DOI:
10.1039/D4CC03535F
(Highlight)
Chem. Commun., 2024,
60, 12787-12802
Recent advances on COF-based single-atom and dual-atom sites for oxygen catalysis
Received
16th July 2024
, Accepted 18th September 2024
First published on 2nd October 2024
Abstract
Covalent organic frameworks (COFs) have emerged as promising platforms for the construction of single-atom and dual-atom catalysts (SACs and DACs), owing to their well-defined structures, tunable pore sizes, and abundant active sites. In recent years, the development of COF-based SACs and DACs as highly efficient catalysts has witnessed a remarkable surge. The synergistic interplay between the metal active sites and the COF has established the design and fabrication of COF-based SACs and DACs as a prominent research area in electrocatalysis. These catalytic materials exhibit promising prospects for applications in energy storage and conversion devices. This review summarizes recent advances in the design, synthesis, and applications of COF-based SACs and DACs for oxygen catalysis. The catalytic mechanisms of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are comprehensively explored, providing a comparative analysis to elucidate the correlation between the structure and performance, as well as their functional attributes in battery devices. This review highlights a promising approach for future research, emphasizing the necessity of rational design, breakthroughs, and in-situ characterization to further advance the development of high-performance COF-based SACs and DACs for sustainable energy applications.
1. Introduction
The demand for sustainable energy has become more urgent. In the face of ecological challenges such as climate change and depletion of fossil fuels, it is essential to discover efficient, economical, and environmentally friendly materials for energy conversion and storage. To address these challenges, researchers have conducted numerous studies on innovative energy devices, such as fuel cells and metal–air batteries.1–5 The fundamental chemical processes for the above devices are the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER). These critical reactions are indispensable in the energy conversion process.6,7 Nevertheless, the sluggish kinetics of these oxygen electrocatalytic reactions often limit the overall efficiency and functionality of energy devices.8,9 Consequently, it is important for advancing energy technologies that we develop highly efficient and robust electrocatalysts with optimized electronic structures and surface properties.10–12
Metal single-atom catalysts (SACs) have emerged as a new generation of electrocatalysts, showing unparalleled catalytic activity and selectivity, which can be attributed to their distinctive electronic and geometric structures.13,14 The isolated metal atoms are uniformly dispersed and anchored in the support matrix, thereby maximizing the atomic utilization efficiency and revealing active sites.15 Dual-atom catalysts (DACs) as an extension of SACs have attracted attention as a new frontier in heterogeneous catalysis recently. DACs are distinguished by having two metal atoms that engage in a synergistic interaction. The synergistic effects between the paired metal atoms in DACs can significantly improvement catalytic activity and selectivity beyond the levels achieved by the single-atom counterparts.16,17 The integration of a second metal atom serves to fine tune the electronic structure of the catalytic site, thereby enhancing the adsorption and activation of reactants, as well as promoting the generation of essential reaction intermediates.18 Additionally, strong metal–support interactions can facilitate the fine-tuning of the electronic structure at the active metal sites, which leads to an improvement in catalytic performance. Despite the above advancements, the synthesis of stable and highly dispersed SACs and DACs remains a significant challenge because the isolated metal atoms tend to aggregate under the severe conditions during catalytic reactions.19
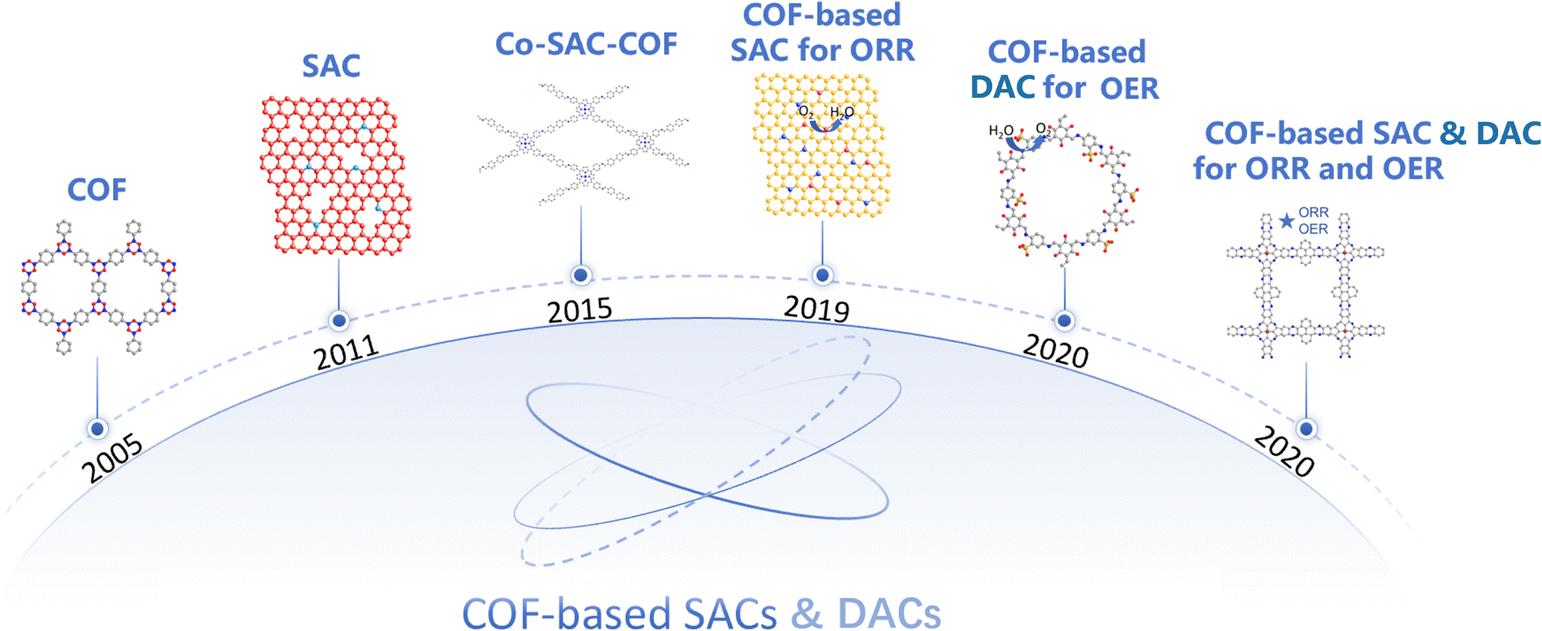
The study of porous materials is undergoing continuous development and transformation. As shown in Fig. 1, since Gilbert N. Lewis's seminal discussion of strong chemical interactions between atoms in 1916, covalent organic chemistry has evolved from discrete molecules to porous organic frameworks.20 The first report of a COF in 2005 demonstrated the possibility of creating highly porous crystalline materials with a tunable functionality. Subsequently, Yaghi et al. pioneered the development of COF-supported single-atom catalysts. Their approach involved the synthesis of COF-366-Co and COF-367-Co via condensation reactions between cobalt porphyrin and two distinct aldehyde linkers.21 Then, others have proposed applications of COF-based SACs and DACs in a broad range of clean energy conversion processes, including the OER and ORR. Thus, the significant research potential of COFs has aroused widespread interest. COFs are assembled from organic motifs connected by strong covalent bonds, which have a large surface area, adjustable pore size, and significant chemical and thermal degradation resistance.22,23 The well-defined structure and abundant functional groups of COFs offer extensive binding sites for metal atoms, which promote the formation of stable and well-dispersed SACs.24 Additionally, the intrinsic porosity of COFs enables efficient transport and sites exposure, further enhancing their catalytic activity.25 Furthermore, the electronic properties of COFs can be customized by selecting different organic building blocks, which can precisely adjust the electronic structure of the introduced metal atoms and subsequently improve catalytic performance.26 Recent years have witnessed the rapid development of COF-based SACs for oxygen electrocatalysis applications.27–29 These materials have demonstrated remarkable activity and robust stability during the OER and ORR, and are expected to surpass traditional noble metals and carbon-based catalysts.30 The investigation of COF-based SACs and DACs for oxygen electrocatalysis has paved the way for innovative designs and advancements of next-generation catalysts. This review aims to summarize recent advancements in the field of COF-based SACs and DACs, elucidating their synthesis, oxygen catalytic activity, catalytic mechanisms, and application in fuel cells and metal-air batteries. In contrast to prior reviews, this study emphasizes the latest breakthroughs in COF-based SAC and DAC research, elucidating their potential utility in oxygen redox applications. In particular, we delve into the catalytic mechanisms of the ORR and OER, providing a comparative analysis of the relationship between the structure and performance, as well as the functional attributes in battery device. A comprehensive understanding of their catalytic behavior and mechanisms is critical for the systematic optimization of clean and efficient energy conversion and storage systems.
