
A diastereoselective strategy for dihydrophenanthrene-fused spirooxindoles via [1,2]-phospha-Brook rearrangement†
Amjad
Ali‡
,
Harish K.
Harit‡
,
Chandana
Behera
and
Ravi P.
Singh
 *
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: ravips@chemistry.iitd.ac.in
First published on 19th July 2024
Abstract
A highly diastereoselective, one-pot strategy for spirooxindoles bearing dihydrophenanthrenes from readily available isatins and p-quinone methides (p-QMs) has been disclosed. Here, a sequential umpolung process via [1,2]-phospha-Brook rearrangement followed by Lewis acid-mediated intramolecular cyclization was employed to furnish the desired spiro product. This protocol provides access to potential medicinally relevant varieties of spirooxindolyl dihydrophenanthrenes in good to excellent yields and diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Integration of one carbocyclic ring with another ring can lead to different scaffolds based on their joining patterns such as fused, bridged bicyclic, and spirocyclic. Spirooxindoles are a unique class of spirocyclic compounds bearing a joining point at the C-3 position of the oxindole core.1 These privileged heterocyclic molecular motifs represent central cores in natural products2 and bioactive compounds3 capable of showing anti-tumor,4 antimalarial,5 and anticancer activities6 (Scheme 1(i)). In the past few decades, several elegant strategies have been developed for stereoselective access of different types of spirooxindoles bearing three-, four-, five- and six-membered unified rings.7 Despite significant advancement in the area of stereoselective synthesis of spirooxindoles, modular and selective approaches for dihydrophenanthrene-fused spirooxindoles are less explored. In 2016, Lautens and Garcia-Lopez independently disclosed two protocols for dihydrophenanthrene-fused spirooxindoles from acrylamides and benzyne precursors using palladium catalysis (Scheme 1(iii)(a) and (iii)(b)).8 Later, Yang et al. also demonstrated a route to access dihydrophenanthrene-fused spirooxindoles via multicomponent coupling of acrylamide and iodobenzene (Scheme 1(iii)(c)).9 Although these reports showcased interesting reaction pathways involving domino Heck-type coupling and intramolecular C–H activation,10 several drawbacks such as high catalyst loading, multi-step synthesis of acrylamide, and harsh reaction conditions limit its practical utility. Apart from these three reports with a Pd-based catalytic system, there are no reports on unifying carbocyclic rings and heterocycles to achieve dihydrophenanthrene-fused spirooxindoles.
