
Enhancing the tumor penetration of multiarm polymers by collagenase modification†
Bo
Yu
a,
Weijie
Wang
a,
Yongmin
Zhang
a,
Ying
Sun
a,
Cheng
Li
a,
Qian
Liu
b,
Xu
Zhen
 *a,
Xiqun
Jiang
*a and
Wei
Wu
*a
*a,
Xiqun
Jiang
*a and
Wei
Wu
*a
aMOE Key Laboratory of High Performance Polymer Materials and Technology, State Key Laboratory of Analytical Chemistry for Life Science, and College of Chemistry & Chemical Engineering, Nanjing University, Nanjing, 210093, P.R. China. E-mail: zhenxu@nju.edu.cn; jiangx@nju.edu.cn; wuwei@nju.edu.cn
bDepartment of Urology, Tianjin First Central Hospital, Tianjin 300192, China
First published on 13th March 2024
Abstract
Tumor penetration is a critical determinant of the therapy efficacy of nanomedicines. However, the dense extracellular matrix (ECM) in tumors significantly hampers the deep penetration of nanomedicines, resulting in large drug-untouchable areas and unsatisfactory therapy efficacy. Herein, we synthesized a third-generation PAMAM-cored multiarm copolymer and modified the polymer with collagenase to enhance its tumor penetration. Each arm of the copolymer was a diblock copolymer of poly(glutamic acid)-b-poly(carboxybetaine), in which the polyglutamic acid block with abundant side groups was used to link the anticancer agent doxorubicin through the pH-sensitive acylhydrazone linkage, and the zwitterionic poly(carboxybetaine) block provided desired water solubility and anti-biofouling capability. The collagenase was conjugated to the ends of the arms via the thiol-maleimide reaction. We demonstrated that the polymer-bound collagenase could effectively catalyze the degradation of the collagen in the tumor ECM, and consequently augmented the tumor penetration and antitumor efficacy of the drug-loaded polymers.
1. Introduction
Nanomedicines can selectively accumulate in tumors via the enhanced permeability and retention (EPR) effect, resulting in improved therapy effectiveness and reduced side effects.1–10 However, after nanomedicines extravasate from tumor vessels, the dense extracellular matrix (ECM) impedes greatly their deep penetration in tumor tissues.11–17 Tumor ECM is a three-dimensional crosslinked network composed of collagen, hyaluronic acid, elastin, fibronectin, and proteoglycans.18–25 To alleviate the unfavorable situation caused by tumor ECM, the ECM modulation strategies involving blocking ECM generation and degrading existing ECM have been developed.26,27 Blocking ECM generation has little impact on the existing ECM, resulting in suboptimal efficiency in nanomedicine diffusion.28–30 In comparison, degrading existing ECM with enzymes such as Collagenase, hyaluronidase, and bromelain is simpler and more operationally feasible.31–34 Collagen is the most abundant protein in tumor ECM and hence becomes the primary target for the degradation of tumor ECM.35–37 Collagenase (Col), a water-soluble matrix metalloproteinase, can specifically catalyze the degradation of collagen within ECM. Col received FDA (the U.S. Food and Drug Administration) approval for Peyronie's disease treatment in 2013.38 However, when administered intravenously as a standalone agent, Col exhibits a short in vivo circulation time and may potentially cause some unwanted side effects such as muscle spasms and vascular occlusions.39,40 Conjugating Col to nanomedicines can overcome these shortcomings to some extent and at the same time augment the tumor penetration of the nanomedicines.Col-modified nanomedicines have been studied in the treatment of liver fibrosis,41 pulmonary fibrosis,42 and tumors.43,44 However, these reported Col-modified nanomedicines are mostly prepared by self-assembly methods. The active sites of the Col might be partially buried by other molecules, leading to reduced enzymatic activity, and the uncontrollability of the self-assembling process would result in undesired batch-to-batch reproducibility of the properties of the Col-modified nanomedicines. In contrast, the unimolecular polymer nanomaterials prepared via controllable synthesis methods have well-defined structures and controlled sizes, and are easy to be modified chemically.45–47 Conjugating Col to the surface of unimolecular polymer nanomaterials through the cysteine residues that are the non-active moieties in Col offers a promising solution to the aforementioned challenges, since in doing so, the Col moieties can stay on the periphery of the nanomaterials and hence can function more effectively, and the controlled structure and size of the unimolecular polymer nanomaterials impart desired batch-to-batch reproducibility.48
In this study, we demonstrated that after conjugated to the surface of a doxorubicin (DOX)-loaded PAMAM-cored multiarm copolymer, Col can significantly improve its tumor penetration and antitumor efficacy. The arms of the copolymer were composed of a diblock copolymer of poly(glutamic acid)-b-poly(carboxybetaine), in which the abundant side groups of the polyglutamic acid (PGA) block were used to link DOX through the pH-sensitive acylhydrazone linkage, and the zwitterionic PCB block provided desired water solubility and anti-biofouling capability. The Col was conjugated to the ends of the arms via the thiol-maleimide reaction. In vitro experiments showed that the polymer-bound Col retained more than 67% activity for catalyzing the degradation of collagen when compared to the free Col.
2. Materials and methods
2.1 Materials
L-glutamic acid-5-tert-butylester (Glu(OtBu)), triphosgene, Cu (0) wire (l = 10 cm, d = 1 mm), tris(2-(dimethylamino)ethyl)amine (Me6TREN), 3-maleimidopropionic acid, propargylamine, 2-(dimethylamino)ethyl methacrylate (DMAEMA), tert-butyl bromoacetate, tert-butyl carbazate, pentamethyldiethylenetriamine (PMDETA), trifluoroacetic acid (TFA), triethylamine (TEA), and 2-bromoisobutyryl bromide (BIBB) were purchased from J&K Scientific Ltd. G3 PAMAM dendrimer, sodium ascorbate, sodium azide, tris(2-carboxyethyl)phosphine hydrochloride (TCEP), glucose gel LH-20, N-hydroxysuccinimide (NHS), Col type I, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCl), 1-hydroxybenzotriazole (HOBT), copper(II) sulfate pentahydrate, doxorubicin hydrochloride (DOX·HCl), fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich. DMAEMA were passed through a basic alumina column to remove the inhibitor before use. Dichloromethane (DCM), dimethylformamide (DMF), and dimethyl sulfoxide (DMSO) were dried and distilled over calcium hydride. Tetrahydrofuran (THF) was dried and distilled over sodium. The solution of PAMAM dendrimer in methanol was distilled to remove the solvent before use. Mouse breast cancer cells (4T1 cells) and mouse colon carcinoma cells (CT26 cells) were purchased from the Shanghai Institute of Cell Biology (Shanghai, China).2.2 Instruments
Nuclear magnetic resonance (NMR) spectra were measured on a Bruker AVANCE III 400 spectrometer. Transmission electron microscopy (TEM) observations were conducted on a JEM-2100 microscope operating at an accelerating voltage of 120 kV. Dynamic light scattering (DLS) measurements were performed on Nanobrook Omni, Brookhaven, USA. Zeta potential measurements were performed on Zetaplus (Brookhaven Instruments Corporation). Confocal laser scanning microscopy (CLSM) images were recorded on LSM-710 (Zeiss Inc., Germany). Flow cytometry was measured on Beckman CytoFlex. UV-vis absorption spectra were obtained by UV spectrometer (UV-5300, Metash, Shanghai).2.3 Synthesis of PAMAM-PGA20-b-PCB37 (DP-1) and PAMAM-PGA20-b-PCB37-DOX (DPD-1)
The DP-1 and DPD-1 were synthesized following the procedures published in our previous work.492.4 Synthesis of PAMAM-PGA20-b-PCB37-N3
PAMAM-PGA20-b-PCB37 (2 g, 5.57 μmol), NaN3 (232 mg, 3.57 mmol), and TEA (364 mg, 3.59 mmol) were dissolved in 20 mL of water and stirred at 70 °C for 3 days. Thereafter, the mixture was dialyzed against water using a dialysis membrane with a molecular weight cutoff of 14 kDa for 2 days. The resulting solution was then freeze-dried to give PAMAM-PGA20-b-PCB37-N3 as a white powder. Yield: 1.68 g (84% conversion rate). 1H NMR (400 MHz, D2O) δ 4.48 (m, 2H), 4.12 (m, 2H), 4.01 (m, 2H), 3.36 (s, 6H), 2.41–2.24 (m, 2H), 2.10–1.92 (m, 2H), 1.21–1.03 (m, 3H).2.5 Synthesis of 2,5-dihydro-2,5-dioxo-N-2-propyn-1-yl-1H-pyrrole-1-propanamide (Mal)
2,5-Dihydro-2,5-dioxo-N-2-propyn-1-yl-1H-pyrrole-1-propanamide (Mal) was synthesized following the published procedures.50 Briefly, 3-maleimidopropionic acid (679 mg, 4.0 mmol), EDCI (920 mg, 4.8 mmol), and HOBT (649 mg, 4.8 mmol) were dissolved in 12 mL of dichloromethane, and then propargylamine (242 mg, 4.4 mmol) dissolved in 3 mL of dichloromethane was added to the above system and stirred at room temperature overnight. Thereafter, the mixture was diluted with dichloromethane (10 mL), washed with saturated sodium carbonate solution (20 mL), 1 N hydrochloric acid solution (20 mL), and saturated sodium chloride solution in sequence, and dried by anhydrous sodium sulfate. The crude product was further purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to obtain 467 mg of pure product Mal in a 52% yield. 1H NMR (400 MHz, chloroform-d) δ 6.72 (s, 2H), 6.16 (s, 1H), 4.03 (s, 2H), 3.85 (t, J = 6.7 Hz, 2H), 2.57 (t, J = 6.8 Hz, 2H), 2.24 (s, 1H).
