mutagenesis.11 Efficient procurement of mutant variants with the desired functionalities may be achieved by constructing smart mutant libraries and employing appropriate experimental or computational high-throughput screening methods.12–14 Rational enzyme design requires detailed knowledge of the enzyme's mechanism of action, e.g. how it binds to substrates and catalyzes reactions, to guide enzyme engineering for improved or altered function. In addition to mutations based on existing natural sequences, the functional enzymes can be designed from scratch through pre-construction of catalytic sites and selection of protein scaffolds, followed by atomistic simulations.15–17Structure-based enzyme design requires the identification of active sites and substrate binding pockets, however, many enzymes of interest lack resolved structures, and their sequences often exhibit low homology with the known proteins with available crystal structures, making homology modeling unsuitable for obtaining reasonable starting structures. In the past few years, deep-learning based protein structure prediction tools such as AlphaFold218 and RoseTTAFold19 have shown great success in predicting protein 3D structures. Ligand binding mode and the dynamic properties of protein complexes can be further explored by using molecular docking and molecular dynamics simulations. The functions and catalytic mechanisms of enzymes are highly intricate, and are dependent on binding affinities of the substrates and the reaction kinetics of the enzymes. Hybrid molecular mechanics and quantum mechanics (QM/MM) enable the prediction of enzyme-catalyzed reaction kinetics. It is worth noting that structure-based enzyme design requires advanced knowledge in molecular modeling and is also computationally prohibitive for screening a large database to identify the enzyme sequences with desirable functions.
1.2 ML-accelerated enzyme design
In the era of big data, enzyme sequence and structural and functional data have been accumulated and shared at an unprecedented pace. This provides a wealth of information resources for machine-learning guided enzyme design by learning the inherent patterns from data to make predictions. However, the surge in data also brings about the challenge of efficiently harnessing the data to generate generalized ML models to make accurate predictions for accelerating the design of enzymes with improved properties.20–22In this review, we summarize the techniques and applications of computer-aided enzyme design using molecular simulation approaches and machine learning techniques. We also provide our perspectives on effective enzyme design through the synergetic combination of molecular simulations, machine learning and experimental validations.
2 Computer-aided enzyme design tools and applications
2.1 Enzyme modelling methods
Compared with the vast protein sequence space in nature (with over 244 million protein sequences in the UniProt database23 as of May 2024), the number of protein structures is much smaller (with over 220 thousand structures in the Protein Data Bank24). Currently characterized structures only account for less than 10% of the total protein sequences, and the capability of structure characterization largely lags behind that of sequence acquisition (Fig. 1a). Experimentally determining the three-dimensional structure of a protein is a costly and time-consuming process and some proteins are highly flexible, which makes structural determination even more challenging. When the 3D structures of proteins are not available, computational methods become powerful tools in predicting protein structures based on their sequences.
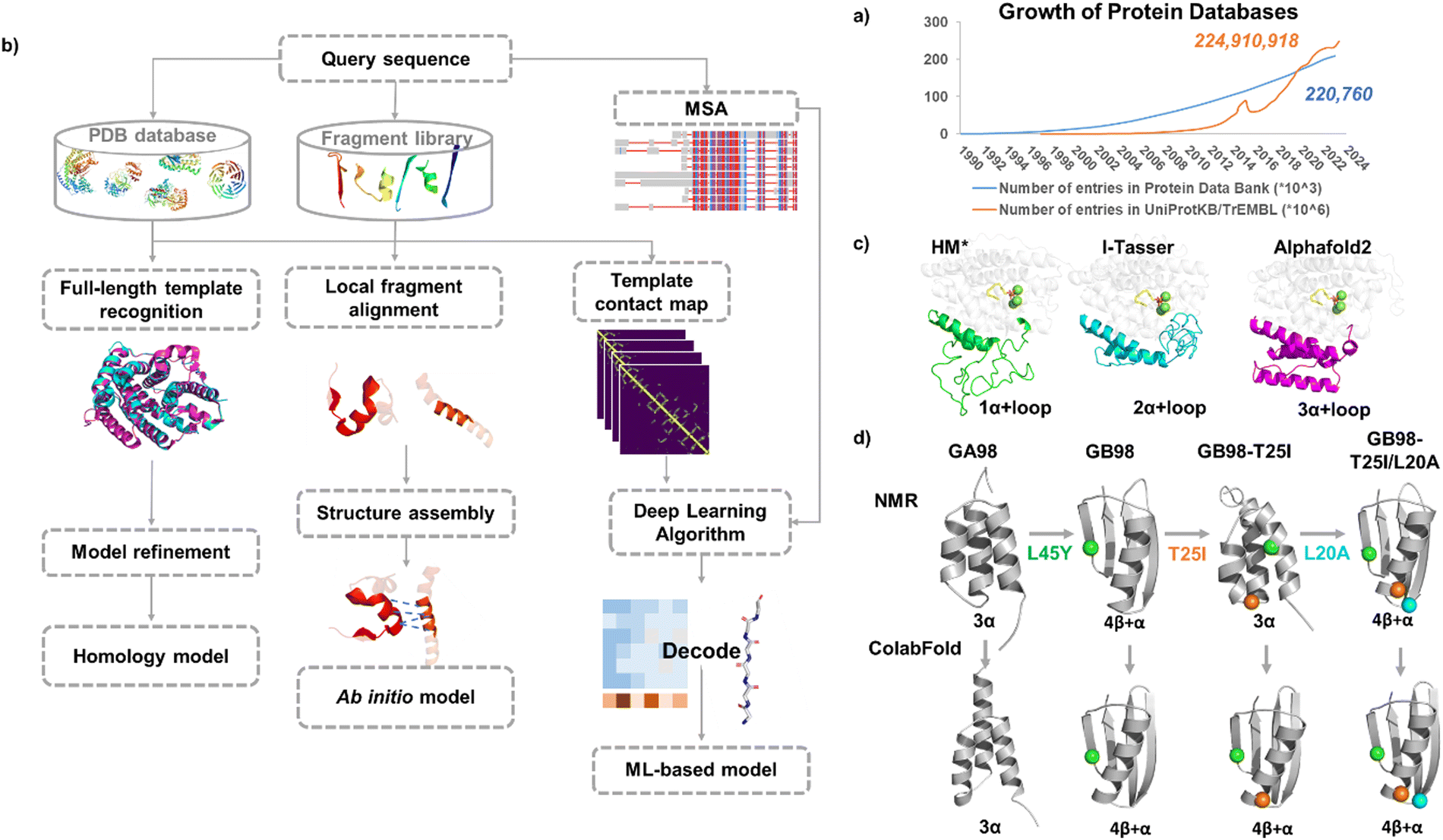 | ||
| Fig. 1 Molecular modeling in enzyme engineering. (a) Growth rate of the data in the Protein Data Bank and UniprotKB/TrEMBL database. (b) Protein modeling approaches. (c) Modelled structures for a new sesquiterpene synthase JeSTS4 using different protein modeling approaches.25 HM*: homology model was built using the crystal structure of the sesquiterpene synthase Copu9 from coniophora puteana (PDB: 7OFL26) as a template (sequence identity: 25%); ab initio models were built using I-TASSER and Alphafold2, respectively. (d) Modelled structures for the Ga98 variants27 with three progressed single mutations using ColabFold. | ||
2.1.1.1 Traditional modeling methods. When 3D structures of proteins are not available, computational methods have shown their power in predicting protein structures based on their sequences.28 Structure prediction approaches can be classified into template-based modeling represented by homology modeling and protein threading, or template-free modeling (ab initio modeling)29 (Fig. 1b).
For sequences that share certain homology with crystal structures, their homology models can be built using tools such as Modeller30 and Swiss-Model.31
For sequences with low sequence identity to known crystal structures, the fold recognition method (e.g. protein threading) can be used to predict structures by matching the query sequence directly onto the 3D structures of other solved proteins.
For sequences with no structural similarity to any solved proteins, ab initio modeling can be used to predict protein structures from scratch.
In principle, the global lowest energy conformation of a protein can be obtained using molecular simulations. In 1998, molecular dynamics simulations (MD simulations) disclosed a marginally stable folded conformation during the folding process of a 36-residue peptide,32 marking the first simulation-based ab initio modeling. Due to the demanding computational cost, it is impractical to predict full length protein structures using simulation-based ab initio modeling.
Currently, most of the ab initio protein structure prediction tools are composite approaches that combine fold recognition, structure assembly, and structure refinement. For example, I-TASSER developed by Zhang lab33 utilizes protein threading to identify similar structural motifs from the structure database, to assemble the well-aligned motifs. For the unaligned regions, Monte Carlo based modeling is used to predict the structure. In Rosetta developed by Baker,34 the target sequence is segmented into a consecutive window of three or nine residues and its structure is predicted by selecting fragments that are then assembled by a Monte Carlo strategy to construct the structure.
2.1.1.2 Deep learning-based structure prediction methods. AlphaFold135 secured the top ranking in the CASP13 free modeling (FM) category.36 AlphaFold1 extracts co-evolutionary information and employs neural networks to generate residue contact maps, which are then used to predict protein structures.
In contrast, AlphaFold219 employs a completely new architecture, differing significantly from previous methods which relied on residue contact maps to indirectly predict protein tertiary structures. The approach to predict protein structures is to learn the three-dimensional structure of proteins directly from their amino acid sequences, a so-called “end-to-end” learning method. AlphaFold2 has significantly advanced the development of “end-to-end” structure prediction, wherein the 3D structures of proteins are directly predicted using the multiple alignment of sequences of homologues as the input. DeepMind's AlphaFold2 achieved remarkable performance in the CASP14 competition,37 showcasing the accuracy and speed in predicting protein structures for the majority of the test cases. It utilized a so-called ‘Evoformer’ neural network block, which allows the exchange of information between the evolutional MSA and the spatial residue pair distances. The Evoformer network is followed by a structure module which produces the coordinates of each composition residue with the iterative refinements of local structures fulfilled by a novel equivariant transformer method. The constructed 3D structures are then relaxed using the OpenMM38 with the Amber99sb force field.39
During the preparation of this review, DeepMind recently released AlphaFold340 and provided a server for structure prediction (https://www.alphafoldserver.com). Compared to AlphaFold2, AlphaFold3 can predict ligand–receptor interactions. It simplifies the Evoformer algorithm and evolved into the Pairformer algorithm (by reducing the number of blocks) and adds a diffusion model after the Pairformer to predict the atom coordinates directly. However, there are still some limitations of AlphaFold3: firstly, the success rate of predicting complex structures with ligands is significantly lower than that of apo-protein; secondly, there is an insufficient accuracy in predicting ligand chirality during benchmark tests; and thirdly, there is a probability of substantial atomic clashing between subunits in multimer structures. Additionally, the AlphaFold3 server currently only supports the prediction of binding sites for dozens of common ligands/co-factors and ions, without support for custom ligands.
Additionally, inspired by AlphaFold2 and also serving as an improvement upon it, ColabFold41 combines the fast homology search function of MMseqs242 with AlphaFold2, and accelerated the prediction speed. AF-cluster43 samples multiple protein conformations on protein energy landscape by clustering MSA based on sequence similarity, which allows exploring the protein functions associated with different conformations.
Another recent implementation of deep learning in protein prediction is RoseTTAfold.19 RoseTTAfold also used the properties extracted from MSA and contact maps as the inputs for “end-to-end” prediction, but it utilized a three-track neural network architecture which allows the information retrieved from 1D sequences, 2D maps and 3D structures communicated via the transformer and attention mechanism and hence achieved accurate prediction of protein structures.
The large language model ESMFold developed by Meta AI is able to predict protein structures one magnitude faster with comparable accuracy, so it can be used for protein structure prediction for metagenomic proteins and it generated ESM Metagenomic Atlas database containing over 600 million proteins.44
The development of Alphafold2 has significantly expanded the reservoir of the 3D protein database. The AlphaFold Protein Structure Database created jointly by DeepMind and EMBL's Bioinformatics institute (EMBL-EBI) contains over 200 million predicted proteins from human proteomes and 47 other proteomes, which are free for public to download individually or via Swiss-Prot interface.
The sequence of a protein determines its structure, which in turn, determines its function. However, sequences lacking similarity may also exhibit similar catalytic sites.45 Benefiting from the above structure prediction tools, the 3D predicted structures in the sequence database have been greatly enriched. Ali Al-Fatlawi et al. showed that AlphaFold2 was able to uncover structures with similar core structural elements, whereas BLAST was unable to identify these similar structural features due to a lack of significant sequence similarity.46,47 Although protein structure search methods have shown great potential, sequence search methods such as BLAST still have advantages. For example, sequence alignment using BLAST is more suitable than structure alignment for structures containing more disordered regions.
Alphafold2 provides a reasonable starting point for enzyme design. For example, for a novel class I terpene synthases from moss Jungermannia exsertifolia,25 the low sequence identification (25%) with the template resulted in an poor homology model, particularly for the prediction of a key loop region 106–201 around the catalytic site, for which the corresponding structure is absent in the template. In contrast, the loop region was better defined by utilizing I-Tasser witch ab initio modeling and was further refined by AlphaFold2 (Fig. 1c).
Mutagenesis in enzyme engineering often only involves single or few mutations but could cause significant impact on enzyme structures and functions. Understanding the impact of structural changes caused by point mutation would accelerate the optimization of enzymes. However, it remains a matter of debate whether ab initio models are sufficiently accurate to pick up the effect of point mutations on local structural change. For instance, the ability of AlphaFold in predicting the effect of single mutations on protein stability (ΔΔG) and function was evaluated and little correlation was observed between the parameters derived from enzyme structures predicted by AlphaFold and the experimentally measured changes in protein stability or fluorescence levels.48 Whereas another research indicated that AlphaFold2 was able to predict the effect of single mutations on local structural deformation for a large range of proteins, using the measure of effective strain (ES).49 AF-cluster43 also demonstrated to be able to predict the conformational transition caused by point mutations in the case of KaiB from Rhodobacter sphaeroides.
These recent deep learning-based protein prediction methods can soon be widely applied in protein structure predictions. An interesting example was for predicting the structures of a designed chameleon protein Ga98 and its three variants with progressed single mutations. The NMR structures of the four proteins have been reported,27 and exhibit transitions between monomeric and folds, so were compared with the predicted structures. Parui et al. utilized ESMFold, AlphaFold2, and ColabFold to predict these structures,50 and ColabFold showed the best performance for the prediction of Ga98 among all, although it failed to predict the correct fold for GB98-T25I (Fig. 1d). The “AF-Cluster” method was able to accurately predict the structure of GB98-T25I but failed to predict the structures of Gb98 and GB98-T25I/L20A correctly.43
Structure prediction tools can serve as initial points for structural and functional analysis of enzymes, however careful inspection has to be conducted for the structure model obtained. Moreover, understanding the subtle mutation effects, particularly single mutations on enzyme properties such as enhanced stability or activity requires more precise structural simulations and sampling.
2.1.2.1 Classical MD simulation method. In structure-based drug discovery, protein targets are usually treated as fixed to allow large scale virtual screening to identify potential hits, by evaluating the binding affinities of small ligands in the binding pocket of the drug target, which can then be processed for bioassay. However, in biocatalysis, due to the promiscuity of enzyme's catalytic pocket induced by mutations or ligand binding, it is inappropriate to neglect the dynamic conformations of enzymes, which cannot be obtained by experimental X-ray, NMR or the ab initio models. Molecular dynamics provides an effective way to describe the dynamic properties of enzymes at the atomic level to interpret their functions.51 The development of molecular dynamics (MD) methodology tailored for biological macromolecules such as GROMACS,52 AMBER,53 CHARMM54 and OpenMM38 and acceleration of simulations by graphics processing units (GPU) on high-performance computing (HPC) has enabled accurate and fast prediction of protein structures as well as the binding modes of protein–ligand or protein–protein interactions.
CHARMM is one of the most widely used MD software packages and the CHARMM force field has been developed along with the software since the 1980s.55 A user-friendly graphic interface CHARMM-GUI56 was developed to prepare the input of simulations interfaced with widely used MD simulation packages such as CHARMM, GROMACS, AMBER and OpenMM. GROMACS52 is known for its highly optimal computing efficiency and open-source code and has become one of the most popular MD software packages for biomacromolecules. It is interfaced with different forcefields including AMBER99SB,39 CHARMM36,57 GROMOS58 and OPLS-AA/M.59,60 Benchmark studies on the commonly used MD simulation packages showed that GROMACS was optimal for biomolecular simulations of medium-sized systems at the microsecond level.61,62 The AMBER package53 includes the AMBER simulation software with the AMBER forcefield. The program assembly package AmberTools is freely accessible and convenient for preparing the input and result analysis. The input filed generated by AmberTools can also be converted by third-party scripts such as ParmEd (https://github.com/ParmEd/ParmEd) and acpype (https://github.com/alanwilter/acpype) so as to be readable by other MD software packages like GROMACS. Other efforts have been reported to automate the process of preparing the AMBER inputs and conducting result analysis.63 OpenMM38 is an open-source MD simulation package with a layered and modular architecture, making it easily integrable with other applications. It is highly extensible, allowing for the implementation of various plugins.
2.1.2.2 Enhanced sampling methods. Depending on the software, hardware and molecular system, the timescale of MD usually ranges from tens to hundreds of nano seconds. It has been demonstrated by a number of MD simulation case studies that the properties of protein–ligand complexes can be captured using simulations at the nano second time scale. However, it is difficult to observe large conformational changes for enzyme complexes e.g. from the reactant to product states of the enzyme by traditional MD simulations, because high energy barriers need to be overcome for the transitions between different conformations to take place, making it challenging to extensively sample free energy landscape.
Potential of mean force (PMF)64 is a modern statistical method commonly used to characterize the energetics of transitions in biomolecules. However, it is impractical to compute PMF directly from MD simulations because of the large configurational space of proteins and also a large energy barrier along the reaction coordinate. Various sampling techniques have been developed to effectively and accurately compute PMF. An effective technique in enhanced sampling to gain large-scale conformational changes is enhanced sampling65 including the umbrella sampling method,66 metadynamic method,67 accelerated molecular dynamics method (AMD)68 and replica exchange molecular dynamics, REMD.69
Umbrella sampling66 is one of the most widely used enhanced sampling methods in MD.70 The conformations between the thermodynamic states are sampled in a set of umbrella windows along the reaction coordinate ξ. At each window ξi (i = 1,2,3…N), MD simulations are conducted with a bias potential (umbrella potential) added to restrain the system around a narrow space around ξi so as to enable more efficient conformational sampling in this region.
The bias potential is usually calculated using a harmonic function
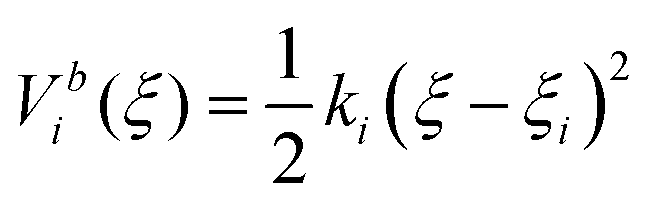 | (1) |
The free energy at the position ξi is calculated with the bias potential added onto the unbiased total energy of the state U(R), which is a function of the coordinate R
| Uib = U(R) + Vib(ξ) | (2) |
For each umbrella window, the probability distribution Pi(ξ) along the reaction coordinate is represented by an umbrella histogram hi(ξ). The weighted histogram analysis algorithm (WHAM) is a widely used technique in umbrella sampling to calculate PMF from the histogram, to resume the unbiased free energy profile by umbrella integration to obtain the complete free energy landscape along the minimum free energy pathway.
Umbrella sampling is traditionally combined with the post-analysis process. Following the MD runs for a number of biased window simulations, the neighbouring overlapping windows are combined, which allows the system to transit from one conformation state to another and generate the free energy over a large range of reaction coordinates. Adaptive umbrella sampling71 constructs a good biasing potential to counterbalance the free energy barrier, so as to allow self-consistently determining the bias potential with less human intervention to achieve a uniform distribution.
Metadynamics is also a bias potential-based method.72,73 Bias potential is placed on the Hamiltonian of the system thus the system would skip the transition barrier provided the growing bias potential counterbalances the transition barrier. This strategy can escape local minimum and allows for navigating free energy landscape as a function of a few collective variables (e.g. bond to be formed or broken, bond angle or dihedral) related to enzyme-catalyzed reactions with accelerated sampling. The choice of independent collective variables is crucial for those reactions for which prior knowledge of reaction coordinates is not available.67
Both umbrella sampling and metadynamics methods require prior knowledge on the degree of freedom for the motion of interest, based on either reaction coordinates or collective variables. The accelerated molecular dynamics method (aMD) does not need prior knowledge of potential energy wells or saddle points to explore the rare events that are related to the reaction. A bias potential is added to the true potential such that it is easier for the system to escape from the potential well and move from one low-energy basin to another. This strategy accelerates the sampling of the conformational landscape while converging to correct probability distribution. Replica exchange molecular dynamics based on a replica-exchange method (REM) also does not need knowledge of reaction coordinates. It generates an ensemble consisting of multiple copies (replicas) at different temperatures, and the copies are exchanged to overcome high-energy barriers so as to effectively explore the transitions among different states and conformational space.
These enhanced sampling methods have largely sped up the conformational sampling, however, they may still be slow processes while sampling irrelevant states so that not suitable to be used to refine the large scale predicted ab initio models. The Bayesian-based modeling employing limited data (MELD)74,75 method applies restraints to incorporate data in MD simulations with coarse physical insight, which harnessed weak information and generated multiple-funnel landscape, and sped up the sampling by up to five orders of magnitude. Recently, MELD combined with REMD (MELD × MD) was employed to predict the ab initio models of Ga98 and its variants (Fig. 1d)50 and accurately predicted all of the four structures.
The advancement of deep learning algorithms has also contributed to the development of enhanced sampling techniques.76,77 For example, Tao et al. developed a deep learning enhanced adaptive sampling method that can predict larger conformational changes efficiently.78 Tiwary et al. developed an enhanced sampling method that combined AlphaFold2 with deep learning enhanced MD to generate a collection of Boltzmann-weighted protein conformations from sequences, using the structures predicted by AlphaFold2 as the initial inputs.79,80 Combining deep learning with statistical mechanics, Noé et al. developed an adaptive sampling method that generated unbiased equilibrium samples of protein conformations using Boltzmann generators initialized by metastable states, without the need of prior knowledge of reaction coordinates.81
2.1.2.3 Binding free energy calculations. The catalytic efficiency of enzyme biocatalysts is dependent on both the thermodynamic binding free energy and reaction kinetic activation energy of the enzymes. The binding affinities of substrates in enzymes can be estimated by binding free energy calculations. The commonly used methods are MM/PB(GB)SA.82–84
In MM/PB(GB)SA, the MD simulation is run for the system solvated in a periodic box with water and counterions. Then the binding free energy between the enzyme and its substrate can be calculated for MD simulated structures processed by stripping the solvent and counterions, according to eqn (3):
| ΔGBinding = GES − GE − GS | (3) |
| ΔGBinding = ΔH − TΔS = ΔEMM + ΔGsol − TΔS | (4) |
Here, ΔH represents the binding enthalpy and −TΔS accounts for the conformational entropy change upon ligand binding. ΔH can be decomposed into different terms: the gas phase free energy contributions ΔEMM (eqn (5)) and the solvation free energy contributions ΔGsol (eqn (6)).
| ΔEMM = ΔEbond + ΔEangle + ΔEdihedral + ΔEele + ΔEvdW | (5) |
In eqn (5), ΔEMM includes the internal energy (ΔEbond, ΔEangle and ΔEdihedral), electrostatic contribution (ΔEele) and van der Waals contribution (ΔEvdW).
| ΔGsol = ΔGpol + ΔGnon-pol = ΔGPB/GB + ΔGnon-pol | (6) |
In eqn (6), the solvation energy can be decomposed into electrostatic term ΔGpol, and non-electrostatic term ΔGnon-pol. The PB and GB models estimate the polar component of the solvation. ΔGPB/GB is calculated with the electrostatic component calculated using the Poisson–Bolzmann equation or the generalized Born model.
The nonpolar free energy ΔGnon-pol is proportional to the molecule's total solvent accessible surface area (SASA), with a proportionality constant γ derived from experimental solvation energies of small non-polar molecules (eqn (7)).
| ΔGnon-pol = γSASA + b | (7) |
To decide the minimum free energy pathways between states of an enzymatic system, the free energy pathway can be explored by umbrella sampling breaking down the distance along the reaction coordinates into a series of very small coupling parameter λ (λ varies from 0 to 1). MD simulations are run at the fixed reaction coordinates along the reaction pathway and then the free energy change at each point is calculated by integrating the mean values of the derivatives (eqn (8)).
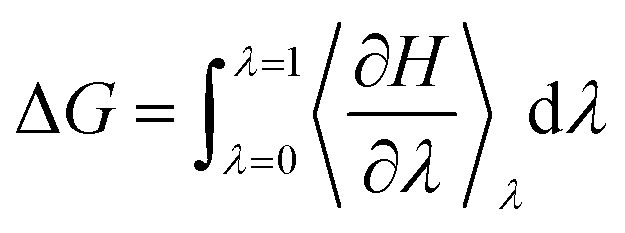 | (8) |
Another class of methods is alchemical methods, where binding free energy is estimated by the statistical analysis of the simulated thermodynamic pathway between two end states. Free perturbation (FEP)85 and thermodynamic integration (TI)64,86 methods are commonly used alchemical methods to explore the enzyme conformation landscape. In free energy perturbation (FEP),85 the free energy difference between two states of a system is calculated using eqn (9).