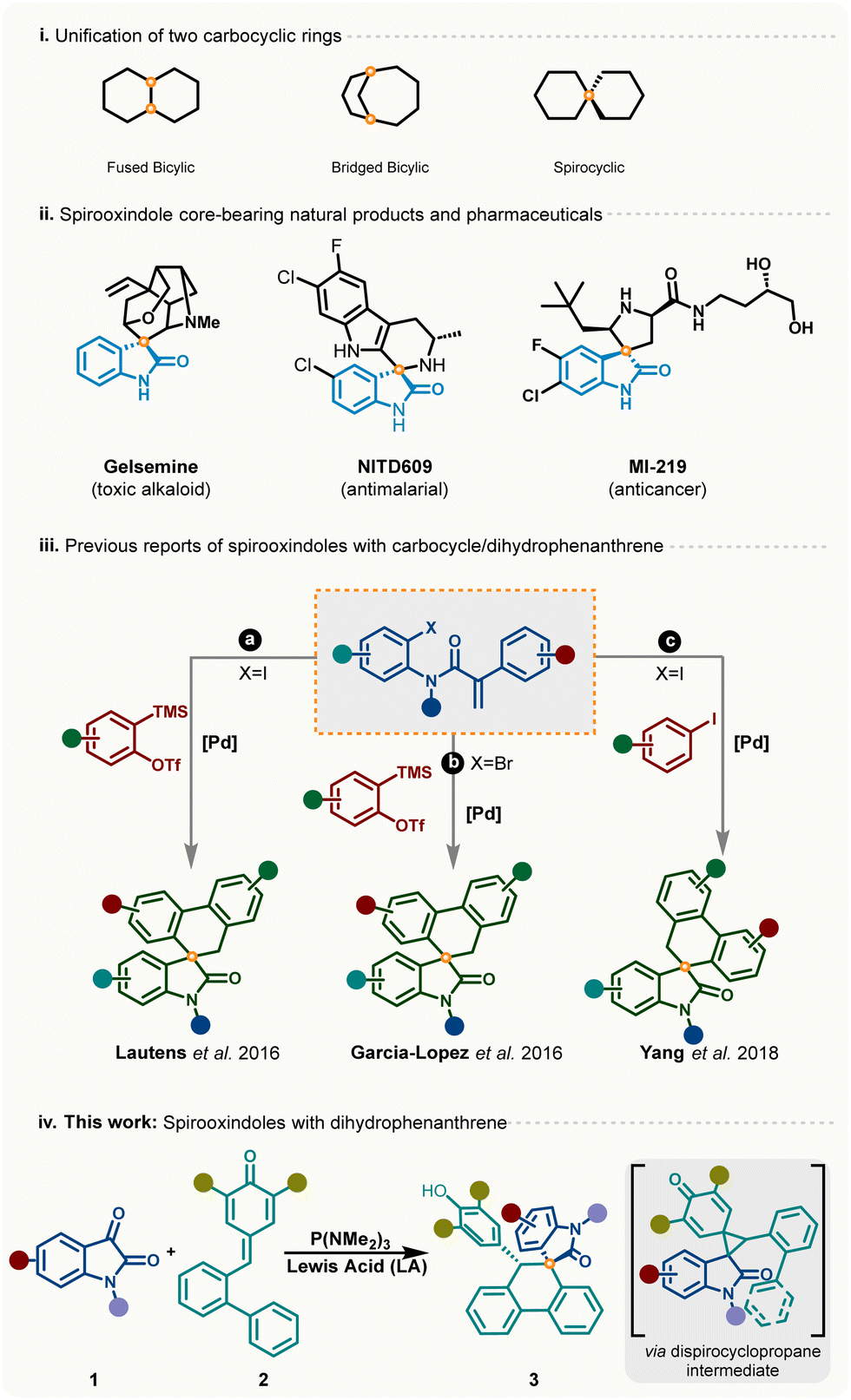 | ||
| Scheme 1 (i) Unification of carbocyclic rings; (ii) representative examples of bioactive molecules containing the spirooxindole core; (iii) recent reports on spirooxindoles with carbocycle/dihydrophenanthrene; (iv) this work. | ||
One strategy (Scheme 1(iv)) for achieving spirooxindole cores can be from commercially available isatin and easily accessible p-QMs11via recently developed unusual [1,2]-phospha-Brook rearrangement guided synthesis.12 In this, it is envisioned that the metal-free synthetic method using isatin and p-QMs would involve two subsequent stepwise rearrangements to deliver the intended outcome (Scheme 1). Based on the literature, mechanistically, it can be proposed that at first P(NMe2)3-mediated [1,2]-phospha-Brook rearrangement of isatin with p-QMs would give the dispirocyclopropane intermediate, which on Lewis acid-mediated selective spirocyclization through the aza-xylylene intermediate would generate the desired product. However, in this strategy, several other possibilities exist making the reaction challenging. For instance, an inefficient ring opening of the cyclopropyl intermediate can lead to other undesired products. In addition, the cyclopropane-adduct can transform into various intermediates via either biaryl cyclization on the proximal cyclopropyl position leading to substituted fluorene or indoyl aryl ring C–C bond migration leading to a disubstituted quinone methide (Scheme 3). Herein, we describe a highly diastereoselective, one-pot strategy for synthesis of spirooxindoles from readily available isatins and p-quinone methides (p-QMs).
We commenced our optimization studies using isatin (1a) and 2-phenyl-p-QMs (2a) as model substrates and keeping in consideration the route to conversion and the formation of the possible intermediates. After a brief reaction condition screening, it was observed that using conditions mediated by P(NMe2)3 at −78 °C to room temperature yielded the dispirocyclopropane adduct 5ain situ (confirmed by NMR and HRMS), followed by BF3·Et2O (2 eq.) mediated spirocyclization, furnishing the desired product 3a in 84% yield (Table 1, entry 1). A controlled experiment without P(NMe2)3 resulted in a lack of intended product formation and the presence of the dispirocyclopropane intermediate (Table 1, entry 2). Additionally, in the absence of BF3·Et2O while the dispirocyclopropane adduct was recovered, the spiro product was not observed (Table 1, entry 3). These two control experiments indicate that both P(NMe2)3 and BF3·Et2O are essential for the transformation.