:1, v/v) to obtain 467 mg of pure product Mal in a 52% yield. 1H NMR (400 MHz, chloroform-d) δ 6.72 (s, 2H), 6.16 (s, 1H), 4.03 (s, 2H), 3.85 (t, J = 6.7 Hz, 2H), 2.57 (t, J = 6.8 Hz, 2H), 2.24 (s, 1H).
2.6 Synthesis of PAMAM-PGA20-b-PCB37-Mal (DPMal)
PAMAM-PGA20-b-PCB37-N3 (1.65 g, 4.60 μmol), PMDETA (26 mg, 0.15 mmol), CuSO4·5H2O (37 mg, 0.15 mmol), and sodium ascorbate (60 mg, 0.30 mmol) were dissolved in 20 mL of water. To the mixture, Mal (92 mg, 0.445 mmol) dissolved in 2 mL of DMF was added. The reaction mixture was then subjected to three cycles of freeze–pump–thaw to remove air and filled with argon. The reaction was allowed to proceed at 30 °C under argon protection for 3 days. After the completion of the reaction, the reaction mixture was concentrated and precipitated three times in diethyl ether after dissolution in a small amount of methanol. The resulting solid was dissolved in water and dialyzed for 2 days using a dialysis bag with a molecular weight cut-off of 14000 Da, followed by freeze-drying. Ultimately, a white product was obtained. Yield: 1.35 g (82% conversion rate). 1H NMR (400 MHz, D2O) δ 4.48 (m, 2H), 4.12 (m, 2H), 4.04 (m, 2H), 3.37 (s, 6H), 2.44 (m, 2H), 2.10–1.92 (m, 2H), 1.19–1.05 (m, 3H).
2.7 Synthesis of PAMAM-PGA20-DOX-b-PCB37-Mal (DPDMal)
DPMal (100 mg, ∼0.12 mmol hydrazide) and DOX·HCl (128 mg, 0.22 mmol) were dissolved in 15 mL of methanol containing a drop of TFA. The reaction mixture was stirred at room temperature for 48 h under dark. The resulting solution was concentrated and passed through a Sephadex LH-20 column with methanol as the eluant to remove free DOX, giving DPDMal. The drug loading content (DLC) is the percentage of DOX in the multiarm copolymer conjugated with DOX.2.8 Synthesis of DPCol and DPDCol
DPMal (5 mg, 14 nmol) and Col type I (10 mg, 439 nmol) were combined in a 50 mL flask, followed by the addition of 25 mL of deionized water for dissolution. Subsequently, 200 μL of a 1 mol L−1 solution of TCEP was added into the system, and the pH of the reaction mixture was adjusted to 7.8. The reaction mixture was stirred at 0 °C under dark for 12 h. Thereafter, the resulting solution was concentrated using a 100 kDa ultrafiltration membrane, followed by purification through a G-100 gel column. After freeze-drying, DPCol was obtained as a white powder. DPDCol was also prepared following the same procedures as stated above. The Col contents in DPCol and DPDCol were quantified using the Bicinchoninic Acid (BCA) protein assay following standard procedures.322.9 General procedures for labeling DP-1 and DPCol with FITC
DP-1 (20 mg) was dissolved in 5 mL of phosphate-buffered saline (PBS, pH = 7.4). To the solution, TEA (10 μL, 0.04 mmol) and FITC (4 mg, 0.01 mmol) were added. The resulting mixture was stirred at room temperature under dark overnight. Thereafter, 5 mL of PBS (pH = 7.4) was added. After 8 cycles of ultrafiltration using a 10 kDa molecular weight cutoff ultrafiltration tube, the resulting solution was freeze-dried to give FITC-labeled DP-1. FITC-labeled DPCol was prepared following the same procedures as stated above.2.10 In vitro drug release
A solution of DPDCol (5 mg) was dissolved in 1 mL of PBS (pH 7.4). Subsequently, the solution was dialyzed against PBS (0.01 M, 5 mL) with different pH (5.0, 6.0, and 7.4) in dialysis bags (MWCO 7000 Da) at 37 °C for 48 h. At regular intervals, the released solution outside the dialysis bag was collected, and an equivalent volume of fresh PBS was replenished to maintain a constant volume. The absorbance at 495 nm of the sampled solutions was measured using a UV-visible spectrophotometer, and the DOX concentrations in the samples were determined by referencing a standard curve obtained from UV-Vis spectrophotometric analysis. The time-dependent release of DOX was plotted to generate a graph illustrating its release behavior and profile.2.11 In vitro enzymatic activity assay of Col
Preliminary determination of enzyme activity was conducted by the following procedures. A gelatin solution was prepared in PBS (pH = 7.4) with a 30 mg mL−1 concentration and co-cultured with the test samples at 37 °C, respectively. The samples include (1) PBS, (2) free DOX, (3) DP-1, (4) DPD-1, (5) Col, (6) DPCol, and (7) DPDCol. The concentrations of DPCol and DPDCol were normalized to 0.1 mg mL−1 Col equivalent, the same as that of free Col. After co-cultured for predetermined periods (1, 4, and 12 h), the samples were placed at 4 °C for 30 min and then photographed by a digital camera.We further quantitatively determined the enzyme activities following published procedures.39 Briefly, 0.3 mL of a 0.2% (w/v) gelatin solution in water was mixed with 0.2 mL of a 150 mM Tris HCl solution containing 12 mM CaCl2 at pH 7.5. Subsequently, 0.1 mL of Col, DPCol, and DPDCol (containing 0.1 mg mL−1 Col equivalent) was added and incubated at 37 °C for 30 min, respectively. The degradation processes were terminated by adding 0.6 mL of 0.1 N HCl. The amounts of the released amino acids were determined by using the ninhydrin method, with glycine as the reference sample. The activity unit was defined as the amount of enzyme required to release 1 μg of amino acids per minute.
