Multi-stimuli sensitive supramolecular hydrogel formed by host–guest interaction between PNIPAM-Azo and cyclodextrin dimers†
Yue Guan,
Hai-Bo Zhao,
Lei-Xiao Yu,
Si-Chong Chen* and
Yu-Zhong Wang*
Center for Degradable and Flame-Retardant Polymeric Materials (ERCEPM-MoE), National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), State Key Laboratory of Polymer Materials Engineering, College of Chemistry, Sichuan University, Chengdu, 610064, P.R. China. E-mail: chensichong@scu.edu.cn; yzwang@scu.edu.cn; Fax: +86-28-85410755; Tel: +86-28-85410755
First published on 12th November 2013
Abstract
We reported here a novel three stimuli sensitive hydrogel that was constructed by the formation of host–guest complexes between poly(N-isopropylacrylamide) (PNIPAM) containing azobenzene groups and cyclodextrin dimers connected by disulfide bonds. The obtained hydrogel gives a smart response to the stimuli of temperature, light, and reduction, manifested in the form of a sol–gel phase transition.
Motivated by the demand for high-performance smart materials in applications such as catalysis, drug delivery, and biomedical materials,1–3 an abundance of developed stimuli-responsive materials have been fabricated and extensively investigated.3–6 These stimuli-responsive materials can generally undergo changes in response to small external changes in environmental conditions,7 such as temperature,8 redox,9 light,10 enzymes,11 pH,12 magnetic fields13 and so on. Meanwhile, smart materials that combine several stimuli-responses are particularly interesting and appealing. Conventionally, more stimuli-responses would generate more flexibility to further the applications of these materials.14,15 Responsive changes often require being sensitive to multiple environmental changes rather than a single signal when the materials are applied in practice, such as biological systems that sometimes need to respond to two different stimulus signals to trigger enzymatic functions.16 Thus, it is important to design and achieve smart biomaterials that are endowed with responsive properties for multiple and diversiform stimuli in a predictable manner, not only for scientific research but also for potential applications.
Hydrogels have attracted great interest for biomaterial applications due to being homogenous materials with physical properties similar to tissues.17 As is well-known, stimuli-response can be found widely in natural biomaterials.18 Thus, synthetic hydrogels that mimic natural biomaterials possessing intriguing biological functions for detecting external stimuli, leading to induced responses in structures and properties, have been explored by some eminent researchers.19,20 So far, plenty of supramolecular hydrogels exhibiting responses to external stimuli such as temperature, redox, light, or pH have been reported.21–24 However, owing to the difficulty in fabricating multi-stimuli soft materials from complicated designs and syntheses,25 most of them deal with response to a single stimulus. Multi-stimuli sensitive hydrogels with diversiform responses are rarely studied26 and are still a real challenge.
Of all the stimulus-responses, temperature, light and redox responses widely exist in the natural world and play important roles in biological systems. Particularly, biological systems usually need to respond to different combinations of these stimuli; for example, light and redox responses in photosynthetic organisms27 or temperature and light responses28 and so on. Thus, the materials possessing temperature, light and redox responses may be excellent mimics of nature and have great potential applications. Here in this work, a novel multi-stimuli sensitive supramolecular hydrogel that responded to changes in temperature, light and reduction potential was prepared based on the host–guest complexes of cyclodextrin (CD) and azobenzene (azo). PNIPAM containing azo groups (PNIPAM-Azo) was synthesized as the guest copolymer, while CD dimers connected by disulfide bonds acted as the host molecules and as a cleavable cross-linking agent. Among these components, PNIPAM has attracted attention due to its sensitivity to temperature, disulfide bonds are ubiquitous in living organisms and used as a redox-responsive switch, and the inherent light-sensitive properties of the host–guest interaction between CDs and azobenzene are well-known.29 Thus, the obtained hydrogel would give a smart response to the stimuli of temperature, light, and reduction; the response being manifested in the form of a sol–gel phase transition via responsive changes being induced in each of the stimuli sensitive groups.
Scheme 1 depicts the chemical structures of the guest copolymer (PNIPAM-Azo) and host molecules (CD dimers). PNIPAM-Azo (Mn = 6.09 × 104 g mol−1, calculated by gel permeation chromatography (GPC)) was prepared in DMF by Reversible Addition-Fragmentation Chain Transfer (RAFT) copolymerization, and the feed ratio of AA-azobenzene (acrylic acid modified azobenzene) to N-isopropylacrylamide (NIPAAM) was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 (mol/mol). The polymers were characterized by 1H NMR spectroscopy and GPC, and the molar composition of PNIPAM-Azo was equal to that of the monomer mixture. The cleavable β-CD dimer was prepared via condensation of bis(2-carboxyethyl) disulfide with 2 equiv. of 6-amino-β-CD catalyzed by N,N′-dicyclohexylcarbodiimide (DCC) and 1-hydroxy-1H-benzotriazole (HOBT) in DMF.30
:20 (mol/mol). The polymers were characterized by 1H NMR spectroscopy and GPC, and the molar composition of PNIPAM-Azo was equal to that of the monomer mixture. The cleavable β-CD dimer was prepared via condensation of bis(2-carboxyethyl) disulfide with 2 equiv. of 6-amino-β-CD catalyzed by N,N′-dicyclohexylcarbodiimide (DCC) and 1-hydroxy-1H-benzotriazole (HOBT) in DMF.30
 | ||
| Scheme 1 Chemical structures of the guest copolymer and host molecules. | ||
The hydrogel was prepared by mixing the PNIPAM-Azo (8 wt%) and CD dimers in an equivalent ratio of the host–guest units in water at 10 °C. The CD dimer plays the role of cross-linker in the formation of the hydrogel, since no hydrogel was generated in the aqueous solutions of the PNIPAM-Azo–β-CD mixture or neat PNIPAM-Azo at 10 °C.
Profiting from the temperature-induced coil–globule transition of PNIPAM, the hydrogel exhibited temperature-responsive swelling–contraction transition behavior. Fig. 1 depicts the photographs of the temperature-responsive swelling–contraction transition of the hydrogel. When heated up to 30 °C, the hydrogel gradually contracted. The responsive gel then underwent a thermally reversible process where it swelled to its original state upon cooling to 10 °C. This temperature dependent transition can be explained as follows: when the temperature is below the lower critical solution temperature (LCST) of the PNIPAM-Azo–CD dimer complexes, PNIPAM is hydrated and swollen. When heated above its LCST, it undergoes a reversible volume phase transition to a collapsed, dehydrated state. The temperature increase caused the polymer to expel water and contract into a more hydrophobic polymer state.31
 | ||
| Fig. 1 Temperature dependent swelling–contraction transitions of the PNIPAM-Azo–CD dimer hydrogel. (a) The original hydrogel at 10 °C; (b) heated up to 30 °C; (c) then cooled down to 10 °C. | ||
Furthermore, to investigate the phase transition temperature of the PNIPAM-Azo copolymer, turbidity measurements of neat PNIPAM-Azo, the PNIPAM-Azo–CD dimer mixture, and the PNIPAM-Azo–β-CD mixture were performed. In order to obtain a clear test solution, the polymer concentration was chosen as 2 mg ml−1, which was dilute enough to form the hydrogel but still could reflect the influence of cross-linking in the LCST.32 For PNIPAM-Azo, the LCST was 13 °C, while the LCST values of the PNIPAM-Azo–β-CD mixture and the PNIPAM-Azo–CD dimer mixture increased to 27 °C and 25 °C, respectively (Fig. 2). The LCST of the PNIPAM-Azo copolymer was significantly lower than that of PNIPAM (35 °C). This phenomenon could be attributed to the existence of hydrophobic AA-azo blocks in the copolymer, which may restrict hydrogen bond formation as well. The PNIPAM-Azo–β-CD and the PNIPAM-Azo–CD dimer mixture exhibited higher LCST values than the neat PNIPAM-Azo solution owing to the inclusion of the hydrophobic azobenzene units by β-CD. Interestingly, the turbidity point of the PNIPAM-Azo–CD dimer was between those for PNIPAM-Azo and PNIPAM-Azo–β-CD. This phenomenon was attributed to the cross-linkage of single polymer chains upon the complexation of the CD dimer, since the mobility and solubility of the resulting supramolecular was restricted.
 | ||
| Fig. 2 Transmittance versus temperature curves from turbidity measurements (Cp = 2 mg ml−1, CCD dimer = 1 mg ml−1, CCD = 2 mg ml−1, where Cp, CCD dimer and CCD are the concentration of the copolymer, CD dimer and CD, respectively). | ||
Next, the response of the hydrogel to changes in light wavelength was investigated. It is known that azobenzene compounds can be applied in light-sensitive materials in view of their peculiar optical properties.33,34 The isomerization of the azobenzene group can be regulated by UV and visible light irradiation, where the former (π–π* transition) achieves trans-to-cis isomerization and the latter (n–π* transition) promotes the reverse isomerization.35 However, β-CDs and their derivatives formed inclusion complexes only with azobenzene compounds in the trans form. Therefore, the hydrogel fabricated by trans-PNIPAM-Azo–β-CD underwent a gel–sol transition upon UV irradiation (λ = 365 nm) when the formed cis-azobenzene groups were excluded from the CD dimer cavities and the cross-linking inclusion complexes were destroyed (Fig. 3). In this process, UV irradiation (λ = 365 nm) of the hydrogel caused a dramatic change in the viscosity, and the η0 value of the hydrogel decreased from 94 Pa s to 12 Pa s after photoirradiation (Fig. 4a and b). By comparison, visible light irradiation (λ = 450 nm) of the sol state recovered the hydrogel, and the η0 value of the system increased to 77 Pa s just by visible light irradiation for 2 h (Fig. 4c). (The fully reversible recovery of the η0 value could be obtained by further increasing the visible light illumination time. However, in consideration of the lighting efficiency, the test was stopped after only 2 hours). Thus, the reversible gel–sol transition could be repeatedly induced simply by photoirradiation with UV and visible light alternately.
 | ||
| Fig. 3 Illustration of the host–guest interactions between the PNIPAM-Azo and CD dimers under irradiation with UV (365 nm) and visible light (450 nm). | ||
 | ||
| Fig. 4 Zero-shear viscosities for (a) original gel, (b) sol after irradiation under 365 nm light, (c) gel after 2 h irradiation under 450 nm light, and (d) gel after reduction (obtained from dynamic rheometry at 10 °C, as shown in Fig. S9†). | ||
Fig. 5 shows the time-resolved absorption spectra for the PNIPAM-Azo–CD dimer mixture (a and c) and PNIPAM-Azo copolymer (b and d) under photo irradiation with UV (a and b) and visible (c and d) light. The photoisomerization process of the azobenzene units in the complex and copolymer was simulated. As expected, the azobenzene groups of the PNIPAM-Azo–CD dimer mixture and PNIPAM-Azo copolymer were isomerized from trans to cis and from cis to trans under irradiation with UV and visible light, respectively. It was noted that the azobenzene units of the PNIPAM-Azo–CD dimer inclusion complexes showed a slower isomerization rate from trans to cis upon UV irradiation than that of neat PNIPAM-Azo, which was attributed to the inclusion of the trans-azobenzene units with the CD dimer. The strong host–guest interaction between the PNIPAM-Azo copolymers and the CD dimer made the isomerization of trans-azobenzene units more difficult. However, compared to the neat PNIPAM-Azo copolymer, the resulting cis-azobenzene groups of the PNIPAM-Azo–CD dimer mixture were more quickly transformed into trans upon visible irradiation because the inclusion of the trans-azobenzene units by the CD dimer promoted the isomerization from cis to trans.
 | ||
| Fig. 5 The time-resolved absorption spectra for the PNIPAM-Azo–CD dimer mixture (a and c) and the PNIPAM-Azo copolymer (b and d) under photo irradiation with UV (a and b) and visible (c and d) light. (Cp = 0.2 mg ml−1, CCD dimer = 0.1 mg ml−1, 25 °C). | ||
Lastly, we investigated the effect of reducing reagents on the phase transition of the supramolecular hydrogel. Disulfide bonds, which can be easily cleaved by using reducing agents within cells (e.g., glutathione), have been widely applied in reduction-sensitive biomaterials such as mesoporous silica nanoparticles36 or polymeric micelles.37,38 In our design, the CD dimers, which act as a noncovalent cross-linker between the guest polymer, were bridged by cleavable disulfide bonds. Thus, during the reaction with the reducing reagents, the cleavage of the CD dimer bond rapidly induced loss of cross-linking in the hydrogel and transformed the gel into the sol (Fig. 6). In this paper, dithiothreitol (DTT) was used as the reductant. As shown in Fig. 6, the hydrogel was transformed into the sol immediately when adding 5 equiv. of DTT, and the viscosity also decreased drastically to 0.7 Pa s (Fig. 4d). Compared to the light-responsiveness, the cross-linking structure of the hydrogel was destroyed more quickly and completely in response to the reduction conditions, because of the immediate and thorough cleavage of chemical bonds. However, the UV irradiation-induced isomerization of the azobenzene was rather slow; moreover, a dynamic equilibrium existed between isomers, in other words, the host–guest interaction cannot be completely eliminated, even after prolonged irradiation. As a result, reduction induced a fast and radical gel–sol phase transition, while the light response was slow and tempered. However, the gel–sol phase transition was not reversible. NaClO or H2O2 (aqueous solution) was chosen as an oxidant to investigate the reversibility of the reduction response.39 Only a small amount of microgel was formed after addition of the oxidant (Fig. S10†), which suggested that very few of the disulfide bonds were re-established. There are some possible reasons that may be responsible for the irreversibility. The thiol groups in our system have much lower mobility and reactivity because the CDs which hold the thiol are locked at the copolymer chain by host–guest interactions with azo groups. The steric hindrance of the CD and azo groups significantly reduced the reactivity towards the re-establishment of the disulfide bonds.
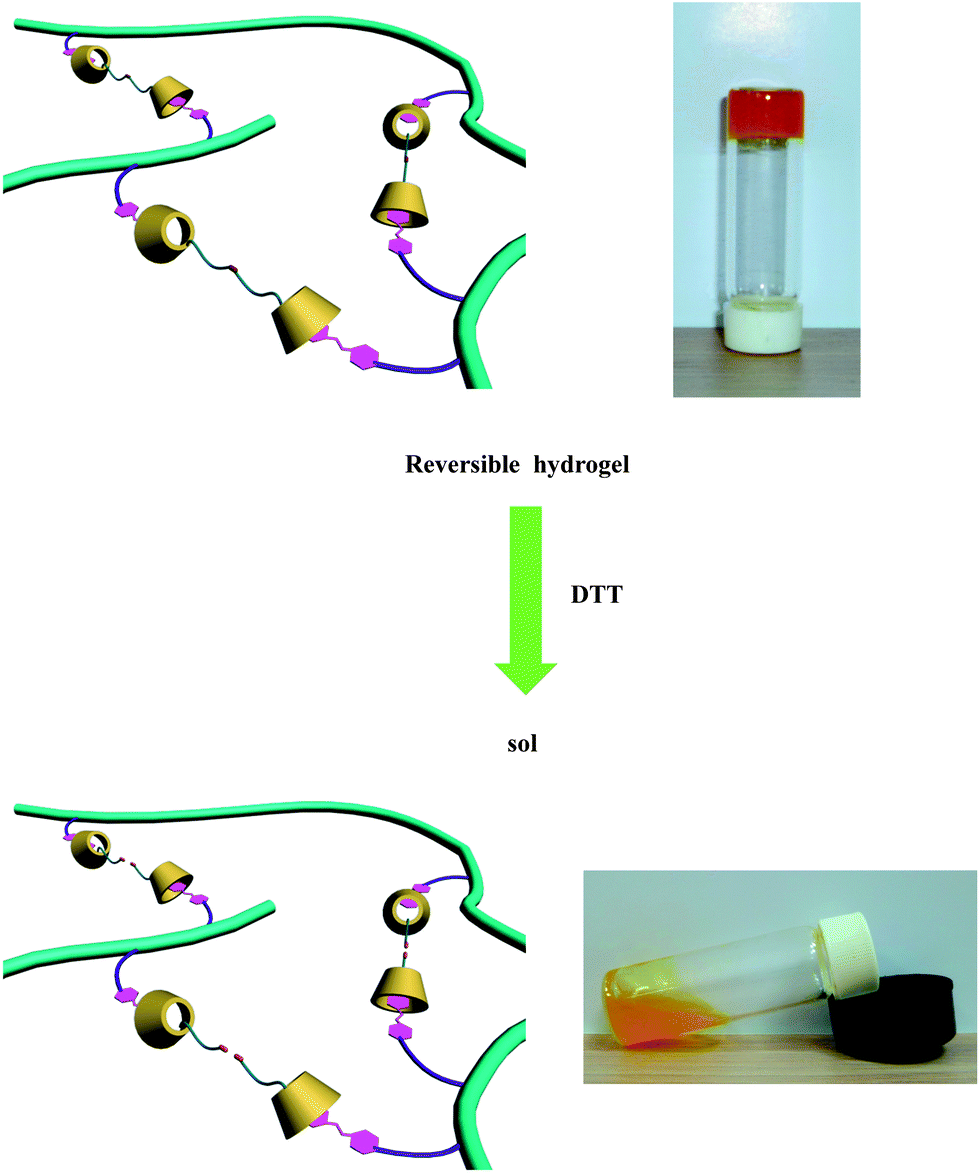 | ||
| Fig. 6 Illustration of the reduction-responsive sol–gel transition experiment. (The reduction experiment was carried out at 10 °C). | ||
Conclusions
In conclusion, a novel triple stimuli sensitive supramolecular hydrogel that responded to changes in temperature, light and reduction potential was successfully designed and synthesized. The responsive hydrogel was constructed by the formation of host–guest complexes between PNIPAM possessing azobenzene groups as the guest and CD dimers decorated by disulfide bonds as the host. The reversible gel–sol phase transition of the hydrogel could be induced by temperature and light, while the reduction potential could induce a quick gel–sol transition. These multiple stimulus-responsive hydrogels have potential applications in drug delivery, catalysis, and biomedical materials.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 51073106 and 51121001), Program for Changjiang Scholars and Innovative Research Team in University of Ministry of Education of China (IRT1026).Notes and references
- T. Terashima, M. Ouchi, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2007, 40, 3581–3588 CrossRef CAS.
- G. Jiang, Y. Wang, R. Zhang, R. Wang, X. Wang, M. Zhang, X. Sun, S. Bao, T. Wang and S. Wang, ACS Macro Lett., 2012, 1, 489 CrossRef CAS.
- H. D. Lu, M. B. Charati, I. L. Kim and J. A. Burdick, Biomaterials, 2012, 33, 2145 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615 CrossRef CAS PubMed.
- J. Wang, L. Lin, Q. Cheng and L. Jiang, Angew. Chem., Int. Ed., 2012, 51, 4676 CrossRef CAS PubMed.
- J. C. Eloi, D. A. Rider, G. Cambridge, G. R. Whittell, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8903 CrossRef CAS PubMed.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- C. Xu, J. S. Ren, L. Y. Feng and X. G. Qu, Chem. Commun., 2012, 48, 3739–3741 RSC.
- H. Yamaguchi, Y. Kobayashi, R. Kobayashi, Y. Takashima, A. Hashidzume and A. Harada, Nat. Commun., 2012, 3, 603 CrossRef PubMed.
- P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2007, 19, 1252–1256 CrossRef.
- C. S. Zhao, S. Q. Nie, M. Tang and S. D. Sun, Prog. Polym. Sci., 2011, 36, 1499–1520 CrossRef CAS.
- Q. Dai and A. Nelson, Chem. Soc. Rev., 2010, 39, 4057–4066 RSC.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315 CrossRef CAS.
- C. Dou, D. Chen, J. Iqbal, Y. Yuan, H. Zhang and Y. Wang, Langmuir, 2011, 27, 6323 CrossRef CAS PubMed.
- J. H. Hurley, J. Biol. Chem., 1999, 274, 7599 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444 CrossRef CAS PubMed.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461 CrossRef CAS PubMed.
- Y. Zheng, A. Hashidzume, Y. Takashima, H. Yamaguchi and A. Harada, ACS Macro Lett., 2012, 1, 1083 CrossRef CAS.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- I. Tomatsu, A. Hashidzume and A. Harada, Macromolecules, 2005, 38, 5223 CrossRef CAS.
- R. Yoshida, Adv. Mater., 2010, 22, 3463 CrossRef CAS PubMed.
- X. Jiang, S. Jin, Q. Zhong, M. D. Dadmun and B. Zhao, Macromolecules, 2009, 42, 8468 CrossRef CAS.
- P. Schattling, F. D. Jochum and P. Theato, Chem. Commun., 2011, 47, 8859 RSC.
- C. Bauer, S. Elsen, L. R. Swem, D. L. Swem and M. Shinji, Philos. Trans. R. Soc., B, 2003, 358, 147 CrossRef CAS PubMed.
- S. Husted and J. K. Schjoerring, Plant Physiol., 1996, 112, 67 CAS.
- X. Liao, G. Chen and M. Jiang, Polym. Chem., 2013, 4, 1733 RSC.
- K. Ohga, Y. Takashima, H. Takahashi, Y. Kawaguchi, H. Yamaguchi and A. Harada, Macromolecules, 2005, 38, 5897 CrossRef CAS.
- J. M. Weissman, H. B. Sunkara, A. S. Tse and S. A. Asher, Science, 1996, 274, 959 CrossRef CAS PubMed.
- O. Kretschmann, S. W. Choi, M. Miyauchi, I. Tomatsu, A. Harada and H. Ritter, Angew. Chem., Int. Ed., 2006, 45, 4361 CrossRef PubMed.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139 CrossRef CAS PubMed.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986 CrossRef CAS PubMed.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, L. Zhao, P. Liu, L. Duan and W. Yang, Angew. Chem., Int. Ed., 2011, 50, 640 CrossRef CAS PubMed.
- W.-F. Dong, A. Kishimura, Y. Anraku, S. Chuanoi and K. Kataoka, J. Am. Chem. Soc., 2009, 131, 3804 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37 CrossRef.
- S. S. Anumolu, A. R. Menjoge, M. Deshmukh, D. Gerecke, S. Stein, J. Laskin and P. J. Sinko, Biomaterials, 2011, 32, 1204 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and Fig. S1–S9. See DOI: 10.1039/c3ra45461d |
| This journal is © The Royal Society of Chemistry 2014 |