
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-dimensional materials for ammonia synthesis
Apabrita
Mallick
 a,
Carmen C.
Mayorga-Martinez
b and
Martin
Pumera
*ac
a,
Carmen C.
Mayorga-Martinez
b and
Martin
Pumera
*ac
aAdvanced Nanorobots and Multiscale Robotics Lab, Faculty of Electrical Engineering and Computer Science, VSB – Technical University of Ostrava, 17. listopadu 2172/15, 70800 Ostrava, Czech Republic. E-mail: martin.pumera@vsb.cz; pumera.research@gmail.com
bSchool of Biomedical Engineering, Peruvian University of Applied Sciences (UPC), Prolongación Primavera 2390, 15023, Lima, Peru
cDepartment of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea
First published on 22nd April 2025
Abstract
Ammonia is an essential chemical due to its immense usage in agriculture, energy storage, and transportation. The synthesis of “green” ammonia via carbon-free routes and renewable energy sources is the need of the hour. In this context, photo- and/or electrocatalysis proves to be highly crucial. Low-dimensional materials (LDMs), owing to their unique properties, play a significant role in catalysis. This review presents a vast library of LDMs and broadly categorizes their catalytic performance according to their dimensionality, i.e., zero-, one-, and two-dimensional catalysts. The rational design of LDMs can significantly improve their catalytic performance, particularly in reducing small molecules like dinitrogen, nitrates, nitrites, and nitric oxides to synthesize ammonia via photo- and/or electrocatalysis. Additionally, converting nitrates and nitrites to ammonia can be beneficial in wastewater treatment and be coupled with CO2 co-reduction or oxidative reactions to produce urea and other valuable chemicals, which are also discussed in this review. This review collates the works published in recent years in this field and offers some fresh perspectives on ammonia synthesis. Through this review, we aim to provide a comprehensive insight into the catalytic properties of the LDMs, which are expected to enhance the efficiency of ammonia production and promote the synthesis of value-added products.
 Apabrita Mallick | Dr Apabrita Mallick earned her PhD in Chemical Science from the Indian Institute of Science Education and Research (IISER), Kolkata, in 2022. Thereafter, she joined as a post-doctoral researcher at the Advanced Nanorobots and Multiscale Robotics Lab, led by Prof. Martin Pumera at VSB–Technical University of Ostrava, Czech Republic. Her current research interests encompass micro/nanorobotics, photocatalysis, low-dimensional materials, environmental remediation, nitrate-to-ammonia conversion, small molecule activation, and the generation of value-added products. |
 Carmen C. Mayorga-Martinez | Dr Carmen Mayorga is currently a professor at the School of Biomedical Engineering, UPC-Peruvian University of Applied Sciences. Previously, she was a senior scientist at VSB–Technical University of Ostrava and led the Kralupy unit of the Centre for Advanced Functional Nanorobots, UCT-Prague. She was a research fellow in the nanobioelectronics and biosensors group/ICN2, Barcelona-Spain, and at Nanyang Technological University, Singapore. Currently, her main research fields include the development of bio/sensors based on 2D-materials platforms functionalized with bioreceptors (enzyme, DNA, and antibodies) as well as micro/nano robotics at different scales and different propulsion modes for biomedical applications and environmental monitoring. Moreover, she is also interested in 2D-materials catalysis for energy applications. |
 Martin Pumera | Professor Martin Pumera is Head of the Advanced Nanorobots and Multiscale Robotics Laboratory at VSB–Technical University Ostrava, Czech Republic. He founded the Center for the Advanced Functional Nanorobots at UCT Prague, where he served as a director (2017–2023). He was a tenured group leader at the National Institute for Materials Science, Japan, in 2006. In 2010, Martin joined Nanyang Technological University, Singapore, where he worked as a tenured associate professor for almost a decade. Prof. Pumera has diverse research interests in nanomaterials and microsystems, in the specific areas of micro and nanomachines, electrochemical energy storage and conversion, machine intelligence, and 3D printing. |
1. Introduction
Ammonia is a valuable chemical because of its diverse applications, such as in fertilizer industries, as a viable precursor to produce fine chemicals, and as a source of clean hydrogen energy storage, making it a potential alternative to fossil fuels.1–3 The current large-scale synthetic industrial technique for ammonia production is the Haber–Bosch process, however, this process demands high energy input. This process consumes about 1–2% of global energy resources per year and contributes to carbon emissions corresponding to ∼1% of global greenhouse gas emissions per year. This has prompted scientists to ponder over alternative, sustainable methods and, thus take a step toward attaining a carbon-neutral economy.4,5 The inspiration for devising such reactions is acquired from the nitrogen cycle occurring in nature, where biological nitrogenase enzymes catalytically reduce dinitrogen to ammonia.6 Consequently, different biohybrid catalysts were engineered to photoreduce nitrogen and produce ammonia. Various materials other than biohybrids evolved with time and participated in ammonia synthesis.7,8 Photo-, electro-, and photoelectrocatalytic processes have been mostly employed for synthesizing ammonia by reducing dinitrogen, nitrates, nitrites, and nitric oxides. The reaction conditions, mechanisms, benefits, and drawbacks of these catalytic processes are discussed in the review.Over recent years, low-dimensional materials (LDMs) have emerged as effective catalysts in photo- and electrocatalysis. LDMs are typically defined as materials smaller than 100 nm in a minimum of one dimension.9 Standard examples of LDMs include 0D spherical nanoparticles, 1D nanorods, nanowires, and 2D nanosheets. In lower dimensional (0D, 1D, and 2D) materials, their quantum-confined structures modify the electronic properties such that the LDMs become more efficient catalysts than their bulk counterparts, offering increased activity and stability.10,11 LDMs have long been studied for their structural and optoelectronic properties, but their utilization in catalysis is quite recent.12 This review explores the current state-of-the-art utilization of LDMs for ammonia synthesis by photo- and/or electroreduction of nitrogen-containing small molecules like dinitrogen, nitrates, nitrites, nitric oxides, etc. For a better understanding of the roles of dimensionalities, the catalytic synthesis of ammonia by LDMs has been categorized according to the dimensionality of the LDM (0D, 1D, and 2D) catalysts used for the photo- or/and electrocatalytic production of ammonia (Fig. 1). These low-dimensional materials present a wide range of physical and chemical characteristics that can boost the catalytic reactions.
 | ||
| Fig. 1 Schematic overview of low-dimensional materials used as catalysts for ammonia synthesis. | ||
In recent years, several reviews have been published, showcasing various techniques for ammonia synthesis. For instance, two reviews by Pang et al. and Ruan et al. focus on two-dimensional materials for nitrogen reduction by electrocatalytic pathways.13,14 Qing and co-workers reviewed recent works on electrocatalytic N2 reduction to produce ammonia,15 and Shi and colleagues provided an account of photocatalytic procedures to generate ammonia.16 Xiong et al. summarized the works on ammonia synthesis via electrochemical nitrate reduction.17 All these reviews on ammonia synthesis primarily focus on either 2D materials for electrocatalytic or photocatalytic synthesis of ammonia. Further elucidation of the novelty of this review compared with some of the existing reviews published in the last few years in the context of ammonia synthesis highlights the catalytic method, materials used, and N-sources used as reactants as presented in Table 1. Based on our current knowledge, a comprehensive review encompassing all low-dimensional materials, including 0D, 1D, and 2D, for ammonia synthesis is still pending. Some reviews also concentrate on LDMs for CO2 reduction or HER.9,18,19 Nevertheless, a review of LDMs for ammonia synthesis is lacking until now. This review presents all three classes of emergent low-dimensional catalysts for photochemical, electrochemical, and photoelectrochemical ammonia synthesis. Further, with the growing importance of ammonia synthesis in the global market and the increasing number of publications in this field, a thorough analysis and discussion on this subject is imperative and has been undertaken in this review.
| Journal | Catalytic approach | Highlights | N-Source | Ref. |
|---|---|---|---|---|
| Nat. Energy | Electrocatalysis | Metal-based catalysts, reactor design | N2 | 20 |
| Nat. Catal. | Electrocatalysis | Reaction mechanism, protocols | NOx | 21 |
| Nat. Catal. | Electrocatalysis | Challenges, protocols, and future perspectives | N2 | 22 |
| Nat. Synth. | Photo- and electrocatalysis | Mechanisms and catalyst design | N2 | 23 |
| Chem. Rev. | Electrocatalysis | Theoretical and experimental N2RR | N2 | 15 |
| Chem. Soc. Rev. | Electrocatalysis | 2D catalysts | N2 | 13 |
| Chem. Soc. Rev. | Photocatalysis | Materials, structure, and reaction engineering of MOx, BiOX, g-C3N4, and organic frameworks | N2 | 16 |
| Chem. Soc. Rev. | Photo-, photoelectro- and photothermocatalysis | Fundamentals and challenges of N2RR, reaction mechanisms, quantification methods, techno-economic applications | N2 | 24 |
| Adv. Mater. | Photo-, electro-, and photoelectrocatalysis | Reaction mechanism, catalyst engineering | N2 | 25 |
| Adv. Mater. | Photo- and electrocatalysis | Reaction and catalyst engineering | N2 | 26 |
| Adv. Mater. | Electrocatalysis | Reaction mechanism, catalyst design | NO3− | 17 |
| Adv. Energy Mater. | Electrocatalysis | Fe-based single-atoms | N2, NO3− | 27 |
| Adv. Funct. Mater. | Electrocatalysis | Active hydrogen in N2RR and NOxRR | N2, NO3−, NO | 28 |
| ACS Energy Lett. | Electrocatalysis | Li and alkaline earth metal catalysts | N2 | 29 |
| ACS Nano | Electrocatalysis | Group VIII-based catalysts | NO3− | 30 |
| ACS Nano | Electrocatalysis | Graphene derivatives | N2 | 31 |
| ACS Catal. | Photocatalysis | Fe-based catalysts | N2 | 32 |
| ACS Catal. | Electrocatalysis, catalysis under temperature and pressure | Ru-based catalysts | N2 | 33 |
| Small | Electrocatalysis | Single-atom catalysts | N2 | 34 |
| Small Methods | Electrocatalysis | Reaction mechanism, catalyst engineering | N2, NO3− | 35 |
| Small Methods | Photo- and photoelectrocatalysis | Reaction mechanism, catalyst engineering | N2 | 36 |
| Chem. Eng. J. | Electrocatalysis | Reaction mechanism, catalyst engineering | NO3− | 37 |
| Chem. Eng. J. | Photo-, electro-, and thermocatalysis | Ru-based single-atoms | N2 | 38 |
| J. Mater. Chem. A | Electrocatalysis | 2D catalysts | N2 | 14 |
| J. Mater. Chem. A | Photo- and photoelectrocatalysis | Three-phase interface heterojunction catalysts | N2 | 39 |
| This Review | Photo-, electro-, and photoelectrocatalysis | 0D, 1D, 2D, and heterostructured catalysts, reaction pathways, applications | N 2 , NO 3 − , NO 2 − , and NO |
The review is divided into eight sections. Section 2 focuses on the different approaches undertaken for synthesizing ammonia over the past years. The most common synthetic strategy of ammonia is the industrial Haber–Bosch process; however, this process is both energy-intensive as well as harmful to environment. Hence, other green strategies have been devised over time, which include enzyme catalysis and photo- and/or electrocatalysis. Here, we will also discuss the reaction pathways for ammonia synthesis.
In Section 3, we explore how the different dimensionalities of the LDMs play significant roles in catalysis. Zero-, one-, and two-dimensional (0D, 1D, and 2D) materials have different optoelectronic properties, impacting the catalytic process for ammonia synthesis. Through this review, we will present the latest advancements in developing LDMs, their characteristics, and the advantages they offer for catalytic ammonia production. The LDMs will be categorically summarized in the review, along with descriptions of the catalytic processes. These LDMs are often integrated to produce heterostructures that form promising catalysts for ammonia formation. Heterostructures render the interfaces with unique properties, leading to improved charge transfer and slowing down the recombination of charge carriers, thereby promoting catalytic ammonia synthesis.40 In the next three sections of the review, we describe up-to-date progress on LDMs in ammonia synthesis and categorize them according to the energy input, i.e.; photocatalysts, electrocatalysts, and photoelectrocatalysts.
Section 4 delves into a comprehensive array of 0D, 1D, and 2D low-dimensional photocatalytic materials used for ammonia synthesis. Structure engineering of the photocatalysts for enhancing catalytic performance via the introduction of defects, construction of heterojunctions,21 and increasing surface area by hybridization with other materials will be explored further.41–43 The role of co-catalysts in boosting the photocatalytic efficiency of ammonia synthesis will also be discussed here.44
Section 5 focuses on electrocatalysts that efficiently convert N-containing species to ammonia. Like photocatalysts, the low-dimensional electrocatalysts (0D, 1D, and 2D) have also been modified for better performance via defect engineering, formation of heterostructures and regulation of crystal planes, and the roles of such modifications will be illustrated with distinct examples.45–47 LDMs have also been used to produce 3D-printed electrodes that can electrocatalytically reduce nitrates and nitrites to form ammonia.48 3D printing techniques can precisely tune the shape, structure, and geometry of LDMs; hence, we will explore these emerging 3D-printed LDM catalysts for enhancing catalytic performance.
Section 6 emphasizes low-dimensional photoelectrocatalysts that combine the advantages of photocatalysts and electrocatalysts. In photoelectrocatalysis the combination of light and electric bias helps improve the separation and migration of electron–hole pairs, reducing recombination losses.49,50 Narrow band gap materials or heterostructures formed by LDMs are mostly used for designing photocathodes, and these LDMs will be explored in detail in this section.51,52
In Section 7 we discuss the advantages and applications of ammonia synthesis reactions. For instance, nitrates are well-known contaminants in groundwater, so reducing nitrates apart from yielding valuable-added ammonia also aids in wastewater treatment.53 The reduction process can also be coupled with valuable oxidation reactions, such as water oxidation, methanol oxidation, glycerol oxidation, and plastic degradation via PET oxidation.54–56 These reactions will also be explored in the review. The reduction of dinitrogen or nitrates, coupled with the co-reduction of CO2 can produce urea via C–N coupling. Urea is the most common fertilizer used in agriculture.57 This process is extremely beneficial as it can help realize carbon/nitrogen neutrality in the environment. The design of catalysts for two co-reduction processes is difficult, but not impossible. Dual-site, bimetallic, single-atom catalysts have been employed to achieve the formation of urea.58–60 The catalysts explored in all these aspects will be discussed in this section.
The final section presents future perspectives and the outlook for ammonia synthesis. All sections in this review are supported and illustrated with representative literature from the last decade (2015–2025). This review aims to present a vast library of emergent low-dimensional materials for ammonia generation. It also provides a comprehensive insight into the choice of materials for catalysts, the strategic designs employed to reach maximum efficiency, and implementations toward next-generation applications.
2. Approaches to make ammonia: methods and reactions of ammonia synthesis
The synthesis of ammonia is important as ammonia has versatile uses, such as zero-carbon fertilizers, energy storage, and the synthesis of value-added products.2,3,61 Global production of ammonia amounts to ∼170 million metric tons per year.62 The industrial synthesis of ammonia mainly involves gaseous nitrogen, abundantly available in the atmosphere. But the process is not very simple. The dissociation of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N requires a very high energy of ∼941.8 kJ mol−1, and the standard enthalpy of formation of ammonia at 25 °C is −46 kJ mol−1.63,64 Hence, the synthesis of ammonia becomes challenging as the reaction involves high temperature, high pressure, or reactive reagents, and also the process is not energy efficient. This section focuses on the most widely used strategies explored till now to synthesize ammonia (Fig. 2).
N requires a very high energy of ∼941.8 kJ mol−1, and the standard enthalpy of formation of ammonia at 25 °C is −46 kJ mol−1.63,64 Hence, the synthesis of ammonia becomes challenging as the reaction involves high temperature, high pressure, or reactive reagents, and also the process is not energy efficient. This section focuses on the most widely used strategies explored till now to synthesize ammonia (Fig. 2).
 | ||
| Fig. 2 Schematic representation of different routes to synthesize ammonia. Industrial Haber–Bosch process, nitrogenase enzyme-catalyzed reaction, reproduced with permission from ref. 65, copyright 2017, Wiley-VCH, photocatalysis, electrocatalysis, and photoelectrocatalysis from the conversion of N2, NOx−, and NOx. | ||
2.1. Haber–Bosch process
To meet the need for ammonia-based fertilizers, in the early twentieth century, two Nobel Prize-winning chemists, Fritz Haber and Carl Bosch, invented a high-pressure reaction to directly combine N2 and H2 to form ammonia (eqn (1)).66 To date, most industries rely on this process for the commercial production of ammonia. The modified Haber–Bosch reaction is typically conducted over Fe- or Ru-based catalysts in the temperature zone of 623–773 K and the pressure region of 15–35 MPa.66 This process is energy-intensive, accounting for about 1–2% of global energy consumption. The H2 required for this process is mostly derived from the combustion of fossil fuels. It releases CO2, contributing to ∼1% of the total greenhouse gas emissions of the world.4 The reaction for ammonia production following the Haber–Bosch process is as follows: | (1) |
2.2. Enzyme-catalyzed reactions
In an attempt to reduce the carbon footprint generated by Haber–Bosch reactions, researchers have taken inspiration from biological nitrogen fixation reactions. In this reaction, nitrogenase, a bacterial enzyme, directly converts atmospheric N2 to NH3 (eqn (2)).67 Instead of H2, this reaction uses protons and electrons to form the N–H bonds in ammonia. Nitrogenase comprises two proteins: Fe-based protein (FeP) and MoFe-based protein (MoFeP).65,67 FeP hydrolyzes ATP to ADP, and the electrons generated in this process are transferred to MoFeP. MoFeP reduces N2 to NH3. Stoichiometrically, six pairs of protons and electrons should be used for the synthesis of NH3 from N2. But the reaction takes up 8 pairs of H+/e−. This apparent wastage of 2e− and 4 ATP molecules contributes to the energy required for thermodynamically driving the reaction. However, excess energy usage leads to a large chemical overpotential of this reaction.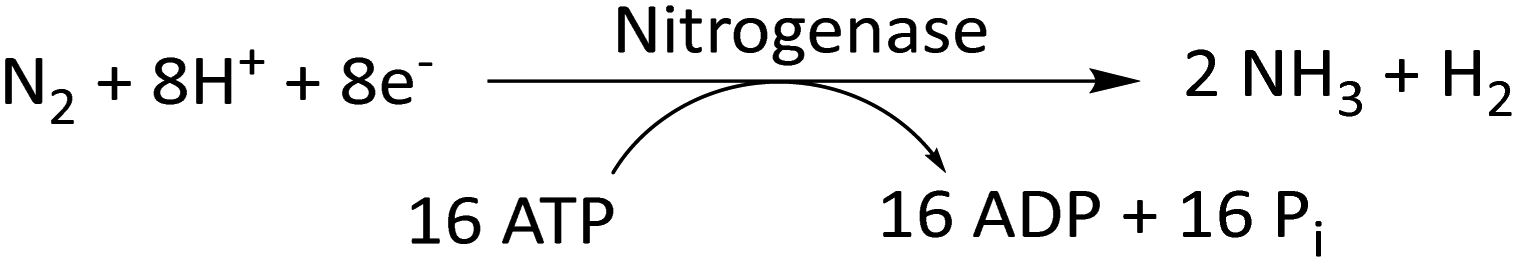 | (2) |
The high demand for ammonia has led researchers to find a sustainable method for its synthesis that will be energy-efficient, cost-effective, and environmentally benign. In recent years, numerous methods, such as plasma, thermochemical, photochemical, electrochemical, and photoelectrochemical techniques have been explored to decarbonize ammonia synthesis. This review will focus on ammonia generation from different nitrogen-containing species (like N2, NO, NO3−, and NO2−) via photocatalysis, electrocatalysis, and photoelectrocatalysis.
2.3. Photocatalysis
Solar energy is a clean and inexhaustible energy source, which has triggered the wide utilization of photocatalysis in the sector of chemical fuels. Literature evidence suggests that over the initial years, it was thought that the photoreduction of N-containing species occurs only in nature. Soil and sand samples were explored to study the effect of minerals on the photoreduction of nitrogen in nature.68,69 Fascinated by the working mechanism of the Fe and FeMo proteins in nitrogenase enzyme reactions, chemists have designed photocatalysts that absorb light to generate electrons and protons. These electrons activate the N-containing species (N2, NOx, and NOx−), which react with protons from water to form ammonia. Brown et al. developed such an analog CdS: FeMo protein biohybrid, which can reduce nitrogen to ammonia upon light activation.7 CdS nanocrystals act as photosensitizers, providing electrons to the FeMo protein for nitrogen reduction. In this work, light is used as the energy source, whereas in the biological nitrogenase system energy is acquired by hydrolysis of ATP. Researchers have extended this know-how to design other photocatalytic systems like metal oxides,70,71 metal sulfides,72 bismuth oxyhalides,73,74 layered double hydroxides,75,76 and carbon-based materials77,78 for the photocatalytic synthesis of ammonia. According to the literature, the photocatalytic N2, NOx, and NOx− reductions by semiconductors occur on photoactivation, when the excited electrons move to the conduction band, leaving holes in the valence band.79 The electrons reduce N-species to ammonia while the holes participate in the water oxidation reaction. The recombination of electrons and holes is one of the significant problems in photocatalytic reactions.80 Thus, hole scavengers are sometimes used as sacrificial agents to prevent recombination. The band gap of the materials and the position of conduction and valence bands play a significant role in photocatalysis. The reduction potentials of N2RR, NOxRR, and water oxidation must lie in between the conduction and valence band positions. NOx− (nitrates and nitrites) are common sources of pollutants in wastewater and the photoreduction of NOx− can help in wastewater treatment, which will be discussed in detail later in Section 7.1. The direct reduction of nitrate and nitrite wastes into ammonia is thus both economically and environmentally important. In some cases, however, the competitive hydrogen evolution reaction (HER) and reduction of NO3− to N2 limits the selectivity of the ammonia produced. A detailed discussion on different classes of photocatalytic materials like metal oxides, metal nanoparticles, single-atoms, quantum dots, and their composites, which can photoreduce N-containing species to ammonia with high selectivity, will be presented in Section 4 of this review.2.4. Electrocatalysis
Electrochemical reduction of N2 or NOx− to ammonia occurs under ambient conditions, i.e., at room temperature and atmospheric pressure at a specific applied voltage. Sometimes, electrochemical reactions are considered to be more energy-efficient than photochemical reactions. This is because, in photocatalysis, some of the absorbed photons remain unutilized due to the variable wavelengths of incident light sources and the recombination of electrons and protons.81 The design of electrocatalysts is quite challenging because the activity, selectivity, efficiency, and stability depend on the choice of materials. The material structure (surface area, porosity, crystal facets) and morphology affect the turnover and yield, while the electronic properties (heteroatom doping, defect engineering) influence the active site of the catalyst.82 The surface properties of the catalyst can affect the adsorption–desorption of N2 or NOx− species, which in turn affects the bond dissociation and the consecutive hydrogenation reactions for ammonia formation. The availability of electrons and protons can also be controlled by the design of the catalyst, which might be able to suppress the competing HER reactions and also help in attaining selectivity of the ammonia, reducing the chance of obtaining lesser reduced products like NO2− or NO from the electroreduction of NO3−.In the electrochemical experiments, initially N2 or NOx− is adsorbed on the surface of the electrode in a simple electrochemical cell. Electrons supplied by the external circuit reduce the N-containing molecule, which undergoes subsequent hydrogenation by the protons to produce ammonia. In most cases, water present in the reaction medium acts as the source of hydrogen for ammonia production. A judicious choice of electrolyte is crucial for electrocatalytic reactions because the electrode reactions of the electrochemical cell are dependent on the electrolyte. In past years, aqueous and non-aqueous electrolytes have been used where protons (H+), hydroxides (OH−), oxides (O2−), and nitrides (N3−) act as mobile charge carriers. The main bottlenecks in electrocatalytic ammonia synthesis are the yield and the Faradaic efficiency (FE). Competing reactions on the surface of electrocatalysts, like water reduction to hydrogen evolution reaction (HER), dominate over N2 or NOx− reduction reactions (N2RR and NOxRR) at higher overpotentials, thus decreasing the selectivity of the product.83 To eliminate the drawbacks, over the past few years, researchers have been working to upgrade electrocatalysis cells, electrocatalysts, electrolytes, and working potentials, which will be discussed in detail in Section 5 of this review.