 |
| | Fig. 1 Timeline of representative COF-based SACs and DACs: COF (COF-121); SAC (single Pt atoms31); Co-SAC-COF (COF-366-Co32); COF-based SAC for the ORR (COF90033); COF-based DAC for the OER ((Cyclen@Ni0.5Fe0.5)@COF-SO334); COF-based SAC & DAC for the ORR and OER (TM-COFs35). | |
2. Covalent organic frameworks
Covalent organic frameworks, a novel type of porous crystalline materials, are characterized by assembly from organic building blocks through strong covalent bonds. COFs were initially introduced in 2005 by Yaghi and colleagues,21 indicating the advent of a rapidly expanding field that currently includes a rich range of variants such as two-dimensional (2D) COFs, three-dimensional (3D) COFs, ionic COFs, macrocycle-based COFs and chiral COFs.36–38 The primary synthesis of COFs involves various condensation reactions, including Schiff base, Spiroboorane, Knoevenagel and imide reactions.39–41 These frameworks can be fabricated via various strategies such as solvothermal, ionothermal, microwave-assisted, atmospheric solution, mechanochemical and interfacial polymerization methods. In 2024, Yang et al. reported a novel technique to fabricate highly robust, tough and elastic films of an emerging class of 2D COFs.21 The refinement of COF's crystallinity and porosity is routinely achieved through the modulation of synthesis parameters, including the selection of solvents, temperature control, reaction duration, and catalyst usage. The porous and crystalline architecture of COFs, combined with their customizable design, has enabled their utilization in a broad range of applications, including catalysis, water treatment, energy conservation, separation technologies, and electronic devices.42–46
COFs have garnered significant attention in the field of electrocatalysis, particularly for the OER and ORR (Fig. 2). During the ORR and OER, the slow kinetics in multi-electron reactions impose significant limitations, including large overpotentials, unsatisfied cycle durability, and decreased energy and power efficiency. COFs possess several potential benefits for oxygen catalysis, which include a large surface area, tunable architecture, porosity, strong stability, abundant intrinsic metal–N–C and metal–N4 sites and synergistic effects between different active sites. The inset image in Fig. 2b depicts the structural features of the COF that facilitate the oxygen redox reaction.26,47 COFs integrated with porphyrin macrocycles are widely employed in oxygen catalysis owing to their unique capability to anchor and stabilize atomic metals. This stabilization is achieved thorough the coordination of four pyrrolic N atoms, which promote the formation of abundant M–N–C units. For instance, a Fe single-atom porphyrinic triazine-based framework with 8.3 wt% of iron exhibited extraordinary ORR activity, which can be ascribed to its abundant Fe–N4 active sites.48 Additionally, the structural integrity of the COF is preserved during ORR operation. On the other hand, recent studies suggest that tailoring the porosity of COFs can effectively improve OER activity by facilitating mass transport and increasing active sites. Zhao et al. engineered a macro-TpBpy-Co COF with a connected hierarchical macro/microporous structure, exhibiting superior OER performance.49 Indeed, the crystallinity of COFs is a critical factor that can improve the electrocatalytic performance in oxygen catalysis. The improvement mechanism includes enhanced electron and ion transport, uniform distribution of active sites, increased structural stability, reduced defect concentrations, optimized pore structures, and synergistic effects. The crystallinity of COFs endows them with exceptional promise as efficient ORR and OER catalysts. For example, COF-derived N-doped carbon nanospheres (N-CNS) revealed that the porous and crystalline nature of the COF precursor, coupled with precisely positioned nitrogen atoms, enables the effective regulation of physical and chemical properties of N-CNS. The surface area, conductivity, active sites, structural defects, and their accessibility in the N-CNS can be tailored by adjusting the pyrolysis temperatures during the synthesis.50 The crystalline COF-C4N catalyst possessed superior OER activity as compared to its amorphous counterpart (CMP-C4N), indicating the pivotal role of crystallinity in COF in terms of the OER.51
 |
| | Fig. 2 (a) COF-based SAC and DAC for the oxygen redox reaction and their application. (Liquid-sate Zn–air battery images: Reproduced with permission from ref. 52, Copyright 2019, American Association for the Advancement of Science; solid-state Zn–air battery images: Reproduced with permission from ref. 53, Copyright 2022 Wiley-VCH; Li–O2 battery images: Reproduced with permission from ref. 54, Copyright 2023, American Association for the Advancement of Science; production of H2O2 images: Reproduced with permission from ref. 55, Copyright 2020 American Chemical Society.) (b) Statistics of publications on COF-based single-sites for electrocatalysis from 2014 to 2024. The data were extracted from Web of Science. Inset is the mechanism of COFs for oxygen redox catalysis. | |
 |
| | Fig. 3 The preparation strategies for COF-based SAC and DAC catalysts. | |
Characterization of COFs is essential for elucidating their functional performance in practical applications. X-ray diffraction (XRD) provides invaluable insights into the crystalline nature of the frameworks, disclosing details about their porosity and structural integrity. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) offer visualizations of COF morphology at multiple length scales, shedding light on the spatial configuration and assembly of the fundamental building blocks that constitute the framework. Thermogravimetric analysis (TGA) discloses the thermal resilience and robustness of COFs, while gas adsorption isotherms elucidate critical insights into the surface area and porosity. Additionally, Fourier-transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR) spectroscopy and X-ray photoelectron spectroscopy (XPS) are instrumental in analyzing the molecular composition and the chemical bonding in COFs. These analytical techniques provide essential information for the design and refinement of COF-based catalysts. For instance, XPS analysis can confirm that the presence of Fe and N elements originates from COF-BTC (benzene-1,3,5-tricarboxylic acid), which is synthesized using a π-conjugated iron phthalocyanine.53 XPS analysis reveals the characteristic peaks associated with the binding energies of Fe 2p and N 1s electrons. The Fe 2p spectrum can provide information about the oxidation state of Fe, while the N 1s spectrum indicates the types of nitrogen-containing functional groups present in COFs. Powder X-ray diffraction (PXRD) is a widely used technique for characterizing the crystallinity and structure of COFs. For example, the PXRD patterns of M-TP-COFs (two-dimensional porphyrin COFs, M = H2, Co, Ni, and Mn) exhibited two prominent diffraction peaks at 4.45 and 8.89 Å, corresponding to the (110) and (220) crystallographic planes, respectively.56 In the case of the H-TP-COF, the NMR spectrum revealed a characteristic peak at 158.06 ppm, attributed to the carbons in the imine bond. Additional peaks at 151.60, 135.59, 128.38, and 119.87 ppm were assigned to the carbons in the aromatic backbones. Beyond the realm of structural characterization, investigators are diligently exploring the functional attributes of COFs to unlock their full potential in diverse applications. Recent research endeavors have focused on improving the stability, electrical conductivity, and catalytic efficiency of COFs through strategic modifications, including the integration of functional groups and the intercalation of metal entities.57–64 These developments open avenues for the utilization of COFs in energy storage and catalysis, highlighting their versatility and adaptability in addressing contemporary challenges.46
3. COF-based SACs for oxygen electrocatalysis
In recent years, COF-based electrocatalysts have attracted increasing attention owing to their special structural features.29 The field of SACs has witnessed significant advancements, offering a promising avenue for enhancing the efficiency of electrocatalytic reactions. Among the various types of SACs, COFs have emerged as a novel platform for hosting single metal atoms to catalyze oxygen electrocatalysis. The preparation strategies for COF-based SACs primarily involve the following approaches (Fig. 3). (1) Post-synthetic modification involves the introduction of metal atoms into a pre-synthesized COF through subsequent chemical treatments.65 This process facilitates the formation of stable catalytic sites within the framework. (2) In situ synthesis is one method where metal precursors are directly incorporated during the synthesis of the COF.52 This approach ensures high dispersion and stability of metal atom sites. (3) Coordination environment design involves the strategic optimization of the organic monomer structure in the COF to create specific coordination sites.66 These sites enable precise positioning of metal atoms to achieve high catalytic activity.
3.1. COF-based SACs for the ORR
The ORR stands as a pivotal electrode process in energy devices (such as fuel cells, metal–air batteries). The ORR predominantly occurs via one of two primary pathways, depending on the electrochemical environment and the nature of the catalyst. The first pathway is a two-electron process, where O2 is partially reduced to hydrogen peroxide (H2O2). This intermediate species can then be further reduced to water (H2O). The overall reaction for this pathway can be summarized as follows: O2 + 2H+ + 2e− → H2O2; H2O2 + 2H+ + 2e− → 2H2O. The alternative pathway is the four-electron process, where O2 is directly and fully reduced to water (H2O) without the formation of H2O2. This direct reduction is represented as O2 + 4H+ + 4e− → 2H2O. Under alkaline conditions, the reactions would involve hydroxide ions (OH−) instead of protons (H+), leading to the formation of OH− as the product. This direct reduction is represented as O2 + 2H2O + 4e− → 4OH−. While these pathways are experimentally validated through electron-transfer number calculations, the inherent sluggish kinetics of multi-electron processes pose significant challenges. Specifically, these challenges manifest as high overpotentials, deteriorated cycling stability, diminished energy efficiency, and limited power density. Traditional catalysts based on expensive noble metals, such as Pt, Ru, and Ir, and their alloys, further exacerbate cost concerns. Non-noble-metal nanocarbon-based materials, although cost-effective, often exhibit inferior performance. An N,P co-doped carbon catalyst with a highly crystalline and two dimensional (2D) COF exhibits remarkable ORR activity in alkaline medium, with an onset potential of 0.87 V and a half-wave potential of 0.81 V. In contrast, N doped carbon showed a lower onset potential of 0.69 V and a half-wave potential of 0.64 V.67 Liu et al. used a dioxin-linked COF as the template to preciously construct different metal–N2 sites along the porous walls.68 The onset potentials for pristine COF-318, Ca-COF-318, Co-COF-318, and Ni-COF-318 were 0.72, 0.75, 0.77, and 0.74 V (vs. RHE), respectively. The half-wave potentials for COF-318, Ca-COF-318, Co-COF-318, and Ni-COF-318 were 0.62, 0.61, 0.61, and 0.63 V (vs. RHE), respectively. The experiments and theoretical calculations confirmed that the Co–N2 site is more suitable for catalyzing the ORR in the 4e− pathway. Hence, a critical need exists to design electrocatalysts and oxygen electrodes capable of mitigating overpotential, enhancing current density, and ensuring durability in demanding environments. COF-based SACs have been demonstrated to be promising in electrocatalysis due to their abundant intrinsic metal–N–C and metal–N4 sites crucial for the ORR, dense and confined earth-abundant metal active sites, and highly ordered structures facilitating mechanistic and structure–property studies. Recent research has demonstrated that the metal–nitrogen–carbon (M–N–C) centers in porphyrin-based COFs display excellent catalytic performance for the ORR. The selection of metal components in these COFs allows for the precise manipulation of their structure and active sites, which in turn influences their catalytic behavior. For example, four distinct two-dimensional porphyrin COFs (M-TP-COF; M = N2, Co, Ni, Mn) exhibited fast kinetics and optimal ORR catalytic activity.56 In addition, introducing redox-active centers or doping with heteroatoms with COFs has also been proven to enhance the catalytic performance.69 CoTAPP-PATA-COF, which incorporates diphenylamine units, exhibits optimized electron transport, a large active surface area, and enhanced catalytic ORR activity. The ORR activity of Co(II) COFs was greatly improved by supplementing the porphyrin unit with benzobisthiazole groups to introduce heteroatoms S and N.70 Various atoms, such as iron, cobalt, and nickel, have been incorporated into COF structures to explore their catalytic capabilities for the ORR, and have achieved remarkable ORR activity, comparable to or even surpassing traditional Pt-based catalysts. The high dispersion of metal atoms in COFs results in enhanced catalytic efficiency and stability. Additionally, the COFs possess a unique structure and local coordination environment, enhancing electrocatalytic ORR performance. Computational modeling techniques, such as Gibbs free energy diagrams, charge density difference analysis, and density of states calculations, coupled with in situ spectroscopic methods, including in situ Raman spectroscopy, in situ FTIR spectroscopy, and in situ X-ray absorption fine structure (XAFS) spectroscopy, have provided valuable insights into the ORR mechanism of SAC-COF materials. These investigations have revealed crucial details regarding reaction pathways and kinetics.