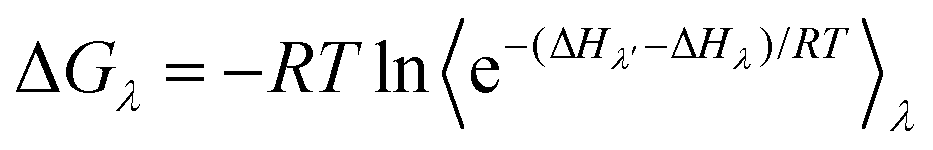 | (9) |
In thermodynamic integration (TI),64 the free energy difference between two states is calculated by the integration of the ensemble average of the derivative of Hamiltonian with respect to λ at different λ values for alchemical reaction pathways.
These robust free energy methods are accurate in principle but require extensive sampling from long MD simulations. They have been combined with conformational sampling techniques such as umbrella sampling and alchemical simulations to speed up the calculations.
2.1.3.1 QM cluster method. In the QM cluster method, the active site of the enzymes is calculated by QM methods most commonly density functional theory and the remainder of the enzyme is fixed and treated using the continuum solvent with dielectric constant ε = 4 to reduce the computing cost. The QM region is usually composed of the substrates, cofactors, metals and interacting residues with side chains truncated. The method is usually applied using different sized models; a smaller model to quickly explore possible reaction pathways, and a larger model to study the environment of the active site.87 With the increasing computing power, QM can contain more than 300 atoms nowadays.88
QM-Cluster methods optimize only truncated active site models, eliminating the degree of freedom of the region beyond the active site and hence reducing the complexity of the sampling problem. However, during the geometry optimization of a QM cluster model, geometric constraints have to be introduced to avoid the deformation of the active site in absence of the full protein environment. Dasgupta et al. proposed to apply a harmonic confining potential to the terminal atoms (“anchor atoms”) of the QM model, rather than using fixed- atom constraints adopted in traditional QM-cluster methods. This approach improved optimization efficiency and robustness in locating the transition states,87 and would be particularly useful for those enzymes with large conformational change during the reaction process involving notable entropic effects.
It is usually impossible to achieve reliable kinetic and thermodynamic results by calculating a small QM cluster model. A “maximal” QM cluster model with a residue interaction network of the entire protein was developed and provided reliable results.89 QM methods have similar computing costs to QM/MM calculations and are popular to those who are only interested in the overall reaction mechanism; however, they may generate different conformations compared to those predicted by QM/MM methods.
2.1.3.2 QM/MM method. Hybrid quantum mechanics/molecular mechanics (QM/MM) methods combine accurate QM methods to study the reactions and classical MM force field methods to capture the conformational energetics and have been widely used to study enzyme-catalyzed reactions.90–99 The starting structures can be obtained either from experimental X-ray or NMR structures or reliable molecular modeling followed by proper sampling from multiple replicas of MD simulations.
Additive QM/MM is a popularly used scheme based on the following equation:
| ETotal = EQM(R,r) + EMM(R) + EQM/MM(R,r) |
The effect of the MM region on the QM region is calculated using either electrostatic embedding or mechanical embedding. For accurate QM/MM studies, the polarization effect of MM estimated using the Drude oscillator (DO) model is insignificant for enzyme systems that involve no significant charge transfer.100 Appropriate choice of the QM region in the QM/MM calculations is crucial for attaining meaningful results.
Bím et al. recommended a mechanism-based practice for predicting the mutation effect on enzyme kinetics,101 which was in good agreement with the experimental value. It combined QM/MM and QM, where QM/MM is used to optimize the geometries of reactants, transition states, intermediates and products and QM is used to estimate the energies.
2.1.3.3 QM/MM MD method. QM cluster and QM/MM methods are suitable for exploring the potential energy surface of reactions. Since the enzymatic reaction process involves conformational dynamics, a combination of QM/MM and MD can be employed to extensively sample the potential energy surface. However, QM/MM MD simulations are computationally very expensive because the QM energy and forces are computed from a converged SCF at every step. For example, a QM/MM MD simulation with a QM region containing 49 atoms, using B3LYP density functional with the 6-31G* basis set and on an NVIDIA V100, can achieve only 1.86 ps per day.102 The scalable QM/MM MD calculation framework MiMiC103 enables running several ps per day in a single simulation using thousands of standard CPU cores.
Alternatively, a less expensive semiempirical method has been adopted in QM/MM MD to reduce the computing cost. For example, the PM3 semiempirical method was employed in a steered QM/MM MD in the hydride transfer mechanism study of zinc-dependent hydrogenase/reductase.104
The steered QM/MM MD method105 has been used to study the enzymatic reactions at an affordable time scale. This method applies harmonic forces on selected atoms to the reaction mechanism along the reaction coordinate and has been used for the design of industrial catalysts such as glycosyltransferases,106 ω-transaminase,107 and MHETase.108
In enzyme engineering, it is useful to know the binding free energy contribution from individual residues. Recently, an ab initio QM/MM109 method was reported to obtain the electrostatic, polarization and van der Waals contributions from each residue to the activation barrier, as well as the contributions from different collective variables along the reaction coordinate to explore the possible reaction mechanism. This was achieved through a mean force integration along the free energy pathway and the reaction coordinate by analyzing the MD simulation trajectories.
For tutorial and practical guidance on the QM cluster, QM/MM and QM/MM MD multiscale simulations on biomolecules, we recommend reading recent reviews.110–112
2.2 Enzyme design applications
There are perennial challenges in enzyme design to identify the active site related to the reaction mechanism and fine-tune enzymes to improve their properties. The enzyme fitness landscape describes the relationship between the enzyme variants and fitness, which measures how well a given enzyme can perform a target function (Fig. 2a). However, the potential protein sequence space is vast, necessitating effective strategies to search through it and identify sequences with desired functions. Common strategies include random mutagenesis, semi-rational design, rational design, and de novo design.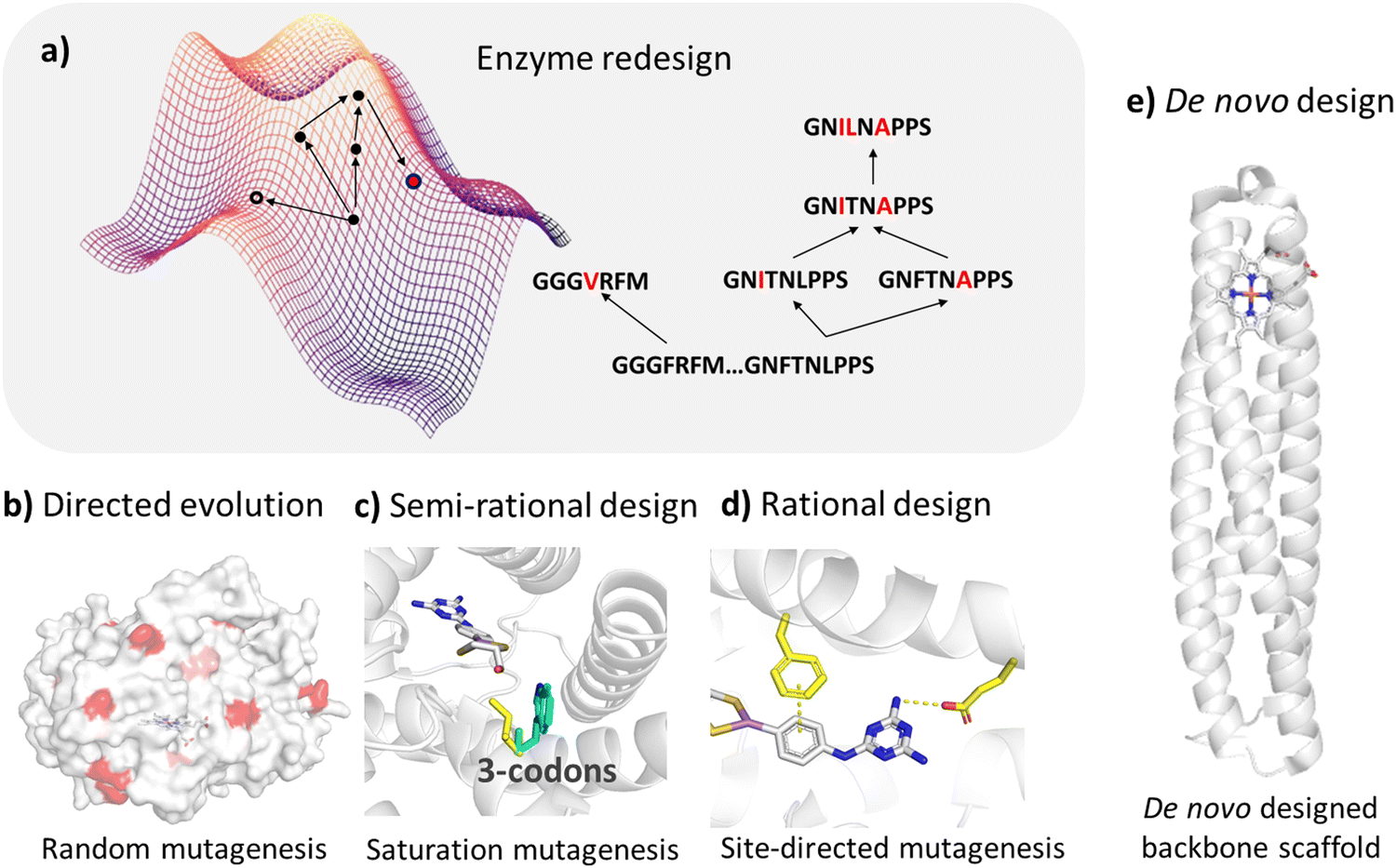 | ||
| Fig. 2 Enzyme design approaches. (a) The fitness landscape map of an enzyme shows the relationship between different variants of an enzyme and their fitness (such as catalytic efficiency, thermal stability, substrate specificity, etc.). Each variant corresponds to a point on the map and the height of the point represents the fitness of the variant. (b) Directed evolution mimics the natural evolution process to improve the function of proteins through multiple rounds of random mutation, screening and selection. (c) In the semi-rational design approach, the key sites identified based on enzyme structures are mutated with saturation mutagenesis to improve the enzyme function. (d) In the rational design approach, the sites identified based on the dynamic structures and catalytic mechanism of enzyme are mutated to improve protein function. (e) De novo design methods are used to construct protein backbones from scratch to generate protein structures with new functions. | ||
Random mutation is conducted when structures are not available and is often combined with high-throughput screening. Hence, we will not discuss this strategy in our review. Compared to high-throughput screening, rational and semi-rational enzyme design strategies demonstrate significant promise due to their reduced cost and efficiency.
The semi-rational design strategy is based on structures and prior knowledge of enzyme functions. It constructs small libraries by performing site-directed mutagenesis on several specific residues, which are identified around the catalytic site of the enzyme.
Rational design strategies typically utilize molecular modeling and structural sampling methods to explore enzyme–substrate binding modes. Additionally, dynamic structures are considered through molecular dynamics simulations and the reaction mechanism is explored by employing quantum mechanical calculations, thereby greatly reducing the search space on the fitness landscape.
Both semi-rational and rational approaches focus on modifying natural enzymes to alter or confer new catalytic functions, while de novo enzyme design strategies aim to generate novel enzymes usually by incorporating the active site of the reaction into a simplified artificial protein scaffold.
There are many structure-based enzyme design/engineering studies. Here we focus on recent computer-aided enzyme design cases that were guided by semi-rational and rational design strategies to improve the enzyme properties, such as enhancing enzyme's activity, controlling regio- or enantio- selectivity preferences, broadening substrate scope and altering enzyme function.
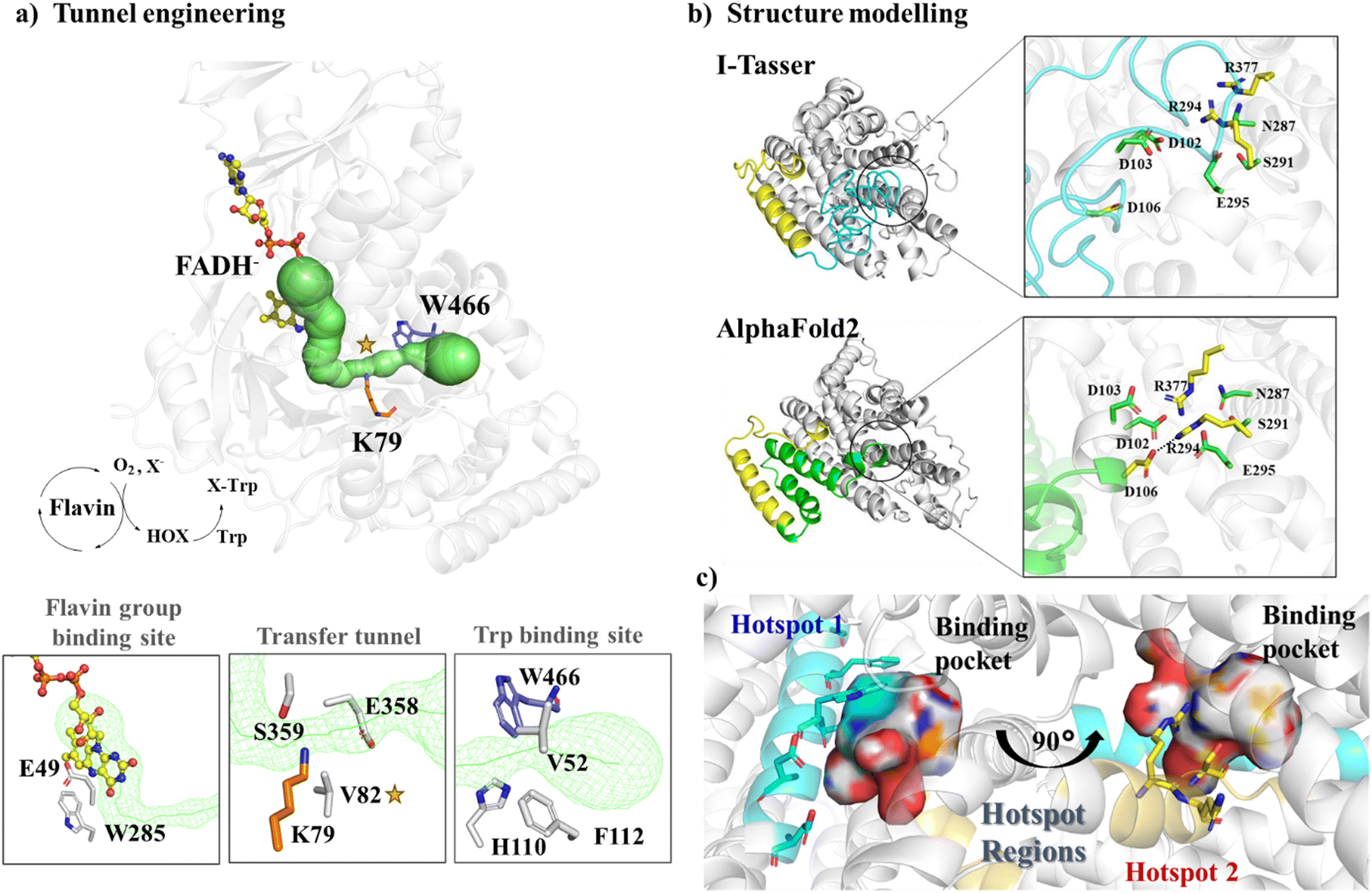 | ||
| Fig. 3 Hotspot region identification in semi-rational design approaches. (a) Engineering the tunnel (shown in green) passing through the FADH− binding site and the tryptophan binding site. The structure is produced based on the crystal structure of flavin-dependent halogenase (FDH) (PDB ID: 7CU2115). (b) Structural modeling of Wild Type JeSTS4 by I-Tasser and AlphaFold2. (c) The two hotspot regions were identified for JeSTS4 by combining coevolution and the structural information obtained from MD simulations. Reproduced with permission.25 Copyright 2022, American Chemical Society. | ||
Multichemical state analysis (MCSA) is an enzyme design method developed for the redesign of enzymes with multiple substrates. Large structure ensembles were abstracted from MD simulation to model each of the chemical states, and library design was performed by sub-designs comprising overlapping subsets of the total designed positions, thus the sequence space was explored effectively. The enzyme sequences were optimized and a ranked list, which is based on Boltzmann-weighted sequence energies averaged over the structural ensembles, was used to generate a position probability matrix (PPM) for each sub-design. Screening a designed small combinatorial library for aminotransferase gave promising variants with up to 200-fold improvement in catalytic efficiency.116
In the absence of a crystal structure, different modeling methods can be used to generate enzyme structures. Qin et al. constructed the structure of L-lysine hydroxylase from Niastella koreensis (NkLH4) through homology modeling and achieved a 24.97-fold increase in activity for L-lysine by employing semi-rational combinatorial active-site saturation test (CAST) on four positions.117
For proteins with low sequence homology with any possible templates, AlphaFold2 offers significant advantages over traditional modeling by using deep learning to predict protein structures. For example, a novel class I terpene synthase discovered from Jungermannia exsertifolia for bicyclogermacrene synthesis shares a low sequence identity with any enzymes. AlphaFold2 outperformed traditional modelling, particularly in loops near the active site25 (Fig. 3b). Guided by structural information along with co-evolution analysis, we identified two hotspot regions (Fig. 3c) and mutations resulted in a significant increase in conversion. Furthermore, based on the structure of glutamate dehydrogenase (GluDH) predicted from AlphaFold2, Yang et al. designed the A145G/P144A/V143A mutant, which expanded the substrate binding pocket and exhibited a remarkable increase in catalytic activity towards bulky substrates.118 In another research, a thermostable P450, CYP175A1 was engineered by tunnel engineering the hot spot residues identified by MD simulations, leading to improvements in hydroxylation activity and regioselectivity of the enzyme.119 Many other successful semi-rational design strategies by reshaping of active sites have been employed to enhance the catalytic efficiency of enzymes, just to name a few ADH enzymes,120,121 P450 enzymes,122,123 and PET hydrolase,124etc.
Rational enzyme design strategies are based on an understanding of enzyme structure–function relationships to predict potential mutations with desired properties. Reasonable reconstruction of the residue interaction network of the active site, including hydrogen bonds, salt bridges, hydrophobic interactions and other interactions formed between the substrate and the enzyme active site residues, can influence the enzyme catalytic processes (substrate binding, transition state stabilization, and product release). Mutation or substrate binding usually induces conformational change of enzymes. In rational design strategies, the dynamic conformations of enzyme should be considered.
Local conformational changes introduced by remote mutations of remote site residues may propagate into the active site so as to affect enzymes’ catalytic efficiency, specificity and substrate scope by reshaping the active site pocket. Mutating a second sphere residue caused the conformational change of adjacent loops as disclosed by MD simulations, which resulted in different preferences of stereo-regio selectivity by the reshaped binding pocket.125 Directed evolution of P450LA1 catalyzed the oxidation of arylalkene to produce ketone products with high activity and enantioselectivity. MD simulations disclosed the distal mutations resulted in a packed and rigid active site compared to the WT with increased dynamic networks, i.e. the dynamic interaction between distal residues and their surrounding residues, which preorganized the active site favourable for the carbocation intermediate.126
Flexible loops are often observed in enzymes serving as the lid of the active site. Manipulating the loop conformational dynamics has become a powerful strategy in enzyme engineering to regulate enzyme functions.127 The effect of distal loop fluctuation on enzyme properties is yet to be known, which brings out the challenge to identify distal loops for enzyme engineering. Recently, a remote flexible loop of a transglutaminase was identified from MD simulations and the mutants were generated by saturation mutagenesis of the residue using Rosetta enzyme design, among which two mutants were identified with increased activity and thermostability.128
Quantum mechanics methods enable precise modeling of the electronic structure of enzyme-catalyzed reactions. Through QM/MM calculations, key information such as catalytic mechanisms, transition state structures, and reaction pathways can be revealed to help understand the functional mechanism of enzymes. Computational simulations of the phosphoryl transfer catalyzed by bimetallic phosphatase of the flavobacterium (PafA) enzyme showed that the mutation of the second-sphere residues modulated binding of the charged substrate rather than the transition state. Additionally, the cumulative mutations modulated the level of hydration of active sites and water-mediated H-bond networks and hence resulted in increased catalytic efficiency.129 From MD simulations followed by QM/MM calculations, we disclosed that the regioselectivity and activity of a P450BM3 variant IV-H4 for the hydroxylation of terpenoid artemisinin were originated from the control of the substrate entrance by a hydrogen bond to adopt an open conformation so that it demonstrated different regioselectivity from other variants.130
For multi-domain enzymes, mutation of interface residues can be guided by the structure of the multimer and it impacts the enzyme's catalytic efficiency and specificity. Based on the crystal structure of β-amino acid dehydrogenases (AADH), the substrate binding pocket is located at the dimeric interface of the enzyme. The E310G mutations combined with A313Y achieved increased enzyme activity by 200-fold in the asymmetric synthesis of (R)-β-homomethionine131,132(Fig. 4).