2.5. Photoelectrocatalysis (PEC)
The general redox reactions for photocatalysis and electrocatalysis are similar. In PEC, these reactions become more favourable because of the combined effects, i.e., electric and light energy.84 In an electrochemical cell, N2RR or NOxRR occur at the cathode while water splitting occurs at the anode. The experimental setup of PEC is very similar to the electrocatalytic set-up; only in PEC, the cathode is replaced with a photocathode. The photocathode utilizes energy from a light source to provide the photovoltage. In this way, the electrical energy required for N2RR and NOxRR either decreases to a certain extent or is completely diminished depending on the Fermi level of the photocathode.49 Generally, in PEC a three-electrode set-up is employed where the catalyst is loaded on the working electrode (photocathode).36 The reduction reactions (N2RR or NOxRR) occur at the photocathode and water splitting at the anode (counter electrode). A proton exchange membrane is placed between the cathode and anode, which ensures only protons are supplied to the cathodic side for ammonia formation, and no other oxidative species can migrate to the cathodic part so that the ammonia produced does not undergo any oxidation. In photocatalysis and electrocatalysis, the competing HER reduces the Faradaic efficiency (FE) of NH3. In PEC, the competing HER can be suppressed using photoelectrodes. For instance, an Au-decorated ordered silicon nanowire array photocathode can perform NO2RR to NH3 with FE as high as 95.6% at 0.2 V vs. RHE.85 This is because the Au and Si surfaces of the electrode are not active for water reduction. The absence of the competing HER makes the FE for NH3 production comparatively higher. The PEC reaction mechanisms of N2RR and NOxRR are similar to photocatalysis and electrocatalysis. The design of materials for PEC follows the same principles as electrocatalysts and photocatalysts. Improvement in the photoelectrocatalytic NH3 yield and selectivity can be achieved by modification of the catalysts such as, doping with heteroatoms, making heterojunctions, defect engineering, and modification of morphology.86–88 Thus, we can infer that the structures and properties of the materials used for designing the catalysts have profound roles in the catalytic reactions and products formed. Section 6 of this review will extensively discuss the emergent low-dimensional materials for photoelectrocatalytic ammonia synthesis.Among the above-mentioned ammonia synthesizing techniques, the most widely used methods in recent literature include photocatalysis, electrocatalysis, and photoelectrocatalysis. These processes are more sustainable and easily achievable and only require catalyst materials and energy sources to produce ammonia. Though all these processes follow the general principles of catalysis, however, the reaction conditions, mechanisms, and catalyst design differ. To get a brief idea about the working principles, catalysts, reaction mechanisms, benefits and drawbacks, and environmental impact, a comparative study of these processes is presented in Table 2 and Fig. 3.
| Factors | Photocatalysis | Electrocatalysis | Photoelectrocatalysis |
|---|---|---|---|
| Energy source | Solar energy (light) | Electrical energy | Light and electrical energy |
| Catalyst materials | Semiconductors, metallic nanoparticles, plasmonic materials, quantum dots, metal oxides, single-atoms, carbon-based materials | Metals, alloys, metallic oxides, single-atoms, carbon-based materials, MXenes, transition metal dichalcogenides | Heterostructures composed of photocatalysts and electrocatalysts |
| Design of catalytic systems | i. Band gap engineering. | i. Good conductivity and corrosion resistance. | i. Heterostructures of photocatalysts and electrocatalysts for simultaneous light and electrical energy utilization. |
| ii. Efficient charge separation. | ii. Electrode surface modification to adsorb reactants and intermediates. | ii. Interface engineering for charge transfer. Catalyst engineering with photo- and electrocatalysts. | |
| iii. Surface area modification for better light absorption and adsorption of N2/NOx. | iii. Electrode porosity for better mass transfer. | iii. Reactor design for efficient light and electrical energy distribution. | |
| iv. Catalyst engineering via the introduction of defects, doping, cocatalysts, heterojunction and heterostructure formation, particle size, and morphology modification. | iv. Catalyst engineering via the introduction of defects, doping, cocatalysts, facets, heterojunction, and heterostructure formation. | ||
| v. Good electrolytes to enhance ionic conductivity. | |||
| Reaction mechanism | i. Light absorption leads to the separation of electron–hole pairs and photoexcitation of electrons from the valence band to the conduction band. | i. Application of electric potential. | i. Light absorption and electric potential generate required charge carriers for photoelectrocatalysis. |
| ii. Electrons in the conduction band reduce N2/NO/NO2−/NO3− to form NH3. | ii. Electrons reduce N2/NO/NO2−/NO3− to NH3 at the cathode. | ii. Synergistic effects enhance electron transfer. | |
| iii. Holes oxidize water or other organic reactants. | iii. Efficient electron transfer at electrode surface facilitated by catalysts. | iii. Optimization of photo- and electrical parameters for efficient reduction of N2/NO/NO2−/NO3− at the photocathode. | |
| iv. The oxidation half-reactions occur at the anode. | |||
| Cost | Low | High | High |
| Efficiency | Low | High | Low |
| Advantages | i. Light energy can be harnessed from abundant solar energy. | i. Ammonia production with high efficiency and greater selectivity. | i. Combined advantages of photo- and electrocatalysts. |
| ii. Low operational cost. | ii. Reaction parameters can be controlled and adjusted easily for electrocatalytic processes. | ii. Potential for synergistic reactions. | |
| iii. Sustainable synthesis of ammonia due to reduced greenhouse gas emissions. | iii. Lower dependency on weather conditions as electric energy can be produced on demand. | iii. Flexible reaction conditions. | |
| Disadvantages | i. For solar-powered photocatalytic reactions, ammonia production is dependent on regions with high solar irradiance. | i. Requires electrical energy, which is sometimes harnessed from non-renewable sources. | i. Complexity of system design. |
| ii. Efficient light absorption and conversion are required. | ii. Electrode degradation with time. | ii. Requires optimization of both light and electrical inputs. | |
| iii. Long-term stability and recyclability of photocatalysts have to be considered. | iii. Higher operational costs due to electricity, maintenance, and replacement of electrochemical setup. | ii. Higher costs of photoelectrochemical setup and maintenance. | |
| Environmental impact | i. Low environmental footprint due to reduced use of fossil fuels for photocatalytic ammonia production. | i. Dependent on electricity, sometimes produced by non-renewable energy sources. | i. Can provide an optimized balance between light and electrical energy sources and reduce environmental impact. |
| ii. Minimal waste generation and greenhouse gas emissions. | ii. Emissions associated with electricity production and potential waste generation from electrode degradation. | ||
| Future applications | i. Solar-powered green ammonia generation. | i. Potential for industrial-scale ammonia productions. | i. Hybrid systems for enhanced sustainable ammonia production under variable reaction conditions. |
| ii. Potential for integration with renewable energy sources. | ii. On-grid applications. | ii. Off-grid and on-grid applications. | |
| iii. Off-grid applications. |
 | ||
| Fig. 3 Comparison of (A) photocatalysis, (B) electrocatalysis, and (C) photoelectrocatalysis methods for ammonia synthesis. | ||
2.6. Reaction mechanisms of N2 reduction to NH3
The reaction of catalytic reduction of dinitrogen (N2) to ammonia (NH3) using natural ingredients like air and water is proposed to be as follows (eqn (3)–(5)):79,89 | (3) |
The half-cell N2 reduction in the acidic and basic media can be depicted as follows:36,90
| Acidic condition: N2 + 6H+ + 6e− → 2NH3 | (4) |
| Basic condition: N2 + 6H2O + 6e− → 2NH3 + 6OH− | (5) |
The reactions (eqn (6)–(9)) for the reduction of N2 to NH3 can change with the reaction medium and charge carriers as follows:15
a. In the presence of H+
| N2 + 6H+ + 6e− → 2NH3 |
| 3H2 → 6H+ + 6e− | (6) |
b. In the presence of OH−
| N2 + 6H2O + 6e− → 2NH3 + 6OH− |
| 3H2 + 6OH− → 6H2O + 6e− | (7) |
c. In the presence of O2−
| N2 + 3H2O + 6e− → 2NH3 + 3O2− |
| 3O2− → 3/2O2 + 6e− | (8) |
d. In the presence of N3−
| N2 + 6e− → 2N3− |
| 3H2 + 2N3− → 2NH3 + 6e− | (9) |
In all reaction conditions, dinitrogen reduction to ammonia is a six-electron reduction reaction, which is a kinetically “uphill” process. The design of photo- and/or electrocatalysts plays a vital role as they provide an alternative lower energy pathway to the production of ammonia. Nitrogen is a highly stable molecule owing to its triple bond and breaking the first bond is considered the rate-determining step. The mechanism of N2 reduction to ammonia is currently being investigated by different research groups and studies are still underway. It has been proposed that there are two possible pathways for N2 reduction to NH3: (i) associative and (ii) dissociative (Fig. 4A).36,91 In the dissociative pathway, the N–N bond breaks before hydrogenation. However, this process is highly energy-demanding and the dissociative pathway is not considered favourable. In the associative mechanism, the first step involves the protonation of N2. Protonation depends on the configuration of N2 and can be either to the distal N atom, forming N2H* species, or to the two N atoms which can alternatively form N2H2* species. In the associative distal pathway, the reaction proceeds via three consecutive hydrogenation steps, forming one NH3 molecule. Then, hydrogenation occurs at the second N atom, forming NH3 after three more steps. In the associative alternating pathway, hydrogenation occurs alternately at both N atoms, and two molecules of ammonia are produced almost simultaneously. Photocatalytic and electrocatalytic reduction of N2 to ammonia can follow any of these pathways, though simple calculations are not enough to predict the exact reduction pathway.
 | ||
| Fig. 4 General reaction pathways of ammonia synthesis. (A) N2 reduction to ammonia via dissociative, associative alternating, and associative distal pathways, (B) NOx− and NOx reductions to ammonia via associative, dissociative, alternating, and distal pathways through N- and O-ends. | ||
2.7. Reaction mechanisms of NOx− and NOx reduction to NH3
As discussed above, the bond dissociation energy of N2 is very high (∼941.8 kJ mol−1). In comparison, the bond dissociation energies for NOx and NOx− species are lower. They can be disintegrated into deoxygenated species with a lower bond dissociation energy (for NO3− ∼204 kJ mol−1); hence, nitrogen oxides can offer a sustainable route to the synthesis of ammonia at a lower energy cost. Also, the valence states of N in nitrogen oxides are higher (+5 in NO3−, +3 in NO2−) than in dinitrogen where the valence state of N is 0; hence, deep reduction reactions are more plausible in the case of nitrogen oxides. The catalytic reactions are similar to that of dinitrogen but require a different number of electrons and protons for each species (eqn (10)–(12)).92 | (10) |
 | (11) |
 | (12) |
Nitrates and nitrites are present in the form of electrolytes for the electrocatalytic reduction, which involves 8e− and 6e− respectively for the conversion to ammonia. The reduction proceeds via the dissociation of NOx− species into deoxygenated species followed by hydrogenation, which is quite similar to the adsorption–desorption mechanism of nitrogen reduction. The reduction of nitrogen oxide species to ammonia is illustrated in Fig. 4B and it exhibits multi-step processes of electron and proton transfer.93,94 Initially, NO3− is adsorbed on the catalyst surface as *NO3−, which is followed by the loss of an electron to form *NO3. This *NO3 intermediate is then converted into *NO2via the transfer of two electrons and two protons. In most cases, the reduction of nitrate to nitrite is the rate-determining step for NO3− reduction. The *NO2 intermediate is further reduced to *NO. The subsequent steps of reduction to ammonia passing through *NO intermediate is the same for NOx− and NO. The next possible steps can follow either dissociative or associative pathways. In the dissociative pathway, the N–O bond undergoes dissociation, forming adsorbed nitrogen (*N) and oxygen (*O) species on the catalyst surface. The *N is then sequentially hydrogenated to yield ammonia. The associative pathway can proceed through any one of the three possible routes depending on the adsorption type of *NO intermediate. The adsorption of *NO on the catalyst surface can be through (i) N-end, (ii) O-end, or (iii) NO-side. The hydrogenation of the N-end and O-end routes can again proceed through either alternating or distal hydrogenation processes. In the case of the distal-O or N hydrogenation pathway, at first one of the O or N atoms is at first fully hydrogenated to form H2O or NH3, followed by the hydrogenation of the other atom. For the case of the alternating-O or N hydrogenation pathway, the O and N atoms are alternately hydrogenated stepwise to form H2O and NH3, respectively.94
3. Low-dimensional materials (LDMs): dimensionality of nanostructures and heterostructures
Nanostructured materials (NSMs) can act as effective catalysts because of their morphologies, electronic properties, and surface characteristics.95 Dimensionality is an important parameter because different dimensional materials of similar chemical composition exhibit separate physical and chemical properties. For instance, carbon has different allotropes: fullerene, carbon nanotube, graphene, and graphite. Fullerene is 0D, carbon nanotube is 1D, graphene is 2D, and graphite is 3D, and all four have different properties. NSMs have building units of size ranging from nanometer to sub-micron scale. The classification of NSMs has been proposed by Gleiter (1995) and Skorokhod (2000); later, Pokropivny and Skorokhod proposed a modified classification.96,97 According to the classification by Pokropivny and Skorokhod, NSMs can be classified as 0D, 1D, or 2D. The structure and morphology of some common low-dimensional materials are illustrated in Fig. 5. In this review, we will explore the implementation of the NSMs and multicomponent heterostructures, which are built from the 0D, 1D, and 2D materials for photocatalytic, electrocatalytic, and photoelectrocatalytic N2RR and NOxRR to produce ammonia. | ||
| Fig. 5 Dimensional classification of low-dimensional materials (LDMs). 0D common structures. (A) Metallic nanoparticles, representative SEM image of bimetallic Au/Ni nanoparticle. Reproduced with permission from ref. 98. Copyright 2019, American Chemical Society. (B) Semiconductor nanoparticles, representative TEM image of CdS semiconductor nanoparticles. Reproduced with permission from ref. 99. Copyright 2020, American Chemical Society. (C) Core–shell structures, representative TEM image of Fe3O4@silica RF core–shell particles. Reproduced with permission from ref. 100. Copyright 2009, Royal Society of Chemistry. (D) Nanocrystals, representative TEM image of maghemite nanocrystals. Reproduced with permission from ref. 101. Copyright 2001, American Chemical Society. (E) Single atoms, representative HAADF-STEM image of Fe single atoms. Reproduced with permission from ref. 102. Copyright 2023, Wiley-VCH. (F) Quantum dots, representative TEM image of carbon quantum dots. Reproduced with permission from ref. 103. Copyright 2023, Royal Society of Chemistry. 1D common structures. (G) Nanowires, representative SEM image of Ag nanowires. Reproduced with permission from ref. 104. Copyright 2020, American Chemical Society. (H) Nanotubes, representative TEM image of carbon nanotube. Reproduced with permission from ref. 105. Copyright 2019, Elsevier. (I) Nanofibers, representative SEM image of Fe2TiO5 nanofibers. Reproduced with permission from ref. 106. Copyright 2022, Wiley-VCH. (J) Nanorods, representative TEM image of g-C3N4 nanorods. Reproduced with permission from ref. 107. Copyright, 2022, Elsevier. (K) Nanobars, representative SEM image of LiNi0.4Co0.2Mn0.4O2 nanobars. Reproduced with permission from ref. 108. Copyright 2012, Royal Society of Chemistry. (L) Nanoribbons, representative TEM image of graphene nanoribbons. Reproduced with permission from ref. 109. Copyright 2015, AIP Publishing. 2D common structures. (M) Nanosheets, representative TEM image of ZnCr-LDH nanosheets. Reproduced with permission from ref. 76. Copyright 2020, Wiley-VCH. (N) Nanoplates, representative TEM image of hexagonal Co(OH)2 nanoplates. Reproduced with permission from ref. 110. Copyright 2013, Royal Society of Chemistry. (O) Nanofilms, representative TEM image of Cu-TCPP MOF ultra-thin nanofilm. Reproduced with permission from ref. 111. Copyright 2013, Royal Society of Chemistry. (P) Nanolayers, representative cross-TEM image of a-ZrTiOx nanolayers. Reproduced with permission from ref. 112. Copyright 2013, Royal Society of Chemistry. (Q) Nanodiscs, representative TEM image of Bi2Se3 nanodisc. Reproduced with permission from ref. 113. Copyright 2012, American Chemical Society. (R) Nanoprisms, representative SEM image of Ag nanoprisms. Reproduced with permission from ref. 114. Copyright 2024, American Chemical Society. | ||
3.1. Zero-dimensional (0D) nanostructured materials
0D materials are mostly nano-dimensional structures with an average size of less than 10 nm along all three dimensions and are isotropic.115 They comprise metal nanoparticles, metal alloy nanoparticles, nanospheres, core–shell structures, yoke–shell structures, quantum dots, single-atoms, nanocrystals, and metal oxides.98–103,116 The fabrication of 0D catalysts has been well explored and various methods, such as thermal evaporation, sputtering, chemical vapour deposition, electrodeposition, solvothermal, sol–gel, annealing, galvanic replacement, gas phase deposition, and template-based methods are quite common fabrication techniques.117,118 The optical and physicochemical properties of these 0D materials can be tuned depending on their size, making them potential catalysts for photo- and electrocatalysis reactions.3.2. One-dimensional (1D) nanostructured materials
1D materials are nanostructured along any two dimensions while the third dimension can be in the microscale range.115 1D catalysts are advantageous over 0D catalysts as they offer a unique anisotropic structure, more density of active sites, and more surface area than 0D materials.119 0D materials are more susceptible to Ostwald ripening due to the high surface energy. In 1D nanostructures, the lower-energy facets are preferentially exposed on the surface, which reduces the surface energy of the whole material, making them more stable.120 The structure of 1D materials enhances mass transport and diffusion, and provides them with superior catalytic properties.121 1D materials comprise nanowires, nanotubes, nanorods, nanoribbons, nanobelts, and nanofibers.104–109 The most common synthetic techniques of 1D materials include hydrothermal, sol–gel, self-assembly, electrospinning, and template-assisted methods.122–124 In this review, we will explore the recently developed 1D catalysts and how their structural and electronic properties aid in the catalytic synthesis of ammonia.3.3. Two-dimensional (2D) nanostructured materials
In 2D materials, only one dimension is confined to the nanoscale regime while the other two dimensions can range from micrometer to centimeter scale.115 Nanosheets, nanoplates, nanodisks, and nanoprisms are the most commonly designed 2D materials for catalysis.76,110–114,125 These 2D nanostructures have exposed surface area, increasing their contact with reactants.126,127 Most of the active sites are present on the surface and are easily accessible for catalysis. Defects and unsaturated sites can be introduced into the 2D structures, which enhances adsorption–desorption and facilitates mass transport.126 In 2D materials, electrons can be trapped within the atomic layers, which brings good conductivity and electron mobility to the structures, thereby facilitating electron transport and charge carrier separation.128According to Sabatier's Principle, an optimized catalytic reaction occurs when the interaction between the reactants and the catalyst surface is optimal, i.e., the adsorption energy is intermediate.129 Thus, it can be said that the catalytic properties of a material are dependent on its surface and electronic properties.130,131 Hence, engineering the band structure of a material proves to be an important tool in catalyst design.132 The electronic structures of low-dimensional nanostructures differ from their bulk counterparts due to the quantum confinement of the electronic density states. As a result, the photon absorption properties of these materials are enhanced, leading to better catalytic activities in these systems.133,134 The electronic properties are also dependent on the morphology of the materials, including kinks, edges, facets, and phases.9 Additionally, the surface area to volume ratio is higher in the low-dimensional catalysts, which ensures the availability of more active sites on the surface of the catalyst.9 The successful utilization of 0D, 1D, and 2D materials for catalysis has also prompted researchers to prepare hybrid structures using these nanomaterials and exploit them in catalysis. Moreover, the integration of two components provides synergistic effects from each counterpart toward catalysis. These individual components of heterostructures are held together mainly via van der Waals forces.9 Heterostructures have emerged as state-of-the-art catalysts as they offer superior-quality interfaces with unique properties compared to normal nanostructures. The newly formed interfaces between the components can lead to improved charge transfer, slowing down the recombination of charge carriers.126 These heterostructures can be 0D–1D, 0D–2D, 1D–2D, 2D–2D, and so on. In the next sections, we will discuss the 0D, 1D, and 2D materials, their heterostructures, and the interplay between the electronic properties and their catalytic activities.
4. Low-dimensional photocatalysts for ammonia synthesis
The design of photocatalysts for the reduction of N-containing species to ammonia requires a few criteria to be met: (i) adsorption of the N-containing species on the surface of the catalyst; (ii) absorption of light by the catalyst; (iii) tuning the band gap corresponding to the wavelength of light absorbed, which can promote the excitation of electrons from the valence band to the conduction band; (iv) position of the valence and conduction bands such that the potentials of the redox reactions are aligned in the gap; and (v) recombination of charge carriers is minimum. These requirements can be fulfilled by choosing the proper catalyst from a wide range of photocatalytic materials, such as semiconductors, metal nanoparticles, metal oxides, metal sulfides, and carbon-based materials. Structural engineering of the photocatalysts via the introduction of defects, construction of heterojunctions, increasing surface area by hybridization with other materials, and the use of cocatalysts can enhance photocatalytic performance.Photosynthesis of ammonia, in principle, requires only water, light, and N2 or NOx− hence, it is quite attractive. However, the photocatalysts developed earlier exhibit low conversion efficiency for ammonia generation. This might be due to the availability of less active sites, limited light absorption properties, and quick recombination of photogenerated charges. Active sites adsorb and activate the N-containing substrate. The activation of N2 is challenging due to the high dissociation energy of NN. Nitrogen has four bonding (2σ and 2π) and four anti-bonding (2σ* and 2π*) orbitals. Activation of N2 requires a donation of electrons to the bonding orbitals and an acceptance of electrons from anti-bonding π* orbitals. The thermodynamic reduction potential of N2 to NH3 is 0.148 V vs. RHE, which is quite close to the hydrogen evolution reaction (0 V vs. RHE), so the competing HER sometimes hinders the N2RR, and this remains a major concern in most cases.135 The drawbacks of the low solubility of N2 in an aqueous medium and the high dissociation energy required for the cleavage of NN can be overcome by replacing the N-containing precursor from N2 with nitrates or nitrites.21 Moreover, high concentrations of nitrates and nitrites in wastewater can be a major health concern; thus, reducing them to ammonia can diminish health risks. However, nitrate reduction to ammonia requires 8e−; hence, the risk of obtaining lesser-reduced products like N2 persists. For achieving higher NH3 selectivity, side reactions of N2 production and HER must be suppressed. This may be achieved by surface modification of catalysts or modification of materials based on the mechanistic pathway of ammonia synthesis.
The principle of photocatalysis relies on harvesting solar energy (Fig. 6). Hence, the band gap of the catalysts should also be tuned for direct utilization of solar light, preferably visible light. However, the band gap tuning sometimes reduces the energy of photogenerated electrons and, thereby, their reduction capability.136 Hence, developing an optimized photocatalyst that effectively absorbs light while maintaining its reduction capability is essential. For activation of N-containing molecules, the active sites must be electron-rich and able to promptly transfer electrons to adsorbed N-containing moieties. Inducing defects and unsaturated sites with abundant localized electrons in the catalysts can address this issue as they effectively transport electrons, and consequently, activate and reduce the N-containing moieties. The synthetic and modification strategies of LDMs are mostly simple and single-step, but structure engineering of the catalysts and the formation of heterostructures sometimes require multi-step fabrication strategies. 0D photocatalysts like metal and metal oxides mostly rely on different reduction methods of metallic salts, like photoreduction, H2 reduction, or chemical reduction, whereas single-atom-based photocatalysts use different deposition techniques for integration with support materials.137–141 Fabrication of 1D materials based on metal oxide, metal oxyhalides, and carbon-based materials mostly uses different thermal treatment procedures like hydrothermal, solvothermal, and annealing.142–144 2D materials based on semiconductor nanosheets, graphitic carbon nitrides, and graphdiynes rely on different thermal synthetic procedures.145–148 The catalytic properties of these materials can be enhanced by defect engineering like the introduction of vacancies,43 doping of heteroatoms,71 formation of heterojunctions,149 heterostructures,150 and use of cocatalyst materials.146 The fabrication strategies of the most widely used 0D, 1D, 2D, and heterostructured photocatalysts are presented in Table 3. This section will explore the emergent low-dimensional photocatalysts (Fig. 6) used recently to convert N2, NOx, or NOx− to NH3.
 | ||
| Fig. 6 Low-dimensional photocatalysts for ammonia synthesis. Classification of photocatalysts into 0D, 1D, and 2D, and schematic representations of few selected photocatalytic LDMs. | ||
| Classification | Materials | Photocatalysts | Synthesis/Modification | Ref. |
|---|---|---|---|---|
| 0D LDMs and heterostructures | Metallic nanoparticles (NPs) | AuNPs–TiO2 | In operando photodeposition | 137 |
| Pd/Cu/Ag–TiO2 | H2 reduction | 138 | ||
| CuPd–TiO2 | Chemical reduction | 139 | ||
| Ru–g-C3N4 | Green tea reduction | 151 | ||
| Ru–ZrO2−x | Impregnation method | 152 | ||
| Ru–K2Ta2O6−x | Thermal decomposition | 153 | ||
| Metal oxide nanocrystals (NCs) | MONCs–TiO2 (M = Mg, Ca, Sr, Ba) | In operando photodeposition | 154 | |
| Cu2ONCs–TiO2 | In operando photodeposition via pseudo-Fehling's reaction | 155 | ||
| Quantum dots (QDs) | TiO2QDs–Fe3S4 | Heterojunction formation | 149 | |
| Bi2Ti2O7 QDs | Defect engineering via hydrothermal method | 156 | ||
| InP QDs | Organometallic reactions, ligand exchange | 157 | ||
| Single-atoms (SAs) | Ru SA–HxMoO3−y | Defect engineering, H-spillover method | 43 | |
| Ru SA–CeO2 | Impregnation-calcination | 158 | ||
| Ru1 SA–d-UiO-66 | Defect engineering, photochemical deposition | 140 | ||
| Pt1 SA–BiOBr | Defect engineering, photochemical deposition | 141 | ||
| 1D LDMs and heterostructures | Carbonaceous materials | g-C3N4 | Defect engineering via molten-salt method | 107 |
| Ti3+–TiO2/HAp-C | Thermal annealing and reduction | 142 | ||
| Metal oxides | MoO3−x NWs | Defect engineering via hydrothermal method | 143 | |
| Cu2O NPs-W18O49 NWs | Defect engineering, in situ reduction, heterojunction formation | 150 | ||
| CdS NPs-WO3 NRs | Heterojunction formation | 159 | ||
| Metal oxyhalide | Bi5O7Br NTs | Defect engineering, thermal treatment | 144 | |
| Pyrochlore | Bi NPs–Bi2Sn2O7 NWs | Defect engineering, heterojunction formation, hydrothermal treatment | 160 | |
| 2D LDMs and heterostructures | Metal oxides | B-TiO2 NS | Hydrothermal treatment, B-doping | 145 |
| C-TiOx NS | Bottom-up substitutional C-doping | 71 | ||
| Ru NPs–W18O49 | Defect engineering via hydrothermal method, chemical reduction | 161 | ||
| Sb–MoO3−x NS | Defect engineering, Sb-doping, heat treatment | 162 | ||
| Metal oxyhalides | M–BiOBr NS (M = Fe, Mo, Ni) | Defect engineering, metal doping, hydrothermal treatment | 73 | |
| Bi NPs–BiOBr nanoplates | Schottky-junction formation, solvothermal reaction | 146 | ||
| Co–BiOCl nanoplatelets | Co-doping, hydrothermal treatment | 44 | ||
| Graphitic carbon nitrides | B-C3N4 NS | B-doping, thermal treatment | 77 | |
| Ni3B NPs-VN-C3N4 NS | Defect engineering, Schottky junction, chemical reduction, electrostatic self-assembly | 163 | ||
| B-C3N5 NS | Defect engineering, B-doping, thermal polymerization | 147 | ||
| Graphdiynes (GDYs) | GDY-Fe3O4 | Modified Glaser–Hay coupling, microwave-hydrothermal approach | 148 | |
| GDY-CoOx QD | In situ growth-deposition | 164 | ||
| Organic framework-based heterostructures | Porous-organic frameworks (POFs) | MPc-POF (M = Fe, Co, Zn) | Schiff base reaction, solvothermal reaction | 165 |
| Covalent-organic frameworks (COFs) | Porphyrin COF-Au SA | Thermal reactions | 166 | |
| DPPCOOH-COF | Schiff base condensation reaction | 167 | ||
| Bi/COF-TaTp | Solvothermal reactions | 168 | ||
| Metal-organic frameworks (MOFs) | MIL-100 (Fe) MOF | Defect engineering of ligands, nonthermal plasma-assisted synthesis | 169 | |
| Ca2+-d-UiO-66 MOF | Ca2+-doping, ligand incorporation, solvothermal reaction | 170 | ||
| MXene-MIL-125(Ti) MOF | In situ etching, delamination heterojunction formation, ligand pre-coupling | 171 | ||
| NH2-MIL-125 MOF-Co(OH)2-ZIF-8 | Heterojunction formation, nanoarchitectonics, solvothermal reactions | 172 |
4.1. 0D photocatalysts and heterostructures
0D photocatalysts for ammonia synthesis mainly consist of three classes of materials: semiconductor nanoparticles, quantum dots, single-atoms, and heterostructures formed with these 0D particles. Due to their plasmonic properties, 0D structures prove to be effective catalysts because the photons can be harvested at lower energy by nanoparticles, and their adsorption properties can be easily tuned by altering their shape and architecture.9 The separation of charge carriers is also possible because of the strong electric fields created at local “hotspots” induced by surface plasmons.173 These properties allow the 0D materials to be incorporated with 1D or 2D materials to create heterostructures that can function as efficient photocatalysts for ammonia synthesis.Metallic nanoparticles have plasmonic properties and their size and shape can be easily tuned, making them important materials for catalysis. Quantum dots also prove to be promising photocatalytic materials owing to their unique characteristics like high absorption coefficient, efficient charge transfer properties, large surface-to-volume ratio, and stability. Single-atoms are another class of materials that have recently emerged as prospective photocatalysts due to their highly efficient atom economy, low coordination environment, unique electronic structural properties, and atomic-level understanding of reaction mechanisms. Metallic nanoparticles, quantum dots, and single-atoms, upon heterogenization with different 1D and 2D support materials, can produce excellent photocatalysts for ammonia synthesis. For instance, the well-explored TiO2 nanosheets can be used as substrates for incorporating metallic nanoparticles, metal oxide nanoparticles, and single-atoms, which can simultaneously modify the interface as well as act as active sites for catalysis. The heterostructures formed are mostly of dimension 0D–2D. Heterostructures of semiconductors like TiO2 and CdS decorated with metallic nanoparticles act as efficient photocatalysts for ammonia generation from N-containing species like N2 and NOx.174–176 Ideally for NO reduction reactions, a high concentration (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) of NO is required for photoproduction of ammonia. However, limited NO conversion and ultra-low solubility of NO in water (1.94 ± 0.03 mmol L−1 at 25 °C) are the major bottlenecks for NO conversion to NH3.177 To address these challenges and for direct photocatalytic conversion of NO, Fe(II)EDTA is employed as the NO chemical absorbent, forming Fe(II)EDTA–NO chelates, and formaldehyde (HCHO) acts as the antioxidant to prevent the formation of Fe(III) from Fe(II) oxidation.137 TiO2 decorated with Au nanoparticles are adorned with active sites to facilitate photogenerated charge separation, thereby promoting ammonia generation. The simultaneous chemical adsorption and photocatalytic reduction system enable continuous NO adsorption, NO reduction, and Fe(II)EDTA regeneration on-site. Notably, the efficiency of NO conversion and the selectivity of ammonia produced remain unaffected even in the presence of H2O, SO2, and metal ions (K+, Ca2+, Cd2+, and Pb2+). In flue gas, the simultaneous presence of NO and SO2 presents certain challenges of removal and recovery. SO2 is soluble and easily oxidized, hence, SO2 acts as a potential reductant in the NO photoreduction reaction. The formation of the SO2–NO redox pair promotes easy conversion of both NO and SO2 in continuous flow.178 Thus, high selectivity is achieved simultaneously for both NO-to-NH3 upcycling (97%) and SO2-to-SO42− purification (92%). Metallic nanoparticles or bimetallic nanoalloys-loaded TiO2, such as Cu/TiO2 and Cu–Pd/TiO2, in the presence of hole scavengers like oxalic acid and methanol, can perform photocatalytic 8e− conversion of NO3− to NH3 without forming N2 while simultaneously producing H2, which lowers the selectivity of NH3.138,139 To address this problem, subnanometric metal oxide nanoparticles like BaO, CaO, and MgO are synthesized in operando on the TiO2 nanosheets.154In operando growth of 0D subnanometric metal oxide nanocrystals (MONC) at the oxygen defect sites of TiO2 nanosheets promotes NO3− reduction to synthesize NH3 selectively. The construction of MONC at defect sites is preferable because, due to the limited number of electron lone pairs, the nanocrystals cannot undergo further agglomeration. These MONC@OVs act as active sites for the 8e− photoreduction of NO3− to NH3. Water splitting, the primary side reaction, produces trace amounts of H2. However, theoretical calculations indicate that the activation energy of NO3− reduction is 1.42 eV less than water splitting, favouring NO3− reduction over the side reaction of water splitting. Compared with the HER, the selectivity of NH4+ has been reported to be 97.67%. The formation of a unique interface between metal oxides and TiO2 promotes charge separation as well as charge transfer properties of the hybrid catalyst. The light absorption property of the catalyst is improved and the conduction band is also elevated, enhancing the photocatalytic performance of NO3− reduction. A similar work along these lines features the in situ formation of dynamic Cu2O sub-nanoclusters on TiO2 nanosheets (TNS) as the ammonia-producing photocatalyst.155 This work introduces a different approach to ammonia formation, utilizing dynamic Cu2O nanoclusters following pseudo-Fehling's reaction. The structure of Cu2O is unstable under photocatalytic working conditions. Hence, dynamic reconstruction of active Cu2O sites is required to perform the photocatalytic reactions using actual Cu2O. The novel active Cu2O sub-nanoclusters are constructed on-site utilizing photoinduced pseudo-Fehling's reaction in a photocatalytic system containing redox pairs of Cu2+ and reducing sugars (formaldehyde and formic acid). The photogenerated electrons (e−) and holes (h+) participate in the Cu2+ to Cu2O reduction and HCHO to HCOOH oxidation for the formation of Cu2O NCs and construction of Cu2O–TNS interface, respectively. The value-added oxidation of HCHO to HCOOH is accompanied by synchronous reduction of different N-sources (NO3−, NO2−, and NO) to value-added NH3 by the active dynamic Cu2O sites. This strategy ensures the formation of realistic dynamic active sites and provides a better understanding of the reaction mechanisms underlying the photocatalytic reactions. The photoactivity of TiO2 semiconductor nanoparticles can also be enhanced by doping with heteroatoms. Co-doping of TiO2 with synergistic transition metals Ce3+/4+-cation and S2−-anion adjusts the band gap of TiO2 such that maximum photoactivity is obtained upon visible light irradiation.179 The ratio of Ce3+/Ce4+ is adjusted with hydrazine to form oxygen vacancies (VO), which helps maintain the charge and lattice electroneutrality of the TiCeOS catalyst. The Ce3+ centers act as the active sites for the adsorption and activation of N2 while the electron-hopping between heterovalent Ce3+/Ce4+ promotes electron transfer for the photoreduction of N2. Simultaneously, the VOs act as the active sites for trapping water molecules and subsequent proton generation from water to protonate N2 to NH3. The introduction of metallic nanoparticles like Ru can tune the band gap of the photocatalytic materials, boost electron transfer, and facilitate charge separation. Ru-modified g-C3N4 in the presence of visible light can selectively photoreduce NO3− to NH3.151 The theoretically calculated activation energy for the rate-determining step of NH3 synthesis is 0.75 eV, which is much less when compared to the activation energies of competing HER (0.98 eV) and N2 synthesis (1.36 eV), making the generation of NH3 more selective. The Ru sites have more Bader charge; hence, the density of electrons is higher on the Ru atoms, making them the active sites for photocatalysis. Ru nanoparticles can also act as cocatalysts for ammonia production when loaded on defective semiconductor-based ZrO2−x nanoparticles with oxygen vacancies (VO).152 The defective ZrO2−x nanoparticles have a narrow band gap, and excellent reducing and electron donation properties, which makes it an outstanding photocatalyst for ammonia generation. The VOs stabilize the dispersed Ru nanoparticles, which act as cocatalysts and pose an upward band bending of ZrO2−x, inducing an interfacial Schottky barrier that promotes the separation of photogenerated charge carriers. The Schottky barrier at the Ru and ZrO2−x interface also provides a unidirectional pathway for photogenerated electron transport. The Ru nanoparticles trap these electrons, thereby ensuring the supply of requisite electrons for N2 reduction to NH3. Ru nanoparticles loaded on defective perovskite and pyrochlore structures can also promote N2 photoreduction to NH3.153 In this work, different perovskite- and pyrochlore-structured
tantalates with low-valent Ta and abundant oxygen vacancies (VO) were fabricated by high-temperature solid-state reduction. These visible light active dark tantalates have narrow band gaps and upon the introduction of Ru nanoparticles, band bending occurs to construct an interfacial Schottky barrier. The Schottky barrier promotes adsorption of N2 molecules and electron transfer to reduce N2 to NH3. Among all the synthesized tantalates, Ru-loaded defective pyrochlore K2Ta2O6−x has the highest electron-donation properties and chemical stability, and therefore is most effective for ammonia photosynthesis in the gas–solid phase at low pressure.