3.1.1. Fe-COF for the ORR.
Iron-based SACs have emerged as one of the most extensively researched classes of electrocatalysts towards the ORR. Several studies have focused on anchoring an isolated iron atom in the porous structure of COFs to achieve efficient ORR activities. Peng et al. employed a facile synthetic approach utilizing a fully π-conjugated iron phthalocyanine (FePc)-COF to fabricate SACs with outstanding electrocatalytic activity for the ORR.52 This method avoids high-temperature pyrolysis and instead leverages intermolecular interactions between the COF network and a graphene matrix to rivet atomically well-designed Fe–N–C centers (Fig. 4a and b). The synthesized pfSAC-Fe-0.2 exhibited enhanced ORR activity with a half-wave potential of 0.91 V and a high kinetic current density (Jk) of 25.86 mA cm−2 at 0.85 V (versus RHE) in 0.1 M KOH, which is four times higher than that of Pt/C (6.49 mA cm−2) in Fig. 4c. The single Fe atoms offered superior catalytic sites for the absorption and electron transfer of oxygen molecules during the ORR (Table 1). Moreover, the highly organized structure of the COF provides the ability to fine-tune the structure of single atom active sites. Wen Da et al. recently designed and fabricated Fe dual-atom catalysts (Fe2DAC) with a Fe2N6C8O2 configuration through one step carbonization of a preorganized COF with bimetallic Fe chelation sites (Fe2COF) (Fig. 4d–f).71 The transition from Fe2COF to Fe2DAC involved the dissociation of the nanoparticles and the capture of atoms by carbon defects. Fe2DAC exhibited outstanding oxygen reduction activity with a half-wave potential of 0.898 V (vs. RHE) in 0.1 M KOH, which is attributed to the enhanced adsorption of OOH* intermediates and the optimized d-band center (Fig. 4g).
 |
| | Fig. 4 (a) Illustration of the synthesis of the pfSAC-Fe catalyst. (b) HAADF-STEM of the pfSAC-Fe catalyst. (c) ORR performance of pfSAC-Fe and Pt/C in 0.1 M KOH electrolyte using a rotation rate of 1600 rpm (reproduced with permission from ref. 52, Copyright 2019, American Association for the Advancement of Science, AAAS). (d) Synthesis process of Fe2COF using P-COF. (e) The crystal structures of Fe2COF. (f) The dynamical evolution process of Fe2N6 in Fe2COF varying with temperature. (g) LSV curves of Fe2DAC, FeSAC, C-P-COF, and Pt/C in 0.1 M KOH solution at a rotation speed of 1600 rpm (reproduced with permission from ref. 71, Copyright 2023, Wiley-VCH). | |
Table 1 A comparison of COF-based single-site catalysts for the ORR
| Catalyst |
Reaction |
Electrolyte |
Onset potential (V) |
E
1/2 (V) |
Tafel slope (mV dec−1) |
Ref. |
| C-Zn/Co800 |
ORR |
0.1 M KOH |
0.98 |
0.86 |
63.7 |
28
|
| Co@COF900 |
ORR |
0.1 M KOH |
0.93 |
0.86 |
N/A |
33
|
| Co-TP-COF |
ORR |
0.1 M KOH |
0.81 |
0.73 |
66 |
56
|
| pfSAC-Fe-0.2 |
ORR |
0.1 M KOH |
N/A |
0.91 |
31.7 |
52
|
| Fe2DAC |
ORR |
0.1 M KOH |
N/A |
0.898 |
57 |
71
|
| N doped COF |
ORR |
0.1 M KOH |
0.69 |
0.64 |
N/A |
67
|
| N, P co-doped COF |
ORR |
0.1 M KOH |
0.87 |
0.81 |
N/A |
67
|
| CoTAPP-PATA-COF |
ORR |
0.1 M KOH |
0.926 |
0.80 |
55 |
72
|
| Co@rhm-PorBTD |
ORR |
0.1 M KOH |
0.89 |
0.83 |
41 |
73
|
| Co@sql-PorBTD |
ORR |
0.1 M KOH |
0.85 |
0.75 |
87 |
73
|
| Co-PorBpy-Co |
ORR |
0.1 M KOH |
N/A |
0.79 |
58.3 |
74
|
| Co-PorBpy-Co/CNT |
ORR |
0.1 M KOH |
N/A |
0.84 |
N/A |
74
|
| 1′′-NP |
ORR |
0.1 M KOH |
N/A |
0.81 |
70 |
75
|
| Fe-COF900 |
ORR |
0.1 M KOH |
0.86 |
0.82 |
97.9 |
76
|
| Fe-ISAS/CN |
ORR |
0.1 M KOH |
N/A |
0.861 |
78 |
77
|
| CoCOF@MOF800 |
ORR |
0.1 M KOH |
1.00 |
0.86 |
39 |
78
|
| CoAC-SAs/N-C@CNT |
ORR |
0.1 M KOH |
0.93 |
0.86 |
68 |
79
|
| COPBTC@Cl-CNTs |
ORR |
0.1 M KOH |
N/A |
0.75 |
64.4 |
80
|
| COF@MOF800-Fe |
ORR |
0.1 M KOH |
0.94 |
0.89 |
80 |
81
|
| COF-318 |
ORR |
0.1 M KOH |
0.72 |
0.62 |
52 |
68
|
| Ca-COF-318 |
ORR |
0.1 M KOH |
0.75 |
0.61 |
33 |
68
|
| Co-COF-318 |
ORR |
0.1 M KOH |
0.77 |
0.61 |
101 |
68
|
| Ni-COF-318 |
ORR |
0.1 M KOH |
0.74 |
0.63 |
77 |
68
|
3.1.2. Co-COF for the ORR.
While it is widely acknowledged that iron exhibits superior ORR activity, other transition metals also possess considerable activity. Among these, cobalt has garnered significant interest as a substitute of Fe-based electrocatalysts due to the durability concerns associated with the degradation caused by Fenton-like reactions. Chang et al. employed COF-SACs featuring a uniform metal coordination environment as model catalysts to explore the intrinsic catalytic activity of diverse metal centers in M–N–C SACs.64 COF-366-Co exhibited superior performance for H2O2 synthesis via the ORR. COF-366-Co demonstrates exceptional selectivity towards hydrogen peroxide (H2O2) production via a two-electron transfer ORR in 0.1 M KOH. This is evidenced by its high hydrogen peroxide yield of 91% and faradaic efficiency of 84%. Furthermore, COF-366-Co maintains high H2O2 selectivity across a broad potential window, achieving a turnover frequency (TOF) of 9.05 s−1 at 0.1 V versus the reversible hydrogen electrode (RHE). Bhunia et al. synthesized two cobalt(II)-based COFs, designated as Co@rhm-PorBTD and Co@sql-PorBTD, by strategically incorporating benzothiadiazole units alongside porphyrinic moieties.73 This integration introduced heteroatoms (sulfur and nitrogen), enhancing the structural diversity of the COFs (Fig. 5a and b). Powder X-ray diffraction (PXRD) patterns and transmission electron microscopy (TEM) images confirmed the highly crystalline nature of these materials. Experimental investigations revealed that these COFs exhibited remarkable catalytic performance for the ORR. Specifically, Co@rhm-PorBTD demonstrated a mass activity (at 0.85 V) that was 5.8 times higher than that of commercial Pt/C (20%), while Co@sql-PorBTD achieved a 1.3-fold enhancement (Fig. 5c). Furthermore, the specific activities of Co@rhm-PorBTD and Co@sql-PorBTD were found to be 10 and 2.5 times greater, respectively, compared to Pt/C (Fig. 5d). Wang et al. innovatively introduced a feasible potassium hydroxide (KOH) etching strategy to simultaneously implant abundant cobalt clusters and cobalt single atoms onto the surface of COF-derived nitrogen-doped carbon nanospheres interwoven with a surrounding network of carbon nanotubes (Fig. 5e).79 This approach involved the strategic disruption of a portion of the Co–N bonds, leading to the reconstruction of Co–Co coordination. The meticulously designed CoAC-SAs/N-C@CNT catalyst exhibited a remarkable ORR performance, and a half-wave potential of 0.86 V in 0.1 M KOH was achieved.
 |
| | Fig. 5 (a) The PXRD pattern and (b) the top view of the structure for the Co@rhm-PorBTD sample. (c) Comparison of mass activities at 0.85 and 0.9 V (vs. RHE) for the catalysts. (d) Comparison of ECSA, turnover frequencies (TOF), overpotential, specific activities, and roughness factors for different samples. (Reproduced with permission from ref. 73, Copyright 2023, American Chemical Society.) (e) An illustration of the synthesis route for CoAC-SAs/N-C@CNT. (Reproduced with permission from ref. 79, Copyright 2023, Beijing Institute of Technology Press.) | |
3.1.3. Other M-COFs for the ORR.
A dioxin-linked COF is employed as a template for the precise construction of diverse metal-N2 sites along the porous walls by Liu et al.68 This innovative approach utilizes the unique structure of the COF to guide the assembly of metal and nitrogen atoms in a coordinated manner. In 0.1 M KOH, COF-318, Ca-COF-318 and Ni-COF-318 have onset potentials of 0.72, 0.75 and 0.74 V, with corresponding diffusion-limiting current densities of 2.53, 2.80 and 2.29 mA cm−2, respectively. Meanwhile, COF-318, Ca-COF-318 and Ni-COF-318 exhibit half-wave potentials (E1/2) of 0.62, 0.61 and 0.63 V, with corresponding Tafel slopes of 52, 33 and 77 mV dec−1, respectively.