|
|
||
|---|---|---|
| Entry | Deviation from standard condition A | Yield (%)a |
| All reactions were performed under an argon atmosphere, 1a (0.1 mmol) and 2a (0.11 mmol) were dissolved in solvent (1 mL) and cooled to −78 °C followed by the addition of P(NMe2)3 (0.1 mmol), and then a Lewis acid was added at rt. Diastereomeric ratios were determined by 1H NMR analysis of crude reaction mixtures.a Isolated yield. | ||
| 1 | None | 84 |
| 2 | Without P(NMe2)3 | n.d. |
| 3 | Without BF3·Et2O | n.d. |
| 4 | In(OTf)3 (0.2 eq.) instead of BF3·Et2O | 25 |
| 5 | In(OTf)3 (0.3 eq.) instead of BF3·Et2O | 33 |
| 6 | BF3·Et2O (0.2 eq.) instead of 2 eq. | 28 |
| 7 | BF3·Et2O (0.2 eq.) instead of 2 eq. | 47 |
| 8 | BF3·Et2O (0.2 eq.) instead of 2 eq. | 60 |
| 9 | TFA (1.0 eq.) instead of 2 eq. | 42 |
| 10 | P(OEt)3, PPh3 instead of P(NMe2)3 | n.d. |

The use of 0.2 eq. of In(OTf)3 as a Lewis acid afforded 3a in 25% yield, whereas increasing the loading of In(OTf)3 showed enhanced reactivity, furnishing 3a in 33% yield (Table 1, entries 4 and 5). Further optimization with an uneconomical high loading of metal triflate was avoided and other Lewis acids were tested. Subsequently, lowering in the equivalents of BF3·Et2O decelerated the reactivity drastically and hampered the product yield (Table 1, entries 6, 7 and 8). In addition to the Lewis acid, the desired reaction took place in the presence of 1 eq. of trifluoroacetic acid albeit in 42% yield after 24 h. An attempt to use PPh3 and P(OEt)3 in place of P(NMe2)3 did not provide 3a as the cyclopropanation adduct was not formed (Table 1, entry 10).
With the optimized conditions in hand, the reactivity of various substituted isatins was examined to check the viability of the reaction conditions. Isatin bearing electron-donating methyl and methoxy groups at the 5-position afforded the corresponding spirooxindoles 3b and 3c in 87% and 89% yields with excellent diastereoselectivity (Table 2). Similarly, isatin having 5,7-di-methyl substitution and 5-phenyl group substitution furnished the intended products 3d and 3e in good yields of 74% and 78%, respectively, with excellent stereoselectivities.
| All reactions were performed under an argon atmosphere, 1a (0.1 mmol) and 2a (0.11 mmol) were dissolved in DCM (1 mL) and cooled to −78 °C followed by the addition of P(NMe2)3 (0.1 mmol), and then BF3·Et2O (0.2 mmol) was added at rt. Diastereomeric ratios were determined by 1H NMR analysis of crude reaction mixtures. Isolated yields for 3a–x. |
|---|

|
Interestingly, the developed reaction conditions tolerate halogen groups such as fluoro, chloro, and iodo at the 5-position of isatin to deliver the desired spirocyclized products 3f–3h in good yields (70–80%) and selectivity. Moreover, 5-nitro isatin was amenable to the described protocol allowing the formation of spirooxindole 3i in 70% yield with >20:1 diastereoselectivity. Additionally, 6-chloroisatin, 5,7-dichloroisatin, and 5,7-dibromoisatin also exhibited good reactivity, furnishing the desired products 3j–3l in 77–75% yields with good selectivity.