000 ppm) of NO is required for photoproduction of ammonia. However, limited NO conversion and ultra-low solubility of NO in water (1.94 ± 0.03 mmol L−1 at 25 °C) are the major bottlenecks for NO conversion to NH3.177 To address these challenges and for direct photocatalytic conversion of NO, Fe(II)EDTA is employed as the NO chemical absorbent, forming Fe(II)EDTA–NO chelates, and formaldehyde (HCHO) acts as the antioxidant to prevent the formation of Fe(III) from Fe(II) oxidation.137 TiO2 decorated with Au nanoparticles are adorned with active sites to facilitate photogenerated charge separation, thereby promoting ammonia generation. The simultaneous chemical adsorption and photocatalytic reduction system enable continuous NO adsorption, NO reduction, and Fe(II)EDTA regeneration on-site. Notably, the efficiency of NO conversion and the selectivity of ammonia produced remain unaffected even in the presence of H2O, SO2, and metal ions (K+, Ca2+, Cd2+, and Pb2+). In flue gas, the simultaneous presence of NO and SO2 presents certain challenges of removal and recovery. SO2 is soluble and easily oxidized, hence, SO2 acts as a potential reductant in the NO photoreduction reaction. The formation of the SO2–NO redox pair promotes easy conversion of both NO and SO2 in continuous flow.178 Thus, high selectivity is achieved simultaneously for both NO-to-NH3 upcycling (97%) and SO2-to-SO42− purification (92%). Metallic nanoparticles or bimetallic nanoalloys-loaded TiO2, such as Cu/TiO2 and Cu–Pd/TiO2, in the presence of hole scavengers like oxalic acid and methanol, can perform photocatalytic 8e− conversion of NO3− to NH3 without forming N2 while simultaneously producing H2, which lowers the selectivity of NH3.138,139 To address this problem, subnanometric metal oxide nanoparticles like BaO, CaO, and MgO are synthesized in operando on the TiO2 nanosheets.154In operando growth of 0D subnanometric metal oxide nanocrystals (MONC) at the oxygen defect sites of TiO2 nanosheets promotes NO3− reduction to synthesize NH3 selectively. The construction of MONC at defect sites is preferable because, due to the limited number of electron lone pairs, the nanocrystals cannot undergo further agglomeration. These MONC@OVs act as active sites for the 8e− photoreduction of NO3− to NH3. Water splitting, the primary side reaction, produces trace amounts of H2. However, theoretical calculations indicate that the activation energy of NO3− reduction is 1.42 eV less than water splitting, favouring NO3− reduction over the side reaction of water splitting. Compared with the HER, the selectivity of NH4+ has been reported to be 97.67%. The formation of a unique interface between metal oxides and TiO2 promotes charge separation as well as charge transfer properties of the hybrid catalyst. The light absorption property of the catalyst is improved and the conduction band is also elevated, enhancing the photocatalytic performance of NO3− reduction. A similar work along these lines features the in situ formation of dynamic Cu2O sub-nanoclusters on TiO2 nanosheets (TNS) as the ammonia-producing photocatalyst.155 This work introduces a different approach to ammonia formation, utilizing dynamic Cu2O nanoclusters following pseudo-Fehling's reaction. The structure of Cu2O is unstable under photocatalytic working conditions. Hence, dynamic reconstruction of active Cu2O sites is required to perform the photocatalytic reactions using actual Cu2O. The novel active Cu2O sub-nanoclusters are constructed on-site utilizing photoinduced pseudo-Fehling's reaction in a photocatalytic system containing redox pairs of Cu2+ and reducing sugars (formaldehyde and formic acid). The photogenerated electrons (e−) and holes (h+) participate in the Cu2+ to Cu2O reduction and HCHO to HCOOH oxidation for the formation of Cu2O NCs and construction of Cu2O–TNS interface, respectively. The value-added oxidation of HCHO to HCOOH is accompanied by synchronous reduction of different N-sources (NO3−, NO2−, and NO) to value-added NH3 by the active dynamic Cu2O sites. This strategy ensures the formation of realistic dynamic active sites and provides a better understanding of the reaction mechanisms underlying the photocatalytic reactions. The photoactivity of TiO2 semiconductor nanoparticles can also be enhanced by doping with heteroatoms. Co-doping of TiO2 with synergistic transition metals Ce3+/4+-cation and S2−-anion adjusts the band gap of TiO2 such that maximum photoactivity is obtained upon visible light irradiation.179 The ratio of Ce3+/Ce4+ is adjusted with hydrazine to form oxygen vacancies (VO), which helps maintain the charge and lattice electroneutrality of the TiCeOS catalyst. The Ce3+ centers act as the active sites for the adsorption and activation of N2 while the electron-hopping between heterovalent Ce3+/Ce4+ promotes electron transfer for the photoreduction of N2. Simultaneously, the VOs act as the active sites for trapping water molecules and subsequent proton generation from water to protonate N2 to NH3. The introduction of metallic nanoparticles like Ru can tune the band gap of the photocatalytic materials, boost electron transfer, and facilitate charge separation. Ru-modified g-C3N4 in the presence of visible light can selectively photoreduce NO3− to NH3.151 The theoretically calculated activation energy for the rate-determining step of NH3 synthesis is 0.75 eV, which is much less when compared to the activation energies of competing HER (0.98 eV) and N2 synthesis (1.36 eV), making the generation of NH3 more selective. The Ru sites have more Bader charge; hence, the density of electrons is higher on the Ru atoms, making them the active sites for photocatalysis. Ru nanoparticles can also act as cocatalysts for ammonia production when loaded on defective semiconductor-based ZrO2−x nanoparticles with oxygen vacancies (VO).152 The defective ZrO2−x nanoparticles have a narrow band gap, and excellent reducing and electron donation properties, which makes it an outstanding photocatalyst for ammonia generation. The VOs stabilize the dispersed Ru nanoparticles, which act as cocatalysts and pose an upward band bending of ZrO2−x, inducing an interfacial Schottky barrier that promotes the separation of photogenerated charge carriers. The Schottky barrier at the Ru and ZrO2−x interface also provides a unidirectional pathway for photogenerated electron transport. The Ru nanoparticles trap these electrons, thereby ensuring the supply of requisite electrons for N2 reduction to NH3. Ru nanoparticles loaded on defective perovskite and pyrochlore structures can also promote N2 photoreduction to NH3.153 In this work, different perovskite- and pyrochlore-structured
tantalates with low-valent Ta and abundant oxygen vacancies (VO) were fabricated by high-temperature solid-state reduction. These visible light active dark tantalates have narrow band gaps and upon the introduction of Ru nanoparticles, band bending occurs to construct an interfacial Schottky barrier. The Schottky barrier promotes adsorption of N2 molecules and electron transfer to reduce N2 to NH3. Among all the synthesized tantalates, Ru-loaded defective pyrochlore K2Ta2O6−x has the highest electron-donation properties and chemical stability, and therefore is most effective for ammonia photosynthesis in the gas–solid phase at low pressure.
0D quantum dots (QDs) also prove to be efficient photocatalysts for ammonia production. TiO2 nanoparticles are one the most widely used semiconductors for photocatalytic applications. Controlling the size of the nanoparticles and reducing them to below 10 nm creates TiO2 QDs with outstanding photocatalytic properties owing to enhanced charge separation, modified textural properties, and altered redox potentials.149 Upon the formation of an S-scheme heterojunction of TiO2 with photoactive Fe3S4 crystalline spinels, the photofixation of N2 to produce NH3 is boosted in the presence of simulated sunlight. This enhanced photocatalytic activity is attributed to the heterojunction formation, which leads to more visible light absorption, accelerated photoinduced charge separation, and enhanced catalyst surface area. Formation of an S-scheme heterojunction between 0D g-C3N4 QDs and 3D macro- and mesoporous TiO2−x with oxygen vacancies (VO) and enhanced charge transfer properties promotes the photo/electrocatalytic reduction of NO to NH3.180 The photogenerated charge carriers are migrated by the S-scheme heterojunction formed in the g-C3N4 QDs/3D-TiO2−x and the accumulated electrons in the conduction band of g-C3N4 quantum dots reduce NO to NH3. Pyrochlore Bi2Ti2O7 QDs with oxygen vacancies (VOs), produced hydrothermally from bismuth nitrate and titanium sulfate also produce NH3 from N2 upon photoirradiation.156 When ammonia production is compared with Bi2Ti2O7 nanosheets, the QDs are found to be more photoactive. Despite possessing the same amount of VOs, the QDs are found to be more photoactive as the synergistic roles of shallow levels arising from VO of Bi2Ti2O7 QDs and the quantum confinement effect promote adsorption and activation of N2 molecules to produce NH3. Ammonia production is also possible from nitrate (NO3−) and nitrite (NO2−) anions using visible-light-active indium phosphide (InP) quantum dots.157 InP QDs have tuneable absorption properties, high charge separation abilities, efficient mobility of charge carriers, and flexibility in modifying the surface chemistry. The photoexcited charge carriers generated by the III–V InP QDs directly induce NO3− reduction to produce NH3. The kinetic experiments from this study confirm that the reduction of NO3− to NO2− is the most energy-demanding rate-determining step in the conversion of NO3− to NH3. The conversion of NO2− to NH3 is faster and almost 100% conversion is achieved in this step. Additionally, in this study, water is used as the source of protons for ammonia production.
Single-atom (SA)-based catalysts are one of the emergent classes of materials currently undergoing widespread investigation. Compared to nanoparticles, single-atoms provide access to a higher density of active sites and contribute to better atomic usage, which in turn escalates the catalytic process.27,43 The high catalytic activity, selectivity, and stability of SA catalysts can also be attributed to homogeneity and the low coordination capability of the single-atoms.181 The Ru SAs can tune the electronic structure of the oxygen vacancies present in TiO2 and improve the adsorption of N2 for photocatalytic N2RR. The synergistic effect of the two active sites, Ru SA and oxygen vacancies (VOs) powers the N2RR, demonstrating that both components of the heterostructures participate in the catalytic reaction. For Ru SAs on CeO2 support, CeO2 generates electron–hole pairs upon photoirradiation and the Ru sites pull the electrons toward them, accumulating photogenerated electrons around the Ru sites (Fig. 7A–E).158 This further modulates the local electron density of the adsorbed N2 molecules on Ru sites, lowers the energy barrier of the rate-limiting step, and, thereby, promotes the hydrogenation of adsorbed N2via the associative distal pathway. Ru SAs have also been implanted on other supports like molybdenum oxides and metal–organic frameworks to photocatalytically synthesize ammonia by N2RR. Here, the active single Ru site anchored on the UiO-66 nodes participates in producing ammonia (Fig. 7F–I).140 Ru SAs can be embedded in TiO2 by electronic metal–support interactions, and this catalytic system eradicates the use of external sacrificial agents for producing ammonia (Fig. 7J–M).182 Pt SAs with tuneable oxidation states photodeposited on a BiOBr support with oxygen vacancies (VOs) can also efficiently produce ammonia upon photoirradiation.141 The electron–metal support interactions (EMSI) between the Pt and BiOBr and the tuneable oxidation state of Pt SA promote charge transfer between the modifiers (VO and Pt SA) and the BiOBr support. The accelerated electron transfer and variation of the local electronic structure of VO by Pt SA leads to selective adsorption and activation of N2 as well as the reduction of the energy barrier of the rate-limiting step, and promote hydrogenation of *N2 intermediate to produce NH3via a multielectron alternating reduction pathway. It is to be noted that though most of the reported SA photocatalysts use noble metals, the metal utilization efficiency of single-atoms in catalysis lowers the cost of noble atoms. However, efforts to prepare a cost-effective, metal-free SA, like a boron-based SA photocatalyst are underway.183 Theoretical studies on single-atom B on graphitic carbon nitride support demonstrate that it can efficiently reduce dinitrogen to selectively produce ammonia-suppressing competitive HER via the “σ donation–π* back-donation” properties of the designed catalyst. The activation barrier and overpotential of dinitrogen reduction are notably less for this metal-free SA catalyst when compared with most existing metallic catalysts.
 | ||
| Fig. 7 Zero-dimensional (0D) photocatalysts for ammonia synthesis. (A) Schematic representation of the isolated 0D Ru sites anchored on CeO2. (B) HAADF-STEM image of Ru–CeO2. The inset features the corresponding simulated STEM image. (C) UV-Vis DRS of CeO2 and Ru–CeO2. (D) PL spectra of Ru–CeO2 with different Ru loadings. With an increase in Ru loading the PL intensity decreases. (E) NH3 yield comparison for CeO2 and Ru–CeO2 catalysts with different Ru loadings. The maximum yield obtained is 18 mmol gcat−1 h−1 for Ru/CeO2-3 where the Ru loading is 1.87%. Reproduced with permission from ref. 158. Copyright 2024, Wiley-VCH. (F) Schematic representation of the 0D Ru single-atom in the Ru1/d-UiO-66 catalyst. (G) Aberration-corrected HAADF-STEM image of the catalyst. (H) UV-Vis DRS of UiO-66, d-UiO-66, and Ru1/d-UiO-66. (I) NH3 yield comparison for UiO-66, d-UiO-66, and Ru1/d-UiO-66 catalysts, and control experiments conducted in the absence of light, N2, and water. Reproduced with permission from ref. 140. Copyright 2023, Wiley-VCH. (J) Schematic representation of the Ru single-atom-bonded TiO2, Ru1/TiO2-VO, prepared by the molten salt method. (K) HADDF-STEM image of Ru1/TiO2-VO, where the Ru single atoms are marked by white circles. (L) UV-Vis DRS of TiO2, TiO2-VO, and Ru1/TiO2-VO. (M) NH3 yield comparison for TiO2, TiO2-VO, and Ru1/TiO2-VO, and control experiments conducted in the absence of light, N2, catalysts, and water. Reproduced with permission from ref. 182. Copyright 2023, Elsevier. | ||
4.2. 1D photocatalysts and heterostructures
1D materials, being microscale along one dimension, provide more surface area and easily accessible catalytic active sites than 0D materials, which enhances their catalytic performance. Various 1D carbonaceous materials have been investigated over the past few years because of their stability, good conductivity, and tuneable electronic structure, which exhibit remarkable catalytic activity.184 The porous structure of the carbonaceous materials also facilitates the adsorption of N-containing species, which enhances the catalytic reaction rate. The onset potential for HER is comparatively more on carbon-based catalysts, which is beneficial for N2RR and NOxRR. The introduction of heteroatoms like B, N, and S, which have different electronegativity than C, results in charge distribution in the structure and induces more active sites in the material for the adsorption of reactants. Graphitic carbon nitrides, g-C3N4, generally form layered two-dimensional structures that can transfer electrons in two dimensions. However, orderly 1D nanostructures of g-C3N4 can constrain charge transfer in one dimension, thus reducing the charge recombination. The introduction of defects, i.e., metal dopant K and –CN, into the g-C3N4 nanorods further enhances their catalytic activities (Fig. 8A–D).107 From theoretical studies, it has been observed that the active site is –CN, which donates electrons while K centres trap electrons. The synergistic effect of dual defects also promotes light absorption, charge separation, and proton adsorption, and enhances the photocatalytic N2RR. Carbon-coated hydroxyapatite (Hap-C) nanorod, another carbon-based material derived from bones, exhibits considerable photoluminescence under ultraviolet light.142 Hydrothermal deposition of Ti3+–TiO2 on the surface of Hap-C promotes the absorption of visible light, increases electron transfer, and reduces agglomeration. Due to these characteristics, the Ti3+-TiO2/HAp-C nanorods can accelerate photocatalytic N2/H2O ammonia synthesis compared to pristine Ti3+-TiO2 and HAp-C nanorods separately.
 | ||
| Fig. 8 One-dimensional (1D) photocatalysts for ammonia synthesis. (A) Schematic representation of the structure of graphitic carbon nitride (CN) and graphitic carbon nitride nanorods (CNNR). (B) TEM image of CNRR. (C) UV-Vis DRS of CN and CNRR. (D) NH3 yield comparison for different carbon nitride photocatalysts. Reproduced with permission from ref. 107. Copyright 2022, Elsevier. (E) Schematic representation of NH3 synthesis by Bi5O7Br nanotubes. (F) TEM image of Bi5O7Br-40 nanotubes prepared at 40 °C. (G) UV-Vis DRS of different Bi5O7Br prepared at various temperatures. (H) Comparison of NH3 evolved by using different Bi5O7Br catalysts prepared at various temperatures. Reproduced with permission from ref. 144. Copyright 2020, American Chemical Society. (I) Schematic representation of the CdS-decorated WO3 nanorods, forming a heterojunction. (J) FE-SEM image of WO3/CdS nanorods. (K) Schematic representation of the electron transfer process at the WO3/CdS heterojunction for photocatalytic “overall N2 fixation”. (L) NH3 yield comparison between CdS, WO3, and WO3/CdS catalysts. Reproduced with permission from ref. 159. Copyright 2022, Wiley-VCH. | ||
Oxygen vacancies, the most frequently formed defect, can act as active sites for the adsorption and activation of precursor molecules, and can also alter the optical and electronic properties of photocatalysts. In the case of photocatalysts like Bi5O7Br nanotubes with oxygen vacancies (VO), the adsorption of N2 can elongate the NN bond and form VO–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, which consequently helps in the activation of N2 (Fig. 8E–H).144 The concentration of VO has to be optimum because a too high amount of VO can entrap the charge carriers to such an extent that it may reduce the rate of photocatalysis. Asymmetric defects induced by VOs can also enhance the segregation of photogenerated electron–hole pairs through charge redistribution and thereby enhance photocatalytic N2 reduction reactions. One such example of asymmetric defects is demonstrated by the MoO3−x nanometric wires fabricated by the hydrothermal process using glycine for inducing defects.143 Mo-based catalysts are quite popular N2 fixation catalysts as the unoccupied d-orbitals of Mo present a strong affinity towards N2. The defects infused by the VOs in the structure of MoO3−x result in charge redistribution and promote N2 adsorption and activation.
N, which consequently helps in the activation of N2 (Fig. 8E–H).144 The concentration of VO has to be optimum because a too high amount of VO can entrap the charge carriers to such an extent that it may reduce the rate of photocatalysis. Asymmetric defects induced by VOs can also enhance the segregation of photogenerated electron–hole pairs through charge redistribution and thereby enhance photocatalytic N2 reduction reactions. One such example of asymmetric defects is demonstrated by the MoO3−x nanometric wires fabricated by the hydrothermal process using glycine for inducing defects.143 Mo-based catalysts are quite popular N2 fixation catalysts as the unoccupied d-orbitals of Mo present a strong affinity towards N2. The defects infused by the VOs in the structure of MoO3−x result in charge redistribution and promote N2 adsorption and activation.
Heterojunction engineering and heteroatom doping are two more approaches besides defects engineering to enhance photocatalytic ammonia synthesis. One such example of a catalyst that has been fabricated using all three approaches is Cu ion-doped W18O49 ultrathin nanowires (Cu-W18O49-x UTNW).150In situ reduction of the Cu-W18O49-x UTNW with ascorbic acid as an antioxidant and NaBH4 as a reducing agent form Cu2O-W18O49-x UTNM, the actual photocatalyst for N2 reduction. This catalyst is adorned with oxygen vacancies (VO) which promote N2 adsorption and activation, and facilitate NN bond dissociation. The in situ-generated Cu2O forms an S-scheme heterojunction with W18O49-x UTNW, which modifies the internal electric field and enhances the separation and transfer of the photogenerated carriers. All these strategies developed for fabricating the nanowires result in efficient photofixation of N2 to form NH3. The formation of a heterojunction between two materials induces a strong electronic coupling that promotes interfacial charge transfer and, at the same time, inhibits the recombination of electron–hole pairs. One such recent work reported the construction of a heterojunction between two redox semiconductors, CdS nanoparticles and WO3 nanorods, forming a 0D–1D-type heterostructure (Fig. 8I–L).159 The reductive-type CdS has a negative conduction band (CB) position and the electrons in CBCdS have a strong reduction ability. The oxidative component WO3 has a higher positive valence band (VB) position and the holes in VBWO3 have a strong oxidation ability. The heterojunction builds an interface and an electric field between CdS and WO3 components. Upon photoexcitation, this built-in electric field promotes the separation of electrons and holes, and migrates them via interfacial charge transfer to CdS and WO3, respectively. These electrons with strong photoreducing ability can efficiently convert N2 to NH3, and the holes can simultaneously oxidize N2 to NO3− under mild conditions, thereby completing the “overall nitrogen fixation” reactions. Another example of such heterojunction formation is Bi/Bi2Sn2O7 nanocomposites where etching of oxygen vacancies (VO) in the Bi–O bonds present in Bi2Sn2O7 pyrochlore is utilized for the in situ preparation of metallic Bi.160 The Schottky junctions formed at the interface of metallic Bi and semiconducting Bi2Sn2O7 lead to the separation of photogenerated charge carriers. Moreover, the contact between the semiconducting Bi2Sn2O7 and metallic Bi facilitates directional electron transfer from Bi2Sn2O7 to Bi, enriching the concentration of photogenerated electrons at the active sites of metallic Bi. These electrons can effectively reduce N2 to form NH3. These results highlight the significance of engineering one-dimensional heterostructured catalysts for photocatalytic production of ammonia.
