
High pressure: a complementary tool for probing solid-state processes
Boris A.
Zakharov
 *abc and
Elena V.
Boldyreva
ab
*abc and
Elena V.
Boldyreva
ab
aBoreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences, Lavrentiev Ave. 5, Novosibirsk, 630090, Russian Federation. E-mail: b.zakharov@yahoo.com
bNovosibirsk State University, Pirogova Str. 2, Novosibirsk, 630090, Russian Federation
cInstitute of Solid State Chemistry and Mechanochemistry, Siberian Branch of the Russian Academy of Sciences, Kutateladze Str. 18, Novosibirsk, 630128, Russian Federation
First published on 17th October 2018
Abstract
Since the early days of X-ray diffraction, researchers have tried to follow the evolution of crystal structures under extremes of pressure. Recently, interest in this area has exploded, attracting scientists from backgrounds across the physical and life sciences. Much of this rapid expansion has been due to the enhancement of diffraction equipment, including detectors, goniometers, and high-pressure cells, and the development of synchrotron radiation and neutron sources. The high-pressure research generally focuses on the direct effects of pressure on the structural evolution of a system. The present contribution describes examples of a less common application: hydrostatic compression of crystals of organic and coordination compounds can be used as a complementary tool in order to rationalise the mechanisms of the transformations in these solids that take place at atmospheric pressure. The data on the compressibility, equations of state, phase changes, or the effects of pressure on intermolecular distances, molecular conformations and chemical bonds can shed light on the factors that are important for thermal and photochemical reactions in the same crystals without an external load as well as on their solid-state or solvent-assisted polymorphic transformations. The manuscript also discusses how structural studies under extreme conditions can help to rationalise the thermo- and photosalient effects that accompany some solid-state transformations. This knowledge is currently of great importance for materials science since mechanically responsive materials have the potential to be used for the design and manufacture of new supramolecular devices.
 Boris A. Zakharov | Dr. Boris A. Zakharov graduated from Novosibirsk State University in 2011. He received his Ph.D. degree in solid-state chemistry in 2013 from the Institute of Solid State Chemistry and Mechanochemistry and is currently employed as a researcher by the Boreskov Institute of Catalysis (Novosibirsk, Russia). He is a member of the Commission on High Pressure of the International Union of Crystallography. His current research interests include intermolecular interactions, structural distortions, and phase transitions at variable temperatures and pressures in different types of crystalline materials. He also teaches a course on crystal structure determination and analysis at Novosibirsk State University. |
 Elena V. Boldyreva | Prof. Elena V. Boldyreva graduated from Novosibirsk State University in 1982. She is the author and coauthor of over 300 research papers and several monographs. She is a leading researcher at the Boreskov Institute of Catalysis (Novosibirsk, Russia) and the head of the Chair of Solid-State Chemistry at the Novosibirsk State University, an honorary professor of the University of Edinburgh, a corresponding member of the Academy of Sciences and Arts of Slovenia, and a member of the Academia Europaea. Her research interests span from photomechanical effects, mechanochemistry, and high-pressure research to crystal growth and design, polymorphism, and physical pharmacy. |
Introduction
X-ray crystallography has been an area of research for over a century, with the structure of NaCl having been determined in 1914.1 In the early days, most crystal structures were determined under ambient conditions. The first experiments at variable temperatures provided a valuable contribution to understanding solid-state transformations, including phase transitions and chemical reactions as well as changes in atomic coordinates, crystal packing and molecular conformations.2–15 Since cooling and heating a crystalline sample is relatively easy, single-crystal determinations at variable temperatures quickly became routine following the first pioneering studies. For several decades, temperature variation remained the main tool to influence crystal structure in crystallographic experiments. The Nobel prize-winning research on various materials accomplished by P. Bridgman16 made it possible to study in situ at high pressure such phenomena as, e.g. volumetric changes, electrical resistivity, viscosity or thermal conductivity. However, both X-ray diffraction and spectroscopy measurements at high pressure were carried out ex situ on samples quench-decompressed to ambient pressures. Applications of in situ X-ray diffraction to study high-pressure phenomena were very rare.17 The situation changed dramatically in the 1950s when different models of the diamond anvil cell (DAC) were developed simultaneously and independently by several research groups (ref. 18, 19 and references therein). This enabled in situ optical observation of pressure-induced phase transitions as well as both spectroscopic and X-ray diffraction measurements.20–22 It became possible to follow in detail the structural response of a crystal to applied pressure at the moment of compression and also to follow the kinetics of the transformations at high-pressures.With the development of crystallography, there has been considerable advancement in the associated technology of diffractometers, detectors (2D instead of a point detector23–26) and high-pressure instrumentation (including DACs with larger opening angles23,27–29). Considerable strides were also made in data processing techniques.30–35 With these developments, it has become possible to follow even subtle variations in structure with pressure. At present one can monitor changes not only in lattice parameters, atomic coordinates and anisotropic thermal displacement parameters, but even in the electron density distribution.36–38
High-pressure research generally focuses on the direct effects of pressure on the structural evolution of a system. Hence, many studies involve monitoring compressibility, equations of state, phase changes, or the effects of pressure on chemical bonds, coordination of ions in the polyhedral structures, molecular conformations, and intermolecular interactions. Numerous results and generalizations concerning the high-pressure behavior of compounds, minerals, and materials have been documented and summarized (see ref. 39–42 and references therein as examples).
Another, albeit less common, application is the use of pressure as a complementary tool in order to investigate solid-state transformations that occur on heating, cooling or irradiation and do not require an external load. The approach was used in the 1980s–1990s to study chemical reactions in solutions under pressure. In particular, the values and the signs of activation volumes made it possible to distinguish between different reaction mechanisms.43–47
In the 1990s, high-pressure IR spectroscopy and X-ray diffraction experiments were first used in order to understand solid-state thermal and photochemical linkage isomerisation in Co(III) crystalline coordination compounds that takes place under atmospheric pressure (ref. 48–50 and references therein). Significant stress and strain are generated by the reaction itself. Pressure was used to probe the mechanical properties of the crystals in order to rationalise the role of mechanical stress and strain in solid-state reactivity.51–55
The above-mentioned linkage isomerisation is not unique in being accompanied by the generation of mechanical stress and strain. The same phenomenon is common for most solid-state transformations. This can have a decisive influence on solid-state reactivity and can determine the kinetics and spatial propagation of the transformation. Further, the stress and strain can influence which products will result and can even be responsible for the very possibility of a reaction to occur.50,56–66 It has been suggested that hydrostatic compression can be used to mimic strain induced by the thermo- or photo-reactions that typically occur under ambient pressures. In this way, pressure was proposed as a method to control solid-state reactions and probe their mechanisms.48,51,52,55,67–69
The aim of the present contribution is to highlight recent high-pressure studies that have provided significant input into understanding solid-state properties and transformations (phase transitions and chemical reactions) of the same solids at atmospheric pressure.
Compressibility: changes in unit cell volume and parameters
Measuring the compressibility of minerals and materials to determine an equation of state (EoS) is common practice.70 This information is necessary to predict the behavior of materials which are used under elevated pressures, or for understanding phenomena that occur in the depths of planets or oceans.71–76 For non-cubic structures, it is not sufficient to measure changes in volume. Instead, it is necessary to consider the material anisotropy, characterized by the strain tensor.77The anisotropy of strain that accompanies variations in temperature has been used to estimate the relative strengths of different types of hydrogen bonds in organic crystals.78–81 A similar approach has been successfully applied for strain induced by hydrostatic compression.55,82–89 The studies that were originally focused on hydrogen bonds were followed by extensive research on various non-covalent interactions. In doing so, the high-pressure investigations offered a means to qualitatively rank the energetic hierarchy of various interactions.90–107 Data on the strain anisotropy of intermolecular interactions have also proven central to the design of empirical potentials. Such potentials have been used to model material properties and predict crystal structures and their transformations under a variety of thermodynamic conditions.108–113
Crystals that can be continuously compressed without undergoing a structural phase transition allow fine-tuning of ferroelectric, optical or magnetic properties, as well as conductivity, and hence provide true insight into the fundamental physics of materials.98,99,114–126 The possibility to measure these properties as a function of selected interatomic distances corresponding to the same crystal structure undergoing a continuous compression makes it possible to verify the models that are used to describe the physical properties of the systems and to optimise the parameters of these models.
For a chemist, continuous compression of a material allows correlating solid-state chemical reactivity and crystallographic parameters including the free volume, size and shape of the reaction cavity, and chemical pressure. To some extent, this approach is analogous to using the value and sign of the activation volume in order to distinguish between intra- and intermolecular mechanisms for chemical reactions in solutions.43–47 However, for reactions in the solid state, interpreting the effects of pressure on the reaction rate, the activation energy, the composition and the structure of the reaction products can be even more difficult than for the same reaction in solution. This is largely because structural strain is anisotropic and most reactions are heterogeneous. That said, it has been proven possible to relate structural strain and chemical reactivity to a qualitative50,56,64,65,69 and quantitative level in certain cases.57–59,62,127–129 The high-pressure experiments play an important role in establishing these relations.
First, the high-pressure experiments provide data that are necessary to estimate the rigidity of the reaction cavity and the ease with which a crystal structure can respond to the perturbations induced by a chemical transformation. Second, if, for example, a photochemical reaction is followed in a hydrostatically compressed crystal, one can monitor the effect of pressure (and in this way – of the size and shape of the reaction cavity) on the photochemical transformation in essentially the same crystalline compound.54,130–137 This allows one to relate the shape and size of the reaction cavity with the reaction kinetics and yield.