2.12 Cellular uptakes of the FITC-labeled DP-1 and DPCol
The cellular uptake behaviors of the FITC-labeled DP-1 and DPCol were studied in 4T1 and CT26 cells. The cells (2 × 105 per well) were seeded in a 6-well plate, and incubated at 37 °C for 24 h. The sample solution (400 μg mL−1, 200 μL in PBS) was added, and the cells were cultured at 37 °C for another 4 h. The cells were washed three times with PBS to remove any uninternalized samples and fixed with 4% paraformaldehyde at room temperature. Then, the cell nuclei were stained with 100 μL of Hoechst 33258 at room temperature. The cells were observed by CLSM. To quantitatively assess the cellular uptake of the polymers, the cells were harvested for flow cytometry measurements.2.13 In vitro cytotoxicity
The in vitro cytotoxicity of the samples against 4T1 and CT26 cells was tested by MTT assay. Briefly, the cells were seeded into a 96-well plate at a density of 5000 cells per well and incubated at 37 °C with 5% CO2 and full humidity for 24 h. Once the cells adhered to the bottom of the wells, the cells were treated with a series of doses of the samples and co-incubated for another 24 h. Thereafter, the culture medium was removed and the cells were washed with PBS 3 times and cultured in the medium containing MTT for 4 h. The culture medium in each well was aspirated, and 150 μL of DMSO was added to dissolve the produced formazan. Cell viability was determined by calculating the ratio of the absorbance of the test wells to that of control wells without any treatment.2.14 Penetration in multicellular spheroids (MCs)
After 7 days of the in vitro cultivation of 4T1 cells, MCs with a diameter of ∼300 μm were obtained. For each experimental group, approximately 20 MCs were selectively picked and transferred to a 15 mL centrifuge tube. A sample solution (400 μg mL−1, 200 μL) was added to the suspension of the MCs, followed by co-culturing at 37 °C in a CO2 incubator for 12 h. The culture medium was then discarded, and the MCs were washed 3 times with PBS. The sample distribution in the MCs was observed by CLSM.2.15 Establishment of the tumor models
All the animal experiments were performed in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Ethical and Welfare Committee of Nanjing University (approval number: IACUC-D2303055). To establish the 4T1 tumor models, 2 × 106 4T1 cells were injected subcutaneously under the armpit of Balb/c mice in 100 μL of PBS. Once the average tumor volume reached ∼200 mm3, in vivo experiments were conducted. All animal experiments were conducted with a single dose administered on the first day.2.16 Tumor tissue immunofluorescence staining and tumor penetration
Tumor tissue immunofluorescence staining was employed to investigate the degradation of collagen in tumors induced by the samples. Certain amounts of saline, DP-1, and DPCol were administered separately into 4T1 tumor-bearing mice via tail vein. At 48 h after the treatments, the mice were euthanized, and the tumors were excised and fixed in 4% paraformaldehyde aqueous solution overnight at 4 °C. Subsequently, the tumors were dehydrated with 30% sucrose solution for 12 h and cut into sections with a thickness of 9 μm after being frozen at optimal cutting temperature (O.C.T.). The tumor sections were hydrated in a 0.1% Triton X-100 PBS solution (pH = 7.4) for 10 min, followed by a 1-hour incubation in a 3% BSA solution at 37 °C. The sections were incubated with a rat anti-mouse antibody CD31 at 37 °C for 1 h, and subsequently incubated with a fluorescent secondary antibody, Alexa Fluor™ 647-conjugated goat anti-rat, at 37 °C in darkness for 40 min. The degradation of collagen in tumor tissues was observed by CLSM and the quantitative analysis of the collagen contents was performed by Image J software.Certain amounts of the FITC-labeled DP-1 and DPCol were administered separately into 4T1 tumor-bearing mice via tail vein. At 48 h after the treatments, the mice were euthanized, and the tumors were excised and fixed in 4% paraformaldehyde aqueous solution overnight at 4 °C. Subsequently, the tumors were dehydrated with 30% sucrose solution for 12 h and cut into sections with a thickness of 9 μm after being frozen at optimal cutting temperature (O.C.T.). The tumor sections were stained with CD31 antibody and Alexa Fluor™ 594-conjugated donkey anti-rat secondary antibody at 37 °C in darkness for 40 min. DAPI was employed for nuclear staining. The stained tumor sections were observed by CLSM.
2.17 Biodistribution
4T1 tumor models were built as described above. The samples of DPD-1 and DPDCol were dissolved in saline and administered to tumor-bearing mice via tail vein at a dose of 4 mg kg−1 DOX equivalent. The mice were euthanized at different intervals, and the hearts, livers, spleens, lungs, kidneys, and tumors were excised, weighed, and documented. The blood samples were collected via eye puncture and centrifuged at 14000 rpm min−1 to obtain serum samples. To both the collected serum samples and excised tissues, 4 mL of 70% ethanol solution containing 0.5 M HCl was added. The samples were vigorously homogenized and extracted at room temperature under dark for 48 h, followed by centrifugation to discard the precipitate. The DOX concentrations in the supernatant were measured by fluorescence techniques with excitation at 480 nm and emission at 590 nm, based on the pre-established calibration curve. The calibration curves were established by adding a known amount of samples to the untreated blood and tissues, followed by the same processes as described above. All pharmacokinetic (PK) parameters were calculated by PK Solver software.
2.18 In vivo tumor inhibition
4T1 tumor models were built as described above. The 4T1 tumor-bearing mice were randomly divided into 6 groups (ten per group). DPD-1, DPDCol, and free DOX were separately administered via tail vein to the 4T1 tumor-bearing mice at a dose of 4 mg kg−1 DOX equivalent. Mice treated with saline, DP-1, and DPCol were designated as negative control groups. Tumor volume (V, V = d2 × D/2, where d represents the shortest diameter of the tumor and D represents the longest diameter of the tumor, respectively) and body weight were measured every other day, and the survival rates were monitored for 60 days. The tumor growth inhibition (TGI) was calculated by the following equation: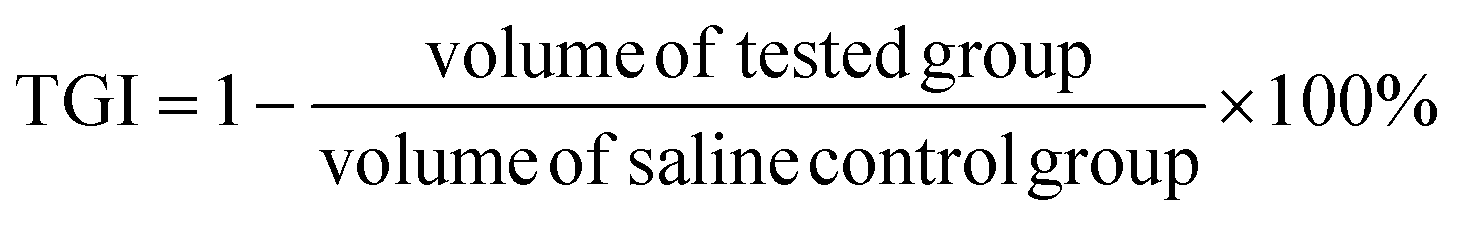
2.19 Histological analysis
Healthy Balb/c mice were randomly divided into three groups (three per group). They were intravenously injected with saline (200 μL), DPD-1, and DPDCol in saline (1 mg mL−1, 200 μL) via tail vein. On the 7th day after the treatments, the mice were euthanized, and the hearts, livers, spleens, lungs, and kidneys were excised. All the organs were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned at 4 μm thickness, stained with hematoxylin and eosin (H&E), and finally observed and captured on an optical microscope (Eclipse Ci-E, Nikon).4T1 tumor models were built as described above. The 4T1 tumor-bearing mice were randomly divided into three groups (three per group). They were intravenously injected with saline (200 μL), DPD-1, and DPDCol in saline (1 mg mL−1, 200 μL) via tail vein. On the 7th day after the treatments, the mice were euthanized, and the tumors were excised. The tumors were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned at 4 μm thickness, stained with hematoxylin and eosin (H&E), and finally observed and captured on an optical microscope (Eclipse Ci-E, Nikon).