4.3. 2D photocatalysts and heterostructures
Due to their light-harvesting properties, semiconductors play a major role in the photocatalytic synthesis of ammonia. When a semiconductor absorbs a photon with energy equal to or greater than its band gap, electrons are excited from the valence band to the conduction band, creating holes in the valence band. These electrons and holes can migrate to the surface of the semiconductor and initiate the corresponding reduction and oxidation reactions. But sometimes, the photogenerated electrons and holes can recombine either on the surface or in bulk, which in turn hampers the photocatalytic redox reactions. The recombination of charge carriers can be slowed down by introducing defects in the lattice or by forming heterostructures with other low-dimensional materials. TiO2, ZnO, CdS, and CdSe are some common semiconductors, but the most widely used semiconductor in photocatalysis is TiO2. TiO2 is cheap, stable, non-toxic, and has band positions aligned to allow water oxidation and reduction reactions to occur simultaneously.185 The semiconductors can be 0D, 1D, or 2D. Among these, 2D semiconductor nanosheets have more surface area and active sites, offering better chances of doping and making them potentially more active than 0D or 1D structures. In TiO2, the most widely explored semiconductor photocatalyst, the oxygen vacancies (VO) have a potential role in regulating its photocatalytic properties. Rutile TiO2(110) structure is composed of alternating rows of Ti4+ and bridging O (Ob) atoms.70 These Ob atoms form the vacant sites and the two excess electrons are transferred to each of the 3d orbitals of the two neighbouring Ti4+, which reduces Ti4+ to Ti3+. The position of the donor level of Ti3+ lies below the conduction band of TiO2. This donor level traps electrons from the conduction band and reduces N2 to NH3, confirming Ti3+ to be the active site for photocatalysis. Hirakawa and co-workers have identified the active sites of TiO2 for the reduction of NO3− to N2 and NH3, respectively.186 They deduced that the Lewis acid sites (Ti4+) of TiO2 can perform non-selective reduction of NO3− to N2 and NH3. The surface defects formed by oxygen vacancies (VOs) can donate electrons to Ti4+ and reduce them to Ti3+, and these Ti3+ sites can selectively reduce NO3− to NH3. So, by maintaining the ratio of surface defects and Lewis acid sites, a higher selectivity of NH3 (97%) can be achieved. The high energy level of LUMO π* makes the donation of electrons to π* orbitals more energy-demanding. The rate-limiting step is the reduction of NO3− to NO2−. Liberation of this NO2− intermediate from Lewis acid sites of TiO2 is mainly responsible for N2 generation. The surface defects of TiO2 can entrap intermediates like NO2− and NO and suppress the formation of N2. Heteroatom doping is another effective strategy for increasing active sites for adsorption and activation of N2. In a typical case, anatase TiO2 doped with B proves to be an excellent visible-light-driven catalyst for ammonia synthesis with a band gap of 1.92 eV and a thermodynamic barrier of 0.44 eV.145 The B non-metal doping narrows the band gap of TiO2 and makes it photoactive in the visible region. The advantages of using B as a dopant are manifold: (i) B forms interband states near the conduction band of TiO2 and reduces the recombination of charge carriers; (ii) the hybrid orbitals of B can form π-back bonding with N to activate N2; (iii) B atoms prohibit binding with H+ and thereby suppress the competing HER; and (iv) B atoms, along with the transition metal, act as active sites for N2 reduction. In another work, C-doping by bottom-up approach from Ti3SiC2 MAX precursor forms two-dimensional C-doped TiOx nanosheets (Fig. 9A–D).71 These C-TiOx nanosheets show enhanced photocatalytic activity under visible light irradiation. C-doping increases the number of Ti3+ centres in the catalyst, induces photoactivity in visible light, and breaks NN, which is the rate-limiting step. Ti3+ centres act as the active sites and the two adjacent Ti3+ centres chemisorb and activate N2 molecules. The optimal ratio of Ti3+/Ti4+ in this TiOx nanosheet is maintained at 72.1% and, with the addition of Ru/RuO2 co-catalyst, charge recombination is reduced. Apart from TiO2, tungsten oxide semiconductors are also well-known photocatalysts. Oxygen vacancy (VO)-rich W18O49 semiconductors with sea-urchin morphology, after low-content Ru modification by NaBH4 reduction get transformed into rough 2D sheets.161 This sheet-like morphology exposes more active sites and facilitates the adsorption of N2 molecules. The VOs enhance the interfacial hydrogen spillover process, thus H* generated from water as the proton source is utilized in the hydrogenation of N2 to NH3. Ru captures electrons from W18O49 and acts as the active site for H2O dissociation. The VOs play multiple roles, such as facilitating H* migration from Ru for efficient hydrogen spillover, promoting the adsorption of N2, acting as active sites for hydrogenation reaction, and lowering the overall energy barrier for NH3 photosynthesis reaction. Plasmonic semiconductors also act as effective photocatalysts for ammonia production. The localized surface plasmon resonance (LSPR) promotes photoconversion of N2 and the surface oxygen vacancies (VO) present in these catalysts adsorb and activate the N2 molecules. However, the VOs can sometimes get oxidized by the photogenerated holes which might diminish the LSPR properties of the catalyst upon heat treatment. To solve this problem, in one of the recent works, plasmonic MoO3−x nanosheets with Sb doping have been developed.162 Though Sb doping does not effectively increase the concentration of VO, the low oxidation state of Sb can help stabilize the LSPR effect of the plasmonic MoO3−x. The uncoupled electrons present in the d orbitals of low-valent Sb help maintain the LSPR in the NIR region, thus producing NH3 from photocatalytic N2 fixation. Bismuth-based semiconductors like bismuth oxyhalides and bismuth-based binary metal oxides are effective photocatalysts in the visible light range and have high chemical stability. However, their weak interactions with N2 limit their use as catalysts. This drawback can be overcome with heteroatom doping, vacancy formation, formation of heterostructures, or modification of exposed crystal facets. Chen and co-workers modified BiOBr nanosheets with transition metals like Ni, Fe, and Mo by hydrothermal method and introduced oxygen vacancies (VOs) via the solvothermal method (Fig. 9E–H).73 The transition metals and VOs tune the band gap of BiOBr and enhance electron transfer to the anti-bonding orbital of N2, thereby triggering adsorption and activation of N2. The simultaneous presence of VOs and transition metals alters the band positions, diminishing the recombination of charge carriers. The modified BiOBr enhances the photoreduction of N2 by six times compared to normal BiOBr, forming 46.1 μmol g−1 h−1 of NH3. Using cocatalysts is another intriguing technique to increase the efficiency of photocatalytic ammonia synthesis. Cocatalysts are generally photocatalytic metallic particles or semiconductors and are used to promote electron transfer processes, inhibit the recombination of charge carriers, and switch to a preferable range of incident light for photocatalysis. In a typical BiOBr semiconductor photocatalyst, the efficacy of solar light-powered N2RR is enhanced 65 times by using Bi nanoparticles as a cocatalyst.146 Bi NPs lower the rate of competing HER and construct a Schottky junction at the Bi/BiOBr interface, which facilitates the interfacial transfer of electrons. The unidirectional transfer of electrons toward the Bi active sites accelerates the solar light N2 conversion efficiency to ammonia. Instead of converting highly pure, air-separated, and expensive N2, aerobic N2 reduction of air (N2/O2) can be used to produce NH3 cost-effectively. The addition of water oxidation cocatalysts like CoOOH with BiOCl nanosheets can also enhance photocatalytic NH3 formation.44 The cocatalyst facilitates the OER half-reaction and, consequently, boosts the N2RR to NH3. Solvothermally prepared trace Bi0-loaded Bi2MO6 (M = W, Mo) can prepare ammonia via aerobic photocatalytic reduction of N2.187 The role of a trace amount of Bi0 is crucial in this reaction. The formation of excess Bi0 is inhibited by the presence of O2 in the reaction medium. The polarization dipole field produced by Bi0 favours an effective separation of photogenerated charge carriers and promotes ammonia synthesis from the photocatalytic reduction of N2. Though the process is an inexpensive route for ammonia production, it has limitations because the O2 present in the reaction medium might react with photogenerated e−s and h+s to create reactive oxygen species like O2˙−, ˙OH, which can oxidize the synthesized ammonia or react with the catalyst itself. Therefore, the aerobic reduction of N2 needs further optimization before it can be effectively used for ammonia synthesis. The problems of aerobic reduction and the expensive nature of pure N2 can be circumvented by using nitrates as the N-source, often present in wastewater systems. In one of the recent works by our group, a BiOI-based “AmmoGen” microrobot has been prepared for the photocatalytic synthesis of NH3 from NO3− upon visible light irradiation.188 The efficiency of the conventional photo/chemical synthetic techniques for ammonia production is limited by intensive mass-transfer processes. To address this challenge, our group has envisioned a novel technology for ammonia generation, where the photocatalytic BiOI particles are hybridized with magnetic Fe3O4 nanoparticles to fabricate an “AmmoGen” microrobot, that can photoreduce nitrate to ammonia using renewable light energy sources. Experiments with “static” particles and “dynamic” microrobots demonstrate that the magnetic propulsion of the “AmmoGen” microrobots significantly improves the mass transfer process, and enhances the photocatalytic ammonia production. This work on microrobots, in principle, can help improve photocatalytic reactions and be utilized in the future for value-added small molecule synthesis. Piezoelectric materials like Bi3TiNbO9189 and BaTiO3@C190 have also emerged recently as efficient photocatalysts for CO2 reduction and organic pollutants degradation. In piezoelectric materials, a polarization-induced electric field is formed by the displacement of the positive and negative charge centres onto opposite sides and the resultant internal electric field (IEF) reduces the recombination of photogenerated electron–hole pairs, enhancing the photocatalytic activity of these materials. Layered bismuth-based piezoelectric SrBi4Ti4O15 nanosheets with oxygen vacancies (VO), formed by low-temperature hydrothermal treatment with glyoxal can effectively photoreduce N2 to produce NH3.191 These nanosheets manifest self-polarization, originating from the [TiO6] octahedral distortion, with the polarization direction parallel to the [Bi2O2]2+ layer. This polarization field and VO synergistically promote N2 adsorption, activation, and reduction to NH3via three electron-transfer pathways. TiO2-decorated layered silicate magadiite piezo-photocatalytic nanosheets also exhibit significant enhancement in NH3 production compared to TiO2 or silicate magadiite separately.192 The TiO2 nanoparticles deposited on silicate magadiite enhance the piezoelectric potential and the polarization-induced internal electric field increases the lifetime of the photogenerated charge carriers in these nanosheets, and the synergistic piezo and photo activities of the catalysts enhance the photocatalytic production of NH3 from N2 reduction.
 | ||
| Fig. 9 Two-dimensional (2D) photocatalysts for ammonia synthesis. (A) Schematic representation of the preparation of 2D C–TiOx nanosheets by thermal oxidation etching at 200 °C. (B) SEM image of C4–TiOx obtained after four hours of thermal oxidation etching. (C) UV-visible absorption spectra of Ti3SiC2, TiO2–air, and C4–TiOx. (D) NH3 yield comparison between Ti3SiC2, TiO2–air, and C–TiOx catalysts obtained after varying durations of thermal etching. Reproduced with permission from ref. 71. Copyright 2021, Wiley-VCH. (E) Schematic representation of the transition metal (TM)-doped BiOBr nanosheets where TM is Fe, Mo, or Ni, obtained by solvothermal reactions. The structure in the dotted box represents the crystal lattice of the TM-doped BiOBr. The best performance is observed for TM = Fe. (F) SEM image of BiOBr-Fe-S-1 catalyst obtained by Fe doping in BiOBr in 1 molar ratio and after solvothermal reaction. (G) The light absorption spectrum of BiOBr-Fe-S-1. (H) NH3 yield comparison between pristine BiOBr and doped BiOBr before and after solvothermal reactions for different molar ratios of Fe doping. Reproduced with permission from ref. 73. Copyright 2021, Elsevier. (I) Schematic representation of photocatalytic N2 reduction to NH3 by B-doped C3N5 nanosheets. (J) TEM image of B-C3N5. (K) UV-Vis DRS of and (L) NH3 yields by g-C3N4, g-C3N5, and B-C3N5x photocatalysts (x is the mass percentage of ammoniumborate to 3-amino-1,2,4-triazole). Reproduced with permission from ref. 147. Copyright 2022, Elsevier. | ||
Another commonly used photocatalytic 2D material is graphitic carbon nitride (g-C3N4). It consists of tri-s-triazine rings interconnected via tertiary amines, which makes it thermally and chemically stable.126 The electronic structure of g-C3N4 makes it a prospective photocatalyst as it can absorb solar light due to its small band gap, unlike graphene. The structure of g-C3N4 bears a close resemblance to graphite due to π conjugation and the two-dimensional structures. Heteroatom doping can modify the electronic properties of g-C3N4 and the vacant active sites can increase the efficiency of the catalytic reactions. For instance, Wang and co-workers prepared B-doped porous g-C3N4 nanosheets by thermal treatment.77 The B-dopant induces a small-scale p–n type heterojunction, which promotes efficient charge separation in the catalyst. B doping changes the electronic structure by inducing localized electron states in the band gap and promotes strong tail absorption in the visible light region. Also, doping passivates the active N atoms of g-C3N4, ensuring that the NH3 produced does not originate from the exposed N atoms. The B atoms act as active sites for the adsorption and activation of N2 and exhibit good photocatalytic activity with 313.9 μmol g−1 h−1 yield rate of NH3. Incorporating N-vacancies (VN) and adding cocatalysts can also improve the photocatalytic performance of g-C3N4 as exhibited by the Ni3B/VN-g-C3N4 catalyst.163 The introduction of VN enhances visible light absorption and electrical conductivity properties of g-C3N4. Noble metal-free cocatalyst Ni3B nanoparticles act as active sites for the adsorption and activation of N2. The Schottky junctions formed between Ni3B and VN-CN also facilitate the separation of photogenerated charge carriers and migration of electrons for photoreduction of N2. Introducing one more N atom to g-C3N4 forms g-C3N5, a two-dimensional material with more π conjugation, which promotes superior charge separation, narrower band gap, and better solar absorption when compared to g-C3N4. Li et al. reported a B-doped g-C3N5 synthesized by a one-step thermal polymerization technique (Fig. 9I–L).147 The five azole rings of g-C3N5 facilitate electron transfer and decrease the adsorption energy of N2. B-doping increases the photocatalytic efficiency of g-C3N5 like in the previous case (B-doped g-C3N4) with B as the active site for photocatalysis and NH3 yield of 421.18 μmol g−1 h−1, which is 1.72 times that of g-C3N5.
Graphdiyne (GDY) is another emergent 2D carbon material, comprising sp and sp2 hybrid states endowed with unique properties like highly conjugated and super-large π structures, infinite number of pores, natural band gap, a hole-transport layer, high charge carrier mobility, electronic conductivity, and stability, which make them excellent photocatalysts.193 Recent studies indicate that GDY can form heterojunctions with different low-dimensional materials to design excellent catalysts for ammonia photosynthesis. Fe site-specific magnetite when incorporated with GDY, the GDY modulates the coordination environment of magnetite to form two selective valence states, namely, tetrahedrally coordinated Fe and octahedrally coordinated Fe.148 The coordination environment and valence charge transition regulate the photocatalytic properties of Fe3O4 and significantly enhance the photocatalytic nitrogen reduction to ammonia. Graphdiyne is also capable of modulating the surface plasmon resonance of quantum dots (QDs), which is evident from the heterojunction-based GDY-CoOx QDs catalysts.164 The natural porous structure, the acetylenic bonds, and the high reduction ability of the GDY incorporate the CoOx QDs and the combined effects of enhanced surface plasmon resonance and modification of valence states of the metal atom enhance the photocatalytic nitrogen fixation reaction to produce ammonia.
4.4. Organic framework-based photocatalysts and heterostructures
Organic framework-based photocatalysts, viz., porous–organic frameworks (POFs), comprising covalent–organic frameworks (COFs), porous–aromatic frameworks (PAFs), covalent–triazine frameworks (CTFs), and metal–organic frameworks (MOFs), are emergent catalysts for photocatalysis owing to their unique physical and optoelectronic properties such as tuneable light absorption, large specific surface area, periodic arrangement of building blocks, precisely controlled pore size, and easy functional modifications. These properties make these materials unique for applications like catalysis, gas absorption and storage, and sensing. Different morphologies varying from 2D to 3D can be designed by tuning the structure and assembly of covalent bonds. This section will describe a few examples of such organic frameworks and their advantages in photocatalytic ammonia generation. Metal phthalocyanine-derived POFs (MPc-POFs (M = Fe, Co, Ni, Cu, Zn)) consist of conjugated planar π-electron organic macrocycles and their optical and electronic properties can be controlled for optimizing photocatalytic N2RR.165 Most photocatalytic N2RR systems proceed via gas-in-solvent (GIS) systems where gaseous N2 is purged in water. However, these systems are limited by certain disadvantages like low solubility of N2 in water, sluggish N2 transfer at the interface, and interaction of water with the catalyst. In an attempt to promote N2 transfer and utilization at the active sites of the catalyst, these POFs use a solvent-in-gas (SIG) system as the reaction medium, where MPc-POFs perform the catalysis in direct N2 gas, and the hydrogenation step is performed in suspended proton source provided as the dispersed phase. Herein, the active sites of the MPc-POFs have access to abundant N2 molecules and can produce ammonia at an ultrafast rate of 1820.7 μmol g−1 h−1, which is approximately eight times higher than that obtained from conventional GIS (226.2 μmol g−1 h−1).2D COFs are porous polymers where any photoactive species can be incorporated into the ordered pores of their frameworks such that they can function as catalysts for different photocatalytic reactions.194,195 The organic units in the 2D π lattices of COF semiconductors can facilitate the separation and transport of photogenerated charge carriers.194 The one-dimensional polygon channels in the porous COFs also help in mass transport. Single-atoms like Au can be easily anchored into porphyrin-based COFs (COFX-Au, X = 1–5) for photocatalytic NH3 synthesis (Fig. 10A–D).166 The performance of the photocatalytic NH3 synthesis can be controlled by tuning the microenvironment of the single-atom Au catalytic centre resulting from the position of different functional groups at the proximal and distal positions of porphyrin units. In the first case, a strong electron-withdrawing group is attached in COF1-Au, which yields NH3 at a rate of 37.0 ± 2.5 mmol gAu−1 h−1. This is 171 times more than that of the porphyrin–Au molecular catalyst. The yield is also 2.8 times higher than COF4-Au, where an electron-donating group is used as the functional group. Thus, it can be deduced that the electron-withdrawing groups can facilitate the separation and transportation of photogenerated electrons within the entire COFX-Au framework. Following this deduction, NH3 production is increased to 61.1 ± 2.7 mmol gAu−1 h−1 by attaching two strong electron-withdrawing groups to the COF. The performance of photocatalytic N2RR can also be enhanced by spatial confinement of N2 near the surface of the COF for better adsorption and activation. This can be achieved by incorporating hydrophilic carboxyl groups (–COOH) into diketopyrrolopyrrole-based COF (DPPCOOH-COF) and forming a localized hydrogen bond network.167 The –COOH groups promote water to form a layered or porous structure that interacts with the N2 molecules and localizes them near the active pyrrole units. This leads to enhanced adsorption and activation of N2 by the DPPCOOH-COF and reduces the energy for photocatalytic NH3 generation. The photocatalytic activity of COFs is sometimes limited by the fast recombination of photogenerated charge carriers and the lack of sufficient active sites. Metal active sites can be easily incorporated into the COFs due to their ordered cavities and large specific surface area. Imine-linked 2D COFs have numerous N atoms, which endow the COFs with strong Lewis acidity, and this property of the imine COF-TaTp facilitates the adsorption of N2, which is a weak Lewis base (Fig. 10E–H).168 Furthermore, the strong interaction of the Bi metal with the imine nitrogen atom and hydroxyl functionalities of the imine COF facilitates the formation of Bi/COF-TaTp composites through N–Bi–O coordination. The incorporated Bi acts as the active site, which promotes the transfer of charge carriers and activation of N2 molecules through the donation and back-donation mode. Additionally, the Bi sites inhibit the competitive HER and thus facilitate the photocatalytic NH3 production from N2RR.
 | ||
| Fig. 10 Organic framework-based photocatalysts for ammonia synthesis. (A) Schematic representation of nitrogen reduction to ammonia by the porphyrin-based covalent organic framework anchoring Au single-atoms (COF5-Au). The structure in the dotted box represents the microenvironment of the Au catalytic centre. (B) Aberration-corrected HAADF-STEM image of COF1-Au, where Au single-atoms are marked by red circles. (C) UV-Vis diffuse reflectance spectra of COFX-Au, where X = 1–5. (D) NH3 yield comparison of COFX-Au catalysts. Reproduced with permission from ref. 166. Copyright 2023, American Chemical Society. (E) Schematic representation of the fabrication of Bi NP-decorated imine linked COF-TaTp (Bi/COF-TaTp). (F) TEM image of 5% Bi/COF-TaTp, (G) UV-Vis diffuse reflectance spectra of COF-TaTp, and 5% Bi/COF-TaTp. (H) NH3 yield comparison between pristine COF and different percentages of Bi NPs. Reproduced with permission from ref. 168. Copyright 2022, Wiley-VCH. (I) Schematic representation of photocatalytic N2 reduction to NH3 by Ca2+-doped UiO-66. (J) SEM image of UN(Zr-0.30Ca)-4. (K) Light absorption spectrum of UN(Zr-0.30Ca)-4. (L) NH3 yield comparisons between UN(Zr), UN(Zr-0.10Ca), UN(Zr-0.30Ca), UN(Zr-0.50Ca), and UN(Zr-0.75Ca) were obtained by feeding ZrCl4 and CaCl2 at Ca:(Zr + Ca) ratios of 0.1, 0.3, 0.5, and 0.75. Reproduced with permission from ref. 170. Copyright 2023, Elsevier. | ||
Metal–organic frameworks (MOFs), another class of organic frameworks composed of metal ions or clusters and organic ligands are also important for photocatalytic applications. Sometimes, the catalytically active metal centres in MOFs are fully coordinated by bridging organic ligands and terminal ligands, and become inaccessible to the reactants. Hence, to address this issue, a defective Fe-based MIL (100) MOF has been designed by partial removal of these ligands, such as bridging organic ligands and terminal inorganic ligands (OH− and H2O), leading to the formation of defective structures.169 The dual defects expose the coordinatively unsaturated Fe sites, which act as the catalytic active centres for adsorbing and activating N2 molecules. The defects also modify the electronic structures, which favours the transfer of d-orbital electrons from Fe sites into the N2 π* antibonding orbital to form the key intermediate *NNH in the photocatalytic N2RR for NH3 formation. As discussed previously, hydrogen evolution reaction (HER) is a major competitive reaction for photocatalytic N2RR processes, which limits the production of NH3. In an attempt to harmonize the competition between HER and N2RR reactions, a core–shell MOF-based catalyst has been prepared by depositing Ni nanoparticles on the polydopamine shell and NH2-MIL125 MOF core.196 Photocatalytic HER occurs at the NH2-MIL125 MOF core and the evolved H2 is captured by the Ni nanoparticles and converted into adsorbed *H species. The produced *H is then transferred back to the NH2-MIL125 core via NiO, which reacts with N2 and produces NH3. This “hydrogen state switching” strategy ensures the supply of requisite protons from HER for hydrogenation of N2 to NH3 formation, thus “transforming-competition-into-cooperation”. Like other photocatalysts, metal atom doping in MOFs can lead to enhanced photoproduction of NH3. This is manifested in Ca2+-doped UiO-66, a Zr-based MOF, where low electronegative Ca2+ increases the electron donation capability of Zr active sites (Fig. 10I–L).170 Furthermore, the incorporated diamino ligand modifies the band gap of Ca2+-UiO-66 to enhance the light absorption range and separation of charge carriers. All these factors contribute to amplified photoreduction of N2 to produce NH3 under the full spectrum of light. The formation of heterojunctions can also enhance the photoactivity of MOFs via inhibition of recombination of charge carriers. An example of such MOF is the heterojunction formation between MXene and MIL-125(Ti) MOF via ligand bridging.171 A ligand pre-coupling strategy is employed in this work to create the ligand-bridged MXene/MIL-125(Ti), where a coordination bond between the terminal oxygen group of MXene is coupled to the carboxyl group of the ligand in MIL-125(Ti). This ligand bridge forms a one-directional transport channel for electron transfer from MIL-125(Ti) to MXene. This strategy diminishes the interfacial charge transfer resistance and reduces the recombination of photogenerated electron–hole pairs, thus boosting the photocatalytic N2 fixation to produce NH3. The design of nanoarchitectonics using S-scheme heterojunction-based MOFs can also facilitate photocatalytic nitrate reduction to ammonia. A nanohouse-like catalyst structure is constructed employing the nanoarchitectonics technology, where NH2-MIL-125 MOF with a nanoplate-like morphology serves as the floor.172 Hollow ZIF-8 cages form the surrounding walls and roof of the nanohouse, and Co(OH)2 nanosheets are locked inside this nanohouse and connected to the ground by forming a heterojunction with NH2-MIL-125 MOF. Each component of this unique sandwich-structured superstructure forming the nanohouse array plays a crucial role in photocatalytic NO3− reduction. The positively charged, hydrophobic, and porous ZIF-8 structure modifies the microenvironment of the NH2-MIL-125/Co(OH)2 heterojunction by increasing the NO3− enrichment, suppressing competitive HER, and promoting NH4+ release. The S-scheme heterojunction between NH2-MIL-125/Co(OH)2 enhances the separation of photogenerated electron–hole pairs and boosts the photo-redox capability of the nanohouse catalyst, promoting NH3 production from NO3−.