The importance of studying the anisotropy of the strain tensor to probe chemical transformation is exemplified by the photochemical linkage isomerisation in [Co(NH3)5NO2]ClNO3. This solid-state transformation is accompanied by the generation and relaxation of stresses and strain. When an elongated rod-shaped crystal is irradiated from one side, the reaction product is distributed non-uniformly in the direction normal to the irradiated surface. This is the result of light being absorbed. Absorption in this manner leads to significant crystal bending.139 Remarkably, the irradiated crystal side is convex, which may seem to suggest that the volume increases on irradiation. However, X-ray diffraction studies demonstrated that the volume change that results from the isomerisation is negative, despite the fact that the crystal structure expands in selected directions.138 The largest expansion occurs along the longest crystallographic axis. On hydrostatic compression, it was found that the same crystal structure is compressed in all directions, albeit to different extents.51 The largest compression coincides with the direction of the largest expansion on photoisomerisation (Fig. 1). This explains why the activation volume of the photoisomerisation is positive (the reaction quantum yield decreases on compression), whereas the change in molar volume is negative. The internal pressure that arises in the crystals on linkage isomerisation was estimated as equivalent to several GPa.51
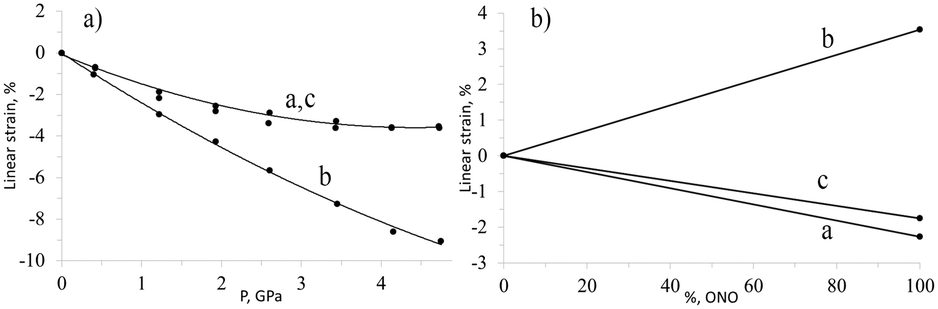 | ||
| Fig. 1 Changes in cell parameters of [Co(NH3)5NO2]ClNO3 on hydrostatic compression (a) and resulting from nitro–nitrito photoisomerization (b). Based on data from ref. 51 and 138. | ||
Almost 20 years after these early studies, the in situ high-pressure single-crystal X-ray diffraction for [Co(NH3)5NO2]ClNO3 allowed the rigidity of selected hydrogen bonds to be followed up to 5 GPa.69 The hydrogen bonds with different geometries formed by the nitro group were shown to have different compressibilities (and thus different rigidities). One of the hydrogen bonds is very long and weak that facilitates the rotation of the nitro group during photochemical isomerisation (Fig. 2).69 The stress and strain imposed on the crystalline environment by the rotation of the nitro group during isomerisation are transferred from one cation to another through an infinite network of flexible hydrogen bonds. The difference in the rigidity of different types of hydrogen bonds as revealed by high-pressure experiments proved to be helpful for understanding the mechanical properties of selected structural motifs. This was particularly important when focused on motifs that play a key role in chemical transformation and are believed to play a key role in the mechanical response, namely in the photosalient effect.
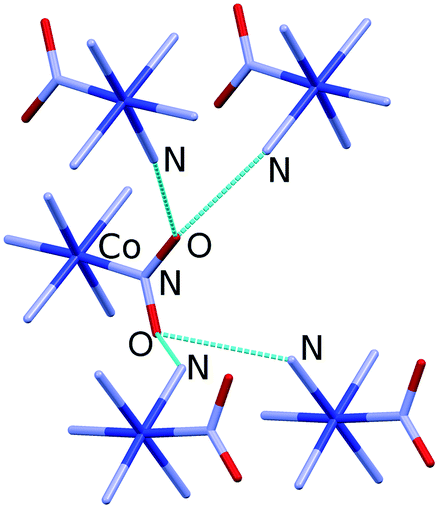 | ||
| Fig. 2 Hydrogen bonds formed by the nitro group in [Co(NH3)5NO2]ClNO3. Hydrogen atoms and Cl− and NO3− anions are omitted for clarity. Structural data were taken from ref. 69. | ||
The continuous structural strain observed on hydrostatic compression can be compared with that on cooling the same crystal in order to get a better insight into the mechanisms of structural strain resulting from the two different actions. Such a comparison is especially informative if the volume decrease is the same in the two cases (Fig. 3).55 Since both temperature and pressure are scalars, the anisotropies of isothermal compressibility and isobaric thermal expansion depend solely on the crystal structure anisotropy and the structural strain mechanism. Hydrostatic compression is related to minimising the free volume, whereas structural contraction on cooling is primarily related to the changes in the anharmonicities of vibrations. Different types of interactions – hydrogen bonds, stacking interactions, van der Waals interactions – make different relative contributions to the changes of the total energy and volume on cooling and on hydrostatic compression. Comparative studies of the anisotropy of strain on cooling and on increasing the pressure thus give insight into the characteristics of different types of intermolecular interactions in the crystal structure. This information is helpful for understanding the behavior of the same crystals also under atmospheric conditions – on crystallisation, dissolution, in the course of polymorphic transitions and chemical reactions. Research in this direction started several decades ago,55,84,89 and it remains a ‘hot’ topic of research today.140–142 Understanding the difference in the crystal structure response to pressure and temperature variations is relevant to the properties of the studied crystalline compounds not only at high but also at ambient pressure. In particular, it can be used to improve the potentials used for model calculations of the crystal structures and polymorph prediction, as well as to rationalise the occurrence of structural phase transitions upon variation in temperature and pressure.99,108–113
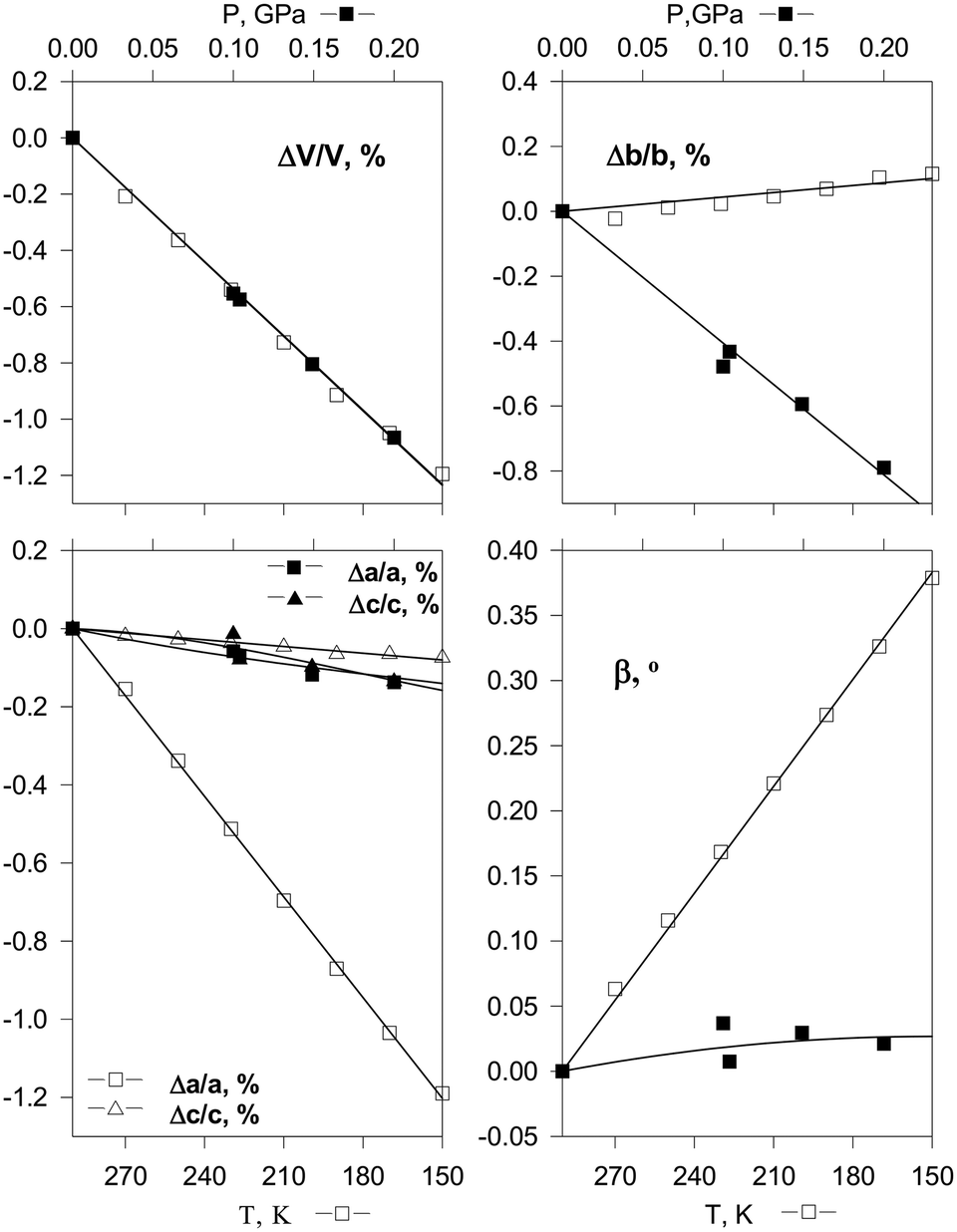 | ||
| Fig. 3 Changes in cell parameters of [Co(NH3)5NO2]Cl2 on hydrostatic compression (top) and on cooling (bottom) corresponding to essentially the same decrease in volume.55 Reproduced with permission from the International Union of Crystallography (https://journals.iucr.org/). | ||
An interesting option is to compare the chemical reaction response to a crystal volume reduction resulting from different perturbations: cooling and pressure. The study of [Co(NH3)5NO2]ClNO3 (ref. 62 and 140) provides a recent example. The quantum yield of photoisomerisation was correlated with crystal bending over the temperature range of 80–363 K. A qualitative correlation was found between the photo-reaction rate and both the cell volume and the volume of the void space around the nitro groups. However, this correlation was non-linear. The effect of the temperature-induced anisotropic structural strain on the photoisomerisation rate was qualitatively different from the effect of strain generated by elastic external loading.
If strain was induced by an external load, the reaction rate correlated linearly with compression along one crystallographic direction, b. In contrast, if strain resulted from temperature variations, then the reaction rate correlated linearly with changes along another (perpendicular) crystallographic direction, a. The relation between lattice strain on hydrostatic compression and the photoisomerisation yield could be rationalised in terms of reducing the voids around the nitro group. The exact mechanism that underpins the effect of the a parameter and the Cl–NO3− (and NO2–NH3) distance on the reaction rate when considering thermal strain requires further study. It was preliminarily interpreted in terms of coupling between selected anharmonic vibrational modes, electronic photoexcitation and the motion of the nitro-ligand, resulting in the change of its orientation in the crystal structure and bonding to the Co atom.62,140 Remarkably, in neither the case of external elastic compression nor thermally induced strain does the reaction rate correlate with lattice strain along the crystallographic c direction. Instead, lattice expansion along c, which takes place on both cooling and on elastic compression of the b axis, corresponds to a reaction rate decrease.