2.20 Biosafety evaluation
Healthy Balb/c mice were randomly divided into 3 groups (3 per group) and injected with saline (200 μL), DPD-1 and DPDCol in saline (1 mg mL−1, 200 μL) via tail vein. On the 14th day after the treatments, all the mice were executed humanitarianly. Blood samples were collected by eye puncture and centrifuged at 3000 rpm for 15 min to collect plasma that was further used for the hematological assessment and blood biochemistry analysis.2.21 Statistical analysis
Statistical analyses were performed by Student's t-test and p-values less than 0.05 were statistically significant.3. Results and discussion
3.1 Syntheses and characterizations of the col-modified multiarm copolymer
The third-generation (G3) PAMAM-cored multiarm copolymer DP-1 was synthesized following the procedures published in our previous work.49 As shown in Scheme 1, to conjugate Col to the multiarm copolymers, we introduced first an azido group at the end of each arm through the reaction with NaN3, and then a linker with an alkynyl group and a maleimide group through Cu(I)-catalyzed alkyne–azide 1,3-dipolar cycloaddition (CuAAC), affording DPMal. The acylhydrazine groups in DPMal were used to link the antitumor drug DOX through the acid-sensitive acylhydrazone linkage, affording DPDMal. Finally, type I Col was conjugated to the ends of the arms of DPDMal and DPMal through the thiol-maleimide reaction, respectively, to give the drug-loaded polymer-Col conjugate DPDCol and the drug-free polymer-Col conjugate DPCol as a control sample. Type I Col can specifically catalyze the degradation of type I collagen that is a primary component of the tumor ECM. The characterization data of the newly synthesized polymers can be found in Fig. S1 and S2.†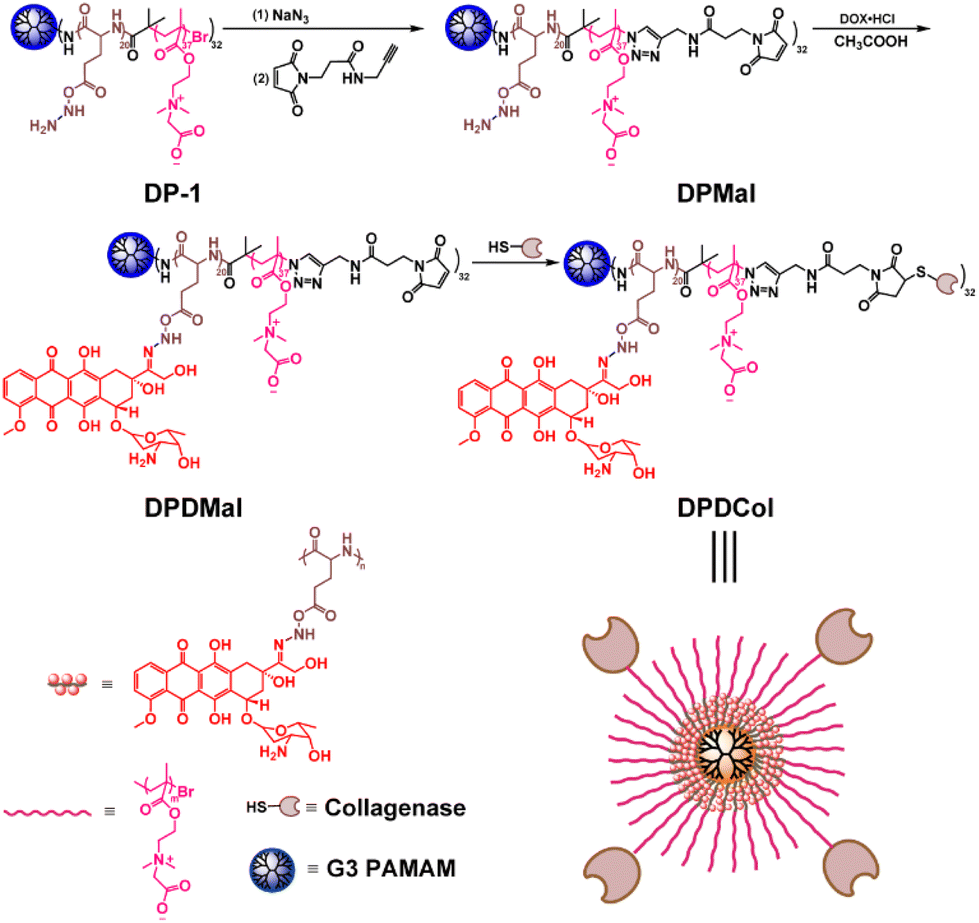 | ||
| Scheme 1 Synthetic route of the Col-modified PAMAM-cored multiarm copolymer. | ||
We measured the loading of Col in DPDMal by BCA protein assay, showing that there are averagely 4 Col moieties in a DPDMal molecule. The drug loading contents of DPDCol and DPD-1 are determined to be about 14.1% and 27.8%, respectively, by fluorescence quantification. The presence of Col moieties lowers the drug loading content of DPDCol when compared to DPD-1. Both DPD-1 and DPDCol exhibit good solubility in aqueous media, including saline and fetal bovine serum (FBS), as illustrated in Fig. S3.† Furthermore, they also have high colloidal stability in PBS and cell culture media, with no significant change in hydrodynamic diameter over two weeks (Fig. S4†).
We examined the morphological structure and size of DPDCol by TEM and DLS and compared with those of DPD-1 (Fig. 1a–c). As observed by TEM, DPDCol exhibits a spherical morphology with an average diameter of 10 nm. The diameter is larger than that of DPD-1 due to the presence of the Col (∼4 nm). The hydrodynamic diameters of DPDCol and DPD-1 in water are about 23 nm and 16.2 nm, respectively, as determined by DLS (Fig. 1c). Due to the existing of Col on the surface, DPDCol has more negative Zeta potential than DPD-1 (Fig. S5†).
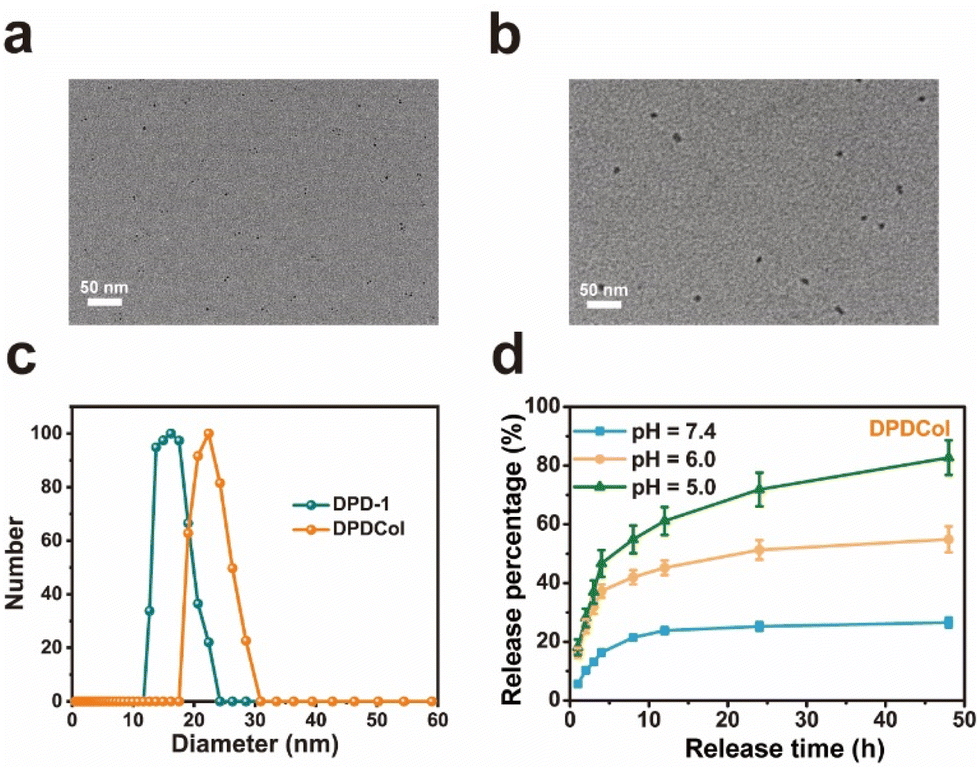 | ||
| Fig. 1 Typical TEM images of DPD-1 (a) and DPDCol (b). Scale bars = 50 nm. (c) Hydrodynamic diameter distributions of DPD-1 and DPDCol. (d) In vitro drug release profiles of DPDMal in PBS with pH 7.4, 6.0, and 5.0 at 37 °C. | ||
3.2 In vitro drug release
We investigated the in vitro drug release profiles of DPDCol in different pH media to simulate its drug release behaviors in various physiological environments, including pH 7.35–7.45 for blood circulation, pH 6.0–6.5 for tumor microenvironment, and pH 4.0–5.0 for lysosome. As shown in Fig. 1d, typical pH-dependent drug release behavior of DPDCol is observed. At pH 7.4, DPDCol exhibits a slow DOX release rate, with only 26% of DOX released within 48 hours. In contrast, at pH 6.0 and 5.0, a pronounced increase in DOX release is observed. At pH 6.0, DPDCol releases 54.9% of DOX within 48 hours, and at pH 5.0, this figure increased to 82.7%. Notably, under identical pH conditions, the DOX release behaviors of DPDCol and DPD-1 are very similar,27 suggesting that the peripheral Col in DPDCol does not impact significantly on DOX release. The pH-responsive drug release behavior holds the potential to minimize premature drug release within the neutral bloodstream, thereby reducing adverse effects. Simultaneously, it enables site-specific drug release within the acidic tumor regions, enhancing the effectiveness of anti-tumor therapy.3.3 Enzymatic activity
To investigate whether DPCol and DPDCol retain enzymatic activity, we conducted gelatin degradation experiments (Fig. 2a). Gelatin can be dissolved in warm water, but it forms a hydrogel at lower temperatures. We employed a pure gelatin solution in water as the control (group 1), and solutions of DOX (group 2), DP-1 (group 3), DPD-1 (group 4), Col (group 5), DPCol (group 6), and DPDCol (group 7) co-cultured with gelatin as the experimental groups. Each sample was incubated at 37 °C for 1, 4, and 12 h, respectively, followed by another 0.5 h incubation at 4 °C. The starting time point was set as 0 h. As shown in Fig. 2a, at 0 h, each sample remains a solution state at room temperature. At 1 h and 4 h after the treatments, only the Col group does not solidify into a hydrogel. At 12 h, the Col, DPCol, and DPDCol groups are in a solution state, while the other four groups solidify into hydrogels. These results indicate that the polymer-bound Col still maintains its enzymatic activity, albeit slightly lower than that of free Col. | ||
| Fig. 2 (a) Photographs of the solutions of co-dissolving gelatin and various samples in distilled water at 37 °C (set as 0 h) and co-incubating gelatin with each sample for different time, followed by storage at 4 °C for 30 minutes. These samples are as follows: (1) pure gelatin, (2) DOX, (3) DP-1, (4) DPD-1, (5) Col, (6) DPCol, (7) DPDCol. (b) Enzymatic activities of Col, DPCol and DPDCol. Data represent mean values ± SD (n = 3). N.S. means no significant difference. | ||
We further quantified the enzyme activities of Col, DPCol, and DPDCol through the ninhydrin method.39 After incubated with gelatin in PBS at 37 °C for 30 min, the enzyme activities of Col, DPCol, and DPDCol were determined by calculating the release of free amino acids. As shown in Fig. 2b, the enzyme activities of Col, DPCol, and DPDCol are measured to be 15.7 U mL−1, 11.8 U mL−1, and 10.6 U mL−1, respectively. The enzyme activities of DPCol and DPDCol can be potentially used to catalyze the degradation of the collagen in tumor ECM and thus enhance their tumor penetration.