5. Low-dimensional electrocatalysts for ammonia synthesis
Electrocatalysts are generally more energy-efficient than photocatalysts because all photons are not effectively utilized in photocatalysis. This inefficiency arises from mismatches between the wavelengths of incident light and the photoactive material's absorption spectrum, as well as from the recombination of photogenerated charge carriers, which reduces the overall efficiency. Electrocatalytic reactions occur under ambient conditions with the external input of electrical energy, which can be easily harnessed from renewable sources like solar, hydro, tidal, or wind energy. The prerequisites for electrocatalysts are: (i) good conductivity; (ii) high surface area and presence of abundant active sites for adsorption of N-containing species; (iii) resistance to corrosion at the working potential range and in the presence of electrolytes; and (iv) long-term stability. The low-dimensional electrocatalytic materials used for ammonia synthesis include transition metal nanoclusters, noble metal-based materials, metal oxides, phosphides, carbides and nitrides, single-atoms, and carbon-based materials. Electrocatalytic materials can be modified by defect engineering, forming heterostructures, or regulating the crystal planes and microenvironment of the catalyst to ensure better electrocatalytic performance. This section will discuss these strategies for designing low-dimensional electrocatalytic materials to synthesize ammonia from N2 or NOx sources (Fig. 11 and Table 4). | ||
| Fig. 11 Low-dimensional electrocatalysts for ammonia synthesis. Classification of electrocatalysts into 0D, 1D, and 2D, and schematic representations of few selected electrocatalytic LDMs. | ||
| Classification | Materials | Electrocatalysts | Synthesis/modification | Ref. |
|---|---|---|---|---|
| 0D LDMs and heterostructures | Metallic nanoparticles (NPs) and nanoclusters (NCs) | Pd NCs | Seed-mediated method, facet engineering | 197 |
| Au NPs | Seed-mediated method | 198 | ||
| Ag NCs | Replacement reaction on Cu foam | 199 | ||
| Aux–Ni NPs | Donor–acceptor construction, galvanic replacement method | 98 | ||
| Hollow PdCu NPs | In situ reduction and nucleation | 200 | ||
| Fragmented Bi0 NPs | In situ reduction and fragmentation of Bi-MOF nanorods under applied potentials | 201 | ||
| Metallic oxides | TiO2@N–C NPs | N-Impregnated carbon coating strategy | 202 | |
| Cu NPs-VO-TiO2 | Defect engineering, chemical reduction | 203 | ||
| Single-atoms (SAs) | Fe1 SA-NC | Thermal modulation | 204 | |
| Fe SA-N/P-C | In situ phosphatizing–adsorption–thermolysis process | 102 | ||
| Cu1 SA-NC/CNT-FEM | Pyrolysis, probe sonication | 205 | ||
| Cu(I) SA-N3C1 | N-doping, wet chemical synthesis, pyrolysis | 206 | ||
| 1D LDMs and heterostructures | Metallic nanowires (NWs) | Cu NWs | Chemical reduction | 207 |
| Ru-CuNW | Cation exchange method, thermal annealing, electrochemical prereduction | 208 | ||
| Rh NCs/SAs-Cu NWs | Electrochemical reduction, galvanic replacement | 209 | ||
| TA-Au NWs | Surface modification with tannic acid (TA) | 210 | ||
| Multimetal alloys | Cu6Sn5 alloy | Co-electrodeposition method | 61 | |
| IrNi, IrRhNi, and IrFeNi alloys | Chemical Co-reduction, solvothermal method | 211 | ||
| Metallic oxides | P-TiO2/TP | P-Doping, hydrothermal growth, ion-exchange, phosphating process | 212 | |
| a-B2.6C@TiO2/Ti | Hydrothermal synthesis, sputter deposition | 213 | ||
| Fe2TiO5 | Defect engineering, electrospinning method | 106 | ||
| Spinel oxides (AB2O4) | NiCo2O4 NWs/CC | Hydrothermal synthesis, thermal annealing | 214 | |
| Mn2–Co3O4 NTs | Hydrothermal synthesis, thermal annealing | 215 | ||
| C-Co3O4 NTs | C-Doping, ‘‘surface locking’’ mechanism, in situ topotactic conversion | 216 | ||
| Carbon nanotubes (CNTs)/nanofibers | MWCNTs/SWCNTs | Calcination and acid treatment | 217 | |
| Ni NPs-N-CNRs | N-Doping, electrospinning, carbonization, selective etching with acid | 218 | ||
| Fe SA-N-carbon nanofibers | N-Doping, C-defects, electrospinning-pyrolysis method | 45 | ||
| 2D LDMs and heterostructures | Graphene | Amorphous graphene | Laser irradiation | 219 |
| Amorphous graphene | Transient laser heating | 220 | ||
| Cu-cis-N2O2/graphene | Breaking of coordination symmetry, thermal annealing | 221 | ||
| Graphdiyne (GDY) | Mo0-GDY | Solvothermal method | 222 | |
| Ru SAs-GDY-graphene | Eglinton coupling reaction, microwave synthesis, impregnation method | 223 | ||
| Cu SAs-GDY | Impregnation method | 224 | ||
| Fe3C-GDY | Pyrolysis, interface engineering, heterojunction formation | 225 | ||
| Carbon nitrides (CN) | B-BCN | Tuning B/N Lewis acid pairs | 226 | |
| BCN | Defect engineering, annealing, tuning frustrated Lewis pairs | 227 | ||
| Metal and metal oxide-based nanosheets (NS) | Ru NS | Hydrothermal method, plasma treatment | 228 | |
| hcp-Co NS | Hydrothermal method | 229 | ||
| CuCo NS | Co-Electrodeposition method | 230 | ||
| Amorphous RuO2 NS | Defect engineering, molten salt synthesis | 231 | ||
| MXenes | Ti3C2O2-VO | Defect engineering, functionalization of terminal group | 232 | |
| OH-terminated Ti3C2 | Etching, exfoliation, functionalization of terminal group | 233 | ||
| B-Ti3C2Tx | Etching, annealing, B-doping, pyrolysis | 234 | ||
| MBenes | FeB2 | Reflux method | 235 | |
| Transition metal dichalcogenides (TMDs) | F-MoS2 | Strain engineering, F-doping, hydrothermal method | 236 | |
| V-MoS2 | Defect engineering, V-doping, hydrothermal method | 237 | ||
| Metastable 1T′′′ MoS2 | Solid state reaction, thermal annealing, deintercalation of K ions | 238 | ||
| Organic framework-based heterostructures | Metal–organic frameworks (MOFs) | Cu-OUC MOF | Hydrothermal method | 239 |
| Fe2M MOF (M = Fe, Co, Ni, Zn) | Solvothermal method | 240 | ||
| NiCoBDC MOF-HsGDY | Interface engineering, dual-template, Glaser-coupling, solvothermal methods | 241 | ||
| PCN-250-Fe3 MOF | Crystallization, thermal activation | 242 | ||
| 3D-Printed electrodes based on LDMs | Metallic NPs | Cu Metallic NPs | Sintering, nanostructuring using electrochemical methods, chemical treatment | 243 |
| Carbonaceous material-based heterostructures | MnOx coated 0D carbon black, 1D carbon nanotubes | Thermal activation, atomic layer deposition (ALD) | 244 | |
| TiO2-coated carbon | Thermal activation, atomic layer deposition (ALD) | 245 |
For the electrocatalytic synthesis of ammonia, the optimal design of an electrocatalytic reactor is pertinent. The electrocatalytic reactor, in principle, consists of the electrocatalytic cell, electrolyte, electrodes, and ion exchange membrane. Electrocatalysts are loaded on the cathode and upon application of a voltage at the electrodes, the electrocatalytic reaction produces ammonia at the cathode by reduction of N2 or NOx. Simultaneously, water is oxidized at the anode to generate oxygen. The electrocatalytic cells can be classified as: (i) back-to-back cells; (ii) proton exchange membrane cells; (iii) single-chamber cells; or (iv) H-type cells.246 A back-to-back cell has two gas diffusion electrodes containing N2 (cathode) and H2O (anode), partitioned by cation/anion exchange membranes. The proton exchange membrane cell has a similar configuration except that the anodic component is in aqueous form. Because the electrolyte and the cathode have no direct contact, HER is limited in these kinds of cells. However, the reaction medium consists of gas and solid, which lowers the efficiency of the catalytic processes. The single-chamber cell, as evident from its name, has cathodic and anodic reactions occurring in the same electrolyte in one chamber. The electrolyte used here is liquid, but the simultaneous presence of the cathode and anode in one chamber can further oxidize the ammonia produced at the cathode. This problem can be overcome by using an H-type cell, the most frequently used electrochemical cell, where the cathodic and anodic compartments are separated by an ion-exchange membrane. The electrolyte is mostly aqueous and, depending on the reaction conditions may be acidic (H2SO4 or HCl), neutral (phosphate buffer or Na2SO4), or alkaline (KOH).247 The three-electrode setup is mostly used with a working electrode (e.g., glassy carbon plate, glassy carbon rotating disk, carbon paper, self-supported), reference electrode (e.g., Ag/AgCl), and counter electrode (e.g., Pt, graphite rod).247 Electrocatalysts are deposited on the working electrodes and proper optimization of the electrocatalysts is pertinent. The most common strategies to improve the efficiency of electrocatalysts for ammonia production include crystal facet engineering, heteroatom doping, and the introduction of vacancies and defects. Taking into account the earlier discussion about the disadvantages of photocatalytic N2 reduction and the advantages of photocatalytic NO3−/NO2− reduction, it can be said that they also hold for electrocatalytic reductions.
While devising catalysts for electrocatalytic N2RR or NOxRR to NH3, the main objectives are to ensure good conductivity of the catalysts, availability of adequate active sites for electroreduction, and restraining the side reactions like HER or formation of by-products like N2 and NO by partial reduction of NOx−. In the following subsections, we will discuss the design of low-dimensional electrocatalysts and a strategy to overcome the bottlenecks, aiming to enhance both the faradaic efficiency and selectivity of the resulting NH3. The most commonly used 0D, 1D, and 2D electrocatalysts use simple one-pot synthetic strategies, whereas their heterostructures rely on multi-step synthesis. Synthesis of 0D metallic and metal oxide nanoparticles or nanocrystals uses strategies like seed-mediated growth, galvanic replacement, or reduction methods.98,197,201,203 Single-atom engineering relies on different thermal modulation techniques like pyrolysis or thermolysis.102,205 1D electrocatalysts like monometallic, bimetallic, multimetal alloys, metal oxides, carbon-based nanowires, nanorods, and nanofibers are prepared by chemical/electrochemical reduction, different deposition techniques, electrospinning methods, and thermal reactions.45,61,106,207,209,213,215 The most commonly used 2D electrocatalysts like graphdiynes, metal, metal oxides, and transition metal dichalcogenides use thermal synthetic strategies like solvothermal, hydrothermal, annealing, or pyrolysis.222,225,227,228,236 The synthesis of MXenes and MBenes relies on etching, thermal treatments, and the functionalization of terminal groups.233–235 The electrocatalytic properties of these materials can be enhanced by defect engineering, heteroatom doping, facet engineering, using cocatalysts, formation of heterojunctions, and heterostructures.197,203,206,225 A more detailed and precise discussion of the different synthetic and modification strategies of ammonia-synthesizing electrocatalysts is presented in Table 4.
5.1. 0D electrocatalysts and heterostructures
Among different 0D materials, metallic nanoparticles have been extensively studied for electrocatalysis over the last few decades. The plasmonic character and possibilities of tuning the size, shape, and structure of nanoparticles have made them significant for catalysis. The packing of metal atoms in a particle can be different: tuning the size of nanoparticles exposes different crystallographic facets, which can provide different types of catalytic sites.248 Several metal nanoparticle-based catalysts have been reported recently; here, we will discuss a few of these and their roles in catalysis. Electrocatalytic denitrification of NOx for simultaneous removal of N-containing oxides and ammonia production proves quite efficient on metal surfaces, particularly transition metals. Among different transition metals, Cu is the most efficient for electrocatalytic NOxRR.249,250 Cu produces NH3 selectively as Cu can bind *NO intermediate and by not binding *H, it provides selectivity against competitive HER.92 Bimetallic nanoparticles are quite common as they offer two different sites for catalysis. Ni–Au nanoparticles can electrochemically reduce N2 to NH3, a reaction facilitated by donor–acceptor couples of Ni and Au (Fig. 12A–D).98 Donor–acceptor pairs can adsorb and activate N2 and also help the desorption of NH3. Bimetallic PdCu nanocrystals can electrosynthesize ammonia selectively from nitrates (Fig. 12E–H).200 For synthesis, C12N–COOH is employed as the template and dimethyl borane as the reducing agent to form the PdCu hollow nanospheres from Pd- and Cu-containing precursors. The hollow structure of PdCu can trap the intermediates and thus promote the nitrate reduction reaction (NO3RR) with an effective FE of 87.3%. Crystal-phase engineering can effectively improve the performance of N2RR and NOxRR. Nanostructures with controlled facets can exhibit high efficiency for the production of NH3. Several researchers have performed extensive studies to uncover the correlation between ammonia production and crystal facets. For instance, body-centered cubic (BCC) PdCu nanoparticles surpass face-centered cubic (FCC) PdCu nanoparticles in terms of N2RR electrocatalysis due to strong d–d orbital interactions between Pd(4d) and Cu(3d) sites.47 A comparative study between exposed (100), (111), and (110) facets of Pd reveals that the (100) facet is most active electrocatalytically as it is extremely stable under electrocatalytic reaction conditions, and the energy barrier for desorption of *NH3 to NH3 is lower than for other facets.197 Pd nanocatalysts show face-dependent reduction capability for NO3− as well as NO2− in the order of Pd(111) > Pd(100) > Pd(hk0) for NO3RR and Pd(100) > Pd(hk0) > Pd(111) for NO2RR in alkaline conditions.251 Cuboctahedral Pd with exposed (111) and (100) facets produce the highest quantity of ammonia. Exposure of the two facets makes the cuboctahedra bifunctional, where the (111) facets catalyse the conversion of NO3− to NO2− and the (100) facets promote the conversion of NO2− to NH3. Theoretical calculations on Cu-based surfaces reveal that among Cu(100), Cu(111), and Cu(110), the best electrocatalytic NO3RR performance is exhibited by Cu(111) in neutral and alkaline conditions, and by Cu(100) in strongly acidic conditions.252 The local coordination environment and the electronic states are different for every exposed facet, and these results indicate that the pH and the exposed facets on the surface of the electrocatalyst affect NO3RR accordingly. Fabrication of Au NPs with multiple high-index facets via a modified seed-mediated method can enhance the adsorption of *NNH intermediate, thus promoting the N2RR reactions.198 The adsorption of H*, an HER intermediate, is less on the high-indexed facets, which demotes the competing HER reaction, resulting in a moderately high FE of 73.32% for N2RR at −0.3 V vs. RHE. Doping with heteroatoms can also impact the electrocatalytic performance. Cu-doped Fe3O4 particles can effectively reduce NO3− to NH3 in a more thermodynamically facile manner with an effective FE ∼100% at −0.6 V vs. RHE.253 Doping the catalyst surface with Cu lowers the energy barrier and strengthens the adsorption of different reaction intermediates. Doping with 0D materials like Ru atoms can also enhance the catalytic properties of emergent materials like MXenes.254 Ru atoms can back-donate electrons to N2, which increases its adsorption and activation. The thermodynamic energy required for the first hydrogenation step is also lowered and these synergistic roles of Ru active sites promote N2RR. Apart from crystal facet engineering and doping, the surface structure of electrocatalysts can also be modulated by defect engineering. Surface defects like oxygen vacancy (VO) can tune the electronic structure of the metal with unsaturated coordination, allowing them to act as active sites to enhance the adsorption of O-containing species like NO3− on the surface of the electrocatalyst. Incorporation of the ultra-low level of Pd in Cu2O induces hierarchical cavity defects and surface defects on the surface of octahedral Cu2O.255 The cavities and VOs impart voids, which results in enhanced mass transport and adsorption of NO3− and H2O reactants on the surface of Pd–Cu2O. Moreover, the VOs weaken the N–O bond of nitrates, and the Pd can act as active sites to generate active Hads from H2O, which helps in protonation to form NH3. It is to be noted that different noble metals like nanocrystalline Pd,256 nanocrystalline Ag,199 and their composites like Ru/g-C3N4257 and bimetallic Cu–Pt258 have been used for catalytic NO3RR. However, considering the scarcity of noble metals and their expensive nature, these are not very cost-effective. For this reason, researchers have shifted their focus to lower-cost transition metal-based electrocatalysts. Several Bi-, Cu-, and Fe-based nanoparticle and single-atom catalysts with efficient and selective ammonia synthesis have been reported till now. One such work demonstrates that Bi-based-MOF, which acts as a precatalyst and, upon electroreduction, produces fragmented Bi0 nanoparticles in situ.201 The stable Bi NPs produced can act as effective N2RR catalysts in both acidic and neutral reaction conditions, though the FEs reported are quite low due to competing HER. Zhou et al. have reported selective electroreduction of NO3− to NH3 by in situ-generated 0D-Cu0 cubes from its oxide (Cu2O precursor) while the Cu/Cu2O favours nitrite production.259 Nitrate reduction at the 0D-Cu cubes is preferred because it has a lower activation energy barrier for nitrates when compared to Cu/Cu2O and Cu microspheres. The intrinsic activity of NO3RR is higher on 0D-Cu cubes than on 0D-Cu microspheres because the 0D-Cu cube surface has more Cu(100) facets, whereas the surface of 0D-Cu microspheres is dominated by Cu(111) facets. Atomically dispersed 0D materials like metallic clusters and single-atoms are emergent catalysts for ammonia synthesis owing to their novel features like unique coordination environment and high atom economy. Atomic dispersed Au nanoclusters (Au-NCs) prove to be effective electrocatalysts for ammonia synthesis when embedded on TiO2 nanosheet support.260 Pristine TiO2 is not a very effective electrocatalyst for ammonia production due to lower charger transport and the absence of abundant catalytically active sites. However, the Au atoms in Au-NC/TiO2 can significantly enhance the adsorption and activation of NO3− and promote the NO3RR-to-NH3 by lowering the energy activation barrier. | ||
| Fig. 12 Zero-dimensional (0D) electrocatalysts for ammonia synthesis. (A) Schematic representation of the donor–acceptor couples of Ni and Au nanoparticles. (B) SEM image of Au and Ni nanoparticles. (C) Schematic representation of the electron transfer between donor–acceptor couple Aux/Ni with different Au loadings. (D) NH3 yields and Faradaic efficiencies using only Ni and Aux/Ni nanoparticles donor–acceptor couple catalysts at −0.14 V vs. RHE. Reproduced with permission from ref. 98. Copyright 2019, American Chemical Society. (E) Schematic of the simulated structure of hollow PdCu (PdCu-H) nanoparticle. (F) TEM image of PdCu-H particle at different magnifications and different locations. (G) NH3 yields using hollow PdCu-H, PdCu-P nanoparticles, and commercial Pd–P. (H) Schematic illustration of NO3− reduction over PdCu-H catalyst. Reproduced with permission from ref. 200. Copyright 2023, Wiley-VCH. (I) Schematic representation of the 0D Fe single atom N, P co-modified carbon catalyst (Fe–N/P–C) obtained on pyrolysis from ZIF-8. (J) Aberration-corrected HAADF-STEM of Fe–N/P–C where the Fe single-atoms are marked by red circles. (K) NH3 yields using N–C (N-modified carbon), N–P–C, Fe–N–C, and Fe–N/P–C catalysts. Reproduced with permission from ref. 102. Copyright 2023, Wiley-VCH. (L) Schematic representation of N2 electroreduction to NH3 using oxygen-doped MoC nanoparticles embedded in graphitized carbon shells (O-MoC@NC). (M) TEM image of O-MoC@NC-800 synthesized at 800 °C annealing temperature. (N) NH3 yields and Faradaic efficiencies after 2 hours of reaction at different potentials. Reproduced with permission from ref. 261. Copyright 2019, American Chemical Society. | ||
Single-atom (SA) confinement also proves to be an effective strategy for devising electrocatalysts that enhance the metal utilization efficiency in catalysis. Nowadays, the use of single-atom catalysts is quite frequent in HER, CO2RR, NO3RR, and metal-based SAs are prolific in electrocatalysis. Fe SAs and Cu SAs are the most widely used single-atom electrocatalysts for ammonia generation reported so far. The coordination environment of the single-atoms is vital because charge localization around the single-atom depends on the coordination. The coordination environment also impacts the electronic structure and geometry of the central single-atom and modulates the adsorption of reactants, which in turn has a direct effect on catalytic efficiency. The most common coordination is N4. Fe–N4 SAs have higher atomic site activity when compared to bulk or nanostructural catalysts due to a lower thermodynamic barrier.262 Fe–N4 coordination is also beneficial because due to the lack of neighbouring metal centres, N–N coupling cannot occur. This prevents the formation of N2 and simultaneously promotes the selectivity of NH3. To elucidate the structure–performance relationship, Liu et al. have prepared Fe active sites with three different coordination environments: square pyramidal Fe–N4–OH, slightly broken square planar Fe–N4, and trigonal pyramidal Fe–N3.204 The interaction between Fe atoms and O atoms of NO3− is determined using DFT, which reveals a strong overlap between the 3d orbitals of Fe and 2p orbitals of O (NO3−) in the case of Fe–N3 as the d orbitals are more localized in N3 coordination when compared with Fe–N4 and Fe–N4–OH. This ensures that the adsorption of NO3− is more favourable on Fe–N3, making Fe–N3 a more active catalyst for NO3RR. Also, the localization of electrons is near the N atoms for Fe–N4 and Fe–N4–OH, whereas for Fe–N3 the electrons are at both the Fe and N sites, enhancing the charge transfer required for NO3RR. Nitrate reduction can also be enhanced by changing the coordination environment from N4 to N2O2.263 The O atoms can regulate the d orbitals of Fe such that the adsorption energy of nitrates decreases on Fe–N2O2 when compared with Fe–N4. The conversion of *NOH to *N is easier on Fe–N2O2. The conductivity and selectivity are also greater, which makes Fe–N2O2 a potentially better electrocatalyst than Fe–N4. Besides N and O atoms, P atoms are also used for the coordination of single-atoms. The P atoms can break the local charge symmetry of the Fe atoms and facilitate the adsorption of nitrate as well as other key reduction reaction intermediates (Fig. 12I–K).102 The atomic interface of the Fe atom is asymmetric, which optimizes the electron density in such a way that it lowers the energy of formation of the NO3RR intermediates and effectively improves the catalytic performance. Cu SAs also play a predominant role in electrocatalytic NO3RR as Cu is electroactive, has tuneable electronic structures, and is also cost-effective. The coordination environment for Cu SAs is also crucial for catalysis like Fe SAs. The most frequently used Cu SAs have Cu–N4 coordination where the symmetry is C4v.205,264,265 Symmetric coordination induces weak polarity, which reduces the attractive power of active sites for NO3− and lowers ammonia production.266 Asymmetric coordination like Cu–N2O2 in cis configuration has more polar active sites, which promotes the adsorption of NO3−.221 Moreover, the cis configuration splits the 3d orbitals of Cu, which reduces the energy of the formation barrier of *ONH intermediate. Modification of the coordination environment to Cu–N3C1 has manifold benefits.206 It increases the energy barrier of competing HER, reduces the desorption of intermediates produced, increases the adsorption of H*, and enhances the electrocatalytic hydrogenation process. However, the structural sensitivity of these metal SA catalysts can sometimes decrease the selectivity of ammonia. For instance, Co-based SAs can more selectively produce hydroxylamine (NH2OH) from the electroreduction of NO, whereas metallic Co in a hexagonal close-packed (hcp) lattice produces NH3 selectively.267 The hcp-Co has excellent electron and proton transfer properties, resulting in superior activity for ammonia production. Co-SA has a positively charged active centre and this modified electronic structure accounts for the exceptional hydroxylamine selectivity. hcp-Co can lead to vertical and strong NO adsorption, whereas moderate adsorption occurs on the Co-SA. Hence, the formation of NOH* intermediate is more favourable over hcp-Co, while HNO* formation is preferable on Co-SA. This local structural difference between hcp-Co and Co-SA leads to the selective formation of NH3 and NH2OH, respectively.
Besides photocatalysis, 0D semiconductor nanoparticles can also perform efficient electrocatalysis owing to their conductive properties. For instance, nitrogen-impregnated carbon increases the conductivity and causes partial oxygen defects on the surface of TiO2.202 This leads to the coexistence of Ti3+ and Ti4+, which increases charge transfer and, thus, facilitates the catalytic reactions. Here, in addition to ammonia, hydrazine is also obtained as a by-product for the electrocatalytic N2RR, which somewhat limits the selectivity of ammonia. The formation of heterostructures can also improve the photocatalytic properties of TiO2. In Cu nanoparticle-loaded oxygen-deficient TiO2 (Cu NP–VO–TiO2), a strong metal support interaction is induced between Cu and TiO2, improving electron density and electron transfer processes.203 This interaction also modifies the local charge distribution properties of the catalyst such that it is asymmetrical, resulting in polarization of the adsorbed N2 and, consequently, improved N2 activation. The experimental results reveal that the oxygen vacancies and Cu NPs act as active sites, and the metal support interaction helps in electron transfer from Cu to the oxygen-deficient TiO2. In N2RR, no hydrazine has been obtained as a by-product, thus confirming the good selectivity of NH3. Further, the absence of hydrazine indicates that the catalytic mechanism proceeds via the associative distal pathway.
Core–shell nanostructures are a class of 0D nanomaterials composed of an inner core and outer shell with a distinct boundary between them. The interface connectivity between the core and shell materials sometimes offers new properties like a tuneable electronic surface and abundant active sites, which are beneficial for catalytic reactions. The core–shell structures have another advantage in catalytic reactions. The thickness of the shells and the composition of the core materials can affect the adsorption–desorption properties of intermediates and products so that the catalytic activity can be tuned easily by modification of the core–shell structures and compositions. Qu et al. synthesized a core–shell structure comprising oxygen-containing molybdenum carbides (O-MoC) and nitrogen-doped carbon layers (N-doped C) by pyrolysis of ammonium heptamolybdate and dopamine, which can electrocatalytically reduce N2 to ammonia (Fig. 12L–N).261 The interaction between O-MoC and N-doped C results in an electronic structure that makes the competing HER negligible. This increases the FE of the catalyst, and no by-products like hydrazine are produced here, which also increases the selectivity of NH3 produced.
5.2. 1D electrocatalysts and heterostructures
The structure of 1D materials has diverse advantages, which makes them prospective in the field of catalysis. The aspect ratio of length: height for 1D structures is always greater than 1. The diameter of the 1D nanowires can be tuned to match the wavelength of photons and the diffusion length of charge carriers, tailored to specific catalytic applications.268 The high surface-to-volume ratio of these materials ensures the availability of enough active sites on the surface for catalysis. These characteristics alter the properties of 1D structures compared to their bulk counterparts, making them advantageous for catalysis. For instance, 1D Au nanowires coated with oxygen-rich tannic acid can electrochemically reduce N2 to NH3.210 Tannic acid is loosely packed on the surface of Au nanowires and has high porosity, which improves access to most of the metal sites on the surface of the catalyst for adsorption and activation of N2. The oxygen richness of the coating at the gold and tannic acid interface enhances N2RR. Hydrazine has not been detected at any potential, thus confirming the selectivity of NH3. Examples of other catalytic nanowires include single-atom-dispersed Cu nanowires where single-atoms (SAs) like Ru and Rh dopants are atomically dispersed in a Cu nanowire matrix to form a 0D–1D type heterostructure.208,209 The Ru/Rh SAs offer diverse atomic and electronic properties that make them suitable for catalytic conversion of nitrates to ammonia. The NO3RR reaction occurs at the Ru/Rh sites while the Cu sites suppress the competitive HER. The selectivity of NO3− to NH3 reaches as high as 99.8%. The authors have also coupled NO3RR to the air-stripping process and successfully obtained NH3 and NH4Cl, thus proving that the system can convert the nitrates present in wastewater to value-added products. Cu materials are widely used for NO3RR reactions because the energies of the d orbital of Cu and the lowest occupied π* orbital of NO3− are similar, which is favourable for NO3RR.269 But Cu materials perform NO3RR at comparatively negative potentials because of the low nucleophilicity and affinity of nitrate on the Cu surface, which makes the competing HER more favourable.270 To address this issue, researchers have modified the electronic structure of Cu-based catalysts using metal heteroatoms or by doping with non-metals. For instance, B-doping in Cu nanowires can transfer electrons from the 3d orbitals of Cu to the empty 2p orbitals of B, thereby inducing electron localization and improving catalytic activity at the Cu sites.271 DFT calculations indicate that boron incorporation also suppresses competing HER, increases the adsorption of intermediates like *NO3, and enhances the conversion of *NO to *HNO. According to a recent study, CO2 molecules enhance the reduction of nitrites to ammonia on Cu nanowire catalysts, achieving a FE of nearly 100% in a broad potential range.207 Reaction mechanism investigations show that CO2 is first reduced to form *CO, which then facilitates the reduction of *NO intermediates to *N. This leads to a decrease in the energy barrier of deoxygenation of *NO intermediates, which is the rate-determining step of the nitrite electroreduction process. Additionally, the adsorbed *CO species can accelerate the hydrogenation of *NH2 intermediates to form NH3 at an enhanced rate. Besides the electroreduction of nitrite, CO2 molecules can also accelerate the reduction of other nitro-containing compounds such as the conversion of nitrate to ammonia and nitrobenzene to aniline.Monometallic nanomaterials with unconventional crystal phases, such as face-centered cubic (fcc) and hexagonal close-packed (hcp) phases exhibit enhanced catalytic activities over their common phase counterparts. However, monometallic materials sometimes lack sufficient active sites for the adsorption and stabilization of multiple intermediates, particularly in multi-step reduction reactions like NOxRR and N2RR. In this attempt, multimetal alloy nanomaterials with unconventional phases have been fabricated which can enhance the electrocatalytic reactions toward ammonia synthesis. One example of such alloy is Cu–Sn-based pine-needle structures that convert NO to ammonia in a flow cell.61 Theoretical investigation implicates that the energy barriers of protonation are low over Cu6Sn5-derived surface structures, which results in enhanced ammonia production. IrNi-based alloy nanobranches (NBs) with unconventional hcp phase demonstrate superior electrocatalytic NO2RR performance toward ammonia synthesis.211 Solvothermally fabricated IrNi, IrRhNi, and IrFeNi alloy NBs consist of a Ni-rich core and an Ir-rich shell. Theoretical studies indicate that the Ir–Ni interactions within the hcp IrNi alloy can accelerate the electron transfer processes for NO2RR. The hcp IrNi alloy surface also produces more active hydrogen, which reduces the energy barriers for the hydrogenation steps for ammonia formation.