The apparently different effects of macroscopic strain on the photoisomerisation rate in the two cases – external elastic compression and thermal strain – were found to be rooted in identical phenomena. This becomes evident if the microscopic stress experienced by the excited state of the complex cation is considered. The macroscopic thermal strain can be suggested to subject the excited state to microscopic stress mainly along the a axis. In contrast, direct external loading of the crystal appears to result in microscopic stress mainly along the b axis. No significant microscopic stress arises along c, presumably since enough free space is present in this direction in the starting crystal structure, and this free space is preserved over the course of isomerisation. Further investigation into the lattice dynamics of this system may shed more light on the microscopic stresses that arise over the course of the reaction at various temperatures and on elastic compression.62,140
Crystallisation and recrystallisation
Another class of pressure-induced phenomena studied since the very first year of high-pressure science is pressure-induced crystallization.16 The low-temperature and the high-pressure phases of the same compound are usually different, with only rare exceptions.106,143,144 A comparison of the phases that are formed on hydrostatic compression with those that crystallise on freezing sheds light on the structure-forming factors as well as on the mechanisms of nucleation and subsequent growth of these nuclei. Further, it offers some insight into the role of kinetics in determining which forms nucleate and dominate growth. From a thermodynamic perspective, the same phase must be obtained at a selected temperature and pressure point, independent of the experimental protocol that has brought the system to this state. In practice, however, a liquid often crystallises in different phases, depending on the exact compression/decompression protocol (the rates of increasing and decreasing pressure, the duration of staying at the same pressure, etc.).145 Additional complexity is added when considering crystallisation or recrystallisation of a solid from its solution. Besides the compression/decompression protocol, one must consider the structure of the starting crystalline phases that were present in the pressure cell as well as the nature of the pressure-transmitting medium. A recent example is provided by a pharmaceutical small-molecule organic compound, chlorpropamide (4-chloro-N-(propylamino-carbonyl)benzenesulfonamide), CPA, which is often used as a model compound in various research projects. This compound is highly prone to polymorphism.146,147 In addition to the α-polymorph, which is thermodynamically stable at atmospheric pressure and room temperature,148 it forms four metastable polymorphs (β, γ, δ, ε).149–151 Once formed, all of these metastable forms can be preserved for a long time. Unfortunately, thermodynamic considerations alone are usually insufficient to predict which polymorphic phase will form under given pressure–temperature conditions for such highly polymorphic compounds. The outcome of crystallisation depends on a complex interplay between the thermodynamic driving force and the kinetics of nucleation and nuclei growth. Both thermodynamics and kinetics of crystallisation depend, among other factors, on the interaction of a certain polymorph with the surrounding (pressure-transmitting) medium. Crystallisation on hydrostatic compression can reveal features which are less pronounced when the same compound crystallises under atmospheric conditions.To study the transformations of different polymorphs of chlorpropamide at high pressures, three different single-crystalline samples of α-, β- and δ-polymorphs were loaded into the same DAC with a pentane–isopentane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture as the pressure-transmitting medium. Although this medium did not visibly dissolve the crystalline samples at the beginning of the experiment, the β-polymorph (which is the least dense of the three forms) recrystallised at a pressure near 0.5 GPa. During recrystallisation, the pressure decreased to 0.3 GPa and thin crystalline needles of the γ-polymorph appeared. At the same time, the crystal of the δ-polymorph was observed to increase in size, while the β-polymorph crystal began to dissolve and the α-form crystal remained unchanged (Fig. 4a).152 If β-chlorpropamide was placed alone in a DAC in the same pressure-transmitting medium, the γ-polymorph was found to grow on recrystallisation, with no other phases appearing (Fig. 4b).152 This was surprising since γ-chlorpropamide is less dense than the α- and δ-polymorphs under ambient pressures and at 0.5 GPa. In fact, the α-form is the most thermodynamically stable, while the δ-form is the densest polymorph. The observed phenomena could be rationalised by assuming that the conformations of both individual molecules and molecular clusters can be preserved on dissolution. The β → γ transformation does not require significant changes in the molecular conformations or packing of molecular H-bonded chains. This suggests a low nucleation barrier, enabling recrystallisation of the β-form into the γ-polymorph, even if no seeds of the latter are present. The β → δ transformation does not require significant changes in the packing of molecular H-bonded chains but requires a large change in the molecular conformation. This transformation can be observed only in the presence of a seed of the densest δ-polymorph. The β → α transformation requires significant changes in both the packing of intermolecular H-bonded chains and in the molecular conformation. This makes the transformation highly unfavourable, even if a seed of the thermodynamically stable α-polymorph is placed in the DAC. The results of this study152 showed that under certain conditions, molecular clusters formed from the parent solid phase can be preserved on dissolution. Thus, the crystallisation product with the lowest kinetic barrier to nucleation is obtained. Surprisingly, in the case of chlorpropamide, this barrier could not be changed by adding seeds of denser and thermodynamically more stable phases. These observations of high-pressure recrystallisation give additional insight into the dissolution and crystallisation of the polymorphs of chlorpropamide also under ambient conditions: they explain why the metastable forms can co-crystallise concomitantly and be preserved as a mixture even in slurries. They also explain why the γ-polymorph crystallises so easily if the β-polymorph is present.
:1) mixture as the pressure-transmitting medium. Although this medium did not visibly dissolve the crystalline samples at the beginning of the experiment, the β-polymorph (which is the least dense of the three forms) recrystallised at a pressure near 0.5 GPa. During recrystallisation, the pressure decreased to 0.3 GPa and thin crystalline needles of the γ-polymorph appeared. At the same time, the crystal of the δ-polymorph was observed to increase in size, while the β-polymorph crystal began to dissolve and the α-form crystal remained unchanged (Fig. 4a).152 If β-chlorpropamide was placed alone in a DAC in the same pressure-transmitting medium, the γ-polymorph was found to grow on recrystallisation, with no other phases appearing (Fig. 4b).152 This was surprising since γ-chlorpropamide is less dense than the α- and δ-polymorphs under ambient pressures and at 0.5 GPa. In fact, the α-form is the most thermodynamically stable, while the δ-form is the densest polymorph. The observed phenomena could be rationalised by assuming that the conformations of both individual molecules and molecular clusters can be preserved on dissolution. The β → γ transformation does not require significant changes in the molecular conformations or packing of molecular H-bonded chains. This suggests a low nucleation barrier, enabling recrystallisation of the β-form into the γ-polymorph, even if no seeds of the latter are present. The β → δ transformation does not require significant changes in the packing of molecular H-bonded chains but requires a large change in the molecular conformation. This transformation can be observed only in the presence of a seed of the densest δ-polymorph. The β → α transformation requires significant changes in both the packing of intermolecular H-bonded chains and in the molecular conformation. This makes the transformation highly unfavourable, even if a seed of the thermodynamically stable α-polymorph is placed in the DAC. The results of this study152 showed that under certain conditions, molecular clusters formed from the parent solid phase can be preserved on dissolution. Thus, the crystallisation product with the lowest kinetic barrier to nucleation is obtained. Surprisingly, in the case of chlorpropamide, this barrier could not be changed by adding seeds of denser and thermodynamically more stable phases. These observations of high-pressure recrystallisation give additional insight into the dissolution and crystallisation of the polymorphs of chlorpropamide also under ambient conditions: they explain why the metastable forms can co-crystallise concomitantly and be preserved as a mixture even in slurries. They also explain why the γ-polymorph crystallises so easily if the β-polymorph is present.
 | ||
| Fig. 4 The crystals of the α-, β-, and δ-polymorphs of chlorpropamide (a) and the crystals of the β-polymorph of chlorpropamide (b) loaded in a DAC immediately after loading and after a certain amount of time. The growth of new needle-shaped crystals (γ-polymorph) and growth of the δ-polymorph (increase in size of originally available crystals) are clearly seen in the presence of α- and δ-phases (a), whereas only the recrystallisation of β- into the γ-polymorph (needles) is clearly seen if the β-phase alone is present (b). The figure is compiled from the original one published in ref. 152 and presented with permission from RSC. | ||
Dissolution and recrystallisation also play a crucial role in the transformations of molecular crystals on different types of mechanical treatment, such as milling, extruding, tableting, or grinding, which are important technological operations in the pharmaceutical industry.153 The solid materials are not merely comminuted during such treatment but also can undergo polymorphic transitions and amorphisation. The role of fluid phases in these transformations remains poorly understood. A liquid is often added to the solid sample on purpose, e.g. in order to facilitate comminution or facilitate a reaction. This is the case for wet milling and liquid-assisted grinding (LAG).153 In many cases, liquid is present inadvertently, either due to trace solvent following up-stream operations or sorbed from the atmosphere. The data on the high-pressure dissolution and recrystallisation in various pressure-transmitting fluids can contribute to understanding the complexity of this problem. This can also be illustrated by an example of the polymorphism of chlorpropamide.
The metastable β-polymorph of chlorpropamide transforms into different polymorphs when subjected to mechanical treatment of different types (restricted impact or grinding) in the presence of different fluids (ethanol, toluene, chloroform or heptane) that have affinity for different fragments of the CPA molecule (Fig. 5).154 The results can be compared with studies on the hydrostatic compression of the β-polymorph in pentane.152 The impact treatment of β-CPA in the presence of heptane, which is expected to interact with the alkyl tail of chlorpropamide molecules in a similar manner to that of pentane, led to near-complete conversion to the δ-phase. Grinding of the β-CPA under atmospheric conditions or in the presence of ethanol, toluene and chloroform led to conversion towards the α-form. Instead, grinding of β-CPA in the presence of heptane led to unmistakable conversion of the material into the γ-form in striking contrast to the effects of impact treatment on the β-form in the presence of the same solvent, which is another example (in addition to those reported before155) of the drastically different effects of grinding and impact on mechanically induced processes. Remarkably, in this case, the effect of hydrostatic compression was similar to that of grinding and different from that imposed by restricted impact. Opposite examples are also known, when the effect of hydrostatic pressure on a solid compound is similar to that of impact and is opposite to the outcome of grinding.156 Thus, high-pressure studies are an important addition to mechanochemical experiments using a variety of mechanoreactors, and they can assist in rationalising the complexity of mechanochemical transformations.