3.2. COF-based SACs for the OER
The OER takes place at the anode during the process of water electrolysis, where H2O is oxidized to produce O2 and H+ or OH−. The OER serves as the counter reaction to the ORR.82,83 In water electrolysis, the OER can be represented by the half-reaction under acidic conditions (2H2O → O2 + 4H+ + 4e−) and under alkaline conditions, the reaction is 2H2O + 4e− → O2 + 4OH−.84 Traditional OER catalysts like RuO2 and IrO2 have been criticized for their low catalytic activity, low selectivity, high cost, and tendency to degrade over time. Concerning metal-free COFs for the OER, COF-C4N prepared by solvothermally reacting triphenylenehexamine (TPHA) and hexaketocyclohexane (HKH) exhibited promising OER activity, achieving a low overpotential of 349 mV and a Tafel slope of 64 mV dec−1.51 TM-COF-C4N materials were synthesized by impregnating COF-C4N in a methanol solution containing the M(OAc)2 precursor. Notably, incorporating nickel or cobalt significantly enhanced the OER activity. Co-COF-C4N exhibited a reduced overpotential of 280 mV compared to the parent COF-C4N. Ni-COF-C4N also demonstrated a notable decrease in overpotential.85 The development of effective catalysts for the OER is critical for improving the overall water electrolysis and metal–air batteries.86,87
Achieving efficient OERs over COFs poses greater challenges compared to the hydrogen evolution reaction (HER), ORR, and carbon dioxide reduction reaction (CO2RR) due to the inherent organic chemical properties of COFs. The structure of COFs plays a crucial role in OER performance. Introducing pyrimidine functionalities or adjusting the coordination geometry within the framework enhances OER activity and the stability of COFs, primarily due to the altered electronic structure. The incorporation of pyrimidine groups introduces new electronic states and potentially enhances the redox properties, while the modification of the coordination geometry can affect the electronic environment of the active sites in COFs. For instance, Fe–NO coordination was constructed using an unusual N and O chelation of Fe-SACs with the ketoenamine unit in COFs.88 This unique Fe–NO coordination results in oxygenated intermediates with lower binding energies, which contributes to outstanding electrocatalytic OER performance. Hence, a few recent studies have demonstrated the potential for an electrocatalytic OER by utilizing COFs as supports for metal catalysts.51,89
3.2.1. Fe-COFs for the OER.
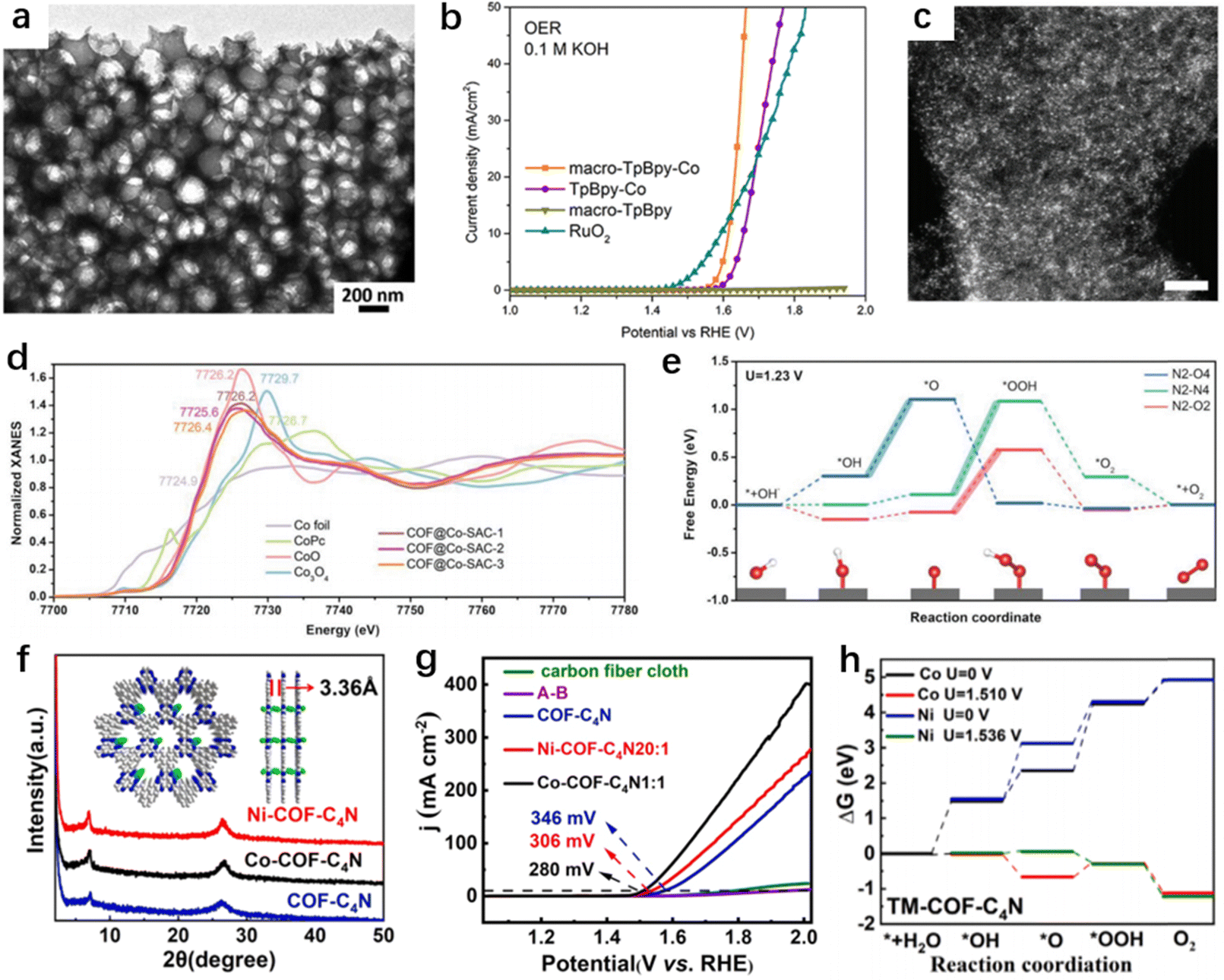
Wang et al. reported the development of a stable incorporation of iron single-atoms coordinated on Tp-Tta COFs with novel and precise coordination modes.88 These unique coordination modes help reduce the need for pyrolysis treatment. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and extended X-ray absorption fine structure (EXAFS) analyses have confirmed the atomic dispersion of iron and the specific coordination environment in the system (Fig. 6a–c). The uncommon coordination configuration of the Fe-SAC@COF catalyst granted the electrocatalyst exceptional OER activity with a low overpotential of 290 mV at a current density of 10 mA cm−2, and a small Tafel slope of 40 mV dec−1 in 1.0 M KOH (Fig. 6d and e).
 |
| | Fig. 6 (a) Schematic illustration of the synthesis process of Fe-SAC@COF. (b) The K-edge XANES spectra, and (c) Fourier transform EXAFS spectra of Fe-SAC@COF. (d) Comparison of TOF values, Tafel slopes, and overpotentials with Fe-based SACs. (e) OER LSV plots of the catalysts. (Reproduced with permission from ref. 88, Copyright 2022, Cell Press.) (f) XRD patterns of H2PcCOF, H2NiPcCOF, and NiPcCOF. (g) Comparison of the OER activities of Ni-containing polymers. (Reproduced with permission from ref. 90, Copyright 2022, Elsevier.) | |