One-dimensional carbon-based nanomaterials like carbon nanotubes (CNTs) and carbon nanorods (CNRs) exhibit excellent catalytic properties toward ammonia production, owing to their electron conductivity, large surface area, and stability. CNTs have a graphite sheet-like structure with sp2 hybridized-C atoms, and the sheets can be rolled to form a cylinder-like structure.272 The inner and outer walls of CNTs provide numerous active sites for catalysis. Single-walled and multi-walled carbon nanotubes (SWCNTs and MWCNTs) are promising 1D materials for efficient electrochemical NO3RR. However, the work of Harmon et al. suggests that when heteroatoms like O and N are introduced into MWCNTs, the efficiency of catalytic NO3RR decreases (Fig. 13A–C).217 This observation proves that the catalytic reaction occurs at the C atoms present on the surface of the carbon nanotubes. The electron-rich O and N dopants reduce the number of active C sites on the surface, thus diminishing the catalytic reaction. Some Fe contaminant from the synthetic counterparts is present in SWCNTs, but not on the surface; hence, it does not affect the NO3RR significantly. The authors also found that the surfaces of SWCNTs are more catalytically active and selective for NO3RR than for MWCNTs. This might be because the SWCNTs have larger curvature than MWCNTs, which modifies the bonding and, consequently, the electronic structure of the active C atoms. For SWCNTs, the FE for NH3 is 90% at −0.85 V vs. RHE. Other by-products like gaseous H2 (FE 6%), liquid NO2− (FE 4%), and NH2OH are also detected. Carbon nanorods (CNRs) are one-dimensional rod-shaped carbon materials with a moderate aspect ratio, high surface area, and good conductivity. The electrocatalytic properties of CNRs can be modulated by doping with heteroatoms like N, and the incorporation of transition metals like Ni.218 Ni-embedded N-doped carbon nanorods (Ni-NCNR) can selectively electroreduce nitric oxide to form ammonia. The Ni atoms act as the active sites for adsorption and activation of NO. N-doping alters the electronic structure and enhances the interaction between the Ni active sites and CNR support; hence Ni incorporation and N-doping synergistically enhance NORR for NH3 production. N-doped 1D carbon nanofibers with carbon defects can also accelerate electrocatalytic N2RR for NH3 synthesis.45 The carbon defects enhance the water-splitting process, generating abundant protons for the protonation of N2 to NH3. Further, the C defects assist the coordination of the Fe atom with four N atoms. The Fe–N4 sites, together with the adjacent C defects, promote the protonation reactions by reducing the energy barrier of the process, benefiting the overall N2RR process.
 | ||
| Fig. 13 One-dimensional (1D) electrocatalysts for ammonia synthesis. (A) Schematic representation of electrochemical NO3− reduction to NH3 by 1D carbon nanotube (CNT). (B) SEM images of single-walled carbon nanotubes (SWCNTs), multi-walled carbon nanotubes (MWCNTs), mildly oxidized MWCNTs (OCNTs), and reduced OCNTs (ROCNTs). (C) Faradaic efficiencies of possible products H2, NO2−, NH2OH, and NH3 obtained from NO3− reduction using SWCNT, MWCNT, OCNT, and ROCNT catalysts. Reproduced with permission from ref. 217. Copyright 2022, American Chemical Society. (D) SEM image of NiCO precursor. (E) SEM image of NiCO2O4 obtained from the precursor upon annealing. (F) TEM image of NiCo2O4 nanowire. (G) NH3 yields and Faradaic efficiencies obtained at different potentials using NiCo2O4/CC nanowire catalyst. (H) Schematic illustration of Zn–NO3− battery using NiCo2O4/CC as the cathode. Reproduced with permission from ref. 214. Copyright 2022, Wiley-VCH. (I) Schematic representation of the fabrication of Mn-incorporated Co3O4 (Co3O4–Mnx) nanotubes. (J) TEM image of Co3O4–Mn2 nanotube, where Mn:Co ratio is 2:1. (K) NH3 yields and Faradaic efficiencies obtained using Co3O4–Mn2 and Co3O4 catalysts at various potentials. Reproduced with permission from ref. 215. Copyright 2023, Elsevier. (L) Schematic representation of the synthetic procedure of Fe2TiO5 nanofibers. (M) SEM image of Fe2TiO5 nanofibers. (N) Faradaic efficiencies of possible products H2, NO2−, and NH3 obtained from NO3− reduction using Fe2TiO5 nanofibers at different potentials. Reproduced with permission from ref. 106. Copyright 2022, Wiley-VCH. | ||
Spinel oxides have a general formula of AB2O4, where A and B are transition metal cations. A is a divalent cation and B is a trivalent cation, occupying tetrahedral and octahedral sites, respectively.273 Due to the distinct electronic structures and reactivities of A and B, their interactions with the reactants also differ significantly. Hence, spinel-type oxides have more catalytic activity compared to single-metal oxides. Bimetallic spinel oxides have a tuneable band gap, better electrical conductivity, and adsorption ability of reactants than single-metal spinel oxides.274 Such a redox-active bimetallic spinel oxide, NiCo2O4, can perform electrocatalytic NO3RR to NH3 under ambient conditions (Fig. 13D–H).214 These spinel oxides have multivalent metals and high electronic conductivity, which are favourable for catalytic reactions. Also, the effect of bimetallic centres Ni and Co result in efficient NO3RR, and the FE reaches a maximum value of ∼99% at −0.3 V vs. RHE, although small amounts of NO2− and N2H4 were obtained as by-products with NH3. Ni2+ acts like a p-type dopant and replaces a Co3+ in Co3O4 to form NiCo2O4. This converts the semiconductor minority spin channel to conducting, and NiCo2O4 possesses half-metal characteristics. The DFT calculations show that the half-metal characteristic of NiCo2O4 facilitates electron transfer. Nitrates are easily adsorbed on the surface of Co3O4 compared to NiCo2O4 but, surprisingly, the NO3RR is more favourable for NiCo2O4. This proves that the Sabatier Principle holds correct, i.e., the adsorption of reactants on the surface should be intermediate, and too weak or too strong interaction disfavours the catalytic process. The catalyst is also used as a cathode in a Zn–NO3− battery with a high yield of 48.5 μmol h−1 cm−2 NH3 and an FE of 96.1%. Another such spinel oxide electrocatalyst, Mn-incorporated Co3O4 (Co3O4–Mn2) nanotubes is synthesized via hydrothermal method and annealing (Fig. 13I–K).215 Mn partially replaces the Co cation in the CoO6 octahedron of Co3O4 to form Co3O4–Mn2. Mn incorporation not only improves conductivity but also suppresses the HER. The surface sites of Co3O4–Mn2 are less active than Co3O4. A Co:Mn ratio 1:2 produces the highest FE of 99.5% at −1.2 V vs. RHE. Doped spinel oxides like carbon-doped cobalt oxide (C/Co3O4) hollow nanotubes also exhibit highly efficient NH3 synthesis from NO2− reduction with FE of almost 100% in the potential range of −0.1 to −0.6 V vs. RHE.216 The C doping facilitates charge transfer by inducing a local electric field and reduces the energy barrier for the *N + e− + H2O → *NH + OH− step, the rate-determining step of the NO2RR process. This electrocatalytic system is further utilized to construct a Zn–NO2− battery that can concurrently degrade NO2−, generate value-added NH3, and create electricity.
Semiconductors are often used extensively as electrocatalysts. However, typical semiconductor materials have less electronic conductivity, which impedes their electrocatalytic activity. The electronic structure of semiconductors can be modified by introducing defects like oxygen vacancies (VO) or by heteroatom doping. Besides modulating the electronic structure, doping TiO2 nanobelts with the P atom induces charge redistribution and generates VOs around the doping sites.212 As a result, P-TiO2 exhibits better NO2RR performance to produce ammonia compared with pristine TiO2. Amorphous boron carbide sputtered on TiO2 (a-B2.6C@TiO2) nanobelts also exhibits selective NH3 synthesis via electrocatalytic NORR.213 Theoretical studies indicate that the B–C bonding in the boron carbide layer effectively injects electrons to the NO π2p* orbital, thus activating NO and ensuring the complete reduction of NO to NH3 by lowering the energy barriers. Employing this electrocatalytic a-B2.6C@TiO2 as the cathode, a Zn–NO battery is assembled to produce ammonia and electricity concurrently. Pseudo-Brookite Fe2TiO5 nanofibers are narrow-band gap semiconductors with TiO2-like atomic and electronic properties (Fig. 13L–N).106 The highly reducing Fe atoms can easily replace the Ti4+ and induce abundant oxygen vacancies (VOs) in the structure. The VOs can reduce the adsorption energy of NO3− and boost the catalytic activity for NH3 production. Iron phosphide (FeP), a transition metal phosphide-based narrow-band gap semiconductor, is also an effective electrocatalyst for NO2RR from wastewater to generate ammonia.275 The NO2− ions bind to the (211) and (011) facets of the two adjacent Fe atoms present in FeP, which are the main active facets for NO2RR. Further, FeP has moderate atomic hydrogen (H*) adsorption capability, which facilitates NH3 formation, whereas excessive H* adsorption leads to competitive HER, reducing the selectivity of NH3.
5.3. 2D electrocatalysts and heterostructures
Two-dimensional carbon-based electrocatalysts are emergent materials for the electrosynthesis of ammonia owing to their diverse benefits such as low cost, easy modification of atomic or molecular structures, and tolerance to acidic and alkaline electrolytes. State-of-the-art 2D carbon-based electrocatalysts for ammonia synthesis include graphene, graphdiyne, and carbon nitrides. These materials also provide superior support for anchoring 0D or 1D nanomaterials to promote their catalytic activity. Engineering their structures by introducing defects or heteroatom doping are various strategies to enhance their catalytic activity. Graphene, one of the most commonly used 2D catalysts, has a nanosheet-like structure with sp2 hybridized carbon atoms, and excellent conductivity and chemical stability.276 The 2D π-conjugated structure of graphene also contributes to the adsorption of N-containing reactants on its surface and facilitates the catalytic reaction. Recently, amorphous graphene has been used extensively for catalytic purposes. The amorphous atomic structural features, as well as the graphene structural features, promote catalytic activity. Such an amorphous graphene electrocatalyst is synthesized by laser irradiation and is composed of disordered four- to eight-membered polygons (Fig. 14A–C).219 The catalytic activity of amorphous graphene has been compared with crystalline reduced graphene oxide (rGO). It is observed that the adsorption properties of amorphous graphene are more due to structural deformations while rGO has lower adsorption properties due to aromaticity. Another graphene-based catalyst with amorphous/crystalline heterophase is synthesized by infrared laser induction.220 This graphene has intermediate crystallinity between amorphous carbon and crystalline graphene, and contains aromatic rings and distorted polygons. The presence of heterophase can modulate the electronic properties of the catalyst as the density of defects and state near the Fermi level increases, resulting in high capacitance. The heterophased graphene has abundant electrons and long-range disorders that favour 8e− NO3RR to NH3. The formation of heterostructures with graphene has certain advantages: (i) promotion of the transfer of electrons to graphene is readily achievable because the Fermi level of graphene is at 0 V vs. NHE, whereas the position of the conduction band of the other component is generally higher than the Fermi level of graphene; (ii) separation of charge carriers is easier due to the rapid electron transfer from the second catalytic component to graphene; (iii) abundant anchoring sites are present in functionalized graphene for the dispersion of nanoparticles or single-atoms. Single-atoms (SAs) are one of the most emerging catalysts in recent times as discussed above. The SAs mostly coordinate to four N atoms, but this induces weak polarity to the SAs, which is unsatisfactory for catalytic processes. If this coordination with N can be partially substituted with heteroatoms like O or S, then the polarity on SA sites will increase, resulting in the adsorption of more NO3−. Functionalized graphene with two N and two O in cis configuration can encapsulate Cu SAs.221 As discussed before, the polarity of Cu sites causes more NO3− adsorption on the electrocatalytic surface and the cis configuration modifies the 3d orbitals of Cu so that it can form a π-complex with *ONH and reduce the energy barrier of the catalytic process. This reveals that functionalized graphene can also moderate the coordination of the SA catalyst and simulate its catalytic activity. | ||
| Fig. 14 Two-dimensional (2D) electrocatalysts for ammonia synthesis. (A) HRTEM images of amorphous graphene obtained by laser induction in air (ox-LIG) at different magnifications. (B) HRTEM image of reduced graphene oxide (rGO) at different magnifications. (C) NH3 yields and Faradaic efficiencies obtained at different time intervals using amorphous graphene obtained by laser induction in air (ox-LIG), in an inert atmosphere (LIG), and reduced graphene oxide (rGO) at a potential of −0.73 V vs. RHE. Reproduced with permission from ref. 219. Copyright 2023, Wiley-VCH. (D) Schematic representation of defective boron carbon nitride (BCN) nanosheets with unsaturated B and N atoms as a frustrated Lewis pair (FLP). The pulling effect of FLPs captures and activates N2, for N2 reduction to NH3. (E) STEM image of BCN. (F) NH3 yields and Faradaic efficiencies obtained using BCN catalyst at different potentials. Reproduced with permission from ref. 227. Copyright 2022, Wiley-VCH. (G) Schematic representation of OH-terminated Ti3C2 MXene, where the hydrogen bonding between –OH groups and NO3− facilitates NO3− reduction to NH3. (H) SEM image of Ti3C2 MXene. (I) Faradaic efficiencies of NH4+ and NO2− obtained at different potentials using Ti3C2 MXene. Reproduced with permission from ref. 233. Copyright 2023, Wiley-VCH. (J) Lattice structures of the 2H, 1T′, and 1T′′ phases of MoS2. (K) Comparison of NH3 yields and Faradaic efficiencies for 1T′′′ and 1T′ MoS2 at a potential of −0.3 V vs. RHE and for 2H MoS2 at a potential of −0.4 V vs. RHE. Reproduced with permission from ref. 238. Copyright 2021, Wiley-VCH. | ||
Graphdiyne (GDY), a carbon allotrope with one-atom-thickness is an emerging 2D carbon material. It comprises sp and sp2 hybridized carbon atoms and has unique properties like high surface area, large cavity structure, large network plane, excellent hole transport properties, uneven surface charge, and multiple active sites, which make it an excellent two-dimensional catalyst.193,277 Recent studies indicate that GDY-based low-dimensional materials exhibit excellent activity, selectivity, and stability toward electrocatalytic ammonia synthesis. Due to the presence of the reductive alkaline bond in the structure of GDY, it can self-reduce Pd2+ to form Pd-GDY.278 The coupling interaction between the Pd, C1, and C2 sites of GDY enhances the electron transfer process, thereby increasing the activity and selectivity of electrocatalytic N2RR to produce ammonia with 100% selectivity in a neutral medium. Individual zero-valent atoms have unique catalytic properties owing to their electronic structure, high activity, and selectivity. One such zero-valent Mo0-GDY catalyst has been prepared based on the incomplete electron transfer properties between the GDY and Mo atom.222 The atomically dispersed Mo0 atoms endow the catalyst with excellent activity toward electrocatalytic nitrogen reduction and hydrogen evolution reactions with highly efficient ammonia and hydrogen production, respectively. The confinement of single-atoms (SAs) like Ru, Rh, and Co in GDY structures also confers excellent properties toward electrocatalytic nitrogen reduction reactions to produce ammonia.271,279 Single-atom (SA) and double-atom (DA)-adorned GDY like Cu2-GDY also exhibits enhanced NORR for ammonia generation.224 This work shows that the d-band centre plays a pivotal role in the adsorption and hydrogenation of NO. The NO molecule activation on SAs and DAs is driven by electron “donation/back-donation” interactions between the metal atom and NO. On the SAs, the thermodynamic process (NH3 and H2O molecule desorption) dominates the entire NORR process, and Cu-GDY exhibits high NORR with selective NH3 formation over H2. In contrast, on the DAs, the electrochemical hydrogenation processes control the NORR, and Cu2-GDY exhibits the highest selectivity toward NH3 among Fe2, Co2, Ni2, and Cu2. These works explore the utilization of hybridized 0D atomically dispersed catalysts on 2D GDY-based structures toward electrocatalytic ammonia generation. Over the past few years, GDY-based heterojunction catalysts have also been developed to enhance electrocatalytic ammonia synthesis. For instance, the Fe3C@GDY heterojunction catalyst has electron-donating triple bonds in GDY and electron-accepting Fe3C, wherein, the sp-carbon–metal–carbon structures at the interface promote charge transfer and electrical conductivity in the catalyst.225 GDY can also regulate the coordination environment of the Fe atoms and thereby improve the adsorption and desorption of the reactants and intermediates, promoting electrocatalytic nitrate reduction to ammonia. The incomplete charge transfer between the donor–acceptor Fe3C@GDY interface also enhances the activity and selectivity of the catalytic ammonia production. Compared to the pristine catalysts, the heterogenized GDY structures have faster electron transfer properties and the integration with GDY also provides enhanced stability to the heterogenized catalysts.
Boron (B)-based carbon 2D materials have also emerged as efficient electrocatalysts for ammonia synthesis. Doping carbon-based materials with boron or nitrogen results in charge redistribution, boosting the chemisorption of reactants.280 The B and N doping also tune the band gap, spin, and charge density of the boron carbon nitride (BCN), promoting N2RR and NOxRR over HER. The empty sp2 orbital of B can interact with the lone pair of N electrons to adsorb and activate the N-species and hinder the binding of H+ to suppress the competitive HER. Surface anion vacancies can enhance the availability of electrons and provide suitable active sites for binding N2 molecules. Defective BCN nanosheets with unsaturated B (e−-deficient) and N (e−-rich) atoms form the frustrated Lewis acid (LA) and base (LB) pairs (Fig. 14D–F).227 These adjacent LA and LB pairs can efficiently adsorb N2 to form a six-membered ring as an intermediate and allow dissociation of NN at much lower energy due to the pull–pull effect by heterolysis. The presence of frustrated Lewis pair B and N as dual active sites increases the electrocatalytic activity of BCN and promotes N2RR. The tunability of the Lewis pairs of B and N has substantial impacts on N2RR.226 The B-enriched BCN exhibits better N2RR activity than the N-enriched BCN. Theoretical studies indicate that the energy of each step of N2RR by B-enriched BCN is relatively lower than that of N-enriched BCN, which results in enhanced N2RR by B-BCN.
2D metal and metal oxide-based nanostructures are extensively used in electrocatalysis due to their high surface-to-volume ratio, unique electronic structures, and abundant exposed active sites. For instance, Ru nanosheets with low coordination numbers can exhibit electrocatalytic properties toward NORR to NH3.228 The low coordination number of the Ru sites promotes the adsorption of NO molecules and lowers the energy barrier of the rate-determining hydrogenation step. The use of expensive noble metals can be circumvented by replacing them with redox-active transition metals, such as Co. Co nanosheets with hexagonal-close-packing (hcp) act as efficient catalysts for NORR compared to the face-centered cubic (fcc) phase of Co nanosheets.229 Enhanced electron donation from the d-π* orbitals of hcp-Co to the adsorbed *NO facilitates NORR. Additionally, the proton diffusion process in the hcp-Co is energetically favourable, ensuring the availability of more protons for the protonation of NO to NH3. Bimetallic catalysts provide more active sites than monometallic catalysts for multistep N2RR and NOxRR and are often used to synthesize ammonia as they mimic bimetallic nitrogen reductase (MoFeP). A bio-inspired CuCo bimetallic nanosheet mimicking Cu–nitrogen reductase is formed by electrodeposition of the corresponding metals and this bimetallic catalyst can electroreduce NOx− to NH3.230 The two active metal centres in the catalyst have separate roles—the Cu centre facilitates the adsorption of NOx− while the Co centres participate in the donation of electrons and protons. DFT calculations show that the adsorption of *NO (the rate-determining step) consumes less energy for CuCo, when compared to Cu and Co metals, separately. The FE for the Cu50Co50 catalyst reaches ∼100% at −0.2 V vs. RHE with a current density of 1035 mA cm−2. Metal oxides with defects and vacancies can also enhance the catalytic activity of N2RR and NOxRR by altering the local coordination environment and electronegativity. Amorphous RuO2 nanosheets exhibit superior NO3RR when compared to their crystalline counterparts.231 This superiority is attributed to atomic disorder in the structure of RuO2, which endows it with numerous oxygen vacancies (VOs). The rate-determining step is calculated to be the conversion of *NH2 to *NH3, which is facilitated on the surface of amorphous RuO2. The formation of by-products like NH2OH and H2 are also suppressed on amorphous RuO2, which boosts the selectivity of NH3.
MXenes formed of carbides and nitrides are emerging 2D materials used in catalysis as they have unique properties like high electrical conductance and capacitance.281,282 The introduction of defects on the surface reduces repulsive electrostatic forces and thus decreases the energy required for the adsorption of N2 or NOx−.283 Functionalization of MXenes suppresses competitive HER and improves the catalytic synthesis of ammonia.232 Functionalized Ti3C2 MXenes with terminal oxygen groups formed by mild calcination reveal the active sites for electrocatalytic NO3RR (Fig. 14G–I).233 Small amounts of N2 and hydrazine by-products are obtained, resulting in moderate selectivity of NH3 with an FE of 90.4% at −1.7 V vs. RHE. Under electrochemical conditions, the oxygen groups are converted to hydroxyl groups in situ to form interfacial hydrogen bonding, thus accelerating the electrocatalytic NO3RR process. These surface hydroxyl groups help the adsorption of NO3− and contribute to hydrogenation, forming NH3; hence, the –OH group functions as both an active site and a reactant. The adsorption of H on the O atoms also diminishes the HER process, increasing the selectivity of NH3. Like other 2D materials, heteroatom doping of MXenes can also effectively increase the rate of NO3RR and enhance the formation of NH3. Doping of B atom into the lattice of B-Ti3C2Tx (Tx depicts the surface terminating group) MXenes can alter their electronic structure and accelerate the NO3RR kinetics compared to the pristine Ti3C2Tx MXenes to produce ammonia with a high current density and at a low working potential.234 The B dopants facilitate the adsorption and activation of NO3RR intermediates, reduce the energy barrier and thereby enhance ammonia production.
MBenes, a newly emerging class of 2D layered materials similar to MXenes, are gaining attention as promising electrocatalysts. Unlike MXenes, MBenes lack passivating surface functional groups, which allows the constituent metal and boron atoms to be fully exposed, enhancing their catalytic activity. The combined effects of metal and boron make MBenes attractive catalytic materials. The metal atoms participate in water splitting and the boron atom activates the N-containing species. A FeB2 MBene synthesized via the reflux method exhibits efficient NO3RR with an FE of 96.8% at −0.6 V vs. RHE.235 Fe acts as the *H donor and B atoms act as the *H acceptor, and this tandem action promotes the NO3RR process. Theoretical studies reveal that the adsorption and activation of NO3− take place at the B sites rather than Fe, confirming B as the active site for catalysis. Water catalysis occurs at Fe and the *H that is generated is transmitted to B through the hydrogen spillover process for subsequent hydrogenation reactions. Another study screens a series of M2B2-type (M = IVB to V transition metals from the periodic table) MBenes for electrocatalytic NORR.284 Among the screened MBenes it has been observed that the Fe2B2, Mn2B2, and Rh2B2 can efficiently convert NO to NH3 with smaller limiting potentials, whereas Nb2B2 and Hf2B2 have low limiting potentials for the NO conversion to NH3. Mechanistic investigations indicate that hydrogenation of *NO to *NOH has lower energy than *HNO; hence, these MBenes have high selectivity for promoting the NORR to NH3 over competitive HER.
Layered transition metal chalcogenides like MoS2 have also been actively used as catalysts for N2 and NOx− reductions. The positive charge on the Mo atom can polarize and activate the adsorbed reactants via Mo–N interactions. Heteroatom doping and defect engineering techniques have been employed to enhance the interaction between Mo and N species. Layered structured MoS2 is a well-known catalyst for HER. Substituting S atoms with F introduces strain in the layered structure and compresses the interlayer spacing in the MoS2 nanosheets.236 Additionally, F is more electronegative than S and these factors can suppress HER and promote N2RR. The introduction of defects in the form of dopants like V can remarkably enhance the efficiency of electrocatalytic NO3RR.237 V is highly conductive and can alter the electronic structure of the MoS2 metalloenzyme. The V-MoS2 electrocatalyst can lower the energy barrier of conversion of NO* to NOH* and also enhance the selectivity of NH4+. Recent works reveal that the metastable phase of MoS2 is more active for performing catalytic reactions and can efficiently reduce N2 to NH3 (Fig. 14J–K).238 Metastable MoS2 provides access to partially filled t2g orbitals, which can simultaneously form σ bonds with N and transfer electrons to N2. The work compares the 2H (stable), 1T′, and 1T′′′ (metastable) phases of MoS2 for the catalytic reactions, and the 1T′′′ phase with maximum electron density was found to be the most suitable for N2RR. Mo–Mo clustering in the metastable phases is responsible for the enhanced electron density in these phases, facilitating activation of N2 and accelerating N2RR, which is almost nine times greater than the stable 2H phase. MoS2 nanosheets deposited on graphite can also produce ammonia from the electroreduction of NO.285 The positively charged Mo-edge sites of MoS2 promote the adsorption and activation of NO via an “acceptance–donation” mechanism, which promotes NORR to NH3 and disfavours the binding of protons and coupling of the N−N bond, ruling out the formation of H2 and N2 and enhancing the selectivity of NH3 produced.