3.4 In vitro cytotoxicity and cellular uptake
To assess the biocompatibility of the drug-free polymer-Col conjugates and the pharmacological activity of the drug-loaded polymer-Col conjugates, we determined the in vitro cytotoxicities of DP-1, DPCol, DPD-1, and DPDCol against 4T1 and CT26 cells by MTT assay, with DOX as the positive control. As shown in Fig. 3a and S6a,† after 48 h of incubation, even at concentrations as high as 800 μg mL−1, DP-1 and DPCol exhibit no significant cytotoxicity in 4T1 and CT26 cells, indicating their good biosafety. All the drug formulations DOX, DPD-1, and DPDCol display dose-dependent cytotoxicity after 24 h incubation with 4T1 or CT26 cells, and their IC50 values follow the order of DOX < DPD-1 ≈ DPDCol (Fig. 3b, c and S6b, c†).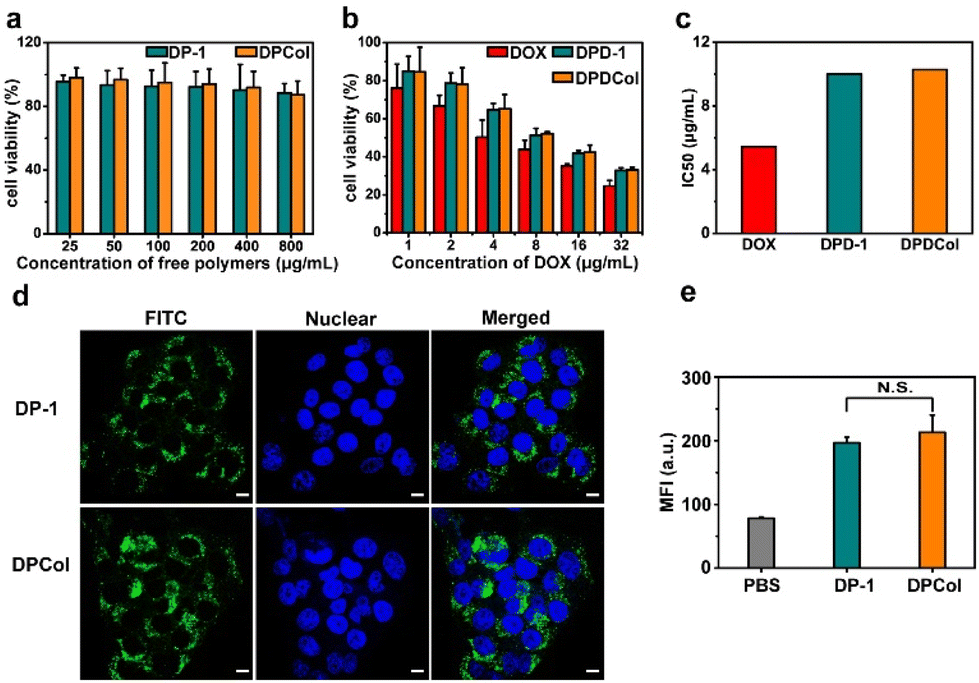 | ||
| Fig. 3 (a) In vitro cytotoxicities of DP-1 and DPCol against 4T1 cells after 48 h of incubation. (b) In vitro cytotoxicities of DOX, DPD-1, and DPDCol against 4T1 cells after 24 h of incubation. Data represent mean ± SD (n = 3). (c) IC50 values calculated from the MTT assay data. Typical CLSM images (d) and mean fluorescence intensities measured by flow cytometry (e) of the 4T1 cells incubated with the FITC-labeled DP-1 and DPCol at 37 °C for 4 h, respectively. Scale bars = 10 μm. Data represent mean ± SD (n = 3). N.S. means no significant difference. | ||
We investigated the cellular uptake behaviors of DP-1 and DPCol in 4T1 cells and CT26 cells by CLSM and flow cytometry after labeling them with FITC. As shown in Fig. 3d, after incubation at 37 °C for 4 h, both the FITC-labeled DP-1 and DPCol can be internalized by the 4T1 cells. The mean fluorescence intensities inside the cells from the labeled DP-1 and DPCol are similar as measured by flow cytometry (Fig. 3e), suggesting that DP-1 and DPCol have comparable cellular uptake behaviors. Similar results are also observed in the case of CT26 cells (Fig. S7†), confirming that the conjugation of Col can not significantly change the cellular uptake behavior of multiarm copolymers.