Furthermore, when evaluating the reactivity of 4-chloro and 4-bromo isatins under the same reaction conditions, they were found to be good reacting partners providing the desired spiro products 3m and 3n in 76% and 79% yields, with moderate diastereoselectivity for 3m but excellent selectivity for 3n (Table 2). Similarly, substituting electron-withdrawing groups such as fluoro and trifluoromethyl at the 7-position of isatin also furnished the corresponding spirooxindoles 3o and 3p in 72% and 76% yields. Unfortunately, 3p gave slightly moderate diastereoselectivity values. Next, the generality of the protocol for various N-protected isatins and p-QMs (Table 2) was examined. For example, methyl-, benzyl-, and MOM-protected isatins smoothly took part in the intended reaction to furnish the corresponding spirooxindoles 3q–3s in 72–80% yields with high selectivity. Moreover, the ester, allylic, and propargylic groups on the N-atom of the isatin were tolerated by the developed protocol to give spiroproducts 3t–3w in good yields (71–82%). Bulky groups such as trityl 3x also worked well under the developed protocol. Additionally, other variations with one nucleophilic site containing 2,4-dimethylaryl and 2-isopropyl gives the desired spiro-adducts 4a and 4b in 77% and 69% yields and excellent selectivity, respectively (Table 3). Additionally, substituting the bulkier benzyloxy group at the ortho-position of the aryl group of p-QM showed good reactivity and selectivity despite having only one nucleophilic centre to afford the adduct 4c in 74% yield. Variation of other substituents such as ortho-naphthyl and 4- and 5-methyl also afforded 4d–4f with good reactivity and selectivity. Unfortunately, the 5-F variation obtained 4g with moderate selectivity but with an excellent yield. The reaction also worked well for o-vinyl substituted p-QMs. Substitution of the internal nucleophile with a vinyl group instead of an aryl group was used to evaluate the suitability of the reaction conditions for generating the corresponding saturated framework. It was observed that, similar to the aryl, the vinyl group was a good reacting partner and afforded the desired product 4h in 71% yield. Unfortunately, ortho-heteroaromatics such as thiophene did not show any reactivity towards the developed protocol 4i. The structure of the spiroadduct was established by the single crystal X-ray analysis of 4h (Table 3; CCDC 2322903) (see the ESI†).
| All reactions were performed under an argon atmosphere, 1 (0.1 mmol) and 2 (0.11 mmol) were dissolved in DCM (1 ml) and cooled to −78 °C followed by the addition of P(NMe2)3 (0.1 mmol), and then BF3·Et2O (0.2 mmol) was added at rt. Diastereomeric ratios were determined by 1H NMR analysis of crude reaction mixtures. Isolated yields for 4a–h. |
|---|

|
Next, in order to expand the product diversity, a similar strategy was implemented to obtain a seven-membered heterocyclic product by introducing an oxygen atom between the two aryl rings of p-QM. A reaction between isatin (1a) and ortho-phenoxy p-QM (2h) under standard conditions led to the formation of quinolinone product 7 instead of the desired spirocyclic adduct. Further treatment of BF3·Et2O (1 eq.) with 7a resulted in spirocyclic product 8 in 69% yield. When dispirocyclopropane 9 was treated with Cu(OTf)2, formation of spirooxindole-oxepine 10 was observed in 78% yield with 1:1 selectivity. Using 5 eq. of AlCl3 in toluene can mono-selectively remove the tert-butyl of 3a to form 11 in 72% yield, whereas increasing the amount of AlCl3 to 10 eq. can deinstall both tert-butyl groups to form 12 in 69% yield (Scheme 2). The reaction developed was thus found to be extremely versatile with broad substrate scope and high selectivity.
 | ||
| Scheme 2 Synthetic transformations (for details see the ESI†). | ||
Supporting the hypothesis of the reaction, it was observed that possible side products relied upon the Lewis acid-mediated selective ring opening of the dicyclopropane 5′. As discussed earlier, the dicyclopropane intermediate 5′ can transform into different products via two competitive pathways (a and b).
In path a, ring-opening leads to the generation of an ortho-aza-xylylene intermediate (6), which undergoes intramolecular spirocyclization to furnish the intended spirooxindoles (Scheme 3). On the other hand, ring opening of the dispiroadduct intermediate (5′) via path b can further lead to two different possible directions and may result in the formation of 2-quinolinone adducts (7via route i) and 3,3-disubstituted oxindole cores (13via route ii). However, during the reactions formation of no undesired products was observed and various control experiments confirmed the preference of path a for the developed strategy, which very efficiently resulted in richly substituted spirooxindoles, thus expanding the molecular library with respect to structural complexity.
 | ||
| Scheme 3 Possible routes to the spirooxindole adduct transformations. | ||
A direct metal-free protocol has been developed to install the spirooxindole-dihydrophenanthrene scaffolds involving the umpolung process via [1,2]-phospha-Brook rearrangement. Here, the sequential addition of phosphine and a Lewis acid was employed to construct the desired scaffold by unifying carbocyclic rings. Broad substrate scopes with substituted oxindoles are demonstrated with good to excellent yields and selectivities. Moreover, synthetic transformations are also carried out to show the utility of the developed reaction.