5.4. Metal–organic framework (MOF)-based electrocatalysts and heterostructures
Metal–organic frameworks (MOFs), composed of metal ions or clusters linked by organic ligands in a three-dimensional arrangement, are one of the most explored emergent materials in catalysis. Their porous structure, high specific surface area, excellent stability, and presence of active sites offer unique features required for catalysis. For instance, a 2D Cu-based MOF, {[Cu(HL)]·H2O}n, (Cu-OUC, H3L = 5-(2′-carboxylphenoxy)isophthalic acid) exhibits efficient electrocatalytic NO-to-NH3 conversion.239 The Cu-OUC MOF activates NO via a two-way charge transfer mechanism of “electron acceptance and donation”, where the *NO formation step is the rate-determining step. Additionally, the Cu-OUC MOF also acts as the cathode for constructing a Zn–NO battery, which besides converting harmful NO into NH3 also generates electricity. The reaction rates of ammonia formation depend on the type of metal centres present in the MOFs. In Fe2M trinuclear cluster-based MOFs, where M = Co, Ni, Fe, Zn, the rates of adsorption and activation of nitrate and protons are in the order of Co > Fe > Ni > Zn (Fig. 15A–C).240 The unsaturated metal sites of the trinuclear complex help in the adsorption and reduction of nitrates while the dinitrogen ligand aids in the interfacial transfer of electrons due to the delocalized conjugation between the lone electron pairs of nitrogen and the π electrons of the ligand's aromatic ring. Ammonium sulfate is obtained by the electroreduction of nitrates and can be directly used as fertilizer for plants without further processing. MOFs have also been used as precursors for deriving metal- and carbon-based catalysts, which inherently have higher reactivity and stability when compared to conventional catalysts. Co–Fe/Fe2O3 has been derived from Co-doped Fe-MOF-74 by pyrolysis, where Co dopant increases conductivity and tunes the electronic properties such that Co–Fe/Fe2O3 shows efficient electrocatalysis.286 The temperature at which pyrolysis is carried out is crucial as experimental evidence suggests that the graphitic carbon produced at 900 °C shows the highest electrocatalytic performance while that produced at 1000 °C shows reduced catalysis due to the collapse of structural properties. The Co center activates NO3− and tunes the energy of the d orbitals of adjacent Fe atoms, facilitating charge transport; thus, both Co and Fe atoms participate in NO3RR. Co also restrains HER, thus increasing the selectivity and FE to 99% and 85.2%, respectively. The conversion of NO3− to NH3 involves deoxygenation and hydrogenation of nitrates with water via proton-coupled electron transfer. This coupling reaction between nitrates and water can be boosted if a multifunctional interface can be produced by incorporating a second component with the MOF. For instance, when the bimetallic Ni/Co-MOF interface is surface-coated with H-substituted graphdiyne, deoxygenation, and hydration reactions are promoted due to the formation of the multifunctional interface (Fig. 15D–F).241 The intersection between the MOF and graphdiyne acts like a bifunctional membrane by facilitating the transport of electrons and reactants to the Ni and Co sites, where the concerted reaction occurs. An integrated-electrocatalysis cell using MOF-based gas diffusion electrodes (GDEs) can electroreduce NO (NORR) and electrooxidize NO (NOOR) simultaneously to produce NH4NO3 under dilute concentrations of NO.287 For preparing the MOFs-GDEs, UIO-66, ZIF-8, and ZIF-67 are modified with Cu nanowires for NORR and Ni/NiO for NOOR to enhance the production of NH4+ and NO3−, respectively. The enhanced NO reduction and oxidation is due to the transfer of adsorbed NO from the adsorption layer to the catalyst layer, which is experimentally verified by the enhanced NO mass transfer from gas to electrolyte across the modified electrode. Modifying MOFs without doping or other functionalization can improve fabrication time efficiency and enhance material properties for ammonia production. One such example is Fe metal center-based MOF, which in its thermally activated form can generate ammonia with FE of ∼90% at −1.0 V vs. RHE, which is much higher when compared with the non-activated pristine form of the MOF (Fig. 15G–I).242 The activation of MOFs is expected to provide access to more active Fe sites for the adsorption of nitrates. The thermal activation process frees the pores of the MOF from trapped components and exposes the high-valent Fe(III) sites for the catalytic reaction. Such activation processes can further open avenues for tailoring the surface structures of desired electrocatalysts like MOFs for energy conversion reactions. | ||
| Fig. 15 Metal–organic framework (MOF)-based electrocatalysts for ammonia synthesis. (A) Schematic representation of NO3− reduction to NH3 by Fe-based trinuclear cluster metal–organic framework (MOF) Fe2M-MOF, where M = Fe, Co, Ni, or Zn. (B) SEM image and element mapping of C, Fe, and Co present in Fe2Co-MOF. (C) NH3 and NO2− yields and the corresponding Faradaic efficiencies using Fe2M-MOFs at a potential of −1.1 V vs. RHE. Reproduced with permission from ref. 240. Copyright 2023, Wiley-VCH. (D) Schematic representation of Ni/Co-MOFs (NiCoBDC) with hydrogen-substituted graphdiyne (HsGDY) nanowire array for electrochemical NO3− reduction to NH3. (E) SEM images of NiCoBDC@HsGDY nanoarray at different magnifications. (F) NH3 yields at different potentials using NiCoBDC, NiCoBDC@HsGDY, CoBDC@HsGDY, and NiBDC@HsGDY. Reproduced with permission from ref. 241. Copyright 2023, American Chemical Society. (G) Schematic representation of the thermal activation of the Fe-MOF from pristine to activated form. (H) SEM images of activated Fe-MOF at different magnifications. (I) Comparison of Faradaic efficiencies for NO3− reduction between pristine and activated Fe-MOFs at different potentials. Reproduced with permission from ref. 242. Copyright 2023, American Chemical Society. | ||
5.5. 3D-printed electrodes based on low-dimensional materials
To design active and stable electrocatalysts for ammonia production, researchers have focused on designing 3D-printed electrodes. Previously, 3D-printed electrodes have been employed in catalytic reactions like carbon dioxide reductions,18 oxygen evolution,288 and hydrogen evolution289 reactions; however, the use of 3D electrodes in N2RR and NOxRR is quite recent and has some advantages over conventionally prepared catalysts. The 3D printing technique enhances the designing of the catalysts and the reaction vessels by automating the prototype and simulations to prepare an optimized catalytic system.48 This eliminates the manual time required for optimization for conventional catalysts, boosting the performance of the process. The 3D printing process allows for precise control over the shape, structure, and geometry of substrates, leading to enhanced catalytic performance. The 3D-printed substrates can be post-modified with desired active materials by sintering, sputter coating, electrodeposition, and atomic layer deposition.290–293 For instance, in a recent study by our group, a Cu electrode is fabricated by fused fabrication filament (FFF) 3D printing (Fig. 16A–I).243 The 3D-printed electrode is subjected to thermal treatment and sintering to induce conductivity and the metallic phase to the electrode. An electrochemical modification is then induced to the electrode followed by NaHCO3 treatment for forming nanostructures on the surface of the electrode with exposed (100) facets. The main composition of the electrode responsible for electrocatalytic NO3RR has been observed to be Cu/Cu2O. These 3D-printed Cu electrodes can perform efficient NO3−-to-NH3 conversion with FE 96.5% at −0.92 V vs. RHE with 95.6% selectivity for NH3. Another work from our group compares the electrocatalytic properties of two 3D-printed electrodes: (i) 0-dimensional porous carbon black and (ii) 1-dimensional porous carbon nanotubes (Fig. 16J–M).244 The 0D carbon black 3D-printed electrode is electrocatalytically inert while the 1D carbon nanotubes actively participate toward NO3RR. The defects and metallic impurities (TiO2, Fe3O4) in 1D CNTs can impact the electrocatalytic reactions. However, the exact content and distribution of the impurities and defects are not quantifiable in industrial catalysts; hence, proper control over electrocatalysis is not always possible over industrial catalysts. To achieve better control, an ultra-thin layer of manganese oxide is deposited onto the 1D carbon nanotubes using the atomic layer deposition technique. This fine-tunes the surface properties of the catalysts and enhances their electrocatalytic performance. The carbon nanotubes are conductive and manganese oxide has electrocatalytic properties, and the synergistic effect from these two components makes the 3D-printed 1D CNT@MnOx efficient toward NO3RR. Surface-modified 3D-printed carbon substrates with atomic layer deposition (ALD) of TiO2 can function as bifunctional platforms for electrocatalytic nitrite oxidation reaction (NO2OR) and nitrite reduction reaction (NO2RR).245 This 3D-printed heterostructure possesses the intrinsic surface properties of carbon nanotubes as well as Ti-dominated metallic impurities, and the formation of interfaces between the conductive carbon and ALD-coated TiO2 enhances its electrocatalytic properties toward both NO2OR and NO2RR. The electrocatalytic properties of the TiO2-coated 1D carbon electrode are associated with the ALD-coated TiO2 layer, which can be tuned by modulating the number of ALD cycles. The 100-TiO2 electrode, formed by non-continuous TiO2 deposition on the 1D carbon framework is endowed with abundant carbon/TiO2 interfaces. These interfaces play a vital function in the electrocatalytic reactions and enhance the formation of NH4+ and NO3− from electro-reduction and oxidation, respectively. | ||
| Fig. 16 3D-printed electrodes designed with low-dimensional materials for ammonia synthesis. (A) Schematic representation of the electrocatalytic cell for NO3− reduction to NH3 using a 3D-printed Cu electrode. (B) SEM image and (C) Cu elemental mapping of the electrode. (D) Schematic representation of the 3D-printed electrode. (E) Pictorial representation of the dimensions of the electrodes, where the length and width are 10 mm each, and the height is 1 mm. (F) Sintering of the electrodes. (G) 3D-printed Cu electrode before (left) and after (right) acid treatment. (H) Faradaic efficiencies of NH3 and NO2− at different potentials using 3D-printed Cu electrodes. (I) NH3 yields at different potentials using 3D-printed Cu electrodes. Reproduced with permission from ref. 243. Copyright 2023, American Chemical Society. (J) Schematic representation of NO3− reduction to NH3 using 1-D@MnOx fabricated by atomic layer deposition of MnOx on a 3D-printed 1D carbon framework. (K) SEM image of 1D carbon framework. (L) HAADF-STEM image of 1D@500-MnOx. (M) NH3 yields at different potentials using 3D-printed 1D carbon and 1D@500-MnOx electrodes. Reproduced with permission from ref. 244. Copyright 2023, Elsevier. | ||
6. Low-dimensional photoelectrocatalysts for ammonia synthesis
Photoelectrocatalysts combine the dual benefits of both photocatalysts and electrocatalysts. Photocatalysts provide light energy for catalysis, and electrocatalysts offer the advantage of the electric bias. In conventional photocatalysts, the efficiency is sometimes limited by the recombination of the charge carriers. In photoelectrocatalysis, the recombination is paused by applying an external potential greater than the onset potential, which draws out charge carriers from the photoelectrode. N2 and NOx− act as effective feedstock for the production of ammonia. Over the last few years, a diverse class of electrocatalysts has been studied for NH3 generation. However, many of these electrocatalysts face a significant drawback: they require high overpotential to achieve considerable Faradaic efficiency (FE) for ammonia production. Photoelectrocatalysis can reduce this potential; the photoelectrodes can harness the light energy to generate a photovoltage and compensate for the reducing potential. The reduction potential for NO3RR is near that of water splitting. Thus, from a thermodynamic viewpoint, the catalysts capable of photoelectrochemical water splitting are also prospective catalysts for NO3RR. But kinetically, NO3RR is more challenging and energy demanding as it requires 8e−s for complete conversion of NO3− to NH3; otherwise, lower-reduced products like NO2−, NO, N2, and NH2OH might be formed. Modification of the photocathode with proper catalysts and cocatalysts leads to active NO3RR and selective production of NH3. Herein lies the advantages of photoelectrocatalysts. The following subsections will discuss the design of emerging low-dimensional photoelectrocatalysts developed over recent years for ammonia generation (Fig. 17). The design of photoelectrocatalysts generally involves multi-step reactions for heterogenization with multiple-component photocatalysts and electrocatalysts. As the photoelectrocatalysts are composed of photocatalysts and electrocatalysts, the synthetic strategies resemble the photo- and electrocatalysts mentioned in Sections 4 and 5 of this review. The synthesis and modification techniques of some selected photoelectrocatalysts are listed in Table 5. | ||
| Fig. 17 Low-dimensional photoelectrocatalysts (PECs) for ammonia synthesis. Classification of photoelectrocatalysts into 0D, 1D, and 2D, and schematic representations of few selected photoelectrocatalytic LDMs. | ||
| Classification | Materials | Photoelectrocatalysts | Synthesis/Modification | Ref. |
|---|---|---|---|---|
| 0D LDMs and heterostructures | Metallic nanoparticles (NPs) | Au NPs-WS2@RGO | Hydrothermal, microwave methods, chemical reduction | 294 |
| Ag NPs-black Si | Etching, deposition methods | 51 | ||
| NiO–Au NPs-TiO2 | Chemical reduction, thin film preparation, deposition methods | 295 | ||
| Quantum dots (QDs) | Bi2S3 QDs-MoS2 | In situ growth of QDs, hydrothermal and solvothermal methods | 296 | |
| Single-atoms (SAs) | Ru SAs–Cu2O | SA embedding by simple mixing | 181 | |
| 1D LDMs and heterostructures | Metal and metal oxide-based heterostructures | MoS2 nanoflakes-La2Zr2O7 nanofibers | Electrospinning, hydrothermal methods | 87 |
| Ni–MoS2/Si nanowires | Ni-Doping, metal-assisted chemical etching, hydrothermal, cast-coating methods | 88 | ||
| Si-α-Fe2O3 nanorods | Hydrothermal, thermal annealing methods | 297 | ||
| B–Bi nanorolls | B-Doping, chemical reduction | 298 | ||
| 2D LDMs and heterostructures | Metal oxide-based heterostructures | TiOx–CdS–Cu2ZnSnS4 | Defect engineering of cocatalysts, chemical bath deposition, spray coating | 299 |
| Carbonaceous materials | CoTiO3 nanorods/N-rGO nanosheets | Interface engineering, N-doping, p–n junction, reflux method | 300 | |
| CuPc–CeO2 | Defect engineering, thin film fabrication, chemical deposition | 301 |
6.1. 0D photoelectrocatalysts and heterostructures
Photoelectrocatalysts for N2RR and NOxRR toward ammonia synthesis encounter similar challenges like photocatalysts and electrocatalysts. The design of stable and selective photoelectrocatalysts for ammonia production using various strategies with improved efficiency is discussed here. Narrow-band gap materials like CuO, Cu2O, and black P are mostly used for photocathodes.49,302 Sometimes, photoelectrocatalysts comprise heterostructures of different low-dimensional materials. Plasmonic nanoparticles like Au loaded on 2D WO3@rGO enhances photoelectrocatalytic performance (Fig. 18A–C).294 The excellent catalytic properties of AuWO3@rGO for N2RR are attributed to its electronic conductivity, porous structure, and high surface area. When low-band gap black silicon is integrated with plasmonic silver (Ag) nanoparticles, the hot electron generation and plasmonic resonant energy transfer properties of Ag, combined with the light absorption capabilities of black silicon, significantly enhance photoelectrocatalytic N2RR.51 The Ag nanoparticles also protect the black silicon from oxidation. The plasmon hot electrons generated from plasmonic Au nanoparticles also activate the NO3RR processes. In a typical NiO/Au plasmon/TiO2 photosystem, NO3− are reduced to ammonia by the plasmon hot electrons at room temperature and neutral pH.295 These hot electrons also suppress H2 formation and the hot holes are converted to atmospheric O2. The plasmon electrons enhance the electrochemical process by achieving optimal efficiency at lower potentials. The lower working potentials confirm the suppression of competing HER, overcoming one of the major drawbacks of electrocatalytic NO3RR. Quantum dots (QDs) also exhibit excellent photoelectrocatalytic properties, e.g., Bi2S3 QDs grown in situ over MoS2 nanoflowers (Fig. 18D–G).296 Upon photoexcitation with visible light, interfacial charge transfer occurs from the QDs to MoS2. This process results in the separation of photogenerated charge carriers, leading to a higher concentration of photogenerated electrons on the MoS2 side. The electron-rich MoS2 then actively participates in the catalytic reduction of N2 to NH3. Single-atoms (SAs) like Ru can also be effective for designing photoelectrocatalysts. Using Ru SAs embedded Cu2O as photocathode and TiO2 as photoanode in two-chamber cells can function as a dual system for ammonia synthesis and degradation of bisphenol A (Fig. 18H–K).181 The electrons supplied by the TiO2 photoanode can reduce N2 to NH3 at the photocathode and, simultaneously, the bisphenol A can be degraded at the photoanode by the ˙OH radicals. This technique is highly prospective in water treatment and energy generation, and further bifunctional systems like this will be discussed in Section 7.1. | ||
| Fig. 18 Zero-dimensional (0D) photoelectrocatalysts for ammonia synthesis. (A) Schematic representation of the 0D plasmonic Au nanoparticles anchored on 2D WS2@rGO. (B) HRTEM image of Au–WS2@rGO; zoomed-in images show d-spacing corresponding to (111) and (002) planes of metallic Au and hexagonal phase of WS2, respectively. (C) NH3 yields and corresponding Faradaic efficiencies using WS2, Au–WS2, and Au–WS2@rGO at a potential of −0.4 V vs. RHE. Reproduced with permission from ref. 294. Copyright 2023, Elsevier. (D) Schematic representation of Bi2S3 quantum dots (QDs) grown over MoS2 nanoflowers. (E) TEM image of MoS2 nanoflower. (F) TEM image of Bi2S3 QDs marked by red circles, grown over MoS2. (G) NH3 yields and the corresponding Faradaic efficiencies using different loadings of Bi2S3:MoS2. Reproduced with permission from ref. 296. Copyright 2022, Elsevier. (H) Schematic representation of the photoelectrochemically driven N2 reduction to NH3, where TiO2 is the photoanode and Ru single-atoms decorated over Cu2O is the photocathode. (I) SEM image of TiO2 photoanode. (J) HRTEM image of Cu2O/Ru photocathode, where the Ru single-atoms are denoted by red circles. (K) NH3 yields and the corresponding Faradaic efficiencies using different photoelectrocatalysts. Reproduced with permission from ref. 181. Copyright 2022, Elsevier. | ||
6.2. 1D photoelectrocatalysts and heterostructures
Creating hybrid heterostructures and leveraging the interactions of the components stands out as one of the most effective strategies in photoelectrocatalysis. Incorporating MoS2 nanoflakes on 1D La2Zr2O7 nanofibers creates a heterostructure that combines their individual properties for effective photoelectrochemical ammonia synthesis.87 MoS2 offers the advantages of semiconductors while La2Zr2O7 introduces interfacial oxygen vacancies, and the combined effects result in an efficient photocathode. The photogenerated electrons from MoS2 are transferred to the vacant oxygen sites of La2Zr2O7, which participates in N2 reduction. The use of doped materials is another effective strategy for designing photoelectrocatalysts. Ni-doped MoS2 coated on silicon nanowires acts as an efficient photocathode for ammonia production in porous water (Fig. 19A–E).88 Combining two materials with limited catalytic properties into a heterostructure can enhance their catalytic performance by creating a unique interface between the components. Si nanowires and MoS2 individually exhibit lower conductance. But when MoS2 is doped with Ni and coupled with Si nanowires to form a heterostructure 1D–2D type electrode, the electron transfer rate is accelerated and so is the efficiency of photoelectrochemical N2RR. Porous water has a high solubility of N2, which removes the solubility constraint. MoS2 and Si nanowires are low-band gap materials that form a type I heterojunction, and doping with Ni provides additional active sites and increases the motility of charge carriers; and all these factors together enhance N2RR. Doping can also benefit the design of photoanodes. For instance, Mo-doped BiVO4 photoanodes can transfer electrons from water to the Si photovoltaic-wired hematite-based photocathode (Fig. 19F–I).297 Upon irradiation, the photocathodes can reduce N2 to NH3. The process is coupled with H2O2-dependent oxyfunctionalization, which produces valuable materials simultaneously at the photocathode and photoanode. The doping of B in 1D Bi nanorolls photocathode also facilitates N2RR by decreasing the energy barrier of the step N2 → *NNH (Fig. 19J–M).298 The surface curvature of the nanorolls improves the adsorption of N2. Upon irradiation, the TiO2 photoanodes supply the necessary photogenerated electrons to the B-doped Bi photocathode N2RR. Ordered Si nanowires, decorated with Au particles as the designed photocathode material, can operate at positive potential (0.2 V vs. RHE) using solar energy.85 The hexagonal, ordered nanowire array of p-type semiconductor, Si allows efficient mass transport and reduces the recombination of charges. The Au particles act as the cocatalyst and aid in the selective formation of NH3 with an FE of 95.6%. | ||
| Fig. 19 One-dimensional (1D) photoelectrocatalysts for ammonia synthesis. (A) Schematic representation of the fabrication of Ni-doped MoS2/Si nanowires (Ni–MoS2/Si NWs) photocathode. (B) SEM image of 1D Si nanowires. (C) TEM image of MoS2. (D) TEM image of Ni–MoS2. (E) NH3 yields obtained using (a) Si nanowires, (b) MoS2/Si NW, (c) Ni–MoS2/Si NW, and (d) Ni–MoS2/Si NW in porous coordinated polymer (PCP) at a potential of 0.25 V vs. RHE. Reproduced with permission from ref. 88. Copyright 2023, American Chemical Society. (F) Schematic illustration of Mo:BiVO4 photoanode oxidizing H2O to produce H2O2in situ, which activates peroxygenase for enantioselective oxyfunctionalization reactions. (G) Schematic illustration of Si-wired α-Fe2O3 photocathode for paired N2 reduction to NH3. (H) SEM image of α-Fe2O3. (I) NH3 yields obtained using the above illustrated photoelectrochemical setup at different potentials. Reproduced with permission from ref. 297. Copyright 2023, Elsevier. (J) Schematic representation of the fabrication of 1D Bi-doped Bi nanorolls from 2D BiOBr nanosheet precursors. (K) TEM image and (L) HRTEM image of B-doped Bi nanorolls. (M) NH3 yields and corresponding Faradaic efficiencies using B-doped Bi nanorolls. Reproduced with permission from ref. 298. Copyright 2021, Elsevier. | ||
6.3. 2D photoelectrocatalysts and heterostructures
2D black phosphorus (P) nanosheets offer significant properties that are beneficial for N2RR. Black P is also a direct band gap material with a wide range of light absorption properties. It has abundant surface and edge sites and weak hydrogen adsorption properties. 2D black P photocathodes can produce N2 effectively and have excellent stability for up to 12 hours (Fig. 20A–D).302 The formation of a strong interfacial junction between p-type perovskite oxide CoTiO3 and N-doped reduced graphene oxide (N-rGO) can also optimize N2RR.300 The interfacial heterojunction can promote the electron transfer encapsulated between the atomic layers on N-rGO by reducing the diffusion path length and the recombination of charge carriers. Additionally, the heterojunction broadens the wavelength window, enabling more efficient light harvesting. Defects and vacancies also boost the photoelectrocatalytic reactions. In an organic–inorganic hybrid like Cu phthalocyanine (CuPc)/CeO2 heterostructure, the Ce3+/Ce4+ pairs modify the oxygen vacancies, which play a predominant role in the adsorption and activation of nitrates.301 The 2D macromolecular CuPc has similar properties like p-type semiconductors, which are active under UV-visible light and can efficiently convert solar charge carriers. Also, the d orbitals of Cu match the energy level of LUMO π* of NO3−, allowing charge transfer between Cu and NO3−. However, CuPc lacks an adequate number of active sites on its surface, necessitating its integration with other catalysts such as CeO2301 or BiVO4303 to perform NO3RR. CeO2 has Ce3+ and Ce4+ oxidation states and abundant oxygen vacancies, which can entrap photogenerated charge carriers and facilitate the adsorption of nitrates. BiVO4 is a p-type semiconductor with a large band gap, which limits its photocatalytic performance, and coupling with CuPc makes BiVO4/CuPc an effective catalyst. Defect-engineered materials like TiOx with oxygen vacancies (VOs) are effective cocatalysts for NO3RR. For the construction of the photocathode, a p–n junction is fabricated by coating Cu2ZnSn4 with the CdS layer (Fig. 20E–H).299 Cu2ZnSn4 has excellent light harvesting properties, and CdS has efficient charge separation properties. TiOx cocatalyst with Ti3+/VO sites allows better adsorption of NO3− and *NO2 intermediate on the photocathode. The efficient electron transfer reduces the work function and extends the carrier lifetime, which results in an FE of 89.1% for ammonia at 0.1 V vs. RHE. The benefit of the photoelectrochemical strategies mentioned above is that they couple photocatalysis and electrocatalysis, and the synergistic effect can enhance the charge separation, reduction capability of electrons, equilibrium of electron distribution, and also reduce the probability of oxidation of ammonia produced.304 | ||
| Fig. 20 Two-dimensional (2D) photoelectrocatalysts for ammonia synthesis. (A) Schematic representation of the black phosphorus (BP) electrode fabrication using layer-by-layer assembly of exfoliated ultra-thin BP nanosheets. (B) Cross-section SEM image of the fabricated BP electrode. (C) NH3 yields and the corresponding Faradaic efficiencies using BP electrodes at different potentials. (D) Schematic representation of the photoelectrochemical N2 reduction to NH3 by the BP electrodes fabricated on ITO. Reproduced with permission from ref. 302. Copyright 2020, Wiley-VCH. (E) Schematic representation of the step-by-step synthesis of TiOx/CdS/CZTS electrodes. (F) Cross-section STEM image of TiOx-250/CdS/CZTS electrodes, synthesized at 250 °C. (G) NH3 yields and the corresponding Faradaic efficiencies using TiOx-250/CdS/CZTS electrodes at different potentials. (H) Schematic representation of the mechanism of photoelectrochemical NO3− reduction to NH3 by the TiOx/CdS/CZTS electrodes. Reproduced with permission from ref. 299. Copyright 2022, Wiley-VCH. | ||
7. Applications
Besides ammonia production by NOxRR and N2RR, these reactions are also important in many aspects. For instance, nitrates and nitrites are common pollutants found in wastewater and their reduction can help in environmental remediation. NOxRR and N2RR, when coupled with CO2RR, C–N coupling can favour the formation of urea. Various relevant and important oxidation reactions can be coupled with these reduction reactions. All these aspects are highlighted in the following sections.7.1. Water purification
Nitrates and nitrites, the precursors of ammonia synthesis, are common sources of pollutants in surface and groundwater. Hence, wastewater containing nitrates and nitrites can act as N-feedstock for ammonia production, helping in wastewater treatment and producing sustainable energy resources. The major sources of nitrate contamination in water originate from fertilizers used in agriculture, industrial waste (including ammonia-producing industries using the Haber–Bosch process), stormwater runoff from metropolitan areas, and sewage water. The concentration of nitrates in wastewater depends on the source of pollution: 1.95 M in low-level nuclear wastewater, 41.6 mM in industrial wastewater, and 7.4 mM in textile wastewater.305 Nitrates can lead to eutrophication and nitrites produced from the reduction of nitrates pose serious health risks to humans and the animal kingdom. Nitrates and their metabolites can also cause blue baby syndrome, hypertension, and cancers. Hence, the U.S. Environmental Protection Agency has set a limit of 10 mg L−1 of N–NO3− and 1 mg L−1 of N–NO2− in drinking water.306 Conventional denitrification approaches reduce nitrates present in water to gaseous nitrogen, avoiding side products like nitrous oxide or ammonia.307–310 However, recent progress in this aspect highlights the benefits of the reduction of nitrates to ammonia as an important precursor to different value-added products, e.g., fertilizer and a prospective hydrogen storage fuel.311 Here, we will discuss a few state-of-the-art catalytic materials to demonstrate the feasibility of the process. | ||
| Fig. 21 Low-dimensional catalysts for ammonia synthesis coupled with water purification. (A) Schematic representation of electrochemical reactor for wastewater purification containing NO3− using self-activated 0D Ni(OH)2 particles formed on Ni substrate, Ni(OH)2@Ni cathode. (B) TEM image of Ni(OH)2@Ni cathode. (C) HRTEM image of the Ni(OH)2@Ni cathode showing the lattice fringes with d-spacing of 0.233 nm and FFT (inset) corresponding to the (101) plane of Ni(OH2). (D) Effects of NO3−-N concentration on the conversion efficiency, Faradaic efficiency of NO3− reduction, and NH4+–N selectivity under the fed-batch conditions. Reproduced with permission from ref. 312. Copyright 2021, American Chemical Society. (E) Schematic representation of the photocatalytic synthesis of value-added ammonia from nitrate-containing wastewater using Ni single-atoms-decorated defective WO3. (F) SEM image of Ni/HxWO3−y catalyst. (G) HRTEM image showing the Ni single-atoms marked by white circles. (H) NH3 yields using Ni/HxWO3−y catalyst and upon addition of different organic, cationic, and anionic pollutants. Reproduced with permission from ref. 41. Copyright 2024, Elsevier. (I) Schematic representation of the electrochemical NO3− to NH4+ by natural hematite electrode. (J) SEM image of α-Fe2O3 deposited on Ni foam at different magnifications. (K) Comparative study of NO3− removal, NO2− selectivity, NH4+ selectivity, and recovery of α-Fe2O3 and γ-Fe2O3. Reproduced with permission from ref. 314. Copyright 2024, Elsevier. | ||
7.2. Urea synthesis
Urea is the most important nitrogen-based fertilizer used in crop production. The current industrial process for urea synthesis uses harsh reaction conditions and leaves a huge carbon footprint. This includes (i) the synthesis of ammonia by the Haber–Bosch process and (ii) the combination of ammonia and CO2 at a high temperature (150–200 °C) and pressure (150–200 bar).318 Hence, a “greener” and more sustainable way to produce ammonia under ambient conditions is required, which would also aid in realizing carbon/nitrogen neutrality in the environment. Co-reduction of CO2 and nitrogenous species like NO3−, NO2−, and N2via electrochemical or photoelectrochemical pathways can lead to the sustainable production of urea by C–N coupling, thus maintaining carbon neutrality and the nitrogen cycle.318,319 However, slow adsorption of substrates, multiple reaction steps, and competitive side reactions impede the catalytic reactions toward urea synthesis.320 Hence, to improve catalytic processes, the design of catalysts is crucial for the co-reduction process. A diverse range of catalysts, such as single-atoms, bimetallic, dual site, oxygen-vacant, and non-metallic materials, have been used in recent years for the co-reduction of nitrogenous species and CO2 to form urea.N* and CO produces the *NCON* intermediate, which acts as a precursor for the formation of urea.