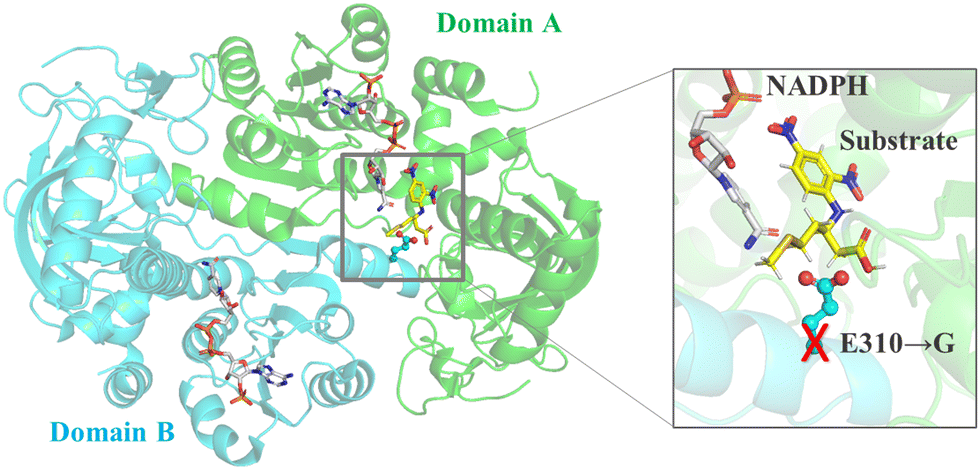 | ||
| Fig. 4 Engineering interface residues for enzymes with multiple domains. Engineering the interface residue E310 into small glycine in β-amino acid dehydrogenase would create additional space, thereby expanding the substrate spectrum. | ||
Ene-reductases are flavin proteins from the old yellow enzyme family (OYEs) that catalyze the asymmetric hydrogenation of alkenes to give chiral products and are of great interest to industry.133 Based on the crystal structure and homology models of variants, the preference toward the admirable (R)-enantioselectivity was achieved for both E- and Z-citral isomers, by only introducing one or two mutations for a NADPH-dependent OYE enzyme OYE3.134 Site-directed mutagenesis based on the crystal structural analysis of two stereocomplementary OYE enzymes GsOYE and BfOYE4 gave stereodivergent products.135
Cytochrome P450 enzymes are a superfamily of enzymes that are important for the synthesis of complex bioactive molecules such as natural products and drug metabolism. Based on the crystal structure, the regioselectivity of P450 BM3 was tailored to give hydroxylated derivatives at different positions of a sesquiterpene lactone compounds parthenolide (PTL) and micheliolide (MCL).136,137 Based on the analysis of the crystal structures of two P450 enzymes IkaD and CftA, it was suggested that the structural difference at the polar moieties of the two enzymes accounts for the regioselectivity and chemselectivity for PoTeM,138 and the regioselectivity of a P450 enzyme IkaD for a polycyclic tetramate macrolactams (PoTeM) ikarugamycin was altered by fine-tuning the catalytic pocket.138
In the search for stereocomplementary serine lipase CALB, all four stereodivergent variants of serine lipase CALB were obtained by only screening an ultra-small variant library constructed based on the MD simulated structures preferable to the four respective stereoisomer products.139 By employing a workflow combining Rosetta enzyme design and MD simulation-based free energy ranking, Delgado-Arciniega et al. introduced 6–8 simultaneous mutations in a ketoreductase and altered the enantioselectivity. They experimentally characterized only four variants and found three variants exhibited inverted enantioselectivity in the reduction of acetophenone-like substrates and an α-keto ester, significantly reducing the experimental screening workload.140
Based on the substrate binding mode of wild type cyclohexanone monooxygenase (WT-CHMO) studied from MD simulations, we found that the substrate is sandwiched between the top or bottom of the binding site featured by two residues F434 and L437 (Fig. 5a). A single mutation at either position led to a complete reversal of enantiopreference towards 4-alkyl and 4-phenyl substituted cyclohexanones.141 However, there is still room for further improvement in reversing the enantioselectivity for cyclohexanone with short substituents like a methyl or ethyl group. Therefore, we designed the F434I/L437A/T435L triple mutation to reconstruct a smaller binding pocket and achieved complete reversal of enantiopreference for cyclohexanone with short substituents.142 Furthermore, we found that replacing F279, located in the second sphere near the active site and forming hydrophobic interactions with F434, with a larger residue like tryptophan, would achieve a marked improvement in enantio- or regioselectivity across a wide range of substrates. Conversely, replacing it with smaller residues would achieve a complete reversal of enantiopreference (Fig. 5b).125
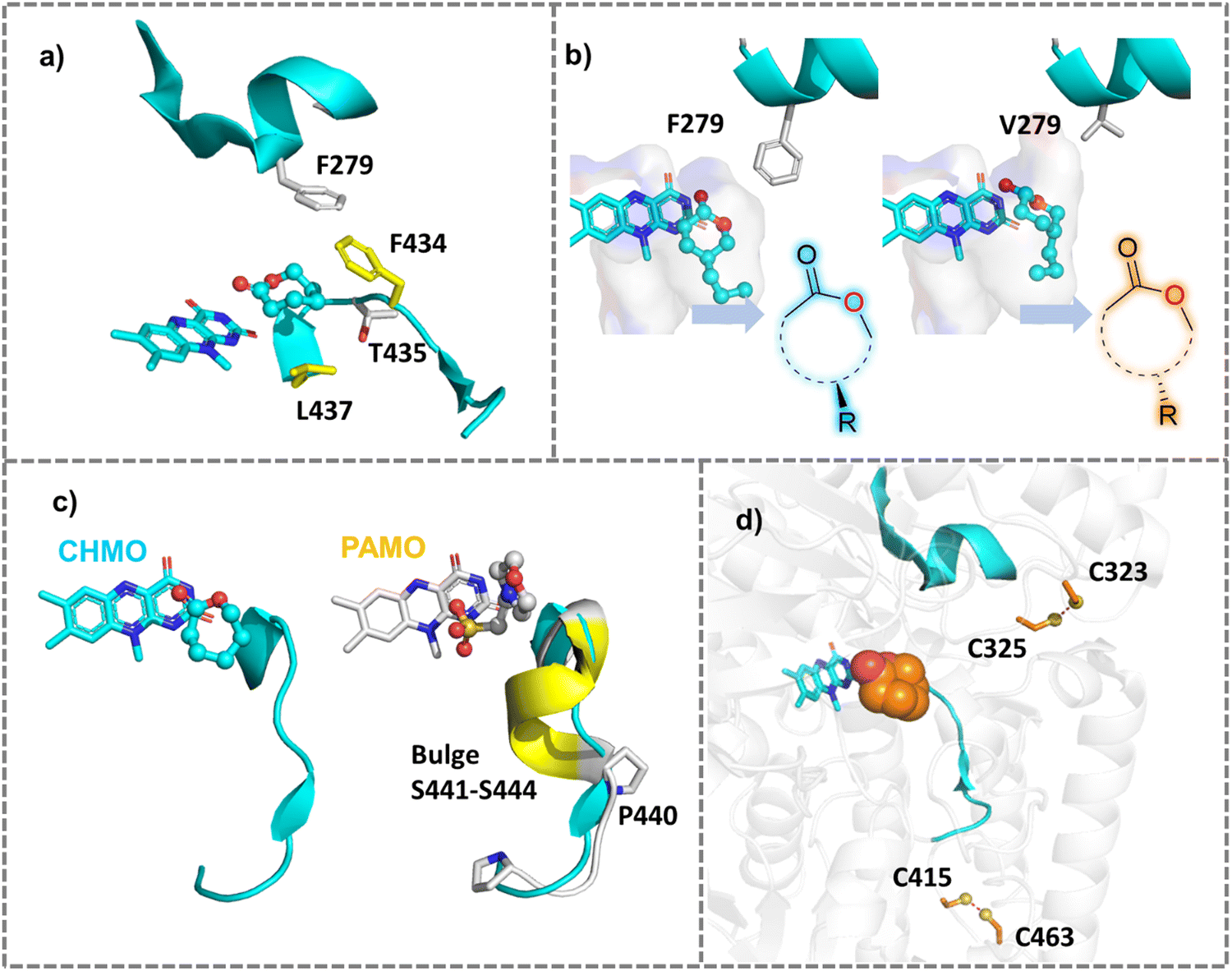 | ||
| Fig. 5 Mutations of Baeyer–Villiger monooxygenases (BVMOs) for improved properties. (a) Single mutation at two active residues F434 or L437 surrounding the substrate reversed the natural enantiopreference of WT-CHMO.141 The crystal structure of CHMO (PDB ID: 4RG3143) was used. (b) Engineering the second sphere residue F279 into smaller residues like Valine reversed the enantioselectivity of CHMO toward diverse substrates.125 (c) Expanding substrate scope of PAMO by engineering the bulge region that is present in PAMO but absent in CHMO.144 (d) Improving the thermal stability of CHMO by creating additional disulfide bonds between two adjacent cysteine residues. | ||
For the design of terpene synthases, the water flow regions identified from MD simulations provided guidance on reshaping the active site of a sesquiterpene synthase to catalyze the synthesis of a valuable terpenoid product while avoiding the hydroxylated product.145 A single mutation of another sesquiterpene synthase, pentalenene synthase, diverted the reaction pathway to give different products, because of the reshaped binding pocket disclosed by molecular docking and MD simulations.146
QM/MM and MD simulations disclosed the reversed regioselectivity of thermostable CHMO (TmCHMO) for 4-phenyl-2-butanone to give the abnormal product attributed to the conformational changes in the Criegee intermediate and transition states in the reaction pathway.147 MD simulations and QM/MM calculations elucidated the catalytic mechanism of PAMO toward its native substrate phenylacetone and the alkyl migration mechanism of the Criegee intermediate decay.148 Furthermore, based on MD simulations of PAMO, we proposed the requirements for a catalytic pocket favourable for non-native linear substrate 2-octanone, which provides structural insight for further engineering the enzyme to accommodate linear substrates.149 QM cluster calculations disclosed that the change in the chirality of the Criegee intermediates and transition states accounts for the regioselectivity so as to give the normal or abnormal products by the WT-TmCHMO and its variants, respectively.150
PAMO is a thermostable enzyme with high industrial value. However, it has a narrow substrate acceptance range compared to CHMO. Structural comparison showed a bulge (S441–S444), which is present in PAMO, but absent in CHMO (Fig. 5c). Deleting the bulge in PAMO turned the enzyme into a phenylcyclohexanonase (PCHMO), which showed a broadened substrate spectrum.144 Saturation mutagenesis of the bulge region in PAMO using codon degeneracy was conducted and variants that accept 2-aryl cyclohexanone were attained.152 Mutating a second sphere residue P440 around the bulge achieved the acceptance of a range of substrates.153 In another work, structure-guided rational design altered the functionality of CHMO to allow it to reduce a range of substituted aromatic α-keto esters. With high catalytic activity and stereoselectivity. The created reductive activity was attributed to shortened reaction coordinates favourable for hydride transfer in the ketoreductase-like variants in comparison with the WT enzyme, as observed from docking and MD simulations.154
The types of tunnels in metalloenzymes catalyze the reductive or oxidative transport and positioning of small gaseous substrates such as H2, N2, NH3, CH4, O2, CO, CO2, etc. dictates the substrate preference, and therefore reshaping the gaseous tunnels would affect substrate selectivity and enzyme functions.155 The substrate tunnel of a soluble methane monooxygenase (sMMO) hydroxylase has been revealed based on different approaches such as crystallography, MD simulations and mutagenesis of the tunnel-lining residues.156
Engineering the composition residues lining the access tunnel of P450Bsβ changed the substrate preference.157 Hotspot identified by MD simulations of haloalkane dehalogenase for the catalytic transformation of linear and branched substrate disclosed the requirements for substrate specificity.158
Ergothioneine sulfoxide synthase from Candidatus Chloracidobacterium (EgtBCth) possesses both EgtB- and Egt1-type activities with the EgtB-type feature more prominent than the Egt1-type; however, the latter is more industrially valuable. By leveraging active site information from EgtBCth crystal structures, EgtBCth variants were designed using Rosetta enzyme design159 and three mutants were tailored to exhibit Egt1-type characteristics.160
Comparison of the key active-site residues in the crystal structures of MPD and MDD that are involved in the bifurcated mevalonate (MVA) pathway, combined with sequence analysis, disclosed the key active-site residues that confer substrate specificity, which facilitated distinguishing enzyme classes involved in two MVA metabolic pathways.161 In another example, sequence comparison and structural analysis of the homology models of two homologous maize terpene synthases TPS4 and TPS10 disclosed the difference in the key active site residues that determined product specificities, and combined mutation of the different residues in the first and second sphere turned TPS4 into TPS10.162
5-Methylene-3,5-dihydro-4H-imidazol-4-one (MIO)-enzyme family comprises two classes of enzymes with different functions, i.e. aromatic amino acid ammonia lyases (ALs) and 2,3-aminomutases (AMs). Based on the crystal structure of an AL, the substrate binding tunnel of AM was engineered, and the resulting variant showed enzyme function of AL.163
Based on the homology model of a sesterterpene synthase SmTS1 and multiple sequence alignment, engineering the substrate binding site residue displayed the function of diterpenes synthase.164 Similarly, in a semi-rational design based on the crystal structure of a diterpene synthase VenA, VenA was changed to a sesterterpene to accommodate larger substrates.165
The surface charge of enzymes also plays a crucial role in determining their pH-activity profile.173–175 For example: the NADH Oxidase from bacillus subtilis exhibits maximum activity at pH 9.0, whereas the pH of its coupled enzyme dehydrogenase is close to 7.0, making the practical industrial application challenging.176 Introducing negatively charged residues on the enzyme surface using Rosetta design lowered the optimal activity pH to 7.0.177 In industrial production, vanillin is produced from waste biomass resources and then vanillin is converted to vanillic acid by vanillin dehydrogenase (VDH) under alkaline conditions; however, VDH displayed poor activity at alkaline pH. By mutating non-conserved, negatively charged surface residues to positively charged arginine, the optimal activity was shifted from pH 7.4 to pH 9.0.171 The comparison of the crystal structures of two SGNH family esterases CrmE10 and AlinE4 showed that the two enzymes have different electrostatic potentials on enzymes’ surfaces. Engineering the charge of CrmE10 surface residues from acid to basic improved the alkaline adaption and therefore increased the enzyme's activities (Fig. 6).178
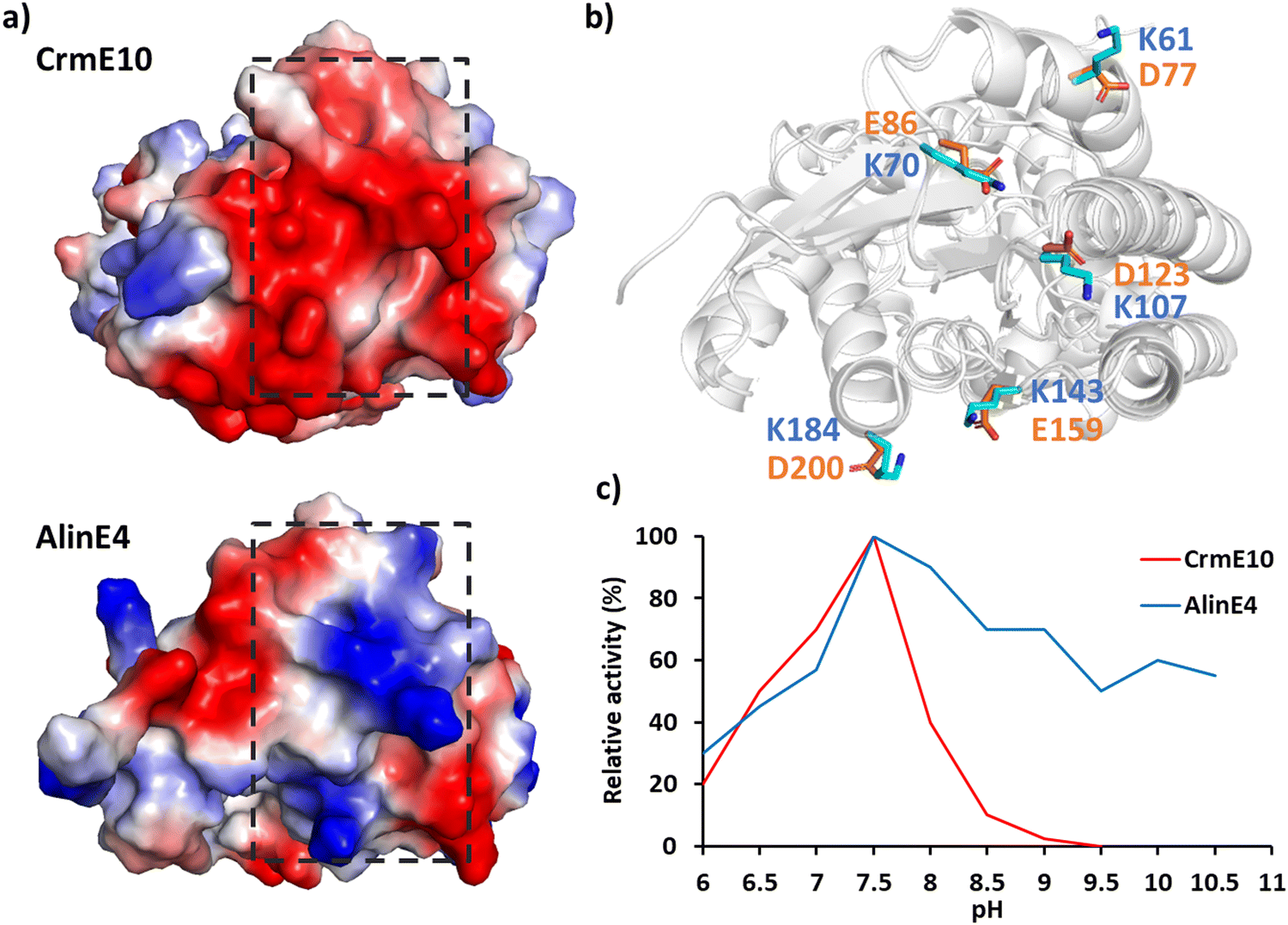 | ||
| Fig. 6 Effect of surface electrostatic potential on activity. (a) Protein surface electrostatic potential of two homologous enzymes of the esterase family CrmE10 (top right, PDB: 7C23178) and AlinE4 (bottom right, PDB: 7C82178). (b) Superimposition of CrmE10 and AlinE4 with the key polar residues on the surface shown in stick mode. (c) The pH/activity profile of CrmE10 and AlinE4. | ||
Highly flexible residues may be responsible for protein unfolding and denaturation, leading to decreased thermostability. The highly flexible residues in levansucrase were identified by root mean square fluctuation (RMSF) for MD simulations of the enzyme crystal structure and these residues were mutated to improve the thermostability.182 The difference in free energy (ΔΔG) between the mutant and wild-type enzyme was calculated to assess the stability of mutants and experimental evaluation shows that the designed K82H/N83R mutant is more thermostable than the wild type. A similar design strategy combining MD simulations and ΔΔG calculations has been used to guide the design of carrageenase,183 lipase,184 and tyrosinase,185 which attained variants with improved thermostability.
Disulfide bonds can reduce the configurational entropy of the unfolded polypeptide to stabilize the structures of protein.186 Disulfide bonds can be introduced at non-catalytic residues using MODIP,187 DbD2188 or BridgeD189 server and the effect of designed disulfide bonds on thermostability can be evaluated by calculating ΔΔG between the designed mutants and the WT enzyme. Two disulfide bonds (S61C–S115C and E190C–E238C) were designed for Rhizopus oryzae lipase (ROL) to rigidify the enzyme, and the thermal stability of the enzyme successfully increased by 5.0 °C and 6.9 °C, respectively.190 The introduction of disulfide bonds near the binding site of divalent cations (e.g. Ca2+, Mg2+) effectively improved the thermostability of polyethylene terephthalate (PET) hydrolase.191 A simultaneous improvement of stability against oxidation of and thermostability of CHMO was achieved by introducing new disulfide bonds guided by a computational study192,193 (Fig. 5d). In some enzymes, cysteine and methionine are liable to be oxidized and therefore hamper enzyme activity. Mutating the cysteine and methionine into non-polar residues or serine may enhance oxidative stability and hence thermal stability.194
3. Machine learning-accelerated enzyme design
Molecular dynamics simulations and the QM/MM method provide valuable insight for atomic level conformational dynamics mechanisms, and the enzymatic reaction mechanism; therefore, they have been widely used to explore conformational space and structure–function relationship. Furthermore, the advances in computer hardware along with the development of accurate force fields and highly efficient sampling methods have enabled employing molecular simulations for enzyme design.195–198 For example, modulating the protein stability guided by MD199 and enzyme engineering for natural product biosynthesis aided by QM/MM.200With the dawn of the big data era, various biological databases have become available and machine learning methods have been applied in enzyme engineering.21,201–205 The advent of a tremendous amount of data from the literature or databases enables us to build machine learning models and implement them into the screening protocol, for example, machine learning guided protocols were reported to predict the properties of mutants so as to reduce the screening demands by traditional experimental high throughput screening.206,207
Machine learning (ML) benefits from molecular modeling and accumulated experimental data. It has been implemented in molecular modeling based on atomistic MD and quantum mechanics and facilitated the effective multiscale or coarse-grained modeling, and therefore enabled exploration of the vast space of functional enzyme sequences speeding up the screening of functional enzyme variants.208–210 The three-pronged atomistic simulations, machine learning and experimental validation, can be synchronized, functioning just like a troika, and would speed up the efficient screening of potential mutants in the enzyme design protocol, with enhanced accuracy in predicting the effect of mutations.
To enable interdisciplinary collaboration between experimentalists and computational scientists, it is essential to understand how computers store and process data in a way that is understandable by both parties to facilitate collaborations.211
In this section, we will introduce the data processing methods, including the methods of generating descriptors from small molecules and proteins, and utilizing various databases as the data resources for machine learning. Model building and evaluation methods will also be introduced. Finally, the latest machine learning research on enzyme engineering will be reviewed.
3.1 Descriptors for small molecules
To retrieve meaningful patterns and rules in machine learning, the databases need to be processed and converted into numerical descriptors. For example, molecular descriptors representing molecular features are developed to predict the biological activities and screen potential lead compounds in QSAR.212 These molecular descriptors are classified as 1D global property, 2D planar features or 3D stereo features.Removing irrelevant descriptors may improve the accuracy of the prediction to develop robust models. Khan et al. reviewed descriptor selection methods in different drug design cases,212 including the filter method that gradually deletes the low-score features by calculating relevance scores of the descriptors and Wrapper method that gradually deletes descriptors guided by the errors in a validation subset using a support vector classifier.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, the number of H-bond donors/acceptors, etc. which are essential properties for drug's pharmacokinetics and hence have been widely used in drug development.214 In addition, atom-type counts, bond-type counts, and molar refractivity are also global descriptors. It should be noted that most of the global descriptors lack information on the molecular structure or atom connectivity.
P, the number of H-bond donors/acceptors, etc. which are essential properties for drug's pharmacokinetics and hence have been widely used in drug development.214 In addition, atom-type counts, bond-type counts, and molar refractivity are also global descriptors. It should be noted that most of the global descriptors lack information on the molecular structure or atom connectivity.
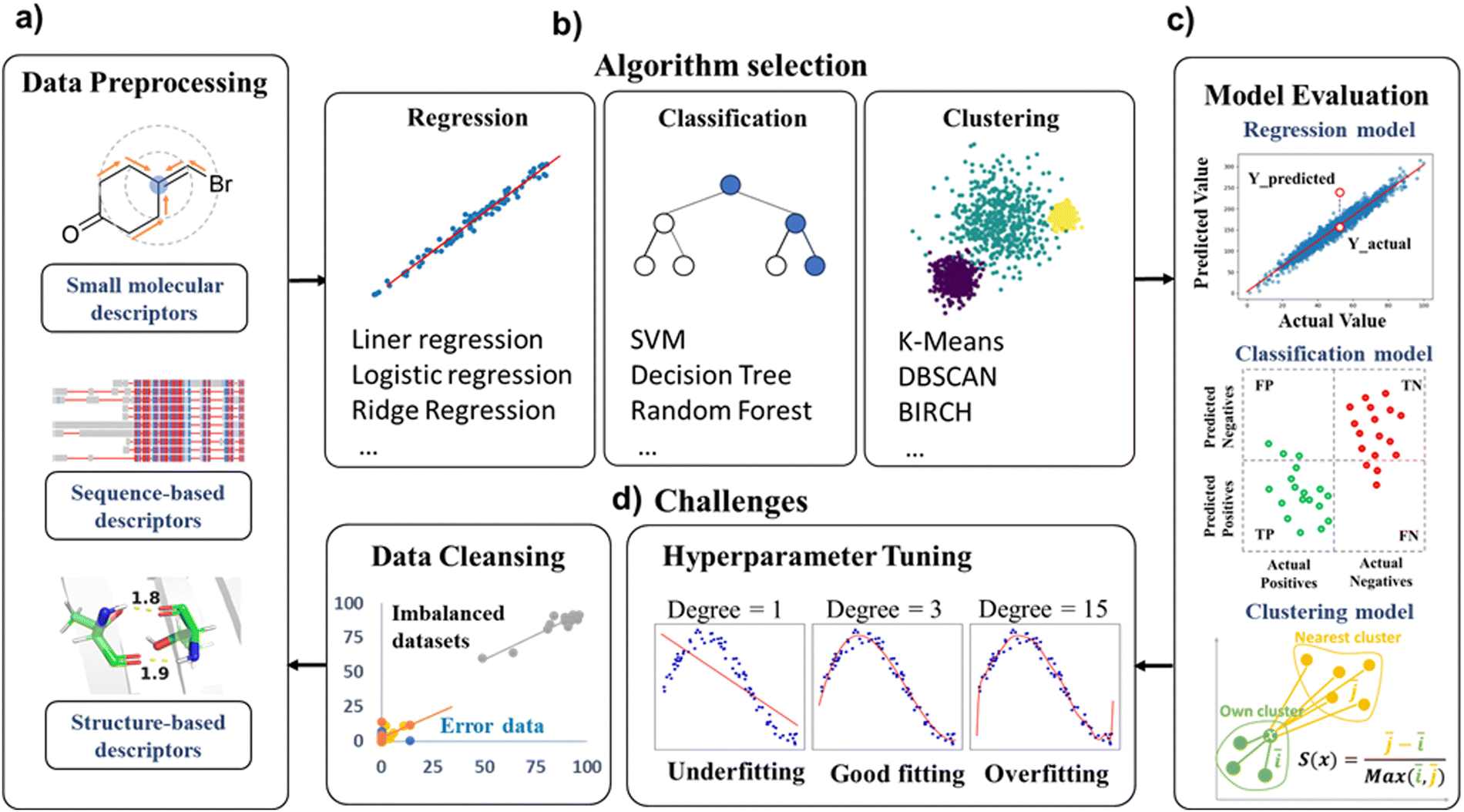 | ||
| Fig. 7 Machine learning for enzyme design. (a) The data used in enzyme engineering modifications mainly consist of small molecules and protein descriptors. (b) Some commonly used algorithms in regression, classification and clustering models. (c) Evaluation metrics in machine learning models. (d) The challenges in achieving a predictive ML model: the imbalanced distribution of data requires manual curation, i.e. some error data must be corrected prior to data preprocessing to assure the quality of prediction models; the issues of model underfitting/overfitting addressed by hyperparameter tuning during model optimization. | ||
Additionally, molecular structures are compressed into library-based 2D representation by a molecular “fingerprint”, which projects the structure information of molecules into binary codes, with each bit representing molecular structure features or the presence/absence of certain structures. The binary representations such as MACCS219 are compatible for data storage and also liable for comparing the similarity among molecules.
In contrast to library-based fingerprint representation, circular fingerprints220 such as Morgan fingerprints, extended-connectivity fingerprints (ECFPs) and functional-class fingerprints (FCFPs) take into consideration of the local environment of molecules to generate a bit vector. For example, the Morgan fingerprint with a radius of 2 considers the connectivity of each atom to other atoms which are linked to the first atom by up to two chemical bonds; it assigns a value of 1 if such a neighborhood is present in the molecule, otherwise, it assigns 0. These fingerprint methods have been implemented in RDKit toolkits.221 The vectors generated by fingerprint methods are high dimensional and sparse, and often bring about the issue of bit collision. Google Inc. compared the quality of word representations in vector space for a very large dataset in a word similarity task and reported two model architectures with promising prediction accuracy and efficiency.222
Convolutional neural network and natural language processing (NLP) techniques have been used in molecular graphic representations. Fuller and Turk et al. reported a Mol2vec algorithm223 to represent the substructures of a molecule as word vectors and the whole molecule as a sentence. Thus each substructure in the molecule can be more efficiently represented.
Molecular structures can also be represented by molecular graphs. With the development of the graph neural network, each atom in a molecule can be considered as the nodes in graphic structures and the connectivity among atoms are defined as edges. The graphic frame can describe the complicated relationship among the substructures by graphs. Utilizing the graph neural network (GNN), molecular graph descriptors have been widely used in predicting drug–target interactions.224–227
To evaluate the catalytic efficiency of enzymes, it is important to estimate the enzyme–substrate interactions as well as enzyme-catalyzed reaction kinetics. Skoraczyński et al. developed binary classification models for predicting the reaction yield using the RDKit descriptors, reaction FP and also chemical-linguistic substructure descriptors as the inputs,228 which showed large error. One of the key reasons was deemed to be the negligence of the subtle difference of molecular structures in the descriptors. To accurately describe the difference, the 3D conformations have to be considered.
