
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable controlled release of bioactive SDF-1α via specific protein interactions within fibrin/nanoparticle composites†
D.
Dutta
,
C.
Fauer
,
H. L.
Mulleneux
and
S. E.
Stabenfeldt
*
School of Biological and Health Systems Engineering, Arizona State University, Tempe, AZ, USA. E-mail: sarah.stabenfeldt@asu.edu
First published on 11th August 2015
Abstract
The chemokine, stromal cell-derived factor 1α (SDF-1α), is a key regulator of the endogenous neural progenitor/stem cell-mediated regenerative response after neural injury. Increased and sustained bioavailability of SDF-1α in the peri-injury region is hypothesized to modulate this endogenous repair response. Here, we describe poly(lactic-co-glycolic) acid (PLGA) nanoparticles capable of releasing bioactive SDF-1α in a sustained manner over 60 days after a burst of 23%. Moreover, we report a biphasic cellular response to SDF-1α concentrations thus the large initial burst release in an in vivo setting may result in supratherapeutic concentrations of SDF-1α. Specific protein–protein interactions between SDF-1α and fibrin (as well as its monomer, fibrinogen) were exploited to control the magnitude of the burst release. Nanoparticles embedded in fibrin significantly reduced the amount of SDF-1α released after 72 h as a function of fibrin density. Therefore, the nanoparticle/fibrin composites represented a means to independently tune the magnitude of the burst phase release from the nanoparticles while perserving a bioactive depot of SDF-1α for release over 60 days.
1. Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability around the world with over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths and an estimated $60 billion in direct and indirect economic costs every year, in the United States alone.1 Although current treatment practices have demonstrated some efficacy in treating its long-term effects, there are no means of directly addressing the underlying pathophysiology of TBI.2 Recent studies have reported the activation of endogenous neural progenitor/stem cell (NPSC)-mediated neurotrophic support and neurogenesis after injury events (e.g. stroke or traumatic brain injury).3–5 NPSCs originating from the two adult neural stem cell niches (subventricular zone, SVZ; and subgranular zone of hippocampus dentate gyrus) selectively migrate to the injury penumbra forming ectopic niches, even within non-neurogenic areas of the brain (such as adult cortical tissues).4,6 The concentration of the chemokine, stromal cell-derived factor-1α (SDF-1α), and the expression of its receptor, CXCR4, increases significantly in the injury penumbra and is implicated as a key regulator of directed NPSC recruitment after neural injury.3,4 However, increased SDF-1α levels subside by 14 days post injury, in most reports, coinciding with a diminishing number of NPSCs (observed at the injured area).3,4,7,8 Building on this inherent injury-stimulated signal, we postulate that increased and sustained bioavailability of SDF-1α locally in the injury penumbra would augment NPSC recruitment and bolster the capacity for endogenous regeneration.
000 deaths and an estimated $60 billion in direct and indirect economic costs every year, in the United States alone.1 Although current treatment practices have demonstrated some efficacy in treating its long-term effects, there are no means of directly addressing the underlying pathophysiology of TBI.2 Recent studies have reported the activation of endogenous neural progenitor/stem cell (NPSC)-mediated neurotrophic support and neurogenesis after injury events (e.g. stroke or traumatic brain injury).3–5 NPSCs originating from the two adult neural stem cell niches (subventricular zone, SVZ; and subgranular zone of hippocampus dentate gyrus) selectively migrate to the injury penumbra forming ectopic niches, even within non-neurogenic areas of the brain (such as adult cortical tissues).4,6 The concentration of the chemokine, stromal cell-derived factor-1α (SDF-1α), and the expression of its receptor, CXCR4, increases significantly in the injury penumbra and is implicated as a key regulator of directed NPSC recruitment after neural injury.3,4 However, increased SDF-1α levels subside by 14 days post injury, in most reports, coinciding with a diminishing number of NPSCs (observed at the injured area).3,4,7,8 Building on this inherent injury-stimulated signal, we postulate that increased and sustained bioavailability of SDF-1α locally in the injury penumbra would augment NPSC recruitment and bolster the capacity for endogenous regeneration.
With this in vivo application in mind, this study focused on developing a drug delivery device for local, sustained release of SDF-1α with the following attributes: (1) injectable, (2) biodegradable, (3) prolonged release well past 14 days and (4) maintenance of SDF-1α levels within the therapeutic concentration range. The most basic form of delivering therapeutics is systemic bolus administration. Drawbacks of this method include a lack of control over biodistribution due to physiological barriers (i.e. endothelial barrier) and rapid systemic clearance.3 Direct, local injection in the target tissue affords control over dosage, but negates temporal control or payload degradation leading to only transient therapeutic benefits. Conventional means for local, sustained delivery to control both dosage and temporal concentration profile involve bulky, invasive minipump systems that are linked to infections, bleeding and neurologic injury.9 Injectable biomaterials for the controlled release of therapeutics (i.e. hydrogels and biodegradable plastics) hold the capacity to overcome the common limitations of drug delivery (i.e. dosage, temporal concentration, biocompatibility & patient compliance).10 Release devices for local and sustained delivery of SDF-1α have been explored in several different physiological applications including neural regeneration, myocardial infarctions, skeletal regeneration and wound healing.11–14 However, many of these previous designs were based on hydrogels (such as, alginate, collagen, gelatin, star PEG–heparin etc.) and provided sustained SDF-1α for less than 14 days. Therefore, we sought to tailor a controlled release system that fits the aforementioned design criterion, ultimately for neural applications.
Poly(lactic-co-glycolic) acid (PLGA), a FDA-approved biodegradable polyester, has long been studied for diverse applications in the central nervous system.15,16 In addition tunable release profiles, a significant benefit of PLGA carriers is insulation of encapsulated cargo from the local microenvironment, limiting specific and non-specific degradation and leading to increased protein half-life.17 With a half-life of 25 min in blood, maintaining SDF-1α bioactivity was an important parameter for this study.18 Sustained delivery of SDF-1α has been achieved with macro-scale PLGA scaffolds and microparticles.19–21 Yet, the utility of such macro-to-micro-scale systems for minimally invasive delivery is limited. As such, novel nanoscale SDF-1α PLGA-based devices are of great interest.
The release profiles of encapsulated cargo from PLGA-based particles vary greatly (zero-order, monophasic, biphasic and triphasic) depending primarily on formulation parameters and typically includes a burst release within the first few hours.22 The PLGA burst phase is problematic since supra-therapeutic concentrations may result in undesired biological consequences. Mechanisms to address the burst issue range multilayer coatings to composite system embedding PLGA particles within hydrogel matrices. The biologically-derived matrix fibrin has been investigated as a carrier for protein delivery.23,24 Numerous studies report engineered fibrin-derivatives crosslinkers to mediate and enhance affinity-based interactions for controlled release of a multitude of biologics (e.g. nerve growth factor (NGF), neurotrophin-3, glial-derived neurotrophic factor, genetic material).25–28 As a natural extracellular matrix (ECM) protein, fibrin possesses inherent ability to bind and sequester soluble signaling factors, predominately through a heparin-like binding domains that mediate immobilization of small, highly basic (and heparin-binding) proteins such as basic fibroblast growth factor (bFGF).29–31 Here in this study, we hypothesized that embedding SDF-1α-loaded NPs within fibrin matrices will modulate the burst release phase due to specific protein–protein interactions between SDF-1α and fibrin. The key objectives for this study were to (1) characterize SDF-1α-loaded NPs, (2) probe the mechanism of SDF-1α/fibrin(ogen) interactions and (3) determine the effect of SDF-1α/fibrin(ogen) interactions on SDF-1α release from PLGA NPs. Collectively, we report a composite fibrin/PLGA system with the capacity to achieve long-term (60 days), bioactive SDF-1α release and the means to independently tune protein release during the burst phase.
2. Methods and materials
2.1 Materials
Poly(lactic-co-glycolic) acid (PLGA; 50:50 ester-terminated; inherent viscosity = 0.55–0.75 dL g−1) was purchased from Lactel (Birmingham, USA). Recombinant mouse stromal cell-derived factor-1α (SDF-1α) was acquired from PeproTech (Rocky Hill, USA). B27 growth supplement, DAPI nuclear stain, tetramethylbenzidine (TMB) substrate and Dulbecco's modified eagle medium were acquired from Life Technologies (Carlsbad, USA). Glucose was obtained from Acros Organics (Geel, Belgium). High-binding 96-well enzyme-linked immunosorbent assay (ELISA) plates were acquired from Greiner Bio-One (Frickenhausen, Germany). The organic solvent ethyl acetate was acquired from Alfa Aesar (Ward Hill, USA) and dimethyl sulfoxide (DMSO) from American bioanalytical (Natick, USA). Human fibrinogen (Plasminogen, von Willebrand Factor and Fibronectin Depleted), human α-thrombin, human factor XIIIa (FXIII) and human plasmin were acquired from Enzyme Research Laboratories (South Bend, USA). All other materials and chemicals were purchased from Sigma-Aldrich (St. Louis, USA) and used without further modification or purification.
2.2 Synthesis of SDF-1α loaded nanoparticles
SDF-1α loaded PLGA nanoparticles were synthesized using a water/oil/water (W/O/W) emulsion technique adapted from a previously published protocol.32 Briefly, the first emulsion (W/O) was obtained by vortexing the oil phase (100 mg mL−1 PLGA in ethyl acetate) with PBS buffer solution (pH = 7.4) containing 20.0 mg mL−1 bovine serum albumin (BSA; 2.0% w/w of PLGA) and 2.0 mg mL−1 SDF-1α (0.2% w/w of PLGA). The above solution was added dropwise to a 3.6× volume excess of an aqueous 5.0% (w/v) D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) and the second emulsion (W/O/W) was produced by ultrasonicating (Omni Ruptor 4000; Omni International; Kennesaw, USA) the solution for two consecutive 15 s periods in an ice bath (120 W power with a 90% duty cycle). The emulsion was then quickly transferred to a stirring (300 RPM) aqueous bath containing 0.5% TPGS + 1.25% (w/v) NaCl (10× volume excess) and left undisturbed for 3 h for solvent evaporation. The particle suspension was washed three times with deionized water by centrifugation at 15000g for 15 min in between rinses (Beckman Counter; Allegra 25R; Pasadena, USA). The particles were supplemented with 25% (w/w) D-(+)-trehalose dihydrate and recovered through lyophilization. Particle size analyses were performed by scanning electron microscope (SEM; Phillips XL-30; San Francisco, USA) using a 3–5 kV electron beam. Lyophilized particle samples were prepared for SEM analysis via gold/palladium sputter coater (108-Auto, Cressington Scientific; Watford, UK) to achieve a 5–10 nm thick layer. A minimum of 7 images were captured per group and were processed using ImageJ to determine the size distributions. At least 85 sampling points were required for each image and thus size distribution histograms are comprised of a minimum of 595 measurement points in total.
2.3 Protein loading & release assays
Total protein loading was determined by complete dissolution of a known amount of particles (10 mg mL−1) in dimethyl sulfoxide (DMSO). The DMSO solution was then diluted 1:15 using 2.5% (w/v) sodium dodecyl sulfate and 0.1 N NaOH in deionized water. The mixture was thoroughly agitated, being careful not to introduce bubbles. Known amounts of soluble BSA added to blank particles (no protein encapsulated; synthesized using identical synthesis protocols) were used to generate calibration curve. Protein quantification was performed using bicinchoninic assay (BCA; G Biosciences; St. Louis, USA) in triplicates following manufacturer's protocols. Encapsulation efficiency and loading capacity were calculated using the following: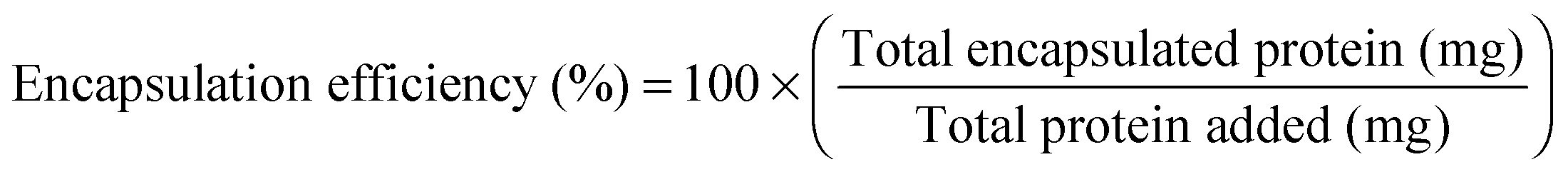
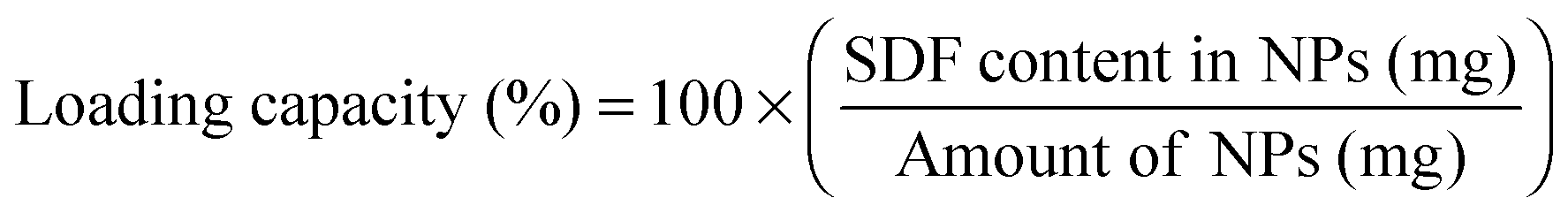
For the release assays, lyophilized particles were resuspended in 1 mL of buffer release media (1× PBS supplemented with 0.01% tween 80 and 0.01% NaN3) at 3.5 mg mL−1 and incubated at 37 °C under constant agitation. At specified time points, the supernatant was collected by centrifuging the particle suspension at 14000g for 15 min, collecting 90% of the supernatant and replenishing with fresh buffer release media. Extracted buffer release media samples were stored at −80 °C for subsequent protein analysis. To specifically determine the SDF-1α content, a known concentration (500 ng mL−1) was incubated alongside the NP suspension as a positive control for subsequent analysis using ELISA-based detection (R&D systems; Minneapolis, USA). NPs with no encapsulated SDF-1α served as the negative control.
2.4 SDF-1α bioactivity assay
000 cells per cm2) on laminin-coated transwell inserts with 12 μm pore diameter (Millipore, Temecula, CA). Growth factor-free NPSC medium (no EGF or bFGF) with 0 or 250 ng mL−1 SDF-1α (negative and positive control, respectively) was added in the bottom chamber. NPSCs were then allowed to undergo chemotaxis for 24 h in an incubator (37 °C and 5% CO2). Subsequently, cells on the top side of the transwell membrane were removed using a cotton swab whereas migrated cells that reached the bottom were fixed, underwent a DAPI nuclear stain and imaged. NPSC nuclei have diameters of approximately 20 μm.35 After intensity thresholding, nuclei count was quantified using a particle count algorithm in ImageJ where stained nuclei 10–30 μm in diameter were counted as individual cells (Fig. S1 and S2, ESI†). Nuclei count was determined by imaging and quantifying whole transwell membranes.
000g for 15 min) to collect the supernatant, and the modified Boyden chamber assay was carried out immediately. NP cell culture release media was diluted 1:10 and 1:2 for day 0–1 and day 20–22 samples, respectively, with growth factor-free NPSC media to achieve a SDF-1α concentration in the NPSC chemotactic range (Fig. 2). The dilution factors were estimated via a preliminary total protein release profile (data not shown). A minimum of 4 replicates per group performed for all migration assays.
2.5 SDF-1α-fibrin(ogen) binding detection ELISAs
SDF-1α-fibrinogen interaction were probed by adapting a previously described modified ELISA.30 SDF-1α, BSA and basic fibroblast growth factor (bFGF) were coated on high-binding ELISA plates by incubating the soluble factors at 100 nM (in 100 mM carbonate buffer; pH 9.6) for 3 h at 37 °C. All growth factor (GF) coated wells were blocked using 2.0% (w/v) fat-free powdered milk in PBS for 1 h at room temperature (RT). Fibrinogen (35 μg mL−1; depleted of fibronectin, plasminogen, and von Willebrand factor) was incubated for 1 h at RT. The primary antibody (rabbit anti-fibrinogen; EMD Millipore; Darmstadt, Germany) was then added for 1 h at RT, followed by the secondary antibody (HRP conjugated goat anti-rabbit IgG; Thermo Scientific; Waltham, USA), also under the same conditions. Detection was carried out TMB substrate following manufacturer's protocols. Four washes were performed in between each of the steps mentioned using PBS with 0.01% Tween-20 (PBS-T). Heparin competition ELISAs were also performed with various concentrations (0.75–200 μg mL−1) of soluble heparin (17–19 kDa), which was supplemented in the fibrinogen solution after the blocking step mentioned above.30 All subsequent steps were kept identical.The next modified ELISA probed SDF-1α interactions with fibrin. A previously described protocol was adapted to generate a thin layer of fibrin network on high-binding ELISA plates.36 In short, fibrinogen (depleted of fibronectin, plasminogen, and von Willebrand factor) was incubated at 100 μg mL−1 for 1 h at RT. After rinsing three times with Tris-buffered solution (10 mM Tris + 150 mM NaCl), the wells were blocked using 1.0% (w/v) BSA for 20 min at RT. After rinsing, human thrombin (2.5 NIH U mL−1) and human FXIIIa (0.5 NIH U mL−1) was added and incubated for 15 min at RT. Fibrin coating was completed after washing and subsequent incubation with fibrinogen (500 μg mL−1), anti-thrombin-III (50 μg mL−1) and heparin (60 μg mL−1) for 1 h at RT. The plates were then washed and stored with 2.0% (w/v) powdered milk in PBS overnight at 4 °C. Control groups included wells that were coated with heparin (70 μg mL−1; positive control) or BSA (1.0 mg mL−1; negative control) in PBS, overnight at 4 °C. Surface-modified wells were then exposed to various concentrations of SDF-1α (0–20 μg mL−1) for 1 h and relative levels of SDF-1α binding were measured using rabbit anti-SDF-1α (Abcam; Cambridge, USA), HRP-conjugated goat anti-rabbit and TMB substrate. A minimum of n = 3 was used for all ELISAs.
2.6 SDF-1α release from fibrin matrices
Release of soluble SDF-1α from fibrin matrices was conducted as described previously.30 In short, fibrin gels were generated with fibrinogen (3 or 25 mg mL−1), human thrombin (5 U mL−1), human FXIIIa (2.5 U mL−1), calcium chloride (5 mM) and SDF-1α (500 ng mL−1) in TBS. The fibrin matrices (150 μL) were polymerized (by combining separate fibrinogen and thrombin solutions) for 1 h at 37 °C under sterile conditions in ultra-low binding 24-well plates. After the polymerization, 700 μL of TBS (with 0.1% w/v BSA) was added to initiate the release assay. The release buffer was extracted, stored in −80 °C and replaced with fresh TBS every 24 h for 7 days. On day 7, the fibrin matrices were digested with plasmin (0.5 U mL−1) to quantify the remaining SDF-1α. A control group consisted of soluble SDF-1α (500 ng mL−1) where 30 μL was extracted for every timepoint and also underwent the plasmin treatment. SDF-1α content was determined via SDF-1α ELISA. All groups were measured in triplicates.2.7 Release assay from SDF-1α-loaded NPs embedded in fibrin
Fibrin gels (3, 10, 25 mg mL−1) were formed using the same procedure as above where free SDF-1α was replaced by PLGA NPs (blank and SDF-1α-loaded NPs) with a final concentration of 1 mg PLGA NPs per 1 mL of fibrin gel. Similar to the previous release assay, fibrin matrices (150 μL) were formed in ultra-low binding 24-well plates for 1 h at 37 °C under sterile conditions, after which 700 μL of TBS (with 0.1% w/v BSA) was added. The release media was extracted, replaced and stored at hours 1, 3, 6, 12, 24 48 and 72 after initiating the release assay. SDF-1α content in all timepoints were quantified via ELISA. A 500 ng mL−1 SDF-1α group as served as the control groups, where similar to before, 30 μL was extracted at every timepoint. All groups were tested in triplicates.2.8 Physical characterization fibrin/NP composites
Fibrin polymerization was monitored with time-dependent optical density measurements (Epoch; Biotek; Winooski, USA). Control (native fibrin clots with no NPs) and NP-embedded gels (0.1, 1 & 10 mg mL−1) were prepared using the same parameters as mentioned earlier. Three different fibrinogen (3, 15 & 30 mg mL−1) groups were tested where optical density (OD) was measured every 30 s for 2 h in triplicate per group. Terminal clot turbidity refers to the OD at the end of 2 h.Percent clottable protein was quantified for control and experimental fibrin gels (with NPs; 0.1, 1 & 10 mg mL−1; n = 3). The fibrinogen content before polymerization was compared to the remaining soluble fibrinogen that remained following fibrin gel formation (40 min).37 Fibrinogen (FBN) content in the remaining solution (clot liquor), taking into account the presence of additional enzymes, was measured using BCA where percent clottable protein (CP) is defined as the following:
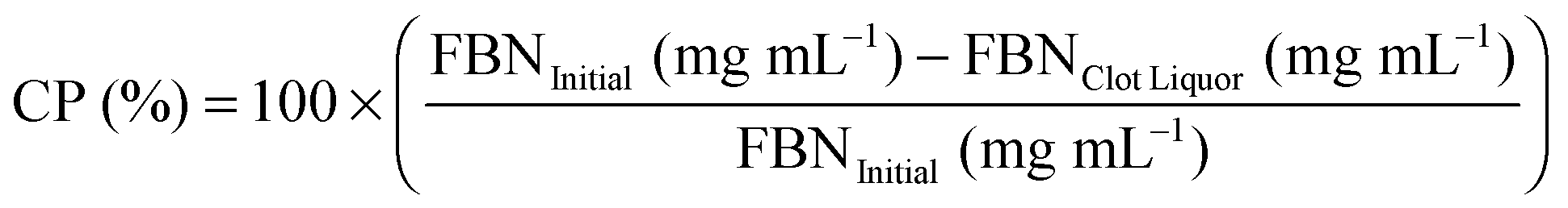
The mechanical properties of the fibrin gels were also analyzed with an oscillatory parallel-plate geometry rheometer (MCR 101; Anton Paar; Ashland, USA). Fibrin clots (3 & 30 mg mL−1) with and without NPs (0.1, 1, 10 mg mL−1) were generated with identical protocols as described above. The fibrin gels (0.4 mL) were polymerized within a 400 μm gap between the top and bottom plates (1 h at 37 °C under high humidity). A strain sweep (0.01 to 100%) was performed to determine the linear viscoelastic regime for all concentrations of fibrin and strain amplitude of 1% was chosen for all subsequent frequency sweep tests (0.01–100 Hz) to determine storage (G′) and loss (G′′) modulus (n = 2).
2.9 Statistics
Statistical analysis was performed on all quantitative assays. All results are depicted as the mean ± one standard deviation, unless otherwise stated. Statistical analyses (GraphPad Prism, La Jolla, CA) evaluated differences between groups using analysis of variance (ANOVA) followed by Tukey post hoc tests to determine statistical significance with p < 0.05 considered significant. Multiplicity adjusted p-values are reported for Tukey post hoc comparisons.3. Results and discussion
3.1 Characterization of SDF-1α loaded NPs
SDF-1α was successfully encapsulated in PLGA NPs using a double emulsion method. Characterization assays included size analyses via SEM micrographs, total protein encapsulation efficiency as well as quantification of SDF-1α release profile (Fig. 1). Resulting NPs were spherical in shape and had smooth surface morphology with an average diameter (± standard error of mean; SE) of 288.9 ± 19.2 nm, comparable to previously reported PLGA NPs.32,38 The reported average NP diameter was an average of 5 separately prepared batches. The NP population distribution for each batch consistently ranged between 100–1500 nm with 90% of the NPs falling within 200–600 nm (Fig. 1C). NP size distributions were statistically consistent between batches and NP yields ranged between 57–65%, indicating minimal batch-to-batch variability. The total protein (BSA + SDF-1α) encapsulation efficiency for the resulting NPs was 61.7% ± 2.8%. Attempts to determine SDF-1α loading using conventional methods requiring dissolution of NPs in an organic solvent followed by protein extraction and quantification via SDF-1α ELISA proved unreliable due to protein denaturation and degradation; this issue was also previously reported by Cross et al.20 As a result, we estimated total encapsulated SDF-1α via cumulative values from the SDF-1α ELISA release profile (Fig. 1D). SDF-1α content measured during the release assay amounted to a total SDF-1α loading capacity of 293 ng of SDF-1α/1 mg of PLGA (i.e. 0.029 ± 0.00076% (w/w) PLGA NPs). Furthermore, the NPs exhibited a tri-phasic release profile, frequently observed in PLGA-based release devices.22 We observed sustained release of SDF-1α for 60 days following an initial burst release and a lag phase approximately between days 2 and 6 (Fig. 1D). In the first day, the NPs release 67 ng SDF-1α for every 1 mg of PLGA NPs, which translates to a 23% burst of total released SDF-1α as determined by ELISA.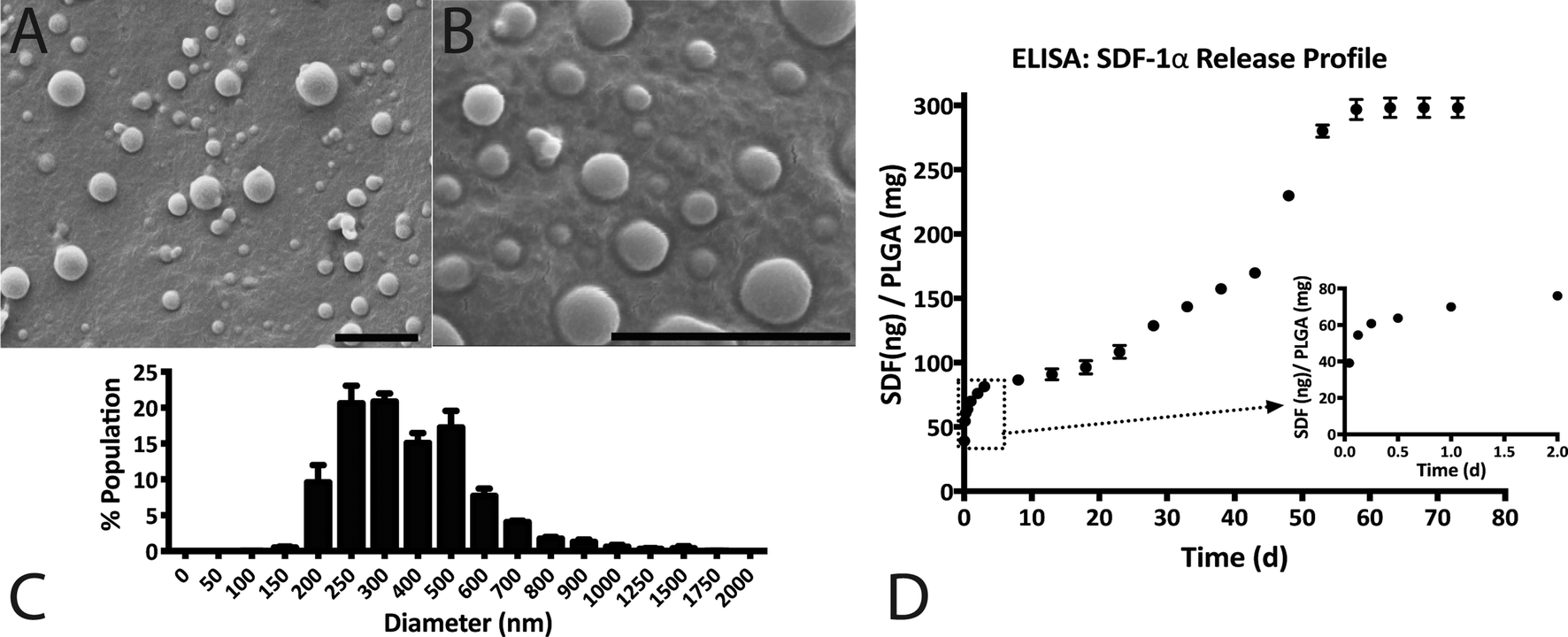 | ||
| Fig. 1 SDF-1α NP characterization: (A and B) representative SEM images of SDF-1α loaded NPs depicting smooth, spherical particles; scale bars = 2 μm. (C) Histogram illustrating the size distribution of NPs with a range between 100–1500 nm and where 90% of the population is between 200–600 nm in diameter. (D) In vitro release assay measured with SDF-1α ELISA demonstrated sustained SDF-1α release for 60 days. The inset illustrates the initial burst release of 23% of total measured SDF-1α within the first 24 h. | ||
PLGA-based release devices are unique since cargo release rate is largely controlled by polymer degradation rate and thus is not purely diffusion-mediated.22 The release profile is a result of various interactions (i.e. cargo/polymer, cargo/cargo etc.) and release mechanisms (i.e. diffusion, bulk erosion etc.) that are relevant to a particular set of particles.9 After the burst release, the mechanism of release for hydrophilic proteins is attributed to the formation, dilation and coalescence of nano-pores (forming mesopores) inside the PLGA matrix.40 The effective protein diffusivity is directly correlated to the properties of the pore network and protein/polymer interactions.39 Thus prolonged release in the later stages is mediated by polymer degradation and erosion as well protein diffusion. As a result, choice of PLGA was a rational decision considering our goal for achieving long-term, sustained and bioactive SDF-1α release.
Previous studies demonstrated the feasibility of encapsulating and releasing bioactive SDF-1α (as tested by in vitro mesenchymal stem cell migration) from PLGA microparticles to achieve controlled release over 40–70 days.19,20 However, both studies report relatively low SDF-1α loading. The PLGA microparticles in one study indicated a loading capacity of approximately 0.0018% (w/w) of PLGA, whereas the other had a theoretical maximum of 0.002% (w/w) SDF-1α relative to PLGA polymer. A low loading capacity equates to a requirement of high amounts of PLGA. Thus achieving adequate, therapeutic levels of SDF-1α may conflict with the accumulation of acidic byproducts that affects the local pH.41 In comparison, we report PLGA nanoparticles with loading capacities of SDF-1α an order of magnitude higher at 0.029% (w/w) PLGA. Additionally, we achieved SDF-1α controlled release for 60 days, meeting our initial design criterion (Fig. 1D).
3.2 NPSC migration assays indicate release of bioactive SDF-1α
Modified-Boyden chamber migration assays were utilized to measure functional SDF-1α bioactivity (Fig. 2A and B). SDF-1α is known to elicit a biphasic migratory response in vitro with a number of cell types (NPSCSs, mesenchymal stem cells; MSCs, leukocytes, hematopoietic cells etc.) over a wide range of concentrations (10–1000 ng mL−1).34,42–46 This biphasic migratory response reportedly relates to the internalization of CXCR4 upon interaction with SDF-1α. Overstimulation from high concentrations of SDF-1α may lead to desensitization to the chemokine.42,47 In our hands, the maximal NPSC migration response occurred at 250 ng mL−1 after 24 h and revealed a biphasic relationship to SDF-1α concentration where 1000 ng mL−1 did not elicit any chemotactic response above the basal media levels (Fig. 2B). The biphasic response of SDF-1α on NSCs suggests control over SDF-1α dosage and its temporal concentration profile is required to achieve a desired biological response.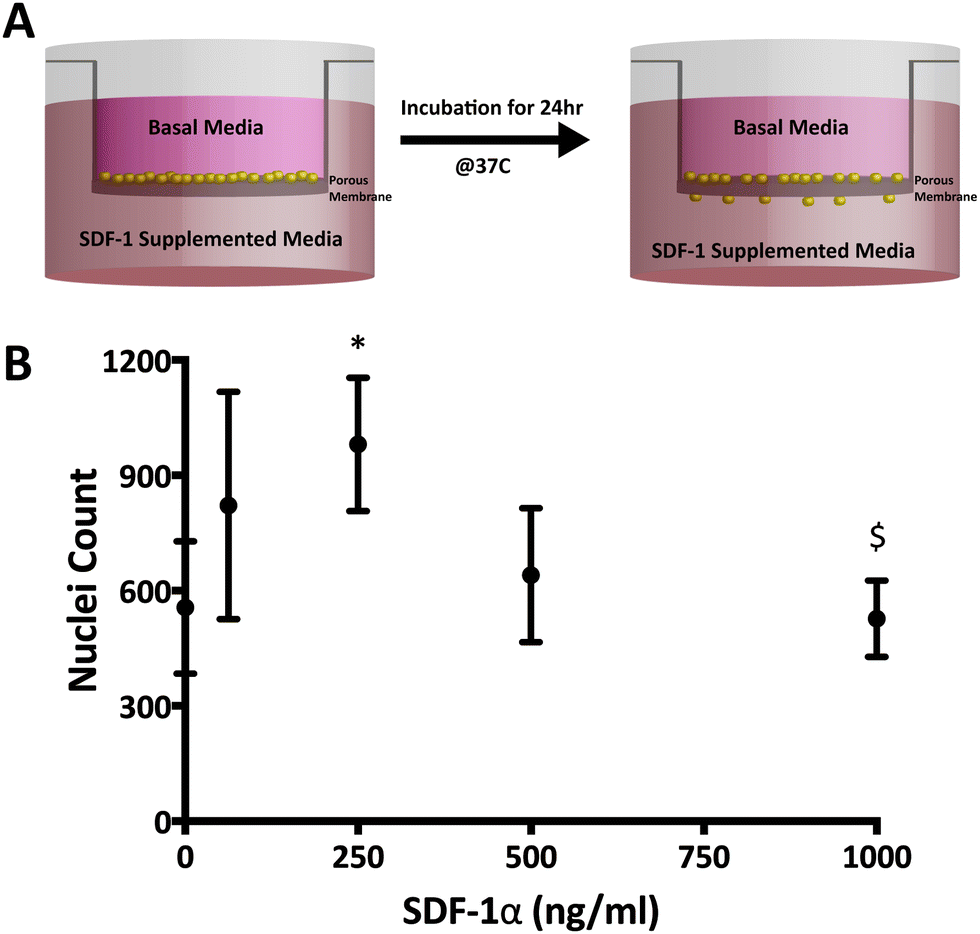 | ||
| Fig. 2 Soluble SDF-1α elicits a biphasic migration response in a Boyden chamber assay. (A) Schematic depicting the Boyden chamber setup where cells that were seeded in the top chamber migrates to reach the bottom side through chemotactic migration after 24 h. (B) NPSC chemotactic migration demonstrates a biphasic response where the 250 ng mL−1 group is significantly different compared to both to baseline and 1000 ng mL−1 SDF-1α group (* represents p < 0.05 relative to 0 ng mL−1 SDF-1α; $ represents p < 0.05 relative to 250 ng mL−1). | ||
The W/O/W double emulsion synthesis for PLGA NPs inherently involve harsh conditions such as water–oil interfaces, ultrasonication, freeze thaw cycles and lyophilization, known to affect the structural integrity and the biological properties of proteins.48 Additionally, detection of SDF-1α via ELISA does not necessarily equate to functional bioactivity since the epitope recognition site for ELISA can vary from the biologically relevant site(s). As a result, the Boyden chamber assay described above was used to measure functional bioactivity of SDF-1α encapsulated in and released from the NPs. For each time interval (day 0–1 & days 20–22), 250 ng mL−1 SDF-1α and 0 ng mL−1 served as the positive and negative controls for NPSC chemotaxis, respectively. In addition, release media from blank NPs were used as a control for confounding affects from PLGA degradation products.
The release media from day 0–1 elicited a robust migratory response that was significantly higher than both the negative control (no SDF-1α; p = 0.0007) and the blank NP group (Fig. 3A and B; p = 0.0201). No adverse effects on NPSC chemotaxis were observed with the blank NP group for the 24 h incubation period, agreeing with previously published results with MSCs.20 The data suggests that bioactivity of SDF-1α is not significantly altered with the NP synthesis protocol. The day 0–1 release media is largely composed of the burst phase that accounts for loosely adsorbed SDF-1α on the NP surface that diffuse out rapidly upon particle hydration. However, preservation of protein bioactivity in the first day does not indicate sustained release of bioactive protein. Local pH within the particles have been reported to be <pH 3 and during release, the cargo can undergo aggregation, non-reversible adsorption and degradation leading to further loss in bioactivity and incomplete release.48 Thus release media from day 20–22 was also evaluated to validate maintenance of bioactive SDF-1α (Fig. 3A and C). Here, NPSC migration significantly increased in the SDF-1α-loaded NP group relative to blank NPs, which controlled for potential confounding affects from the acidic PLGA degradation products (p < 0.05). A similar trend was observed in the pairwise comparison to the negative control, though not statistically significant (p = 0.0599). In summary, encapsulation of SDF-1α in the NPs maintained the long-term bioactivity of SDF-1α. Moreover, encapsulated cargo is insulated from biological proteolytic factors in future in vivo applications. Thus we postulate this device has the potential to improve SDF-1α half-life (25 min in blood) as shown with other proteins in vitro and in vivo.17,48
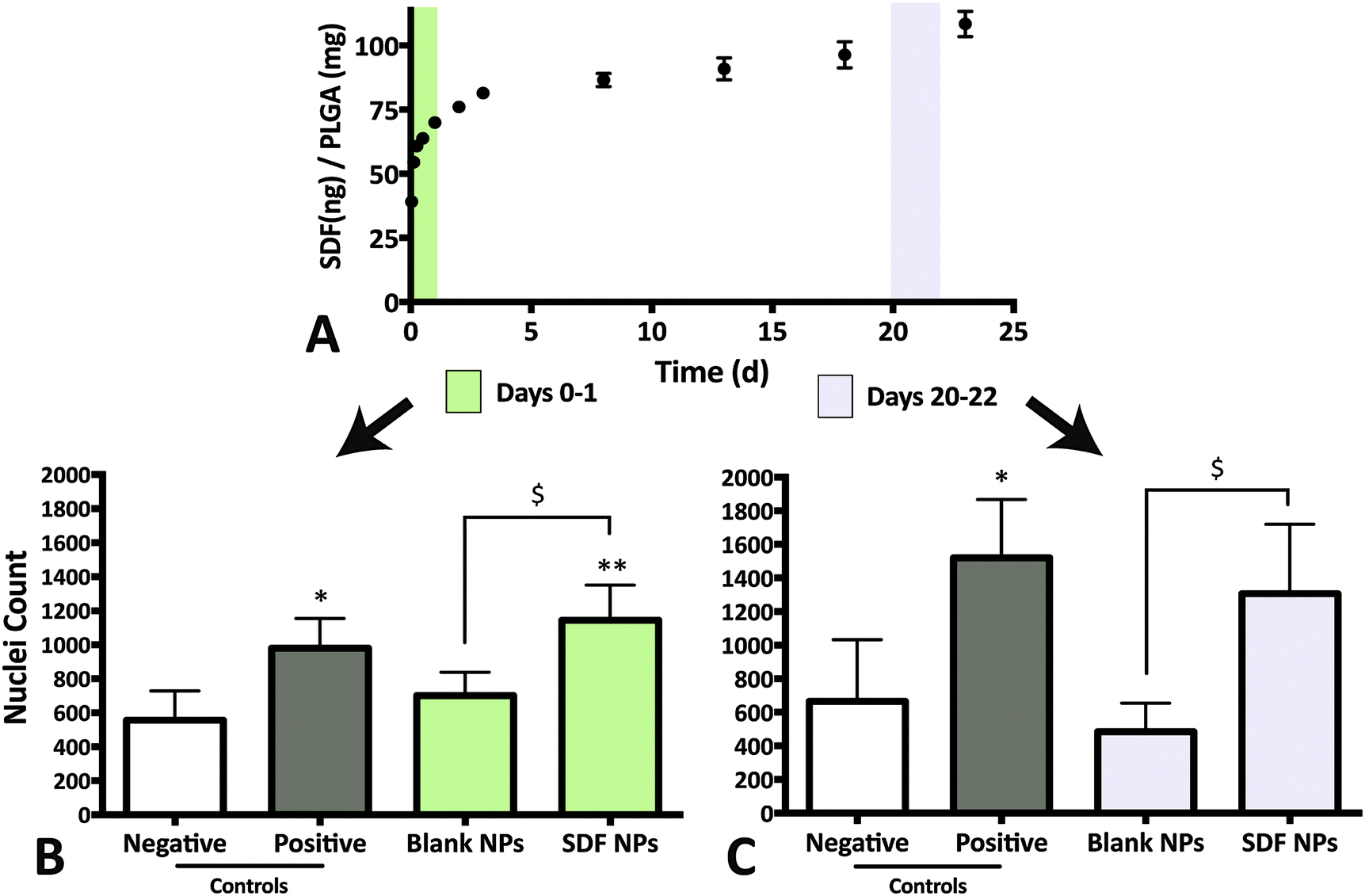 | ||
| Fig. 3 Bioactive SDF-1α released from NPs. (A) Release media from day 0–1 and 20–22 were evaluated for bioactivity via NPSC chemotaxis assay. (B) Chemotactic response from NPSCs incubated with release media from day 0–1. SDF-1α-loaded NPs (‘SDF NP’) elicits a significant increase in nuclei count relative to both blank NPs and the negative control. (C) NPSC migration in response to release media from days 20–22. Although SDF-1α-loaded NPs were able to increase nuclei count significantly relative to blank NPs, the difference was not statistically significant in comparison to the negative control (* & ** represents p < 0.05 and p < 0.001 respectively, relative to negative control and $ represents p < 0.05 relative to blank NPs). | ||
3.3 Protein–protein interactions between SDF-1α and fibrin(ogen)
The high burst release from the NPs within the first hours is a concern considering biphasic response to SDF-1α (i.e. decreased NPSC migration; Fig. 2). Moreover, high SDF-1α concentrations in vivo reportedly initiate systemic immune cell recruitment and infiltration.49 Modest decrease of NP burst release magnitude may be achieved through alterations of the NP formulation parameters.50,51 However, changes in synthesis conditions create complex, multifaceted interactions that affect several particle properties at once. For example, changing polymer concentration not only affects encapsulation efficiency and loading capacity, but it also influences particle size and porosity, key factors determining release rate and duration.51,52 Therefore, we pursued a composite system as it affords the ability to independently tune the release profile without compromising the desired attributes already attained by the NPs (sustained release of bioactive SDF-1α).Composite biomaterials with drug carriers embedded in hydrogels has been explored for a number of applications.53–55 One relevant example includes basic fibroblast growth factor (bFGF)-loaded nanoparticles embedded in fibrin to achieve tunable, zero-order release with the ability to reduce/eliminate the burst phenomenon.55 The presence of heparin-like binding domains in the structure of native fibrin and its monomer, fibrinogen, is correlated to specific binding with highly basic, heparin-binding proteins such as bFGF, vascular endothelial growth factor (VEGF), placenta growth factor-2 (PlGF-2) and insulin-like growth factor-binding protein-3.30,31,56 Due to the similarities in molecular weight, isoelectric point and heparin binding capacities between known fibrin-binding proteins such as, bFGF, and SDF-1α, we probed the existence of fibrin(ogen)/SDF-1α interactions.
Fibrin(ogen)/SDF-1α interactions were determined using three modified ELISAs that probed both fibrin/fibrinogen affinity as well as, mechanistic competition with soluble heparin (Fig. 4). First, the fibrinogen binding assay consisted of investigating soluble fibrinogen binding to adsorbed soluble factors (BSA, negative control; bFGF, positive control; or SDF-1α). The results demonstrated a significant increase in fibrinogen binding/retention on SDF-1α-coated wells relative to BSA-coated wells (Fig. 4A and B; p < 0.05). More importantly, the fibrinogen retention levels for SDF-1α were comparable to the positive control bFGF-coated wells, suggesting a binding interaction between SDF-1α and fibrinogen (Fig. 4A and B).
 | ||
| Fig. 4 Specific protein–protein interactions exist between SDF-1α and fibrin(ogen). (A) Schematic depicting fibrinogen binding assay that consisted of high-binding ELISA plates coated with the soluble factors, bovine serum albumin (BSA; negative control), bFGF (positive control) and SDF-1α ELISA setup used to measure fibrinogen binding affinity. (B) Signal from SDF-1α coated wells were significantly higher compared to negative controls (BSA) and comparable to the positive control (bFGF) suggesting a specific interaction between SDF-1α and fibrinogen. (C) Increase in signal from fibrinogen binding is attenuated for bFGF and SDF-1α-coated wells in a dose-dependent manner due to competition from soluble heparin. In contrast, BSA-coated wells do not exhibit heparin-dependant change in signal. (D) ELISA designed to probe fibrin interactions with SDF-1α where high-binding ELISA plates were coated with a thin layer of fibrin, BSA (negative control), and heparin (positive control). (E) A concentration-dependent increase in signal was observed for the fibrin-coated wells, similar to the trend observed for the positive control suggesting SDF-1α interactions persist in the polymerized form of fibrinogen, fibrin (** represents p < 0.001 relative to negative BSA control). | ||
The second ELISA assay probed the mechanism of SDF-1α/fibrinogen interaction. The heparin-like binding domain in fibrinogen is located in the first 66 amino acid residues of the Bβ chain.30,57 The high concentration of arginine and lysine residues in that region allows promiscuous binding to both soluble factors and heparin.30 Thus, a competition ELISA was performed where known amounts of soluble heparin (0.75–200 μg mL−1) was added to compete with the heparin-like binding domains on fibrinogen. The results indicate significant attenuation of signal with increasing heparin concentration for both bFGF and SDF-1α-coated plates (Fig. 4C). Conversely, signal from the BSA-coated wells do not exhibit a similar dependence on heparin content. The trends observed in Fig. 4B and C agree with data from similar assays reported for other known fibrin-binding proteins.30 Thus, the observed fibrinogen/SDF-1α interaction cannot be attributed to non-specific interactions, but most likely due to specific SDF-1α/fibrinogen interactions via the heparin-binding domain.
The final ELISA probed SDF-1α interactions with the insoluble, polymerized fibrin after a thin layer was deposited on ELISA plates.36 Concentration dependent retention of SDF-1α on both, heparin-coated (positive control) and fibrin-coated wells relative to BSA-coated negative controls, indicate that specific SDF-1α/fibrinogen interactions are maintained when fibrinogen polymerizes to form fibrin (Fig. 4D and E). Collectively, the results from these modified ELISA assays indicate specific protein–protein interactions between SDF-1α and fibrin(ogen) exists primarily via SDF-1α interactions with the heparin binding domain.
3.4 Fibrin sequesters free SDF-1α
Affinity-based interactions via the heparin-like binding domains on fibrin(ogen) is hypothesized to play a major role in determining growth factor release profile. For example, soluble heparin binding factors such as PlGF-2, bFGF and VEGF exhibit prolonged release and sequestration in native fibrin matrices while, VEGF121 (isoform lacking the heparin-binding domain) and NGF are released in a more diffusion-limited manner.24,30,31 Although fibrin density, fiber length and fiber aspect ratio are important determinants of release profile, evidence of immobilization/sequestration (slow or no protein release) is a key indicator for affinity-based interactions playing a dominant role in determining release rate. For example, PlGF-2-loaded fibrin gels release ∼15% of cumulative cargo in the first 2 days, whereas the remaining cargo was sequestered in the fibrin matrix for at least 7 days.30 In contrast, NGF (low fibrinogen binding affinity) encapsulated in an identical fibrin matrix exhibited 100% cumulative release within 2 days.30 Additionally, Wong et al. reported a similar release profile for bFGF and concluded that diffusion-only release mechanism cannot account for cargo sequestration observed.31Building on the SDF-1α-fibrin(ogen) ELISA assays (Fig. 4), we conducted SDF-1α release assays from fibrin gels to evaluate ability of the protein–protein interactions to sequester SDF-1α within three-dimensional fibrin matrices. We also evaluated the effects of fibrin density on SDF-1α release profile with the hypothesis that altering the number of available binding sites will dictate the maximal amount of sequestered SDF-1α within fibrin matrices.58 We must note that altering fibrin density also modifies various matrix morphological properties and thus diffusion-limited cargo release profile; however, our experiment was designed to focus specifically on the sequestration of SDF-1α within a short 7 day release period (Fig. 5). Thus, two concentrations of fibrin were evaluated, 3 mg mL−1 (physiologically relevant concentration) and 25 mg mL−1 gels loaded with a constant mass of SDF-1α (500 ng mL−1). The release study indicates that 90% of the encapsulated SDF-1α was released after 2 days in the 3 mg mL−1 group whereas only 20% of the SDF-1α was released from the 25 mg mL−1 group within that same time frame (Fig. 5A). More interestingly, SDF-1α release between days 2–7 was undetectable using ELISA, suggesting sequestration of residual SDF-1α not immediately released within the first 2 days. After 7 days, fibrin clots were digested with plasmin, a serine protease, to liberate immobilized SDF-1α for quantification. Cumulative detection of released SDF-1α in combination with SDF-1α recovered after fibrin digestion was similar to the initial payload for both groups (Fig. 5B). Thus, the plateau in the SDF-1α release profile after the first 2 days strongly suggests that SDF-1α was captured and sequestered in fibrin matrices. The amount of SDF-1α sequestered was proportional to fibrin density as expected, where the 3 mg mL−1 group was only able to sequester roughly 10% of the encapsulated SDF-1α. Additionally, The SDF-1α release profile from fibrin was comparable to previously reported for release kinetics for soluble bFGF from fibrin.30,31
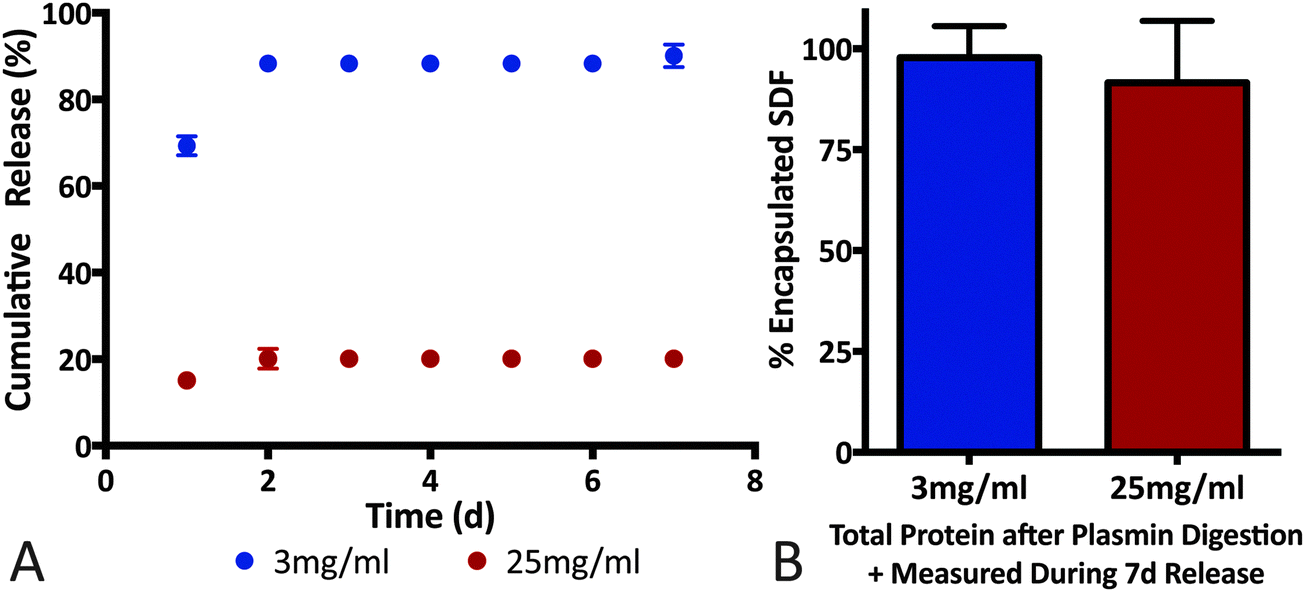 | ||
| Fig. 5 SDF-1α sequestered in fibrin matrices. (A) Release profile of free SDF-1α was significantly altered by encapsulation in different densities of fibrin. The 25 mg mL−1 group showed a release of 20% after 2 days, with no detectable SDF-1α released up to day 7. In contrast, the 3 mg mL−1 released 90% of its cargo within the first 2 days. (B) To ensure that the differences seen in the release profile were not due to problems with SDF-1α detection, the fibrin matrices were digested using plasmin and the total protein detected was similar to the initial payload for both groups. | ||
3.5 Fibrin modulates SDF-1α burst release from PLGA NPs
Given the interaction uncovered between SDF-1α and fibrin(ogen), we evaluated potential modulation of NP release profile. Specifically, we aimed to determine the effect of embedding SDF-1α-loaded NPs in different fibrin densities on the burst release in an idealized in vitro release assay. This experiment only focused on the first 72 h to fully capture the kinetics of the burst phenomena. The same concentration of NPs (1 mg of PLGA/1 mL of fibrin) was embedded within three different fibrin clot densities (3, 10 & 25 mg mL−1). The resulting SDF-1α release profiles were strongly dependent on the fibrin concentrations (Fig. 6A). Specifically, NPs embedded in 25 mg mL−1 clots reduced the amount of detected SDF-1α by ∼55% after 24 h compared to NPs freely suspended in buffer. The total cumulative released SDF-1α was significantly reduced in the 10 mg mL−1 (p < 0.01) and 25 mg mL−1 (p < 0.01) groups after 72 h in comparison to free NPs (Fig. 6B). Additionally, difference in total protein released between the 25 mg mL−1, and both 3 mg mL−1 (p < 0.01) and 10 mg mL−1 (p < 0.01) groups was also statistically significant. Although, total SDF-1α detected in the 3 mg mL−1 group was not significantly different (compared to free NPs) after 72 h, cumulative SDF-1α dosage was 15.8% lower after the first 10 h upon addition of fibrin. Therefore, these data suggest the following, (1) amplitude of the NP burst may be tuned by exploiting SDF-1α/fibrin interactions and (2) embedding the NPs within fibrin gels of varying concentrations controls the amount of SDF-1α released within the first 72 h in vitro, under idealized conditions.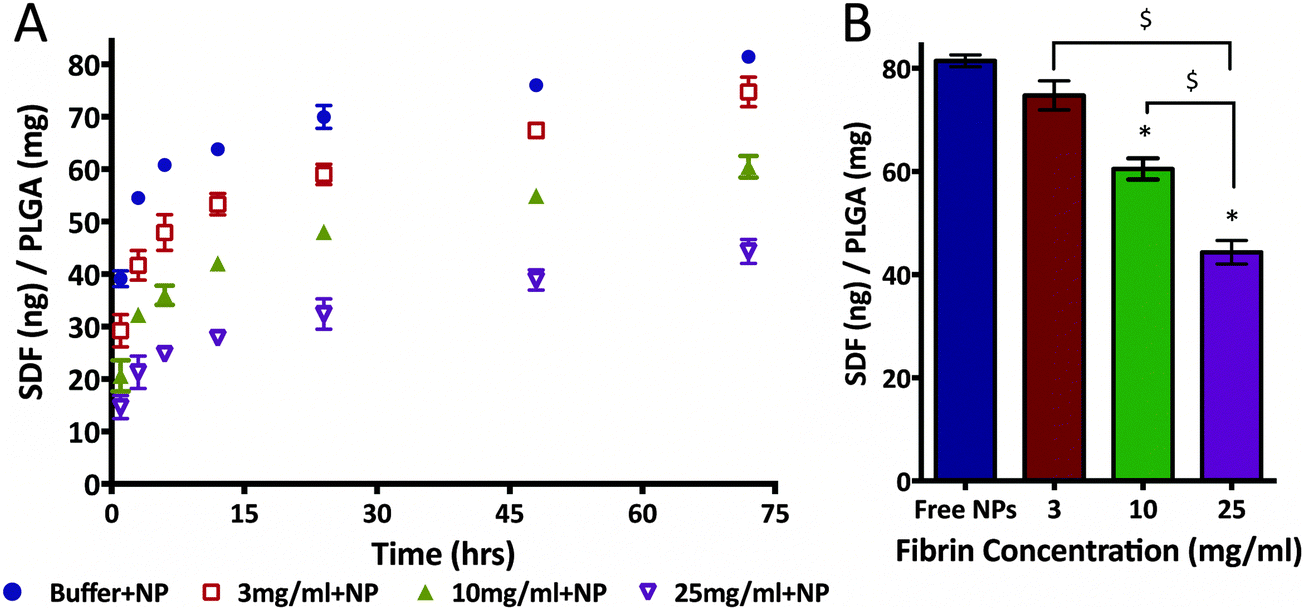 | ||
| Fig. 6 Magnitude of burst release from SDF-1α-loaded NPs was modulated with fibrin. (A) SDF-1α release from first three days was significantly altered by embedding NPs in different concentrations of fibrin. After 24 h, the 25 mg mL−1 group was able to reduce SDF-1α detected in the release media by 60% relative to free NPs. (B) Total SDF-1α detected was significantly different between the free NPs and 10 mg mL−1, as well as the 10 and 25 mg mL−1 groups. Although the total SDF-1α detected after 72 h in the 3 mg mL−1 group was not significantly different, at 24 h cumulative SDF-1α dosage is decreased by 15.8% in the fibrin group (* represents p < 0.05 relative to free NPs; $ represents p < 0.05 relative to 25 mg mL−1 fibrin group). | ||
3.6 PLGA NPs do not significantly alter fibrin polymerization
Fibrin (with and without NPs) matrices were assessed by measuring clottability of fibrinogen monomer, end-point turbidity and rheological characterization of viscoelastic properties. Our data suggests that the presence of NPs (up to 10 mg mL−1) does not significantly disrupt the formation of fibrin (Fig. 7A). As a result, end-point turbidity of fibrin clots were also similar between native and NP-embedded gels for the 15 & 30 mg mL−1 fibrin groups (Fig. 7B). The differences observed for the 3 mg mL−1 group was due to the presence of the NPs themselves (data not shown). Rheological studies further supports that fibrin clot integrity was maintained upon addition of NPs (Fig. 7C and D). The low-frequency plateau in the storage modulus (G′) of both 3 & 30 mg mL−1 fibrin groups indicate polymerization of fibrinogen to form viscoelastic fibrin (Fig. 7C).59 Additionally, the overall characteristics of the storage and loss moduli was not altered upon adding 10 mg mL−1 NPs in either 3 or 30 mg mL−1 fibrin matrices (Fig. 7C). The overall strength of fibrin was not dependent on NP content for the 3 mg mL−1 fibrin group. Although statistically significant changes in G′ was observed for 30 mg mL−1 fibrin group with 1 & 10 mg mL−1 NP (Fig. 7D), the two-way ANOVA indicates that overall, NP concentration is not a significant determinant of fibrin storage modulus (p = 0.086). Overall values for G′ acquired for the fibrin groups were similar to other reports in literature for native fibrin.59–61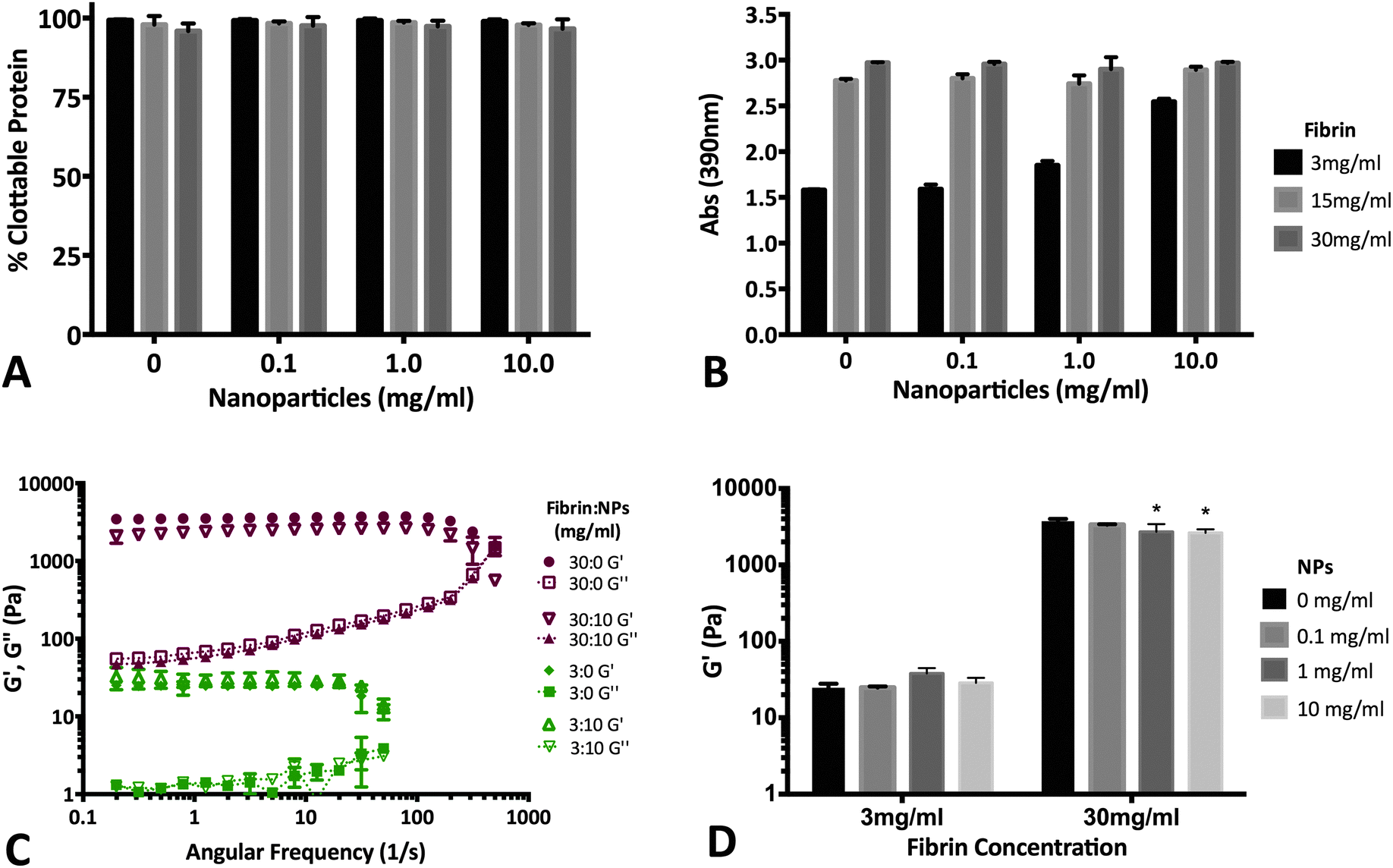 | ||
| Fig. 7 NP encapsulation does not significantly affect fibrin clot properties. (A) Percent clottable protein is not a function of NP encapsulation (up to 10 mg mL−1). Presence of NPs do not affect the ability of fibrinogen to interact with other monomers to form the insoluble fibrin clot. (B) Terminal turbidity of fibrin clots is not affected by NP concentrations up to 10 mg mL−1. The increase in turbidity seen in the 3 mg mL−1 fibrinogen group with 10 mg mL−1 NPs is likely due to the presence of the NPs themselves. (C) Storage (G′) and loss (G′′; dotted plots) moduli of 3 mg mL−1 (green) and 30 mg mL−1 (red) fibrin clots. Native fibrin (no NPs) and fibrin with 10 mg mL−1 NP illustrate that the presence of NPs cause minimal changes in fibrin mechanical properties. (D) Overall strength of 3 mg mL−1 fibrin gels are not a function of NP content. Although 30 mg mL−1 gels did show significant differences at 1 & 10 mg mL−1 NP groups, two-way ANOVA analysis indicates NPs do not play a statistically significant role in fibrin storage modulus (p = 0.086) (* represent p < 0.05 relative to 30 mg mL−1 fibrin with 0 mg mL−1 NPs). | ||
4. Conclusion
Here, we report successful encapsulation of SDF-1α within PLGA NPs to achieve controlled release over 60 days. Functional bioactivity of encapsulated and released SDF-1α was demonstrated through in vitro NPSC chemotactic migration assays. However, careful control over time-dependent SDF-1α concentration is crucial in eliciting desired therapeutic outcome. We determined that SDF-1α was successfully sequestered in fibrin clots and that NPs embedded in different concentrations of fibrin controlled the magnitude of the burst release profile without negatively affecting fibrin matrix properties. These results are significant in potentially obtaining local and sustained release of SDF-1α in a neural injury site to amplify and/or sustain NPSC-mediated endogenous repair response.Acknowledgements
The authors acknowledge Dr Rachael Sirianni of Barrow Neurological Institute for technical training and support with the PLGA NP synthesis, Dr Vikram Kodibagkar of ASU for his assistance with the ultrasonicator, Caroline Addington as well as Christine Pauken for cell culture expertise and the John M. Cowley Center for High Resolution Electron Microscopy (CHREM) for assistance with the SEM. The following funding sources are acknowledged: NIH (1DP2HD084067; SES), NSF (1454282; SES), and ASU Start-up Funds (SES).Notes and references
- V. G. Coronado, L. C. McGuire, K. Sarmiento, J. Bell, M. R. Lionbarger, C. D. Jones, A. I. Geller, N. Khoury and L. Xu, J. Saf. Res., 2012, 43, 299–307 CrossRef PubMed.
- M. Faul, M. M. Wald, W. Rutland-Brown, E. E. Sullivent and R. W. Sattin, J. Trauma: Inj., Infect., Crit. Care, 2007, 63, 1271–1278 CrossRef PubMed.
- C. P. Addington, A. Roussas, D. Dutta and S. E. Stabenfeldt, Biomarker Insights, 2015, 43 CAS.
- T. Itoh, T. Satou, H. Ishida, S. Nishida, M. Tsubaki, S. Hashimoto and H. Ito, Neurol. Res., 2009, 31, 90–102 CrossRef CAS PubMed.
- S. Li, M. Wei, Z. Zhou, B. Wang, X. Zhao and J. Zhang, Brain Res., 2012, 1444, 76–86 CrossRef CAS PubMed.
- N. L. Sundholm-Peters, H. K. C. Yang, G. E. Goings, A. S. Walker and F. G. Szele, J. Neuropathol. Exp. Neurol., 2005, 64, 1089–1100 CrossRef PubMed.
- S. Chen, J. D. Pickard and N. G. Harris, Exp. Neurol., 2003, 182, 87–102 CrossRef CAS PubMed.
- C. Moon, M. Ahn, S. Kim, J.-K. Jin, K.-B. Sim, H.-M. Kim, M.-Y. Lee and T. Shin, Brain Res., 2004, 1028, 238–242 CrossRef CAS PubMed.
- D. Aprili, O. Bandschapp, C. Rochlitz, A. Urwyler and W. Ruppen, Anesthesiology, 2009, 111, 1346–1355 CrossRef PubMed.
- D. J. Overstreet, D. Dutta, S. E. Stabenfeldt and B. L. Vernon, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 881–903 CrossRef CAS.
- T. C. Lim, S. Rokkappanavar, W. S. Toh, L.-S. Wang, M. Kurisawa and M. Spector, FASEB J., 2013, 27, 1023–1033 CrossRef CAS PubMed.
- L. Baumann, S. Prokoph, C. Gabriel, U. Freudenberg, C. Werner and A. G. Beck-Sickinger, J. Controlled Release, 2012, 162, 68–75 CrossRef CAS PubMed.
- M. Fujio, A. Yamamoto, Y. Ando, R. Shohara, K. Kinoshita, T. Kaneko, H. Hibi and M. Ueda, Bone, 2011, 49, 693–700 CrossRef CAS PubMed.
- P. W. Henderson, S. P. Singh, D. D. Krijgh, M. Yamamoto, D. C. Rafii, J. J. Sung, S. Rafii, S. Y. Rabbany and J. A. Spector, Wound Repair Regen., 2011, 19, 420–425 CrossRef PubMed.
- R. W. Sirianni, M.-Q. Zheng, T. R. Patel, T. Shafbauer, J. Zhou, W. M. Saltzman, R. E. Carson and Y. Huang, Bioconjugate Chem., 2014, 25, 2157–2165 CrossRef CAS PubMed.
- J. Zhang and M. Saltzman, Chem. Eng. Prog., 2013, 109, 25–30 CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- P. Misra, D. Lebeche, H. Ly, M. Schwarzkopf, G. Diaz, R. J. Hajjar, A. D. Schecter and J. V. Frangioni, J. Nucl. Med., 2008, 49, 963–969 CrossRef CAS PubMed.
- M. Zamani, M. P. Prabhakaran, E. S. Thian and S. Ramakrishna, J. Colloid Interface Sci., 2015, 451, 144–152 CrossRef CAS PubMed.
- D. P. Cross and C. Wang, Pharm. Res., 2011, 28, 2477–2489 CrossRef CAS PubMed.
- P. T. Thevenot, A. M. Nair, J. Shen, P. Lotfi, C.-Y. Ko and L. Tang, Biomaterials, 2010, 31, 3997–4008 CrossRef CAS PubMed.
- S. Fredenberg, M. Wahlgren, M. Reslow and A. Axelsson, Int. J. Pharm., 2011, 415, 34–52 CrossRef CAS PubMed.
- A. S. Pandit, D. J. Wilson and D. S. Feldman, J. Biomater. Appl., 2000, 14, 229–242 CrossRef CAS PubMed.
- P. P. Spicer and A. G. Mikos, J. Controlled Release, 2010, 148, 49–55 CrossRef CAS PubMed.
- M. D. Wood, G. H. Borschel and S. E. Sakiyama-Elbert, J. Biomed. Mater. Res., Part A, 2009, 89, 909–918 CrossRef PubMed.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 69, 149–158 CrossRef CAS PubMed.
- K. Vulic and M. S. Shoichet, Biomacromolecules, 2014, 15, 3867–3880 CrossRef CAS PubMed.
- A. Breen, T. O’Brien and A. Pandit, Tissue Eng., Part B, 2009, 15, 201–214 CrossRef CAS PubMed.
- G. S. Schultz and A. Wysocki, Wound Repair Regen., 2009, 17, 153–162 CrossRef PubMed.
- M. M. Martino, P. S. Briquez, A. Ranga, M. P. Lutolf and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4563–4568 CrossRef CAS PubMed.
- C. Wong, E. Inman, R. Spaethe and S. Helgerson, Thromb. Haemostasis, 2003, 89, 573–582 CrossRef CAS.
- R. L. McCall and R. W. Sirianni, J. Visualized Exp., 2013 DOI:10.3791/51015.
- B. A. Reynolds, W. Tetzlaff and S. Weiss, J. Neurosci., 1992, 12, 4565–4574 CAS.
- C. P. Addington, C. M. Pauken, M. R. Caplan and S. E. Stabenfeldt, Biomaterials, 2014, 35, 3263–3272 CrossRef CAS PubMed.
- T. Aoto, N. Saitoh, T. Ichimura, H. Niwa and M. Nakao, Dev. Biol., 2006, 298, 354–367 CrossRef CAS PubMed.
- T. Riedel, E. Brynda, J. E. Dyr and M. Houska, J. Biomed. Mater. Res., Part A, 2009, 88, 437–447 CrossRef PubMed.
- S. E. Stabenfeldt, M. Gourley, L. Krishnan, J. B. Hoying and T. H. Barker, Biomaterials, 2012, 33, 535–544 CrossRef CAS PubMed.
- W. Asghar, M. Islam, A. S. Wadajkar, Y. Wan, A. Ilyas, K. T. Nguyen and S. M. Iqbal, IEEE Trans. Nanotechnol., 2012, 11, 546–553 CrossRef.
- D. J. Hines and D. L. Kaplan, Crit. Rev. Ther. Drug Carrier Syst., 2013, 30, 257–276 CrossRef CAS PubMed.
- R. P. Batycky, J. Hanes, R. Langer and D. A. Edwards, J. Pharm. Sci., 1997, 86, 1464–1477 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- A. M. Robin, Z. G. Zhang, L. Wang, R. L. Zhang, M. Katakowski, L. Zhang, Y. Wang, C. Zhang and M. Chopp, J. Cereb. Blood Flow Metab., 2006, 26, 125–134 CrossRef CAS PubMed.
- J. Imitola, K. Raddassi, K. I. Park, F.-J. Mueller, M. Nieto, Y. D. Teng, D. Frenkel, J. Li, R. L. Sidman, C. A. Walsh, E. Y. Snyder and S. J. Khoury, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18117–18122 CrossRef CAS PubMed.
- T. Ponomaryov, A. Peled, I. Petit, R. S. Taichman, L. Habler, J. Sandbank, F. Arenzana-Seisdedos, A. Magerus, A. Caruz, N. Fujii, A. Nagler, M. Lahav, M. Szyper-Kravitz, D. Zipori and T. Lapidot, J. Clin. Invest., 2000, 106, 1331–1339 CrossRef CAS PubMed.
- S. Bhakta, P. Hong and O. Koc, Cardiovasc. Revasc. Med., 2006, 7, 19–24 CrossRef PubMed.
- R. S. Klein, J. B. Rubin, H. D. Gibson, E. N. DeHaan, X. Alvarez-Hernandez, R. A. Segal and A. D. Luster, Development, 2001, 128, 1971–1981 CAS.
- N. Signoret, J. Oldridge, A. Pelchen-Matthews, P. J. Klasse, T. Tran, L. F. Brass, M. M. Rosenkilde, T. W. Schwartz, W. Holmes, W. Dallas, M. A. Luther, T. N. C. Wells, J. A. Hoxie and M. Marsh, J. Cell Biol., 1997, 139, 651–664 CrossRef CAS PubMed.
- G. Zhu, S. R. Mallery and S. P. Schwendeman, Nat. Biotechnol., 2000, 18, 52–57 CrossRef CAS PubMed.
- M. Kucia, K. Jankowski, R. Reca, M. Wysoczynski, L. Bandura, D. J. Allendorf, J. Zhang, J. Ratajczak and M. Z. Ratajczak, J. Mol. Histol., 2004, 35, 233–245 CrossRef CAS PubMed.
- A. Giteau, M. C. Venier-Julienne, A. Aubert-Pouëssel and J. P. Benoit, Int. J. Pharm., 2008, 350, 14–26 CrossRef CAS PubMed.
- Y. Yeo and K. Park, Arch. Pharmacal Res., 2004, 27, 1–12 CrossRef CAS.
- N. S. P. Edith and J. A. M. Schlicher, Int. J. Pharm., 1997, 153, 235–245 CrossRef.
- J. C. Stanwick, M. D. Baumann and M. S. Shoichet, J. Controlled Release, 2012, 160, 666–675 CrossRef CAS PubMed.
- D.-H. Kim and D. C. Martin, Biomaterials, 2006, 27, 3031–3037 CrossRef CAS PubMed.
- O. Jeon, S.-W. Kang, H.-W. Lim, J. Hyung Chung and B.-S. Kim, Biomaterials, 2006, 27, 1598–1607 CrossRef CAS PubMed.
- P. G. Campbell, S. K. Durham, J. D. Hayes, A. Suwanichkul and D. R. Powell, J. Biol. Chem., 1999, 274, 30215–30221 CrossRef CAS PubMed.
- M. W. Mosesson, J. Thromb. Haemostasis, 2005, 3, 1894–1904 CrossRef CAS PubMed.
- I. Catelas, J. F. Dwyer and S. Helgerson, Tissue Eng., Part C, 2008, 14, 119–128 CrossRef CAS PubMed.
- J. M. Zuidema, C. J. Rivet, R. J. Gilbert and F. A. Morrison, J. Biomed. Mater. Res., Part B, 2014, 102, 1063–1073 CrossRef PubMed.
- J. Wedgwood, A. J. Freemont and N. Tirelli, Macromol. Symp., 2013, 334, 117–125 CrossRef CAS.
- E. A. Ryan, L. F. Mockros, J. W. Weisel and L. Lorand, Biophys. J., 1999, 77, 2813–2826 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb00935a |
| This journal is © The Royal Society of Chemistry 2015 |