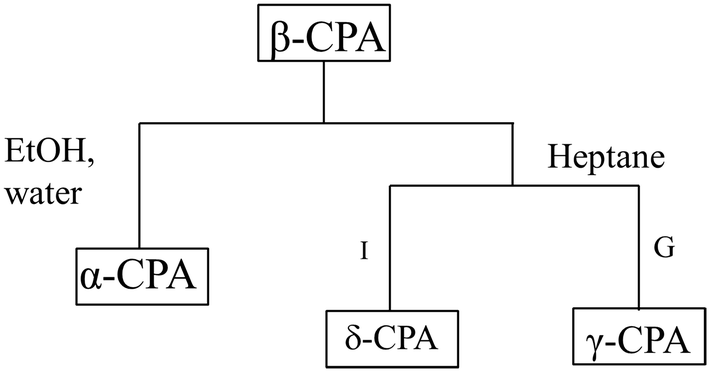 | ||
| Fig. 5 Schematic summary of the mechanically induced transformations of β-CPA. Conversion to the α-CPA in the presence of moisture, ethanol, chloroform or toluene (all types of treatment, left link). Specific reactivity under restricted impact (I), or grinding (G) for LAG with heptane (right link). Based on data from ref. 154. | ||
High-pressure polymorphism of chlorpropamide can also be affected by non-dissolving pressure-transmitting media.157 On hydrostatic compression β-chlorpropamide gives different high-pressure phases depending on the choice of “inert” pressure-transmitting fluid. The compression of this polymorph in three different pressure-transmitting media (paraffin, neon and helium) results in a series of phase transitions. None of these fluids can dissolve chlorpropamide under ambient conditions; two of them are inert gases. The different polymorphic transformations resulting from hydrostatic compression in different media can be rationalised by possible penetration of one of the fluids (helium) into the solid, and by likely surface interactions of the solid (alkyl tails of chlorpropamide) with the fluid (paraffin). Both phenomena have notable effects on the mechanical properties of the material being compressed. The high-pressure experiments, therefore, suggest that chlorpropamide can interact with “inert” fluids, stimulating new experiments also under atmospheric pressure aimed at characterizing the surface of these crystals (different faces of different polymorphs) and possible surface complexes formed with adsorbed molecules.
Solid-state structural transformations
One of the most common types of high-pressure studies involves looking for pressure-induced phase transitions and solving structures of the high-pressure phases. If a high-pressure phase can be quenched on decompression, then the new polymorph can be preserved under ambient conditions as a metastable form. These metastable forms have the potential as new materials or pharmaceutical forms with improved properties.158–160Pressure-induced polymorphism is often kinetically controlled.93,101,145,156,161–164 This means that the forms that are obtained on compression are often metastable forms rather than thermodynamically stable states. These metastable phases may remain kinetically trapped under pressure due to large nucleation barriers. Hence, the thermodynamically stable phase may not even be accessible in practice.
Recent decades have provided many examples of transformations at high pressure that were sensitive to the compression–decompression protocol, the size of particles (a single crystal or a powder sample with submicron particles), the choice of pressure-transmitting fluids and the presence of seeds of other phases. Even if these phenomena are observed for transformations at high pressures, they can contribute to our general understanding of the properties of the solid phase: they reveal the factors that are important for the nucleation and growth of new phases and suggest the mechanisms of the transformations. In particular, a comparison of the phases formed on hydrostatic compression with those that are formed on cooling (or sometimes on heating) is helpful to understand the microscopic mechanisms of solid-state transformations and their driving forces.
As a recent example, we can consider the role of kinetic factors in the polymorphic transformations in β-alanine. For this compound, a series of poorly reproducible phase transitions were reported to depend on the compression–decompression protocol.
Different polymorphs were formed if the sample was compressed quickly (reaching 8 GPa in a few hours) or slowly (reaching the same pressure within several days). Under quick compression, form II results on initial compression, transforming to phase III at higher pressures. In contrast, on the slow compression, phase IV is obtained, and it does not convert to II or III.164
Further experiments were carried out with this compound. A crystal of the ambient-pressure orthorhombic phase I of β-alanine was placed in a DAC and the pressure was increased to 6.3 GPa. This resulted in the phase transition to the orthorhombic high-pressure form II. When the sample was held at this pressure for 15 hours, the formation of a new monoclinic high-pressure phase V was detected.163 This phase appeared to be metastable. After holding at ∼6 GPa for approximately 30 hours, it transformed into a new disperse polycrystalline phase, which may correspond to the phase IV described in ref. 164. In an additional experiment, the pressure was increased directly to 8 GPa. The crystal of the initial form I underwent a phase transition to the high-pressure orthorhombic phase II, which was preserved for at least five months at this pressure163 without transformation to phase V. All of the observed routes of the high-pressure transformations of β-alanine are shown in Fig. 6. The abovementioned results suggest that the transformation of β-alanine II into the monoclinic polymorph V, while preserving the sample as a single crystal, is kinetically hindered.
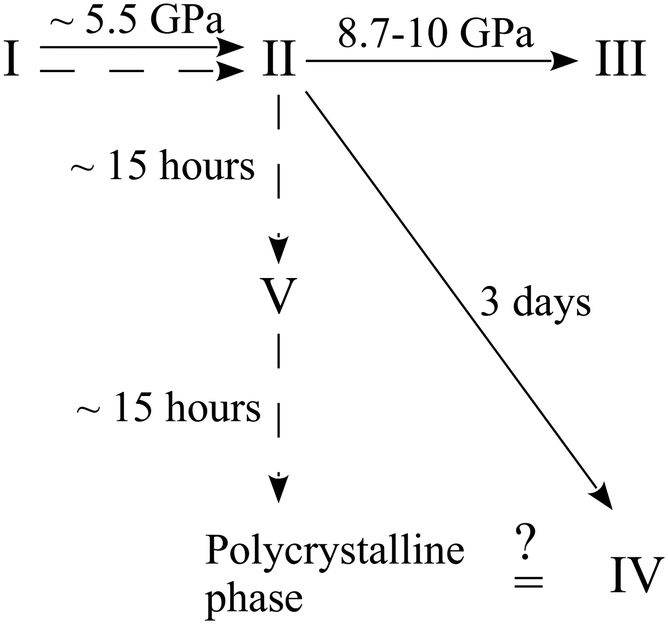 | ||
| Fig. 6 Phase transformation routes in β-alanine at high pressures according to ref. 163 and 164. The figure is reproduced from ref. 163 with permission from RSC. | ||
This is despite the fact that the molecular volume of polymorph V at ∼6 GPa is lower than that of polymorph II at 8 GPa. Polymorph V displays a molecular packing and hydrogen-bond network that is unique as compared to those in polymorphs I and II, which are closely related to each other. Polymorph II can even be considered as a strongly distorted and compressed form of phase I. Therefore, an immediate pressure increase to 8 GPa allows one to bypass the phase transition to phase V, even on very prolonged storage at this pressure, since the kinetic barrier of a dramatic rearrangement of the hydrogen-bonded network and molecular packing is too large under higher pressures.163
A similar behavior has also been observed for 2-fluorophenylacetylene165 and L-serine.162 For these compounds, the pressure increase rate had a strong influence on the structure of the high-pressure phases. High-pressure crystallisation of 2-fluorophenylacetylene yields three different polymorphs depending on the rate of compression.165 A slow and continuous compression of L-serine favors phase IV, whereas phase II can be obtained on rapid compression.162 Remarkably for L-serine, there is no observed direct interconversion route between the two possible high-pressure phases. Other examples of kinetically hindered transformations are also provided by paracetamol85,156 and L-alanine solvates,161 where low-pressure phases could be “over-pressurised” on fast compression and converted into the high-pressure phases on decompression. Studies that correlate the compression rates and resulting crystal structures provide crucial insight into the processes proceeding during the polymorph interconversion under non-ambient conditions. They also allow clear confirmation that kinetic factors and sample compression history should be taken into account during polymorph screening.
By studying pressure-induced phase transitions one can obtain better insight into the mechanisms of thermal structural transformations, photochemical solid-state reactions or solid-state dehydration that are observed at atmospheric pressure. This can be illustrated using several recent examples.
1,2,4,5-Tetrabromobenzene (TBB) is an extensively studied compound that provides a nice example of a strong mechanical response on heating: the thermosalient effect.59 This response is related to the phase transition from the low-temperature β- to the high-temperature γ-phase and results in crystal jumping and/or cracking.166–173 Initially, the thermosalient phenomenon in TBB was proposed to occur in two stages: (1) accumulation of strain as a result of the phase transition and (2) sudden release of strain, resulting in ballistic crystal displacement.171 The progression of this structural rearrangement throughout the crystal in a preferred direction was believed to make the crystal move, jumping across distances significantly exceeding its own size.171 Based on Brillouin light scattering spectroscopy,173 the lowest transverse acoustic mode was shown to exhibit a substantial softening while approaching the phase transition on heating. This was suggested to indicate that the transition is driven by the elastic instability of this soft acoustic mode. This hypothesis seems to be very attractive. However, it does not explain why such a strong mechanical response is observed on transformation between the two phases, which are structurally very similar (Fig. 7) and require only subtle molecular rearrangements to interconvert.