3.5 Penetration in 4T1 multicellular spheroids (MCs)
Three-dimensional multicellular spheroids (MCs) have been frequently used to simulate tumor tissues in tumor penetration studies since the location and quantity of fluorescence materials in MCs can be precisely determined by CLSM. In our work, we compared the penetration behaviors of the FITC-labeled DP-1 and DPCol in the MCs prepared from 4T1 cells. From Fig. 4, it can be clearly seen that after 12 h incubation, the FITC-labeled DPCol penetrates much deeper in the MCs than the FITC-labeled DP-1, indicating that the modification with Col enhances significantly the penetration of the polymers in the MCs. This is likely associated with the DPCol-catalyzed degradation of collagen within MCs.51 | ||
| Fig. 4 Typical Z-stack images of the 4T1 MCs incubated with the FITC-labeled DP-1 and DPCol for 12 h. Scale bars = 100 μm. 2.5 D model images at the depth of 80 μm of MCs. | ||
3.6 Tumor penetration
To further assess the ability of the polymer-bound Col to catalyze the degradation of collagen in vivo, we injected intravenously DPCol into 4T1 tumor-bearing mice with saline and DP-1 as the negative controls. We measured the contents of type I collagen in the tumors by immunofluorescence staining and semi-quantitative analysis (Fig. 5a and b). As shown in Fig. 5a and b, the fluorescence signal intensities of the tumors treated with saline and DP-1 do not show a significant difference, in comparison, the signal intensity of the tumors treated with the DPCol group is much weaker. The mean fluorescence intensity in the DPCol-treated tumors is about 51% of that in the saline-treated tumor (Fig. 5b), signifying that DPCol can effectively catalyze the degradation of collagen and reduce the collagen contents in tumor tissues.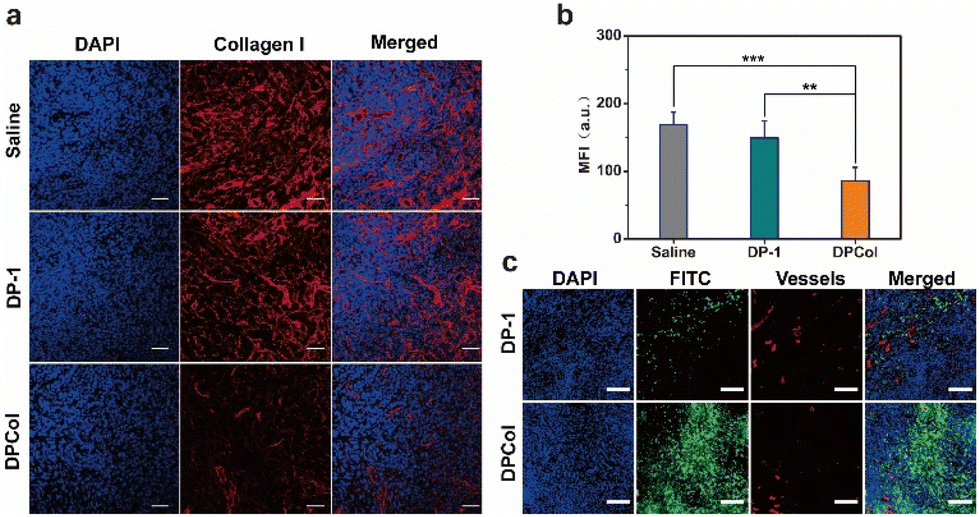 | ||
| Fig. 5 (a) Immunofluorescence collagen I staining images of the frozen sections of the 4T1 tumor tissues at 48 h after tail-vain injection of saline, DP-1, or DPCol. Cell nuclei were stained by DAPI (blue) and collagen I was stained by Alexa Fluor™ Plus 647 conjugated anti-collagen I antibody (red). Scale bars = 50 μm. (b) Quantitative analysis of collagen I levels in tumor tissues at 48 h after tail-vain injection of saline, DP-1, or DPCol (**P < 0.01, ***P < 0.001, n = 3). (c) CLSM images of the frozen sections of the 4T1 tumor tissues at 48 h after tail-vain injection of FITC-labeled DP-1 and DPCol, respectively. Scale bars = 100 μm. | ||
To investigate whether the degradation of collagen in tumors can enhance the permeability of the nanomedicines after extravasating from tumor vessels, we injected intravenously the FITC-labeled DP-1 and DPCol into 4T1 tumor-bearing mice and assessed their distributions in tumors by immunofluorescence staining. As shown in Fig. 5c, the red fluorescence generated by the CD31 antibodies indicates the location of blood vessels, and the green fluorescence indicates the position of the FITC-labeled samples. At 48 h post-injection (p.i.), it can be observed that DPCol distributes much more uniformly in the tumor than DP-1, signifying that the collagen reduction induced by Col is indeed helpful for the tumor penetration of nanomedicines.
3.7 Biodistribution
We evaluated the in vivo drug delivery properties of DPD-1 and DPDCol by determining the DOX concentrations in different tissues after intravenous injection with free DOX as control. The samples were injected into 4T1 tumor-bearing mice via tail vein at a dose equivalent to 4 mg kg−1 DOX. At predetermined time points, the mice were euthanized, different organs and tissues including the blood, tumor, heart, liver, spleen, lung, and kidney were collected, and DOX was extracted from the tissue homogenates under acid conditions. The DOX concentrations in different tissues were determined via fluorescence quantification and are presented as the percentage of injected dose per gram (%ID g−1) of wet tissues (the data for blood are presented as %ID mL−1) (Fig. 6). The blood concentration-time data for both DPD-1 and DPDCol exhibit a good fit to the two-compartment model (Fig. 6b). Accordingly, the elimination half-lives for DPD-1 and DPDCol are calculated to be about 34.6 and 36.6 h, respectively, significantly longer than that of free DOX (0.6 h). As shown in Fig. 6g, compared to free DOX, both DPD-1 and DPDCol exhibit pronounced tumor accumulation and reach their peak DOX concentrations of 3.6 %ID g−1 and 4.6 %ID g−1 in the tumor at 4 h, respectively. In contrast, free DOX reaches its peak concentration at 1 h, with a value of 1.2 %ID g−1. Furthermore, we assessed their tumor enrichment abilities by calculating the tumor area-under-the-curve (AUC) based on DOX concentration over time. (Fig. 6h). The results indicate that DPDCol exhibits a greater tendency for tumor enrichment than DPD-1 and free DOX, as reflected by the tumor AUC values of approximately 124.0 %ID h g−1, 94.0 %ID h g−1, and 27.6 %ID h g−1, respectively.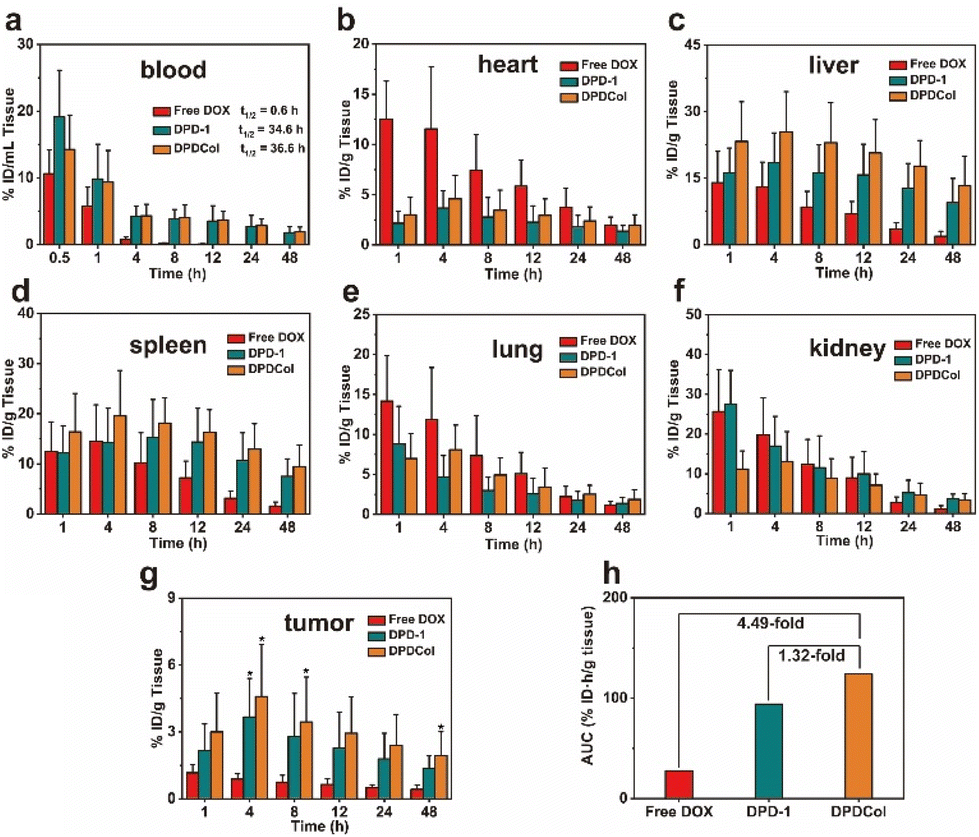 | ||
| Fig. 6 DOX concentrations in the blood (a), heart (b), liver (c), spleen (d), lung (e), kidney (f), and tumor (g) of the 4T1 tumor-bearing mice at different time points after tail-vein injection of free DOX, DPD-1, and DPDCol (*P < 0.05 compared with the free DOX group, n = 3). Data are presented as mean ± SD (n = 3). (h) AUC of DOX accumulation in the tumor for free DOX, DPD-1, and DPDCol. | ||
The maximum DOX concentrations and their corresponding appearance time points in various tissues (heart, liver, spleen, lungs, kidneys, and tumor) for both DPD-1 and DPDCol are presented in Table S1.† It can be found that compared to DPD-1, DPDCol exhibits higher uptake in the liver and spleen, comparable uptake in the lungs and heart, and lower uptake in the kidneys, suggesting that DPDCol is more readily captured by the mononuclear phagocyte system (MPS), while DPD-1, due to its smaller size, is more prone to accumulate in the kidney.