Because it is time-consuming to obtain minimized 3D conformations of molecules, geometry-based methods were developed, e.g. the extended three-dimensional fingerprint (E3FP), which encodes the 3D substructures of small molecules, was used to describe molecular 3D conformations and showed a better performance in predicting bioactivity similarity compared to the 2D extended connectivity fingerprint (ECFP), which is based on the 2D Morgan fingerprint.236 A geometry-enhanced molecular representation learning method (GEM), which is composed of a geometry-based GNN, was proposed and then self-supervised tasks were designed to learn from large-scale 3D structures.237 Pan et al. predicted molecular properties by implementing algebraic graph-based fingerprints (AG-FPs) into bidirectional transformer-based fingerprints (BT-FPs).238 Zeng et al. predicted molecular properties based on the 3D representations of molecules which were obtained by grid-based 3D Convolutional Neural Network (3D CNN) descriptors derived from the original SMIILES databases.239
Compared to 2D representations, 3D structural descriptors contain more information. Interestingly, the model based on 3D structural descriptors of ligands performed similarly to that based on 2D molecular fingerprints in predicting protein–ligand binding affinities, whereas the model based on the 3D information of protein–ligand complexes outperformed those based on the 2D fingerprint of complexes.240 Because the induced conformational flexibility of the catalysts is crucial for the catalytic capability, the conformational flexibility upon substrate–catalyst binding throughout the catalytic cycle needs to be considered.241
Isayev et al. developed an Auto3D package using SMILES as the input, to generate low-energy conformations of molecules.243 They also developed an AIMNet-NSE model using the conformations sampled from MD simulations, to construct conformational ensembles related to chemical reactions by passing the expensive QM calculations.244 Zhu et al. benchmarked the deep learning models with 1D, 2D, 3D and conformer ensemble representations and found those with conformational ensembles showed improved performance.245
The descriptors incorporating conformation ensembles have showed improved performance in the prediction of molecular properties and hence in the applications of chiral catalyst selection246 and drug discovery.247
3.2 Descriptors for enzymes
Different from small molecules, enzymes have significantly larger molecular weights. While small molecules typically have molecular weights ranging from tens to hundreds of Daltons, enzymes have molecular weights that usually range from thousands to hundreds of thousands of Daltons. This makes it unrealistic to derive descriptors through quantum chemistry calculations or represent them using molecular fingerprints. Therefore, descriptors related to enzymes are often derived from the enzymes' amino acid sequences or three-dimensional structures. The common descriptors for enzymes are listed in Table 1.| Descriptors | Feature type | Features | Ref. |
|---|---|---|---|
| Sequence-based descriptor | Natural language processing (NLP) | One-hot encoding | J. Chem. Inf. Model., 60 (6), 2773–2790, (2020)248 |
| N-gram encoding | Protein Sci., 1 (5), 667–677, (1992)249 | ||
| Homologous information | PSSM | Bioinformatics,33 (17), 2756–2758, (2017)250 | |
| Physical and chemical properties | zScales | J. Med. Chem., 41 (14), 2481–2491, (1998)251 | |
| sScales | Protein Eng. Des. Sel., 2 (3), 185–191, (1988)252 | ||
| TScales | J. Mol. Struct., 830 (1–3), 106–115, (2017)253 | ||
| stScales | Amino Acids, 38, 805–816, (2010)254 | ||
| vhseScales | Pept. Sci., 80 (6), 775–786, (2005)255 | ||
| protFP | J. Cheminf., 5 (1), 1–11,(2013)256 | ||
| AA-Index | Nucleic Acids Research., 36 (1), D202–D205, (2007)257 | ||
| Structure-based descriptor | Planar features | Residue contact map | PLoS Comput. Biol., 13 (1), e1005324, (2017)258 |
| Stereo features | Geometric vector | arXiv:2009.01411, (2020)259 |
On the other hand, a number of approaches have been developed for retrieving sequence-based descriptors from amino acid composition (Fig. 7). A commonly adopted natural language processing (NLP) method is one-hot encoding. One-hot encoding represents the sequence by an array of a binary vector (0 or 1) to indicate the presence of a certain type of 20 amino acids at each position of the sequence. Another NLP method is n-gram encoding, where a protein sequence is broken into segments of size n to represent the local combinations of amino acids. These segments are then stored in an “n-gram” dictionary, which can be used to calculate the similarity among mutant strains. Other language embedding models like ProtVec also treat protein amino acid sequences as a series of “words” and map each amino acid to a vector representation in a high-dimensional space.260 ProtVec can be easily combined with the aforementioned Mol2vec.223
The above vectors capture the similarity and functional relevance among amino acids. Another method position-specific scoring matrix (PSSM)261 considers homology information among sequences, where each element represents the frequency of a certain amino acid (or base) at a given position across different sequences. These frequencies are calculated through multiple sequence alignments and are then converted into scores or probabilities. PSSM embodies information on conservation and variation of specific amino acids at particular positions in the sequences.
Certain overall physicochemical properties are related to enzyme functions, but their interpretability needs scrutiny. Descriptors based on protein sequences typically reveal fundamental characteristics of enzymes. The amino acid composition indicates the relative proportions and frequencies of different amino acid types, which are related to enzyme diversity and specificity. Conservation describes amino acid residue conservation across different species, which reflects the enzyme's evolutionary history and functional conservation. These descriptors could be used in machine learning for different tasks such as predicting substrate scope, enzyme functions, and classifying enzymes according to their properties.
The research of using structure-based descriptors in machine learning for enzyme engineering is relatively limited compared to that of the sequence-based approaches. Geometric descriptors such as atomic distances, angles, and dihedrals264 can be used to describe the spatial relationships among active site residues that are functionally important. These features can be represented by distance matrices and used as the inputs to construct machine learning models (e.g. sPairs, an AA-index-based aa pairwise contact potential248 and residue-residue contact map265,266). In addition, enzyme structure representations by space filling curves (SFCs) were reported in classification tasks for evaluating substrate selectivity.267
Structure-based features can reflect the substrate–enzyme interaction information. However, it is worth noting that replacing sequence-based features by structure-based features would not necessarily lead to improved predictive performance. In practical applications, these enzyme-related descriptors are often combined to construct comprehensive models to predict enzyme properties. For instance, sequence-based descriptors can be combined with structure-based descriptors to enhance model accuracy for predicting enzyme activities. Protein sequence descriptors have been combined with small molecule structural descriptors to model the interactions between compounds and proteins.268,269 Incorporating protein structural features in the models may further improve the accuracy and interpretability of the predictive models in the design and optimization of biocatalysts.
3.3 Databases
Machine learning algorithms highly rely on the quality of the training dataset (Fig. 7). It is important to resource the enzyme databases for the applications of machine learning. There are numerous publicly available databases online with vast amounts of data. The commonly used databases related to enzyme engineering are summarized in Table 2, encompassing protein sequence database, structure database, protein–ligand interaction database, reaction mechanism database, enzyme property database, etc.| Database | Database Size | Properties | Website | |
|---|---|---|---|---|
| Comprehensive enzyme databases | BRENDA | 8423 different enzymes | Enzyme EC number, structure, isolation and preparation information, reaction mechanism, substrate specificity, functional parameters, mutation, application, and related diseases. It also supports small molecule structure similarity query, and the corresponding enzyme can be searched by the structure of the substrate, product, or inhibitor | https://brenda-enzymes.org/oldstart.php |
| Protein sequence databases | UniProt | 248 million sequences | A vast collection of protein sequences and functional annotations | https://uniprot.org/ |
| Protein structure databases | PDB bank | 209 thousand structures | Experimentally determined three-dimensional structures of biological macromolecules, including proteins, nucleic acids, and complex assemblies | https://rcsb.org/pdb |
| AlphaFold protein structure database | 200 million predicted protein structures | The structures predicted with varying levels of confidence and should be inspected carefully | https://alphafold.ebi.ac.uk/ | |
| Protein–ligand interaction databases | STITCH | Proteins from 630 organisms and over 74000 different chemical |
Protein–ligand interactions from metabolic pathways, crystal structures, binding experiments and drug–target relationships | https://stitch.embl.de/ |
| Enzyme reaction mechanism databases | ExplorEnz | 8077 different enzymes | The reaction mechanism of enzymes, including substrates, products, and cofactors | https://enzyme-database.org/ |
| EMBL-EBI M-CSA | 694 detailed mechanisms | The catalytic mechanisms of enzymes. It focuses on elucidating the molecular mechanisms through which enzymes facilitate specific chemical reactions, including information about catalytic residues, substrate binding sites, and the overall reaction pathways | https://ebi.ac.uk/thornton-srv/m-csa/ | |
| Enzyme properties and mutation databases | PDBbind-CN | 23496 complex structures |
Complex structures and the corresponding experimentally measured binding affinity data | https://pdbbind.org.cn |
| KENDA | ∼13000 kinetic values |
KENDA is a supplement to the BRENDA database, providing enzyme functional kinetic data including KM, Ki, kcat, Vmaxetc. | https://www.brenda-enzymes.org/search_result.php?a=55 | |
| FireProt DB | 13274 entries |
Protein mutations and thermodynamic data | https://loschmidt.chemi.muni.cz/fireprot | |
| ProTherm | Over 7000 mutation data | Protein mutations and thermodynamic data | https://web.iitm.ac.in/bioinfo2/prothermdb | |
| eSOL | 788 protein entries | Protein mutations and enzyme solubility data | https://tanpaku.org/tp-esol/ | |
| SoluProtMut DB | 17392 mutation data |
Protein mutations and enzyme solubility data | https://loschmidt.chemi.muni.cz/soluprotmutdb/ | |
| D3DistalMutation | 7201 mutation data | The effects of distal mutations on enzyme activities | https://www.d3pharma.com/D3DistalMutation/ | |
| PiSite | 147817 PDB bank entries |
A database based on the PDB bank used for searching for protein interaction sites. | https://pisite.hgc.jp | |
| PhosphoSitePlus | 58477 protein entries |
Protein site modification includes phosphorylation, methylation, acetylation, ubiquitination, etc. | https://www.phosphosite.org/ | |
| dbPTM | 2.7 Million post-translational modification information | Protein post-translational modification information. | https://awi.cuhk.edu.cn/dbPTM/ |
It is crucial to craft a dataset to tailor it for specific research objectives and computational resources. For instance, AlphaFold2 achieves high accuracy in predicting protein structures, yet current predictions are limited to the apoprotein and do not include interactions with ligands, cofactors, or metal ions. It is reported that the AlphaFill algorithm270 can be used to transplant cofactors and ions from experimentally determined structures into the prediction models by AlphaFold2, based on the sequences and structural similarity. However, direct transportation of the ligands or metal ions from known structures has limitation for certain enzymes for which the crystal structures have not been resolved and the sequence identity among species is very low, e.g. terpene synthases, an important enzyme family to industry for the biosynthesis of natural products.
Typically, the effect of mutations around the catalytic sites of enzymes is predicted by analyzing their configurations. However, the effect of mutating remote site residues on enzymes’ function remains largely elusive. The D3DistalMutation database271 documents the impact of distal mutations (mutations more than 10 Å away from the active site) on enzyme activities. It should be noted that the research was focused on the mutation of enzymes’ activities and the substrate–enzyme interactions were not considered and its applications would be limited to disease-related mechanisms or drug discovery tasks.
Customized databases can be constructed by collecting data from literature data or experiments. The cleaned data would improve data quality and provide comparable data to achieve accurate models for engineering specific enzymes. In practical applications, despite the data resources and the acquirement approach, emphasis should be on data quality, timeliness, and legality to ensure model accuracy and reliability.
The quality of training data is the foundation for constructing accurate models. However, there are inevitably noise and imbalances in experimental data. A million disorganized data is inferior to a hundred clean data. Prior to training, preprocessing the data, such as removing outliers and balancing data distribution, is necessary to enhance the stability and generalization ability of ML models (Fig. 7).
3.4 Machine learning model construction
Statistical analysis can help to retrieve the enzyme sequence–function relationship and hence guide the enzyme evolution. For example, in predicting the substrate selectivity of the ene-reductase enzyme,272 the predictivity of ML models was evaluated by forward-stepwise multivariate linear regression (MLR) of the predicted properties versus the experimentally measured values for the training/test datasets. In another study, partial least square statistical analysis of protein sequence activity relationship (ProSAR) was conducted for bacterial dehalogenase and the beneficial mutations were identified.273
In machine learning, datasets are usually divided into the training set and the testing set and are focused on identifying the generalizable patterns. The model learns from the training dataset and adjusts its internal parameters to make accurate predictions. The testing set is used to evaluate the model's performance and generalization ability. It contains data that the model has not seen during training, so that the model can make predictions on new and unseen data, for example, in the applications of enzyme evolution or discovery of new enzymes.274,275 Classic machine learning models include Naive Bayes, decision trees, random forests, support vector machines, and others.
Deep learning is a subset of machine learning that evolved from neural networks, includes models such as the convolutional neural network (CNN), graph neural network (GNN), recurrent neural network (RNN), variational autoencoder (VAE), generative adversarial network (GAN), transformer, etc.
Machine learning can be divided into supervised learning and unsupervised learning according to the purpose of tasks. Supervised learning fits the data that are labelled based on experimental measurements or manual denotation. The training set is used to train the model by feeding it with input data and their corresponding output labels. Depending on the purpose of prediction, supervised learning can be used for regression and classification tasks. e.g. to predict the effect of mutation on enzyme's activity, the model can be used for predicting the numerical activity value by regression models or binary classification. On the other hand, unsupervised learning can discern the pattern of the unlabelled data and is mainly used for clustering tasks. For example, to search for the subset of sequences with similar functions from sequence-function relationship studies.
Hybrid semi-supervised learning combining supervised and unsupervised approaches can also be used, employing a small portion of experimentally labelled data and with a large amount of remaining data unlabelled. A semi-supervised method ProteinNPT was reported for predicting protein properties and design, and the model was trained on a large number of unlabelled protein sequences. The usage of a MSA transformer enables reflecting the evolutional and structural information of proteins.276 Recently, semi-supervised deep transfer learning techniques were used for small datasets, which showed promising results compared to other methods.277
There are many good reviews or benchmark studies on machine learning algorithms. Just to name a few, Raschka discussed model evaluation/selection and algorithm selection.278 Jones et al. reviewed the key machine learning concepts, how different ML techniques would be selected for different types of biological data and also discussed some best practices.279 Xu and Johnston et al. benchmarked the performance of different machine learning methods and protein descriptors, using various evaluation approaches.248 We benchmarked the performance of our deep-learning based ALDELE toolkits comprising different combined representations of enzymes and substrates, on a range of biocatalyst datasets comprising 150 to 23000 compound–protein pairs.280
Despite high accuracy on training data during evaluation, there are still challenges to overcome to achieve a predictive and generalized ML model. This is because models may lack generalization capabilities on unseen data. For example, data points can be increasingly fitted with the increasing degree of x in polynomial regression. Excessive dimensions may lead to overfitting, as the model would capture the pattern from the noise in the training data resulting in poor generalization capabilities. The issues of underfitting or overfitting necessitate hyperparameter tuning during model optimization (Fig. 7).
3.5 ML-accelerated enzyme design applications
It is challenging to explore all the mutant landscape using structure-based rational design and directed evolution, due to the cost brought about by combinatorial explosion, and also easily trapped local minima. Machine learning has been widely used to explore the sequence landscape in enzyme design. In the past few years, successful machine learning-accelerated enzyme designs have emerged. We summarised some of the latest representative research in Table 3.| Task | Dataset | Model and framework | Availability | Citation |
|---|---|---|---|---|
| Improve the activity of a transpeptidase and investigate the effects of a highly positive variant in training data | Sequences of 80 variants | Gaussian process (GP) | N/A | ACS Catal., 2021, 11(23), 14615–14624 |
| Improve the hydrolytic activity and thermostability of PET hydrolases | Over 19000 sructures from the Protein Data Bank |
3D Convolutional neural network (3D-CNN) | N/A | Nature, 2022, 604(7907), 662–667 |
| Improve the substrate specificity of XylM for the 2,6-xylenol degradation pathway | Sequences of 126 variants | Bayesian optimization | N/A | ACS Synth. Biol., 2023, 12(2), 572–582 |
| Improve the substrate channel flexibility and catalytic performance of P450 CYP116B31 | Sequences of 165 variants | Partial least squares (PLS) regression and multi-layer perceptron (MLP) | N/A | Catalysts, 2023, 13(8), 1228 |
| Improve the enantioselectivity of an epoxide hydrolase | Sequences of 37 variants | PLS | N/A | Sci. Rep., 2018, 8, 16757 |
| Predict the outcomes of hydrolase-catalyzed kinetic resolution | Complex structures of 672 hydrolase–substrate pairs | MLP | Code available | Chem. Sci., 2023, 14(43), 12073–12082 |
| Predict protease enzyme specificity | Cleavage information of HCV and TEV protease variants | Graph convolutional network (GCN) | N/A | Proc. Natl. Acad. Sci. U. S. A., 2023, 120(39), e2303590120 |
| Identify residues for cofactor specificity conversion in enzymes | Sequences of 952 malic enzymes with different cofactor specificities | Logistic regression | N/A | ACS Synth. Biol., 2022, 11(12), 3973–3985 |
| Predict reactivity without dynamic simulations | QM/MM trajectories of 27 reactive and 27 almost-reactive ensembles | Least absolute shrinkage and selection operator (LASSO) | N/A | J. Am. Chem. Soc., 2019, 141, 4108–4118 |
| Enhance the methanol tolerance of lipase | Sequences of 266 variants | Multi different regression models | N/A | Syst. Microbiol. Biomanuf., 2023, 3(3), 427–439 |
| Predict enzyme–substrate relationship | 18351 Annotated enzyme–substrate pairs |
XGBoost | Code available | Nat. Commun., 2023, 14, 2787 |
| Identify new enzymes for mycotoxin degradation | Enzyme–substrate pairs from five databases, over 600000 positive and 6.4 million unlabeled data |
PU-EPP, a deep learning model that combines GNN, continuous bag-of-words, and multi-head attention mechanisms. | Code available | ACS Catal., 2024, 14, 3336–3348 |
| Predict enzyme Kcat value | 16838 Entries from the BRENDA database and the SABIO-RK database |
Graph neural network (GNN) + convolutional neural network (CNN) | Code available | Nat. Catal. 2022, 5, 662–672 |
| Predict enzyme Kcat value | 16838 Entries from the BRENDA database, UniProt database and the SABIO-RK database |
XGBoost | Code available | Nat. Commun., 2023, 14(1), 4139 |
| Predict enzyme KM value | The input dataset is derived from the BRENDA database and the SABIO-RK database, which comprises 8375 entries invloves sequences, substrate name, EC number, UniProt ID of the enzyme, and PubMed ID. | Graph neural network (GNN) + convolutional neural network (CNN) | Code available | PLOS Biol. 2021, 19 e30014022021 |
| Predict enzyme Kcat value | Preprocessed DLKcat dataset with 11923 enzyme-substrate pairs and protein 3D structures. |
DeepEnzyme, a deep learning model that combines Transformer and GCN | Code available | 2013, bioRxiv:2023.12.09.570923 |
| Predict enzyme Kcat value | 4 Different datasets including DLKcat dataset, pH and temperature datasets, Km dataset and kcat/Km dataset | Language model | Code available | Nat. Commun., 2013, 14(1), 8211 |
| Predict enzyme optimal catalytic temperatures and protein melting temperatures | 3 Million enzyme sequences with OGT labels | Residual neural network (ResNet) | Code available | Protein Sci., 2022, 31(12), e4480 |
| Predict enzyme optimum pH | 125 Amino acid sequences in GH11 family with optimal pH labels | Support vector regression (SVR) | Code available | BMC Bioinf., 2020, 21(1), 512 |
| Predict enzyme optimum pH | 7 Million enzyme sequences with pH(opt) labels | Multiple different regression models | Code available | 2023, bioRxiv:2023.06.22.544776 |
| Predict Sequence EC number | EC numbers and reaction information for 38320 enzymatic reactions from BRENDA and KEGG |
Support vector regression (SVM), random forest (RF), k-nearest neighbors (kNN) and multi-layer perceptron (MLP) | N/A | J. Chem. Inf. Model., 2020, 60(3), 1833–1843 |
| Predict sequence EC number | Sequences from the UniProt database | CLEAN, a contrastive learning model using ESM-1b embeddings | Code/Web server available | Science, 2023, 379, 1358–1363 |
| Generate protein sequences for specific reactions | 16898 sequences from the UniProt database |
Generative adversarial network (GAN) | Code available | Nat. Mach. Intell., 2021, 3, s324–333 |
| Generate protein sequences for specific reactions | 281 million sequences from the UniParc, UniprotKB, Pfam and NCBI taxonomy databases | Transformer | Code available | Nat. Biotechnol., 2023, 41, 1099–1106 |
| Generate protein sequences for specific reactions | Luciferase-like protein sequences from InterPro | Variational autoencoder (VAE) | Code available | PLoS Comput. Biol., 2021, 17(2), e1008736 |
| Generate protein sequences for specific reactions | MSAs for different protein families generated by PFAM and HMMER | VAE | Code available | Nat. Commun., 2023, 14(1), 2222 |
The applications of machine learning in enzyme design can be classified into the improvement of properties for specific enzymes or the development of general predictive models. Considering the interest of different readers, we will discuss these two types of tasks respectively in the following section.
3.5.1.1 The predictivity of ML for small datasets. Building a reliable machine learning model requires a large amount of data. However, for specific enzymes, usually only a limited number of experimental data points are available such that the predictivity of the ML model developed from the small database is insufficient.
By iteratively evaluating ML-predicted sequences and feeding the new experimental data points with improved properties into the training set, the predictivity of the model can be improved. For example, Ohnishi et al. used Bayesian optimization to screen a Xylene monooxygenase (XylM) variant library. The library was generated by codon-randomization at five residue positions located close to the ion coordination site at the catalytic site.281 The iterative predictions after two rounds gave a mutant that increased 3-methylsalicylic acid production by 15 fold compared to that of WT-XylM. In another work, Liu et al. disclosed that five positions located at the substrate access channel of the P450 enzyme are critical for the loop stability and hence enzyme's activity.282 165 variants were created by simultaneous saturation mutagenesis on these five positions for machine learning. The representation of the sequences was generated by AAindex. The best mutant A86T/T91H/M108G/A109M/T111P showed a 15-fold improvement in the activity compared to the WT.283
Transfer learning starts with pre-training a model on a large dataset, then the model is fine-tuned to generate a new model for a smaller dataset, thus the knowledge learned from the large dataset is transferred to improve predictivity performance of the new model for the small dataset. For example, Engqvist et al. constructed a dataset of 3 million optimal growth temperatures (OGTs) from the BRENDA database to train a model called DeepET.284 DeepET is based on a residual neural network using the fast one-hot encoding method to retrieve enzyme thermal adaptation features from enzyme sequences. Transfer learning was then employed to predict two temperature properties related to the thermal stability, i.e. optimal catalytic temperature Topt and melting temperatures (Tm) for small datasets. DeepET showed more accurate predictions on these two datasets compared to other feature extraction methods like iFeature285 and UniRep.286 However, it is worth noting that if the small dataset is very divergent from the large dataset, the knowledge transferred may not be relevant, leading to poor performance.
In the case of an extremely small data set (9 single mutation variants). Frédéric Cadet et al. reported a sequence–activity relationship method called innov’SAR and improved the enantioselectivity of an epoxide hydrolase guided by machine learning. This method numerically encodes the protein sequence by AAindex and then applies the Fast Fourier transform to convert the encoded sequence into protein spectra. The protein spectra and protein activity are used as learning datasets to build a partial least squares regression (PLS regression) model to predict the activity.287 In addition, by adding 28 mutants into the training set, the prediction model includes some information about the epistasis between mutations, thereby improving the accuracy of the prediction model.
3.5.1.2 Impact of dataset construction on predictivity. Machine learning can guide directed evolution in exploring sequence space. Would the composition of training data affect the predicted results of machine learning?
Guided by ML models, two series of directed evolution for Sortase A were performed. The dataset for one ML model contained a highly positive variant 5 M, whereas the other excluded 5 M.274 The ML models were trained using the Bayesian optimization method and used to evaluate the probability of a variant being positive and promising variants (with activity 2.2–2.5 times higher than that of 5 M) were predicted by both ML models. However, it is worth noting that the regions for advantageous mutation on the sequence fitness landscape identified by the two ML models are different, indicating that diverse positive variants may be attained by using divergent datasets.
3.5.1.3 Structure-based machine learning. Structure-based representations of proteins have been developed to describe the substrate–enzyme interactions. For example, Ran et al. reported a deep learning framework EnzyKR, which used complex structures constructed from docking to encode the hydrolase–substrate interactions between hydrolases and the enantiomer products,288 and showed good performance in predicting activation free energy as well as in predicting enantiomeric excess ratios. Using structure-based ML enzyme engineering, Alper et al. obtained a PET hydrolase variant that was generated by combining the triple mutation predicted by ML and double mutation from the scaffold, which showed promising hydrolytic activity and thermostability.289 The machine learning architecture used in the study employed the 3D CNN method MutCompute290 proposed by Ross Thyer.