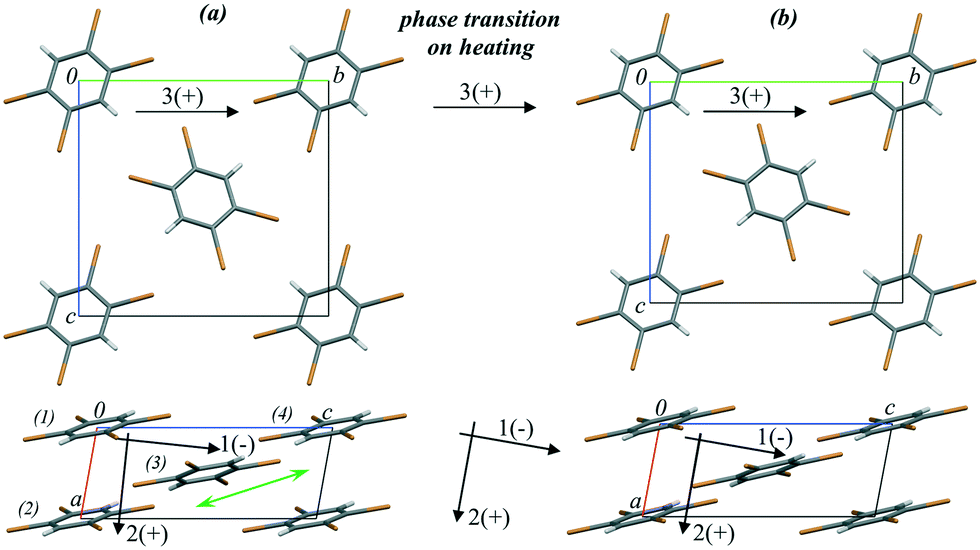 | ||
| Fig. 7 Crystal structure fragments of TBB (a) before (303 K) and (b) after (313 K) the phase transition from the β- to the γ-phase on heating. The arrows show the directions of the principal axes of the strain ellipsoid before, during and after the phase transition. (+) and (−) signs indicate positive and negative deformations on heating, respectively. The green double arrow shows the direction of the relative shift of the TBB layers during the phase transition. The figure is reproduced from ref. 166 with the permission of RSC. | ||
In a further study the changes in the atomic coordinates and the lattice strain anisotropy for both the low- and the high-temperature phases were followed by single-crystal X-ray diffraction at multiple temperature points on heating. Strain on heating as well as that induced by the phase transition was calculated.166 Raman spectroscopy and computational techniques were used to follow the vibrational spectra, lattice energies and non-covalent interactions in relation to the thermosalient phase transition.166 The structure was found to soften on heating before the phase transition, in agreement with the Brillouin light scattering spectroscopy results.173 The phase transition on further heating was accompanied by a significant anisotropic lattice deformation. The crystal structure became more rigid on further temperature increase after the phase transition.166 Therefore, both the low- and the high-temperature phases were shown to be the softest at temperatures close to the phase transition point. The results of phonon calculations indicated a reorientation of the phonon polarisation axes for softening modes across the phase transition. The delay of mechanical response on phase transition171 was suggested to be related to the coexistence of β- and γ-TBB domains near the phase transition point.166 The phase boundaries accumulated structural stress, which could not be expected to be too large judging from the subtle structural differences alone. However, in this particular case a set of perpendicularly polarised lattice modes introduce a dynamic stress to this boundary. These modes are responsible for considerable stress within the newly formed γ-TBB phase, particularly as they are associated with drastic compression along the principal strain axis across the phase transition.166 Based on this information it was possible to suggest that rapid release of this stress in a concerted manner on further heating (leading to completion of the phase transition) results in a large mechanical response. Thus, the thermosalient effect is not a direct result of the transition itself, but a by-product of it.166 The martensitic nature of this phase transition also suggested an explanation for the delay of the mechanical response with respect to the structural transformation. The transformation appears complete when studied by single-crystal X-ray diffraction due to the similarity of the phases. However, some inclusions of the parent β-phase still remain un-transformed. The model based on the detailed variable-temperature study of the structural changes in the TBB crystals also suggests that the transformation can be hindered and even completely prevented by hydrostatic compression. The conclusions from these studies of TBB can be applicable to other thermosalient compounds.
A structural phase transition observed in [Co(NH3)5NO2]Br2 between 6 and 7 GPa (ref. 174) adds to our understanding of the mechanism of the nitro–nitrito isomerisation in this compound without external load. This high-pressure phase transition is accompanied by a change of crystal system from monoclinic to orthorhombic. However, no discontinuities have been noticed in the dependence of the cell volume on pressure. The main structural rearrangement is related to the discontinuous rotation of the NO2 ligands.174 The most interesting feature of this pressure-induced phase transition is that the crystal structure of the resulting high-pressure phase of the compound is very similar to that of the product of the light-induced linkage isomerisation. Packing of complex cations and bromide anions in the high-pressure and photoisomerised phases is effectively the same. The only difference is in the coordination of the nitro group (the N-coordinated NO2 in the high-pressure phase and the O-coordinated ONO in the isomerisation product) (Fig. 8).51,68,138 Accounting for the effect of pressure on the rate of thermal isomerisation in [Co(NH3)5ONO]Br2,54 one can expect that pressure should have a pronounced effect on the yield of the linkage photoisomerisation in [Co(NH3)5NO2]Br2, which has in fact been confirmed in a recent study.175
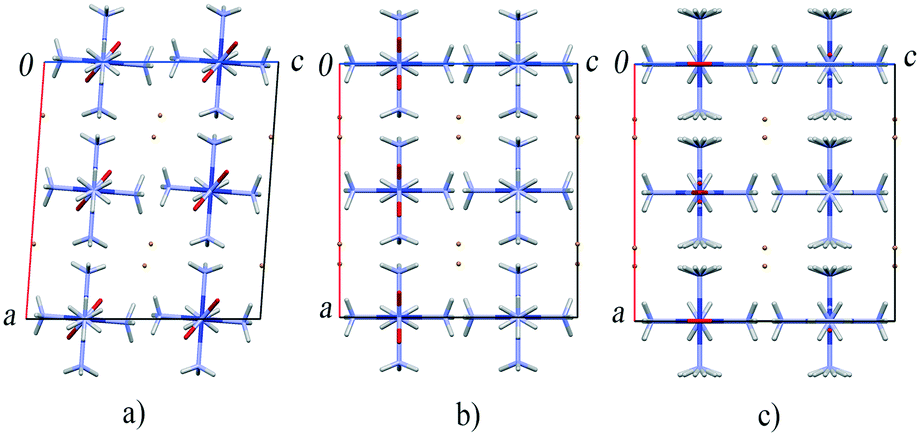 | ||
| Fig. 8 The fragments of the crystal structure of [Co(NH3)5NO2]Br2 at ambient pressure (a), after a high-pressure phase transition above 6 GPa (b) and after the photochemical transformation into [Co(NH3)5ONO]Br2 isomer (c).51,68,138,174 | ||
As a final example, we chose a study in which a high-pressure structural transformation was used to rationalise the product of a solid-state dehydration of the same compound that takes place at atmospheric pressure. Y(III) and Sm(III) form isostructural compounds with the general composition (REE)2(C2O4)3·10H2O, which undergo dehydration on heating to give hexahydrates (REE)2(C2O4)3·6H2O as intermediate products.176 Although the initial decahydrate compounds Sm2(C2O4)3·10H2O and Y2(C2O4)3·10H2O are isostructural, their dehydration products have different crystal structures, even if the water content of the product is the same.177,178 Sm2(C2O4)3·10H2O gives a solely monoclinic hexahydrate on thermal dehydration, but the behavior of Y2(C2O4)3·10H2O appears to be more complex. If Y2(C2O4)3·10H2O dehydrates in air at temperatures around 40–50 °C, the triclinic crystalline polymorph of the hexahydrate is formed.177 Heating to higher temperatures (∼85 °C) in a closed container or in silicon oil results in the formation of a monoclinic hexahydrate polymorph, which is isostructural to Sm2(C2O4)3·6H2O.177,179 This example clearly shows that the size of the cation can influence the dehydration mechanism and the structure of the product.
The abovementioned crystals undergo a significant shape change on dehydration. The difference in the crystal structures of the reaction products can therefore be sought in the different mechanical stresses that arise over the course of the transformation. The structural changes accompanying a solid-state transformation are influenced strongly by the mechanical properties of the sample. High-pressure X-ray diffraction studies appeared to be one of the most efficient techniques to study these properties, to follow the anisotropy of lattice strain and to measure the bulk compressibility. X-ray diffraction techniques also permitted monitoring of structural transformations and changes in the atomic coordinates. Isostructural Sm2(C2O4)3·10H2O, Y2(C2O4)3·10H2O, and their 1:1 solid solution SmY(C2O4)3·10H2O were studied on hydrostatic compression up to ∼6 GPa.179 Despite the similarities of the crystal structures of the parent crystals, high-pressure experiments revealed that the anisotropy of lattice strain on hydrostatic compression of these compounds is different. The analysis of the changes in the atomic coordinates and the crystal structure voids at different pressures allowed us to propose different mechanisms of crystal structure compression for the Sm- and Y-based salts. In the Sm-based salt, compression of the crystal structure is explained by a relatively soft deformation of the coordination polyhedra in the metal-oxalate layer. In the Y-based salt the coordination polyhedra are stressed due to shorter distances between oxygen atoms. Therefore, deformation of the polyhedra in the metal-oxalate layers is difficult. The overall reduction in volume that occurs on increased pressure is caused by ordering of the outer-sphere water molecules. The different mechanisms of dehydration and the formation of the monoclinic and triclinic crystalline products of Sm2(C2O4)3·6H2O and Y2(C2O4)3·6H2O can therefore be explained by the significant differences in the anisotropic response of the isostructural compounds on hydrostatic compression caused by anisotropy of the lattice deformation.179 The fact that the dehydration mechanisms of Sm2(C2O4)3·10H2O and Y2(C2O4)3·10H2O are different can also be confirmed by an unusual observation. The crystals of Sm2(C2O4)3·10H2O and SmY(C2O4)3·10H2O remained intact on compression to 6 GPa. However, a single-crystalline inclusion appeared in the sample of Y2(C2O4)3·10H2O at a pressure as low as 1 GPa (Fig. 9). No further transformation occurred within the remaining parts of the initial crystal. The diffraction experiment revealed that this phase could be indexed in the monoclinic crystal system. Together with the orientation of this phase in relation to the edges of the initial crystal (Fig. 9) and the similarity of lattice parameters, this suggested that the structure of the newly formed inclusion should correspond to the monoclinic polymorph of Y2(C2O4)3·6H2O.177,179 According to the phase diagram of water, the dehydration of Y2(C2O4)3·10H2O is in principle possible at pressures above 1 GPa, where the formation of ice VI provides an overall reduction in volume.179 The fact that Sm2(C2O4)3·10H2O and SmY(C2O4)3·10H2O do not dehydrate on increasing pressure suggests that the dehydration process in Y2(C2O4)3·10H2O is triggered by a higher strain of the Y-based metal-oxalate framework.179 In this case, the application of high-pressure X-ray diffraction studies revealed different mechanical properties of the isostructural hydrated salts formed by different metals. This information offered an explanation as to why the dehydration products have different crystal structures, even though the parent phases were isostructural.