3.2.2. Co-COF for the OER.
Transition metals like Co(II) have garnered attention due to their ability to exist in multiple redox states. Zhao et al. incorporated atomic Co(II) into COFs via bipyridine sites, leveraging the active centers of Co-TpBpy (Bpy = 2,2′-bipyridine-5,5′-diamine) and the inherent microporosity and mesopores to achieve high OER activity (Fig. 7a).49 By employing polystyrene spheres as hard templates during the Schiff condensation of 1,3,5-triformylphloroglucinol (Tp) and 2,2′-bipyridine-5,5′-diamine (Bpy), followed by template etching, they obtained macro-TpBpy-Co. In 0.1 M KOH, Macro-TpBpy-Co exhibited outstanding OER performance with an overpotential of 380 mV at a current density of 10 mA cm−2, significantly lower than that of microporous TpBpy-Co (Fig. 7b). The superior performance of macro-TpBpy-Co can be ascribed to the enhanced mass diffusion characteristics within its hierarchically porous architecture, coupled with a higher number of available active sites. This facile synthesis method can be further extended to produce COFs with a range of microporous sizes by employing various polymer templates or COFs utilizing different amine linkers (Table 2). Chi et al. utilized a bipyridine-rich COF to synthesize the out-of-plane coordination of Co-SAC. The bipyridine nitrogen atoms on the COF serve as the anchoring sites for metal centers.91 This arrangement leads to a coordination that extends outside of the framework plane. Consequently, the cobalt atoms are able to bind to other molecular fragments in a way that extends beyond the conventional two-dimensional limits of the COF plane, potentially endowing the SACs with unique catalytic properties and expanded functional capabilities. Isolated Co sites, which can be obviously observed as different bright spots, exhibit uniform distribution (Fig. 7c). XAS results confirmed Co–N bonds with immobilized bipyridine sites and multiple additional ligands. The theoretical overpotentials of the N2–O4, N2–N4, and N2–O2 models are 0.80, 0.97, and 0.65 V (Fig. 7d and e), respectively. To generate a current density of 10 mA cm−2, COF@Co-SAC-3 requires a low overpotential of 387 mV and demonstrates a Tafel slope of 61.2 mV dec−1 in 1.0 M KOH. COF-C4N has optimal N-edge cavities that serve as anchoring sites for transition metal centers, achieving SACs with higher OER activity. In addition, Co-COF-C4N exhibits excellent OER activity with an overpotential of 280 mV at 10 mA cm−2, and is more active than most previously reported OER electrocatalysts (Fig. 7f–h).85
 |
| | Fig. 7 (a) TEM image of macro-TpBpy. (b) OER LSV curves of macro-TpBpy-Co. (Reproduced with permission from ref. 49, Copyright 2019 American Chemical Society.) (c) High-magnification HAADF-STEM image of COF@Co-SAC, scale bar: 2 nm. (d) Co K-edge normalized XANES of the catalysts. (e) Free energy diagram for the OER of three COF@Co-SAC modes. (Reproduced with permission from ref. 91, Copyright 2022, Wiley-VCH.) (f) The XRD curves of COF-C4N, Ni-COF-C4N, and Co-COF-C4N. (g) OER LSV plots in a 1.0 M KOH solution. (h) The free energy curves of the OER pathway of Ni-COF-C4N and Co-COF-C4N. (Reproduced with permission of from ref. 85, Copyright 2023, Elsevier.) | |
Table 2 A comparison of COF-based single-site catalysts for the OER
| Catalyst |
Reaction |
Electrolyte |
Tafel slope (mV dec−1) for the OER |
Overpotential (mV) |
Ref. |
| Co@COF-SO3 |
OER |
1.0 M KOH |
85 |
363 |
34
|
| Macro-TpBpy-Co |
OER |
0.1 M KOH |
54 |
380 |
49
|
| TpBpy-Co |
OER |
0.1 M KOH |
58 |
430 |
49
|
| COF-C4N |
OER |
1.0 M KOH |
64 |
349 |
51
|
| Fe-SAC@COF |
OER |
1.0 M KOH |
40 |
290 |
88
|
| NiPc |
OER |
1.0 M NaOH |
75 |
410 |
90
|
| COF@Co-SAC-3 |
OER |
1.0 M KOH |
61.2 |
387 |
91
|
| Co-COF-C4N |
OER |
1.0 M KOH |
64 |
280 |
85
|
| Ni-COF-C4N |
OER |
1.0 M KOH |
65 |
307 |
85
|
| COF-366-2/3Co@CNT |
OER |
1.0 M KOH |
50 |
378 |
92
|
| COF-366-Co@CNT-0.4 |
OER |
1.0 M KOH |
62 |
358 |
92
|
| Co0.5V0.5@COF-SO3 |
OER |
1.0 M KOH |
62 |
318 |
93
|
| Ni0.5Fe0.5@COF-SO3 |
OER |
1.0 M KOH |
83 |
308 |
94
|
| Co-PDY/CF |
OER |
1.0 M KOH |
99 |
270 |
95
|
3.2.3. Other M-COFs for the OER.
Jarju et al. reported a synthetic strategy to fabricate a series of imine-linked metal-phthalocyanine (Pc)-containing COFs with varying metal content.90 These catalysts were subjected to an in-depth investigation of their electrocatalytic activity towards the OER under alkaline conditions. COFs prepared with NiPc exhibit superior OER performance, achieving a current density of 10 mA cm−2 at an overpotential of 410 mV and a Tafel slope of 75 mV dec−1. Moreover, this electrocatalyst displays excellent long-term stability over 100 hours (Fig. 6f and g). Additionally, Gao et al. presented a novel strategy to synthesize Ru embedded in a carbon nitride matrix, which facilitates efficient HER and OER in alkaline solution.96 This approach involves the synthesis of a two-dimensional COF structure, designated as CIN-1, featuring a coordinated RuII center. Subsequent pyrolysis of CIN-1 results in the formation of Ru oxide nanoparticles with low-valent Ru sites, which are arranged as nanowires between the graphitic carbon nitride layers. The synthesized electrocatalyst exhibits a lower OER overpotential of 280 mV and a Tafel slope value of 49.5 mV dec−1, inferior to commercial RuO2.
4. COF-based DACs for the ORR and OER
In addition, Co-based COFs are capable of catalyzing both ORR and OER reactions (Table 3). Tang et al. demonstrated the fabrication of a large-area acylhydrazone-based free-standing COF film utilizing cobalt porphyrin and thioaldehyde as building blocks through a liquid-air interface approach (Fig. 8a–c).53 This CoP-TOB catalyst exhibited high activities in both the ORR and OER, and was successfully integrated into a rechargeable Zn–air battery, achieving a specific capacity of 740 mA h g−1. Similarly, Ke et al. developed a cobalt porphyrin-based COF incorporating a metal bis(dithiolene) structure, resulting in a Li–O2 battery with a bimetallic Ni/Co-COF-based cathode showing a minimal discharge/charge potential gap of 1.0 V and a remarkable discharge capacity of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 104 mA h g−1.63 Metal bis(dithiolene) was chosen for its π-conjugated structure, rich redox activity, and electron-withdrawing effect, while the porphyrin units provided M–N4 active centers crucial for the ORR/OER. The conductivity of the bimetallic Ni-TAPP-Co COF measured at 1.18 × 10−4 S m−1 surpassed that of its monometallic or nonmetal counterparts significantly. This enhanced conductivity is attributed to the S–S interaction and the presence of cobalt porphyrin units, which are suggested to establish conducting pathways within the material. Subsequently, Ni-TAPP-Co was utilized as an electrode alongside the other three COFs in a Li–O2 cell, where their electrocatalytic performance in the ORR/OER was assessed. Among them, the bimetallic cathode exhibited the potential gap of 1.0 V and demonstrated stable cyclic charging/discharging for up to 200 cycles, outperforming the others. Through comparative analyses and DFT calculations, it was observed that both the Ni and Co centers displayed significant reaction activity, elucidating the superior battery performance achieved (Fig. 8d–f). Liao et al. reported constituent-tunable metal-phthalocyanine-based covalent organic polymers as self-sacrificial precursors for the fabrication of mono- (Fe or Ni) and bi-metallic (Fe–Ni) heteroatoms/carbon catalysts toward both the ORR and OER (Fig. 8g and h).97 FeNi-COP-800 exhibited an onset potential of 0.955 V and a half-wave potential of 0.803 V in 0.1 M KOH. Regarding OER performance, FeNi-COP-800 achieved a comparable potential of 1.63 V at 10 mA cm−2 to the benchmark IrO2/C catalyst in 1.0 M KOH, as shown in Fig. 8i.
104 mA h g−1.63 Metal bis(dithiolene) was chosen for its π-conjugated structure, rich redox activity, and electron-withdrawing effect, while the porphyrin units provided M–N4 active centers crucial for the ORR/OER. The conductivity of the bimetallic Ni-TAPP-Co COF measured at 1.18 × 10−4 S m−1 surpassed that of its monometallic or nonmetal counterparts significantly. This enhanced conductivity is attributed to the S–S interaction and the presence of cobalt porphyrin units, which are suggested to establish conducting pathways within the material. Subsequently, Ni-TAPP-Co was utilized as an electrode alongside the other three COFs in a Li–O2 cell, where their electrocatalytic performance in the ORR/OER was assessed. Among them, the bimetallic cathode exhibited the potential gap of 1.0 V and demonstrated stable cyclic charging/discharging for up to 200 cycles, outperforming the others. Through comparative analyses and DFT calculations, it was observed that both the Ni and Co centers displayed significant reaction activity, elucidating the superior battery performance achieved (Fig. 8d–f). Liao et al. reported constituent-tunable metal-phthalocyanine-based covalent organic polymers as self-sacrificial precursors for the fabrication of mono- (Fe or Ni) and bi-metallic (Fe–Ni) heteroatoms/carbon catalysts toward both the ORR and OER (Fig. 8g and h).97 FeNi-COP-800 exhibited an onset potential of 0.955 V and a half-wave potential of 0.803 V in 0.1 M KOH. Regarding OER performance, FeNi-COP-800 achieved a comparable potential of 1.63 V at 10 mA cm−2 to the benchmark IrO2/C catalyst in 1.0 M KOH, as shown in Fig. 8i.