Lu et al. represented a structure-based graph convolutional network that denotes the protein–ligand interaction energetics (generated using Rosetta159) and successfully predicted the specificity of proteases for two noncanonical substrates.291
The dynamic properties of enzymes are crucial for their activities. Tidor et al. selected descriptors from 68 geometric parameters including atom distances, planar angles, dihedral angles, etc. that represent the local conformation of the active site and accurately predicted the reactivity of ketol-acid reductoisomerase (KARI).292
3.5.2.1 Predicting enzyme–substrate pairs. Machine learning has been widely used for predicting protein–ligand interactions based on the datasets constructed from various databases. However, most databases only contain the active substrates that enzymes can catalyze (positive instances) and lack data on non-active substrates (negative instances). The imbalance of the dataset can lead to models with poor generalization ability. To address this issue, Alexander Kroll et al. reported an enzyme–substrate pair prediction model by constructing a database composed of the experimentally validated enzyme–substrate pairs derived from the gene ontology (GO) annotation database and randomly generated negative samples similar to the real substrates using data augmentation.293 They constructed a gradient-boosted decision tree model for predicting enzyme–substrate pairs. By only using the sequence-based representation for enzymes and GNN-generated fingerprints for small molecules, a general model was achieved with high accuracy that can be applied across enzyme families and a broad range of small molecules.
Hu et al. designed a dataset comprised of 606555 corresponding enzyme-substrate pairs, with the ratio of negative data to positive data around 10 times. They developed a positive unlabeled learning-based enzyme promiscuity prediction (PU-EPP) model for predicting the substrate promiscuity and specificity by extracting substrate features using GNNs and encoding protein sequences using continuous bag-of-words. The model showed good robustness on the test set and successfully identified 15 new enzymes for Mycotoxin Detoxification. It also allowed us to identify the important key residues attributed to the catalytic activity of the enzyme.294
3.5.2.2 Predicting catalytic efficiency. Binding free energy and reaction energy barrier are closely related to enzymes’ catalytic efficiency. However, the intricate and diverse catalytic mechanisms of enzymes pose challenges, especially for those with unknown structures particularly, the experimental and computational simulations methods such as QM/MM or QM/MM MD on these properties are expensive, which has limited their applications in evaluating the large-scale mutations to screen highly efficient enzymes.
K M (Michaelis constant) and kcat (turnover number) are two important parameters directly related to the catalytic efficiency of enzymes and therefore it is important to predict these properties to evaluate the effect of mutations.
Nielsen et al. reported a deep learning package DLKcat for predicting genome-scale kcat, using an enzyme-constrained metabolic model which is solely based on substrate structures and protein sequences, combining a graph neural network (GNN) for substrate molecule graphs and a convolutional neural network (CNN) for extracting protein n-gram properties.268 DLKcat has been used to predict the enzyme activity of β-ketothiolase295 and thiolase.296
Kroll et al. predicted KM using molecular fingerprints as a numerical representation of substrate molecules.297 They further predict kcat values for natural reactions of wild-type enzymes taking into consideration of numerical fingerprints for substrates and products, representing enzymes using transformer networks.298 The model is able to make meaningful predictions for enzymes that are less than 40% homologous to the data in the training set.
Wang et al. presented a model DeepEnzyme for predicting kcat values based on the 3D structures of the proteins. By leveraging the features from both sequences and 3D structures, the DeepEnzyme model achieved improved prediction than DLKcat on the performance for those with low homology with the training set.299
We recently developed a deep learning-based workflow ALDELE.280 This workflow includes five toolkits (Fig. 8): (1) NN representation of substrates based on whole compound properties, (2) GNN representation of substrates based on molecular graphs, (3) CNN representation of proteins based on N-Gram vectors, (4) CNN representation of proteins based on PSSM, and (5) CNN representation of protein structure-based features. The comprehensive toolkits allow customized combination of the physicochemical and graphic properties of substrates, with the sequence, evolutionary and structural information of enzymes, for predicting the interactions between enzymes and substrates. Benchmark studies for multiple datasets including a kcat dataset comprising 16838 enzyme–substrate interaction pairs show the accuracy of ALDELE for predicting the biocatalytic activities of enzymes.
 | ||
| Fig. 8 Workflow of ALDELE. The model architecture takes protein sequences and substrate SMILES as inputs and processes them through five toolkits to produce features. These toolkits include RDKit and SMILES for compound inputs, “Words” for protein sequences, PSSM for protein sequences, and protein structure-based features. A two-phase attention NN is applied to extract two sets of vectors representing the protein sequence or ligand. A multi-layer perceptron (MLP) is used for prediction. Reproduced with permission.280 Copyright 2024, American Chemical Society. | ||
3.5.2.3 Annotating enzyme function. Protein function prediction is critical for discovering and developing new biocatalysts. Following the sequence–structure–function paradigm, the protein sequence dictates the spatial structure and functions of proteins.300 Protein sequences, structures, functions, and protein–ligand interactions have been deposited in many databases such as Uniprot,301 however, the functions for a large number of newly discovered sequences have not been denoted.
Enzymes can be classified by the Enzyme Commission (EC) number, using a coding system consisting of four digits representing the reaction type, substrate type, reaction type, and specific enzyme order. Estimating the EC number of a new sequence allows predicting the function of the enzyme. Several machine learning models have been developed to predict EC numbers, as well as to predict the related substrates and products, among which the random forest and k-nearest neighbour-based model combining the enzyme sequence and the structural information of substrates and products was shown to be able predict almost all types of reactions.302
The contrastive learning-enabled enzyme annotation (CLEAN) method was trained on the UniProt database to assign enzymes’ EC number and functions.303 The method used the Euclidean distance as a metric to reflect the similarity in enzymes’ functions by embedding sequences into numerical vectors and was further validated in the uncharacterized halogenase database.
Additionally, a comprehensive review on the protein representation learning methods by language models since 2015 was reported by Doğan et al. They evaluated their performance in identifying protein functions by four benchmarking tasks.304e.g. The function of proteins is annotated using gene ontology (GO),305 which contains the information on molecular function, cellular components and biological processes.
Critical assessment of functional annotation (CAFA) is a project where the performance of different learning and representation methods for predicting the GO-annotated functions of target proteins are benchmarked by the accuracy compared with later acquired protein functions.306 NetGO 2.0 utilizes protein information from various resources to predict the function of annotated proteins, and achieved top performance in CAFA4.307 Furthermore, NetGO 3.0 integrates self-supervised protein language model (ESM)-1b embedding to represent protein sequences combined with logistic regression (LR-ESM) and has shown improved capability in predicting protein functions.308
Protein function prediction was initially based on the assumption that homologous proteins would share similar functions. However, this approach has obvious limitation with the presence of abundant distant and orphan proteins. Hence data-driven machine learning and deep learning techniques based on various representations of sequence, structure and interaction features have emerged. Dhanuka et al. presented a comprehensive review and compared feature-based machine learning and algorithm-based methods for protein function prediction.309
3.5.2.4 Generating functional sequences. Based on known functional proteins, mutagenesis and selection are commonly employed techniques for generating novel sequences with admirable functions. However, due to the vast sequence space of proteins, it remains challenging to predict sequences with new functions, which necessitates directly generating new sequences with admirable functions from the raw sequences.
Insights gained from sequence variations would provide insights on directed evolution. The advances in machine learning techniques enable deep generative models such as generative adversarial networks (GANs), transformers and variational autoencoders (VAEs) to be used to explore protein sequence space efficiently so as to generate protein sequences with specific functions.310
The ProteinGAN model, based on self-attention GAN, generates new protein sequences with natural-like functions.311 It has been successfully applied to generate soluble and catalytically active sequences of malate dehydrogenase, demonstrating the ability of the neural network architecture to generate highly diverse sequences by learning intricate evolutionary dependencies between amino acids and generalizing across the protein sequence space.
Furthermore, the language model ProGen312 was developed, trained on 280 million protein sequences using a transformer-based self-attention neural network architecture. The generated artificial sequences showed similar functions to natural proteins from diverse families, as demonstrated in the cases of chorismite mutase and malate dehydrogenase.
VAE models trained on 70000 oxidoreductases were used to generate new bacterial luciferase. The comparison of VAE models trained on aligned sequences and raw sequences showed that both models are able to capture the amino acid pattern of the enzyme family, whereas the former is able to better capture the long-distance features inferring their constraints on the protein functions.313
Latent generative landscape (LGL) was created using VAE sequence space, enabling flexible exploration of diverse protein functional space without labeling, guiding generative protein design and providing insights into evolutionary fitness and functional diversification.314
3.5.2.5 De novo design of artificial enzymes. In contrast to traditional enzyme design by directed evolution of native enzymes, the de novo design of enzymes with new functions from scratch is still in the infant stage, and new methods are rapidly emerging.15,315,316
The Rosetta de novo enzyme design protocol has been widely used in generating protein scaffolds since it was first reported over a decade ago.159,317–319 These design cases mostly rely on existing protein scaffold templates from nature. By transplanting the natural enzyme active sites into other unrelated protein structures and redesigning the amino acid sequence around the substrate, the goal is to stabilize the conformational energy of the enzyme's reaction intermediate state. However, due to limitations in energy functions and design accuracy, the designed enzymes often do not match the activity of natural enzymes.320
To address the fitness issue of the designed scaffold, Sarel Fleishman et al. proposed a CADENZ approach where the structural fragments from homologous but structurally divergent enzymes were recombined to generate diverse protein scaffolds while preserving enzyme catalytic function.321
An enumerative algorithm was developed by Baker et al. for generating scaffolds, where enzyme pocket scaffolds were constructed by Monte Carlo assembly of secondary structure folds, thus the possible combination of the structural parameters associated with the folds was enumerated. The approach was successfully applied to generate nuclear transport factor 2 (NTF2-like) protein structures with diverse pockets to accommodate diverse active sites.322 Recently, Baker et al. developed a deep-learning based ‘family-wide hallucination’ approach to generate a large number of NTF2-like scaffolds with diverse binding pockets and introduce the luciferin substrate of luciferases into scaffolds. The designed artificial luciferases exhibited high activity and specificity toward substrates of natural luciferase.323
4. Summary and perspectives
In this paper, we reviewed the enzyme design methods guided by computational simulations, as well as the revolution brought to traditional computational modeling by integrating machine learning. We also reviewed enzyme design directed by machine learning.The development of machine learning-based AlphaFold2 has demonstrated its great success in predicting the protein 3D structures from sequences and hence significantly expanded the size of the protein structure database for structure-based enzyme design. Ligand-bound conformations now can be generated from apo-protein structures directly.324 MD simulations and multiscale QM/MM calculations are used to explore protein conformational landscape to guide the site-specific mutagenesis. However, these simulation methods are not suitable for high-throughput screening of a large designed sequence database due to the high computing demands. It is important to balance between the extensive sampling and calculation accuracy. The conformational space can be sampled extensively employing multiple short, parallel MD simulations325 and enhanced QM/MM sampling would allow the exploration of enzyme free energy surface more efficiently.326,327
Machine learning methods may be combined with the traditional simulations to sample equilibrium states and rare events76,328,329 and even sample the catalytically relevant conformations in catalytic reaction space.330 Furthermore, exploring protein fitness would benefit from a fully automated process,10 which will largely reduce the efforts compared to the traditional screening. In an automated device, integrating site-specific mutagenesis and machine learning,331 the data generated from high-throughput screening were used as the input for the ML model to automatically explore the sequence fitness landscape.
High-quality datasets are crucial for the predictivity and generalization of the ML-model used for enzyme design. Numerous ML-guided enzyme design was based on the datasets extracted from the commonly used databases and substantial efforts are required to collect the specific data such as enzyme sequences, structures, substrate specificity, thermostability, kinetic properties, etc. from diverse databases. Additionally, many of these databases contain redundant data and irrelevant information, and the data are usually not standardized. Therefore, additional effort involves cleaning the data to construct a high-quality dataset. Furthermore, usually only the enzyme variants with improved properties were reported in the literature, so that the databases generated are biased toward positive samples. To improve predictivity of ML, artificially constructing negative data sets may be a practical strategy.
On the other hand, the dataset available for machine learning in enzyme engineering is usually small, therefore, it is necessary to develop algorithms tailored to improve the predictivity of ML models for these small datasets. The traditional deep learning models like GNN and RNN were originally designed for large datasets for text recognition and image recognition so they may not attain satisfactory results on small datasets.332 Transfer learning that transfers the “knowledge” learned from a large dataset to the model for a small dataset holds promise for enhancing the predictivity of ML models.
Data availability statement
The data that support this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Z. acknowledges the financial support of Queen's University Belfast (QUB) and Chinese Scholarship Council. The authors are grateful for the computing resources from QUB high performance computing Tier2 computing resource funded by EPSRC (EP/T022175).References
- R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, From nature to industry: Harnessing enzymes for biocatalysis, Science, 2023, 382(6673), eadh8615 CrossRef CAS PubMed.
- H. Chen, F. Y. Dong and S. D. Minteer, The progress and outlook of bioelectrocatalysis for the production of chemicals, fuels and materials, Nat. Catal., 2020, 3(3), 225–244 CrossRef.
- A. I. Benitez-Mateos, D. Roura Padrosa and F. Paradisi, Multistep enzyme cascades as a route towards green and sustainable pharmaceutical syntheses, Nat. Chem., 2022, 14(5), 489–499 CrossRef CAS PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Biocatal. Nat. Rev. Methods Primers, 2021, 1(1), 46 CrossRef CAS.
- G. P. Pinto, M. Corbella, A. O. Demkiv and S. C. L. Kamerlin, Exploiting enzyme evolution for computational protein design, Trends Biochem. Sci., 2022, 47(5), 375–389 CrossRef CAS PubMed.
- D. Yi, T. Bayer, C. P. S. Badenhorst, S. Wu, M. Doerr, M. Hohne and U. T. Bornscheuer, Recent trends in biocatalysis, Chem. Soc. Rev., 2021, 50(14), 8003–8049 RSC.
- A. Sharma, G. Gupta, T. Ahmad, S. Mansoor and B. Kaur, Enzyme Engineering: Current Trends and Future Perspectives, Food Rev. Int., 2021, 37(2), 121–154 CrossRef CAS.
- P. Notin, N. Rollins, Y. Gal, C. Sander and D. Marks, Machine learning for functional protein design, Nat. Biotechnol., 2024, 42(2), 216–228 CrossRef CAS PubMed.
- Y.-F. Ao, M. Dörr, M. J. Menke, S. Born, E. Heuson and U. T. Bornscheuer, Data-Driven Protein Engineering for Improving Catalytic Activity and Selectivity, ChemBioChem, 2024, 25(3), e202300754 CrossRef CAS PubMed.
- J. Yang, F.-Z. Li and F. H. Arnold, Opportunities and Challenges for Machine Learning-Assisted Enzyme Engineering, ACS Cent. Sci., 2024, 10(2), 226–241 CrossRef CAS PubMed.
- L. Alejaldre, J. N. Pelletier and D. Quaglia, Methods for enzyme library creation: Which one will you choose? A guide for novices and experts to introduce genetic diversity, BioEssays, 2021, 43(8), e2100052 CrossRef PubMed.
- R. A. Chica, N. Doucet and J. N. Pelletier, Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design, Curr. Opin. Biotechnol., 2005, 16(4), 378–384 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, Streamlining Design, Engineering, and Applications of Enzymes for Sustainable Biocatalysis, ACS Sustainable Chem. Eng., 2021, 9(24), 8032–8052 CrossRef CAS.
- M. Wittmund, F. Cadet and M. D. Davari, Learning Epistasis and Residue Coevolution Patterns: Current Trends and Future Perspectives for Advancing Enzyme Engineering, ACS Catal., 2022, 12(22), 14243–14263 CrossRef CAS.
- T. Kortemme, De novo protein design—From new structures to programmable functions, Cell, 2024, 187(3), 526–544 CrossRef CAS PubMed.
- K. Nam, Y. Shao, D. T. Major and M. Wolf-Watz, Perspectives on Computational Enzyme Modeling: From Mechanisms to Design and Drug Development, ACS Omega, 2024, 9(7), 7393–7412 CAS.
- S. L. Lovelock, R. Crawshaw, S. Basler, C. Levy, D. Baker, D. Hilvert and A. P. Green, The road to fully programmable protein catalysis, Nature, 2022, 606(7912), 49–58 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millan, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Accurate prediction of protein structures and interactions using a three-track neural network, Science, 2021, 373(6557), 871–876 CrossRef CAS PubMed.
- V. Marx, Biology: The big challenges of big data, Nature, 2013, 498(7453), 255–260 CrossRef CAS PubMed.
- S. Mazurenko, Z. Prokop and J. Damborsky, Machine Learning in Enzyme Engineering, ACS Catal., 2020, 10(2), 1210–1223 CrossRef CAS.
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Machine learning-assisted directed protein evolution with combinatorial libraries, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(18), 8852–8858 CrossRef CAS PubMed.
- T. U. Consortium, UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Res., 2022, 51(D1), D523–D531 CrossRef PubMed.
- S. K. Burley, C. Bhikadiya, C. Bi, S. Bittrich, H. Chao, L. Chen, P. A. Craig, G. V. Crichlow, K. Dalenberg, J. M. Duarte, S. Dutta, M. Fayazi, Z. Feng, J. W. Flatt, S. Ganesan, S. Ghosh, D. S. Goodsell, R. K. Green, V. Guranovic, J. Henry, B. P. Hudson, I. Khokhriakov, C. L. Lawson, Y. Liang, R. Lowe, E. Peisach, I. Persikova, D. W. Piehl, Y. Rose, A. Sali, J. Segura, M. Sekharan, C. Shao, B. Vallat, M. Voigt, B. Webb, J. D. Westbrook, S. Whetstone, J. Y. Young, A. Zalevsky and C. Zardecki, RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning, Nucleic Acids Res., 2022, 51(D1), D488–D508 CrossRef PubMed.
- X. Yan, J. Zhou, J. Ge, W. Li, D. Liang, W. Singh, G. Black, S. Nie, J. Liu, M. Sun, J. Qiao and M. Huang, Computer-Informed Engineering: A New Class I Sesquiterpene Synthase JeSTS4 for the Synthesis of an Unusual C10-(S)-Bicyclogermacrene, ACS Catal., 2022, 12(7), 4037–4045 CrossRef CAS.
- M. Ringel, N. Dimos, S. Himpich, M. Haack, C. Huber, W. Eisenreich, G. Schenk, B. Loll and T. Brück, Biotechnological potential and initial characterization of two novel sesquiterpene synthases from Basidiomycota Coniophora puteana for heterologous production of δ-cadinol, Microb. Cell Fact., 2022, 21(1), 64 CrossRef CAS PubMed.
- Y. He, Y. Chen, P. A. Alexander, P. N. Bryan and J. Orban, Mutational tipping points for switching protein folds and functions, Structure, 2012, 20(2), 283–291 CrossRef CAS PubMed.
- B. Wallner and A. Elofsson, All are not equal: A benchmark of different homology modeling programs, Protein Sci., 2005, 14(5), 1315–1327 CrossRef CAS PubMed.
- Q. Wuyun, Y. Chen, Y. Shen, Y. Cao, G. Hu, W. Cui, J. Gao and W. Zheng, Recent Progress of Protein Tertiary Structure Prediction, Molecules, 2024, 29(4), 832 CrossRef CAS PubMed.
- B. Webb and A. Sali, Comparative Protein Structure Modeling Using MODELLER, Curr. Protoc. Bioinformatics, 2016, 54, 5.6.1–5.6.37 Search PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. Gallo Cassarino, M. Bertoni, L. Bordoli and T. Schwede, SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information, Nucleic Acids Res., 2014, 42(W1), W252–W258 CrossRef CAS PubMed.
- Y. Duan and P. A. Kollman, Pathways to a Protein Folding Intermediate Observed in a 1-Microsecond Simulation in Aqueous Solution, Science, 1998, 282(5389), 740–744 CrossRef CAS PubMed.
- A. Roy, A. Kucukural and Y. Zhang, I-TASSER: a unified platform for automated protein structure and function prediction, Nat. Protoc., 2010, 5(4), 725–738 CrossRef CAS PubMed.
- K. T. Simons, C. Kooperberg, E. Huang and D. Baker, Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions, J. Mol. Biol., 1997, 268(1), 209–225 CrossRef CAS PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Improved protein structure prediction using potentials from deep learning, Nature, 2020, 577(7792), 706–710 CrossRef CAS PubMed.
- M. AlQuraishi, AlphaFold at CASP13, Bioinformatics, 2019, 35(22), 4862–4865 CrossRef CAS PubMed.
- A. Kryshtafovych, T. Schwede, M. Topf, K. Fidelis and J. Moult, Critical assessment of methods of protein structure prediction (CASP)-Round XIV, Proteins, 2021, 89(12), 1607–1617 CrossRef CAS PubMed.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L. P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks and V. S. Pande, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics, PLoS Comput. Biol., 2017, 13(7), e1005659 CrossRef PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Comparison of multiple Amber force fields and development of improved protein backbone parameters, Proteins, 2006, 65(3), 712–725 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O’Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Accurate structure prediction of biomolecular interactions with AlphaFold3, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- M. Mirdita, K. Schutze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, ColabFold: making protein folding accessible to all, Nat. Methods, 2022, 19(6), 679–682 CrossRef CAS PubMed.
- M. Steinegger and J. Soding, MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets, Nat. Biotechnol., 2017, 35(11), 1026–1028 CrossRef CAS PubMed.
- H. K. Wayment-Steele, A. Ojoawo, R. Otten, J. M. Apitz, W. Pitsawong, M. Homberger, S. Ovchinnikov, L. Colwell and D. Kern, Predicting multiple conformations via sequence clustering and AlphaFold2, Nature, 2023, 625, 832–839 CrossRef PubMed.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y. Shmueli, A. dos Santos Costa, M. Fazel-Zarandi, T. Sercu, S. Candido and A. Rives, Evolutionary-scale prediction of atomic-level protein structure with a language model, Science, 2023, 379(6637), 1123–1130 CrossRef CAS PubMed.
- J. Koehler Leman, P. Szczerbiak, P. D. Renfrew, V. Gligorijevic, D. Berenberg, T. Vatanen, B. C. Taylor, C. Chandler, S. Janssen, A. Pataki, N. Carriero, I. Fisk, R. J. Xavier, R. Knight, R. Bonneau and T. Kosciolek, Sequence-structure-function relationships in the microbial protein universe, Nat. Commun., 2023, 14(1), 2351 CrossRef CAS PubMed.
- A. Al-Fatlawi, M. Menzel and M. Schroeder, Is Protein BLAST a thing of the past?, Nat. Commun., 2023, 14(1), 8195 CrossRef CAS PubMed.
- A. Al-Fatlawi, M. Schroeder and A. F. Stewart, The Rad52 SSAP superfamily and new insight into homologous recombination, Commun. Biol., 2023, 6(1), 87 CrossRef CAS PubMed.
- M. A. Pak, K. A. Markhieva, M. S. Novikova, D. S. Petrov, I. S. Vorobyev, E. S. Maksimova, F. A. Kondrashov and D. N. Ivankov, Using AlphaFold to predict the impact of single mutations on protein stability and function, PLoS One, 2023, 18(3), e0282689 CrossRef CAS PubMed.
- J. M. McBride, K. Polev, A. Abdirasulov, V. Reinharz, B. A. Grzybowski and T. Tlusty, AlphaFold2 Can Predict Single-Mutation Effects, Phys. Rev. Lett., 2023, 131(21), 218401 CrossRef CAS PubMed.
- S. Parui, E. Brini and K. A. Dill, Computing Free Energies of Fold-Switching Proteins Using MELD x MD, J. Chem. Theory Comput., 2023, 19(19), 6839–6847 CrossRef CAS PubMed.
- S. A. Hollingsworth and R. O. Dror, Molecular Dynamics Simulation for All, Neuron, 2018, 99(6), 1129–1143 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, V. W. D. Cruzeiro, T. A. Darden and R. E. Duke, Amber 2021, University of California, San Francisco, 2021 Search PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, CHARMM: the biomolecular simulation program, J. Comput. Chem., 2009, 30(10), 1545–1614 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. A. Swaminathan and M. Karplus, CHARMM: a program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 1983, 4(2), 187–217 CrossRef CAS.
- S. Jo, T. Kim, V. G. Iyer and W. Im, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem., 2008, 29(11), 1859–1865 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. Lopes, J. Mittal, M. Feig and A. D. MacKerell Jr, Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles, J. Chem. Theory Comput., 2012, 8(9), 3257–3273 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6, J. Comput. Chem., 2004, 25(13), 1656–1676 CrossRef CAS PubMed.
- M. J. Robertson, J. Tirado-Rives and W. L. Jorgensen, Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field, J. Chem. Theory Comput., 2015, 11(7), 3499–3509 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236 CrossRef CAS.
- H. H. Loeffler and M. Winn Large biomolecular simulation on hpc platforms III. AMBER, CHARMM, GROMACS, LAMMPS and NAMD. Technical report, STFC Daresbury Laboratory, Warrington WA4 4AD, UK, 2012.
- A. Sedova, J. D. Eblen, R. Budiardja, A. Tharrington and J. C. Smith, In High-performance molecular dynamics simulation for biological and materials sciences: Challenges of performance portability, 2018 IEEE/ACM International Workshop on Performance, Portability and Productivity in HPC (P3HPC), IEEE, 2018, 1–13 Search PubMed.
- Z. W. Zhang and W. C. Lu, AmberMDrun: A Scripting Tool for Running Amber MD in an Easy Way, Biomolecules, 2023, 13(4), 635 CrossRef CAS PubMed.
- J. G. Kirkwood, Statistical Mechanics of Fluid Mixtures, J. Chem. Phys., 2004, 3(5), 300–313 CrossRef.
- Y. I. Yang, Q. Shao, J. Zhang, L. Yang and Y. Q. Gao, Enhanced sampling in molecular dynamics, J. Chem. Phys., 2019, 151(7), 070902 CrossRef PubMed.