 | ||
| Fig. 9 Dehydration of Y2(C2O4)3·10H2O in silicon oil (a) and on hydrostatic compression at 1 GPa (b). The streaks correspond to the monoclinic phase of Y2(C2O4)3·6H2O formed on partial dehydration. The figure is compiled from the original ones published in ref. 179 and presented with the permission of De Gruyter. | ||
Conclusions
Information regarding the changes that occur in a crystal structure as a function of pressure is of great importance for chemistry, geology, physics and materials science. Many phenomena observed at high pressures are interesting in themselves. However, the information gained by application of pressure can also offer invaluable insight into phenomena that are observed without applying any external load to the sample, under atmospheric pressure, e.g. transformations on heating, cooling, or on irradiation with light. This is particularly true when these transformations are accompanied by stress and strain generation. Analogy can be made to human psychology: probing a human under extreme conditions helps to reveal some features that may be less evident in “normal life”, but nonetheless exist and may influence a person's “normal-life behavior”. High-pressure investigations probe deeper into the mechanisms of crystallisation, phase transitions, and temperature- and light-induced solid-state chemical reactions as well as mechanochemical transformations on grinding. Structural studies under extreme conditions can help to rationalise the thermo- and photosalient effects that accompany solid-state transformations. High-pressure research is therefore not a narrow research field of interest for a limited group of people but is of general interest for solid-state chemists and materials scientists.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. A. Z. acknowledges the support by a grant from the Russian Foundation for Basic Research (RFBR), research project No. 16-33-60093 mol_a_dk. E. V. B. acknowledges the support by Russian Ministry of Science and High Education. B. A. Z and E. V. B. thank Mrs. Evgeniya Kolesnik, Mr. Nikita Bogdanov for technical assistance and Mr. Adam Michalchuk for valuable advice and language polishing.Notes and references
- W. L. Bragg, Proc. R. Soc. London, Ser. A, 1914, 89, 468–489 CrossRef CAS.
- R. L. McFarlan, J. Chem. Phys., 1936, 4, 253–259 CrossRef CAS.
- E. D. Eastman, J. Am. Chem. Soc., 1924, 46, 917–923 CrossRef CAS.
- F. Rinne, Berichte Verein Sächsischer Akademie der Wissenschaften. Mathematik – Physik, 1917, 69, 57–62 Search PubMed.
- A. S. John, Proc. Natl. Acad. Sci. U. S. A., 1918, 4, 193–197 CrossRef CAS.
- D. M. Dennison, Phys. Rev., 1921, 17, 20–22 CrossRef CAS.
- A. W. Hull, Phys. Rev., 1917, 10, 661–696 CrossRef CAS.
- E. G. Cox, Nature, 1928, 122, 401–401 CrossRef CAS.
- A. R. Nichols and J. H. Walton, J. Am. Chem. Soc., 1942, 64, 1866–1870 CrossRef CAS.
- H. Kersten, Physics, College Park Md, 1932, vol. 2, pp. 276–279 Search PubMed.
- W. L. Baun, J. Phys. Chem., 1961, 65, 2122–2126 CrossRef CAS.
- F. C. Todd, Phys. Rev., 1933, 44, 787–793 CrossRef CAS.
- A. de Bretteville and J. W. McBain, J. Chem. Phys., 1943, 11, 426–429 CrossRef CAS.
- A. de Bretteville, Rev. Sci. Instrum., 1942, 13, 481–483 CrossRef.
- D. F. Clifton, Rev. Sci. Instrum., 1950, 21, 339–342 CrossRef.
- P. W. Bridgman, Physics of high pressure, Bell, London, 1949 Search PubMed.
- W. M. Cohn, Phys. Rev., 1933, 44, 326–327 CAS.
- R. M. Hazen, The Diamond Makers, Cambridge University Press, Cambridge, 1999 Search PubMed.
- S. M. Stishov, L. G. Khvostantsev, V. N. Slesarev, S. V. Popova, V. V. Brazhkin, T. I. Dyuzheva, L. N. Dzhavadov, E. L. Gromnitskaya, G. N. Stepanov, Y. A. Timofeev, E. M. Dizhur, V. A. Ventsel, A. N. Voronovskiy, V. N. Ryzhov, A. F. Barabanov, M. V. Magnitskaya and E. E. Tareeva, Phys.-Usp., 2008, 51, 1055–1083 CrossRef.
- A. Van Valkenburg, Rev. Sci. Instrum., 1962, 33, 1462–1462 CrossRef.
- C. E. Weir, E. R. Lippincott, A. Van Valkenburg and E. N. Bunting, J. Res. Natl. Bur. Stand., Sect. A, 1959, 63, 55–62 CrossRef.
- J. C. Jamieson, A. W. Lawson and N. D. Nachtrieb, Rev. Sci. Instrum., 1959, 30, 1016–1019 CrossRef CAS.
- H. Ahsbahs, Z. Kristallogr., 2004, 219, 305–308 CAS.
- L. Dubrovinsky, T. Boffa-Ballaran, K. Glazyrin, A. Kurnosov, D. Frost, M. Merlini, M. Hanfland, V. B. Prakapenka, P. Schouwink, T. Pippinger and N. Dubrovinskaia, High Pressure Res., 2010, 30, 620–633 CrossRef CAS.
- I. Kantor, V. Prakapenka, A. Kantor, P. Dera, A. Kurnosov, S. Sinogeikin, N. Dubrovinskaia and L. Dubrovinsky, Rev. Sci. Instrum., 2012, 83, 125102 CrossRef CAS PubMed.
- A. Dawson, D. R. Allan, S. Parsons and M. Ruf, J. Appl. Crystallogr., 2004, 37, 410–416 CrossRef CAS.
- H. Sowa and H. Ahsbahs, J. Appl. Crystallogr., 2006, 39, 169–175 CrossRef CAS.
- R. Boehler, Rev. Sci. Instrum., 2006, 77, 115103 CrossRef.
- S. A. Moggach, D. R. Allan, S. Parsons and J. E. Warren, J. Appl. Crystallogr., 2008, 41, 249–251 CrossRef CAS.
- E. V. Boldyreva, B. A. Zakharov, S. V. Rashchenko, Y. V. Seryotkin and N. A. Tumanov, Studying Solid-State Transformations Using In Situ X-Ray Diffraction Studies at High-Pressures, Publishing House of Siberian Branch of Russian Academy of Sciences, Novosibirsk, 2016 Search PubMed.
- A. Katrusiak, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 135–148 CrossRef CAS PubMed.
- A. Katrusiak, Z. Kristallogr., 2004, 219, 461–467 CAS.
- P. Dera, K. Zhuravlev, V. Prakapenka, M. L. Rivers, G. J. Finkelstein, O. Grubor-Urosevic, O. Tschauner, S. M. Clark and R. T. Downs, High Pressure Res., 2013, 33, 466–484 CrossRef CAS.
- N. Casati, P. Macchi and A. Sironi, J. Appl. Crystallogr., 2007, 40, 628–630 CrossRef CAS.
- R. Angel and J. Gonzalez-Platas, J. Appl. Crystallogr., 2013, 46, 252–254 CrossRef CAS.
- J. Veciana, M. Souto, M. C. Gullo, H. Cui, N. Casati, F. Montisci, H. O. Jeschke, R. Valentí, I. Ratera and C. Rovira, Chem. – Eur. J., 2018, 24, 5500–5505 CrossRef PubMed.
- N. Casati, A. Genoni, B. Meyer, A. Krawczuk and P. Macchi, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 584–597 CrossRef CAS PubMed.
- N. Casati, A. Kleppe, A. P. Jephcoat and P. Macchi, Nat. Commun., 2016, 7, 10901 CrossRef PubMed.
- R. M. Hazen and L. W. Finger, Comparative Crystal Chemistry: Temperature, Pressure, Composition, and the Variation of Crystal Structure, Wiley, New York, 1982 Search PubMed.
- R. Fourme, E. Girard, R. Kahn, A.-C. Dhaussy and I. Ascone, Annu. Rev. Biophys., 2009, 38, 153–171 CrossRef CAS PubMed.
- High-Pressure Crystallography, ed. A. Katrusiak and P. McMillan, Springer Netherlands, Dordrecht, 2004 Search PubMed.
- High-Pressure Crystallography, ed. E. Boldyreva and P. Dera, Springer Netherlands, Dordrecht, 2010 Search PubMed.
- T. Asano and W. J. Le Noble, Chem. Rev., 1978, 78, 407–489 CrossRef CAS.
- J. Dibenedetto and P. C. Ford, Coord. Chem. Rev., 1985, 64, 361–382 CrossRef CAS.
- M. Kotowski and R. van Eldik, Coord. Chem. Rev., 1989, 93, 19–57 CrossRef CAS.
- R. Van Eldik and A. E. Merbach, Comments Inorg. Chem., 1992, 12, 341–378 CrossRef CAS.
- G. Stochel, Coord. Chem. Rev., 1992, 114, 269–295 CrossRef CAS.
- E. Boldyreva, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1994, 242, 17–52 CrossRef.
- E. V. Boldyreva, Russ. J. Coord. Chem., 2001, 27, 297–323 CrossRef CAS.
- Reactivity of Molecular Solids, Molecular Solid State Series, ed. E. V. Boldyreva and V. V. Boldyrev, Wiley, Chichester, vol. 3, 1999 Search PubMed.
- E. V. Boldyreva, H. Ahsbahs and H. Uchtmann, Ber. Bunsen-Ges., 1994, 98, 738–745 CrossRef CAS.
- E. V. Boldyreva, L. P. Burleva, E. B. Burgina, V. P. Baltachinov, H. Ahsbahs, H. Uchtmann and V. E. Doulepov, Ber. Bunsen-Ges., 1992, 96, 931–937 CrossRef CAS.
- E. V. Boldyreva, S. L. Kuzmina and H. Ahsbahs, J. Struct. Chem., 1998, 39, 343–349 CrossRef CAS.
- E. V. Boldyreva, S. L. Kuzmina and H. Ahsbahs, J. Struct. Chem., 1998, 39, 762–773 CrossRef CAS.
- E. V. Boldyreva, D. Y. Naumov and H. Ahsbahs, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 798–808 CrossRef.
- A. P. Chupakhin, A. A. Sidel'nikov and V. V. Boldyrev, React. Solids, 1987, 3, 1–19 CrossRef CAS.
- T. Luty and C. J. Eckhardt, J. Am. Chem. Soc., 1995, 117, 2441–2452 CrossRef CAS.
- N. M. Peachey and C. J. Eckhardt, J. Phys. Chem., 1993, 97, 10849–10856 CrossRef CAS.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed.
- E. V. Boldyreva, Solid State Ionics, 1997, 101–103, 843–849 CrossRef CAS.
- V. M. Tapilin, N. N. Bulgakov, A. P. Chupakhin, A. A. Politov and A. G. Druganov, J. Struct. Chem., 2010, 51, 635–641 CrossRef CAS.
- S. Chizhik, A. Sidelnikov, B. Zakharov, P. Naumov and E. Boldyreva, Chem. Sci., 2018, 9, 2319–2335 RSC.
- V. V. Boldyrev, Russ. Chem. Rev., 1973, 42, 515 CrossRef.
- V. V. Boldyrev, Russ. Chem. Bull. Int. Ed., 2018, 67, 933–948 CrossRef CAS.
- J. M. McBride, Acc. Chem. Res., 1983, 16, 304–312 CrossRef CAS.
- J. M. McBride, B. E. Segmuller, M. D. Hollingsworth, D. E. Mills and B. A. Weber, Science, 1986, 234, 830–835 CrossRef CAS PubMed.
- E. V. Boldyreva, in Reactivity of solids: past, present and future, ed. V. V. Boldyrev, Blackwell Science Cambridge, 1996, pp. 141–184 Search PubMed.
- N. Masciocchi, A. Kolyshev, V. Dulepov, E. Boldyreva and A. Sironi, Inorg. Chem., 1994, 33, 2579–2585 CrossRef CAS.
- P. Naumov, S. C. Sahoo, B. A. Zakharov and E. V. Boldyreva, Angew. Chem., Int. Ed., 2013, 52, 9990–9995 CrossRef CAS PubMed.
- J. Gonzalez-Platas, M. Alvaro, F. Nestola and R. Angel, J. Appl. Crystallogr., 2016, 49, 1377–1382 CrossRef CAS.
- K. S. Scheidl, A. Kurnosov, D. M. Trots, T. Boffa Ballaran, R. J. Angel and R. Miletich, J. Appl. Crystallogr., 2016, 49, 2129–2137 CrossRef CAS.
- J. Buchen, H. Marquardt, T. Boffa Ballaran, T. Kawazoe and C. McCammon, Am. Mineral., 2017, 102, 2494–2504 CrossRef.
- Z. Mao, F. Wang, J.-F. Lin, S. Fu, J. Yang, X. Wu, T. Okuchi, N. Tomioka, V. B. Prakapenka, Y. Xiao and P. Chow, Am. Mineral., 2017, 102, 357–368 CrossRef.
- H. Grüninger, K. Armstrong, D. Greim, T. Boffa-Ballaran, D. J. Frost and J. Senker, J. Am. Chem. Soc., 2017, 139, 10499–10505 CrossRef PubMed.
- K. Schulze, J. Buchen, K. Marquardt and H. Marquardt, High Pressure Res., 2017, 37, 159–169 CrossRef CAS.
- R. J. Angel, M. Alvaro and F. Nestola, Phys. Chem. Miner., 2018, 45, 95–113 CrossRef CAS.
- J. F. Nye, Physical properties of crystals: their representation by tensors and matrices, Oxford University Press, 1985 Search PubMed.
- J. M. Robertson and A. R. Ubbelohde, Proc. R. Soc. London, Ser. A, 1939, 170, 222–240 CrossRef CAS.
- A. R. Ubellohde, Proc. R. Soc. London, Ser. A, 1939, 173, 417–427 CrossRef.
- A. R. Ubbelohde and I. Woodward, Proc. R. Soc. London, Ser. A, 1946, 185, 448–465 CrossRef CAS.
- K. J. Gallagher, A. R. Ubbelohde and I. Woodward, Acta Crystallogr., 1955, 8, 561–566 CrossRef CAS.
- A. Katrusiak, Cryst. Res. Technol., 1991, 26, 523–531 CrossRef CAS.
- E. Boldyreva, J. Kivikoski and J. A. K. Howard, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 394–404 CrossRef.
- E. Boldyreva, J. Kivikoski and J. A. K. Howard, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 405–414 CrossRef.
- E. V. Boldyreva, T. P. Shakhtshneider, M. A. Vasilchenko, H. Ahsbahs and H. Uchtmann, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 299–309 CrossRef.
- E. V. Boldyreva, T. P. Shakhtshneider, H. Ahsbahs, H. Uchtmann, E. B. Burgina and V. P. Baltakhinov, Pol. J. Chem., 2002, 76, 1333–1346 CAS.
- E. V. Boldyreva, Cryst. Eng., 2003, 6, 235–254 CrossRef CAS.
- E. V. Boldyreva, Phase Transitions, 2009, 82, 303–321 CrossRef CAS.
- E. V. Boldyreva, T. N. Drebushchak and T. P. Shakhtshneider, ARKIVOC, 2004, 12, 128–155 Search PubMed.
- I. Orgzall, F. Emmerling, B. Schulz and O. Franco, J. Phys.: Condens. Matter, 2008, 20, 295206 CrossRef.
- M. Podsiadło and A. Katrusiak, J. Phys. Chem. B, 2008, 112, 5355–5362 CrossRef PubMed.
- W. Marczak, M. A. Varfolomeev, I. T. Rakipov, P. Lodowski, K. Kowalska-Szojda, M. Łężniak, L. Almásy and A. Len, J. Phys. Chem. B, 2017, 121, 3070–3086 CrossRef CAS PubMed.
- A. Y. Fedorov, D. A. Rychkov, E. A. Losev, B. A. Zakharov, J. Stare and E. V. Boldyreva, CrystEngComm, 2017, 19, 2243–2252 RSC.
- S. Nuzzo, B. Twamley and R. J. Baker, J. Chem. Crystallogr., 2017, 47, 182–186 CrossRef CAS.
- S. G. Patra, N. Mandal, A. Datta and D. Datta, Comput. Theor. Chem., 2017, 1114, 118–124 CrossRef CAS.
- D. Paliwoda, M. Szafrański, M. Hanfland and A. Katrusiak, J. Mater. Chem. C, 2018, 6, 7689–7699 RSC.
- A. S. Batsanov, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2018, 74, 570–574 CrossRef CAS PubMed.
- G. A. Craig, A. Sarkar, C. H. Woodall, M. A. Hay, K. E. R. Marriott, K. V. Kamenev, S. A. Moggach, E. K. Brechin, S. Parsons, G. Rajaraman and M. Murrie, Chem. Sci., 2018, 9, 1551–1559 RSC.
- E. V. Boldyreva, in Understanding Intermolecular Interactions in the Solid State, ed. D. Chopra, RSC Publishing, 2018, pp. 32–97 Search PubMed.
- G. Mínguez Espallargas, L. Brammer, D. R. Allan, C. R. Pulham, N. Robertson and J. E. Warren, J. Am. Chem. Soc., 2008, 130, 9058–9071 CrossRef PubMed.
- R. Gajda, A. Katrusiak and J. Crassous, CrystEngComm, 2009, 11, 2668–2676 RSC.
- B. A. Zakharov, B. A. Kolesov and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 275–286 CrossRef CAS PubMed.
- I. F. Bruce-Smith, B. A. Zakharov, J. Stare, E. V. Boldyreva and C. R. Pulham, J. Phys. Chem. C, 2014, 118, 24705–24713 CrossRef CAS.
- E. V. Boldyreva, Z. Kristallogr. - Cryst. Mater., 2014, 229, 236–245 CAS.
- W. Cai and A. Katrusiak, Nat. Commun., 2014, 5, 4337 CrossRef CAS PubMed.
- G. Resnati, E. Boldyreva, P. Bombicz and M. Kawano, IUCrJ, 2015, 2, 675–690 CrossRef CAS PubMed.
- M. Podsiadło, A. Olejniczak and A. Katrusiak, Cryst. Growth Des., 2017, 17, 2218–2222 CrossRef.
- A. V. Dzyabchenko and E. V. Boldyreva, Acta Crystallogr., Sect. A: Found. Crystallogr., 2000, 56(Supplement), s215 Search PubMed.
- E. V. Boldyreva, H. Ahsbahs, V. V. Chernyshev, S. N. Ivashevskaya and A. R. Oganov, Z. Kristallogr., 2006, 221, 186–197 CAS.
- M. V. Aleinikova, Y. N. Zhuravlev and D. V. Korabelnikov, Russ. Phys. J., 2012, 55, 495–500 CrossRef CAS.
- K. Adhikari, K. M. Flurchick and L. Valenzano, Comput. Theor. Chem., 2015, 1062, 90–98 CrossRef CAS.
- J. Nyman, O. S. Pundyke and G. M. Day, Phys. Chem. Chem. Phys., 2016, 18, 15828–15837 RSC.
- D. A. Rychkov, J. Stare and E. V. Boldyreva, Phys. Chem. Chem. Phys., 2017, 19, 6671–6676 RSC.
- C. L. Hobday, C. H. Woodall, M. J. Lennox, M. Frost, K. Kamenev, T. Düren, C. A. Morrison and S. A. Moggach, Nat. Commun., 2018, 9, 1429 CrossRef PubMed.
- E. Tailleur, M. Marchivie, J.-P. Itié, P. Rosa, N. Daro and P. Guionneau, Chem. – Eur. J., 2018, 24, 14495–14499 CrossRef CAS PubMed.
- X. Jiang, S. Luo, L. Kang, P. Gong, W. Yao, H. Huang, W. Li, R. Huang, W. Wang, Y. Li, X. Li, X. Wu, P. Lu, L. Li, C. Chen and Z. Lin, Adv. Mater., 2015, 27, 4851–4857 CrossRef CAS PubMed.
- Y. Yang, X. Jiang, P. Gong, M. S. Molokeev, X. Li, Y. Li, X. Wu, Y. Wu and Z. Lin, RSC Adv., 2017, 7, 2038–2043 RSC.
- X. Jiang, Y. Yang, M. S. Molokeev, P. Gong, F. Liang, S. Wang, L. Liu, X. Wu, X. Li, Y. Li, S. Wu, W. Li, Y. Wu and Z. Lin, Adv. Mater., 2018, 30, 1801313 CrossRef PubMed.
- K. V. Kamenev, M. R. Lees, G. Balakrishnan, D. M. Paul, W. G. Marshall, V. G. Tissen and M. V. Nefedova, Phys. Rev. Lett., 2000, 84, 2710–2713 CrossRef CAS PubMed.