Table 3 A comparison of COF-based single-site catalysts for the OER and ORR
| Catalyst |
Reaction |
Onset potential of the ORR (V) |
E
1/2 of the ORR (V) |
Tafel slope (mV dec−1) for the ORR/OER |
Overpotential of the OER (mV) |
Ref. |
| CoP-TOB |
OER/ORR |
N/A |
0.818 |
44/89 |
450 |
53
|
| CoTAPP-PATA-COF |
OER/ORR |
N/A |
0.80 |
N/A |
420 |
69
|
| FeNi-COP-800 |
OER/ORR |
0.955 |
0.803 |
91/103 |
400 |
97
|
| PCL113-NiFe |
OER/ORR |
N/A |
N/A |
36.2/66.9 |
N/A |
98
|
| COPBTC-Co |
OER/ORR |
0.945 |
0.864 |
35.7/133.2 |
397 |
99
|
 |
| | Fig. 8 (a) The pressing measurement of a CoP-TOB film. (b) ORR LSV and Tafel slope of CoP-TOB. (c) OER LSV and Tafel slope of CoP-TOB. (Reproduced with permission from ref. 53, Copyright 2023, Wiley-VCH.) (d) Molecular structures of Ni-TAPP-Co. Free energy diagrams of battery reactions on the surface of the (e) Co site in TTF-TAPP-Co, and (f) Co site in Ni-TAPP-Co. (Reproduced with permission from ref. 54, Copyright 2023, Wiley VCH.) (g) Schematic illustration of the preparation of FeNi-COP-800. (h) Solid-state 13C CP MAS NMR spectra of FeNi-COP. (i) The overall LSV curves of Fe-COP-800, FeNi-COP-800 and Ni-COP-800. Inset: ORR Tafel plots (left) and OER Tafel plots (right). All the LSV curves were tested in O2-saturated 0.1 M KOH solution at scan and rotation rates of 5 mV s−1 and 1600 rpm, respectively. (Reproduced with permission from ref. 97, Copyright 2018, Elsevier.) | |
5. COF-SACs for liquid and solid-sate Zn–air batteries
The transition towards renewable energy sources, coupled with the demand for advanced energy storage technologies, have propelled substantial research efforts into the development of next-generation batteries. Zn–air batteries (ZABs) have garnered significant attention owing to their high theoretical energy density, safety, and cost-effectiveness.100–105 A typical ZAB is composed of a zinc anode, an oxygen-permeable cathode, and an alkaline electrolyte. Zn–air batteries possess a high theoretical specific energy of 1218 W h kg−1, and a large volumetric energy density of 6136 W h L−1.106,107 One of the primary challenges in the development of efficient ZABs is the sluggish kinetics of the OER and ORR, which hinder performance during charge and discharging cycles. Traditional catalysts, which are primarily composed of precious metals such as platinum for the ORR and iridium oxide for the OER, although effective, are plagued by high costs, scarcity, and insufficient operational stability. Hence, there is an urgent demand for alternative materials that not only possess cost-effectiveness and abundance but also display superior catalytic activity and stability. This is a juncture where the advent of COF-based SACs presents a groundbreaking solution. COFs provide a robust scaffold where single atoms can be precisely immobilized, thereby enabling the fabrication of SACs with extraordinary catalytic characteristics. The uniform distribution of single-atom active sites within the COF matrix maximizes atom utilization and provides a highly controlled environment, which is crucial for enhancing the ORR and OER performance in ZABs (Table 4).
Table 4 A comparison of the performance of zinc–air batteries
| Catalyst |
Electrolyte |
Open circuit voltage (V) |
Cycle time@Jk |
Power density (mW cm−2) |
Ref. |
| pfSAC-Fe-0.2 |
8 M KOH + 0.5 M ZnO |
1.41 |
300 h@5 |
123.43 |
52
|
| CoP-TOB |
0.2 M ZnCl2 + 6.0 M KOH |
1.36 |
10 h@2 |
N/A |
53
|
| Co@rhm-PorBTD |
6 M KOH + 0.2 M Zn(OAc)2 |
1.53 |
33 h@5 |
138 |
73
|
| Co@sql-PorBTD |
6 M KOH + 0.2 M Zn(OAc)2 |
1.48 |
33 h@5 |
117 |
73
|
| Co-PorBpy-Co/CNT |
N/A |
1.451 |
80 h@10 |
159.4 |
74
|
| CoAC-SAs/N-C@CNT |
6 M KOH + 0.2 M ZnCl2 |
N/A |
139 h@10 |
90.12 |
77
|
| FeNi-COP-800 |
8 M KOH + 0.2 M ZnO |
N/A |
175 h@5 |
64 |
97
|
| PCL113-NiFe |
6 M KOH + 0.2 M Zn(Ac)2 |
N/A |
200 h@10 |
187.7 |
98
|
| COPBTC-Co |
6.0 M KOH + 0.20 M Zn (CH3COO)2 |
N/A |
100 h@10 |
157.7 |
99
|
| CoSA@NC |
6 M KOH + 0.2 M Zn(Ac)2 |
1.445 |
80 h@10 |
116.7 |
108
|
| CoPOF@CNT |
6 M KOH |
N/A |
1000 h@2 |
141 |
109
|
| 640 h@5 |
| CoFe2O4@CN |
6 M KOH + 0.2 M Zn(Ac)2 |
1.472 |
180 h@5 |
189 |
110
|
| COFBTC-Fe |
8 M KOH + 0.5 M ZnO |
N/A |
80 h@10 |
N/A |
111
|
Bhunia et al. reported two covalent organic frameworks (Co@rhm-PorBTD and Co@sql-PorBTD), which are efficient SAC systems for the ORR. Rhm-PorBTD and sql-PorBTD can be utilized as a coating layer on carbon cloth and subsequently integrated into a ZAB, with the zinc plate as the anodic electrode (Fig. 9a).73 The Co@rhm-PorBTD-based cathode exhibited a stable open-circuit voltage (OCP) of 1.53 V, while the Co@sql-PorBTD cathode demonstrated an OCP of 1.48 V (Fig. 9b). Long-term galvanostatic charge/discharge testing was conducted for each system, with no obvious deterioration detected following 1000 cycles over a duration of 33 hours (Fig. 9c). In contrast, the Pt/C electrode exhibited a significant decrease in performance after only 100 cycles, highlighting the superior stability of the COF-based cathodes compared to the commercial Pt/C catalyst. Wang et al. assembled a liquid primary ZAB using the as-synthesized CoAC-SAs/N-C@CNT catalyst as the air cathode.108 The ZAB equipped with the CoAC-SAs/N-C@CNT electrode exhibits sustained operation for 139 hours (Fig. 9d). This excellent performance demonstrates that the CoAC-SAs/N-C@CNT catalyst possesses outstanding long-term durability and substantial potential for practical application in ZABs. In solid-state Zn–air batteries, Tang et al. synthesized a CoP-TOB catalyst to assemble a rechargeable ZAB, which possesses a specific capacity of 740 mA h g−1 at 20 mA cm−2 and a charge–discharge voltage gap of 0.91 V at 2 mA cm−2 (Fig. 9e and f).53 They further synthesized a large-scale metalloporphyrin-based COF film and utilized the film as the air electrode to assemble a flexible all-solid-state ZAB (Fig. 8g). The ZAB exhibits a good charge–discharge cycling performance at 1.0 mA cm−2 under different bending conditions (Fig. 9h), indicating its robust performance even under mechanical stress. A blue LED was successfully lit using the flexible battery at various bending angles (Fig. 9i), showing the potential of CoP-TOB in flexible batteries systems. The integration of COF-based single-atom catalysts in both liquid and solid-state ZAB represents a significant advancement in the field of energy conversion and storage. The distinctive characteristics of COFs, in conjunction with the superior catalytic performance of SACs, effectively address several limitations inherent to traditional catalysts, thereby facilitating the realization of more efficient, stable, and cost-effective energy devices.
 |
| | Fig. 9 (a) Schematic illustration of the Zn–air battery (ZAB). (b) The open circuit curves of the ZAB using Co@sql-PorBTD and Co@rhm-PorBTD. (c) The discharge/charge cycling measurement of ZAB using Co@sql-PorBTD, Co@rhm-PorBTD, and 20 wt% Pt/C. (Reproduced with permission of ref. 73, Copyright 2023 American Chemical Society.) (d) Charge–discharge cycling performance of ZAB using CoAC-SAs/N-C@CNT and Pt/C. (Reproduced with permission of ref. 77, Copyright 2023, Beijing Institute of Technology Press.) (e) Discharge capacity of a ZAB using CoP-TOB. (f) Galvanostatic discharge–charge cycling measurement of ZAB using CoP-TOB at 2 mA cm−2. (g) Schematic diagram of a flexible solid-state ZAB. (h) Discharge–charge polarization plots under different bent conditions. (i) Photographs of a LED lit by the ZAB under bent conditions. (Reproduced with permission of ref. 53, Copyright 2023, Wiley VCH.) | |
6. Conclusion and perspectives
The past few years have witnessed a rapid upsurge in the development of COF-based SACs and DACs as highly efficient catalysts. These materials exhibit promising applications in energy storage and conversion devices due to the synergistic interplay between the metal active sites and the COF framework. The superior catalytic performance of COF-based SACs and DACs can be attributed to their inherent properties, which include the precise engineering of the framework and functional groups, the inherent flexibility and predictable properties of COF structures, the precise tailoring of metal coordination environments, and post-synthetic modifications that can further expand their physical and chemical properties. Despite the significant advancements achieved in the past few years, COF-based single-site electrocatalysts still encounter a plethora of drawbacks and challenges.
(a) The abundant metal binding sites in COFs enable substantial metal loadings and resistance to atomic metal aggregation, representing a significant advantage over conventional SAC supports. It is critical that the improvement in electrical conductivity and the number of metal sites be further explored to achieve more efficient activities. In addition, the optimized metal microenvironment demonstrates remarkable flexibility and designability through the regulation of metal types and binding sites, integrating heteroatoms and functionalities, and implementing bimetallic strategies.
