DOI:
10.1039/D4GC05740F
(Paper)
Green Chem., 2025,
27, 1410-1422
The sustainable and catalytic synthesis of N,N-alkylated fatty amines from fatty acids and esters†
Received
10th November 2024
, Accepted 30th December 2024
First published on 3rd January 2025
Abstract
The reductive amination of fatty acids (FAs) and fatty acid methyl esters (FAMEs) has been identified as a green and effective method to produce N,N-dimethylalkylamines (ADMAs). With current technology, this reaction requires at least two reaction steps. Here, we report a heterogeneous catalytic system for the one-pot synthesis of ADMAs from FA(ME)s, utilizing solely H2 and methylamines (i.e. di- and trimethylamine). The reaction requires two recyclable catalysts: ortho-Nb2O5 for the amidation of FA(ME)s and PtVOx/SiO2 for the hydrogenation of the in situ generated fatty amide to ADMAs. The developed system has a wide range of applicability: it is able to convert all natural FAs to ADMAs (yields up to 90%) and also other tertiary amines were synthesized. Aside from the development of a sustainable and industrially applicable process (e.g. utilizing benign solvents or performing solventless reactions), a kinetic model was developed that describes the reaction rate's relationship with key process parameters such as the H2 pressure and water content. By tuning the reaction conditions, different ratios of primary, secondary and tertiary fatty amines can be obtained.
Green foundation
1. They successfully demonstrated that their method is more efficient regarding energy cost, reactor cost and product isolation. The compounds are fully characterized. The supporting information has all the data necessary to validate the work. The research is conceptually new and interesting, fits well the scope of the journal.
2. Achievement of a process with (A) environmentally friendly conditions (in CPME or solventless conditions) (B) a limited energy input (C) recyclable catalysts (D) a wide substrate scope and range of applicability (yields >90%).
3. Actual industrial implementation or application of this work in other fields/similar reactions.
|
Introduction
Fatty amines are nitrogen-containing molecules, which in most cases are derived from natural fats and oils, typically with a chain length of 8 to 22 carbons. A large portion of the fatty amine market is made up of tertiary fatty amines of which alkyldimethylamines (ADMAs) with an alkyl chain length from C8 to C22 are the most important ones in terms of market volume (200 kton per year) followed by shorter chain C8 to C10 dialkylmethylamines (DAMAs) (100 kton per year).1,2 Over the past few years, the production of these compounds as precursors for amphoteric and cationic surfactants (e.g. quaternized fatty amines, betaines and amine N-oxides) rapidly increased; they are used in a variety of personal and home care formulations. The synthesis of ADMAs from natural oils or fats, however, requires a multistep process, as visualized in Fig. 1.
 |
| | Fig. 1 Common industrial synthesis routes for the production of alkyldimethylamines in contrast to the synthesis method in this manuscript. | |
Classically, natural oils and fats are converted with water or an alcohol, typically methanol, liberating the fatty acid (FA) or fatty acid methyl ester (FAME) respectively. From these platform building blocks, tertiary fatty amines are produced via either the nitrile process (Fig. 1, bottom) or the alcohol process (Fig. 1, top).2 In the nitrile process, a fatty acid is reacted with ammonia to an ammonium carboxylate. This ammonium salt is dehydrated (first to the amide, then to the nitrile) at a temperature above 250 °C and in the presence of an oxide catalyst, e.g. Al2O3 or ZnO. Next the nitrile is hydrogenated with a Ni or Co catalyst, at a temperature around 130 °C, 1–30 bar H2 and a large amount of NH3 to steer the selectivity towards the primary amine (up to 96% yield).3,4 Finally, the primary amine is methylated with methanol or formaldehyde.5,6 Although the output of this process is an amine of high quality and purity, it is clear that such a multistep process is very energy intensive, requires an extensive work-up and a complex (and therefore expensive) reactor setup. Alternatively, in the alcohol process, FAMEs are first hydrogenated to fatty alcohols. This hydrogenation is typically performed with a Cu-based catalyst (promoted with Cr or Zn), at a temperature between 250–350 °C and a high pressure of up to 200 bar H2.2,7 In more recent years, newer catalysts have been proposed, e.g. supported RuSn, that can operate between 150 °C to 250 °C and 50 bar H2.8,9 After hydrogenation, a wide variety of metal catalysts can be used for the amination of the fatty alcohol with dimethylamine resulting in the same ADMA, in either a gas-phase or liquid-phase process.2,10,11 This alcohol process has the advantage that it requires only two separate reaction steps. However, during hydrogenation towards the alcohol, a significant fraction of up to 10% is lost due to defunctionalisation reactions. Additionally, fatty alcohols are valuable molecules with many uses and the global capacity for FAME hydrogenation is limited, resulting in a limited availability of these alcohols.
An interesting alternative to these processes would be the direct one-pot synthesis of ADMAs from relatively cheap and readily available FAs or FAMEs (e.g. palmitic acid costs around 1.3 € per kg).12 However, reports on the direct synthesis of amines from carboxylic acids using reusable heterogeneous catalysts and H2 gas are extremely limited. One of the only reports is the work of Barrault et al., where they converted dodecanoic acid and its corresponding methyl ester directly into a mixture of amines using Cu-based catalysts.13–16 Although the corresponding ADMA could be generated directly, the scope was limited to only C12 FA(ME) and only with methyl laurate high yields of the corresponding N,N-dimethyl laurylamine were obtained (i.e. up to 93%). Additionally, reactions were performed in the vapour phase, limiting the potential applicability to only short- and medium chain FAs to the extent that they can be vapourised. In more recent years, we reported two catalytic systems for the one-pot synthesis of amines from aliphatic and aromatic carboxylic acids, respectively.17,18 Although both systems were highly efficient, with yields >90%, they were specifically designed for the synthesis of primary amines with NH3. Efforts to produce tertiary amines were unsuccessful.19
In this work, we report a new catalytic system for the reductive amination of FAs and FAMEs directly to tertiary fatty amines, e.g. ADMAs. Such a reaction proceeds via an initial amidation of the FA(ME) with a secondary amine, e.g. dimethylamine (DMA), to a tertiary fatty amide intermediate, followed by a hydrogenation. Both the amidation and the hydrogenation were studied in detail and robust heterogeneous catalysts were identified. For the amidation of FA(ME)s, we report an ortho-Nb2O5 catalyst and provide insightful data on the kinetics and thermodynamical equilibrium that is virtually absent in existing literature.17,19,20,24–30 For the hydrogenation of amides, an effective PtVOx/SiO2 catalyst was developed. Over the past two decennia, similar bimetallic amide hydrogenation catalysts have been proposed.17,19,24–30 However, these catalysts lack activity in the presence of protic compounds. Especially the presence of water, generated in the reaction, seems to be highly problematic. Commonly, water is captured with drying zeolites or an enormously high loading of noble metal is used to mitigate the issue (e.g. up to 29 mol% Rh).28,29 In this report we propose a kinetic model which allows to describe the inhibitory effect of water.
Amidation of fatty acids and FAMEs
Catalyst evaluation
As a start, we hypothesized that a one-pot conversion of fatty acids (FAs) and fatty acid methyl esters (FAMEs) to amines could be achieved through the formation of an intermediate tertiary amide.21–23 For this initial amidation, several metal oxides were evaluated as catalysts (Fig. 2). Reactions were performed with palmitic acid or methyl palmitate as the model compounds.
 |
| | Fig. 2 Influence of the catalyst on the amidation of methyl palmitate. Reaction conditions: methyl palmitate (5 mmol), 200 °C, 0.8 g dimethylamine (DMA), 0.25 g catalyst, dodecane standard (100 μL), CPME (cyclopentyl methyl ether, 18.5 mL), 5 h reaction time, batch setup. (com) = commercial catalyst. HAP = hydroxyapatite. | |
For the preliminary catalyst screening methyl palmitate was selected as the model substrate. Such an alkyl ester is generally regarded as a relatively inert substance that typically requires harsh reaction conditions to facilitate amidation (in contrast to the corresponding carboxylic acid).21 Under the intended reaction conditions, the highest conversion and yield for the desired tertiary amide were obtained with niobium oxides (Nb2O5) and zirconia (ZrO2; 44–47% yield). Apparently, a reaction temperature of 200 °C is sufficiently high to induce some acid-catalyzed disproportionation of dimethylamine (DMA) to mono- and trimethylamine (MMA and TMA). This leads to the generation of the corresponding secondary amide, i.e. N-methylpalmitamide, which was observed in small quantities as the main side product (up to 3%). Both ortho-Nb2O5 and ZrO2 contain a variety of Brønsted and Lewis acidic sites. However, a reaction with Na+/Nb2O5, i.e. ortho-Nb2O5 in which the acid protons were exchanged with sodium cations, led to an analogous result. Thus, this experiment illustrates that mainly Lewis acid sites accelerate the amidation reaction. Materials with only basic sites, e.g. hydroxyapatite (HAP), or with a limited number of acid sites, e.g. pyrogenic silica or y-Al2O3, are poor catalysts for this reaction. The commercially obtained Nb2O5 possesses only a fraction of the catalytic activity displayed by a self-synthesized ortho-Nb2O5. Such a commercial Nb2O5 was calcined at a much higher temperature (i.e. ≥550 °C vs. 400 °C) resulting in the loss of most of the Lewis acid surface sites.31ortho-Nb2O5 was selected as the best catalyst for its excellent activity and selectivity. An extensive characterization of this ortho-Nb2O5 amidation catalyst is available in a previous report (see also ESI†).32
Optimization of the reaction conditions and rate
Next, the reaction conditions were optimized, starting with the ideal DMA content (Fig. 3 and S4†). The DMA concentration has a strong influence on the reaction rate (and equilibrium). Analogous findings were observed for both Nb2O5 and ZrO2 as amidation catalysts (Fig. 3 and S4† respectively). A clear optimum in DMA concentration was observed at 1 g DMA per 20 mL solvent (approximately 4× excess of DMA over substrate) for the amidation of FAMEs (e.g. methyl palmitate). This resulted in a methyl palmitate conversion of 63% to mainly N,N-dimethylpalmitamide (61% yield). We hypothesize that with a decreasing DMA concentration (<1 g DMA per 20 mL) the reduced availability of DMA limits the reaction. On the other hand, at concentrations >2 g DMA per 20 mL, DMA inhibits the reaction. Most of the Lewis acid sites on the catalyst surface are then occupied by DMA making it difficult for substrate molecules to interact with the surface. Reactions with FAs (i.e. palmitic acid) lead to almost complete conversion to N,N-dimethylpalmitamide under all tested conditions.
 |
| | Fig. 3 Influence of dimethylamine (DMA) concentration on the amidation of methyl palmitate and palmitic acid. Reaction conditions: substrate (5 mmol), 200 °C, variable amount of DMA, 0.25 g Nb2O5, dodecane standard (100 μL), CPME (18.5 mL), 5 h reaction time, batch setup. | |
In a consecutive series of experiments, the reaction time and temperature were varied. Catalytic reactions with a reaction time of only 5 h provided information about the reaction rate (Fig. 4), whereas those with a 16 h reaction time gave insights about the thermodynamic equilibrium (Fig. 5). Noteworthy differences were observed when comparing the amidation of FAs with the amidation of FAMEs. While FAME amidation requires a catalyst, the impact of a catalyst on the amidation of fatty acids (FAs) is rather small. Whereas we only observe a 6% methyl palmitate conversion after an uncatalyzed reaction of 16 h at 200 °C (4× excess of DMA), under similar reaction conditions a reaction with the corresponding FA already yields 69% N,N-dimethylpalmitamide (same conversion, Fig. 5). After the addition of a limited amount of Nb2O5, the conversions of reactions with methyl palmitate and palmitic acid rose to 74% and 96% respectively (with 92% selectivity for N,N-dimethylpalmitamide in both cases). In the range from 170 °C to 225 °C, the reaction equilibrium for the amidation of FAs is always at high conversions (>90%). With increasing reaction temperatures, however, the disproportionation of dimethylamine (DMA) to NH3, methylamine (MMA) and trimethylamine (TMA) is increasingly dominant. This leads to side reactions with N-methylpalmitamide as the main by-product.
 |
| | Fig. 4 Amidation of palmitic acid or methyl palmitate after a reaction time of 5 h. Reaction conditions: substrate (5 mmol), 1 g DMA, if a catalyst was used: 0.25 g Nb2O5, dodecane standard (100 μL), CPME (18.5 mL), 5 h. | |
 |
| | Fig. 5 Amidation of palmitic acid or methyl palmitate after a reaction time of 16 h. Reaction conditions: substrate (5 mmol), 1 g DMA, if a catalyst was used: 0.25 g Nb2O5, dodecane standard (100 μL), CPME (18.5 mL), 16 h reaction time, batch setup. | |
Choosing the right reaction solvent is a delicate task. The solvent helps to dissolve reagents and remove products from the catalyst but must be easily recoverable and must not adversely affect the reaction (e.g. by competitive adsorption on the catalyst). Mainly in the following sections (hydrogenation of amides), the reaction solvent will exert an important influence; the hydrogenation of tertiary amides is usually only possible in aprotic solvents such as ethers and alkanes.19 Since the ultimate goal was to establish a one-pot fatty acid (methyl ester)-to-amine reaction, the influence of the reaction solvent on the amidation reaction of fatty acid methyl esters was studied (Fig. S5†). Although mild fluctuations are observed in conversion depending on the exact reaction solvent, the amidation can be performed with high selectivity (i.e. >95%) in a wide range of ether solvents that vary widely in boiling point (e.g. MeTHF: 80 °C versus tetraglyme: 275 °C). This allows, based on the FA(ME) employed, to select a solvent that is easily separable from the final product (e.g. by distillation).
Hydrogenation of fatty amides
Catalyst evaluation
After selecting the best metal oxides for the amidation of fatty acids, mono- and bimetallic hydrogenation catalysts were synthesized for the hydrogenation of fatty amides, e.g. N,N-dimethylpalmitamide to N,N-dimethyl-palmitylamine (Fig. 6). In order to establish a one-pot fatty acid-to-amine reaction, the catalyst must tolerate methylamines. For this reason, DMA was added to the reaction mixture as well, although no net DMA is consumed during this hydrogenation itself. Non-promoted catalysts show little hydrogenation activity. Among these monometallic catalysts, the best result was obtained with Pt/Nb2O5 (1 wt% Pt) leading to a yield of 17% N,N-dimethylpalmitylamine. The catalytic activity can be improved significantly by adding a Lewis acid metal oxide promotor (to the catalyst), with optimal results when utilizing vanadium oxide. Ideally, the best metal oxide catalysts for the amidation of FAs and FAMEs (i.e. Nb2O5 and ZrO2) could be employed as carrier materials for the hydrogenation of the corresponding amide. However, supports with acidic sites generally lead to catalysts with a low hydrogenation activity. Additionally, such materials also catalyze the formation of secondary amides and amines, i.e. N-methylpalmitamide and N-methylpalmitylamine. This is particularly the case for ZrO2-based materials (e.g. with Pd/ZrO2 and Pt/ZrO2). It is likely that the reduced catalytic activity of these materials is associated, at least in part, with the formation of these side products (in comparison with catalysts supported on neutral or basic materials). The best hydrogenation catalysts were obtained when Pt and V were deposited on a basic or neutral support material. Such catalysts lead to virtually no N-methylpalmitamide as a by-product. Addition of an amidation catalyst, leading to a physical mixture of both catalysts, would still allow a one-pot fatty acid-to-amine reaction and did not generate a substantial quantity of side-products (Fig. 6, see PtVOx/HAP + Nb2O5). Hydrogenation catalysts supported on hydroxyapatite and BaAl2O4 were not considered suitable because of limited stability in corrosive mixtures of dimethylammonium carboxylates.17 Therefore, PtVOx/SiO2 (7 wt% Pt, with Pt–V ratio of 1) was selected as the optimal hydrogenation catalyst. A catalytic reaction under non-optimized conditions already resulted in a conversion of 56% with an overall amine selectivity of 93% (i.e. yields of 43% N,N-dimethylpalmitylamine, 10% N-methylpalmitylamine and <1% palmitylamine). Small quantities of the defunctionalized product are observed as well (i.e. hexadecane; 6% selectivity).
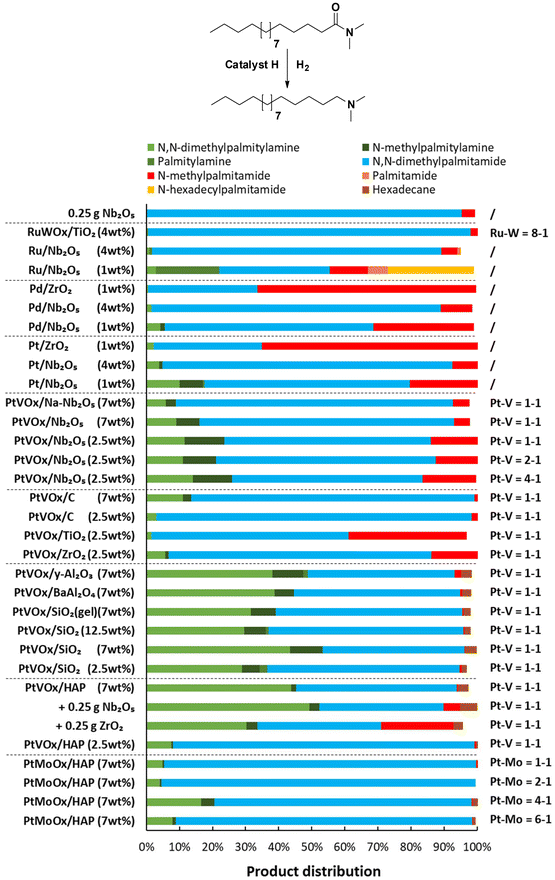 |
| | Fig. 6 Catalyst evaluation for the hydrogenation of N,N-dimethylpalmitamide. Reaction conditions: N,N-dimethylpalmitamide (5 mmol), 200 °C, 2 g DMA, 40 bar H2, 5 mg noble metal (loaded on a support material), dodecane standard (100 μL), CPME (18.5 mL), reaction time of 16 h, batch setup. | |
Optimization of the reaction conditions
In a subsequent series of experiments, the reaction conditions were optimized, starting with the DMA concentration and hydrogen pressure (Fig. 7 and S6†). The influence of DMA is twofold. On the one hand, an increasing concentration of DMA increases the selectivity for amine formation over defunctionalization. On the other hand, polar components such as DMA interact strongly with the active sites of the hydrogenation catalyst. As a result, the catalytic activity decreases with increasing DMA concentration. For this reason, 1 g DMA per 20 mL solvent was selected as the optimum. This concentration falls within the optimum range for the amidation of FA(ME)s to tertiary fatty amides as well (Fig. 3). Elevating the hydrogen pressure has a negligible impact on the product selectivity but exerts a positive influence on the reaction rate (Fig. S6†).
 |
| | Fig. 7 Variation of DMA concentration during the hydrogenation of N,N-dimethylpalmitamide to N,N-dimethylpalmitylamine. Conditions: N,N-dimethyl-palmitamide (5 mmol), 200 °C, DMA, 40 bar H2, 0.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), dodecane standard (100 μL), CPME (18.5 mL), reaction time of 16 h. | |
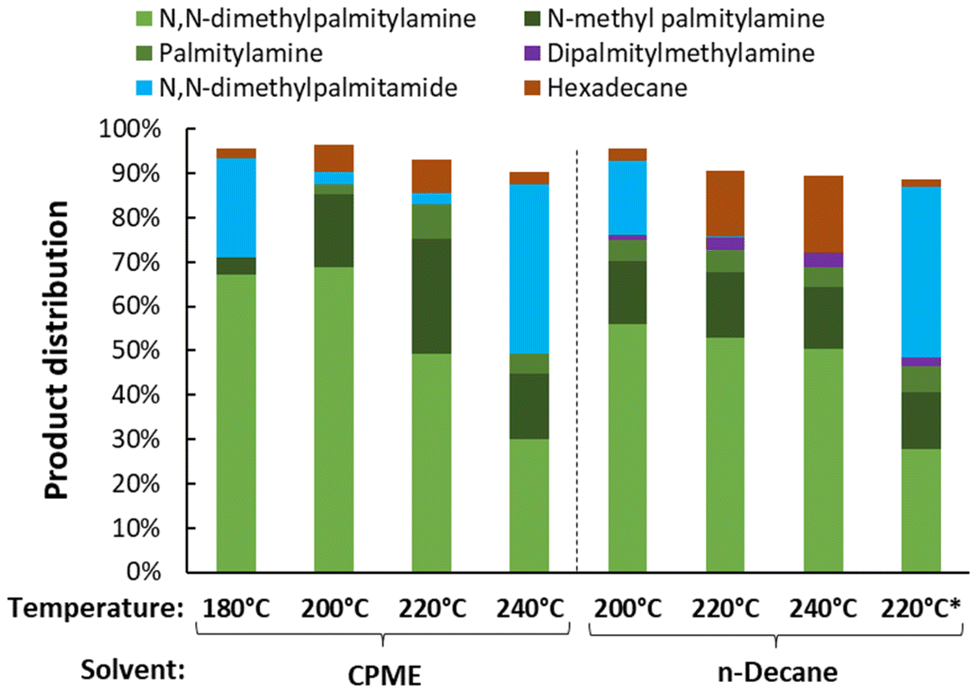
As previously mentioned, the selection of reaction solvent plays a crucial role in the effectiveness of amide hydrogenation.17,19,25–30 Polar (and protic) compounds more easily desorb amine products and water from the catalyst, leading to less consecutive side reactions (e.g., hydrogenolysis and transalkylation). However, especially protic compounds tend to preferentially adsorb on the active sites of the catalyst, drastically lowering the amide hydrogenation rate. Additionally, they have a limited H2 solubility in contrast to apolar solvents. Therefore, ethers are often selected as ideal candidates: they are aprotic, yet able to undergo H-bonding with e.g. water; and dissolve the reactants and the formed amine products rather well. This was confirmed in a solvent screening (Fig. S7†). The choice of solvent also depends on the employed or required reaction temperature (Fig. 8). When increasing the reaction temperature, for example to 220 °C, the hydrogenation rate decreases in CPME (i.e. cyclopentyl methyl ether). This counterintuitive observation can be attributed to the degradation of CPME into alcohols (i.e. cyclopentanol and MeOH) or even water. Such an inhibition is not observed in n-decane. However, working in this non-polar solvent is suboptimal due to impaired removal of the amine product from the catalytic surface to the liquid phase. This, in turn, leads to an increased susceptibility to complete defunctionalization through C–N hydrogenolysis to n-hexadecane. Elevating the reaction rate also increases the rate of transalkylation of the desired N,N-dimethylpalmitylamine product into its corresponding secondary or even primary alkylamine. Striking a balance between activity and selectivity, an optimal reaction temperature of 200 °C (in CPME) was identified. Under these conditions, we achieve a near complete conversion within 16 h, utilizing only 0.5 mol% Pt and obtaining a global amine selectivity of 92%, with specific amine yields of 69% for N,N-dimethylpalmitylamine, 16% for N-methylpalmitylamine and 2% for palmitylamine.
 |
| | Fig. 8 The influence of the reaction temperature on the hydrogenation of N,N-dimethylpalmitamide. Reaction conditions: N,N-dimethylpalmitamide (5 mmol), 180–240 °C, 1 g DMA, 60 bar H2, 0.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), dodecane standard (100 μL), solvent (18.5 mL), 16 h. *2 g DMA instead of 1 g. | |
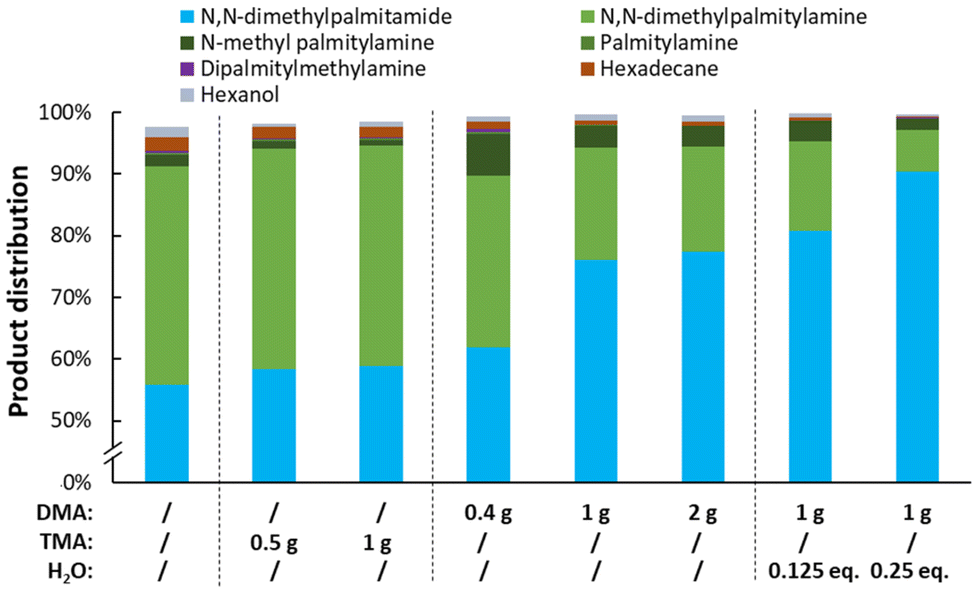
As mentioned previously, the water (or MeOH) content has an enormous effect on the amide hydrogenation rate (Fig. 9). While after a normal reaction run only 4% amide substrate remains after 16 h, the reaction nearly completely halts upon addition of 1 equivalent of water along with 1 equivalent of methanol at the start of the reaction. Interestingly, MeOH has a stronger inhibitory effect compared to water. To deal with this issue, in most reports either water is removed continuously by drying zeolites (working at a low temperature) or large amounts of noble metal catalyst are utilized, e.g. 29 mol% Rh.28,32–34 On an industrial level, neither approaches are feasible or practical.
 |
| | Fig. 9 The influence of water and methanol on the hydrogenation of N,N-dimethylpalmitamide. Reaction conditions: N,N-dimethylpalmitamide (5 mmol), 200 °C, 1 g DMA, 60 bar H2, 0.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), dodecane standard (100 μL), CPME (18.5 mL), H2O and/or methanol (0–2 eq.), 16 h. | |
A kinetic model with competitive adsorption
Although the general consensus is that water has a strongly negative influence on the amide hydrogenation rate, to date a kinetic model has never been established. The experimental data for the hydrogenation of amides do not fit with simple 1st or 2nd order kinetics in amide concentration (Fig. S9†). For this reason, we propose a model for the hydrogenation of fatty amides to fatty amines with competitive adsorption of amines and water to the catalytic surface, based on a Langmuir–Hinshelwood adsorption–desorption mechanism.35 The reaction rate could be reduced to eqn (1) (a complete mathematical derivatization is provided in the ESI†).| |  | (1) |
The equation resembles a regular second order rate equation but has an additional term in the denominator. This (simplified) equation is valid if 2 important conditions are met; firstly, water and the ADMA product must be generated in equimolar amounts and stay within the reactor. Secondly, and more importantly, no secondary products are generated with significantly different adsorption/desorption rate constants. This last condition is not always met. For instance, transalkylation reactions occur more prominently at lower H2 pressures (Fig. S17†). Such transalkylations yield primary amines (i.e. MMA) which adsorb much more strongly to the catalytic surface than secondary and tertiary amines, causing a significant deviation between experimental and theoretical values. Nevertheless, the model fits perfectly with the experimental data (Fig. 10 and S10–S12†). Remarkably, the denominator comprises a [H2O]3 term, uncovering the extreme sensitivity of the reaction to water (or other protic molecules, Fig. 10). Additionally, the reaction displays a second order in H2 pressure. This is at sharp contrast with the results of Mitsudome et al., who hydrogenated tertiary amides with a H2 pressure of 1 bar, but needed a drying zeolite to remove the water.28 We were able to confirm that drying zeolites can be used to enhance the reaction. However, we opted for an intermediate drying of the reaction mixture (Fig. 11). This does not limit us to perform the hydrogenation below 150 °C. In addition, such an approach is easier to adopt industrially.
 |
| | Fig. 10 Kinetic profile for the hydrogenation of N,N-dimethylpalmitamide with competitive adsorption of water and amines, with external addition of water (and N,N-dimethylpalmitylamine). Orange dots: experimental data. Black line: fit with the model. Error bars represent the standard deviation. Experimental conditions: N,N-dimethylpalmitamide (5 mmol; 0.25 M), 200 °C, 1 g DMA, 60 bar H2, 0.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), dodecane (100 μL), CPME (18.5 mL), variable reaction time and variable, but equimolar addition of water and N,N-dimethylpalmitylamine. | |
 |
| | Fig. 11 Hydrogenation of N,N-dimethylpalmitamide to N,N-dimethylpalmitylamine with and without intermediate drying of the reaction mixture with molecular sieves. Reaction conditions: N,N-dimethylpalmitamide (5 mmol), 200 °C, 1 g DMA, 60 bar H2, 0.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), dodecane standard (100 μL), CPME (18.5 mL), batch setup. In case of intermediate drying: after a reaction time of 1 h the reaction was stopped; 0.5 g of 3A zeolite were added to the supernatant for 12 h; next, the reaction was continued for another h. | |
This intermediate drying of the reaction mixture significantly increased the conversion from 76% to 95%, with an N,N-dimethylpalmitylamine yield of 83% (compared to an initial yield of only 70%). The kinetic model with competitive adsorption, also allows us to calculate the apparent activation energy (Ea) for the hydrogenation of N,N-dimethylalkylamides. Ea appears to be 103 kJ mol−1, which is a very typical value for amide hydrogenations in general.19
Previous observations and equations are not only valid in diluted solutions (in CPME). As a proof of concept, short solventless reactions were also conducted (Fig. 12). Once again, addition of water drastically decreased the overall hydrogenation rate. Addition of DMA, another protic compound, had a similar effect but to a lesser extent. Upon addition of TMA (non protic molecule) the reaction rate remains essentially the same in comparison with the reference reaction, but with the added benefit of an increase selectivity for the ADMA product.
 |
| | Fig. 12 Solventless hydrogenation of N,N-dimethylpalmitamide. Reaction conditions: N,N-dimethylpalmitamide (9.47 g), 200 °C; DMA, TMA and/or water; 60 bar H2, 0.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), dodecane (333 μL), 1 hour reaction time. | |
Recyclability and catalyst characterization
Next, the PtVOx/SiO2 catalyst was extensively characterized. Synthesizing a material with well-dispersed Pt crystallites is not sufficient to create an effective amide hydrogenation catalyst (e.g. Pt/C). CO chemisorption experiments indicate that both carbon and niobia supported Pt catalysts exhibit significantly greater Pt dispersion (i.e. between 25% and 35%) compared to the best performing catalyst, PtVOx/SiO2, which has a dispersion of only 9% (Table S4†). Experimental results also indicate that the mere presence of Lewis acid sites on the carrier material, like in Pt/Nb2O5, is not adequate to obtain a performant catalyst (Fig. 6). High angle annular dark field scanning transmission electron microscopy experiments (HAADF-STEM) clearly confirm that while there is no perfect overlap between the regions containing Pt and V, the major portion of V is concentrated near or even on Pt particles (Fig. 13). Overall, Pt and V are distributed uniformly on the SiO2 support, with small Pt crystallites averaging 1.70 nm in size. The partial coverage of Pt by V also explains the relatively low Pt dispersion as observed in CO chemisorption measurements.
 |
| | Fig. 13 HAADF-STEM imaging and EDX elemental mapping of a fresh PtVOx/SiO2 catalyst (7 wt% Pt, molar Pt–V ratio of 1–1). Elemental mapping of Si, O, Pt and V. | |
A recycling test confirmed that the PtVOx/SiO2 catalyst can be reused multiple times (Fig. 14). Despite a decline in catalytic performance following the initials runs, the catalytic activity seems to stabilize after three uses. A mild calcination at 300 °C between the 4th and 5th runs minimally improved the overall reaction conversion and product yield. Thus, the decrease in catalytic activity is not attributed to catalytic deactivation via coke formation but rather to the sintering of small Pt nanoparticles (Fig. 15). HAAFD-STEM analysis indicates that following the first reaction run, the average Pt particle size increases from 1.7 nm to 2.1 nm, with very few particles exceeding 4 nm.
 |
| | Fig. 14 Recycling test of PtVOx/SiO2 for the hydrogenation of N,N-dimethylpalmitamide. Reaction conditions: N,N-dimethylpalmitamide (5 mmol), 200 °C, 1 g DMA, 60 bar H2, 0.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), dodecane standard (100 μL), CPME (18.5 mL), 16 h. | |
 |
| | Fig. 15 Size distribution of Pt nanoparticles of PtVOx/SiO2 catalysts (7 wt% Pt, Pt–V ratio of 1), determined by TEM analysis. Size distributions based on 150 particles for each sample. Mean Pt diameter sizes of 1.70 nm, 2.14 nm and 2.28 nm for fresh, 1× used and 5× used catalyst respectively. | |
Subsequently, after 5 reaction runs, the average size increases only slightly to 2.3 nm (once again with few particles exceeding 4 nm). The data suggest that Pt nanoparticles of ≥2 nm remain (virtually) stable and do not sinter to large Pt aggregates. TEM analysis confirms that after 5 runs, both Pt and V remain well dispersed over the SiO2 support (Fig. S28†). In addition, the reactions were performed with only 0.5 mol% Pt. Aside from the catalyst's excellent recyclability, very little Pt is required to generate a large quantity of fatty amines from fatty amides.
Reductive amination of FAs and FAMEs
Compatibilization of amidation with hydrogenation.
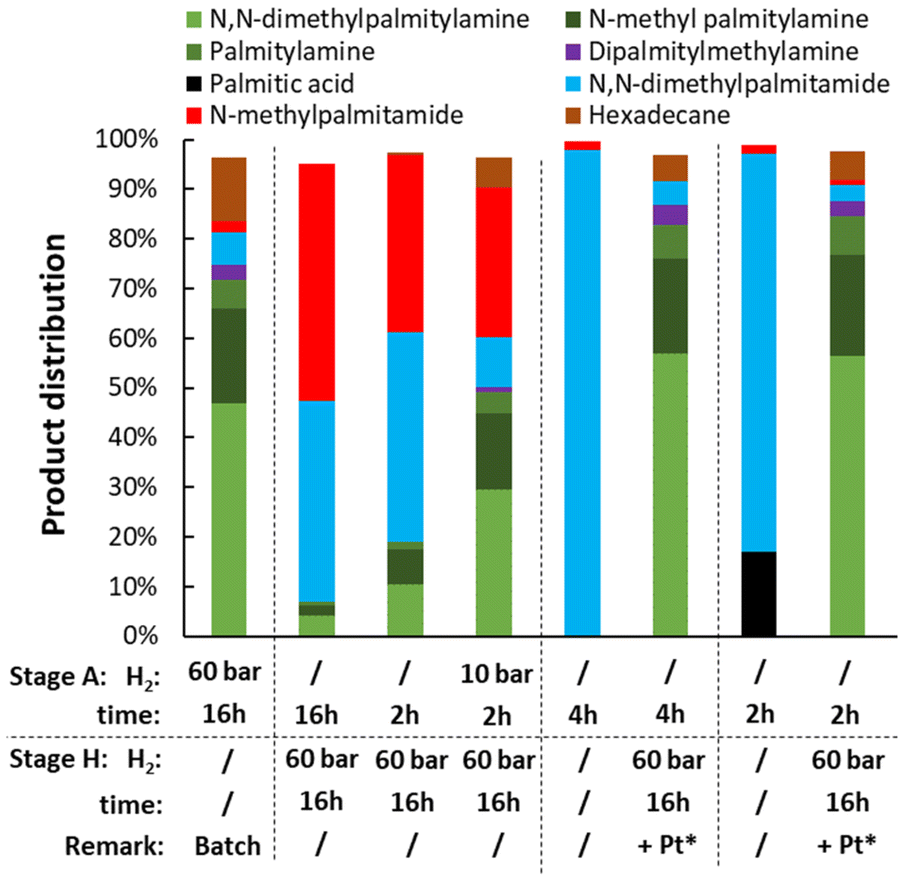
After performing the amidation and hydrogenation separately, both reactions were combined into one reductive amination step. Different reaction protocols were evaluated as summarized in Fig. 16. In a first run, the amidation of palmitic acid and the hydrogenation of the in situ generated amide were performed simultaneously by combining the Nb2O5 and PtVOx/SiO2 catalysts in one batch reaction. A lot more defunctionalized product, i.e. n-hexadecane, is observed than in the hydrogenation of N,N-dimethylpalmitamide. Most likely, the FA is hydrogenated directly to the corresponding fatty alcohol, which is more prone to defunctionalization (Fig. 17). In an initial attempt to minimize the formation of hexadecane, the coupled reaction was performed with intermediate addition of H2. Such a procedure consists of a stage A (i.e. amidation) in the absence of H2, or with limited H2, followed by a stage H (i.e. hydrogenation) in which H2 is added to the reaction mixture. However, under the initial H2-deficient conditions, disproportionation of DMA to MMA and TMA is significant. This metal-catalyzed reaction follows a hydrogen borrowing mechanism on the Pt catalyst (Fig. S16–S19†). The formed MMA not only reacts with FAs to form a more stable secondary amide (in comparison with a tertiary amide) but also preferentially adsorbs onto the hydrogenation catalyst, greatly limiting the hydrogenation rate. In an alternative approach, the PtVOx catalyst was added together with H2, without intermediate removal of Nb2O5 in between the two steps. A significant improvement is observed when compared with the initial batch reaction. Even with partial amidation of the FA after stage A (e.g. 17% palmitic acid remaining), limited defunctionalization is observed. Presumably, the acid is sufficiently stabilized in an ion pair by the amines present, which gradually increase in concentration.
 |
| | Fig. 16 Variation of the reaction protocol for the reductive amination of palmitic acid. Each run consists of a stage A (amidation) and stage H (hydrogenation), except for the bar at the left, which is one-step reaction. Reaction conditions (unless stated otherwise): palmitic acid (5 mmol), 200 °C, 1 g DMA, 2.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), 0.2 g Nb2O5, dodecane standard (100 μL), CPME (18.5 mL). Pt*: PtVOx catalyst added in stage H. | |
 |
| | Fig. 17 Reaction network for the reductive amination of a fatty acid (or fatty acid methyl ester) with dimethylamine. | |
Based on previous experiments, we were able to construct a reaction network (Fig. 17). Starting from a FA (1) or FAME (2), the molecule is amidated with DMA to the corresponding N,N-dimethylalkylamide (3). FAs can also form an organic salt with secondary (or primary) amines (although this salt is in equilibrium with its free amine-acid form). Ideally, the N,N-dimethylalkylamide (3) is hydrogenated to the corresponding amine (4), but alongside C–O cleavage, a small amount of C–N cleavage also occurs, leading to an alcohol (5). This alcohol can also be formed by direct hydrogenation of (1) or (2) and is much more susceptible to complete defunctionalization (to (6)) than the N,N-dimethylalkylamine (ADMA). Alongside the tertiary fatty amine, we also observe the formation of secondary (7) or even primary fatty amines (8). This is (mainly) the result of transalkylation (or disproportionation) reactions (see also Fig. S16–S19†). The disproportionation of DMA leads to the formation of TMA and MMA; the latter in turn reacts further to a less reactive N-methylalkylamide (9). Although it is unlikely that this secondary amide is hydrogenated directly, the molecule can be transamidated or hydrolysed (to (3) and (1) respectively).19
Range of applicability
After establishing a proper fed-batch reductive amination protocol, we checked the range of applicability. Virtually all natural FAs were converted to the corresponding amines (Fig. 18 & Table S1†). For all saturated fatty acids, very similar product distributions were observed at the end of each run, including a stage A and H, of approximately 8% alkylamine; 21% N-methylalkylamine; 55% N,N-dimethylalkylamine; 4% N,N-dimethylalkylamide; 6% alkane and 3% N-methyldialkylamine. The hydrogenation rate is slightly lower for unsaturated FAs, since the catalyst also has to hydrogenate the double bond. The reaction also works for FAs containing hydroxyl groups (e.g. ricinoleic acid, not in figure) although approximately half of the hydroxyl groups are removed, leading to a complex product mixture. A reductive amination was also performed with a FAME (i.e. methyl palmitate). Interestingly, this reaction led to an increased ADMA yield (i.e. 62%), most likely due to the alkylation of secondary amine with MeOH. However, leftover FAME is more readily converted towards the corresponding fatty alcohol, which in turn leads to an increased amount of alkane product (10% yield). Aside from varying the FA(ME) substrate, reductive aminations were also performed with different amine reactants (Table S1,† entries 13–16). Secondary amines that contain carbon chain ≥2 are worse nucleophiles for the amidation of FAMEs than their C1 counterparts (e.g. reactivity: DMA > N-methylbutylamine > diethylamine), leading to a decreased amidation rate. Although every consecutive hydrogenation can be considered successful, the products and reactants are more prone to hydrogenolysis and transalkylation side reactions.
 |
| | Fig. 18 Fed-batch reductive amination of natural FA(ME)s with dimethylamine (DMA). Each run consists of a stage A (amidation) and stage H (hydrogenation). General reaction conditions: FA(ME) (5 mmol), 200 °C, 1 g DMA, 0.2 g Nb2O5, dodecane or nonane standard (100 μl, nonane only for the C10 and C12 substrate), CPME (18.5 mL). Stage A: reaction time of 2 h. Stage H: follows directly after stage A, addition of 60 bar H2, 2.5 mol% Pt (PtVOx/SiO2, 7 wt% Pt), reaction time of 16 h. *5 mol% Pt. | |
Improving the selectivity with TMA
After proving the viability of the direct reductive amination of FA(ME)s, further improvements were made. In previous hydrogenation reactions, a significant fraction of the products consisted of primary and secondary fatty amines. Although these products are valuable as well, in this work, we intended to maximize the ADMA yield. To improve the selectivity for this tertiary amine, additional experiments were conducted in which DMA is (partially) replaced with TMA. Both the amidation of FAs and hydrogenation of fatty amides were investigated separately (Fig. 19). For the amidation reaction (Fig. 19, left), the reaction rate drops with increasing content of TMA (and decreasing content of DMA). Nevertheless, a significant conversion is still observed with only 1 equivalent of DMA. Identical conditions were also evaluated for the hydrogenation of N,N-dimethylpalmitamide (Fig. 19, right). While all reactions reach nearly full conversion, an increased amount of TMA increases the overall yield of N,N-dimethylpalmitylamine to up to 90% in both THF and CPME. This beneficial effect of TMA can be attributed to its effect on the transalkylation equilibrium. When secondary fatty amines are generated (e.g., via N–CH3 hydrogenolysis or transalkylation with DMA), they can still be converted to ADMAs via transalkylations with TMA. Such transalkylation reactions can also be used to cleave high-boiling tertiary amines, e.g., N-methyldipalmitylamine, into ADMAs (Fig. S18 & S19†). Next, direct reductive aminations of FAs were performed with the addition of TMA as well (Fig. S14†). Although the addition of TMA greatly minimizes the formation of primary amines, the fed-batch protocol is still necessary (see Fig. 16). It is clear that also for the direct acid-to-amine conversion, replacing a major part of DMA with TMA results in an increased selectivity for the ADMA product.
 |
| | Fig. 19 The influence of trimethylamine (TMA) on the FA amidation (left) and fatty amide hydrogenation (right). Reaction conditions: substrate (5 mmol), 200 °C, 1 g of methylamines; dodecane standard (100 μL), THF (18.5 mL, unless stated otherwise). Left: aside from the 1 g methylamines, for the majority of reactions, an additional 1 eq. DMA was added as well (≈0.2 g DMA). | |
Conclusion
In conclusion, the reductive amination of FA(ME)s is a viable approach to synthesize (mostly tertiary) fatty amines, such as ADMAs. Aside from DMA and H2, this reaction requires a physical mixture of two catalysts: a Nb2O5 catalyst, containing Lewis acid sites, for the conversion of FA(ME) to the corresponding fatty amide, and, a PtVOx/SiO2 catalyst for the conversion of the amide to the corresponding amine. In order to minimize transalkylations and transamidation side-reactions, a high H2 pressure must be maintained. The product ratios of primary–secondary–tertiary fatty amine can be customized by adjusting the reaction conditions (i.e. adjusting the DMA to TMA ratio). Nevertheless, in a one-pot fed-batch reaction at 200 °C, with 1 g methylamines, 60 bar H2 and 20 mL CPME as the benign reaction solvent, an amine yield of >90% is obtained. The reaction can be carried out with all natural fatty acids and is not limited to the synthesis of ADMAs (i.e. with methyl substituents). Fatty amines with longer alkyl substituents can also be prepared (e.g. N-methyl-N-butyl-palmitylamine). By studying the kinetics of the amide hydrogenation, we were able to construct a kinetic model. The model revealed that the hydrogenation rate is correlate to [H2 pressure]2 and [water content]−3. These findings confirm the necessity of drying zeolites in other reports on amide hydrogenation (an approach to continuously remove water). The hydrogenation of fatty amides can be carried out without the need for a solvent, using a very low PtVOx/SiO2 catalyst loading of only 0.5 mol% Pt. A recycling test and comprehensive characterization showed that the Pt catalyst can be reused multiple times and, aside from an initial decrease in catalytic activity due to sintering of Pt crystallites < 2 nm during the first run, remains reasonably stable in later consecutive runs.
Author contributions
Under the supervision of D.E.D.V., R.C. and T.C. were responsible for the concept and interpretation of the experiments. N.C. and S.B. characterized the catalysts via TEM analysis. T.C. provided expert knowledge into the methylamine & fatty amine manufacture and market. Additionally, the catalyst synthesis and design of the experiments were performed by R.C. as well. All authors contributed to writing the manuscript.
Data availability
The crude data for this article is partially available in the ESI† and stored locally at the cMACS. It is available upon request after approval by Prof. Dirk De Vos and Eastman Chemical Company (Taminco division).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This manuscript was made possible thanks to the research funding from VLAIO (Agentschap Innoveren & Ondernemen). R. C. thanks Paul Van der Aerschot and Sam Van Minnebruggen for technical support and CO chemisorption experiments respectively.
References
- Fatty amines market – Market estimates & trend analysis [online], Grand View Research. 2016 Available from: https://www.grandviewresearch.com.
-
K. Visek, Amines Fatty, in Kirk–Othmer Encyclopedia of Chemical Technology, Wiley, 2003, Vol. 4, pp. 4250–4257 Search PubMed
 .
.
-
N. Waddleton, Hydrogenation of nitriles to primary amines. Patent GB1321981A, 1973 Search PubMed .
- W. Rupilius, Fatty amines from palm oil and palm kernel oil, J. Oil Palm Res., 2011, 23, 1222–1226 CAS .
-
J. G. Erickson, Methyl fatty tertiary amines, Patent US2776314, 1957 Search PubMed .
- M. A. R. Jamil, A. S. Touchy, M. N. Rashed, K. W. Ting, S. M. A. H. Siddiki, T. Toyao, Z. Maeno and K. Shimizu,
N-Methylation, of Amines and Nitroarenes with Methanol Using Heterogeneous Platinum Catalysts, J. Catal., 2019, 371(12), 47–56 CrossRef CAS .
-
G. Demmering, H. Schütt and H. Rutzen, Singly unsaturated higher fatty alcohol production by catalytic hydrogenation of higher fatty acid (esters) at high temp and pressure, Patent DE2513377A1, 1975 Search PubMed .
- A. Ali, B. Li, Y. Lu and C. Zhao, Highly Selective and Low-Temperature Hydrothermal Conversion of Natural Oils to Fatty Alcohols, Green Chem., 2019, 21(11), 3059–3064 RSC .
- M. A. Sánchez, G. C. Torres, V. A. Mazzieri and C. L. Pieck, Selective Hydrogenation of Fatty Acids and Methyl Esters of Fatty Acids to Obtain Fatty Alcohols-a Review, J. Chem. Technol. Biotechnol., 2017, 92(1), 27–42 CrossRef .
- H. Kimura, K. Matsutani, S. I. Tsutsumi, S. Nomura, K. Ishikawa, Y. Hattori, M. Itahashi and H. Hoshino, Basic Function of Cu/Ni-Based Catalyst in a Colloidal State for One-Pot Animation Alcohols to N,N-Dimethyl Tertiary Amines, Catal. Lett., 2005, 99(3–4), 119–131 CrossRef CAS .
- D. Ruiz, A. Aho, P. Mäki-Arvela, N. Kumar, H. Oliva and D. Y. Murzin, Direct Amination of Dodecanol over Noble and Transition Metal Supported Silica Catalysts, Ind. Eng. Chem. Res., 2017, 56(45), 12878–12887 CrossRef CAS .
- IHS Market reports, Natural Fatty Acids, 2021.
-
J. Barrailt, M. Seffen and C. Forquy, Process for the production of long chain alkylamines and dimethylalkylamines and catalysts therefor catalysts therefor, Patent US4935546A, 1990 Search PubMed .
- J. Barrault and Y. Pouilloux, Synthesis of Fatty Amines. Selectivity Control in Presence of Multifunctional Catalysts, Catal. Today, 1997, 37(2), 137–153 CrossRef CAS .
- J. Barrault, S. Brunet, N. Suppo-Essayem, A. Piccirilli and C. Guimon, Selectivity Control in Substituted Fatty Amines Synthesis from Esters or Nitriles in the Presence of Bifunctional Catalysts, J. Am. Oil Chem. Soc., 1994, 71(11), 1231–1238 CrossRef CAS .
- J. Barrault, G. Delahay, N. Essayem, Z. Gaizi, C. Forquy and R. Brouard, Synthesis of Dimethylalkylamines from Acids and Esters over Promoted Copper Catalysts, Stud. Surf. Sci. Catal., 1991, 59, 343–350 CrossRef CAS .
- R. Coeck and D. E. De Vos, One-Pot Reductive Amination of Carboxylic Acids: A Sustainable Method for Primary Amine Synthesis, Green Chem., 2020, 22(15), 5105–5114 RSC .
- R. Coeck, J. Meeprasert, G. Li, T. Altantzis, S. Bals, E. A. Pidko and D. E. De Vos, Gold and, Silver-Catalyzed Reductive Amination of Aromatic Carboxylic Acids to Benzylic Amines, ACS Catal., 2021, 11(13), 7672–7684 CrossRef CAS .
- R. Coeck, S. Berden and D. E. De Vos, Sustainable, Hydrogenation of Aliphatic Acyclic Primary Amides to Primary Amines with Recyclable Heterogeneous Ruthenium–Tungsten Catalysts, Green Chem., 2019, 21(19), 5326–5335 RSC .
-
M. Terasaka and T. Fukushima, Process for producing aliphatic carboxylic acid amide, Patent US2010/298604A1, 2010 Search PubMed .
- T. Ohshima, Y. Hayashi, K. Agura, Y. Fujii, A. Yoshiyama and K. Mashima, Sodium Methoxide: A Simple but Highly Efficient Catalyst for the Direct Amidation of Esters, Chem. Commun., 2012, 48(44), 5434 RSC .
- M. A. R. Jamil, S. M. A. H. Siddiki, A. S. Touchy, M. N. Rashed, S. S. Poly, Y. Jing, K. W. Ting, T. Toyao, Z. Maeno and K. Shimizu, Selective Transformations of Triglycerides into Fatty Amines, Amides, and Nitriles by Using Heterogeneous Catalysis, ChemSusChem, 2019, 12(13), 3115–3125 CrossRef CAS PubMed .
- M. A. Ali, S. M. A. H. Siddiki, W. Onodera, K. Kon and K. Shimizu, Amidation of Carboxylic Acids with Amines by Nb2O5 as a Reusable Lewis Acid Catalyst, ChemCatChem, 2015, 7(21), 3555–3561 CrossRef CAS .
- A. M. Smith and R. Whyman, Review of Methods for the Catalytic Hydrogenation of Carboxamides, Chem. Rev., 2014, 114(10), 5477–5510 CrossRef CAS PubMed .
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, Selective Hydrogenation of Amides Using Rh/Mo Catalysts, J. Catal., 2010, 269(1), 93–102 CrossRef CAS .
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, Selective Hydrogenation of Amides Using Bimetallic Ru/Re and Rh/Re Catalysts, J. Catal., 2011, 278(2), 228–238 CrossRef CAS .
- R. Burch, C. Paun, X. M. Cao, P. Crawford, P. Goodrich, C. Hardacre, P. Hu, L. McLaughlin, J. Sá and J. M. Thompson, Catalytic Hydrogenation of Tertiary Amides at Low Temperatures and Pressures Using Bimetallic Pt/Re-Based Catalysts, J. Catal., 2011, 283(1), 89–97 CrossRef CAS .
- T. Mitsudome, K. Miyagawa, Z. Maeno, T. Mizugaki, K. Jitsukawa, J. Yamasaki, Y. Kitagawa and K. Kaneda, Mild Hydrogenation of Amides to Amines over a Platinum-Vanadium Bimetallic Catalyst, Angew. Chemie, 2017, 129(32), 9509–9513 CrossRef .
-
W. Y. Hernandez Enciso and S. Streiff, Process for converting amide to amine, Patent WO2021109109A1, 2021 Search PubMed .
- K. Sakoda, S. Yamaguchi, K. Honjo, Y. Kitagawa, T. Mitsudome and T. Mizugaki, Reductive Amination of Carboxylic Acids under H2 Using a Heterogeneous Pt–Mo Catalyst, Green Chem., 2024, 26, 2571–2576 RSC .
- T. Murayama, J. Chen, J. Hirata, K. Matsumoto and W. Ueda, Hydrothermal Synthesis of Octahedra-Based Layered Niobium Oxide and Its Catalytic Activity as a Solid Acid, Catal. Sci. Technol., 2014, 4(12), 4250–4257 RSC .
- R. Coeck, A. De Bruyne, T. Borremans, W. Stuyck and D. E. De Vos, Ammonolytic Hydrogenation of Secondary Amides: An Efficient Method for the Recycling of Long-Chain Polyamides, ACS Sustainable Chem. Eng., 2022, 10(9), 3048–3056 CrossRef CAS .
- A. Pennetier, W. Y. Hernandez, B. T. Kusema and S. Streiff, Efficient Hydrogenation of Aliphatic Amides to Amines over Vanadium-Modified Rhodium Supported Catalyst, Appl. Catal., A, 2021, 624, 118301 CrossRef CAS .
-
W. Y. Hernandez and S. Streiff, Process for converting amide to amine, Patent WO2021/109109A1, 2021 Search PubMed .
-
M. E. Davis and R. J. Davis, Heterogeneous Catalysis in Fundamentals of Chemical Reaction Engineering, McGraw Hill Higher Education, New York, 2002, pp. 133–183 Search PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional experimental data, characterization data, GC-MS and/or NMR spectra of all compounds (PDF). See DOI: https://doi.org/10.1039/d4gc05740f |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Nathalie
Claes
a,
Nathalie
Claes