- G. M. Torrie and J. P. Valleau, Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling, J. Comput. Phys., 1977, 23(2), 187–199 CrossRef.
- B. Ensing, M. De Vivo, Z. Liu, P. Moore and M. L. Klein, Metadynamics as a tool for exploring free energy landscapes of chemical reactions, Acc. Chem. Res., 2006, 39(2), 73–81 CrossRef CAS PubMed.
- D. Hamelberg, J. Mongan and J. A. McCammon, Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules, J. Chem. Phys., 2004, 120(24), 11919–11929 CrossRef CAS PubMed.
- Y. Sugita and Y. Okamoto, Replica-exchange molecular dynamics method for protein folding, Chem. Phys. Lett., 1999, 314(1–2), 141–151 CrossRef CAS.
- B. Roux, The Calculation of the Potential of Mean Force Using Computer-Simulations, Comput. Phys. Commun., 1995, 91(1–3), 275–282 CrossRef CAS.
- M. Mezei, Adaptive Umbrella Sampling - Self-Consistent Determination of the Non-Boltzmann Bias, J. Comput. Phys., 1987, 68(1), 237–248 CrossRef.
- A. Laio and M. Parrinello, Escaping free-energy minima, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(20), 12562–12566 CrossRef CAS PubMed.
- A. Barducci, M. Bonomi and M. Parrinello, Metadynamics, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1(5), 826–843 CAS.
- J. L. MacCallum, A. Perez and K. A. Dill, Determining protein structures by combining semireliable data with atomistic physical models by Bayesian inference, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(22), 6985–6990 CrossRef CAS PubMed.
- A. Perez, J. L. MacCallum and K. A. Dill, Accelerating molecular simulations of proteins using Bayesian inference on weak information, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(38), 11846–11851 CrossRef CAS PubMed.
- P. Gkeka, G. Stoltz, A. Barati Farimani, Z. Belkacemi, M. Ceriotti, J. D. Chodera, A. R. Dinner, A. L. Ferguson, J.-B. Maillet, H. Minoux, C. Peter, F. Pietrucci, A. Silveira, A. Tkatchenko, Z. Trstanova, R. Wiewiora and T. Lelièvre, Machine Learning Force Fields and Coarse-Grained Variables in Molecular Dynamics: Application to Materials and Biological Systems, J. Chem. Theory Comput., 2020, 16(8), 4757–4775 CrossRef CAS PubMed.
- D. E. Kleiman, H. Nadeem and D. Shukla, Adaptive Sampling Methods for Molecular Dynamics in the Era of Machine Learning, J. Phys. Chem. B, 2023, 127(50), 10669–10681 CrossRef CAS PubMed.
- H. Tian, X. Jiang, S. Xiao, H. La Force, E. C. Larson and P. Tao, LAST: Latent Space-Assisted Adaptive Sampling for Protein Trajectories, J. Chem. Inf. Model., 2023, 63(1), 67–75 CrossRef CAS PubMed.
- B. P. Vani, A. Aranganathan, D. Wang and P. Tiwary, AlphaFold2-RAVE: From Sequence to Boltzmann Ranking, J. Chem. Theory Comput., 2023, 19(14), 4351–4354 CrossRef CAS PubMed.
- J. M. L. Ribeiro, P. Bravo, Y. Wang and P. Tiwary, Reweighted autoencoded variational Bayes for enhanced sampling (RAVE), J. Chem. Phys., 2018, 149(7), 072301 CrossRef PubMed.
- F. Noé, S. Olsson, J. Köhler and H. Wu, Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning, Science, 2019, 365(6457), eaaw1147 CrossRef PubMed.
- J. Srinivasan, T. E. Cheatham, P. Cieplak, P. A. Kollman and D. A. Case, Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices, J. Am. Chem. Soc., 1998, 120(37), 9401–9409 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models, Acc. Chem. Res., 2000, 33(12), 889–897 CrossRef CAS PubMed.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. H. Zhang and T. Hou, End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design, Chem. Rev., 2019, 119(16), 9478–9508 CrossRef CAS PubMed.
- P. Kollman, Free-Energy Calculations - Applications To Chemical and Biochemical Phenomena, Chem. Rev., 1993, 93(7), 2395–2417 CrossRef CAS.
- P. A. Bash, U. C. Singh, R. Langridge and P. A. Kollman, Free energy calculations by computer simulation, Science, 1987, 236(4801), 564–568 CrossRef CAS PubMed.
- S. Dasgupta and J. M. Herbert, Using Atomic Confining Potentials for Geometry Optimization and Vibrational Frequency Calculations in Quantum-Chemical Models of Enzyme Active Sites, J. Phys. Chem. B, 2020, 124(7), 1137–1147 CrossRef CAS PubMed.
- X. Sheng and F. Himo, The Quantum Chemical Cluster Approach in Biocatalysis, Acc. Chem. Res., 2023, 56(8), 938–947 CrossRef CAS PubMed.
- Q. Cheng and N. J. DeYonker, The Glycine N-Methyltransferase Case Study: Another Challenge for QM-Cluster Models?, J. Phys. Chem. B, 2023, 127(43), 9282–9294 CrossRef CAS PubMed.
- M. C. Mooney, Y. T. Xu, J. McClory and M. L. Huang, A disappearing act performed by magnesium: the nucleotide exchange mechanism of Ran GTPase by quantum mechanics/molecular mechanics studies, Theor. Chem. Acc., 2016, 135(8), 197 Search PubMed.
- M. Huang, K. Wei, X. Li, J. McClory, G. Hu, J. W. Zou and D. Timson, Phosphorylation Mechanism of Phosphomevalonate Kinase: Implications for Rational Engineering of Isoprenoid Biosynthetic Pathway Enzymes, J. Phys. Chem. B, 2016, 120(41), 10714–10722 CrossRef CAS PubMed.
- J. McClory, D. J. Timson, W. Singh, J. Zhang and M. Huang, Reaction Mechanism of Isopentenyl Phosphate Kinase: A QM/MM Study, J. Phys. Chem. B, 2017, 121(49), 11062–11071 CrossRef CAS PubMed.
- J. McClory, C. Hui, J. Zhang and M. Huang, The phosphorylation mechanism of mevalonate diphosphate decarboxylase: a QM/MM study, Org. Biomol. Chem., 2020, 18(3), 518–529 RSC.
- J. McClory, J. T. Lin, D. J. Timson, J. Zhang and M. Huang, Catalytic mechanism of mevalonate kinase revisited, a QM/MM study, Org. Biomol. Chem., 2019, 17(9), 2423–2431 RSC.
- J. McClory, G. X. Hu, J. W. Zou, D. J. Timson and M. Huang, Phosphorylation Mechanism of N-Acetyl-l-glutamate Kinase, a QM/MM Study, J. Phys. Chem. B, 2019, 123(13), 2844–2852 CrossRef CAS PubMed.
- J. McClory, J. T. Lin, D. J. Timson, J. Zhang and M. Huang, Water-mediated network in the resistance mechanism of fosfomycin, Phys. Chem. Chem. Phys., 2018, 20(33), 21660–21667 RSC.
- W. Singh, M. Bilal, J. McClory, D. Dourado, D. Quinn, T. S. Moody, I. Sutcliffe and M. Huang, Mechanism of Phosphatidylglycerol Activation Catalyzed by Prolipoprotein Diacylglyceryl Transferase, J. Phys. Chem. B, 2019, 123(33), 7092–7102 CrossRef CAS PubMed.
- W. Singh, D. Quinn, T. S. Moody and M. Huang, Reaction Mechanism of Histone Demethylation in alphaKG-dependent Non-Heme Iron Enzymes, J. Phys. Chem. B, 2019, 123(37), 7801–7811 CrossRef CAS PubMed.
- Y. Cen, W. Singh, M. Arkin, T. S. Moody, M. Huang, J. Zhou, Q. Wu and M. T. Reetz, Artificial cysteine-lipases with high activity and altered catalytic mechanism created by laboratory evolution, Nat. Commun., 2019, 10(1), 3198 CrossRef PubMed.
- A. Ganguly, E. Boulanger and W. Thiel, Importance of MM Polarization in QM/MM Studies of Enzymatic Reactions: Assessment of the QM/MM Drude Oscillator Model, J. Chem. Theory Comput., 2017, 13(6), 2954–2961 CrossRef CAS PubMed.
- D. Bim, M. Navratil, O. Gutten, J. Konvalinka, Z. Kutil, M. Culka, V. Navratil, A. N. Alexandrova, C. Barinka and L. Rulisek, Predicting Effects of Site-Directed Mutagenesis on Enzyme Kinetics by QM/MM and QM Calculations: A Case of Glutamate Carboxypeptidase II, J. Phys. Chem. B, 2022, 126(1), 132–143 CrossRef CAS PubMed.
- M. Manathunga, H. M. Aktulga, A. W. Götz and K. M. Merz, Quantum Mechanics/Molecular Mechanics Simulations on NVIDIA and AMD Graphics Processing Units, J. Chem. Inf. Model., 2023, 63(3), 711–717 CrossRef CAS PubMed.
- B. Raghavan, M. Paulikat, K. Ahmad, L. Callea, A. Rizzi, E. Ippoliti, D. Mandelli, L. Bonati, M. De Vivo and P. Carloni, Drug Design in the Exascale Era: A Perspective from Massively Parallel QM/MM Simulations, J. Chem. Inf. Model., 2023, 63(12), 3647–3658 CrossRef CAS PubMed.
- G. V. Dhoke, M. D. Davari, U. Schwaneberg and M. Bocola, QM/MM Calculations Revealing the Resting and Catalytic States in Zinc-Dependent Medium-Chain Dehydrogenases/Reductases, ACS Catal., 2015, 5(6), 3207–3215 CrossRef CAS.
- D. Platero-Rochart, T. Krivobokova, M. Gastegger, G. Reibnegger and P. A. Sánchez-Murcia, Prediction of Enzyme Catalysis by Computing Reaction Energy Barriers via Steered QM/MM Molecular Dynamics Simulations and Machine Learning, J. Chem. Inf. Model., 2023, 63(15), 4623–4632 CrossRef CAS PubMed.
- J. Del Arco, A. Perona, L. Gonzalez, J. Fernandez-Lucas, F. Gago and P. A. Sanchez-Murcia, Reaction mechanism of nucleoside 2'-deoxyribosyltransferases: free-energy landscape supports an oxocarbenium ion as the reaction intermediate, Org. Biomol. Chem., 2019, 17(34), 7891–7899 RSC.
- D. P. Gavin, F. J. Reen, J. Rocha-Martin, I. Abreu-Castilla, D. F. Woods, A. M. Foley, P. A. Sanchez-Murcia, M. Schwarz, P. O'Neill, A. R. Maguire and F. O'Gara, Genome mining and characterisation of a novel transaminase with remote stereoselectivity, Sci. Rep., 2019, 9(1), 20285 CrossRef CAS PubMed.
- A. V. Pinto, P. Ferreira, R. P. P. Neves, P. A. Fernandes, M. J. Ramos and A. L. Magalhães, Reaction Mechanism of MHETase, a PET Degrading Enzyme, ACS Catal., 2021, 11(16), 10416–10428 CrossRef CAS.
- X. Pan, R. Van, J. Pu, K. Nam, Y. Mao and Y. Shao, Free Energy Profile Decomposition Analysis for QM/MM Simulations of Enzymatic Reactions, J. Chem. Theory Comput., 2023, 19(22), 8234–8244 CrossRef CAS PubMed.
- C. M. Clemente, L. Capece and M. A. Marti, Best Practices on QM/MM Simulations of Biological Systems, J. Chem. Inf. Model., 2023, 63(9), 2609–2627 CrossRef CAS PubMed.
- W. Meelua, T. Wanjai, N. Thinkumrob, R. Friedman and J. Jitonnom, Multiscale QM/MM Simulations Identify the Roles of Asp239 and 1-OH. Nucleophile in Transition State Stabilization in Arabidopsis thaliana Cell-Wall Invertase 1, J. Chem. Inf. Model., 2023, 63(15), 4827–4838 CrossRef CAS PubMed.
- S. Ahmadi, L. B. Herrera, M. Chehelamirani, J. Hostas, S. Jalife and D. R. Salahub, Multiscale modeling of enzymes: QM-cluster, QM/MM, and QM/MM/MD: A tutorial review, Int. J. Quantum Chem., 2018, 118(9), e25558 CrossRef.
- T. Wu, Y. Wang, N. Zhang, D. Yin, Y. Xu, Y. Nie and X. Mu, Reshaping Substrate-Binding Pocket of Leucine Dehydrogenase for Bidirectionally Accessing Structurally Diverse Substrates, ACS Catal., 2023, 13(1), 158–168 CrossRef CAS.
- K. Prakinee, A. Phintha, S. Visitsatthawong, N. Lawan, J. Sucharitakul, C. Kantiwiriyawanitch, J. Damborsky, P. Chitnumsub, K. H. Van Pée and P. Chaiyen, Mechanism-guided tunnel engineering to increase the efficiency of a flavin-dependent halogenase, Nat. Catal., 2022, 5(6), 534–544 CrossRef CAS.
- A. Phintha, K. Prakinee, A. Jaruwat, N. Lawan, S. Visitsatthawong, C. Kantiwiriyawanitch, W. Songsungthong, D. Trisrivirat, P. Chenprakhon, A. Mulholland, K.-H. van Pée, P. Chitnumsub and P. Chaiyen, Dissecting the low catalytic capability of flavin-dependent halogenases, J. Biol. Chem., 2021, 296, 100068 CrossRef CAS PubMed.
- A. D. St-Jacques, M. E. C. Eyahpaise and R. A. Chica, Computational Design of Multisubstrate Enzyme Specificity, ACS Catal., 2019, 9(6), 5480–5485 CrossRef CAS.
- F. Wang, M. Zhu, Z. Song, C. Li, Y. Wang, Z. Zhu, D. Sun, F. Lu and H.-M. Qin, Reshaping the Binding Pocket of Lysine Hydroxylase for Enhanced Activity, ACS Catal., 2020, 10(23), 13946–13956 CrossRef CAS.
- Z. Wang, H. Zhou, H. Yu, Z. Pu, J. Xu, H. Zhang, J. Wu and L. Yang, Computational Redesign of the Substrate Binding Pocket of Glutamate Dehydrogenase for Efficient Synthesis of Noncanonical l-Amino Acids, ACS Catal., 2022, 12(21), 13619–13629 CrossRef CAS.
- M. Taher, K. D. Dubey and S. Mazumdar, Computationally guided bioengineering of the active site, substrate access pathway, and water channels of thermostable cytochrome P450, CYP175A1, for catalyzing the alkane hydroxylation reaction, Chem. Sci., 2023, 14(48), 14316–14326 RSC.
- Y. Nie, S. Wang, Y. Xu, S. Luo, Y.-L. Zhao, R. Xiao, G. T. Montelione, J. F. Hunt and T. Szyperski, Enzyme Engineering Based on X-ray Structures and Kinetic Profiling of Substrate Libraries: Alcohol Dehydrogenases for Stereospecific Synthesis of a Broad Range of Chiral Alcohols, ACS Catal., 2018, 8(6), 5145–5152 CrossRef CAS.
- G.-C. Xu, Y. Wang, M.-H. Tang, J.-Y. Zhou, J. Zhao, R.-Z. Han and Y. Ni, Hydroclassified Combinatorial Saturation Mutagenesis: Reshaping Substrate Binding Pockets of KpADH for Enantioselective Reduction of Bulky–Bulky Ketones, ACS Catal., 2018, 8(9), 8336–8345 CrossRef CAS.
- L. R. Rapp, S. M. Marques, E. Zukic, B. Rowlinson, M. Sharma, G. Grogan, J. Damborsky and B. Hauer, Substrate Anchoring and Flexibility Reduction in CYP153AM.aq Leads to Highly Improved Efficiency toward Octanoic Acid, ACS Catal., 2021, 11(5), 3182–3189 CrossRef CAS.
- R.-J. Li, K. Tian, X. Li, A. R. Gaikaiwari and Z. Li, Engineering P450 Monooxygenases for Highly Regioselective and Active p-Hydroxylation of m-Alkylphenols, ACS Catal., 2022, 12(10), 5939–5948 CrossRef CAS.
- Q. Yin, J. Zhang, S. Ma, T. Gu, M. Wang, S. You, S. Ye, R. Su, Y. Wang and W. Qi, Efficient polyethylene terephthalate biodegradation by an engineered Ideonella sakaiensis PETase with a fixed substrate-binding W156 residue, Green Chem., 2024, 26, 2560–2570 RSC.
- Y. Hu, W. Xu, C. Hui, J. Xu, M. Huang, X. Lin and Q. Wu, The mutagenesis of a single site for enhancing or reversing the enantio- or regiopreference of cyclohexanone monooxygenases, Chem. Commun., 2020, 56(65), 9356–9359 RSC.
- S. Gergel, J. Soler, A. Klein, K. H. Schülke, B. Hauer, M. Garcia-Borràs and S. C. Hammer, Engineered cytochrome P450 for direct arylalkene-to-ketone oxidation via highly reactive carbocation intermediates. Nat., Catal., 2023, 6(7), 606–617 CAS.
- M. Corbella, G. P. Pinto and S. C. L. Kamerlin, Loop dynamics and the evolution of enzyme activity, Nat. Rev. Chem., 2023, 7(8), 536–547 CrossRef CAS PubMed.
- P. Yang, X. Wang, J. Ye, S. Rao, J. Zhou, G. Du and S. Liu, Enhanced Thermostability and Catalytic Activity of Streptomyces mobaraenesis Transglutaminase by Rationally Engineering Its Flexible Regions, J. Agric. Food Chem., 2023, 71(16), 6366–6375 CrossRef CAS PubMed.
- J. Deng and Q. Cui, Second-Shell Residues Contribute to Catalysis by Predominately Preorganizing the Apo State in PafA, J. Am. Chem. Soc., 2023, 145(20), 11333–11347 CrossRef CAS PubMed.
- C. Hui, W. Singh, D. Quinn, C. Li, T. S. Moody and M. Huang, Regio- and stereoselectivity in the CYP450(BM3)-catalyzed hydroxylation of complex terpenoids: a QM/MM study, Phys. Chem. Chem. Phys., 2020, 22(38), 21696–21706 RSC.
- N. Liu, L. Wu, J. Feng, X. Sheng, J. Li, X. Chen, J. Li, W. Liu, J. Zhou, Q. Wu and D. Zhu, Crystal Structures and Catalytic Mechanism of l-erythro-3,5-Diaminohexanoate Dehydrogenase and Rational Engineering for Asymmetric Synthesis of beta-Amino Acids, Angew. Chem., Int. Ed., 2021, 60(18), 10203–10210 CrossRef CAS PubMed.
- D. Zhang, X. Chen, R. Zhang, P. Yao, Q. Wu and D. Zhu, Development of β-Amino Acid Dehydrogenase for the Synthesis of β-Amino Acids via Reductive Amination of β-Keto Acids, ACS Catal., 2015, 5(4), 2220–2224 CrossRef CAS.
- A. Scholtissek, D. Tischler, A. H. Westphal, W. J. H. van Berkel and C. E. Paul, Old Yellow Enzyme-Catalysed Asymmetric Hydrogenation: Linking Family Roots with Improved Catalysis, Catalysts, 2017, 7(5), 130 CrossRef.
- T. Wang, R. Wei, Y. Feng, L. Jin, Y. Jia, D. Yang, Z. Liang, M. Han, X. Li, C. Lu and X. Ying, Engineering of Yeast Old Yellow Enzyme OYE3 Enables Its Capability Discriminating of (E)-Citral and (Z)-Citral, Molecules, 2021, 26(16), 5040 CrossRef CAS PubMed.
- M. S. Robescu, L. Cendron, A. Bacchin, K. Wagner, T. Reiter, I. Janicki, K. Merusic, M. Illek, M. Aleotti, E. Bergantino and M. Hall, Asymmetric Proton Transfer Catalysis by Stereocomplementary Old Yellow Enzymes for C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C Bond Isomerization Reaction, ACS Catal., 2022, 12(12), 7396–7405 CrossRef CAS.
C Bond Isomerization Reaction, ACS Catal., 2022, 12(12), 7396–7405 CrossRef CAS. - J. N. Kolev, K. M. O'Dwyer, C. T. Jordan and R. Fasan, Discovery of potent parthenolide-based antileukemic agents enabled by late-stage P450-mediated C–H functionalization, ACS Chem. Biol., 2014, 9(1), 164–173 CrossRef CAS PubMed.
- H. Alwaseem, S. Giovani, M. Crotti, K. Welle, C. T. Jordan, S. Ghaemmaghami and R. Fasan, Comprehensive Structure-Activity Profiling of Micheliolide and its Targeted Proteome in Leukemia Cells via Probe-Guided Late-Stage C–H Functionalization, ACS Cent. Sci., 2021, 7(5), 841–857 CrossRef CAS PubMed.
- P. Jiang, H. Jin, G. Zhang, W. Zhang, W. Liu, Y. Zhu, C. Zhang and L. Zhang, A Mechanistic Understanding of the Distinct Regio- and Chemoselectivity of Multifunctional P450s by Structural Comparison of IkaD and CftA Complexed with Common Substrates, Angew. Chem., Int. Ed., 2023, 62(51), e202310728 CrossRef CAS PubMed.
- J. Xu, Y. Cen, W. Singh, J. Fan, L. Wu, X. Lin, J. Zhou, M. Huang, M. T. Reetz and Q. Wu, Stereodivergent Protein Engineering of a Lipase To Access All Possible Stereoisomers of Chiral Esters with Two Stereocenters, J. Am. Chem. Soc., 2019, 141(19), 7934–7945 CrossRef CAS PubMed.
- E. Delgado-Arciniega, H. J. Wijma, C. Hummel and D. B. Janssen, Computationally Supported Inversion of Ketoreductase Stereoselectivity, ChemBioChem, 2023, 24(9), e202300032 CrossRef CAS PubMed.
- Y. Hu, J. Wang, Y. Cen, H. Zheng, M. Huang, X. Lin and Q. Wu, “Top” or “bottom” switches of a cyclohexanone monooxygenase controlling the enantioselectivity of the sandwiched substrate, Chem. Commun., 2019, 55(15), 2198–2201 RSC.
- Y. Hu, J. Xu, Y. Cen, D. Li, Y. Zhang, M. Huang, X. Lin and Q. Wu, Customizing the Enantioselectivity of a Cyclohexanone Monooxygenase by a Strategy Combining “Size-Probes” with in silico Study, ChemCatChem, 2019, 11(20), 5085–5092 CrossRef CAS.
- B. J. Yachnin, M. B. McEvoy, R. J. D. MacCuish, K. L. Morley, P. C. K. Lau and A. M. Berghuis, Lactone-Bound Structures of Cyclohexanone Monooxygenase Provide Insight into the Stereochemistry of Catalysis, ACS Chem. Biol., 2014, 9(12), 2843–2851 CrossRef CAS PubMed.
- M. Bocola, F. Schulz, F. Leca, A. Vogel, M. W. Fraaije and M. T. Reetz, Converting Phenylacetone Monooxygenase into Phenylcyclohexanone Monooxygenase by Rational Design: Towards Practical Baeyer–Villiger Monooxygenases, Adv. Synth. Catal., 2005, 347(7–8), 979–986 CrossRef CAS.
- P. L. Srivastava, A. M. Escorcia, F. Huynh, D. J. Miller, R. K. Allemann and M. W. van der Kamp, Redesigning the Molecular Choreography to Prevent Hydroxylation in Germacradien-11-ol Synthase Catalysis, ACS Catal., 2021, 11(3), 1033–1041 CrossRef CAS PubMed.
- H. Liu, S. Fang, L. Zhao, X. Men and H. Zhang, A Single Active-Site Mutagenesis Confers Enhanced Activity and/or Changed Product Distribution to a Pentalenene Synthase from Streptomyces sp. PSKA01, Bioengineering, 2023, 10, 3 Search PubMed.
- G. Li, M. Garcia-Borras, M. Furst, A. Ilie, M. W. Fraaije, K. N. Houk and M. T. Reetz, Overriding Traditional Electronic Effects in Biocatalytic Baeyer-Villiger Reactions by Directed Evolution, J. Am. Chem. Soc., 2018, 140(33), 10464–10472 CrossRef CAS PubMed.
- A. T. P. Carvalho, D. Dourado, T. Skvortsov, M. de Abreu, L. J. Ferguson, D. J. Quinn, T. S. Moody and M. Huang, Catalytic mechanism of phenylacetone monooxygenases for non-native linear substrates, Phys. Chem. Chem. Phys., 2017, 19(39), 26851–26861 RSC.
- A. T. P. Carvalho, D. Dourado, T. Skvortsov, M. de Abreu, L. J. Ferguson, D. J. Quinn, T. S. Moody and M. Huang, Spatial requirement for PAMO for transformation of non-native linear substrates, Phys. Chem. Chem. Phys., 2018, 20(4), 2558–2570 RSC.
- Y. Dong, T. Li, S. Zhang, J. Sanchis, H. Yin, J. Ren, X. Sheng, G. Li and M. T. Reetz, Biocatalytic Baeyer–Villiger Reactions: Uncovering the Source of Regioselectivity at Each Evolutionary Stage of a Mutant with Scrutiny of Fleeting Chiral Intermediates, ACS Catal., 2022, 12(6), 3669–3680 CrossRef CAS.