- Y. Sun, J. Kamarad, Z. Arnold, Z. Kou and Z. Cheng, Appl. Phys. Lett., 2006, 88, 102505 CrossRef.
- P. J. Byrne, P. J. Richardson, J. Chang, A. F. Kusmartseva, D. R. Allan, A. C. Jones, K. V. Kamenev, P. A. Tasker and S. Parsons, Chem. – A Eur. J., 2012, 18, 7738–7748 CrossRef CAS PubMed.
- J. A. Gould, M. J. Rosseinsky and S. A. Moggach, Dalton. Trans., 2012, 41, 5464–5467 RSC.
- L. Egan, K. Kamenev, D. Papanikolaou, Y. Takabayashi and S. Margadonna, J. Am. Chem. Soc., 2006, 128, 6034–6035 CrossRef CAS PubMed.
- Ł. Lindner, M. Zdanowska-Frączek, A. Pawłowski, Z. J. Frączek and T. Masłowski, J. Appl. Phys., 2017, 122, 035105 CrossRef.
- N. Bajaj, H. Bhatt, K. K. Pandey, H. K. Poswal, A. Arya, P. S. Ghosh, N. Garg and M. N. Deo, CrystEngComm, 2018, 20, 3728–3740 RSC.
- G. G. Levchenko, L. V. Berezhnaya, G. G. Filimonov and W. Han, J. Phys. Chem. B, 2018, 122, 6846–6853 CrossRef CAS PubMed.
- E. V. Boldyreva and A. A. Sidelnikov, Izv. Sib. Otd. Akad. Nauk SSSR, Ser. Khim. Nauk, 1987, 5, 139–145 Search PubMed.
- L. Zhu, F. Tong, R. O. Al-Kaysi and C. J. Bardeen, in Photomechanical Materials, Composites, and Systems: Wireless Transduction of Light into Work, ed. T. J. White, John Wiley & Sons, 2017, pp. 233–274 Search PubMed.
- M. Kim, J.-H. Yun and M. Cho, Sci. Rep., 2017, 7, 967 CrossRef PubMed.
- P. Guionneau and E. Collet, in Spin-Crossover Materials, John Wiley & Sons Ltd, Oxford, UK, 2013, pp. 507–526 Search PubMed.
- J. Bąkowicz and I. Turowska-Tyrk, J. Photochem. Photobiol., A, 2012, 232, 41–43 CrossRef.
- K. Konieczny, J. Bąkowicz and I. Turowska-Tyrk, CrystEngComm, 2015, 17, 7693–7701 RSC.
- K. Konieczny, J. Bąkowicz and I. Turowska-Tyrk, J. Photochem. Photobiol., A, 2016, 325, 111–115 CrossRef CAS.
- J. Bąkowicz and I. Turowska-Tyrk, CrystEngComm, 2016, 18, 8898–8905 RSC.
- T. Galica, J. Bąkowicz, K. Konieczny and I. Turowska-Tyrk, CrystEngComm, 2016, 18, 8871–8879 RSC.
- K. Konieczny, J. Bąkowicz, T. Galica, R. Siedlecka and I. Turowska-Tyrk, CrystEngComm, 2017, 19, 3044–3050 RSC.
- T. Galica, J. Bąkowicz, K. Konieczny and I. Turowska-Tyrk, Cryst. Growth Des., 2018, 18, 1636–1644 CrossRef CAS.
- E. V. Boldyreva, N. V. Podberezskaya, A. V. Virovets, L. P. Burleva and V. E. Dulepov, J. Struct. Chem., 1993, 34, 128–138 CAS.
- E. V. Boldyreva, A. A. Sidelnikov, A. P. Chupakhin, N. Z. Lyakhov and V. V. Boldyrev, Dokl. Akad. Nauk SSSR, 1984, 277, 893–896 CAS.
- A. A. Sidelnikov, S. A. Chizhik, B. A. Zakharov, A. P. Chupakhin and E. V. Boldyreva, CrystEngComm, 2016, 18, 7276–7283 RSC.
- J. Marciniak and A. Katrusiak, J. Phys. Chem. C, 2017, 121, 22303–22309 CrossRef CAS.
- A. J. Brock, J. J. Whittaker, J. A. Powell, M. C. Pfrunder, A. Grosjean, S. Parsons, J. C. McMurtrie and J. K. Clegg, Angew. Chem., Int. Ed., 2018, 57, 11325–11328 CrossRef CAS PubMed.
- E. V. Boldyreva, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 218–231 CrossRef CAS PubMed.
- R. Lee, J. A. K. Howard, M. R. Probert and J. W. Steed, Chem. Soc. Rev., 2014, 43, 4300–4311 RSC.
- E. Boldyreva, Cryst. Growth Des., 2007, 7, 1662–1668 CrossRef CAS.
- A. P. Ayala, M. W. C. Caetano, S. B. Honorato, J. Mendes Filho, H. W. Siesler, S. N. Faudone, S. L. Cuffini, F. T. Martins, C. C. P. da Silva and J. Ellena, J. Raman Spectrosc., 2012, 43, 263–272 CrossRef CAS.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed.
- V. A. Drebushchak, T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, J. Therm. Anal. Calorim., 2008, 93, 343–351 CrossRef CAS.
- T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o4393–o4395 CrossRef CAS.
- T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o355–o357 CrossRef CAS PubMed.
- T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o623–o625 CrossRef CAS PubMed.
- B. A. Zakharov, S. V. Goryainov and E. V. Boldyreva, CrystEngComm, 2016, 18, 5423–5428 RSC.
- E. Boldyreva, Curr. Pharm. Des., 2016, 22, 4981–5000 CrossRef CAS PubMed.
- N. Bouvart, R.-M. Palix, S. G. Arkhipov, I. A. Tumanov, A. A. L. Michalchuk and E. V. Boldyreva, CrystEngComm, 2018, 20, 1797–1803 RSC.
- I. A. Tumanov, A. F. Achkasov, S. A. Myz, E. V. Boldyreva and V. V. Boldyrev, Dokl. Chem., 2014, 457, 154–159 CrossRef CAS.
- E. V. Boldyreva, T. P. Shakhtshneider, H. Ahsbahs, H. Sowa and H. Uchtmann, J. Therm. Anal. Calorim., 2002, 68, 437–452 CrossRef CAS.
- B. A. Zakharov, Y. V. Seryotkin, N. A. Tumanov, D. Paliwoda, M. Hanfland, A. V. Kurnosov and E. V. Boldyreva, RSC Adv., 2016, 6, 92629–92637 RSC.
- M. A. Neumann, J. van de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef CAS PubMed.
- F. P. A. Fabbiani and C. R. Pulham, Chem. Soc. Rev., 2006, 35, 932–942 RSC.
- I. D. H. Oswald, I. Chataigner, S. Elphick, F. P. A. Fabbiani, A. R. Lennie, J. Maddaluno, W. G. Marshall, T. J. Prior, C. R. Pulham and R. I. Smith, CrystEngComm, 2009, 11, 359–366 RSC.
- N. A. Tumanov, E. V. Boldyreva, B. A. Kolesov, A. V. Kurnosov and R. Quesada Cabrera, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 458–471 CrossRef CAS PubMed.
- M. Fisch, A. Lanza, E. Boldyreva, P. Macchi and N. Casati, J. Phys. Chem. C, 2015, 119, 18611–18617 CrossRef CAS.
- B. A. Zakharov, N. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2015, 17, 2074–2079 RSC.
- E. V. Boldyreva, S. V. Goryainov, Y. V. Seryotkin, H. Ahsbahs and V. P. Dmitriev, Proceed. NSU, 2007, 2, 30–35 Search PubMed.
- J. Ridout, L. S. Price, J. A. K. Howard and M. R. Probert, Cryst. Growth Des., 2014, 14, 3384–3391 CrossRef CAS.
- B. A. Zakharov, A. A. L. Michalchuk, C. A. Morrison and E. V. Boldyreva, Phys. Chem. Chem. Phys., 2018, 20, 8523–8532 RSC.
- D. Mondieig, M. A. Cuevas-Diarte and Y. Haget, J. Therm. Anal., 1989, 35, 2491–2500 CrossRef CAS.
- F. B. Johnson, Nature, 1956, 178, 590–590 CrossRef CAS.
- H. F. Lieberman, R. J. Davey and D. M. T. Newsham, Chem. Mater., 2000, 12, 490–494 CrossRef CAS.
- K. Schaum, K. Schaeling and F. Klausing, Justus Liebigs Ann. Chem., 1916, 411, 161–195 CrossRef CAS.
- S. C. Sahoo, S. B. Sinha, M. S. R. N. Kiran, U. Ramamurty, A. F. Dericioglu, C. M. Reddy and P. Naumov, J. Am. Chem. Soc., 2013, 135, 13843–13850 CrossRef CAS PubMed.
- S. C. Sahoo, M. K. Panda, N. K. Nath and P. Naumov, J. Am. Chem. Soc., 2013, 135, 12241–12251 CrossRef CAS PubMed.
- J.-H. Ko, K.-S. Lee, S. Chandra Sahoo and P. Naumov, Solid State Commun., 2013, 173, 46–50 CrossRef CAS.
- B. A. Zakharov, A. S. Marchuk and E. V. Boldyreva, CrystEngComm, 2015, 17, 8812–8816 RSC.
- A. S. Marchuck, Influence of high pressures on [Co(NH3)5X]Br2 (X = NO2−, ONO−) complexes and their interconversions, Thesis, Chair of Solid State Chemistry, Novosibirsk State University, 2018 Search PubMed.
- M. J. Fuller and J. Pinkstone, J. Less-Common Met., 1980, 70, 127–142 CrossRef CAS.
- P. A. Gribov, A. A. Matvienko, B. A. Zakharov, S. A. Chizhik and A. A. Sidelnikov, Mater. Today Proc., 2017, 4, 11470–11475 CrossRef.
- A. A. Matvienko, D. V. Maslennikov, B. A. Zakharov, A. A. Sidelnikov, S. A. Chizhik and E. V. Boldyreva, IUCrJ, 2017, 4, 588–597 CrossRef CAS PubMed.
- B. A. Zakharov, P. A. Gribov, A. A. Matvienko and E. V. Boldyreva, Z. Kristallogr. - Cryst. Mater., 2017, 232, 751–757 CAS.
| This journal is © The Royal Society of Chemistry 2019 |