(b) The utilization of advanced characterization methodologies, including in situ techniques, should be pursued to further elucidate the differences between various single-atom coordination environments and to evaluate the critical reaction intermediates by proposing specific reaction pathways. Then, the precise identification of metal active sites and the comprehension of their catalytic behaviors represent a pivotal task that might be accomplished through the application of in situ characterization techniques.
(c) There is a lack of investigation regarding COF-based SAC and DAC catalysts towards oxygen catalysis under a large-current density. The porosity, crystallinity, electronic configuration and stability of COF-based SACs and DACs can be fine-tuned, but achieving high current density catalytic efficiency and long-term stability remains a major challenge. In addition, numerous post-synthetic modifications can introduce unexpected side effects on the structure and composition.
Future research endeavors should integrate experimental and theoretical approaches to gain a comprehensive understanding of the preparation and reaction mechanisms that govern COF-based single-atom and dual-atom electrocatalysts. The in-depth examination of the synergistic interaction between the structural and functional characteristics of COFs and the single-atom active centers is essential for the optimization of the electrocatalytic performance. It is vital to clarify the specific reaction mechanisms through in situ characterization techniques and theoretical calculations which serve as powerful tools for clarifying electron transfer properties and the local coordination environment around the single-site active center within the COF framework.
Author contributions
This manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Data availability
The authors confirm that the data supporting the findings of this study are available within the review.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, Grant No. 52303359), the Basic and Applied Basic Research Foundation of Guangdong Province (2023A1515011511, 2022A1515110848, 2023B1515040027), the Natural Science Foundation of Guangdong Province (Excellent Youth Team Project, 2023B1515040027), the Open program of State Key Laboratory of Optoelectronic Materials and Technologies (OEMT-2022-KF-03), and the Basic and Applied Basic Research Project of Guangzhou (202201011442).
References
- N. Logeshwaran, S. Ramakrishnan, S. S. Chandrasekaran, M. Vinothkannan, A. R. Kim, S. Sengodan, D. B. Velusamy, P. Varadhan, J.-H. He and D. J. Yoo, Appl. Catal., B, 2021, 297, 120405 CrossRef.
- J. Fu, R. Liang, G. Liu, A. Yu, Z. Bai, L. Yang and Z. Chen, Adv. Mater., 2019, 31, 1805230 CrossRef PubMed.
- R. Haider, Y. Wen, Z.-F. Ma, D. P. Wilkinson, L. Zhang, X. Yuan, S. Song and J. Zhang, Chem. Soc. Rev., 2021, 50, 1138–1187 RSC.
- J. Zhong, Z. Liang, N. Liu, Y. Xiang, B. Yan, F. Zhu, X. Xie, X. Gui, L. Gan, H. B. Yang, D. Yu, Z. Zeng and G. Yang, ACS Nano, 2024, 18, 5258–5269 Search PubMed.
- Z. Chen, J. Jiang, M. Jing, Y. Bai, X. Zhang, W. Deng, Y. Wu, F. Chen, M. Yi, M. Yang, X. Xu, T. Wu, Y. Zhang and X. Wang, Carbon Neutralization, 2024, 3, 689–699 CrossRef.
- S. Zheng, Z. Ouyang, M. Liu, S. Bi, G. Liu, X. Li, Q. Xu and G. Zeng, Carbon Neutralization, 2024, 3, 415–422 CrossRef.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef PubMed.
- A. Vazhayil, L. Vazhayal, J. Thomas, S. Ashok C and N. Thomas, Appl. Surf. Sci. Adv., 2021, 6, 100184 CrossRef.
- N. Liu, Z. Liang, F. Yang, X. Wang, J. Zhong, X. Gui, G. Yang, Z. Zeng and D. Yu, ChemSusChem, 2023, 16, e202202192 CrossRef PubMed.
- Z. Liang, W. Guo, R. Zhao, T. Qiu, H. Tabassum and R. Zou, Nano Energy, 2019, 64, 103917 CrossRef.
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Adv. Energy Mater., 2020, 10, 1902844 CrossRef.
- D. Kong, W. Lv, R. Liu, Y.-B. He, D. Wu, F. Li, R. Fu, Q.-H. Yang and F. Kang, Energy Mater. Dev., 2023, 1, 9370017 Search PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef PubMed.
- H. Xia, G. Qu, H. Yin and J. Zhang, J. Mater. Chem. A, 2020, 8, 15358–15372 RSC.
- J. Liu, B. Bunes, L. Zang and C. Wang, Environ. Chem. Lett., 2017, 16, 477–505 CrossRef.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef.
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef PubMed.
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2021, 31, 2007423 CrossRef CAS.
- J. Li, Z. Yang, Y. Li and G. Zhang, J. Hazard. Mater., 2022, 429, 128285 CrossRef PubMed.
- G. N. Lewis, J. Am. Chem. Soc., 1916, 38, 762–785 CrossRef.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2015, 310, 1166–1170 CrossRef PubMed.
- M. Li, F. Zeng, S. Li, S. Hu, Q. Liu, T. Zhang, J. Zhou and C. Han, Energy Mater. Dev., 2023, 1, 9370007 Search PubMed.
- D.-G. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC.
- Z. Alsudairy, N. Brown, A. Campbell, A. Ambus, B. Brown, K. Smith-Petty and X. Li, Mater. Chem. Front., 2023, 7, 3298–3331 RSC.
- T. F. Machado, M. E. S. Serra, D. Murtinho, A. J. M. Valente and M. Naushad, Polymers, 2021, 13, 970 CrossRef CAS PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- Y. Liang, T. Xia, Z. Wu, Y. Yang, Y. Li, Z. Sui, C. Li, R. Fan, X. Tian and Q. Chen, Mater. Today Chem., 2022, 24, 100777 CrossRef CAS.
- X. Yang, X. Li, M. Liu, S. Yang, Q. Xu and G. Zeng, Small, 2024, 20, 2306295 CrossRef CAS PubMed.
- X. Cui, S. Lei, A. C. Wang, L. Gao, Q. Zhang, Y. Yang and Z. Lin, Nano Energy, 2020, 70, 104525 CrossRef CAS.
- Q. Zhang and J. Guan, J. Power Sources, 2020, 471, 228446 CrossRef.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef PubMed.
- Q. Xu, H. Zhang, Y. Guo, J. Qian, S. Yang, D. Luo, P. Gao, D. Wu, X. Li, Z. Jiang and Y. Sun, Small, 2019, 15, 1905363 CrossRef PubMed.
- X. Feng, Z. Gao, L. Xiao, Z. Lai and F. Luo, Inorg. Chem. Front., 2020, 7, 3925–3931 RSC.
- J. Wang, J. Wang, S. Qi and M. Zhao, J. Phys. Chem. C, 2020, 124, 17675–17683 CrossRef.
- N. Lv, Q. Li, H. Zhu, S. Mu, X. Luo, X. Ren, X. Liu, S. Li, C. Cheng and T. Ma, Adv. Sci., 2023, 10, 2206239 Search PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- X. Guan, Q. Fang, Y. Yan and S. Qiu, Acc. Chem. Res., 2022, 55, 1912–1927 CrossRef CAS PubMed.
- C. Zhao, H. Lyu, Z. Ji, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 14450–14454 CrossRef CAS PubMed.
- J. Sun, Y. Xu, A. Li, R. Tian, Y. Fei, B. Chen and X. Zhou, ACS Appl. Nano Mater., 2022, 5, 15592–15599 CrossRef CAS.
- J. Maschita, T. Banerjee, G. Savasci, F. Haase, C. Ochsenfeld and B. V. Lotsch, Angew. Chem., Int. Ed., 2020, 59, 15750–15758 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- B. Sun, C.-H. Zhu, Y. Liu, C. Wang, L.-J. Wan and D. Wang, Chem. Mater., 2017, 29, 4367–4374 CrossRef.
- X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 5050–5091 CrossRef PubMed.
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef PubMed.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef PubMed.
- C. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef PubMed.
- J.-D. Yi, R. Xu, Q. Wu, T. Zhang, K.-T. Zang, J. Luo, Y.-L. Liang, Y.-B. Huang and R. Cao, ACS Energy Lett., 2018, 3, 883–889 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed.
- T. Jiang, W. Jiang, Y. Li, Y. Xu, M. Zhao, M. Deng and Y. Wang, Carbon, 2021, 180, 92–100 CrossRef CAS.
- C. Yang, Z.-D. Yang, H. Dong, N. Sun, Y. Lu, F.-M. Zhang and G. Zhang, ACS Energy Lett., 2019, 4, 2251–2258 CrossRef CAS.
- P. Peng, L. Shi, F. Huo, C. Mi, X. Wu, S. Zhang and Z. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef CAS PubMed.
- J. Tang, Z. Liang, H. Qin, X. Liu, B. Zhai, Z. Su, Q. Liu, H. Lei, K. Liu, C. Zhao, R. Cao and Y. Fang, Angew. Chem., Int. Ed., 2023, 135, e202214449 CrossRef.
- S.-W. Ke, W. Li, Y. Gu, J. Su, Y. Liu, S. Yuan, J.-L. Zuo, J. Ma and P. He, Sci. Adv., 2023, 9, eadf2398 CrossRef CAS PubMed.
- C. Liu, H. Li, F. Liu, J. Chen, Z. Yu, Z. Yuan, C. Wang, H. Zheng, G. Henkelman, L. Wei and Y. Chen, J. Am. Chem. Soc., 2020, 142, 21861–21871 CrossRef CAS PubMed.
- J.-Y. Yue, Y.-T. Wang, X. Wu, P. Yang, Y. Ma, X.-H. Liu and B. Tang, Chem. Commun., 2021, 57, 12619–12622 RSC.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, G. Tian, J. Schmidt and A. Thomas, Chem. Mater., 2019, 31, 3274–3280 CrossRef CAS.