- K. Balke, A. Beier and U. T. Bornscheuer, Hot spots for the protein engineering of Baeyer-Villiger monooxygenases, Biotechnol. Adv., 2018, 36(1), 247–263 CrossRef CAS PubMed.
- M. T. Reetz and S. Wu, Greatly reduced amino acid alphabets in directed evolution: making the right choice for saturation mutagenesis at homologous enzyme positions, Chem. Commun., 2008, 5499–5501 RSC.
- M. T. Reetz and S. Wu, Laboratory Evolution of Robust and Enantioselective Baeyer−Villiger Monooxygenases for Asymmetric Catalysis, J. Am. Chem. Soc., 2009, 131(42), 15424–15432 CrossRef CAS PubMed.
- J. Xu, Y. Peng, Z. Wang, Y. Hu, J. Fan, H. Zheng, X. Lin and Q. Wu, Exploiting Cofactor Versatility to Convert a FAD-Dependent Baeyer-Villiger Monooxygenase into a Ketoreductase, Angew. Chem., 2019, 131(41), 14641–14645 CrossRef.
- S. Singh and R. Anand, Tunnel Architectures in Enzyme Systems that Transport Gaseous Substrates, ACS Omega, 2021, 6(49), 33274–33283 CrossRef CAS PubMed.
- R. Banerjee and J. D. Lipscomb, Small-Molecule Tunnels in Metalloenzymes Viewed as Extensions of the Active Site, Acc. Chem. Res., 2021, 54(9), 2185–2195 CrossRef CAS PubMed.
- S. Meng, R. An, Z. Li, U. Schwaneberg, Y. Ji, M. D. Davari, F. Wang, M. Wang, M. Qin, K. Nie and L. Liu, Tunnel engineering for modulating the substrate preference in cytochrome P450BsβHI, Bioresources Bioprocess., 2021, 8(1), 26 CrossRef PubMed.
- S. Kaushik, S. M. Marques, P. Khirsariya, K. Paruch, L. Libichova, J. Brezovsky, Z. Prokop, R. Chaloupkova and J. Damborsky, Impact of the access tunnel engineering on catalysis is strictly ligand-specific, FEBS J., 2018, 285(8), 1456–1476 CrossRef CAS PubMed.
- F. Richter, A. Leaver-Fay, S. D. Khare, S. Bjelic and D. Baker, De novo enzyme design using Rosetta3, PLoS One, 2011, 6(5), e19230 CrossRef CAS PubMed.
- N. Naowarojna, S. Irani, W. Hu, R. Cheng, L. Zhang, X. Li, J. Chen, Y. J. Zhang and P. Liu, Crystal Structure of the Ergothioneine Sulfoxide Synthase from Candidatus Chloracidobacterium thermophilum and Structure-Guided Engineering To Modulate Its Substrate Selectivity, ACS Catal., 2019, 9(8), 6955–6961 CrossRef CAS PubMed.
- S. T. Thomas, G. V. Louie, J. W. Lubin, V. Lundblad and J. P. Noel, Substrate Specificity and Engineering of Mevalonate 5-Phosphate Decarboxylase, ACS Chem. Biol., 2019, 14(8), 1767–1779 CrossRef CAS PubMed.
- T. G. Köllner, J. Degenhardt and J. Gershenzon, The Product Specificities of Maize Terpene Synthases TPS4 and TPS10 Are Determined Both by Active Site Amino Acids and Residues Adjacent to the Active Site, Plants, 2020, 9(5), 552 CrossRef PubMed.
- Z. Bata, Z. Molnár, E. Madaras, B. Molnar, E. Sánta-Bell, A. Varga, I. Leveles, R. Z. Qian, F. Hammerschmidt, C. Paizs, B. G. Vértessy and L. Poppe, Substrate Tunnel Engineering Aided by X-ray Crystallography and Functional Dynamics Swaps the Function of MIO-Enzymes, ACS Catal., 2021, 11(8), 4538–4549 CrossRef CAS.
- A. Hou and J. S. Dickschat, Targeting active site residues and structural anchoring positions in terpene synthases, Beilstein J. Org. Chem., 2021, 17, 2441–2449 CrossRef CAS PubMed.
- Z. Li, L. Zhang, K. Xu, Y. Jiang, J. Du, X. Zhang, L. H. Meng, Q. Wu, L. Du, X. Li, Y. Hu, Z. Xie, X. Jiang, Y. J. Tang, R. Wu, R. T. Guo and S. Li, Molecular insights into the catalytic promiscuity of a bacterial diterpene synthase, Nat. Commun., 2023, 14(1), 4001 CrossRef CAS PubMed.
- H. Li and O. Turunen, Effect of acidic amino acids engineered into the active site cleft of Thermopolyspora flexuosa GH11 xylanase, Biotechnol. Appl. Biochem., 2015, 62(4), 433–440 CrossRef CAS PubMed.
- S. Anbarasan, T. Timoharju, J. Barthomeuf, O. Pastinen, J. Rouvinen, M. Leisola and O. Turunen, Effect of active site mutation on pH activity and transglycosylation of Sulfolobus acidocaldarius β-glycosidase, J. Mol. Catal. B: Enzym., 2015, 118, 62–69 CrossRef CAS.
- T. Kim, E. J. Mullaney, J. M. Porres, K. R. Roneker, S. Crowe, S. Rice, T. Ko, A. H. Ullah, C. B. Daly and R. Welch, Shifting the pH profile of Aspergillus niger PhyA phytase to match the stomach pH enhances its effectiveness as an animal feed additive, Appl. Environ. Microbiol., 2006, 72(6), 4397–4403 CrossRef CAS PubMed.
- C.-H. Wang, X.-L. Liu, R.-B. Huang, B.-F. He and M.-M. Zhao, Enhanced acidic adaptation of Bacillus subtilis Ca-independent alpha-amylase by rational engineering of pKa values, Biochem. Eng. J., 2018, 139, 146–153 CrossRef CAS.
- A. Hirata, M. Adachi, S. Utsumi and B. Mikami, Engineering of the pH Optimum of Bacillus cereus β-Amylase: Conversion of the pH Optimum from a Bacterial Type to a Higher-Plant Type, Biochemistry, 2004, 43(39), 12523–12531 CrossRef CAS PubMed.
- L. Xu, M. Z. Nawaz, H. R. Khalid, H. Waqarul, H. A. Alghamdi, J. Sun and D. Zhu, Modulating the pH profile of vanillin dehydrogenase enzyme from extremophile Bacillus ligniniphilus L1 through computational guided site-directed mutagenesis, Int. J. Biol. Macromol., 2024, 263, 130359 CrossRef CAS PubMed.
- M. V. Ushasree, J. Vidya and A. Pandey, Replacement P212H Altered the pH–Temperature Profile of Phytase from Aspergillus niger NII 08121, Appl. Biochem. Biotechnol., 2015, 175(6), 3084–3092 CrossRef CAS PubMed.
- A. J. Russell and A. R. Fersht, Rational modification of enzyme catalysis by engineering surface charge, Nature, 1987, 328(6130), 496–500 CrossRef CAS PubMed.
- A. Tomschy, R. Brugger, M. Lehmann, A. Svendsen, K. Vogel, D. Kostrewa, F. Lassen Søren, D. Burger, A. Kronenberger, P. G. M. van Loon Adolphus, L. Pasamontes and M. Wyss, Engineering of Phytase for Improved Activity at Low pH, Appl. Environ. Microbiol., 2002, 68(4), 1907–1913 CrossRef CAS PubMed.
- D. W. Cockburn and A. J. Clarke, Modulating the pH-activity profile of cellulase A from Cellulomonas fimi by replacement of surface residues, Protein Eng., Des. Sel., 2011, 24(5), 429–437 CrossRef CAS PubMed.
- T. Yang, L. Pan, W. Wu, X. Pan, M. Xu, X. Zhang and Z. Rao, N20D/N116E Combined Mutant Downward Shifted the pH Optimum of Bacillus subtilis NADH Oxidase, Biology, 2023, 12(4), 522 CrossRef CAS PubMed.
- K. W. Kaufmann, G. H. Lemmon, S. L. DeLuca, J. H. Sheehan and J. Meiler, Practically useful: what the Rosetta protein modeling suite can do for you, Biochemistry, 2010, 49(14), 2987–2998 CrossRef CAS PubMed.
- Z. Li, L. Li, Y. Huo, Z. Chen, Y. Zhao, J. Huang, S. Jian, Z. Rong, D. Wu, J. Gan, X. Hu, J. Li and X. W. Xu, Structure-guided protein engineering increases enzymatic activities of the SGNH family esterases, Biotechnol. Biofuels, 2020, 13(1), 107 CrossRef CAS PubMed.
- G. Abrusan and J. A. Marsh, Alpha Helices Are More Robust to Mutations than Beta Strands, PLoS Comput. Biol., 2016, 12(12), e1005242 CrossRef PubMed.
- P. Y. Chou and G. D. Fasman, Empirical Predictions of Protein Conformation, Annu. Rev. Biochem., 1978, 47(1), 251–276 CrossRef CAS PubMed.
- Z. Zhou and X. Wang, Rational design and structure-based engineering of alkaline pectate lyase from Paenibacillus sp. 0602 to improve thermostability, BMC Biotechnol., 2021, 21(1), 32 CrossRef CAS PubMed.
- M. Klaewkla, R. Pichyangkura, T. Charoenwongpaiboon, K. Wangpaiboon and S. Chunsrivirot, Computational design of oligosaccharide producing levansucrase from Bacillus licheniformis RN-01 to improve its thermostability for production of levan-type fructooligosaccharides from sucrose, Int. J. Biol. Macromol., 2020, 160, 252–263 CrossRef CAS PubMed.
- Y. Sang, X. Huang, H. Li, T. Hong, M. Zheng, Z. Li, Z. Jiang, H. Ni, Q. Li and Y. Zhu, Improving the thermostability of Pseudoalteromonas Porphyrae κ-carrageenase by rational design and MD simulation, AMB Express, 2024, 14(1), 8 CrossRef CAS PubMed.
- R. Wang, S. Wang, Y. Xu and X. Yu, Enhancing the thermostability of Rhizopus chinensis lipase by rational design and MD simulations, Int. J. Biol. Macromol., 2020, 160, 1189–1200 CrossRef CAS PubMed.
- Z. Li, C. Zhao, D. Li and L. Wang, Enhancing the thermostability of Streptomyces cyaneofuscatus strain Ms1 tyrosinase by multi-factors rational design and molecular dynamics simulations, PLoS One, 2023, 18(7), e0288929 CrossRef CAS PubMed.
- M. Matsumura, G. Signor and B. W. Matthews, Substantial increase of protein stability by multiple disulphide bonds, Nature, 1989, 342(6247), 291–293 CrossRef CAS PubMed.
- R. Sowdhamini, N. Srinivasan, B. Shoichet, D. V. Santi, C. Ramakrishnan and P. Balaram, Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis, Protein Eng., Des. Sel., 1989, 3(2), 95–103 CrossRef CAS PubMed.
- D. B. Craig and A. A. Dombkowski, Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins, BMC Bioinf., 2013, 14, 1–7 CrossRef PubMed.
- J. L. Pellequer and S. W. W. Chen, Multi-template approach to modeling engineered disulfide bonds, Proteins: Struct., Funct., Bioinf., 2006, 65(1), 192–202 CrossRef CAS PubMed.
- J. Huang, S. Dai, X. Chen, L. Xu, J. Yan, M. Yang and Y. Yan, Alteration of Chain-Length Selectivity and Thermostability of Rhizopus oryzae Lipase via Virtual Saturation Mutagenesis Coupled with Disulfide Bond Design, Appl. Environ. Microbiol. J. Homepage, 2023, 89(1), e0187822 CrossRef PubMed.
- F. Kawai, The Current State of Research on PET Hydrolyzing Enzymes Available for Biorecycling, Catalysts, 2021, 11(2), 206 CrossRef CAS.
- S. Schmidt, M. Genz, K. Balke and U. T. Bornscheuer, The effect of disulfide bond introduction and related Cys/Ser mutations on the stability of a cyclohexanone monooxygenase, J. Biotechnol., 2015, 214, 199–211 CrossRef CAS PubMed.
- H. L. van Beek, H. J. Wijma, L. Fromont, D. B. Janssen and M. W. Fraaije, Stabilization of cyclohexanone monooxygenase by a computationally designed disulfide bond spanning only one residue, FEBS Open Bio, 2014, 4(1), 168–174 CrossRef CAS PubMed.
- D. J. Opperman and M. T. Reetz, Towards Practical Baeyer–Villiger-Monooxygenases: Design of Cyclohexanone Monooxygenase Mutants with Enhanced Oxidative Stability, ChemBioChem, 2010, 11(18), 2589–2596 CrossRef CAS PubMed.
- S. F. Sousa, A. J. M. Ribeiro, R. P. P. Neves, N. F. Brás, N. M. F. S. A. Cerqueira, P. A. Fernandes and M. J. Ramos, Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms, WIREs Comput. Mol. Sci., 2017, 7(2), e1281 CrossRef.
- D. J. Huggins, P. C. Biggin, M. A. Dämgen, J. W. Essex, S. A. Harris, R. H. Henchman, S. Khalid, A. Kuzmanic, C. A. Laughton, J. Michel, A. J. Mulholland, E. Rosta, M. S. P. Sansom and M. W. van der Kamp, Biomolecular simulations: From dynamics and mechanisms to computational assays of biological activity, WIREs Comput. Mol. Sci., 2019, 9(3), e1393 CrossRef.
- R. C. Bernardi, M. C. R. Melo and K. Schulten, Enhanced sampling techniques in molecular dynamics simulations of biological systems, Biochim. Biophys. Acta, 2015, 1850(5), 872–877 CrossRef CAS PubMed.
- T. Schlick and S. Portillo-Ledesma, Biomolecular modeling thrives in the age of technology, Nat. Comput. Sci., 2021, 1(5), 321–331 CrossRef PubMed.
- M. C. Childers and V. Daggett, Insights from molecular dynamics simulations for computational protein design, Mol. Syst. Des. Eng., 2017, 2(1), 9–33 RSC.
- F. Zhang, T. Zeng and R. Wu, QM/MM Modeling Aided Enzyme Engineering in Natural Products Biosynthesis, J. Chem. Inf. Model., 2023, 63(16), 5018–5034 CrossRef CAS PubMed.
- Y. Jiang, X. Ran and Z. J. Yang, Data-driven enzyme engineering to identify function-enhancing enzymes, Protein Eng., Des. Sel., 2022, 36 Search PubMed.
- R. Feehan, D. Montezano and J. S. G. Slusky, Machine learning for enzyme engineering, selection and design, Protein Eng., Des. Sel., 2021, 34 Search PubMed.
- K. K. Yang, Z. Wu and F. H. Arnold, Machine-learning-guided directed evolution for protein engineering, Nat. Methods, 2019, 16(8), 687–694 CrossRef CAS PubMed.
- G. Li, Y. Dong and M. T. Reetz, Can Machine Learning Revolutionize Directed Evolution of Selective Enzymes?, Adv. Synth. Catal., 2019, 361(11), 2377–2386 CrossRef CAS.
- R. Vanella, G. Kovacevic, V. Doffini, J. Fernandez de Santaella and M. A. Nash, High-throughput screening, next generation sequencing and machine learning: advanced methods in enzyme engineering, Chem. Commun., 2022, 58(15), 2455–2467 RSC.
- T. Clark, V. Subramanian, A. Jayaraman, E. Fitzpatrick, R. Gopal, N. Pentakota, T. Rurak, S. Anand, A. Viglione, R. Raman, K. Tharakaraman and R. Sasisekharan, Enhancing antibody affinity through experimental sampling of non-deleterious CDR mutations predicted by machine learning, Commun. Chem., 2023, 6(1), 244 CrossRef PubMed.
- K. Köchl, T. Schopper, V. Durmaz, L. Parigger, A. Singh, A. Krassnigg, M. Cespugli, W. Wu, X. Yang, Y. Zhang, W. W.-S. Wang, C. Selluski, T. Zhao, X. Zhang, C. Bai, L. Lin, Y. Hu, Z. Xie, Z. Zhang, J. Yan, K. Zatloukal, K. Gruber, G. Steinkellner and C. C. Gruber, Optimizing variant-specific therapeutic SARS-CoV-2 decoys using deep-learning-guided molecular dynamics simulations, Sci. Rep., 2023, 13(1), 774 CrossRef PubMed.
- N. E. Jackson, B. M. Savoie, A. Statt and M. A. Webb, Introduction to Machine Learning for Molecular Simulation, J. Chem. Theory Comput., 2023, 19(14), 4335–4337 CrossRef CAS PubMed.
- Z. Song, F. Trozzi, H. Tian, C. Yin and P. Tao, Mechanistic Insights into Enzyme Catalysis from Explaining Machine-Learned Quantum Mechanical and Molecular Mechanical Minimum Energy Pathways, ACS Phys. Chem. Au, 2022, 2(4), 316–330 CrossRef CAS PubMed.
- X. Pan, J. Yang, R. Van, E. Epifanovsky, J. Ho, J. Huang, J. Pu, Y. Mei, K. Nam and Y. Shao, Machine-Learning-Assisted Free Energy Simulation of Solution-Phase and Enzyme Reactions, J. Chem. Theory Comput., 2021, 17(9), 5745–5758 CrossRef CAS PubMed.
- M. Cechova, Ten simple rules for biologists initiating a collaboration with computer scientists, PLoS Comput. Biol., 2020, 16(10), e1008281 CrossRef CAS PubMed.
- M. Danishuddin and A. U. Khan, Descriptors and their selection methods in QSAR analysis: paradigm for drug design, Drug Discovery Today, 2016, 21(8), 1291–1302 CrossRef PubMed.
- K. V. Chuang, L. M. Gunsalus and M. J. Keiser, Learning Molecular Representations for Medicinal Chemistry, J. Med. Chem., 2020, 63(16), 8705–8722 CrossRef CAS PubMed.
- M. Staszak, K. Staszak, K. Wieszczycka, A. Bajek, K. Roszkowski and B. Tylkowski, Machine learning in drug design: Use of artificial intelligence to explore the chemical structure–biological activity relationship, WIREs Comput. Mol. Sci., 2022, 12(2), e1568 CrossRef.
- M. Karelson, V. S. Lobanov and A. R. Katritzky, Quantum-Chemical Descriptors in QSAR/QSPR Studies, Chem. Rev., 1996, 96(3), 1027–1044 CrossRef CAS PubMed.
- S. Tortorella, E. Carosati, G. Sorbi, G. Bocci, S. Cross, G. Cruciani and L. Storchi, Combining machine learning and quantum mechanics yields more chemically aware molecular descriptors for medicinal chemistry applications, J. Comput. Chem., 2021, 42(29), 2068–2078 CrossRef CAS PubMed.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS.
- S. Heller, A. McNaught, S. Stein, D. Tchekhovskoi and I. Pletnev, InChI—the worldwide chemical structure identifier standard, J. Cheminf., 2013, 5(1), 7 CAS.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, Reoptimization of MDL Keys for Use in Drug Discovery, J. Chem. Inf. Comput. Sci., 2002, 42(6), 1273–1280 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50(5), 742–754 CrossRef CAS PubMed.
- G. Landrum, RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling, Greg Landrum, 2013, 8, 31 Search PubMed.
- T. Mikolov, K. Chen, G. Corrado and J. Dean, Efficient estimation of word representations in vector space, arXiv, 2013, preprint, arXiv:1301.3781 DOI:10.48550/arXiv.1301.3781.
- S. Jaeger, S. Fulle and S. Turk, Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition, J. Chem. Inf. Model., 2018, 58(1), 27–35 CrossRef CAS PubMed.
- W. Chen, G. Chen, L. Zhao and C. Y.-C. Chen, Predicting Drug–Target Interactions with Deep-Embedding Learning of Graphs and Sequences, J. Phys. Chem. A, 2021, 125(25), 5633–5642 CrossRef CAS PubMed.
- Q. Yin, X. Cao, R. Fan, Q. Liu, R. Jiang and W. Zeng, DeepDrug: A general graph-based deep learning framework for drug-drug interactions and drug-target interactions prediction, Quant. Biol., 2023, 11(3), 260–274 CrossRef CAS.
- A. S. Rifaioglu, E. Nalbat, V. Atalay, M. J. Martin, R. Cetin-Atalay and T. Doğan, DEEPScreen: high performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations, Chem. Sci., 2020, 11(9), 2531–2557 RSC.
- A. Dalkıran, A. Atakan, A. S. Rifaioğlu, M. J. Martin, R. Ç. Atalay, A. C. Acar, T. Doğan and V. Atalay, Transfer learning for drug–target interaction prediction, Bioinformatics, 2023, 39(Supplement_1), i103–i110 CrossRef PubMed.
- G. Skoraczyński, P. Dittwald, B. Miasojedow, S. Szymkuć, E. P. Gajewska, B. A. Grzybowski and A. Gambin, Predicting the outcomes of organic reactions via machine learning: are current descriptors sufficient?, Sci. Rep., 2017, 7(1), 3582 CrossRef PubMed.
- Y. L. Liao and T. Smidt: Equiformer: Equivariant graph attention transformer for 3d atomistic graphs, arXiv, 2022, preprint, arXiv:2206.11990 DOI:10.48550/arXiv.2206.11990.
- Y.-L. Liao; B. Wood; A. Das and T. Smidt, EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations. arXiv, 2023, preprint, arXiv:2306.12059 Search PubMed.
- Y. Liu; L. Wang; M. Liu; X. Zhang; B. Oztekin and S. Ji, Spherical message passing for 3d graph networks. arXiv, 2021, preprint, arXiv:2102.05013 Search PubMed.
- L. Wang, Y. Liu, Y. Lin, H. Liu and S. Ji, ComENet: Towards complete and efficient message passing for 3D molecular graphs, Adv. Neural Inform. Process. Syst., 2022, 35, 650–664 Search PubMed.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K.-R. Müller, Schnet – a deep learning architecture for molecules and materials, J. Chem. Phys., 2018, 148, 241722 CrossRef PubMed.
- J. Gasteiger; J. Groß and S. Günnemann, Directional message passing for molecular graphs. arXiv, 2020, preprint, arXiv:2003.03123 Search PubMed.
- J. Gasteiger, F. Becker and S. Günnemann, Gemnet: Universal directional graph neural networks for molecules, Adv. Neural Inform. Process. Syst., 2021, 34, 6790–6802 Search PubMed.
- S. D. Axen, X.-P. Huang, E. L. Cáceres, L. Gendelev, B. L. Roth and M. J. Keiser, A Simple Representation of Three-Dimensional Molecular Structure, J. Med. Chem., 2017, 60(17), 7393–7409 CrossRef CAS PubMed.
- X. Fang, L. Liu, J. Lei, D. He, S. Zhang, J. Zhou, F. Wang, H. Wu and H. Wang, Geometry-enhanced molecular representation learning for property prediction, Nat. Machine Intelligence, 2022, 4(2), 127–134 CrossRef.
- D. Chen, K. Gao, D. D. Nguyen, X. Chen, Y. Jiang, G.-W. Wei and F. Pan, Algebraic graph-assisted bidirectional transformers for molecular property prediction, Nat. Commun., 2021, 12(1), 3521 CrossRef CAS PubMed.
- C. Li, J. Wang, Z. Niu, J. Yao and X. Zeng, A spatial-temporal gated attention module for molecular property prediction based on molecular geometry, Briefings Bioinf., 2021, 22(5), bbab078 CrossRef PubMed.
- K. Gao, D. D. Nguyen, V. Sresht, A. M. Mathiowetz, M. Tu and G.-W. Wei, Are 2D fingerprints still valuable for drug discovery?, Phys. Chem. Chem. Phys., 2020, 22(16), 8373–8390 RSC.
- J. M. Crawford and M. S. Sigman, Conformational dynamics in asymmetric catalysis: is catalyst flexibility a design element?, Synthesis, 2019, 1021–1036 CAS.
- K. V. Chuang, L. M. Gunsalus and M. J. Keiser, Learning Molecular Representations for Medicinal Chemistry, J. Med. Chem., 2020, 63(16), 8705–8722 CrossRef CAS PubMed.
- Z. Liu, T. Zubatiuk, A. Roitberg and O. Isayev, Auto3D: Automatic Generation of the Low-Energy 3D Structures with ANI Neural Network Potentials, J. Chem. Inf. Model., 2022, 62(22), 5373–5382 CrossRef CAS PubMed.
- R. Zubatyuk, J. S. Smith, B. T. Nebgen, S. Tretiak and O. Isayev, Teaching a neural network to attach and detach electrons from molecules, Nat. Commun., 2021, 12(1), 4870 CrossRef CAS PubMed.
- Y. Zhu, J. Hwang, K. Adams, Z. Liu, B. Nan, B. Stenfors, Y. Du, J. Chauhan, O. Wiest and O. Isayev, Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks, arXiv, 2023, preprint, arXiv:2310.00115 DOI:10.48550/arXiv.2310.00115.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning, Science, 2019, 363(6424), eaau5631 CrossRef CAS PubMed.
- S. Axelrod and R. Gomez-Bombarelli, Molecular machine learning with conformer ensembles, Mach. Learn.: Sci. Technol., 2023, 4(3), 035025 Search PubMed.
- Y. Xu, D. Verma, R. P. Sheridan, A. Liaw, J. Ma, N. M. Marshall, J. McIntosh, E. C. Sherer, V. Svetnik and J. M. Johnston, Deep dive into machine learning models for protein engineering, J. Chem. Inf. Model., 2020, 60(6), 2773–2790 CrossRef CAS PubMed.
- C. Wu, G. Whitson, J. McLarty, A. Ermongkonchai and T. C. Chang, Protein classification artificial neural system, Protein Sci., 1992, 1(5), 667–677 CrossRef CAS PubMed.
- J. Wang, B. Yang, J. Revote, A. Leier, T. T. Marquez-Lago, G. Webb, J. Song, K.-C. Chou and T. Lithgow, POSSUM: a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles, Bioinformatics, 2017, 33(17), 2756–2758 CrossRef CAS PubMed.
- M. Sandberg, L. Eriksson, J. Jonsson, M. Sjöström and S. Wold, New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids, J. Med. Chem., 1998, 41(14), 2481–2491 CrossRef CAS PubMed.
- V. Biou, J.-F. Gibrat, J. Levin, B. Robson and J. Garnier, Secondary structure prediction: combination of three different methods, Protein Eng., Des. Sel., 1988, 2(3), 185–191 CrossRef CAS PubMed.
- F. Tian, P. Zhou and Z. Li, T-scale as a novel vector of topological descriptors for amino acids and its application in QSARs of peptides, J. Mol. Struct., 2007, 830(1–3), 106–115 CrossRef CAS.
- L. Yang, M. Shu, K. Ma, H. Mei, Y. Jiang and Z. Li, ST-scale as a novel amino acid descriptor and its application in QSAM of peptides and analogues, Amino Acids, 2010, 38, 805–816 CrossRef CAS PubMed.
- H. Mei, Z. H. Liao, Y. Zhou and S. Z. Li, A new set of amino acid descriptors and its application in peptide QSARs, Peptide Sci.: Orig. Res. Biomol., 2005, 80(6), 775–786 CrossRef CAS PubMed.
- G. J. van Westen, R. F. Swier, J. K. Wegner, A. P. IJzerman, H. W. van Vlijmen and A. Bender, Benchmarking of protein descriptor sets in proteochemometric modeling (part 1): comparative study of 13 amino acid descriptor sets, J. Cheminf., 2013, 5(1), 1–11 Search PubMed.
- S. Kawashima, P. Pokarowski, M. Pokarowska, A. Kolinski, T. Katayama and M. Kanehisa, AAindex: amino acid index database, progress report 2008, Nucleic Acids Res., 2007, 36(suppl_1), D202–D205 CrossRef PubMed.
- S. Wang, S. Sun, Z. Li, R. Zhang and J. Xu, Accurate de novo prediction of protein contact map by ultra-deep learning model, PLoS Comput. Biol., 2017, 13(1), e1005324 CrossRef PubMed.
- B. Jing; S. Eismann; P. Suriana; R. J. Townshend and R. Dror, Learning from protein structure with geometric vector perceptrons. arXiv, 2020, preprint, arXiv:2009.01411 Search PubMed.
- E. Asgari and M. R. Mofrad, Continuous distributed representation of biological sequences for deep proteomics and genomics, PLoS One, 2015, 10(11), e0141287 CrossRef PubMed.
- M. Gribskov, A. D. McLachlan and D. Eisenberg, Profile analysis: detection of distantly related proteins, Proc. Natl. Acad. Sci., 1987, 84(13), 4355–4358 CrossRef CAS PubMed.
- J. Durairaj, D. de Ridder and A. D. J. van Dijk, Beyond sequence: Structure-based machine learning, Comput. Struct. Biotechnol. J., 2023, 21, 630–643 CrossRef CAS PubMed.
- A. Glielmo, B. E. Husic, A. Rodriguez, C. Clementi, F. Noé and A. Laio, Unsupervised Learning Methods for Molecular Simulation Data, Chem. Rev., 2021, 121(16), 9722–9758 CrossRef CAS PubMed.
- A. Lodola, J. Sirirak, N. Fey, S. Rivara, M. Mor and A. J. Mulholland, Structural Fluctuations in Enzyme-Catalyzed Reactions: Determinants of Reactivity in Fatty Acid Amide Hydrolase from Multivariate Statistical Analysis of Quantum Mechanics/Molecular Mechanics Paths, J. Chem. Theory Comput., 2010, 6(9), 2948–2960 CrossRef CAS PubMed.
- U. Doshi, M. J. Holliday, E. Z. Eisenmesser and D. Hamelberg, Dynamical network of residue–residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation, Proc. Natl. Acad. Sci., 2016, 113(17), 4735–4740 CrossRef CAS PubMed.
- Y. Li, Y. Fang and J. Fang, Predicting residue–residue contacts using random forest models, Bioinformatics, 2011, 27(24), 3379–3384 CrossRef CAS PubMed.
- D. Rappoport and A. Jinich, Enzyme Substrate Prediction from Three-Dimensional Feature Representations Using Space-Filling Curves, J. Chem. Inf. Model., 2023, 63(5), 1637–1648 CrossRef CAS PubMed.
- F. Li, L. Yuan, H. Lu, G. Li, Y. Chen, M. K. M. Engqvist, E. J. Kerkhoven and J. Nielsen, Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction, Nat. Catal., 2022, 5(8), 662–672 CrossRef.
- M. Tsubaki, K. Tomii and J. Sese, Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences, Bioinformatics, 2019, 35(2), 309–318 CrossRef CAS PubMed.
- M. L. Hekkelman, I. de Vries, R. P. Joosten and A. Perrakis, AlphaFill: enriching AlphaFold models with ligands and cofactors, Nat. Methods, 2023, 20(2), 205–213 CrossRef CAS PubMed.
- X. Wang, X. Zhang, C. Peng, Y. Shi, H. Li, Z. Xu and W. Zhu, D3DistalMutation: a Database to Explore the Effect of Distal Mutations on Enzyme Activity, J. Chem. Inf. Model., 2021, 61(5), 2499–2508 CrossRef CAS PubMed.
- H. D. Clements, A. R. Flynn, B. T. Nicholls, D. Grosheva, S. J. Lefave, M. T. Merriman, T. K. Hyster and M. S. Sigman, Using Data Science for Mechanistic Insights and Selectivity Predictions in a Non-Natural Biocatalytic Reaction, J. Am. Chem. Soc., 2023, 145(32), 17656–17664 CrossRef CAS PubMed.
- R. J. Fox, S. C. Davis, E. C. Mundorff, L. M. Newman, V. Gavrilovic, S. K. Ma, L. M. Chung, C. Ching, S. Tam, S. Muley, J. Grate, J. Gruber, J. C. Whitman, R. A. Sheldon and G. W. Huisman, Improving catalytic function by ProSAR-driven enzyme evolution, Nat. Biotechnol., 2007, 25(3), 338–344 CrossRef CAS PubMed.
- Y. Saito, M. Oikawa, T. Sato, H. Nakazawa, T. Ito, T. Kameda, K. Tsuda and M. Umetsu, Machine-Learning-Guided Library Design Cycle for Directed Evolution of Enzymes: The Effects of Training Data Composition on Sequence Space Exploration, ACS Catal., 2021, 11(23), 14615–14624 CrossRef CAS.
- G. Li, K. S. Rabe, J. Nielsen and M. K. M. Engqvist, Machine Learning Applied to Predicting Microorganism Growth Temperatures and Enzyme Catalytic Optima, ACS Synth. Biol., 2019, 8(6), 1411–1420 CrossRef CAS PubMed.
- P. Notin; R. Weitzman; D. S. Marks and Y. Gal, ProteinNPT: Improving Protein Property Prediction and Design with Non-Parametric Transformers. bioRxiv, 2023, preprint, 2023.12.06.570473 Search PubMed.
- J. A. Barbero-Aparicio, A. Olivares-Gil, J. J. Rodríguez, C. García-Osorio and J. F. Díez-Pastor, Addressing data scarcity in protein fitness landscape analysis: A study on semi-supervised and deep transfer learning techniques, Information Fusion, 2024, 102, 102035 CrossRef.
- S. Raschka, Model evaluation, model selection, and algorithm selection in machine learning. arXiv, 2018, preprint, arXiv:1811.12808 Search PubMed.
- J. G. Greener, S. M. Kandathil, L. Moffat and D. T. Jones, A guide to machine learning for biologists, Nat. Rev. Mol. Cell Biol., 2022, 23(1), 40–55 CrossRef CAS PubMed.
- X. Wang, D. Quinn, T. S. Moody and M. Huang, ALDELE: All-Purpose Deep Learning Toolkits for Predicting the Biocatalytic Activities of Enzymes, J. Chem. Inf. Model., 2024, 64(8), 3123–3139 CrossRef CAS PubMed.
- Y. Ogawa, Y. Saito, H. Yamaguchi, Y. Katsuyama and Y. Ohnishi, Engineering the Substrate Specificity of Toluene Degrading Enzyme XylM Using Biosensor XylS and Machine Learning, ACS Synth. Biol., 2023, 12(2), 572–582 CrossRef CAS PubMed.
- Z. Li, S. Meng, K. Nie, U. Schwaneberg, M. D. Davari, H. Xu, Y. Ji and L. Liu, Flexibility Regulation of Loops Surrounding the Tunnel Entrance in Cytochrome P450 Enhanced Substrate Access Substantially, ACS Catal., 2022, 12(20), 12800–12808 CrossRef CAS.
- Y. Liu, Z. Li, C. Cao, X. Zhang, S. Meng, M. D. Davari, H. Xu, Y. Ji, U. Schwaneberg and L. Liu, Engineering of Substrate Tunnel of P450 CYP116B3 though Machine Learning, Catalysts, 2023, 13(8), 1228 CrossRef CAS.
- G. Li, F. Buric, J. Zrimec, S. Viknander, J. Nielsen, A. Zelezniak and M. K. M. Engqvist, Learning deep representations of enzyme thermal adaptation, Protein Sci., 2022, 31(12), e4480 CrossRef PubMed.
- Z. Chen, P. Zhao, F. Li, A. Leier, T. T. Marquez-Lago, Y. Wang, G. I. Webb, A. I. Smith, R. J. Daly and K.-C. Chou, iFeature: a python package and web server for features extraction and selection from protein and peptide sequences, Bioinformatics, 2018, 34(14), 2499–2502 CrossRef CAS PubMed.
- E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi and G. M. Church, Unified rational protein engineering with sequence-based deep representation learning, Nat. Methods, 2019, 16(12), 1315–1322 CrossRef CAS PubMed.
- F. Cadet, N. Fontaine, G. Li, J. Sanchis, M. Ng Fuk Chong, R. Pandjaitan, I. Vetrivel, B. Offmann and M. T. Reetz, A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes, Sci. Rep., 2018, 8(1), 16757 CrossRef PubMed.
- X. Ran, Y. Jiang, Q. Shao and Z. J. Yang, EnzyKR: a chirality-aware deep learning model for predicting the outcomes of the hydrolase-catalyzed kinetic resolution, Chem. Sci., 2023, 14(43), 12073–12082 RSC.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Machine learning-aided engineering of hydrolases for PET depolymerization, Nature, 2022, 604(7907), 662–667 CrossRef CAS PubMed.
- R. Shroff, A. W. Cole, D. J. Diaz, B. R. Morrow, I. Donnell, A. Annapareddy, J. Gollihar, A. D. Ellington and R. Thyer, Discovery of Novel Gain-of-Function Mutations Guided by Structure-Based Deep Learning, ACS Synth. Biol., 2020, 9(11), 2927–2935 CrossRef CAS PubMed.
- C. Lu, J. H. Lubin, V. V. Sarma, S. Z. Stentz, G. Wang, S. Wang and S. D. Khare, Prediction and design of protease enzyme specificity using a structure-aware graph convolutional network, Proc. Natl. Acad. Sci., 2023, 120(39), e2303590120 CrossRef CAS PubMed.
- B. M. Bonk, J. W. Weis and B. Tidor, Machine Learning Identifies Chemical Characteristics That Promote Enzyme Catalysis, J. Am. Chem. Soc., 2019, 141(9), 4108–4118 CrossRef CAS PubMed.
- A. Kroll, S. Ranjan, M. K. M. Engqvist and M. J. Lercher, A general model to predict small molecule substrates of enzymes based on machine and deep learning, Nat. Commun., 2023, 14(1), 2787 CrossRef CAS PubMed.
- D. Zhang, H. Xing, D. Liu, M. Han, P. Cai, H. Lin, Y. Tian, Y. Guo, B. Sun, Y. Le, Y. Tian, A. Wu and Q.-N. Hu, Discovery of Toxin-Degrading Enzymes with Positive Unlabeled Deep Learning, ACS Catal., 2024, 3336–3348 CrossRef CAS.
- L. Liu, S. Zhou and Y. Deng, Rational Design of the Substrate Tunnel of β-Ketothiolase Reveals a Local Cationic Domain Modulated Rule that Improves the Efficiency of Claisen Condensation, ACS Catal., 2023, 13(12), 8183–8194 CrossRef CAS.
- L. Liu, S. Liu, X. Hu, S. Zhou and Y. Deng, Enhancing the activity and succinyl-CoA specificity of 3-ketoacyl-CoA thiolase Tfu_0875 through rational binding pocket engineering, Synth. Syst. Biotechnol., 2024, 9(3), 558–568 CrossRef CAS PubMed.
- A. Kroll, M. K. M. Engqvist, D. Heckmann and M. J. Lercher, Deep learning allows genome-scale prediction of Michaelis constants from structural features, PLoS Biol., 2021, 19(10), e3001402 CrossRef CAS PubMed.
- A. Kroll, Y. Rousset, X.-P. Hu, N. A. Liebrand and M. J. Lercher, Turnover number predictions for kinetically uncharacterized enzymes using machine and deep learning, Nat. Commun., 2023, 14(1), 4139 CrossRef CAS PubMed.
- W. Tong; X. Guangming; H. Siwei; S. Liyun; Y. Xuefeng and L. Hongzhong, DeepEnzyme: a robust deep learning model for improved enzyme turnover number prediction by utilizing features of protein 3D structures. bioRxiv, 2023, preprint, 2023.12.09.570923 Search PubMed.
- C. B. Anfinsen, Principles that Govern the Folding of Protein Chains, Science, 1973, 181(4096), 223–230 CrossRef CAS PubMed.
- C. UniProt, UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Res., 2023, 51(D1), D523–D531 CrossRef PubMed.
- N. Watanabe, M. Murata, T. Ogawa, C. J. Vavricka, A. Kondo, C. Ogino and M. Araki, Exploration and Evaluation of Machine Learning-Based Models for Predicting Enzymatic Reactions, J. Chem. Inf. Model., 2020, 60(3), 1833–1843 CrossRef CAS PubMed.
- T. Yu, H. Cui, J. C. Li, Y. Luo, G. Jiang and H. Zhao, Enzyme function prediction using contrastive learning, Science, 2023, 379(6639), 1358–1363 CrossRef CAS PubMed.
- S. Unsal, H. Atas, M. Albayrak, K. Turhan, A. C. Acar and T. Doğan, Learning functional properties of proteins with language models, Nat. Machine Intelligence, 2022, 4(3), 227–245 CrossRef.
- The Gene Ontology Consortium, The Gene Ontology Resource: 20 years and still GOing strong, Nucleic Acids Res., 2019, 47(D1), D330–D338 CrossRef PubMed.
- N. Zhou, Y. Jiang, T. R. Bergquist, A. J. Lee, B. Z. Kacsoh, A. W. Crocker, K. A. Lewis, G. Georghiou, H. N. Nguyen, M. N. Hamid, L. Davis, T. Dogan, V. Atalay, A. S. Rifaioglu, A. Dalkıran, R. Cetin Atalay, C. Zhang, R. L. Hurto, P. L. Freddolino, Y. Zhang, P. Bhat, F. Supek, J. M. Fernández, B. Gemovic, V. R. Perovic, R. S. Davidović, N. Sumonja, N. Veljkovic, E. Asgari, M. R. K. Mofrad, G. Profiti, C. Savojardo, P. L. Martelli, R. Casadio, F. Boecker, H. Schoof, I. Kahanda, N. Thurlby, A. C. McHardy, A. Renaux, R. Saidi, J. Gough, A. A. Freitas, M. Antczak, F. Fabris, M. N. Wass, J. Hou, J. Cheng, Z. Wang, A. E. Romero, A. Paccanaro, H. Yang, T. Goldberg, C. Zhao, L. Holm, P. Törönen, A. J. Medlar, E. Zosa, I. Borukhov, I. Novikov, A. Wilkins, O. Lichtarge, P. H. Chi, W. C. Tseng, M. Linial, P. W. Rose, C. Dessimoz, V. Vidulin, S. Dzeroski, I. Sillitoe, S. Das, J. G. Lees, D. T. Jones, C. Wan, D. Cozzetto, R. Fa, M. Torres, A. Warwick Vesztrocy, J. M. Rodriguez, M. L. Tress, M. Frasca, M. Notaro, G. Grossi, A. Petrini, M. Re, G. Valentini, M. Mesiti, D. B. Roche, J. Reeb, D. W. Ritchie, S. Aridhi, S. Z. Alborzi, M. D. Devignes, D. C. E. Koo, R. Bonneau, V. Gligorijević, M. Barot, H. Fang, S. Toppo, E. Lavezzo, M. Falda, M. Berselli, S. C. E. Tosatto, M. Carraro, D. Piovesan, H. Ur Rehman, Q. Mao, S. Zhang, S. Vucetic, G. S. Black, D. Jo, E. Suh, J. B. Dayton, D. J. Larsen, A. R. Omdahl, L. J. McGuffin, D. A. Brackenridge, P. C. Babbitt, J. M. Yunes, P. Fontana, F. Zhang, S. Zhu, R. You, Z. Zhang, S. Dai, S. Yao, W. Tian, R. Cao, C. Chandler, M. Amezola, D. Johnson, J. M. Chang, W. H. Liao, Y. W. Liu, S. Pascarelli, Y. Frank, R. Hoehndorf, M. Kulmanov, I. Boudellioua, G. Politano, S. Di Carlo, A. Benso, K. Hakala, F. Ginter, F. Mehryary, S. Kaewphan, J. Björne, H. Moen, M. E. E. Tolvanen, T. Salakoski, D. Kihara, A. Jain, T. Šmuc, A. Altenhoff, A. Ben-Hur, B. Rost, S. E. Brenner, C. A. Orengo, C. J. Jeffery, G. Bosco, D. A. Hogan, M. J. Martin, C. O'Donovan, S. D. Mooney, C. S. Greene, P. Radivojac and I. Friedberg, The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens, Genome Biol., 2019, 20(1), 244 CrossRef CAS PubMed.
- S. Yao, R. You, S. Wang, Y. Xiong, X. Huang and S. Zhu, NetGO 2.0: improving large-scale protein function prediction with massive sequence, text, domain, family and network information, Nucleic Acids Res., 2021, 49(W1), W469–W475 CrossRef CAS PubMed.
- S. Wang, R. You, Y. Liu, Y. Xiong and S. Zhu, NetGO 3.0: Protein Language Model Improves Large-scale Functional Annotations, Genomics, Proteomics Bioinf., 2023, 21(2), 349–358 CrossRef PubMed.
- R. Dhanuka, J. P. Singh and A. Tripathi, A comprehensive survey of deep learning techniques in protein function prediction, IEEE/ACM Trans. Comput. Biol. Bioinf., 2023 Search PubMed.
- C. Hsu, C. Fannjiang and J. Listgarten, Generative models for protein structures and sequences, Nat. Biotechnol., 2024, 42(2), 196–199 CrossRef CAS PubMed.
- D. Repecka, V. Jauniskis, L. Karpus, E. Rembeza, I. Rokaitis, J. Zrimec, S. Poviloniene, A. Laurynenas, S. Viknander, W. Abuajwa, O. Savolainen, R. Meskys, M. K. M. Engqvist and A. Zelezniak, Expanding functional protein sequence spaces using generative adversarial networks, Nat. Machine Intelligence, 2021, 3(4), 324–333 CrossRef.
- A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos, C. Xiong, Z. Z. Sun, R. Socher, J. S. Fraser and N. Naik, Large language models generate functional protein sequences across diverse families, Nat. Biotechnol., 2023, 41(8), 1099–1106 CrossRef CAS PubMed.
- A. Hawkins-Hooker, F. Depardieu, S. Baur, G. Couairon, A. Chen and D. Bikard, Generating functional protein variants with variational autoencoders, PLoS Comput. Biol., 2021, 17(2), e1008736 CrossRef CAS PubMed.
- C. Ziegler, J. Martin, C. Sinner and F. Morcos, Latent generative landscapes as maps of functional diversity in protein sequence space, Nat. Commun., 2023, 14(1), 2222 CrossRef CAS PubMed.
- W. M. Dawson, G. G. Rhys and D. N. Woolfson, Towards functional de novo designed proteins, Curr. Opin. Chem. Biol., 2019, 52, 102–111 CrossRef CAS PubMed.
- D. N. Woolfson, A Brief History of De Novo Protein Design: Minimal, Rational, and Computational, J. Mol. Biol., 2021, 433(20), 167160 CrossRef CAS PubMed.
- C. E. Tinberg, S. D. Khare, J. Dou, L. Doyle, J. W. Nelson, A. Schena, W. Jankowski, C. G. Kalodimos, K. Johnsson, B. L. Stoddard and D. Baker, Computational design of ligand-binding proteins with high affinity and selectivity, Nature, 2013, 501(7466), 212–216 CrossRef CAS PubMed.
- M. J. Bick, P. J. Greisen, K. J. Morey, M. S. Antunes, D. La, B. Sankaran, L. Reymond, K. Johnsson, J. I. Medford and D. Baker, Computational design of environmental sensors for the potent opioid fentanyl, eLife, 2017, 6, e28909 CrossRef PubMed.
- J. Dou, L. Doyle Jr, P. Greisen, A. Schena, H. Park, K. Johnsson, B. L. Stoddard and D. Baker, Sampling and energy evaluation challenges in ligand binding protein design, Protein Sci., 2017, 26(12), 2426–2437 CrossRef CAS PubMed.
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Röthlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, De Novo Computational Design of Retro-Aldol Enzymes, Science, 2008, 319(5868), 1387–1391 CrossRef CAS PubMed.
- R. Lipsh-Sokolik, O. Khersonsky, S. P. Schroder, C. de Boer, S. Y. Hoch, G. J. Davies, H. S. Overkleeft and S. J. Fleishman, Combinatorial assembly and design of enzymes, Science, 2023, 379(6628), 195–201 CrossRef CAS PubMed.
- B. Basanta, M. J. Bick, A. K. Bera, C. Norn, C. M. Chow, L. P. Carter, I. Goreshnik, F. Dimaio and D. Baker, An enumerative algorithm for de novo design of proteins with diverse pocket structures, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(36), 22135–22145 CrossRef CAS PubMed.
- A. H. Yeh, C. Norn, Y. Kipnis, D. Tischer, S. J. Pellock, D. Evans, P. Ma, G. R. Lee, J. Z. Zhang, I. Anishchenko, B. Coventry, L. Cao, J. Dauparas, S. Halabiya, M. DeWitt, L. Carter, K. N. Houk and D. Baker, De novo design of luciferases using deep learning, Nature, 2023, 614(7949), 774–780 CrossRef CAS PubMed.
- W. Lu, J. Zhang, W. Huang, Z. Zhang, X. Jia, Z. Wang, L. Shi, C. Li, P. G. Wolynes and S. Zheng, DynamicBind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model, Nat. Commun., 2024, 15(1), 1071 CrossRef CAS PubMed.
- P. Wang, J. Zhang, S. Zhang, D. Lu and Y. Zhu, Using High-Throughput Molecular Dynamics Simulation to Enhance the Computational Design of Kemp Elimination Enzymes, J. Chem. Inf. Model., 2023, 63(4), 1323–1337 CrossRef CAS PubMed.
- D. Ray, S. Das and U. Raucci, Kinetic View of Enzyme Catalysis from Enhanced Sampling QM/MM Simulations, J. Chem. Inf. Model., 2024, 64(9), 3953–3958 CrossRef CAS PubMed.
- J. Lameira, I. Kupchencko and A. Warshel, Enhancing Paradynamics for QM/MM Sampling of Enzymatic Reactions, J. Phys. Chem. B, 2016, 120(9), 2155–2164 CrossRef CAS PubMed.
- Y. Wang; L. Wang; Y. Shen; Y. Wang; H. Yuan; Y. Wu and Q. Gu, Protein Conformation Generation via Force-Guided SE (3) Diffusion Models. arXiv, 2024, preprint, arXiv:2403.14088 Search PubMed.
- I. Poltavsky and A. Tkatchenko, Machine Learning Force Fields: Recent Advances and Remaining Challenges, J. Phys. Chem. Lett., 2021, 12(28), 6551–6564 CrossRef CAS PubMed.
- S. Das, U. Raucci, R. P. Neves, M. J. Ramos and M. Parrinello, Correlating Enzymatic Reactivity for Different Substrates using Transferable Data-Driven Collective Variables, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-1xhm0.
- J. T. Rapp, B. J. Bremer and P. A. Romero, Self-driving laboratories to autonomously navigate the protein fitness landscape, Nat. Chem. Eng., 2024, 1(1), 97–107 CrossRef PubMed.
- B. Dou, Z. Zhu, E. Merkurjev, L. Ke, L. Chen, J. Jiang, Y. Zhu, J. Liu, B. Zhang and G. W. Wei, Machine Learning Methods for Small Data Challenges in Molecular Science, Chem. Rev., 2023, 123(13), 8736–8780 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |