
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of unknown 6,7-dihydrofuro[3,4-c]pyridines and 3,4-diaryloylpyridines from N-homopropargylic β-enaminones†
Elif Serel
Yilmaz
 a,
Kerem
Kaya‡
b and
Metin
Zora
*a
a,
Kerem
Kaya‡
b and
Metin
Zora
*a
aDepartment of Chemistry, Faculty of Arts and Science, Middle East Technical University, 06800 Ankara, Turkey. E-mail: zora@metu.eddu.tr
bDepartment of Chemistry, Faculty of Science, Istanbul Technical University, 34469 Istanbul, Turkey
First published on 15th January 2025
Abstract
In this paper, we have uncovered a new reaction of N-homopropargylic β-enaminones, i.e. N-(4-phenyl-3-butynyl)-β-enaminones. When subjected to a reaction with excess molecular iodine or N-iodosuccinimide in the presence of cesium carbonate, N-homopropargylic β-enaminones afford 6,7-dihydrofuro[3,4-c]pyridines in low to moderate yields. The generation of two new C/O–C bonds during the reaction leads to the construction of unknown heterobicyclic 5,6-fused ring systems. In some reactions, 3,4-diaryloylpyridines are also observed in low yields. During the formation of 3,4-diaryloylpyridines, a new carbonyl (ketone) group is generated. The synthesized 6,7-dihydrofuro[3,4-c]pyridines and 3,4-diaryloylpyridines may be of use in pharmaceutical and medicinal chemistry as new and novel molecular entities and structural leads.
Introduction
Isobenzofurans (1) are recognized as an interesting class of reactive intermediates in organic synthesis since they generally behave as reactive dienes and participate in Diels–Alder reactions, leading to the formation of a variety of polycyclic ring systems including natural products of biological importance (Scheme 1).1 On the other hand, heteroanalogues of isobenzofurans have received much less attention.2 For instance, the parent furo[3,4-c]pyridine (2) was reported in 1977 as a white crystalline solid that is stable only at low temperature and undergoes fast polymerization around room temperature (Scheme 1).3 Although not isolated, substituted furo[3,4-c]pyridines (2) have also been identified as reactive intermediates/dienes, similar to isobenzofurans (1), in the synthesis of hetero-polyaromatic compounds by tandem Hamaguchi–Ibata4 and Diels–Alder reactions.5 More recently, the Sarkar research group used their reactivity to prepare conformationally restricted analogues of nicotine and anabasine via intramolecular Diels–Alder reactions.6 Notably, the utility of heteroisobenzofurans as potential building blocks for the construction of polycyclic heteroaromatics is critically dependent on their availability. So far, furo[3,4-c]pyridines have been synthesized by (i) retro-Diels–Alder reactions of 1,4-epoxides via flash vacuum thermolysis (FVT) (Scheme 2a),3 (ii) lithiation and subsequent o-silylation of pyridine-phthalides (Scheme 2b),7 and (iii) the Hamaguchi–Ibata reaction4 of o-aminodiazocarbonyl precursors (Scheme 2c).6,8 Sometimes, dihydro derivatives hold greater importance in terms of their biological activity and material properties as compared to their parent compounds. 1,3-Dihydrofuro[3,4-c]pyridines (3) are known (Scheme 1)9,10 and they are commonly synthesized by the metal-catalyzed [2 + 2 + 2] cycloaddition between dipropargyl ethers and nitriles (Scheme 2d).10 In contrast, to the best of our knowledge, 6,7-dihydrofuro[3,4-c]pyridines (4) are not known (Scheme 1). Therefore, the potential value of 6,7-dihydrofuro[3,4-c]pyridines, particularly in medicinal chemistry and materials science, is not known. Obviously, the development of new methodologies for the synthesis of these compounds may increase their significance in related fields.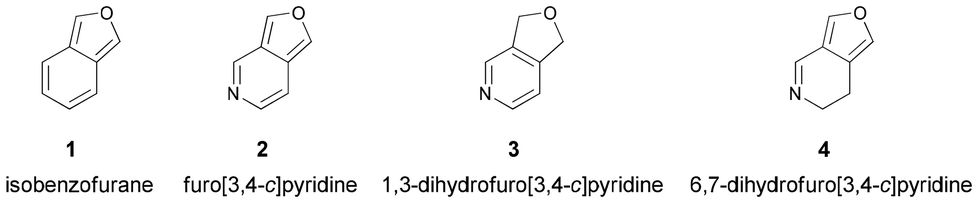 | ||
| Scheme 1 Structures of isobenzofuran, furo[3,4-c]pyridine, 1,3-dihydrofuro[3,4-c]pyridine and 6,7-dihydrofuro[3,4-c]pyridine. | ||
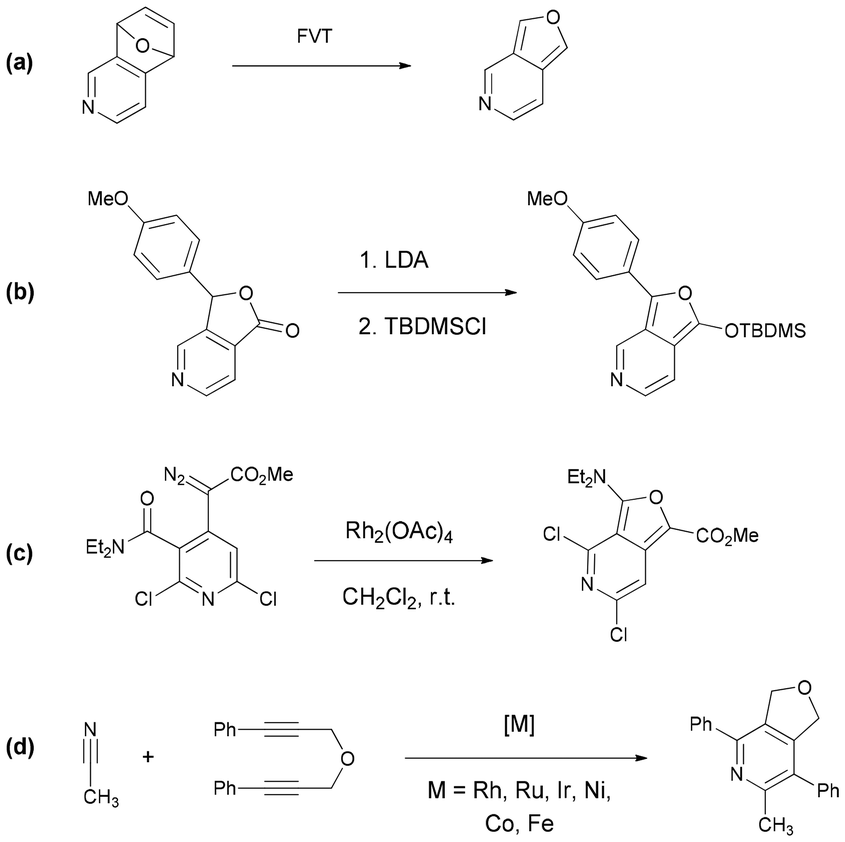 | ||
| Scheme 2 Strategies for the synthesis of furo[3,4-c]pyridines (a–c) and 1,3-dihydrofuro[3,4-c]pyridines (d). | ||
Pyridines are one of the most studied families of heterocyclic compounds due to their widespread presence in the structures of medicines, vitamins, food flavorings, dyes, adhesives, insecticides and herbicides.11 Notably, pyridines, due to their diverse physiochemical properties such as water solubility, weak basicity, chemical stability, hydrogen bond-forming ability, protein-binding capacity and cell permeability, have gained significant importance in drug design and development. In fact, there are many FDA approved pharmaceuticals that stem from a pyridine or dihydropyridine core.12 Abiraterone acetate and crizotinib (cancer), delavirdine (HIV/AIDS), isoniazid and ethionamide (tuberculosis), nilvadipine (hypertension) and tacrine (Alzheimer's) are some examples of such pharmaceuticals.13 An ever-growing aspect of these studies is to find novel pyridine molecules, which may provide a new mode of action for the treatment of a specific disease. To the best of our knowledge, 3,4-diaryloylpyridines (3,4-dibenzoylpyridines) are very limited and only a few examples of 3,4-diaryloyl-containing monocyclic (unfused) pyridine derivatives in particular are known.14,15 So the value of these derivatives is underrepresented in medicinal chemistry due to limitations in existing synthetic approaches.
Recently, enaminones have emerged as valuable substrates in organic synthesis.16 In particular, N-propargylic β-enaminones have been commonly employed as important starting precursors and/or intermediates for the synthesis of new heterocyclic ring systems (Scheme 3).17 When treated with suitable reagents, these substances can easily produce five-, six- and seven-membered heterocyclic compounds via one-pot cyclization. Pyrroles,18 2-acetylpyrroles,19 pyridines,18 iodo-substituted pyridines,20 1,4-oxazepines21 and 1,4-thiazepines22 are the main products obtained from these reactions (Scheme 3).23
 | ||
| Scheme 3 Representative reactions of N-propargylic β-enaminones. | ||
Although N-propargylic β-enaminones have been extensively explored, little is known about the reactions of N-(3-butynyl)-β-enaminones, generally called N-homopropargylic β-enaminones, which feature one more carbon in their N-substitution as compared to N-propargylic β-enaminones. When N-(3-butynyl)-β-enaminones are employed in these reactions, elementary considerations suggest that their reactions should produce heterocyclic ring systems with an additional carbon atom. To the best of our knowledge, there is only one study in this regard. The Stratakis research group reported that N-homopropargylic β-enaminones 7, generated in situ from the reaction of allenones 5 with 3-butynylamine (6), underwent 6-exo-dig cyclization, followed by dehydrogenation (aromatization), in the presence of Au nanoparticles on TiO2 (Au/TiO2), to yield 3-ketopyridines or 4-picolines 8 in good yields (Scheme 4a).24 An isolated derivative of β-enaminones 7 also furnished the corresponding 4-picoline 8 under the same conditions (Au/TiO2). However, when 4-aryl-3-butynyl amines containing an internal alkyne functionality were employed under the same reaction conditions, the in situ generated N-homopropargylic β-enaminones were entirely unreactive and did not produce any cyclization products, which was attributed to steric effects. Our previous studies prompted us to investigate the reactions of N-homopropargylic β-enaminones 9 with iodinating reagents in the presence of a base (Scheme 4b).20 Unfortunately, our initial studies showed that β-enaminones 9 did not give the expected products, presumably due to conformational and electronic effects; instead, they afforded two new products, bicyclic 6,7-dihydrofuro[3,4-c]pyridines 10 and/or 3,4-diaryloylpyridines 11 (Scheme 4b). To the best of our knowledge, the formation of such heterocyclic molecules from β-enaminones is without any precedent. In this paper, we report the preliminary results of this study.
 | ||
| Scheme 4 Reactions of N-homopropargylic β-enaminones. | ||
Results and discussion
After preparing homopropargylamines according to known literature procedures (see the ESI†),24,25 we synthesized N-homopropargylic β-enaminones, as shown in Scheme 5. First of all, we prepared the parent N-homopropargylic β-enaminone 14a in 70% yield by conjugate addition of 3-butynylamine (6) to α,β-alkynic ketones 12a in refluxing methanol.18,20 Similarly, we synthesized phenyl-substituted N-homopropargylic β-enaminones 9 as well. Conjugate addition of 4-phenylbut-3-yn-1-amine (13) to α,β-alkynic ketones 12 in refluxing methanol afforded the desired β-enaminones 9.18,20 We prepared 16 derivatives of β-enaminones 9, the yields of which ranged between 18–84% (Scheme 5). | ||
| Scheme 5 Synthesis of N-homopropargylic β-enaminones. All the presented yields are isolated yields. | ||
Afterward, we studied the reactions of N-homopropargylic β-enaminones 9 with iodinating reagents such as molecular iodine (I2) or N-iodosuccinimide (NIS). In order to test the reaction and optimize the conditions, we first examined the reaction of N-homopropargylic β-enaminone 9a, as shown in Table 1. In the beginning, we chose molecular iodine as the iodinating reagent since it is a low cost, mild and eco-friendly electrophile, which catalyzes or mediates a variety of organic reactions with high efficiency and selectivity.26 In the first phase of the study, we tested the reaction of 9a in the presence of varying amounts of molecular iodine and sodium bicarbonate in refluxing acetonitrile (Table 1, entries 1–3). These reactions produced an iodo-substituted azepine derivative with the formula C25H18INO, determined from the HRMS analysis, in 9–25% yields, the structure of which could not be fully assigned. Then we employed cesium carbonate as the base and repeated the reaction with different amounts of I2 and Cs2CO3 (Table 1, entries 4–6). Surprisingly, two new products, 1,3,4-triphenyl-6,7-dihydrofuro[3,4-c]pyridine (10a) and 3,4-dibenzoyl-2-phenylpyridine (11a), were obtained from these reactions in 13–30% and 10–23% yields, respectively. The structure of 10a was unambiguously identified by X-ray crystal analysis. The ORTEP view of 10a is given in Table 1. When using 5.0 equiv. of I2, the yield (19%) of 10a was reduced (Table 1, entry 7). In short, the highest yield (30%) of 10a was obtained with 4.0 equiv. of I2 and 2.5 equiv. of Cs2CO3 (Table 1, entry 5). Thus we continued the reaction with these equivalencies. When the reaction time was prolonged to the maximum such as 48 h, no product was isolated, possibly due to decomposition (Table 1, entry 8). On the other hand, when the reaction was carried out under dilute conditions or in a pressure tube, only 6,7-dihydrofuro[3,4-c]pyridine 10a was formed but in low yields (11 and 21%, respectively) (Table 1, entries 9 and 10). In the absence of molecular iodine or a base, no cyclized products were observed (Table 1, entries 11 and 12) but, in the first reaction, the starting β-enaminone 9a was recovered in 97% yield. Potassium carbonate and silver carbonate were also tested in these reactions (Table 1, entries 13 and 14), but only the latter gave a product (11a) in 16% yield. The reaction was also conducted in N,N-dimethylformamide and ethanol (Table 1, entries 15 and 16), producing 6,7-dihydrofuro[3,4-c]pyridine 10a and 3,4-dibenzoylpyridine 11a in 6–28% and 10–15% yields, respectively. In the case of 1,2-dichloroethane, no product formation was observed (Table 1, entry 17). When the reaction was performed in dioxane, only 6,7-dihydrofuro[3,4-c]pyridine 10a was formed but in a low yield (11%). Clearly, other solvents were not as efficient as acetonitrile. In general, N-iodosuccinimide is better than molecular iodine in terms of the iodinating property.27 When the reactivity of I2 is insufficient, NIS is frequently employed. Thus, we carried out the reaction with N-iodosuccinimide as well as in acetonitrile and N,N-dimethylformamide (Table 1, entries 19 and 20). From these reactions, 6,7-dihydrofuro[3,4-c]pyridine 10a was isolated in 28–29% yields, very close to the highest yield (30%) produced by molecular iodine, while 3,4-dibenzoylpyridine 11a was obtained in 10–29% yields, slightly higher than that (23%) with molecular iodine. In short, the results obtained by using both reagents (I2 and NIS) were quite similar, and no dramatic change in yields was observed with NIS, especially with regard to 10a. Finally, we tested the reaction of β-enaminone 14a, i.e. the terminal alkyne analog of 9a (Table 1, entry 21). Unfortunately, the reaction of 14a with molecular iodine in refluxing acetonitrile ended up with no product formation. It should be mentioned that the optimization reactions were generally not so clean, resulting in some unidentifiable products, but in low amounts, which made purification difficult. In summary, the highest yields of 1,3,4-triphenyl-6,7-dihydrofuro[3,4-c]pyridine (10a) were obtained with 4.0 equiv. of I2 or NIS and 2.5 equiv. of Cs2CO3 in refluxing acetonitrile, as shown in entries 5 and 19 in Table 1. So the generality of the reaction and the scope of the substrates were explored under these conditions.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Enaminone | Iodinating reagent (equiv.)b | Base (equiv.)b | Solvent | Temp. (°C) | Time (h) | Yield of 10a (%) | Yield of 11a (%) |
| a Isolated yields. b Equivalency is given according to that of the starting β-enaminone 9a or 14a. c The starting β-enaminone 9a was recovered in 25% yield. d An iodo-substituted azepine derivative with the formula C25H18INO was isolated in 9% yield, the structure of which could not be fully elucidated. e The starting β-enaminone 9a was recovered in 10% yield. f An iodo-substituted azepine derivative with the formula C25H18INO was isolated in 25% yield, the structure of which could not be fully elucidated. g An iodo-substituted azepine derivative with the formula C25H18INO was isolated in 18% yield, the structure of which could not be fully elucidated. h This reaction was performed under dilute conditions by using 30 mL of solvent at the scale of 0.1 mmol of 9a. i This reaction was performed in a pressure tube by using 2 mL of solvent at the scale of 0.1 mmol of 9a. j The starting β-enaminone 9a was recovered in 97% yield. | ||||||||
| 1c,d | 9a | I2 (1.5) | NaHCO3 (1.5) | MeCN | 82 | 4.0 | — | — |
| 2e,f | 9a | I2 (2.5) | NaHCO3 (2.5) | MeCN | 82 | 4.0 | — | — |
| 3g | 9a | I2 (4.0) | NaHCO3 (4.0) | MeCN | 82 | 4.0 | — | — |
| 4 | 9a | I2 (2.5) | Cs2CO3 (2.5) | MeCN | 82 | 4.0 | 13 | 23 |
| 5 | 9a | I2 (4.0) | Cs2CO3 (2.5) | MeCN | 82 | 4.0 | 30 | 10 |
| 6 | 9a | I2 (4.0) | Cs2CO3 (4.0) | MeCN | 82 | 3.5 | 15 | 16 |
| 7 | 9a | I2 (5.0) | Cs2CO3 (2.5) | MeCN | 82 | 3.0 | 19 | — |
| 8 | 9a | I2 (4.0) | Cs2CO3 (2.5) | MeCN | 25 | 48.0 | — | — |
| 9h | 9a | I2 (4.0) | Cs2CO3 (2.5) | MeCN | 82 | 3.0 | — | 11 |
| 10i | 9a | I2 (4.0) | Cs2CO3 (2.5) | MeCN | 82 | 2.5 | — | 21 |
| 11j | 9a | — | Cs2CO3 (2.5) | MeCN | 82 | 4.0 | — | — |
| 12 | 9a | I2 (4.0) | — | MeCN | 82 | 1.5 | — | — |
| 13 | 9a | I2 (4.0) | K2CO3 (2.5) | MeCN | 82 | 3.0 | — | — |
| 14 | 9a | I2 (4.0) | Ag2CO3 (2.5) | MeCN | 82 | 4.0 | — | 16 |
| 15 | 9a | I2 (4.0) | Cs2CO3 (2.5) | DMF | 110 | 3.5 | 28 | 15 |
| 16 | 9a | I2 (4.0) | Cs2CO3 (2.5) | EtOH | 78 | 9.0 | 6 | 10 |
| 17 | 9a | I2 (4.0) | Cs2CO3 (2.5) | DCE | 84 | 3.5 | — | — |
| 18 | 9a | I2 (4.0) | Cs2CO3 (2.5) | Dioxane | 101 | 3.0 | 11 | — |
| 19 | 9a | NIS (4.0) | Cs2CO3 (2.5) | MeCN | 82 | 6.0 | 29 | 13 |
| 20 | 9a | NIS (4.0) | Cs2CO3 (2.5) | DMF | 110 | 6.0 | 28 | 29 |
| 21 | 14a | I2 (4.0) | Cs2CO3 (2.5) | MeCN | 82 | 4.0 | — | — |

We next investigated the generality and substrate scope of the reaction of N-homopropargylic β-enaminones 9, as illustrated in Scheme 6. As an iodinating reagent, we mostly preferred to use molecular iodine for the reasons mentioned above. However, in some cases, N-iodosuccinimide was also used in addition to molecular iodine. A large variety of N-homopropargylic β-enaminones 9 bearing aromatic and heteroaromatic groups with electron-donating and electron-withdrawing substituents were utilized in these reactions, which afforded 6,7-dihydrofuro[3,4-c]pyridines 10 and/or 3,4-diaryloylpyridines 11. In fact, all reactions afforded the expected 6,7-dihydrofuro[3,4-c]pyridine derivatives 10 in 11–55% yields. Apparently, the use of NIS, instead of I2, increased the yields of four products (10g, i, m and o) to some extent (Scheme 6). The highest yields were observed for 6,7-dihydrofuro[3,4-c]pyridines 10g (50%) and 10k [55%]. Six derivatives of 3,4-diaryloylpyridines 11 were also obtained from these reactions, as shown in Scheme 6, the yields of which ranged between 9–29%. Notably, in some reactions, 3,4-diaryloylpyridines 11 did not form or were formed in very low yields, as a result of which they could not be isolated. The highest yields for 3,4-diaryloylpyridines were obtained for 11a [29%]. Thiophenes are highly important precursors and intermediates for the synthesis of a diverse range of pharmaceuticals and agrochemicals and in materials science.28 Thus, we synthesized two derivatives (10l and p) of 4-(thiophen-3-yl)-substituted 6,7-dihydrofuro[3,4-c]pyridines and one derivative (11l) of 2-(thiophen-3-yl)-substituted 3,4-dibenzoylpyridines, as shown in Scheme 6. Many drugs and drug candidates in clinical trials are halogenated compounds because halogen bonds improve the drug-target binding affinity.29 In particular, fluorinated drugs occupy a special place in the pharmaceutical industry since fluorination has a positive effect on absorption, distribution, metabolism and excretion.30 Overall, we synthesized seven derivatives (10b, c, g, h and m–o) of halogen-containing 6,7-dihydrofuro[3,4-c]pyridines and one derivative (11h) of halogen-bearing 3,4-aryloylpyridines (Scheme 6). The reaction of β-enaminone 9a with I2 was carried out on a relatively larger scale (by using 1.99 mmol of 9a) as well. This reaction afforded the expected products 10a and 11a in 29 and 13% yields, respectively (Scheme 6).
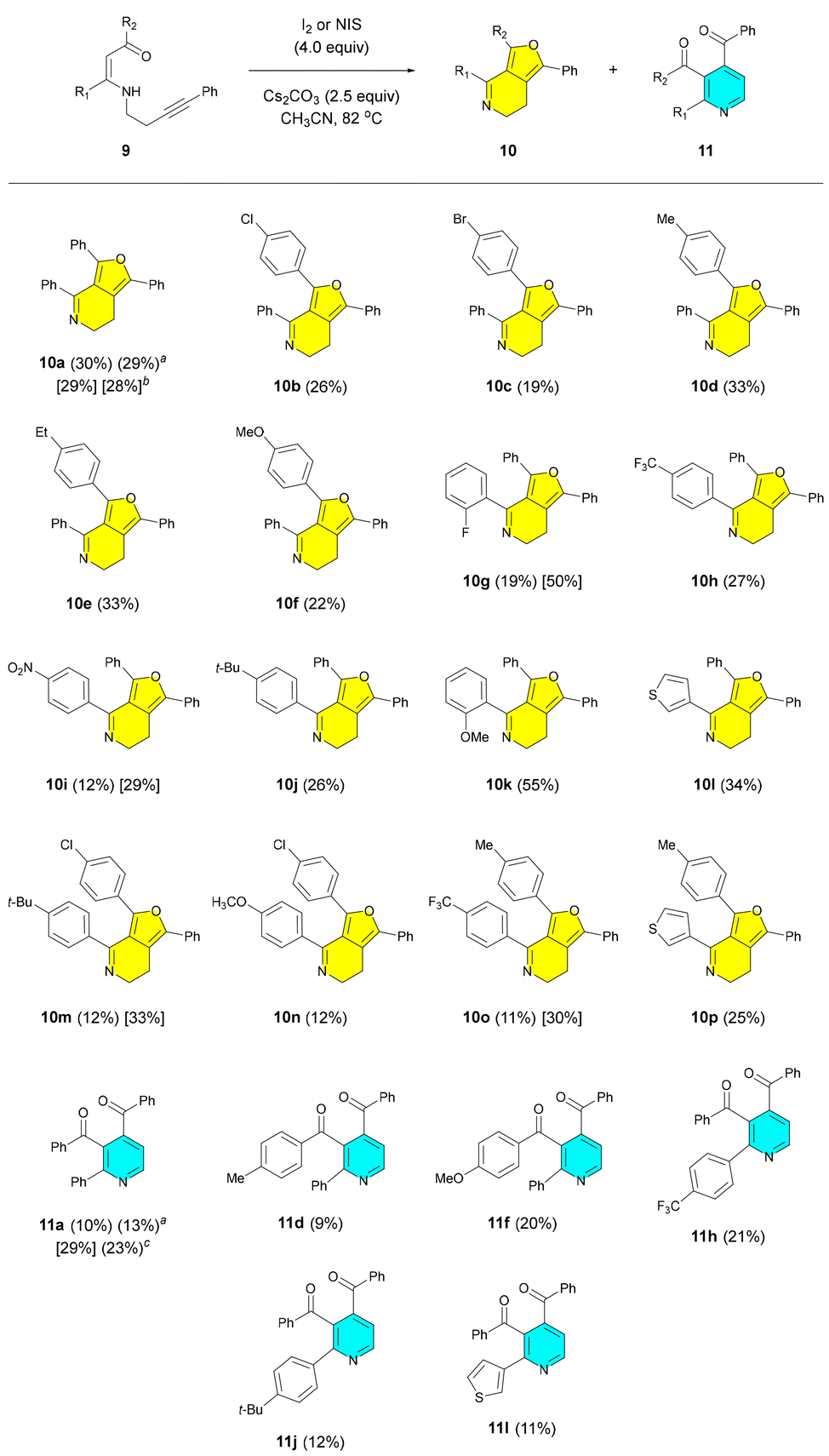 | ||
Scheme 6 Scope of the cyclization of N-homopropargylic β-enaminones 9. All the presented yields are isolated yields. Yields obtained with I2 are given in parentheses while those obtained with NIS are shown in square brackets. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yield obtained from the reaction performed at the 1.99 mmol scale of 9a. bYield obtained from the reaction performed in DMF at 110 °C. cYield obtained from the reaction carried out by using 2.5 equiv. of I2. Yield obtained from the reaction performed at the 1.99 mmol scale of 9a. bYield obtained from the reaction performed in DMF at 110 °C. cYield obtained from the reaction carried out by using 2.5 equiv. of I2. | ||
It is noteworthy that all the isolated 3,4-diaryloylpyridine derivatives in this study are new compounds although 3,4-diaryloylpyridines are known. The synthesis of pyridines from N-propargylic and N-homopropargylic β-enaminones is well known,18,20,23,24 but, to the best of our knowledge, the formation of 3,4-diaryloylpyridine derivatives from such β-enaminones is not known. Since our focus in this study has been mainly on the synthesis of the unknown bicyclic 6,7-dihydrofuro[3,4-c]pyridine ring systems, we plan to investigate the synthesis of 3,4-diaryloylpyridines in detail from N-homopropargylic β-enaminones in a separate study.
The proposed mechanism for the synthesis of 6,7-dihydrofuro[3,4-c]pyridines 10 and 3,4-diaryloylpyridines 11 is shown in Scheme 7. First, the reaction of molecular iodine with enaminone 9 produces the iodonium ion 15, which initiates the nucleophilic addition of the alkyne moiety to give vinyl cation 16. Then this vinyl cation is trapped by the ketone functionality, resulting in the formation of the cyclized intermediate 17. The abstraction of α-hydrogen by the base furnishes tetrahydrofuro[3,4-c]pyridine 18. Finally, the elimination of iodine generates the 6,7-dihydrofuro[3,4-c]pyridine derivative 10 (Scheme 7). Possibly, the vinyl cation intermediate 16 could also be trapped by trace amounts of water present in the reaction, forming intermediate 19. Removal of one hydrogen from the water moiety yields enol 20, the tautomerization of which affords 3,4-diaryloylpiperidine 21. Elimination of hydrogen iodide with a base gives the tetrahydropyridine derivative 22. In the presence of molecular iodine and a base, α-iodination takes place to yield intermediate 23, which, upon hydrogen iodide elimination, produces dihydropyridine 24. Keto–enol tautomerization of 24 first generates in situ enol 25; then its reaction with molecular iodine furnishes iodo-substituted dihydropyridine 26. Finally, elimination of hydrogen iodide with a base affords the 3,4-diaryloylpyridine derivative 11 (Scheme 7). It is noteworthy that the reaction does not stop with the formation of tetrahydropyridine 22, presumably due to the presence of a carbonyl group at the 4-position of the molecule. In the presence of excess molecular iodine, this carbonyl group triggers iodination, setting the stage for hydrogen iodide elimination from the molecule by the base. So, this oxidative aromatization continues until a pyridine ring forms. Notably, iodine-catalyzed or mediated oxidative aromatization is well known in the literature.31
 | ||
| Scheme 7 Proposed mechanism for the formation of 6,7-dihydrofuro[3,4-c]pyridines and 3,4-diaryloylpyridines. | ||
Conclusions
In summary, we have disclosed a new reaction of N-homopropargylic β-enaminones with molecular iodine and N-iodosuccinimide, leading to the formation of 6,7-dihydrofuro[3,4-c]pyridines and/or 3,4-diaryloylpyridines. When treated with excess molecular iodine in the presence of cesium carbonate, N-homopropargylic β-enaminones produced bicyclic 6,7-dihydrofuro[3,4-c]pyridines in low to moderate yields. During the course of the reaction, two new C/O–C bonds formed, giving rise to the formation of new five- and six-membered fused heterobicyclic molecules. Importantly, the skeletal diversity of the synthesized heterobicyclic frame may provide many new nitrogen- and oxygen-based heterobicyclic hybrid systems for drug discovery and development. Some reactions afforded 3,4-diaryloylpyridines in low yields as well. During the formation of 3,4-diaryloylpyridine derivatives, a new carbonyl (ketone) group formed. Notably, the presence of a dibenzoyl group in the pyridine unit offers the potential for further functionalization of these molecules, given that this functionality can be converted into pyrroles, furans, thiophenes and pyridazines. In fact, the reaction could be a potentially powerful method for the synthesis of 6,7-dihydrofuro[3,4-c]pyridines and 3,4-diaryloylpyridines; however, at present, the scope and limitations exhibited by these systems are less than optimal, which need to be improved. Further investigation of the mechanism, scope and limitations of this methodology is currently underway and will be reported in due course.Experimental
General procedure for the synthesis of N-homopropargylic β-enaminones 9 and 14 (Scheme 5)
To a stirred solution of the appropriate α,β-alkynic ketone 12 (1.94 mmol) in MeOH (10 mL) was added 3-butynylamine (6) (1.94 mmol) or 4-phenylbut-3-yn-1-amine (13) (1.94 mmol), and the resulting mixture was then refluxed until α,β-alkynic ketone 12 was completely consumed, as monitored by routine TLC. After the reaction was over, the solvent was removed using a rotary evaporator, and ethyl acetate (50 mL) and a saturated NH4Cl solution (50 mL) were added. After the layers were separated, the aqueous layer was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were dried over MgSO4 and evaporated using a rotary evaporator to give the crude product, which was purified by flash chromatography on silica gel using hexane/ethyl acetate (9:1 followed by 4:1) as the eluent to afford the corresponding N-homopropargylic β-enaminone 9 or 14.
General procedure for the cyclization of N-homopropargylic β-enaminones 9 leading to 6,7-dihydrofuro[3,4-c]pyridines 10 and/or 3,4-diaryloylpyridines 11 (Scheme 6)
The appropriate N-homopropargylic β-enaminone 9 (0.28 mmol) in acetonitrile (4.0 mL) was stirred at room temperature under argon and then molecular iodine (1.12 mmol) and Cs2CO3 (0.70 mmol) were added to the reaction mixture. The resulting mixture was then refluxed until N-homopropargylic β-enaminone 9 was completely consumed, as monitored by routine TLC. After the reaction was over, the solvent was removed using a rotary evaporator, and ethyl acetate (40 mL) and a saturated aqueous solution of Na2S2O3 (15 mL) were added. After the layers were separated, the aqueous layer was extracted with ethyl acetate (2 × 30 mL). The combined organic layers were dried over MgSO4 and evaporated using a rotary evaporator to give the crude product, which was purified by flash chromatography on silica gel using 4:1 hexane/ethyl acetate as the eluent to afford the corresponding 6,7-dihydrofuro[3,4-c]pyridine 10 and/or 3,4-diaryloylpyridine 11.
Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for 10a have been deposited at the CCDC under 2379026.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Scientific and Technological Research Council of Turkey [TUBITAK, Grant No. 123Z511] for financial support of this study. We also thank undergraduate student Barış Korkmaz for technical support.References
- (a) A. Padwa, C. O. Kappe, J. E. Cochran and J. P. Synder, Studies Dealing with the Cycloaddition/Ring Opening/Elimination Sequence of 2-Amino-Substituted Isobenzofurans, J. Org. Chem., 1997, 62, 2786–2797 CrossRef CAS PubMed; (b) W. Friedrichsen, Recent Advances in the Chemistry of Benzo[c]furans and Related Compounds, Adv. Heterocycl. Chem., 1999, 73, 1–96 CrossRef CAS.
- S. Basak, S. K. Ghosh and T. K. Sarkar, Progress in the chemistry of heteroisobenzofurans, J. Indian Inst. Sci., 2001, 81, 431–452 CrossRef CAS.
- U. E. Wiersum, C. D. Eldred, P. Vrijhof and H. C. van der Plas, Preparative flash vacuum thermolysis. Synthesis of furo[3,4-c]pyridine by retro-Diels-Alder reaction, Tetrahedron Lett., 1977, 18, 1741–1742 CrossRef.
- (a) M. Hamaguchi and T. Ibata, Reaction of o-acylphenyldiazomethane I. Formation of 1-alkoxyisobenzofurans by intramolecular carbenic reaction, Chem. Lett., 1976, 287–288 CrossRef CAS; (b) For giving this reaction a named reaction status, see: O. Peters and W. Friedrichsen, Intramolecular cycloadditions with isobenzofurans – X. 1 a novel synthesis of annulated hydroquinolines, Tetrahedron Lett., 1995, 36, 8581–8700 CrossRef CAS.
- (a) T. K. Sarkar, S. K. Ghosh and T. J. Chow, Synthesis and Reactions of a Stable o-Quinoid 10-π-Electron System, Furo[3,4-c]pyridine, J. Org. Chem., 2000, 65, 3111–3115 CrossRef CAS PubMed; (b) T. K. Sarkar, N. Panda and S. A. Basak, Sequential Pummerer-Diels-Alder Route for the Generation and Trapping of Furo[3,4-c]pyridines: Synthesis of Heterocyclic Analogues of 1-Arylnaphthalene Lignans, J. Org. Chem., 2003, 68, 6919–6927 CrossRef CAS PubMed.
- T. K. Sarkar, S. Basak, Z. Slanina and T. J. Chow, Studies on Intramolecular Diels-Alder Reactions of Furo[3,4-c]pyridines in the Synthesis of Conformationally Restricted Analogues of Nicotine and Anabasine, J. Org. Chem., 2003, 68, 4206–4214 CrossRef CAS PubMed.
- M. Iwao, H. Inoue and T. Kuraishi, Generation and Diels-Alder reaction of 1-siloxy-3-arylisobenzofurans from 3-arylphthalides, Chem. Lett., 1984, 1263–1266 CrossRef CAS.
- T. K. Sarkar, S. K. Ghosh, S. K. Nandy and T. J. Chow, Methyl 4,6-dichloro-3-diethylaminofuro[3,4-c]pyridine-1-carboxylate: Synthesis of the first stable azaisobenzofuran by Hamaguchi-Ibata reaction, Tetrahedron Lett., 1999, 40, 397–398 CrossRef CAS.
- A. E. Frissen, A. T. M. Marcelis, D. G. Buurman, C. A. M. Pollmann and H. C. van der Plas, Synthesis of 1,3-Dihydrofuro[3,4-d]pyridines and 5,7-Dihydrofuro[3,4-b]pyridines by Intramolecular Diels-Alder Reactions of Pyrimidines. Investigation of the Effect of Steric Interactions on the Reaction Rate, Tetrahedron, 1989, 45, 5611–5620 CrossRef CAS.
- (a) D. D. Young and A. A. Deiters, General Approach to Chemo- and Regioselective Cyclotrimerization Reactions, Angew. Chem., Int. Ed., 2007, 46, 5187–5190 CrossRef CAS PubMed; (b) J. S. Wang, J. Ying and X. F. Wu, Cobalt-catalyzed regioselective cycloaddition of unsymmetric diynes and nitriles to form substituted pyridines, Mol. Catal., 2021, 516, 111956 CrossRef CAS; (c) I. Y. Cho, W. G. Kim, J. H. Jeon, J. W. Lee, J. K. Seo, J. Seo and S. Y. Hong, Nickelocene as an Air- and Moisture-Tolerant Precatalyst in the Regioselective Synthesis of Multisubstituted Pyridines, J. Org. Chem., 2021, 86, 9328–9343 CrossRef CAS PubMed.
- (a) J. B. Harper, K. S. S. McHale, E. F. Lopes, D. S. Lüdtke, A. V. Moro, A. C. Mantovani, M. W. Paixao, R. N. Lima, P. M. Matos, M. Hapke, C. H. McAteer, R. Murugan and J. H. Yamamoto, in Comprehensive Heterocyclic Chemistry IV, ed. D. S. Black, J. Cossy, C. V. Stevens and N. Maulide, Elsevier, 2022, ch 7.01–7.06, vol. 7, pp. 1–242 Search PubMed; (b) Recent Developments in the Synthesis and Applications of Pyridines, ed. P. Singh, Elsevier, 2022 Search PubMed; (c) Exploring Chemistry with Pyridine Derivatives, ed. S. Pal, IntechOpen, 2023, Ebook. DOI:10.5772/intechopen.100792.
- (a) Y. Ling, Z. Y. Hao, D. Liang, C. L. Zhang, Y. F. Liu and Y. Wang, The Expanding Role of Pyridine and Dihydropyridine Scaffolds in Drug Design, Drug Des., Dev. Ther., 2021, 15, 4289–4338 CrossRef CAS PubMed; (b) R. G. Yousef, I. H. Eissa, A. Elwan and M. A. El-Zahabi, Pyridine Derivatives as Anticancer Agents: FDA-Approved Drugs and Promising Reported Compounds, Az. J. Pharm. Sci., 2023, 68, 64–81 Search PubMed.
- (a) N. D. Tsopelas and D. B. Marin, Cholinergic Treatments of Alzheimer's Disease, in Functional Neurobiology of Aging, Academic Press, 2001, ch. 32, pp. 475–486 Search PubMed; (b) S. Oudard, Progress in emerging therapies for advanced prostate cancer, Cancer Treat. Rev., 2013, 39, 275–289 CrossRef CAS PubMed; (c) W. M. Rashed, C-MET, as a potential target therapy toward personalized therapy in some pediatric tumors: An overview, Crit. Rev. Oncol. Hematol., 2018, 131, 7–15 CrossRef PubMed; (d) H. Devarbhavi, H. L. Bonkovsky, M. Russo and N. Chalasani, Drug-Induced Liver Injury, In Zakim and Boyer's Hepatology (A Textbook of Liver Disease), Elsevier, 7th edn, 2018, ch. 56, pp. 844–890 Search PubMed; (e) J. Popovic-Djordjevic, C. Quispe, R. Giordo, A. Kostic, J. S. K. Stankovic, P. V. T. Fokou, K. Carbone, M. Martorell, M. Kumar, G. Pintus, J. Sharifi-Radl, A. O. Docea and D. Calina, Natural products and synthetic analogues against HIV: A perspective to develop new potential anti-HIV drugs, Eur. J. Med. Chem., 2022, 233, 114217 CrossRef CAS PubMed; (f) A. Pourmohammadi, R. Riahi, S. M. Hosseini and I. Adibi, Pharmacological treatment of tremor in multiple sclerosis; a systematic review, Mult. Scler. Relat. Disord., 2022, 60, 103722 CrossRef PubMed; (g) P. Nagar, P. Sharma and D. HariKrishnaReddy, Endoplasmic reticulum stress in Alzheimer's disease: Molecular mechanisms and therapeutic prospects, Life Sci., 2023, 330, 121983 CrossRef CAS PubMed.
- (a) S. Mataka, K. Takahashi and M. Tashiro, Preparation of Hexa-, Penta- and Tetraphenyl-2,6- and -2,7-napthyridines, J. Heterocycl. Chem., 1983, 20, 971–974 CrossRef CAS; (b) S. Mataka, M. Tashiro, O. Misumi, W. H. Lin, K. Takahashi and A. Torii, Substituent effects on the spectra of fluorescent aryl-substituted N-methylpyrrolo[3,4-c]pyridines, Dyes Pigm., 1992, 20, 83–96 CrossRef CAS; (c) X. Li, Q. Pang, Y. Zhang, Y. Li, Q. Q. Yang, X. Lin, X. Xie and W. Huang, Metal-free assembly of diverse polysubstituted pyridines via an efficient cascade approach using tertiary enaminones and α,β-unsaturated sulfonylketimines, Org. Chem. Front., 2024, 11, 2607–2612 RSC.
- For 3,4-diacetylpyridine, see: (a) M. Sainsbury, B. Webb and R. Schinazi, Improved synthesis of 6H-pyrido[4,3-b]carbazole derivatives, J. Chem. Soc., Perkin Trans. 1, 1975, 3, 289–298 RSC; (b) F. Maiolo, L. Testaferri, M. Tiecco and M. Tingoli, Selectivity of radical ipso attack. Alkyldeacylation reactions by dioxanyl radicals in unprotonated acetylpyridines, Tetrahedron Lett., 1981, 22, 2023–2024 CrossRef CAS; (c) Z. Tian, D. Lv, P. Shen, Z. Liu, X. Zhang, S. Wang and S. Yin, Preparation method of water electrolysis oxygen evolution non-noble metal catalyst, China Patent, CN107326393A, 2017 Search PubMed.
- For recent use of enaminones in organic synthesis, see: (a) C. Yang, X. Zhang and X. Fan, Synthesis of homophthalimide spironaphthalenones through [5 + 1] spiroannulation of aryl/alkenyl enaminones with diazo homophthalimides, Org. Chem. Front., 2023, 10, 4282–4288 RSC; (b) W. Zuo, Y. Cheng, Z. Zhu, L. Zuo, X. Geng, Z. Li and L. Wang, Visible-Light-Induced Domino Cyclization to Access Pyrido[2,3-d]pyrimidine-2,4-diones via a Radical-Polar Crossover Reaction, Chin. J. Chem., 2024, 42, 2346–2350 CrossRef CAS; (c) M. Wang, Y. Gao, X. J. Zhao, L. Gao and Y. He, Electrochemical multicomponent [2 + 2 + 1] cascade cyclization of enaminones and primary amines towards the synthesis of 4-acylimidazoles, Chem. Commun., 2024, 60, 2677–2680 RSC; (d) H. Feng, J. Huo, X. Mu, R. Zheng, X. Geng and L. Wang, BPO-promoted [4 + 2] cyclization of enaminones and o-phenylenediamines to 2-acyl quinoxalines via a cascade transamination and C–H amination, Org. Biomol. Chem., 2024, 22, 4067–4071 RSC; (e) L. Fu, H. Huang, Y. Jiang, X. Liu, H. Chen and J. P. Wan, Copper(II)-Catalyzed [2 + 2 + 2] Annulation of Enaminones with Maleimides Using a Traceless Directing Group Strategy, Adv. Synth. Catal., 2024, 366, 4139–4144 CrossRef CAS; (f) S. Fan, W. Wu, Y. Su, X. Han, Z. Wang and J. Zhu, Co(III)-Catalyzed Coupling of Enaminones with Oxadiazolones for Imidazole Synthesis, Org. Lett., 2024, 26, 7620–7625 CrossRef CAS PubMed; (g) Z. C. Yu, K. L. Zheng, X. Shen, Y. Zhou, X. L. Chen, L. S. Wang, Y. D. Wu and A. X. Wu, I2–Induced Umpolung: Synthesis of a 1,6-Dihydrofuro[3,2-b]pyrazolo[3,4-e,][1,4]thiazine Skeleton via an Unconventional 1,4-Dithiane-2,5-diol Reaction Mode, Org. Lett., 2024, 26, 7891–7896 CrossRef CAS PubMed; (h) Q. Wang, X. Yuan, H. Sun, J. Yang, X. Yang, J. Lin and Y. Jin, Iron(III)-Catalyzed Amine-Release Triple Condensation of Enaminones to C3-Alkenylated Dihydroquinolinones, J. Org. Chem., 2024, 89, 10538–10550 CrossRef CAS PubMed; (i) X. Zhang, L. Song, Y. Jin and K. Luo, Metal-Carbenoid-Mediated Selective Transformation: Experimental and DFT Studies of Ag, Pd, and Rh with Enaminones and Diazoesters, ACS Catal., 2024, 14, 13509–13519 CrossRef CAS.
- (a) S. Arshadi, E. Vessally, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, N-Propargylic β-enaminocarbonyls: powerful and versatile building blocks in organic synthesis, RSC Adv., 2017, 7, 13198–13211 RSC; (b) T. Farghaly, A. M. A. Alnaja and M. R. Shaaban, Recent Advances in Synthesis and Reactions of β-Amino-α,β-Enones (Enaminones), Curr. Org. Synth., 2021, 18, 639–684 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi and E. Filisti, N-Propargylic β-Enaminones: Common Intermediates for the Synthesis of Polysubstituted Pyrroles and Pyridines, Org. Lett., 2008, 10, 2629–2632 CrossRef CAS PubMed.
- N. Kanova, B. A. Dundar, Y. Kelgokmen and M. Zora, One-Pot Synthesis of 2-Acetyl-1H-pyrroles from N-Propargylic β-Enaminones via Intermediacy of 1,4-Oxazepines, J. Org. Chem., 2021, 86, 6289–6304 CrossRef CAS PubMed.
- S. Karabiyikoglu, Y. Kelgokmen and M. Zora, Facile synthesis of iodopyridines from N-propargylic β-enaminones via iodine-mediated electrophilic cyclization, Tetrahedron, 2015, 71, 4324–4333 CrossRef CAS.
- (a) K. Goutham, D. A. Kumar, S. Suresh, B. Sridhar, R. Narender and G. V. Karunakar, Gold-Catalyzed Intramolecular Cyclization of N-Propargylic β-Enaminones for the Synthesis of 1,4-Oxazepine Derivatives, J. Org. Chem., 2015, 80, 11162–11168 CrossRef CAS PubMed; (b) Y. Kelgokmen, Y. Cayan and M. Zora, Zinc Chloride Mediated Synthesis of 1,4-Oxazepines from N-Propargylic β-Enaminones, Eur. J. Org. Chem., 2017, 7167–7178 CrossRef CAS.
- Y. Kelgokmen and M. Zora, Synthesis of 1,4-Thiazepines, J. Org. Chem., 2018, 83, 8376–8379 CrossRef CAS PubMed.
- For other reactions of N-propargylic β-enaminones leading to five-, six- and seven-membered heterocyclic compounds, see: (a) A. Saito, T. Konishi and Y. Hanzawa, Synthesis of Pyrroles by Gold(I)-Catalyzed Amino−Claisen Rearrangement of N-Propargyl Enaminone Derivatives, Org. Lett., 2010, 12, 372–374 CrossRef CAS PubMed; (b) K. Goutham, N. S. V. M. Rao Mangina, S. Suresh, P. Raghavaiah and G. V. Karunakar, Gold-catalysed cyclisation of N-propargylic β-enaminones to form 3-methylene-1-pyrroline derivatives, Org. Biomol. Chem., 2014, 12, 2869–2873 RSC; (c) G. Cheng, Y. Weng, X. Yang and X. Cui, Base-Promoted N-Pyridylation of Heteroarenes Using N-Propargyl Enaminones as Equivalents of Pyridine Scaffolds, Org. Lett., 2015, 17, 3790–3793 CrossRef CAS PubMed; (d) J. Shen, X. Yang, F. Wang, Y. Wang, G. Cheng and X. Cui, Base-mediated regiospecific cascade synthesis of N,-(2-pyridyl)pyrroles from N-propargylic β-enaminones, RSC Adv., 2016, 6, 48905–48909 RSC; (e) M. Zora, E. Dikmen and Y. Kelgokmen, One-pot synthesis of iodine-substituted 1,4-oxazepines, Tetrahedron Lett., 2018, 59, 823–827 CrossRef CAS; (f) E. Karadeniz and M. Zora, Synthesis of 1-Azaspiro[4.5]deca-1,3-dienes from N-Propargylic β-Enaminones in Basic Medium, Synthesis, 2019, 51, 2157–2170 CrossRef CAS; (g) E. Karadeniz and M. Zora, One-Pot Synthesis of Spiro-2H-pyrroles from N-Propargylic β-Enaminones, Synlett, 2019, 1231–1236 CAS; (h) Y. Kelgokmen and M. Zora, A new strategy for the synthesis of pyridines from N-propargylic β-enaminothiones, Org. Biomol. Chem., 2019, 17, 2529–2541 RSC; (i) E. S. Yilmaz and M. Zora, A New Strategy for the Synthesis of 4-Propargyl-Substituted 1H-Pyrroles from N,-(5-phenyl-2,4-pentadiynyl) β-Enaminones, ChemistrySelect, 2019, 4, 11043–11047 CrossRef CAS; (j) E. Korkmaz and M. Zora, Synthesis of 3-[(4-Nitrophenyl)thio]-Substituted 4-Methylene-1-pyrrolines from N-Propargylic β-Enaminones, J. Org. Chem., 2020, 85, 4937–4950 CrossRef CAS PubMed; (k) O. Ibis and M. Zora, A facile synthesis of 6-chloro-2-methylene-2,3-dihydro-1,4-oxazepines from N-propargylic β-enaminones, Tetrahedron, 2020, 76, 131650 CrossRef CAS; (l) A. R. Bena, E. G. Bakalbassis, M. M. Sigalas and I. N. Lykakis, One-Pot Synthetic Approach to 3-Carboxyl- and 3-Ketopyridines in Aqueous Media, J. Org. Chem., 2023, 88, 8055–8068 CrossRef CAS PubMed.
- M. Fragkiadakis, M. Kidonakis, L. Zorba and M. Stratakis, Synthesis of 3-Keto Pyridines from the Conjugated Allenone – Alkynylamine Oxidative Cyclization Catalyzed by Supported Au Nanoparticles, Adv. Synth. Catal., 2020, 362, 964–968 CrossRef CAS.
- S. Shen, M. Hadley, K. Ustinova, J. Pavlicek, T. Knox, S. Noonepallas, M. T. Tavares, C. A. Zimprich, G. Zhang, M. B. Robers, C. Barinka, A. P. Kozikowski and A. Villagra, Discovery of a New Isoxazole-3-hydroxamate-Based Histone Deacetylase 6 Inhibitor SS-208 with Antitumor Activity in Syngeneic Melanoma Mouse Models, J. Med. Chem., 2019, 62, 8557–8577 CrossRef CAS PubMed.
- (a) B. Gabriele, R. Mancuso and R. C. Larock, Recent Advances in the Synthesis of Iodoheterocycles via Iodocyclization of Functionalized Alkynes, Curr. Org. Chem., 2014, 18, 341–358 CrossRef CAS; (b) T. Aggarwal, S. Kumar and A. K. Verma, Iodine-mediated synthesis of heterocycles via electrophilic cyclization of alkynes, Org. Biomol. Chem., 2016, 14, 7639–7653 RSC; (c) P. M. Jadhav, A. B. Rode, L. Kotai, R. P. Pawar and S. U. Tekale, Revisiting applications of molecular iodine in organic synthesis, New J. Chem., 2021, 45, 16389–16425 RSC; (d) Monika, Chander, S. Ram and P. K. Sharma, A Review on Molecular Iodine Catalyzed/Mediated Multicomponent Reactions, Asian J. Org. Chem., 2023, 12, e202200616 CrossRef CAS.
- H. Taguchi, Iodinating Reagents, in Iodine Chemistry and Applications, ed. T. Kaiho, John Wiley and Sons, 2015, ch. 15, pp. 251–276 Search PubMed.
- (a) D. Gramec, L. P. Masic and M. S. Dolenc, Bioactivation Potential of Thiophene-Containing Drugs, Chem. Res. Toxicol., 2014, 27, 1344–1358 Search PubMed; (b) Thiophenes, in Topics in Heterocyclic Chemistry, ed. J. A. Joule, Springer, Berlin, 2015, vol. 39, pp. 1–298 Search PubMed; (c) Z. C. Smith, D. M. Meyer, M. G. Simon, C. Staii, D. Shukla and S. W. Thomas III, Thiophene-Based Conjugated Polymers with Photolabile Solubilizing Side Chains, Macromolecules, 2015, 48, 959–966 CrossRef CAS.
- (a) M. Hernandes, S. M. Cavalcanti, D. R. Moreira, W. de Azevedo Junior and A. C. Leite, Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design, Curr. Drug Targets, 2010, 11, 303–314 CrossRef CAS PubMed; (b) N. K. Shinada, A. G. de Brevern and P. Schmidtke, Halogens in Protein−Ligand Binding Mechanism: A Structural Perspective, J. Med. Chem., 2019, 62, 9341–9356 CrossRef CAS PubMed.
- (a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of Fluorine in Medicinal Chemistry, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- (a) M. J. Mphahlele, Molecular Iodine—An Expedient Reagent for Oxidative Aromatization Reactions of α,β-Unsaturated Cyclic Compounds, Molecules, 2009, 14, 5308–5322 CrossRef CAS PubMed; (b) V. Domingo, C. Prieto, L. Silva, J. M. L. Rodilla, J. F. Q. del Moral and A. F. Barrero, Iodine, a Mild Reagent for the Aromatization of Terpenoids, J. Nat. Prod., 2016, 79, 831–837 CrossRef CAS PubMed; (c) S. K. Wang, M. T. Chen, D. Y. Zhao, X. You and Q. L. Luo, Iodine-Catalyzed Oxidative Aromatization: A Metal-Free Concise Approach to meta-Substituted Phenols from Cyclohex-2-enones, Adv. Synth. Catal., 2016, 358, 4093–4099 CrossRef CAS; (d) X. Tuo, S. Chen, P. Jiang, P. Ni, X. Wang and G. J. Deng, Iodine-catalyzed convergent aerobic dehydro-aromatization toward benzazoles and benzazines, RSC Adv., 2020, 10, 8348–8351 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and copies of NMR spectra for starting materials and products (PDF). CCDC 2379026 (10a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01884b |
| ‡ Author to whom inquiries concerning the X-ray analysis should be directed. |
| This journal is © The Royal Society of Chemistry 2025 |