3.8 Antitumor efficacy and biosafety evaluation
We further evaluated the antitumor performance of DPD-1 and DPDCol by using 4T1 tumor-bearing mice as the model animals. The negative control groups were treated with DP-1, DPCol, and saline, and the positive control group was treated with DOX. After a single intravenous injection on the first day, the tumor volumes were measured every other day over the next 15 days (Fig. 7a). As shown in Fig. 7a, at all the defined time points, there are no significant differences in the relative tumor volumes among the DP-1, DPCol, and saline negative control groups, indicating that DP-1 and DPCol do not possess antitumor activity. For the three DOX formulations DPD-1, DPDCol, and free DOX, both DPD-1 and DPDCol exhibit higher inhibitory effects on tumor growth than free DOX. On the 15th day, for the DOX, DPD-1, and DPDCol groups, the relative tumor volumes are 13.9, 6.1, and 1.9, and the tumor growth inhibition (TGI) values are 43.5%, 73.8%, and 91.8%, respectively, indicating that the order of antitumor activity is DPDCol > DPD-1 > free DOX. The necrosis of the tumor cells induced by the therapeutic agents was evaluated on the 7th day after the treatments. The images of the tumor slices stained by hematoxylin and eosin (H&E) are shown in Fig. S8.† As can be seen, the DPDCol group shows the highest necrosis rate, comfirming the superior treatment efficacy of DPDCol. We also monitored the body weights of all the test mice (Fig. S9†). When compared to the saline group, there are no significant differences in body weights of the DPD-1, DPDCol, DP-1, and DPCol groups, suggesting that the drug-free and drug-loaded polymers do not cause significant acute toxicity to the experimental mice. The survival time of the test mice is shown in Fig. 7b. All the mice in the negative control and DOX groups died within 55 days after the treatments. By contrast, on the 60th day, there are still 20% and 80% of the mice surviving in the DPD-1 and DPDCol groups, respectively, signifying that the high tumor permeability of DPDCol can significantly augment its therapy efficacy.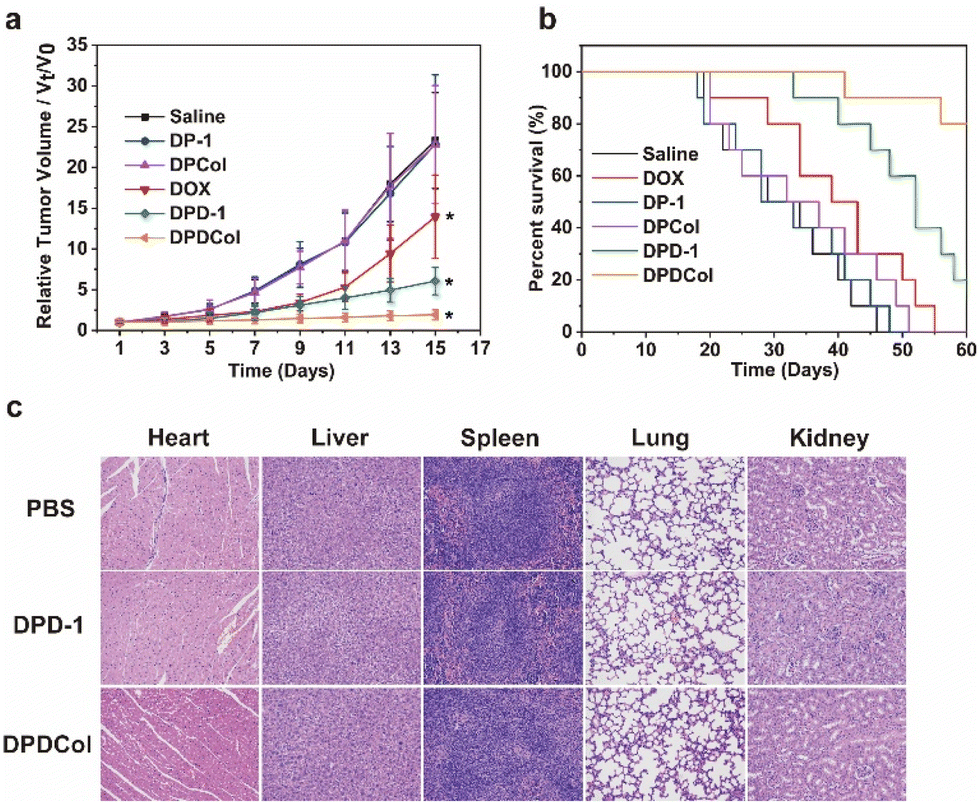 | ||
| Fig. 7 Tumor growth curves (a) and survival rates (b) of the 4T1 tumor-bearing mice after different treatments. Data are presented as mean ± SD (n = 10). *P < 0.05 (DOX versus DPDCol from day 5, DPD-1 versus DPDCol from day 7). (c) H&E stained tissues of different major organs obtained from healthy BALB/c mice administrated with PBS, DPD-1, and DPDCol on the 7th day (all tissues: ×200). | ||
We assessed the biosafety of DPD-1 and DPDCol by histological analysis after tail-vein injection into healthy mice with the PBS-treated mice as the negative control. The histological slices stained by H&E show that the mice in the DPD-1 and DPDCol groups do not have significant pathological changes in major organs compared to the control group, signifying the negligible toxicity of DPD-1 and DPDCol (Fig. 7c). Meanwhile, as shown in Fig. S10 and S11,† the hematological and biochemical parameters of the DPD-1 and DPDCol treatment groups are within normal ranges, indicating that neither of them induced inflammation or impaired liver and kidney function in mice.
4. Conclusions
We synthesized a G3 PAMAM-cored multiarm copolymer and modified the polymer with Col to enhance its tumor penetration. Each arm of the polymer was a diblock copolymer of PGA-b-PCB, in which the PGA block was used to link DOX through the pH-sensitive acylhydrazone linkage, and the zwitterionic PCB block provided desired water solubility and anti-biofouling capability. The Col was conjugated to the ends of the arms via the thiol-maleimide reaction. The degradation effect of the polymer-bound Col on the collagen in the tumor ECM augmented significantly the tumor penetration of the drug-loaded polymers and consequently high antitumor efficacy was attained.Author contributions
X. Zhen, X. Jiang, W. Wu and B. Yu conceived the idea and designed the experiments. B. Yu, W. Wang, Y. Zhang, Y. Sun, C. Li and Q. Liu conducted the experiments. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All the animal experiments were performed in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Ethical and Welfare Committee of Nanjing University (approval number: IACUC-D2303055). This research was supported by the National Natural Science Foundation of China (No. 52173129 and 51973092) and funded by Tianjin Key Medical Discipline (Specialty) Construction Project.References
- T. Li, D. Wang, Z. Guo, L. Lin, M. Meng, C. Liu, K. Hao, X. Pang, H. Tian and X. Chen, Biomater. Sci., 2023, 11, 6524–6536 RSC.
- J. Wang, Y. Li and G. Nie, Nat. Rev. Mater., 2021, 6, 766–783 CrossRef CAS.
- Y. Wang, Z. Wang, C. Xu, H. Tian and X. Chen, Biomaterials, 2019, 197, 284–293 CrossRef CAS PubMed.
- Z. Zhang, Q. Deng, C. Xiao, Z. Li and X. Yang, Acc. Chem. Res., 2022, 55, 526–536 CrossRef CAS PubMed.
- Y. Cao, W. Song and X. Chen, Biomater. Sci., 2023, 11, 2620–2638 RSC.
- P. Zhang, Y. Zhang, X. Ding, C. Xiao and X. Chen, Biomater. Sci., 2020, 8, 3052–3062 RSC.
- S. Zhou, C. Li, Y. Yuan, L. Jiang, W. Chen and X. Jiang, Biomater. Sci., 2022, 10, 7032–7041 RSC.
- A. S. Hoffman and J. J. Lai, Adv. Drug Delivery Rev., 2020, 158, 2–3 CrossRef CAS PubMed.
- X. Zhen, R. Qu, W. Chen, W. Wu and X. Jiang, Biomater. Sci., 2021, 9, 285–300 RSC.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS.
- M. Souri, M. Soltani, F. M. Kashkooli and M. K. Shahvandi, J. Controlled Release, 2022, 341, 227–246 CrossRef CAS.