Generous financial support from SERB-INDIA (CRG/2021/006502 and SERB/SCP/2022/000599) is greatly acknowledged. A. A., H. K. H. and C. B. are grateful to the UGC, CSIR India, and MHRD for the fellowship.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Bariwal, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3831–3848 RSC; (b) M. Xia and R.-Z. Ma, J. Heterocycl. Chem., 2014, 51, 539–554 CrossRef CAS; (c) A. R. Liandi, A. H. Cahyana, D. N. Alfariza, R. Nuraini, R. W. Sari and T. P. Wendari, Green Synth. Catal., 2024, 5, 1–13 CrossRef CAS.
- (a) H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36–51 CrossRef CAS PubMed; (b) T. J. Greshock, A. W. Grubbs, P. Jiao, D. T. Wicklow, J. B. Gloer and R. M. Williams, Angew. Chem., Int. Ed., 2008, 47, 3573–3577 CrossRef CAS PubMed; (c) K. A. Miller, S. Tsukamoto and R. M. Williams, Nat. Chem., 2009, 1, 63–68 CrossRef CAS PubMed.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed.
- (a) Y. Zhao, S. Yu, W. Sun, L. Liu, J. Lu, D. McEachern, S. Shargary, D. Bernard, X. Li, T. Zhao, P. Zou, D. Sun and S. Wang, J. Med. Chem., 2013, 56, 5553–5561 CrossRef CAS PubMed; (b) Y. Zhao, L. Liu, W. Sun, J. Lu, D. McEachern, X. Li, S. Yu, D. Bernard, P. Ochsenbein, V. Ferey, J.-C. Carry, J. R. Deschamps, D. Sun and S. Wang, J. Am. Chem. Soc., 2013, 135, 7223–7234 CrossRef CAS PubMed.
- M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175–1180 CrossRef CAS PubMed.
- (a) S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933–3938 CrossRef CAS PubMed; (b) B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- (a) D. Cheng, Y. Ishihara, B. Tan and C. F. I. Barbas, ACS Catal., 2014, 4, 743–762 CrossRef CAS; (b) G.-J. Mei and F. Shi, Chem. Commun., 2018, 54, 6607–6621 RSC; (c) A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026–1084 RSC; (d) N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165–5181 RSC.
- (a) H. Yoon, A. Lossouarn, F. Landau and M. Lautens, Org. Lett., 2016, 18, 6324–6327 CrossRef CAS PubMed; (b) M. Pérez-Gómez and J.-A. García-López, Angew. Chem., Int. Ed., 2016, 55, 14389–14393 CrossRef PubMed.
- X. Luo, Y. Xu, G. Xiao, W. Liu, C. Qian, G. Deng, J. Song, Y. Liang and C. Yang, Org. Lett., 2018, 20, 2997–3000 CrossRef CAS PubMed.
- (a) I. Franzoni, H. Yoon, J.-A. García-López, A. I. Poblador-Bahamonde and M. Lautens, Chem. Sci., 2018, 9, 1496–1509 RSC; (b) M. Sickert, H. Weinstabl, B. Peters, X. Hou and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 5147–5151 CrossRef CAS PubMed.
- (a) J.-Y. Wang, W.-J. Hao, S.-J. Tu and B. Jiang, Org. Chem. Front., 2020, 7, 1743–1778 RSC; (b) C. G. S. Lima, F. P. Pauli, D. C. S. Costa, A. S. de Souza, L. S. M. Forezi, V. F. Ferreira and F. de Carvalho da Silva, Eur. J. Org. Chem., 2020, 2650–2692 CrossRef CAS.
- (a) R. Kaur and R. P. Singh, J. Org. Chem., 2023, 88, 10325–10338 CrossRef CAS PubMed; (b) A. Ali, H. K. Harit, M. Devi, D. Ghosh and R. P. Singh, J. Org. Chem., 2022, 87, 16313–16327 CrossRef CAS PubMed; (c) R. Kaur, D. Singh and R. P. Singh, J. Org. Chem., 2021, 86, 15702–15711 CrossRef CAS PubMed; (d) A. Ali, C. Behera and R. P. Singh, J. Org. Chem., 2024, 89, 7644–7655 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2322903. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02215g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |