
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of multi-vinyl taxogen dimensions on high molecular weight soluble polymer synthesis using transfer-dominated branching radical telomerisation†
Oliver B.
Penrhyn-Lowe
,
Sean
Flynn
,
Savannah R.
Cassin
,
Samuel
Mckeating
,
Sarah
Lomas
,
Stephen
Wright
,
Pierre
Chambon
and
Steve P.
Rannard
 *
*
Materials Innovation Factory & Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: srannard@liv.ac.uk
First published on 22nd October 2021
Abstract
Complex macromolecular architectures offer innovation opportunities through the formation of new polymers with novel physical and chemical behaviour. Optimally existing commercial feedstocks are used as the introduction of new small molecule chemistries may extend regulatory requirements at high expense. In a highly orthogonal synthesis strategy, we recently utilised telomerisation approaches, conventionally targeting small molecule dimerisation, to generate high molecular weight branched polymers. Transfer-dominated Branching Radical Telomerisation (TBRT) employs radical reactions, but yields branched polymers with extended backbones resembling those conventionally formed via step-growth polymerisation. Here we examine the impact of taxogen dimensions on the outcomes of TBRT using multi-vinyl taxogens ethylene glycol dimethacrylate (EGDMA), 1,6-hexanediol dimethacrylate (HDMA) and 1,12-dodecanediol dimethacrylate (LDMA). Marked differences in gelation behaviours were correlated to the dimensions of the taxogen, notably due to the shifting balance of intermolecular and intramolecular branching processes occuring during TBRT.
Introduction
Directing polymer architecture has enabled numerous advances in materials chemistry and the creation of novel technologies ranging from block copolymers as surface active materials or elastomeric materials,1,2 to statistical copolymers used as ionomeric coatings,3 and branched polymers as DNA transfection agents.4 The academic and industrial synthesis of high molecular weight branched polymers has intrigued chemists for many decades and has inspired a range of elegant polymerisation technologies.5–7 Generally speaking, branched polymers are synthesised via a relatively small number of strategies:8,9 (a) step-growth polymerisations, utilising ABn (n ≥ 2) monomers (including reactions employing Bn, A2 + Bn monomer combinations and A–B monomers) are used to form ideally branched or statistical hyperbranched architectures with linking chemistries such as esters, amides, carbonates and urethanes;10 (b) graft, star and comb copolymer architectures containing predominantly C–C backbones may be formed via the inclusion of unsaturated macromonomers;11 (c) multiple distinct chain-growth polymerisation functionalities may be combined into single bespoke structures, such as “inimers” and “transmers”, to offer branching akin to that of AB2 monomers;12 and (d) introduction of low concentrations of multi-vinyl monomers into linear chain-growth copolymerisations forms branched structures of conjoined primary linear chains, again containing predominantly C–C backbones.7,13,14Control of direct multi-vinyl monomer homopolymerisation, or the inclusion of multi-vinyl monomers at relatively high concentrations in copolymerisations with monovinyl monomers, has proved elusive and high vinyl group conversion is hard to achieve. Gelation, caused by extended interchain crosslinking, occurs even at very low vinyl group conversion when employing multi-vinyl monomers, leading to insoluble gel fractions of nominally ‘infinite molecular weight’.13,15 This was first discussed in detail and predicted by Flory–Stockmayer theory, which utilised a theoretical ideal scenario to determine that the gel point is associated with the creation of ≥1 multi-vinyl monomer derived crosslink per linear primary chain.16,17 In recent years, several researchers have achieved multi-vinyl monomer homopolymerisation to relatively high conversion (reaction of <70% of available vinyl functionality) by employing deactivation-enhanced atom transfer radical polymerisation (DE-ATRP) or reversible addition–fragmentation chain transfer (RAFT) polymerisation.18,19 These novel approaches led to gelled materials at high vinyl conversion as also predicted by modifications to the Flory–Stockmayer assumptions and detailed Monte Carlo simulation of conventional reversible-deactivation radical polymerisation (RDRP) chemistries.20,21
As an orthogonal chemistry, telomerisation has developed from its initial research focus of forming very short vinyl polymer chains, by limiting propagation within conventional free radical polymerisation (FRP), to the formation of dimers and trimers from sustainable, often biosourced, unsaturated starting materials. Telomerisation terminology was introduced >70 years ago,22–24 and a telomer is defined as a chain of reacted vinyl monomers denoted by the general formula X(Q)nY, where n is a number average value <5, and X–Y is a telogen that limits the propagation of an unsaturated taxogen, denoted as Q. Telogen chemistry has been reviewed several times over the last 65 years24–26 and encompasses numerous functional telogen reagents including alcohols, water, amines, CO2, CO and various mixtures.27 Thiols are convenient telogens for free radical telomerisation reactions as the RS–H bond is highly susceptible to free radical chain transfer; under FRP conditions thiols are classed as chain transfer agents.
In a very recent report, we introduced the concept of TBRT that applies free radical telomerisation conditions to the reaction of multi-vinyl taxogens (MVTs; termed multi-vinyl monomers under polymerisation conditions) with thiol-based telogens to form high molecular weight branched polymers at complete vinyl conversion and in the absence of gelation (Fig. 1).28 TBRT conditions utilise an excess of telogen relative to MVT, as is common in conventional telomerisation, and ensures that the number average telomer length is <2 vinyl groups. This dramatically limits chain propagation and the resultant primary telomers are very short; however, the distribution of telomers is conjoined via the MVT linking chemistry and very high molecular weight structures are thus readily available.
 | ||
| Fig. 1 Comparison of telomer formation via (A) the thiol-mediated free radical telomerisation of monovinyl taxogens versus (B) the TBRT of multi-vinyl taxogens to form branched polymer structures. | ||
The chemical structure and molecular dimensions of different MVTs are expected to have an impact on the course of a TBRT reaction. Our first reports have utilised ethylene glycol dimethacrylate (EGDMA) as the taxogen and 1-dodecanethiol (DDT) as the telogen,28,29 although TBRT is applicable to a large variety of MVT and telogen structures. Systematic studies are therefore required to identify the key parameters that define and control TBRT products, in order to provide clear guidance for future branched polymer design and development. Here, MVT structure has been selected as the only chemical variable within TBRT reactions using EGDMA, 1,6-hexanediol dimethacrylate (HDMA) and 1,12-dodecanediol dimethacrylate (lauryl dimethacrylate, LDMA). We have normalised other chemical parameters such as telogen selection (DDT), reaction solvent and free radical source to eliminate other contributions to the variation in polymerisation behaviour. Although full conversion of vinyl groups and soluble high molecular weight branched polymers were achievable with all MVTs, the aliphatic chain length within the MVT led to very different taxogen/telogen feedstock ratios that may be used within successful TBRT reactions to avoid gelation. Detailed investigation suggests that although the rate of vinyl group consumption during telomerisation does vary, different chain transfer rates between the telogen and each taxogen appears to be the dominant reaction. The resulting distribution of telomer chain lengths, however, does not appear to determine the gelation behaviour alone and we propose a ‘radical lifetime sphere’ model to describe variations in intermolecular branching efficiency due to differing MVT dimensions.
Results and discussion
Comparison of TBRT using varying MVTs at 50 wt% solids
Following our initial reports, TBRTs of EGDMA and LDMA were conducted using DDT as the telogen in ethyl acetate at a concentration of 50 weight percent (wt%) solids (Scheme 1). 2,2′-Azobis(2-methylpropionitrile) (AIBN) was used as a source of radicals at 1.5 mole percent (mol%) equivalents with respect to MVT vinyl bonds. A taxogen/telogen feedstock ratio ([MVT]0/[DDT]0) of 0.80 was selected to mirror earlier reports; the reaction medium was analysed by 1H NMR spectroscopy to confirm this ratio prior to initiation (ESI Fig. S1†). The telogen is a key structural component of the products of TBRT and nominal repeat units within the complex branched polymer architecture ideally contains a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MVT/telogen adduct.
:1 MVT/telogen adduct.
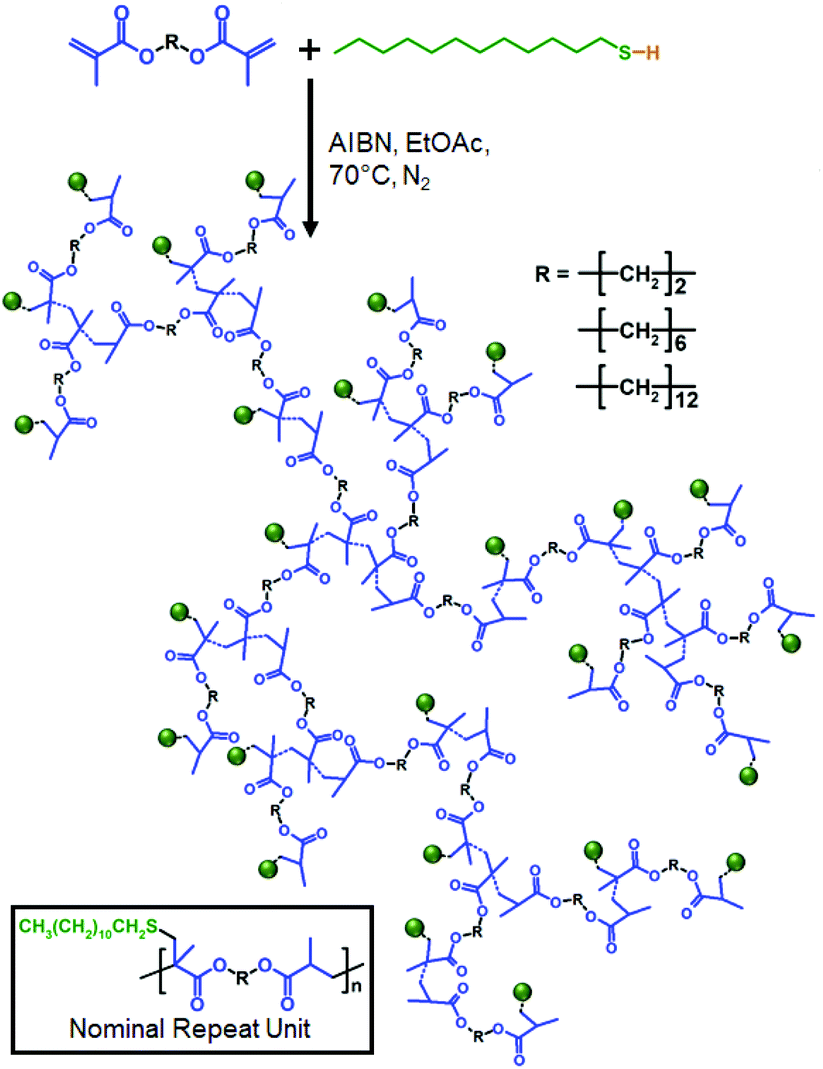 | ||
| Scheme 1 TBRT synthesis of branched polymers using EGDMA, HDMA and LDMA with DDT at fixed solvent concentration. | ||
As expected, soluble branched polymer was obtained from the TBRT of EGDMA; however, insoluble crosslinked gel was observed after only 1.5 hours when using LDMA under these conditions. A wide range of [MVT]0/[DDT]0 ratios was subsequently employed to identify the limiting [MVT]0/[DDT]0 gel point ratios for each MVT. TBRT requires a taxogen/telogen ratio that ensures transfer to telogen dominates the reaction mechanism and limits propagation to <2 vinyl groups. Limiting [MVT]0/[DDT]0 gel point ratios were determined as 0.88 and 0.64 for EGDMA and LDMA respectively (ESI Table T1†). 1H NMR spectroscopy of each crude reaction confirmed >99% vinyl group conversion (ESI Fig. S2† (EGDMA example)), as indicated by no detectable vinyl group resonances. Triple-detection size exclusion chromatography (TD-SEC) of the purified polymers showed high molecular weight products for both p(DDT1.00-EGDMA0.85) and p(DDT1.00-LDMA0.60) (weight average molecular weight (Mw) values = 1208 kg mol−1 and 800 kg mol−1 respectively), accompanied by large dispersities (Đ) of 88.64 and 47.70. Broad molecular weight distributions are a characteristic of TBRT polymers synthesised close to the limiting [MVT]0/[DDT]0 gel point ratios. Mark–Houwink analyses gave alpha exponents (α) ranging between 0.297–0.444, strongly indicating compact branched structures in all reactions.
Comparison of TBRT at fixed MVT concentration
The variation in observed outcomes when undertaking TBRT at a fixed 50 wt% solids was surprising given the use of a single telogen (DDT) and methacrylic MVTs (EGDMA and LDMA). However, variation of [MVT]0/[DDT]0 ratios was conducted using a fixed mass of MVT (1 g) and varying DDT, necessitating different solvent volumes to achieve the target 50 wt% solids. The significant molecular weight difference between EGDMA and LDMA will impact the moles of vinyl groups at fixed MVT mass and generate significant vinyl group concentration differences when adjusting solvent.To provide a clearer comparison and standardise vinyl group concentration, further reactions were conducted at a fixed molar concentration of MVT (10 mmol MVT in 51.07 mmol of ethyl acetate) with thiol varying to achieve different [MVT]0/[DDT]0 ratios; this approach inevitably creates differing solids contents for each reaction but the number of reacting functional groups is fixed for each [MVT]0/[DDT]0 ratio. A further MVT, HDMA, was added to the investigation to enable trends relating to MVT dimensions to be confidently determined. TBRT reactions were conducted for each of the EGDMA, HDMA and LDMA MVTs, Scheme 1, with the [MVT]0/[DDT]0 ratio systematically increased until the limiting gel point ratios were observed. In all instances, vinyl conversions exceeded 99% as analysed by 1H NMR spectroscopy; TD-SEC analysis indicated dense branched structures through Mark–Houwink α values ranging between 0.256–0.497 (Table 1).
| MVT | 1H NMR (CDCl3) | TD-SEC (THF/TEA)d | ||||||
|---|---|---|---|---|---|---|---|---|
| Reaction [MVT]0/[DDT]0a | Conversionb (%) | Polymer [MVT]f/[DDT]fc | M w (g mol−1) | M n (g mol−1) | Đ | α | dn/dc | |
| a Determined for sample taken at t = 0 (ESI Fig. S4†). b Determined for crude sample taken at t = 24 hours, referenced against sample analysed at t = 0 (ESI Fig. S5†). c Determined for sample after purification and drying in vacuo (ESI Fig. S6†). d Determined by TD-SEC using a 2% v/v TEA/THF eluent system. e Sample gave strong resistance to filtration. | ||||||||
| EGDMA | 0.91 | Gel | Gel | — | — | — | — | — |
| EGDMA | 0.88 | Microgele | Microgele | — | — | — | — | — |
| EGDMA | 0.85 | >99 | 1.03 | 1019000 |
6530 | 156.01 | 0.497 | 0.0998 |
| EGDMA | 0.83 | >99 | 1.03 | 510445 |
7100 | 71.89 | 0.323 | 0.0941 |
| EGDMA | 0.80 | >99 | 1.00 | 287515 |
3670 | 77.95 | 0.333 | 0.0905 |
| EGDMA | 0.75 | >99 | 1.01 | 134475 |
3060 | 43.92 | 0.398 | 0.0979 |
| EGDMA | 0.70 | >99 | 1.05 | 75420 |
3230 | 23.36 | 0.306 | 0.0910 |
| EGDMA | 0.65 | >99 | 0.99 | 44325 |
2750 | 16.10 | 0.264 | 0.0959 |
| EGDMA | 0.60 | >99 | 1.04 | 30145 |
2450 | 12.31 | 0.256 | 0.0880 |
| HDMA | 0.75 | Gel | Gel | — | — | — | — | — |
| HDMA | 0.73 | Microgele | Microgele | — | — | — | — | — |
| HDMA | 0.70 | >99 | 0.99 | 720010 |
5770 | 124.94 | 0.366 | 0.0877 |
| HDMA | 0.68 | >99 | 1.00 | 215015 |
3930 | 54.75 | 0.347 | 0.0889 |
| HDMA | 0.65 | >99 | 0.98 | 100565 |
2450 | 41.02 | 0.345 | 0.0868 |
| HDMA | 0.59 | >99 | 0.96 | 39415 |
1935 | 20.39 | 0.325 | 0.0876 |
| HDMA | 0.55 | >99 | 0.91 | 21150 |
2970 | 7.12 | 0.321 | 0.0859 |
| HDMA | 0.49 | >99 | 0.89 | 11875 |
1470 | 8.08 | 0.300 | 0.0845 |
| LDMA | 0.60 | Gel | Gel | — | — | — | — | — |
| LDMA | 0.58 | Microgele | Microgele | — | — | — | — | — |
| LDMA | 0.54 | >99 | 0.82 | 275065 |
3225 | 84.89 | 0.373 | 0.0819 |
| LDMA | 0.53 | >99 | 0.86 | 123590 |
2500 | 49.41 | 0.358 | 0.0843 |
| LDMA | 0.49 | >99 | 0.81 | 37915 |
1720 | 22.01 | 0.361 | 0.0822 |
| LDMA | 0.45 | >99 | 0.76 | 15110 |
1960 | 7.71 | 0.363 | 0.0809 |
| LDMA | 0.40 | >99 | 0.74 | 8300 | 1895 | 4.38 | 0.359 | 0.0812 |
| LDMA | 0.35 | >99 | 0.69 | 5150 | 1655 | 3.11 | 0.363 | 0.0803 |
The limiting [MVT]0/[DDT]0 gel point ratios of EGDMA, HDMA and LDMA reactions were found to occur at 0.88, 0.73 and 0.58 respectively under these conditions. Note: these values are different to those obtained when using a fixed 50 wt% reaction mixture. Again, this represents a considerable variation for MVTs with chemically comparable methacrylate functionality. As with the reactions conducted at a fixed 50 wt% solids, dramatic increases in Mw were observed for ratios closely approaching the respective limiting gel points (Fig. 2).
 | ||
| Fig. 2 Development of Mw with varying [MVT]0/[DDT]0 for branched polymers derived from EGDMA (filled red circles), HDMA (open blue diamonds) and LDMA (open black circles) generated via TBRT. Limiting gel point ratios are highlighted by vertical lines for each multi-vinyl telogen. | ||
The rapid increase in Mw is indicative of extended macromolecular structure formation through high levels of intermolecular branching. Decreasing the [MVT]0/[DDT]0 ratios led to corresponding decreases in Mw, which plateaued for ratios markedly lower than the respective limiting gel point values. Interestingly, the increase of Mw for the EGDMA series of TBRT polymerisations rises smoothly towards the limiting gel point ratio; however, more abrupt increases were observed for HDMA and LDMA polymerisations under these controlled conditions. The maximum achievable Mw for each MVT also varied significantly, with the highest Mw values achieved for EGDMA and decreasing systematically with MVT molar mass.
Investigating the impact of molecular structure on telomer length distributions
The observations above suggest an influence of the MVT molecular dimensions on the course of the TBRT reaction. Alternatively, the decreasing trend in limiting [MVT]0/[DDT]0 gel point ratios with increasing MVT molar mass may also be viewed as indicating a variation in reactivity of the vinyl functionality within each MVT as each reaction series was conducted at fixed vinyl group concentration. The limiting gel point ratios represent the reaction conditions where the telomer distribution has reached a DPn ≥ 2 units and suggest that LDMA telomers are less regulated by the transfer dominated reaction mechanism than EGDMA, with HDMA being an intermediate case. The propagation of the MVTs would, therefore, appear to follow a trend of LDMA > HDMA > EGDMA. The apparent order may be impacted by the varying aliphatic chain length of each MVT, which might be influencing propagation through inductive and/or steric effects to provide a combination of enhanced reactivity and greater vinyl group accessibility. Importantly, homogeneous reaction mixtures were observed for all TBRT reactions, with no signs of polymerisation-induced phase separation during the reactions that may also impact reactivity.30,31Variation of alkyl substituents is known to influence the propagation rate of monofunctional monomers under FRP conditions; the common consensus rationalises higher propagation rates from increasing pendant length due to inductive effects.32,33 To establish a relative rate of propagation for the EGDMA, HDMA and LDMA MVTs, a series of model kinetic FRP experiments using analogous monofunctional methacrylates (methyl methacrylate (MMA), hexyl methacrylate (HMA) and lauryl methacrylate (LMA)) was conducted at 70 °C using AIBN (1.5 mol% equivalent) and ethyl acetate (50 wt% with respect to monomer). Samples were taken until the homogeneous reactions became too viscous for sampling (2–3 hours); vinyl conversions were estimated using 1H NMR spectroscopy and molecular weight data was obtained by TD-SEC (ESI Table T2 and Fig. S7, 8†). Semilogarithmic analysis of the vinyl conversion with time demonstrated comparable rates of propagation for LMA and HMA, but a markedly slower rate for MMA (Fig. 3a). This suggests that, under these conditions, inductive effects are important for short pendant chains but have little additional impact above a critical alkyl chain length.
 | ||
| Fig. 3 Analyses of various radical reactions (A) semilogarithmic analysis of conventional FRP using MMA, HMA and LMA. (B) Evolution of vinyl conversion during TBRT kinetic studies of EGDMA, HDMA and LDMA at the highest respective [DVM]0/[DDT]0 ratios without triggering gelation. (C) Mayo Plot for the determination of chain transfer coefficients, CT, during linear polymerisations of MMA, HMA and LMA with DDT. | ||
It is reasonable to assume that this effect would translate to the TBRT reactions and that LDMA and HDMA would also telomerise at similar rates but significantly faster than EGDMA. Kinetic studies of EGDMA, HDMA and LDMA TBRT reactions were therefore conducted (ESI Table T3 and Fig. S9†) at [MVT]0/[DDT]0 ratios of 0.85, 0.70 and 0.54 in order to obtain sufficiently high molecular weight polymers for comparable analyses. These chosen ratios represented the highest respective values achievable without triggering gelation (Table 1) under conditions with fixed moles of MVT and solvent. The rate of vinyl consumption was found to increase in the order EGDMA < HDMA < LDMA (Fig. 3b). This is demonstrated clearly by comparing the vinyl conversions after 1 hour where >61% of vinyl groups had been consumed during the formation of p(DDT1.00-LDMA0.54) compared to 43% and 32% for the TBRT of p(DDT1.00-HDMA0.70) and p(DDT1.00-EGDMA0.85) respectively. Moreover, the TBRT of LDMA achieved >99% vinyl conversion after only 2.5 hours, representing a significant rate enhancement over the HDMA and EGDMA polymerisations, which achieved the same conversion at >4 hours. Although HDMA appears to closely mirror EGDMA under TBRT conditions, LDMA was again shown to react significantly faster, and the general rate trend observed for monofunctional monomers was maintained for the MVTs. This study suggests that the Mw trend with respect to varying [MVT]0/[DDT]0 ratios for each MVT (Fig. 2) may be rationalised purely by an increasing DPn within the telomer distribution under identical ratios, following the trend of EGDMA < HDMA < LDMA.
It is important to note that the kinetic analyses described above (Fig. 3A & B) do not provide unequivocal evidence of an increased DPn within the telomer distribution on progression from EGDMA to LDMA under TBRT conditions. The chain transfer behaviour during TBRT is the dominant mechanism and must also be considered, therefore, Mayo plots were constructed to ascertain the impact of alkyl chain length on chain transfer coefficients (CT = kct/kp) for FRP reactions with MMA, HMA and LMA (ESI Table T4†). DDT was incrementally varied to provide ratios of DDT to monomer ([DDT]0/[M]0) ranging between 3.0 × 10−3–11.5 × 10−3 and employed AIBN (1.5 mol%) and ethyl acetate (50 wt% solids) as in previous reactions. The polymerisations were terminated at monomer conversions below 10% to minimise deviation from initial [DDT]0/[M]0 ratios.34 Vinyl conversions were estimated using 1H NMR spectroscopy, referenced against a corresponding 1H NMR spectrum taken prior to initiation, and number average molecular weight (Mn) values were determined using TD-SEC. As expected, small variations in DDT led to considerable decreases in measured Mn for all monomers studied (ESI Table T4 and Fig. S10†) and the observed Mn values were used to determine DPn for recovered polymers at each [DDT]0/[M]0 ratio. The subsequent Mayo plots (Fig. 3C) gave CT values of 0.4730, 0.7599 and 0.8927 for MMA, HMA and LMA respectively.
Surprisingly, the trend in CT suggests a more rapid chain transfer for LMA than MMA, again with HMA showing intermediate behaviour. A rapid chain transfer would be expected to result in more enhanced control of propagation and chains with reduced DPn. The relative propagation rates indicate MMA to be polymerising slower than both LMA and HMA (Fig. 3A); therefore, suggesting that chain transfer may be dominating the consumption of vinyl groups within the analogous MVT reactions. To investigate this further, reactions of MMA, HMA and LMA were conducted under telomerisation conditions that relate to the TBRT of the corresponding EGDMA, HDMA and LDMA. The monomer concentrations were fixed and a [DDT]0/[M]0 ratio of 1:2 was utilised for each telomerisation, thereby standardising the molar quantities of monomer, telogen, initiator and solvent used. The telomers generated were characterised by matrix-assisted laser desorption ionisation–time of flight (MALDI-TOF) mass spectrometry, which revealed significant differences between the molecular weight distributions of the telomers formed (Fig. 4).
 | ||
| Fig. 4 Comparison of telomeric chain lengths for linear telomers of MMA (red), HMA (blue) and LMA (black) with DDT as telogen by MALDI-TOF mass spectrometry. Peak intensities have been normalised to the DP of highest intensity in each mass spectrum and m/z is shown as number degree of polymerisation. | ||
Each spectrum showed incremental peak-to-peak separation in accordance with the monomer repeat unit molecular weight (ESI Fig. S11–13†). The p(MMA) distribution exhibited short chains with DP ≤ 15 monomer units; however, a trend of decreasing chain length was seen with increasing alkyl side chain. The telomerisation of HMA contained detectable chains of DP ≤ 11, whilst LMA telomerisation displayed a maximum observable chain length of just 8 repeat units, representing a value that was approximately 50% of that seen for MMA. Utilising the MALDI-TOF data, telomer DPn values of 5.96, 3.89 and 2.30 were determined for p(MMA), p(HMA) and p(LMA) respectively, under telomerisation conditions.
The MALDI-TOF analysis clearly correlates with the trend in CT observed during Mayo analysis with appreciable differences in telomer chain lengths under comparable telomerisation conditions across these different monomers. To avoid misinterpretation of MALDI-TOF data due to variation in ionisation between samples, further analysis of the telomers by SEC, utilising oligomer columns, was also conducted (Fig. 5).
 | ||
| Fig. 5 SEC (oligomer columns) refractive index chromatograms for linear telomers of MMA (red), HMA (blue) and LMA (black) with DDT as telogen. DP units are annotated above corresponding resolved peaks. | ||
The resolution of peaks corresponding to telomers of DP values of 4, 5 and 6 was possible for p(LMA), p(HMA) and p(MMA) respectively, with unresolved fractions containing higher chain lengths that appear to indicate a broader distribution for p(MMA) samples than either p(HMA) or p(LMA). MALDI-TOF and SEC analyses appear to indicate a trend of decreasing telomer chain lengths with increasing pendant alkyl group and support the dominant role of chain transfer over propagation under linear telomerisation conditions.
Studies utilising MMA, HMA and LMA provide insights to enable the fundamental understanding of LDMA, HDMA and EGDMA TBRT. Under TBRT conditions where stoichiometric equivalents of telogen are employed, telomer DPn across the three MVTs is likely to be influenced considerably by the range of chain transfer rates that have been identified for the monofunctional monomers. The telomer chain length distribution within the branched TBRT macromolecules would be expected to decrease in the order EGDMA > HDMA > LDMA.
Within our initial reports of TBRT using EGDMA, we identified resonances within the 1H NMR spectra that were assigned to different degrees of polymerisation and allowing the quantification of DP1 structures relative to vinyl residues within longer telomer substructures. Close examination of the 1H NMR spectra of purified p(DDT1.00-EGDMA0.85), p(DDT1.00-HDMA0.70) and p(DDT1.00-LDMA0.54) showed broad resonances (3.5–5.5 ppm) indicating the overlap of numerous methylene microenvironments adjacent to the backbone esters derived from DP variation within the telomer substructures (Fig. 6). As previously described, the more resolved resonance shouldering this broad signal may be assigned to DP1 structures which have not propagated and have solely undergone reaction with the DDT telogen.28,29
 | ||
| Fig. 6 Comparison of α-ester hydrogen resonances by 1H NMR spectroscopy for branched polymers of p(DDT1.00-EGDMA0.85), p(DDT1.00-HDMA0.70), and p(DDT1.00-LDMA0.54) synthesised using TBRT. | ||
The resolution of the methylene resonance shoulder increases sequentially from being difficult to distinguish, for p(DDT-EGDMA) analysis, to being dominant in p(DDT-LDMA) spectrum. This would strongly suggest an increasing prevalence of DP1 telomer units within the polymers formed. The overall resonance attributed to the methylenes adjacent to backbone esters also decreases is breadth across the series of TBRT polymers, suggesting fewer hydrogen microenvironments within p(DDT1.00-LDMA0.54) or, essentially, a narrower distribution of telomer substructures than either the HDMA or EGDMA derived branched polymers. This qualitative assessment is consistent with the conclusions of the chain transfer behaviour, Mayo analysis and MALDI-TOF studies described above.
Exploring the factors influencing TBRT outcomes from structurally varying MVTs
Whilst strongly supporting a dominance of chain-transfer within the TBRT of these three different methacrylate MVTs, the studies described above also point to a variation in chain transfer coefficients within the chemically similar MVTs. DDT appears to show a substrate structure dependence on its observed chain transfer, and this would be expected to be mirrored when selecting other telogens. Whilst the combined studies support this clear conclusion, they also present a contradiction; if the DPn of the telomer substructures decrease in the order EGDMA > HDMA > LDMA, the limiting [MVT]0/[DDT]0 gel point ratios would be expected to increase in the order EGDMA < HDMA < LDMA. Essentially, EGMDA would be expected to reach a critical telomer DPn = 2 at lower [MVT]0/[DDT]0 ratios than HDMA which, in turn, would be lower than LDMA. This is the exact opposite of what was observed in our study (Fig. 2).To account for this apparent contradiction, it is assumed that the extended intermolecular branching between telomeric substructures is controlled by factors other than the relative rates of chain transfer or propagation and telomeric primary chain length. The probability of gelation within homo-polymerisations of multi-vinyl monomers, and co-polymerisations containing low concentrations of multi-vinyl species, is conventionally governed by two key factors, namely the number of multi-vinyl species per primary chain and the efficiency of intermolecular branching.35–40 TBRT limits the number of vinyl functional groups available for intermolecular branching by ensuring the telomer distribution is maintained below a DPn = 2 units. It is reasonable to expect that gelation will be primarily influenced by the efficiency of intermolecular branching given the control of telomer length; it is also reasonable to assume that the molecular dimensions of each MVT will impact access to the available vinyl groups and influence the ability of the growing branched polymers to undergo intermolecular reactions. To explain the apparent contradictions, we introduce a model that explores branching behaviour from the perspective of a thiyl radical formed during the TBRT mechanism (Fig. 7).
 | ||
| Fig. 7 Schematic representation of the ‘lifetime sphere’ surrounding newly formed thiyl radicals (yellow sphere). (Ai) in the absence of MVT; (Aii) initial TBRT stages – small lifetime sphere; (B) during TBRT – decreasing telogen concentration allows lifetime sphere growth (note: branched polymer bearing unreacted vinyl groups); (C) high conversion – large lifetime sphere at reduced MVT and telogen concentration. | ||
TBRT relies upon rapid chain transfer that irreversibly caps propagating vinyl chains. This necessitates a continuous and dominant thiyl-telogen chain transfer mechanism, that regenerates thiyl radical and telogen, with rapid carbon centred radical-telogen transfer being ensured by the excess of telogen. This second step also regenerates thiyl radicals (Scheme 2). This is unlike other reported branching radical polymerisations where the key radical species are either carbon centred or in a dynamic equilibrium.
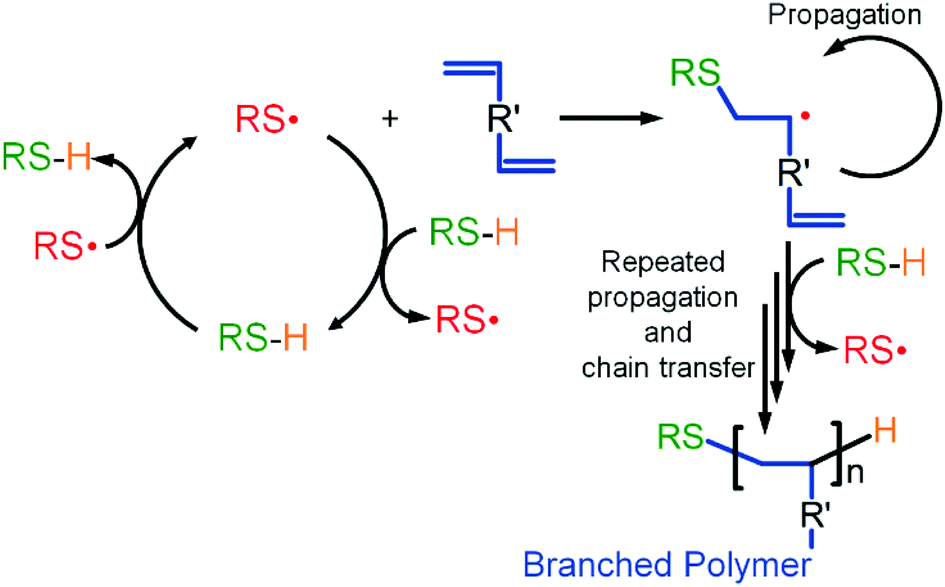 | ||
| Scheme 2 TBRT reaction mechanism showing chain transfer to telogen (thiol) as the dominant process. | ||
It is important to focus on the behaviour of a newly formed thiyl radical; after formation through the ongoing chain transfer reactions, a thiyl radical will have a finite lifetime during which it will diffuse in 3 dimensions throughout the reaction medium (Fig. 7A). In the absence of MVT, the thiyl radical will undergo chain transfer via reaction with telogen, and the distance travelled prior to chain transfer can be depicted as an average radius of a spherical volume of reaction medium (Fig. 7Ai). In the presence of MVT molecules, but under TBRT conditions where telogen is in excess, the radius may be considered as representing the average radical lifetime from the formation of the thiyl radical through to a chain transfer event but incorporating creation of carbon-centred radicals, telomer formation and subsequent chain transfer (Fig. 7Aii) to form C–H bonds. For simplicity, we have depicted this radius within Fig. 7A as being constant in the presence or absence of MVT. This model can be considered as a radical lifetime sphere where the outer limits represent a chain transfer perimeter or the most probable distance from thiyl formation to chain transfer. Within this conceptual framework, termination processes are assumed to be negligible during the lifetime of the polymerisation; this has been established experimentally under TBRT conditions.28
The dimensions of the radical lifetime sphere will be dependent on several factors including the telogen concentration, solvent, temperature and chain transfer coefficients. As transfer to telogen is the key mechanistic step under consideration (Scheme 2), the dimensions of the radical lifetime sphere are assumed to not be significantly influenced by MVT propagation kinetics. Under ideal conditions, TBRT consumes the telogen in a 1:1 ratio with MVT, therefore both telogen and MVT concentration will decrease during the formation of polymer. The excess of telogen will ensure that chain transfer dominates throughout the full conversion of MVT. This is, again, unlike other branching vinyl polymerisations where catalyst concentrations or key functional additives that mediate the reaction, such as RAFT agents, are maintained at near constant concentrations. Within the DE-ATRP of EGDMA, a ‘growth boundary’ concept was postulated to explain the observed deviation from Flory–Stockmayer theory; essentially, a consistently high concentration of deactivator ensured a low kinetic chain length.18 Unlike the previously proposed ‘growth boundary’, the radical lifetime sphere within TBRT will increase in dimensions as MVT and telogen consumption leads to the formation of vinyl-functional branched polymer species (Fig. 7B & C). Initially, local telogen concentrations are at their highest resulting in very rapid consumption of radicals by chain transfer. The decrease in telogen concentration during the formation of telomeric substructures and larger polymeric macrostructures, will consequently allow radical diffusion through a greater volume of reaction medium before chain transfer.
When considering the three MVTs studied here, the higher CT values expected for LDMA will lead to more rapid consumption of telogen during TBRT and a more rapid increase in radical lifetime sphere dimensions; equally, EGDMA would be expected to demonstrate the slowest change in sphere dimensions (ESI Fig. S14†). The observed variation in limiting [MVT]0/[DDT]0 gel point ratios may, therefore, be manifested in how effectively unreacted polymer-bound vinyl groups can populate the radical lifetime sphere.
The concentration of polymeric species within the radical lifetime sphere will be dependent on the relative size of the branched macromolecular species, which in turn is influenced directly by the dimensions of the constituent MVT. To investigate the dimensions of the TBRT branched polymers derived from EGDMA, HDMA and LDMA, TD-SEC analysis of hydrodynamic radii (RH) was conducted across the molecular weight distributions (Fig. 8A).
 | ||
| Fig. 8 Investigation of branched polymer dimensions: (A) TD-SEC derived analysis of the hydrodynamic radius across the molecular weight distributions for TBRT branched polymers of p(DDT1.00-EGDMA0.85) (red), p(DDT1.00-HDMA0.70) (blue) and p(DDT1.00-LDMA0.54) (black); (B) 3D molecular models of branched polymer structures containing 20 MVTs and 21 DDT taxogens of (i) p(DDT-EGDMA), (ii) p(DDT-HDMA) and (iii) p(DDT-LDMA) after energy minimization. | ||
At all comparative molecular weights, the corresponding RH of p(DDT1.00-LDMA0.54) polymers were found to be the largest followed sequentially by those of p(DDT1.00-HDMA0.70) and p(DDT1.00-EGDMA0.85). Additionally, TD-SEC analysis of samples taken during the TBRT of LDMA, HDMA and EGDMA showed RH development throughout the reaction and the same trend of higher RH for polymers derived from LDMA at all equivalent Mw values, with HDMA and EGDMA showing sequentially lower values (ESI Fig. S15†). As these polymer samples were synthesised using different [MVT]0/[DDT]0 ratios, three ideal branched polymer structures containing 20 MVT units and 21 DDT residues were modelled for EGDMA, HDMA and LDMA MVTs using Spartan 18 software. All energy minimisation calculations were conducted in a polar solvent environment, and globular conformations were seen upon approaching the estimated ground state (Fig. 8B). The theoretical volume of the geometrically equilibrated structures was found to increase with increasing MVT dimensions; the EGDMA, HDMA and LDMA derived structures occupied volumes of 7978 Å3, 9338 Å3 and 11296 Å3 respectively. These correspond to approximately 1.42-fold and 1.17-fold volume increases for LDMA and HDMA structures respectively over the equivalent EGDMA polymer. Importantly, these theoretical calculations followed the same trend as those obtained experimentally via TD-SEC analysis.
The increased RH values for p(DDT-LDMA) at an equal concentration of macromolecules and equivalent conversion will, therefore, occupy a greater volume fraction of the reaction medium than p(DDT-EGDMA) branched polymers, with p(DDT-HDMA) having an intermediate value (Fig. 9A & B).
 | ||
| Fig. 9 Schematic representation of the TBRT of EGDMA in comparison to LDMA. Model depicts a theoretical scenario where conversion is identical, resulting in the same concentration of telogen and MVT with a corresponding identical radius of radical lifetime sphere (yellow spheres). Branched polymers of the same DP and concentration are also shown: (A) shorter MVT structures lead to branched polymers occupying a low volume fraction of the reaction medium; (B) large MVTs occupy higher volume fraction of medium and are able to bridge between radical lifetime spheres. (Ai) Short MVTs lead to probability that two or more vinyl groups from one branched polymer are present within a single radical lifetime sphere leading to cyclisation; (Aii+Bi) DP1 formation if only one pendant group is present within the radical lifetime sphere; (Bii) two pendant vinyl groups from different branched polymers within the radical lifetime sphere lead to a probability of intermolecular reaction. | ||
At an equivalent and relatively high MVT conversion, the radical lifetime spheres in both cases would occupy near-identical volumes, despite the observed variation in CT, as thiyl-telogen transfer would be dominant. Within this scenario, p(DDT-LDMA) polymers would be expected to have a higher probability of populating more than one radical lifetime sphere than the polymers derived from shorter MVTs due to their increased volume fraction of polymer (Fig. 9). When considering p(DDT-EGDMA) structures, the proximity of two pendant vinyl bonds on a single branched macromolecule may also increase the probability of both functional groups being present within the same radical lifetime sphere, especially at higher conversions when the sphere may have increased to significant volumes. In this case, intramolecular reaction will lead to cycle formation (Fig. 9Ai); if only one vinyl group occupies the radical lifetime sphere, a DP1 terminal structure will result (Fig. 9Aii). For p(DDT-LDMA), it is also highly probable that only one pendant group occupies any given radical lifetime sphere (Fig. 9Bi) with subsequent terminal group formation. However, the potential to bridge between radical lifetime spheres will increase the probability of the presence of pendant vinyl groups from different polymer structures within a single sphere and, therefore, enhance intermolecular reaction. The obvious consequence is gel formation for LDMA at lower MVT concentrations than EGDMA and HDMA unless the radical lifetime spheres are maintained at much smaller dimensions by increased telogen concentration.
To further investigate the validity of this model, the presence of cycles within the high molecular weight regions of the different TBRT distributions was studied using recently reported approaches.29 The p(DDT1.00-LDMA0.54), p(DDT1.00-HDMA0.70) and p(DDT1.00-EGDMA0.85) polymers were each subjected to solvent fractionation to isolate the high molecular weight fractions of each distribution. TD-SEC analysis of the solvent fractionated samples showed considerable increases in Mn values and narrowing of dispersity (Mn = 621226 g mol−1, 102485 g mol−1 and 36582 g mol−1 for samples derived from EGDMA, HDMA and LDMA respectively), (ESI Table T5 and Fig. S16†). 1H NMR characterisation of the fractionated materials provided [MVT]/[DDT] ratios within the purified polymers of 1.20, 1.17 and 1.04 for the p(DDT-EGDMA), p(DDT-HDMA) and p(DDT-LDMA) polymers respectively (ESI Fig. S17–S19†). As presented in our initial reports of TBRT, the [MVT]/[telogen] ratio follows an n/(n + 1) relationship with increasing MVT (n = number of MVT structures within the branched polymer).28,29 This can be readily understood by considering the smallest possible adduct of one MVT plus two telogens in the absence of any propagation – i.e. the formation of DP1 structures only. This adduct would have the lowest possible MVT/telogen ratio = 0.5 (Fig. S20†). As additional MVTs are included in larger structures, adducts with two MVTs would have a value of 0.66 whilst those with three MVTs would a value 0.75, progressing to values that approach 1 at high MVT incorporation whilst always staying below 1 (Fig. S20†). Equally, the formation of cycles requires one less telogen per cycle formed in any given branched polymer structure (Fig. S21†), as established in our recent report that quantified branching and the presence of cycles within TBRT polymers derived from EGDMA.29 The relationship is modified to [MVT]/[telogen] = n/[(n + 1) − c], where c is the number cycles within each macromolecule and an appreciable number of cycles will cause the ratio will tend to values >1.29 It is, therefore, possible to estimate the percentage of MVT vinyl bonds that have reacted intramolecularly to form cycles within the fractionated samples. For the polymers studied here, the ratios strongly indicate 17%, 15% and 6% cyclisation for p(DDT-EGDMA), p(DDT-HDMA) and p(DDT-LDMA) respectively, under these conditions. This is also in agreement with the measured [MVT]f/[DDT]f values determined for the full unfractionated samples (Table 1), where the longer LDMA MVT yielded values <1 whilst HDMA polymers approach 1 and polymers derived from EGDMA are generally >1, indicating a higher susceptibility to cyclisation with decreasing MVT dimensions.
Analysis of the cumulative weight fraction versus molecular weight of these polymers also shows a considerable difference in the mass of the LDMA-derived and EGDMA-derived polymer samples prepared at the limiting [MVT]0/[DDT]0 gel point ratios. It is clear that a larger fraction of the mass of the LDMA-derived sample is formed by lower molecular weight species which would clearly lead to an impact on the observed [MVT]f/[DDT]f values as described above.
Conclusions
Transfer-dominated Branching Radical Telomerisation is a new approach to forming complex branched polymer architectures with high molecular weight. It is unlike other reports that aim to polymerise multi-vinyl monomers, known as multi-vinyl taxogens in this case, as it employs telomerisation conditions and conventional free radical chemistries. TBRT is fundamentally different to the CTA-mediated free radical branched polymer synthesis known as the “Strathclyde” approach, and modifications of that approach that use RDRP techniques.13,14 These widely reported techniques modify a linear polymerisation of monovinyl monomer, often targeting appreciable primary chain lengths, by adding less than 1 branching multivinyl monomer per chain. As such the chemistry of the backbone is dictated by the monovinyl monomer and the overall architecture is determined by repeated propagation of vinyl bonds. TBRT chemistry utilises vinyl feedstocks, but the resulting polymers are controlled by telomerisation conditions to prevent extended vinyl propagation and the backbone chemistry is dictated by the preformed linking chemistry within the MVT. Also, the final polymer incorporates, on average, one telogen per MVT and the nominal repeat unit, Scheme 1, is created during telomerisation. The telogen becomes an integral feature of the repeating chemistry and not a trivial chain end as in the “Strathclyde” or conventional linear approaches. To be clear, the polymerisable vinyl groups within a “Strathclyde” reaction are predominantly derived from monovinyl monomer and the vinyl functionality/CTA ratios are often >30, although values as low as 6 have also been reported, as would be expected for linear homopolymerisations.41,42 TBRT utilises vinyl group/telogen starting feed ratios <2; this feature mirrors the rationale for all small molecule telomerisations to ensure the creation of new structures from telogen + taxogen reactions with limited taxogen + taxogen coupling. Although the use of thiols, chain transfer reactions, free radical initiators and vinyl functional feedstocks appears familiar in the context of “Strathclyde” chemistry, TBRT is novel and generates polymers that are not available through this well-established strategy. When introducing new chemistries, it is important to establish the principles that govern successful outcomes. The diversity of MVT structures that are commercially available, or can be envisaged, is considerable but it is clear from the presented work that simply replacing one MVT with another that bears the same class of reactive functionality (in this case methacrylates) requires exploration of reaction conditions, even when utilising the same telogens and solvent.In addition to the different rates of chain transfer and consumption of vinyl group, it is clear that MVT size impacts the mechanism of branched polymer formation. Intermolecular branching is favoured by larger MVTs and intramolecular cyclisation is significantly reduced. Both features appear to scale across EDMA, HDMA and LDMA, thereby lending further support to the interpretation of the accumulated data and the presented models.