- C. Lin, L. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2017, 29, 1606635 CrossRef PubMed.
- H. Zhang, M. Zhu, O. G. Schmidt, S. Chen and K. Zhang, Adv. Energy Sustainability Res., 2021, 2, 2000090 CrossRef CAS.
- Y. Zhu, S. Jiang, X. Jing and X. Feng, Trends Chem., 2022, 4, 128–141 CrossRef CAS.
- H. Li, J. Chang, S. Li, X. Guan, D. Li, C. Li, L. Tang, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 13324–13329 CrossRef CAS PubMed.
- Y. Yue, H. Li, H. Chen and N. Huang, J. Am. Chem. Soc., 2022, 144, 2873–2878 CrossRef CAS PubMed.
- Y. Wu, D. Yan, Z. Zhang, M. M. Matsushita and K. Awaga, ACS Appl. Mater. Interfaces, 2019, 11, 7661–7665 CrossRef CAS PubMed.
- Y. Zhang, S. N. Riduan and J. Wang, Chem. – Eur. J., 2017, 23, 16419–16431 CrossRef CAS PubMed.
- X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Nat. Commun., 2018, 9, 1294 CrossRef PubMed.
- Z. Zhang, Z. Zhang, C. Chen, R. Wang, M. Xie, S. Wan, R. Zhang, L. Cong, H. Lu, Y. Han, W. Xing, Z. Shi and S. Feng, Nat. Commun., 2024, 15, 2556 CrossRef PubMed.
- C. Yang, S. Maenosono, J. Duan and X. Zhang, ChemNanoMat, 2019, 5, 957–963 CrossRef.
- M. Liu, S. Yang, S. Liu, Q. Miao, X. Yang, X. Li, Q. Xu and G. Zeng, Small, 2022, 18, 2204757 CrossRef PubMed.
- M. Liu, S. Liu, C. Cui, Q. Miao, Y. He, X. Li, Q. Xu and G. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202213522 CrossRef PubMed.
- W. Niu, J. He, B. Gu, M. Liu and Y. Chueh, Adv. Funct. Mater., 2021, 31, 2103558 CrossRef.
- W. Zhang, L. Zhou, Y. Shi, Y. Liu, H. Xu, X. Yan, Y. Zhao, Y. Jiang, J. Zhang and Z. Gu, Angew. Chem., Int. Ed., 2023, 62, e202304412 CrossRef PubMed.
- S. Huang, B. Zhang, H. Sun, H. Hu, J. Wang, F. Duan, H. Zhu, M. Du and S. Lu, Chem. Commun., 2023, 59, 10424–10427 RSC.
- S. Bhunia, A. Peña-Duarte, H. Li, H. Li, M. F. Sanad, P. Saha, M. A. Addicoat, K. Sasaki, T. A. Strom, M. J. Yacamán, C. R. Cabrera, R. Seshadri, S. Bhattacharya, J.-L. Brédas and L. Echegoyen, ACS Nano, 2023, 17, 3492–3505 CrossRef PubMed.
- J. Li, P. Liu, J. Yan, H. Huang and W. Song, Adv. Sci., 2023, 10, 2206165 CrossRef PubMed.
- C. Yang, S. Tao, N. Huang, X. Zhang, J. Duan, R. Makiura and S. Maenosono, ACS Appl. Nano Mater., 2020, 3, 5481–5488 CrossRef.
- S. Yang, X. Li, T. Tan, J. Mao, Q. Xu, M. Liu, Q. Miao, B. Mei, P. Qiao, S. Gu, F. Sun, J. Ma, G. Zeng and Z. Jiang, Appl. Catal., B, 2022, 307, 121147 CrossRef.
- S. Wei, Y. Wang, W. Chen, Z. Li, W.-C. Cheong, Q. Zhang, Y. Gong, L. Gu, C. Chen, D. Wang, Q. Peng and Y. Li, Chem. Sci., 2020, 11, 786–790 RSC.
- S. Yang, Y. Wei, X. Li, J. Mao, B. Mei, Q. Xu, X. Li and Z. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202313029 CrossRef PubMed.
- P. Wang, R. Zhang, K. Wang, Y. Liu, L. Zhang, X. Wang, H. Li, Y. He and Z. Liu, Energy Mater. Adv., 2023, 4, 0042 CrossRef CAS.
- X. Li, T. Chen, B. Yang and Z. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202215441 CrossRef CAS PubMed.
- Q. Miao, S. Yang, Q. Xu, M. Liu, P. Wu, G. Liu, C. Yu, Z. Jiang, Y. Sun and G. Zeng, Small Struct., 2022, 3, 2100225 CrossRef CAS.
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- R. Zhang, W. Liu, F.-M. Zhang, Z.-D. Yang, G. Zhang and X. C. Zeng, Appl. Catal., B, 2023, 325, 122366 CrossRef CAS.
- Z. Ma, Y. Zhang, S. Liu, W. Xu, L. Wu, Y.-C. Hsieh, P. Liu, Y. Zhu, K. Sasaki, J. N. Renner, K. E. Ayers, R. R. Adzic and J. X. Wang, J. Electroanal. Chem., 2018, 819, 296–305 CrossRef.
- F. Hess, Curr. Opin. Electrochem., 2023, 41, 101349 CrossRef.
- X. Wang, L. Sun, W. Zhou, L. Yang, G. Ren, H. Wu and W.-Q. Deng, Cell Rep. Phys. Sci., 2022, 3, 100804 CrossRef.
- J. Tang, C. Su and Z. Shao, Small Methods, 2021, 5, 2100945 CrossRef PubMed.
- J. J. Jarju, A. M. Díez, L. Frey, V. Sousa, E. Carbó-Argibay, L. P. L. Gonçalves, D. D. Medina, O. I. Lebedev, Yu. V. Kolen’ko and L. M. Salonen, Mater. Today Chem., 2022, 26, 101032 CrossRef.
- K. Chi, Y. Wu, X. Wang, Q. Zhang, W. Gao, L. Yang, X. Chen, D. Chang, Y. Zhang, T. Shen, X. Lu, Y. Zhao and Y. Liu, Small, 2022, 18, 2203966 CrossRef PubMed.
- Z. Gan, S. Lu, L. Qiu, H. Zhu, H. Gu and M. Du, Chem. Eng. J., 2021, 415, 127850 CrossRef.
- Z. Gao, Z. Yu, Y. Huang, X. He, X. Su, L. Xiao, Y. Yu, X. Huang and F. Luo, J. Mater. Chem. A, 2020, 8, 5907–5912 RSC.
- Z. Gao, L. L. Gong, X. Q. He, X. M. Su, L. H. Xiao and F. Luo, Inorg. Chem., 2020, 59, 4995–5003 CrossRef PubMed.
- H. Huang, F. Li, Y. Zhang and Y. Chen, J. Mater. Chem. A, 2019, 7, 5575–5582 RSC.
- T. Gao, K. S. Kumar, Z. Yan, M. Marinova, M. Trentesaux, M. A. Amin, S. Szunerits, Y. Zhou, V. Martin-Diaconescu, S. Paul, R. Boukherroub and V. Ordomsky, J. Mater. Chem. A, 2023, 11, 19338–19348 RSC.
- Z. Liao, Y. Wang, Q. Wang, Y. Cheng and Z. Xiang, Appl. Catal., B, 2019, 243, 204–211 CrossRef.
- X. Li, D. Liu, Q. Liu and Z. Xiang, Small, 2022, 18, 2201197 CrossRef PubMed.
- C. Mi, H. Yu, L. Han, L. Zhang, L. Zhai, X. Li, Y. Liu and Z. Xiang, Adv. Funct. Mater., 2023, 33, 2303235 CrossRef CAS.
- J. Pan, Y. Y. Xu, H. Yang, Z. Dong, H. Liu and B. Y. Xia, Adv. Sci., 2018, 5, 1700691 CrossRef PubMed.
- J. Lee, S. Tai Kim, R. Cao, N. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- Y. Zhang, Y.-P. Deng, J. Wang, Y. Jiang, G. Cui, L. Shui, A. Yu, X. Wang and Z. Chen, Energy Storage Mater., 2021, 35, 538–549 CrossRef.
- X. Cai, L. Lai, J. Lin and Z. Shen, Mater. Horiz., 2017, 4, 945–976 RSC.
- L. Li and A. Manthiram, Adv. Energy Mater., 2016, 6, 1502054 CrossRef.
- Y. Huang, Y. Wang, C. Tang, J. Wang, Q. Zhang, Y. Wang and J. Zhang, Adv. Mater., 2019, 31, 1803800 CrossRef PubMed.
- S. Kim, J.-W. Jung, D. Song, S.-H. Cho, J. Kim, J. K. Kim, D. Oh, H. Sun, E. Cho, I.-D. Kim and W. Jung, Appl. Catal., B, 2022, 315, 121553 CrossRef CAS.
- Z. Chen, H. Zheng, J. Zhang, Z. Jiang, C. Bao, C.-H. Yeh and N.-C. Lai, J. Colloid Interface Sci., 2024, 670, 103–113 CrossRef CAS PubMed.
- H. Zhang, Z. Qu, H. Tang, X. Wang, R. Koehler, M. Yu, C. Gerhard, Y. Yin, M. Zhu, K. Zhang and O. G. Schmidt, ACS Energy Lett., 2021, 6, 2491–2498 CrossRef CAS.
- H. Kang, B. Su and Z. Lei, ACS Appl. Mater. Interfaces, 2024, 16, 16261–16270 CrossRef PubMed.
- P. Peng, L. Shi, F. Huo, S. Zhang, C. Mi, Y. Cheng and Z. Xiang, ACS Nano, 2019, 13, 878–884 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Xuchun
Gui
a,
Xuchun
Gui