- W. Ni, J. Wu, Y. Feng, Y. Hu, H. Liu, J. Chen, F. Chen and H. Tian, Biomater. Sci., 2022, 10, 5596–5607 RSC.
- Y. Liu, J. Zhou, Q. Li, L. Li, Y. Jia, F. Geng, J. Zhou and T. Yin, Adv. Drug Delivery Rev., 2021, 172, 80–103 CrossRef CAS.
- Z. Guo, Y. Hu, M. Zhao, K. Hao, P. He, H. Tian, X. Chen and M. Chen, Nano Lett., 2021, 21, 3721–3730 CrossRef CAS PubMed.
- Z. Li, X. Shan, Z. Chen, N. Gao, W. Zeng, X. Zeng and L. Mei, Adv. Sci., 2020, 8, 2002589 CrossRef PubMed.
- G. Cao, W. He, Y. Fan and X. Li, Biomater. Sci., 2021, 9, 7895–7910 RSC.
- J. X. Ding, J. J. Chen, L. Q. Gao, Z. Y. Jiang, Y. Zhang, M. Q. Li, Q. C. Xiao, S. S. Lee and X. S. Chen, Nano Today, 2019, 29, 100800–100818 CrossRef CAS.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS.
- S. M. S. Shahriar, N. S. Polavoram, S. M. Andrabi, Y. Su, D. Lee, H. Q. Tran, S. J. Schindler and J. Xie, BMEMat, 2023, 1, e12046 CrossRef.
- J. Song, L. Li, L. Fang, E. Zhang, Y. Zhang, Z. Zhang, P. Vangari, Y. Huang, F. Tian, Y. Zhao, W. Chen and J. Xue, BMEMat, 2023, 1, e12046 CrossRef.
- S. Amorim, D. S. da Costa, I. Pashkuleva, C. A. Reis, R. L. Reis and R. A. Pires, Biomater. Sci., 2021, 9, 252–260 RSC.
- J. Winkler, A. Abisoye-Ogunniyan, K. J. Metcalf and Z. Werb, Nat. Commun., 2020, 11, 5120 CrossRef CAS PubMed.
- T. Xue, C. Xu, Y. Wang, Y. Wang, H. Tian and Y. Zhang, Biomater. Sci., 2019, 7, 4615–4623 RSC.
- T. Zuo, J. Zhang, J. Yang, R. Xu, Z. Hu, Z. Wang, H. Deng and Q. Shen, Biomater. Sci., 2021, 9, 1872–1885 RSC.
- R. Baghban, L. Roshangar, R. Jahanban-Esfahlan, K. Seidi, A. Ebrahimi-Kalan, M. Jaymand, S. Kolahian, T. Javaheri and P. Zare, Cell Commun. Signaling, 2020, 18, 59 CrossRef.
- X. He, Y. Yang, L. Li, P. Zhang, H. Guo, N. Liu, X. Yang and F. Xu, Drug Discovery Today, 2020, 25, 1727–1734 CrossRef CAS PubMed.
- Y. Niu, J. Zhu, Y. Li, H. Shi, Y. Gong, R. Li, Q. Huo, T. Ma and Y. Liu, J. Controlled Release, 2018, 277, 35–47 CrossRef CAS PubMed.
- X.-Y. Yang, J.-G. Zhang, Q.-M. Zhou, J.-N. Yu, Y.-F. Lu, X.-J. Wang, J.-P. Zhou, X.-F. Ding, Y.-Z. Du and R.-S. Yu, J. Nanobiotechnol., 2022, 20, 524 CrossRef CAS.
- A. V. V. V. R. Kiran, G. K. Kumari, P. T. Krishnamurthy and R. R. Khaydarov, Biomater. Sci., 2021, 9, 7667–7704 RSC.
- K. Fernando, L. G. Kwang, J. T. C. Lim and E. L. S. Fong, Biomater. Sci., 2021, 9, 2362–2383 RSC.
- A. Zinger, L. Koren, O. Adir, M. Poley, M. Alyan, Z. Yaari, N. Noor, N. Krinsky, A. Simon, H. Gibori, M. Krayem, Y. Mumblat, S. Kasten, S. Ofir, E. Fridman, N. Milman, M. M. Lubtow, L. Liba, J. Shklover, J. Shainsky-Roitman, Y. Binenbaum, D. Hershkovitz, Z. Gil, T. Dvir, R. Luxenhofer, R. Satchi-Fainaro and A. Schroeder, ACS Nano, 2019, 13, 11008–11021 CrossRef CAS.
- S. Yin, Y. Gao, Y. Zhang, J. Xu, J. Zhu, F. Zhou, X. Gu, G. Wang and J. Li, ACS Appl. Mater. Interfaces, 2020, 12, 18273–18291 CrossRef CAS.
- J. Li, C. Xie, J. Huang, Y. Jiang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3995–3998 CrossRef CAS.
- J. Son, S. Parveen, D. MacPherson, Y. Marciano, R. H. Huang and R. V. Ulijn, Biomater. Sci., 2023, 11, 6457–6479 RSC.
- J. Wang, Q. Wu, Y. Wang, L. Xiang, J. Feng, Z. Zhou, Q. Fu and L. Zhang, Nanoscale, 2021, 13, 9402–9414 RSC.
- S. Ray, H. Adelnia and H. T. Ta, Biomater. Sci., 2021, 9, 5714–5731 RSC.
- K. Dey, E. Roca, G. Ramorino and L. Sartore, Biomater. Sci., 2020, 8, 7033–7081 RSC.
- P. Cioni, E. Gabellieri, B. Campanini, S. Bettati and S. Raboni, Curr. Med. Chem., 2022, 29, 411–452 CrossRef CAS PubMed.
- H. Yao, X. Guo, H. Zhou, J. Ren, Y. Li, S. Duan, X. Gong and B. Du, ACS Appl. Mater. Interfaces, 2020, 12, 20214–20227 CrossRef CAS PubMed.
- J. Luo, Z. Zhang, Y. Zeng, Y. Dong and L. Ma, Carbohydr. Polym., 2021, 263, 117964 CrossRef CAS PubMed.
- Q. Q. Fan, C. L. Zhang, J. B. Qiao, P. F. Cui, L. Xing, Y. K. Oh and H. L. Jiang, Biomaterials, 2020, 230, 119616 CrossRef CAS PubMed.
- M.-Y. Yang, Y.-J. Lin, M.-M. Han, Y.-Y. Bi, X.-Y. He, L. Xing, J.-H. Jeong, T.-J. Zhou and H.-L. Jiang, J. Controlled Release, 2022, 351, 623–637 CrossRef CAS PubMed.
- T. Yin, H. Chen, A. Ma, H. Pan, Z. Chen, X. Tang, G. Huang, J. Liao, B. Zhang, M. Zheng and L. Cai, Biomaterials, 2023, 293, 121992 CrossRef CAS PubMed.
- M. Ding, Y. Zhang, J. Li and K. Pu, Nano Converg., 2022, 9, 7 CrossRef CAS.
- G. Chen, Y. Wang, R. Xie and S. Gong, Adv. Drug Delivery Rev., 2018, 130, 58–72 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, J. Zhao, C. Li, W. Wu and X. Jiang, Biomater. Sci., 2019, 7, 5124–5131 RSC.
- C. Yin, P. Xiao, M. Liang, J. Li, Y. Sun, X. Jiang and W. Wu, Biomater. Sci., 2022, 10, 3236–3244 RSC.
- A. Scomparin, H. F. Florindo, G. Tiram, E. L. Ferguson and R. Satchi-Fainaro, Adv. Drug Delivery Rev., 2017, 118, 52–64 CrossRef CAS.
- B. Yu, Y. Sun, P. Xiao, C. Li, K. Yuan, L. Zhang, X. Jiang and W. Wu, ACS Appl. Bio Mater., 2023, 6, 2426–2434 CrossRef CAS PubMed.
- G. Dong, M. Bao, X. Xie, S. Jia, W. Hu and X. Xu, Angew. Chem., Int. Ed., 2021, 60, 1992–1999 CrossRef CAS PubMed.
- T. T. Goodman, P. L. Olive and S. H. Pun, Int. J. Nanomed., 2007, 2, 265–274 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm02123h |
| This journal is © The Royal Society of Chemistry 2024 |