Each MVT utilised in future applications of TBRT will require an analysis of limiting [MVT]0/[DDT]0 gel point ratios and optimisation of reaction conditions as dictated by variation in solvent, reaction concentration and telogen selection. It is apparent that variations in the rate of chain transfer between telogen and MVT outweighs differences in propagation kinetics. Although an apparent increase in relative rate of MVT propagation, measured by vinyl group consumption, would suggest the formation of a longer average telomer length, the study of monovinyl taxogens would suggest the opposite due to the dominant and enhanced chain transfer.
The increasing relative size of the TBRT polymers with larger MVTs is intuitive and the measured RH values, supported by the 3D modelling, does provide evidence for this. It was not obvious that this would have such a dramatic effect on intermolecular branching and molecular weight evolution; however, our studies here do provide a rationale for the observed behaviour and trends.
Conflicts of interest
SRC, PC and SPR are co-inventors on patents that protect the TBRT chemistry; these patents have been licensed to Scott Bader and form the basis of Polymer Mimetics Ltd (Company number 12598928).Acknowledgements
This manuscript is based on work supported by the Engineering & Physical Sciences Research Council (EPSRC) through grant EP/R010544/1 and the co-authors are grateful for the funding award. OBPL is grateful for studentship funding from EPSRC and the University of Liverpool. SRC is grateful for financial support from Itaconix. SM is grateful for studentship funding from EPSRC and Unilever. The authors would like to thank the Materials Innovation Factory for access to analytical equipment.References
- K. Mortensen, Colloids Surf., A, 2001, 183–185, 277–292 CrossRef CAS.
- B. Adhikari and S. Maiti, Prog. Polym. Sci., 2000, 25, 909–948 CrossRef CAS.
- B. M. Culbertson, Prog. Polym. Sci., 2001, 26, 577–604 CrossRef CAS.
- W. Wang, D. Zhou, L. Cutlar, Y. Gao, W. Wang, J. O'Keeffe-Ahern, S. McMahon, B. Duarte, F. Larcher, B. J. Rodriguez and U. Greiser, Sci. Adv., 2016, 2, 1–15 Search PubMed.
- M. H. Jones, H. W. Melville and W. G. P. Robertson, Nature, 1954, 174, 78 CrossRef CAS.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- Y. Gao, D. Zhou, J. Lyu, A. Sigen, Q. Xu, B. Newland, K. Matyjaszewski, H. Tai and W. Wang, Nat. Rev. Chem., 2020, 4, 194–212 CrossRef CAS.
- M. Jikei and M. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233–1285 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- D. Wang, Y. Jin, X. Zhu and D. Yan, Prog. Polym. Sci., 2017, 64, 114–153 CrossRef CAS.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- X. Wang and H. Gao, Polymers, 2017, 9, 1–22 CrossRef CAS.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- F. L. Hatton, P. Chambon, T. O. McDonald, A. Owen and S. P. Rannard, Chem. Sci., 2014, 5, 1844–1853 RSC.
- C. Degoulet, T. Nicolai, D. Durand and J. P. Busnel, Macromolecules, 1995, 28, 6819–6824 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- W. H. Stockmayer, J. Chem. Phys., 1944, 12, 125–131 CrossRef CAS.
- T. Zhao, Y. Zheng, J. Poly and W. Wang, Nat. Commun., 2013, 4, 1873 CrossRef.
- M. L. Koh, D. Konkolewicz and S. Perrier, Macromolecules, 2011, 44, 2715–2724 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- J. Lyu, Y. Gao, Z. Zhang, U. Greiser, P. Polanowski, J. K. Jeszka, K. Matyjaszewski, H. Tai and W. Wang, Macromolecules, 2018, 51, 6673–6681 CrossRef CAS.
- M. D. Peterson and A. G. Weber, EI Du Pont de Nemours and Co., US Pat., 2395292, 1946 Search PubMed.
- W. E. Hanford, EI Du Pont de Nemours and Co., US Pat., 2396786, 1946 Search PubMed.
- E. Müller, Angew. Chem., Int. Ed. Engl., 1952, 64, 233–247 CrossRef.
- R. K. Freidlina and A. B. Terent'ev, Acc. Chem. Res., 1977, 10, 9–15 CrossRef CAS.
- B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3235–3243 CrossRef CAS.
- T. A. Faßbach, A. J. Vorholt and W. Leitner, ChemCatChem, 2019, 11, 1153–1166 CrossRef.
- S. R. Cassin, P. Chambon and S. P. Rannard, Polym. Chem., 2020, 11, 7637–7649 RSC.
- S. R. Cassin, S. Flynn, P. Chambon and S. P. Rannard, RSC Adv., 2021, 11, 24374–24380 RSC.
- H. Yao, Y. Ning, C. P. Jesson, J. He, R. Deng, W. Tian and S. P. Armes, ACS Macro Lett., 2017, 6, 1379–1385 CrossRef CAS.
- R. Takahashi, S. Miwa, F. H. Sobotta, J. H. Lee, S. Fujii, N. Ohta, J. C. Brendel and K. Sakurai, Polym. Chem., 2020, 11, 1514–1524 RSC.
- R. A. Hutchinson, S. Beuermann, D. A. Paquet and J. H. McMinn, Macromolecules, 1997, 30, 3490–3493 CrossRef CAS.
- R. Vinu and G. Madras, Macromol. React. Eng., 2009, 3, 556–567 CrossRef CAS.
- M. K. Donald and S. A. F. Bon, Polym. Chem., 2020, 11, 4281–4289 RSC.
- F. Isaure, P. A. G. Cormack, S. Graham, D. C. Sherrington, S. P. Armes and V. Bütün, Chem. Commun., 2004, 4, 1138–1139 RSC.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, Macromolecules, 2004, 37, 2096–2105 CrossRef CAS.
- F. Y. Lin, M. Yan and E. W. Cochran, Macromolecules, 2019, 52, 7005–7015 CrossRef CAS.
- J. Poly, J. D. Wilson, M. Destarac and D. Taton, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5313–5327 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145–2156 CrossRef CAS.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS.
- P. Besenius, S. Slavin, F. Vilela and D. C. Sherrington, React. Funct. Polym., 2008, 68, 1524–1533 CrossRef CAS.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 1455–1460 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, full experimental details and additional characterisation. See DOI: 10.1039/d1py01103k |
| This journal is © The Royal Society of Chemistry 2021 |