 | ||
| Fig. 22 Low-dimensional catalysts for urea synthesis from co-reduction of NO3− and CO2. (A) Schematic representation of urea synthesis by dual active Fe(a)@C and Fe3O4 on carbon nanotubes (CNTs). (B) TEM image of Fe(a)@C-Fe3O4/CNT. (C) HRTEM image showing the lattice fringes with d-spacings of 0.25 nm and 0.294 nm corresponding to the (113) and (022) planes of Fe3O4 nanoparticles, respectively. (D) Urea yields and the corresponding Faradaic efficiencies using Fe(a)@C-Fe3O4/CNT at different potentials. Reproduced with permission from ref. 60. Copyright 2023, Wiley-VCH. (E) Schematic representation of the fabrication of core–shell Cu@Zn nanowires and catalytic formation of urea over these nanowires. (F) TEM image of Cu@Zn nanowire. (G) HRTEM image showing the lattice fringes with d-spacings of 0.23 nm and 0.21 nm corresponding to the Zn(100) and Cu(111) planes, respectively. (H) Urea yields at different applied potentials using the Cu@Zn nanowires. Reproduced with permission from ref. 326. Copyright 2022, American Chemical Society. (I) Schematic representation of grain boundary-rich Bi nanosheets reconstructed from Bi2Se3 nanosheets, utilized for the synthesis of urea from NO3− and CO2. (J) TEM and (K) HRTEM images of grain boundary-rich Bi nanosheets showing lattice fringes with d-spacing of 0.328 nm corresponding to the (012) plane of Bi. (L) Comparison of urea yields by grain boundary-rich Bi, low grain boundary Bi, and bulk Bi at different applied potentials. Reproduced with permission from ref. 332. Copyright 2024, Wiley-VCH. | ||
7.3. Coupled oxidation reactions
An overall reaction consists of an oxidation half-reaction and a reduction half-reaction. Generally, the reduction reactions of N2 and NOx− are accompanied by water-splitting, leading to oxygen evolution reaction (OER), and the formation of active hydrogen.339–341 The active hydrogen produced from water splitting helps in the protonation process and promotes the formation of ammonia. However, the active hydrogen (H*) produced should be consumed immediately by the nitrogen intermediates, which is difficult as the interaction between two H* forms H2, giving rise to competitive HER. The active H* can be successfully utilized by catalysts, such as Co(OH)2 and CoP, where the water dissociation process is facilitated on the Co catalysts’ surface.342,343 However, the OER reaction is sluggish and has a high thermodynamic potential of 1.23 V vs. RHE. As an outcome, this hampers the N2RR and NOxRR and slows down the entire redox process. Hence, efforts have been made to replace the OER with lower-potential oxidation reactions, like the oxidation of urea, hydrazine, methanol, ethanol, benzyl alcohol, glycerol, and PET, which can yield some value-added products. This section will explore different low-dimensional catalysts, focusing on this approach. | ||
| Fig. 23 Low-dimensional catalysts for ammonia synthesis via nitrate reduction coupled with oxidation reactions. (A) Schematic representation showing nitrate reduction (NO3RR) coupled hydrazine oxidation (HzOR) by tungsten phosphide (WP) nanowires deposited on Ni foam (WP/NF) electrodes coupled with perovskite solar cell. (B) Low and (C) high magnification SEM images of WP nanowires. (D) Potential profiles comparing the cathodic and anodic nitrate reduction-coupled hydrazine oxidation reaction (NORR-HzOR) and nitrate reduction-coupled oxygen evolution reaction (NORR-OER). Reproduced with permission from ref. 55. Copyright 2023, Wiley-VCH. (E) Schematic representation of co-electrolysis of NO3− and PET plastic at the low-crystalline CoOOH-based cathode and Pd nanothorn-based anodes to produce NH3 and glycolic acid simultaneously. (F) SEM image of the low-crystalline CoOOH grown on cobalt foam. (G) SEM image of Pd nanothorns grown on Ni foam. (H) Faradaic efficiencies of NH3 and glycolic acid produced at different potentials. Reproduced with permission from ref. 54. Copyright 2023, American Chemical Society. (I) Schematic representation of paired electrochemical refining, where NiCu-based electrodes convert nitrate and glycerol to value-added ammonia and formate, respectively. (J) HRTEM image of reconstructed NiCu–OH cathode showcasing amorphous Ni(OH)2 marked by red circles and lattice fringes with d-spacing of 0.20 nm corresponding to (111) planes of crystalline Cu, inset showing the corresponding FFT pattern. (K) STEM image of the reconstructed NiCuO anode showing lattice fringes with d-spacings of 0.209 nm (blue square) corresponding to the (200) of cubic NiO phase and 0.252 nm (red square), corresponding to the (111) plane of the monoclinic CuO phase. (L) NH3 yields and the corresponding Faradaic efficiencies at the reconstructed NiCu–OH cathode at different voltages. Reproduced with permission from ref. 352. Copyright 2022, Royal Society of Chemistry. | ||
8. Conclusions and perspectives
Over the past decade, the development of novel low-dimensional materials in electrocatalysis and photocatalysis has not only advanced the efficient synthesis of various value-added products but has also deepened our understanding of the complex mechanisms behind catalytic reactions, simplifying the design of more effective catalysts. The electronic properties of these materials can be easily tuned and their interfaces modulated for better adsorption and availability of catalytic sites, thereby making them suitable for photocatalysis and electrocatalysis. The combined effects of the materials and unique interfacial properties present in the heterostructures make them state-of-the-art catalysts in this field. Doping, defects, facet engineering, and formation of heterojunctions boost the catalytic properties of the low-dimensional materials. These catalytic materials can lead to selective production of green ammonia in a sustainable fashion by the reduction of N-containing species like N2, NOx, or NOx−. The enzymes present in the nitrogen cycle act as the inspiration for fabricating active low-dimensional materials as catalysts. In this review, we have outlined the recent 0D, 1D, and 2D low-dimensional materials used for the photo- and/or electrocatalytic production of ammonia. These low-dimensional materials present an array of physical and chemical characteristics for boosting the catalytic reactions. For instance, the alignment of the band positions of the photocatalysts and the redox potentials of the desired reactions is crucial for performing the photocatalytic reactions. Low-dimensional materials offer the opportunity to design heterostructures such that the band positions can be engineered to align with the redox potentials of N2RR and NOxRR. The important features of low-dimensional materials like light absorption properties, charge carrier properties, and lower recombination of charges further make them ideal photocatalysts. Most state-of-the-art photocatalysts developed in the lab use simulated light sources. Direct harvest of solar light for the effective generation of ammonia still poses a challenge. Though few reports of solar light-harvesting photocatalysts have been reported, their efficiency is unsatisfactory. Hence, increasing the solar-to-fuel efficiency of the photocatalysts for the production of ammonia remains a major concern. Quantum materials with plasmonic structures have better light absorption properties and can be used to design efficient photocatalysts. The band structures of quantum materials are optimized to provide the electrons required for the catalytic reactions and, thus they act as electron reservoirs. The quantum states can provide better conductivity, transportation of charges, improved lifetime, and enhanced surface properties, which make them prospective materials for energy-converting catalytic reactions. Quantum materials can transfer charge and electron spin, which might boost the catalytic activity of reduction reactions of N-containing species to ammonia. Generally, the production of ammonia originates from the reduction of dinitrogen or nitrates, which requires 6 and 8 electrons, respectively, and, in principle, the reactions require high overpotentials to surpass the high energy barrier. In electrocatalysis, the low-dimensional materials are supposed to provide lower overpotential compared with ideal thermodynamic potentials. However, the overpotentials reported for most electrocatalysts developed for ammonia production are still much higher than the thermodynamic potential, which requires further initiatives for designing heterostructures from experimental and theoretical aspects. Determination of the active sites of the catalysts through theoretical calculations is also pertinent for assessing and comparing the performance of various catalytic materials. To effectively utilize electrocatalytically synthesized ammonia in fertilizers, it is crucial to select appropriate electrolytes. Electrolytes should be non-toxic for plants and also should not affect their growth. NaCl, KOH, and Na2SO4 are mostly used as electrolytes for this purpose. For large-scale practical applications, designing flow cells appears to be a promising strategy, and some progress has been made in this area. However, significant optimizations are still required to enhance efficiency, increase yield, and improve the stability of catalysts before they can be used for the mass production of ammonia. The stability of catalysts is often hindered due to side reactions and can be modulated by driving the flow of charge carriers to the active sites for the reaction. The input of two energy sources, photo and electrical energy for generating ammonia by photocathodes has also been explored. However, state-of-the-art photoelectrocatalysts developed at the lab-scale report a low efficiency of ammonia production. Coupling with an electric grid or battery to increase the current density might lead to a higher yield of ammonia.Over the past years, the constant progress in ammonia synthesis has necessitated the establishment of a rigorous protocol for the measurement and quantification of NH3. Though diverse and well-established quantification techniques exist in the literature, contamination from different nitrogen-containing molecules like NOx or a non-negligible amount of ammonia from solvents or electrolytes, nitrogen or hydrogen leaching, and non-catalytic ammonia production can lead to errors in the quantification of ammonia. Additionally, ammonia can be in the form of ammonium ion (NH4+) or un-ionized ammonia (NH3) in liquid reaction media, depending on the pH and the temperature. At higher temperatures and pH (>11), ammonia predominantly remains in the gaseous state.355 Hence, in alkaline solutions, gaseous ammonia is present in the head space and the liquid phase as dissolved gas. This further complicates the quantitative analysis and preserving the samples for measurements. Reviews and protocols describing the correct method for ammonia measurement and quantification are present in the literature, following which correct protocols for ammonia quantification can be established in the lab.5,15,24,356,357 Among the existing techniques, the spectroscopic methods for ammonia quantification are the most common, less expensive and widely used. However, these methods depend on the pH of the reaction medium and the concentration of ammonia to be measured. For instance, Nessler's reagent method works in both alkaline and acidic solutions, over a concentration range of 0–8 mg L−1.357 However, the high alkalinity of Nessler's reagent, the toxicity due to the presence of mercury, and the short lifetime somewhat limit the use of this method. The indophenol blue method, another spectroscopic technique, works in alkaline media and detects low-concentration ammonia ranging from 0–0.6 mg NH3–N L−1.15 Nevertheless, the sample preparation for this method is lengthy and the presence of organic nitrogen-containing molecules can perturb the quantification. The salicylate spectroscopic method despite being a stable method suffers from major drawbacks like lower sensitivity and higher cost. Ion chromatography is also widely used for the quantification of ammonia and it offers many advantages over the spectroscopic methods, including efficiency, reproducibility, and high sensitivity toward NH4+ detection, and covers a wide detection range of 0.02–40 mg NH3–N L−1.136 However, this process is comparatively expensive, requires complex instrumentation, proper selection of columns and eluents, and is sometimes incompatible with acidic or basic solutions, and organic solvents. Ammonia can also be detected by fluorescence upon reaction with o-phthaldialdehyde and sulfite, but the detection limit is only up to 1 nmol L−1.358 Ammonium ion-selective electrode (ISE) is another efficient technique for the measurement of ammonia with concentrations ranging 0.03–1400 mg NH3–N L−1, however, the accuracy of this method is poor for concentrations less than 0.5 mg NH3–N L−1.136 Extensive background control experiments are always necessary to check and confirm the presence of any non-synthesized ammonia contaminants. In this regard, nuclear magnetic resonance (NMR) techniques with isotope labeling experiments play a crucial role. Catalytic experiments with 15N-labeled N-sources as reactants analyzed by 1H and 15N NMR methods can provide conclusive proof of the catalytic production of ammonia. The use of 15N-isotope-labeled reactant generates 15NH4+, which can be confirmed from 1H NMR spectra showing the chemical shifts of triplet coupling of 14N and doublet coupling for 15N.5 In the case of non-aqueous systems, the presence of organic solvents sometimes interferes with the detection of the NH3 signal, which can be resolved using solvent signal suppression methods. The accurate concentration of ammonia can be measured by calibration curves with standard solutions containing ammonia. Isotope-labeling experiments can also be performed with liquid chromatography–mass spectrometry (LC–MS) techniques. An LC-MS coupled Berthelot assay can detect 14NH3 and 15NH3 by analyzing 14N-indophenol (m/z: 198) and 15N-indophenol (m/z: 199) formed by Berthelot reactions.359 Although many quantification techniques are available, based on the reaction conditions, reactants, solvents, and the concentration of ammonia produced, the appropriate method has to be chosen for the accurate quantification of ammonia.
Despite the vast ongoing research on ammonia production, certain obstacles persist, necessitating additional progress. Currently, researchers are focusing on addressing these challenges and a few advancements have been achieved, and further upgradation in these aspects is required in the future. Despite being energy-intensive and leaving a significant carbon footprint, the Haber–Bosch (H–B) approach remains the industrial technology for ammonia synthesis. Recent efforts are now directed toward modification of the H–B plants with CO2 capture facilities to reduce the carbon footprint. Different electrolyzers have been developed in recent years to increase the energy efficiency of the ammonia synthesis process. For instance, the Topsoe green ammonia demonstration uses a high-temperature solid oxide electrolyzer cell to power the ammonia synthesis process.360 This electrolyzer can split water to generate green hydrogen and separate nitrogen from the air, thus reducing energy input intake and increasing overall efficiency. The small-scale green ammonia demonstrator by the ThyssenKrupp industries applies the chlor-alkaline water electrolyzer cell for ammonia synthesis.361 Polymer electrolyte membrane (PEM)-based electrolyzers are often used for industrial-scale ammonia synthesis. The green ammonia plant planned by Yara proposes to use a PEM electrolyzer produced by ITM Power.362 The wind power-to-ammonia demonstrator at Harwell, Oxford, United Kingdom, in association with Siemens, uses a PEM water electrolyzer to produce H2 and N2 from air separation over a Johnson Matthey Fe-based catalyst.363 In an attempt to develop decentralized and energy-efficient technologies for ammonia generation, Europe has also proposed over 20 projects by 2030 for green ammonia generation.364 Green ammonia-generating plants are now being proposed to be constructed or currently are constructed in different parts of the world, such as the Australian Renewable Energy Agency (ARENA) in western Australia, Kapsom in north-eastern India, and Fertiberia in Spain.365–367 The Yuri Renewable Hydrogen to Ammonia Project, led by Yara Pilbara Fertilisers Pty. Ltd (Yara), a fertilizer company, and ENGIE, a leader in low-carbon energy and services, with the support of ARENA, ENGINE, and Mitsui & Co. Ltd (Mitsui), was started in 2022, and is expected to be complete in 2028. This project proposes to industrially scale up the production of green ammonia using off-grid intermittent renewable H2 obtained via electrolysis. This project also aims to build a 10 MW electrolyzer powered by 18 MW solar PV and supported by an 8 MW battery energy storage system.365 In the same year (2022), Fertiberia, a Spanish fertilizer producer with support from the Green H2F project also constructed a green ammonia plant in Spain with Iberdrola and Spain's National Hydrogen Center. This plant project has a 20 PEM MW electrolyzer powered by a 100 MW PV with a capacity of 3000 tH2 year−1. An additional 800 MW capacity is also under development and will be complete by 2027.368 Although the polymer electrolyte membrane (PEM) electrolyzers are quite promising H2 production is somewhat limited by the low temperature. High-temperature electrolyzers including solid oxide or alkaline electrolyzers have also been considered but the higher cost limits their use for large-scale applications.23 Different literature studies indicate that for the complete industrial production of decarbonized, sustainable production of green ammonia further developments are required to construct highly efficient electrolyzers (>80%).369 For entirely decarbonizing the Haber–Bosch approach to ammonia production, the upcoming goal is to generate ammonia from water, air, and renewable energy sources. In this regard, several photo- and/or electrocatalysts have been designed; still, they suffer from a few limitations like the cost and production of electrical energy and the low yield of ammonia produced in the range of mmol h−1 g−1. The yield of ammonia in the millimolar range is quite inferior compared to industrial standards. Hence, the photo- and electrocatalytic approaches developed so far are not sufficient for the lab to industrial-level scale-up of the technology. Further investigations in this regard are necessary and would require cumulative expertise from the fields of materials science, catalysis, reactor engineering, and the use of artificial intelligence and robotization.24
Ammonia is highly water soluble due to the hydrogen bond between ammonia and water; hence, it is difficult to separate from aqueous medium. Most photocatalytic and electrocatalytic reactions are performed in an aqueous media, and the extraction of ammonia from aqueous phase remains an obstacle in most cases. Air and steam stripping are sometimes used to separate ammonia from aqueous medium, but these processes are energy-intensive.208 For air stripping of ammonia, maintenance of high airflow, temperature, and pressure of the packed bed are required to separate ammonia from water and convert it to the gaseous phase, eventually consuming a significant amount of energy.370 In the case of steam stripping, steam generation requires high temperatures consuming energy, while coupling heat recovery systems to improve the energy efficiency of this process can be complex and expensive.371 Another effective way of ammonia separation is upscaling the produced ammonia to salts like ammonium sulfate or ammonium chloride. This process helps in easy ammonia retrieval and directly converts ammonia to fertilizers.53,208,316 The development of membrane-based electrolyzers can also promote the separation of ammonia from the reaction medium. Recently, several lab-scale electrolyzers have been constructed for the electro/photoelectrocatalytic synthesis of ammonia. The electrolyzers have certain benefits over conventional catalytic setups. For instance, in conventional electrocatalysis electricity is sometimes harnessed from fossil fuels and produces greenhouse gases. Instead, the electrolyzers are powered by renewable energy sources like solar power or wind to produce electricity for the electrocatalytic reactions, thus making ammonia synthesis green and reducing the carbon footprint. Additionally, the electrolyzer concurrently couples electrosynthesis and gaseous product separation, which minimizes the undesired redox reaction between NH3 and the oxidized products. Thus, the electrolyzers can produce highly pure ammonia with high yield rates and minimal loss. The electrolyzers can also participate in redox reactions and extract the oxidized and reduced products simultaneously. For instance, NH3 and Cl2 can be electrosynthesized simultaneously and separated by incorporating gas-extraction electrodes into a flow-type membrane-free electrolyzer, coupled with ammonia and chlorine trap channels and a waste stream channel.372 Electrolyzers can be either membrane-based or membrane-free. The membrane-based electrolyzers can be proton exchange membranes (PEM), anion exchange membranes (AEM), or membrane–electrode assemblies (MEA). Membranes play a crucial role in determining the performance and scalability of ammonia-producing electrolyzers. In non-solid electrochemical/photoelectrochemical one-chamber cells, oxidation and reduction reactions occur in the single chamber and the ammonia produced upon reduction of N2 or NOx is susceptible to oxidation. To avoid this possibility, an electrolyzer consisting of a double-chamber cell or H-cell, separated by a proton exchange membrane (PEM) or an anion exchange membrane (AEM) is preferred. During the electrochemical reaction, the electrons generated at the anode-chamber can pass through the membrane to the cathode-chamber and participate in the N2RR or NOxRR to produce ammonia. However, the ammonia produced as NH4+ in aqueous solutions cannot cross the membrane, thus avoiding oxidation and product loss. Hence, the choice of membranes that can limit NH4+ crossover is extremely crucial. The most commonly used cation exchange membranes, Nafion 212 and Nafion 112 allow the transport of NH4+; therefore they prove inefficient for ammonia generation electrolyzers.367,373 Among anion exchange membranes, AEM PiperION-A80 exhibits negligible NH4+ crossover in both acidic and neutral electrolytes and can be used for the electrosynthesis of ammonia in neutral or acidic reaction media. However, in basic electrolytes, AEM PiperION-A80 proves inefficient as it is permeable to NH3 under basic conditions.373 Additionally, membranes allow the freedom of using two different electrolytes in the cathodic and anodic chambers.374 Anion exchange membranes are highly permeable to nitrates, hence, AEMs have to be avoided for NO3RR systems. Instead, a proton-exchange membrane electrode assembly (PEMEA) constructed with Nafion 117 and coupled with electrocatalysts like oxide-derived Cu nanoneedle cathodes can efficiently produce ammonia from nitrates.375 The production of ammonia can be scaled up from the millimolar range obtained from conventional electrocatalytic reactions by copper–tin alloy catalyst to molar range by transferring to a membrane electrode assembly (MEA) electrolyzer using renewable electricity and the cathode and anode separated by anion-exchange quaternary ammonium poly(N-methylpiperidine-co-p-terphenyl) (QAPPT) membrane.322 To meet the real application and industrial standards of ammonia production via the NOxRR process, electrocatalysis experiments are now performed in electrolyzers using various electrolytes to obtain ampere-level current density.230,376,377 Achieving long-term stability of ion-exchange membranes is still a challenge in many works, thus limiting long operating time for catalysis and high yield of ammonia. The construction of a 3D physically interlocked interface bipolar membrane can increase water dissociation sites, ionic transfer, and interfacial stability. By combining a Co nanoarray cathode with the bipolar membrane reactor, electrosynthesis of ammonia with increased yield can be achieved at 1000mAcm−2 for 100 hours of operation.378 MEA electrolyzers composed of cathodic CuZn ribbons, a proton exchange membrane (Nafion117), and an anodic IrO2–Ti mesh, exhibit stable operation at 500mAcm−2 for 220hours for producing ammonia.379 The electrolytes in the electrolyzers also have a significant impact on ammonia synthesis. The design of a flow electrolyzer with chain-ether-based electrolyte and gas diffusion electrodes can demonstrate 300 hours of continuous operation under ambient conditions.2 The flow electrolyzers can also help separate synthesized ammonia by constructing a ‘two-in-one” flow cell electrolyzer that integrates the two chambers of NO3RR electrolysis, and NH3 capture through a commercial gas diffusion electrode.380 Apart from synchronized NH3 production and capture, this electrolyzer rapidly transports NH3 molecules away from the reaction interfaces, thereby promoting NO3RR to NH3. With such rapid development in catalyst and membrane design, electrolyzer device optimization; green, sustainable, carbon-free, low-cost, large-scale industrial methods for ammonia production are expected soon. A multidisciplinary approach is needed to upgrade lab-scale ammonia production into full-scale industrial ammonia fertilizer production. This includes expertise in photocatalysis, electrocatalysis, chemical separation, agricultural science, and technical engineering. These combined skills are essential to developing an efficient alternative to the Haber–Bosch process and addressing the current global demand for sustainable ammonia production. Exploring and identifying the most efficient methods for ammonia production is a critical challenge this field must address in the coming years. Advancing these technologies will be the key to driving growth in both the agriculture-based economy and the energy sectors that rely on ammonia as a fuel source.
Author contributions
M. P., A. M., and C. C. M. conceived and designed the review. A. M. wrote the initial draft. A. M., C. C. M., and M. P. reviewed, edited, and wrote the manuscript. M. P. supervised the entire review. All the authors contributed to the discussion and manuscript preparation.Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was co-funded by the European Union under the REFRESH – Research Excellence For REgion Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition. AM acknowledges the funding from the project MSCA CZ fellowship at VSB-TUO with project number CZ.02.01.01/00/22_010/0006136.References
- R. F. Service, Science, 2018, 361, 120–123 CrossRef CAS PubMed.
- S. Li, Y. Zhou, X. Fu, J. B. Pedersen, M. Saccoccio, S. Z. Andersen, K. Enemark-Rasmussen, P. J. Kempen, C. D. Damsgaard, A. Xu, R. Sažinas, J. B. V. Mygind, N. H. Deissler, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Nature, 2024, 629, 92–97 CrossRef CAS PubMed.
- K. Zhang, A. Cao, L. H. Wandall, J. Vernieres, J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, Science, 2024, 383, 1357–1363 CrossRef CAS PubMed.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- J. Kim and D. C. Rees, Biochemistry, 1994, 33, 389–397 CrossRef CAS PubMed.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- Z. Chen, G. Quek, J.-Y. Zhu, S. J. W. Chan, S. J. Cox-Vázquez, F. Lopez-Garcia and G. C. Bazan, Angew. Chem., Int. Ed., 2023, 62, e202307101 CrossRef CAS PubMed.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- C. Katan, N. Mercier and J. Even, Chem. Rev., 2019, 119, 3140–3192 CrossRef CAS PubMed.
- T. Edvinsson, R. Soc. Open Sci., 2018, 5, 180387 CrossRef CAS PubMed.
- Q. Shao, P. Wang, T. Zhu and X. Huang, Acc. Chem. Res., 2019, 52, 3384–3396 CrossRef CAS PubMed.
- Y. Pang, C. Su, G. Jia, L. Xu and Z. Shao, Chem. Soc. Rev., 2021, 50, 12744–12787 RSC.
- Y. Ruan, Z.-H. He, Z.-T. Liu, W. Wang, L. Hao, L. Xu, A. W. Robertson and Z. Sun, J. Mater. Chem. A, 2023, 11, 22590–22607 RSC.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- Y. Shi, Z. Zhao, D. Yang, J. Tan, X. Xin, Y. Liu and Z. Jiang, Chem. Soc. Rev., 2023, 52, 6938–6956 RSC.
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, 2304021 CrossRef CAS PubMed.
- A. K. K. Padinjareveetil and M. Pumera, Adv. Mater. Interfaces, 2023, 10, 2201734 CrossRef.
- B. Wang, S. Chen, Z. Zhang and D. Wang, SmartMat, 2022, 3, 84–110 CrossRef CAS.
- S. Li, X. Fu, J. K. Nørskov and I. Chorkendorff, Nat. Energy, 2024, 9, 1344–1349 CrossRef CAS.
- J. John, D. R. MacFarlane and A. N. Simonov, Nat. Catal., 2023, 6, 1125–1130 CrossRef.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- D. Ye and S. C. E. Tsang, Nat. Synth., 2023, 2, 612–623 CrossRef CAS.
- L. Collado, A. H. Pizarro, M. Barawi, M. García-Tecedor, M. Liras and V. A. D. L. P. O'Shea, Chem. Soc. Rev., 2024, 53, 11334–11389 RSC.
- Y. Feng, L. Jiao, X. Zhuang, Y. Wang and J. Yao, Adv. Mater., 2025, 37, 2410909 CrossRef CAS PubMed.
- W.-Q. Li, M. Xu, J.-S. Chen and T.-N. Ye, Adv. Mater., 2024, 36, 2408434 CrossRef CAS PubMed.
- R. Nittoor-Veedu, X. Ju and M. Pumera, Adv. Energy Mater., 2024, 2402205 CrossRef.
- G. Gan, G. Hong and W. Zhang, Adv. Funct. Mater., 2024, 2401472 CrossRef.
- Y. Zhou, X. Fu, I. Chorkendorff and J. K. Nørskov, ACS Energy Lett., 2025, 10, 128–132 CrossRef CAS PubMed.
- S. Yin, Z. Guan, Y. Zhu, D. Guo, X. Chen and S. Wang, ACS Nano, 2024, 18, 27833–27852 CrossRef CAS PubMed.
- M. Majumder, H. Saini, I. Dědek, A. Schneemann, N. R. Chodankar, V. Ramarao, M. S. Santosh, A. K. Nanjundan, S. Kment, D. Dubal, M. Otyepka, R. Zbořil and K. Jayaramulu, ACS Nano, 2021, 15, 17275–17298 CrossRef CAS PubMed.
- T. Hu, X. Cheng, J. Luo, Y. Yan, Q. Zhang and Y. Li, ACS Catal., 2024, 14, 14539–14563 CrossRef CAS.
- H. Fang, D. Liu, Y. Luo, Y. Zhou, S. Liang, X. Wang, B. Lin and L. Jiang, ACS Catal., 2022, 12, 3938–3954 CrossRef CAS.
- X. Long, F. Huang, Z. Yao, P. Li, T. Zhong, H. Zhao, S. Tian, D. Shu and C. He, Small, 2024, 20, 2400551 CrossRef CAS PubMed.
- Y. Li, Q. Zhang, Z. Mei, S. Li, W. Luo, F. Pan, H. Liu and S. Dou, Small Methods, 2021, 5, 2100460 CrossRef CAS PubMed.
- K. Ithisuphalap, H. Zhang, L. Guo, Q. Yang, H. Yang and G. Wu, Small Methods, 2019, 3, 1800352 CrossRef.
- N. H. Truong, J.-S. Kim, J. Lim and H. Shin, Chem. Eng. J., 2024, 495, 153108 CrossRef.
- S. M. Ghoreishian, K. Shariati, Y. S. Huh and J. Lauterbach, Chem. Eng. J., 2023, 467, 143533 CrossRef CAS.
- A. H. Pizarro, J. Fermoso, M. García-Tecedor, M. Barawi, V. A. D. L. P. O'Shea and L. Collado, J. Mater. Chem. A, 2024, 12, 16987–17001 RSC.
- C.-W. Tsao, M.-J. Fang and Y.-J. Hsu, Coord. Chem. Rev., 2021, 438, 213876 CrossRef CAS.
- Y. Wang, H. Yin, X. Zhao, Y. Qu, A. Zheng, H. Zhou, W. Fang and J. Li, Appl. Catal., B, 2024, 341, 123266 CrossRef CAS.
- J. Qin, X. Hu, X. Li, Z. Yin, B. Liu and K.-H. Lam, Nano Energy, 2019, 61, 27–35 CrossRef CAS.
- H. Yin, Z. Chen, Y. Peng, S. Xiong, Y. Li, H. Yamashita and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202114242 CrossRef CAS PubMed.
- M. Mohebinia, X. Xing, G. Yang, D. Wang, C. Solares-Bockmon, Z. Ren and J. Bao, Mater. Today Chem., 2022, 24, 100993 CrossRef CAS.
- Y. Kong, L. Wu, X. Yang, Y. Li, S. Zheng, B. Yang, Z. Li, Q. Zhang, S. Zhou, L. Lei, G. Wu and Y. Hou, Adv. Funct. Mater., 2022, 32, 2205409 CrossRef CAS.
- D. Yin, D. Chen, Y. Zhang, W. Wang, Q. Quan, W. Wang, Y. Meng, Z. Lai, Z. Yang, S. Yip, C.-Y. Wong, X. Bu, X. Wang and J. C. Ho, Adv. Funct. Mater., 2023, 33, 2303803 CrossRef CAS.
- W. Tong, B. Huang, P. Wang, L. Li, Q. Shao and X. Huang, Angew. Chem., Int. Ed., 2020, 59, 2649–2653 CrossRef CAS PubMed.
- A. K. K. Padinjareveetil, J. V. Perales-Rondon and M. Pumera, Adv. Mater. Technol., 2023, 8, 2202080 CrossRef CAS.
- Y. J. Jang, A. E. Lindberg, M. A. Lumley and K.-S. Choi, ACS Energy Lett., 2020, 5, 1834–1839 CrossRef CAS.
- L. P. Camargo, P. R. C. da Silva, A. Batagin-Neto, V. Klobukoski, M. Vidotti and L. H. Dall'Antonia, Appl. Mater. Today, 2022, 28, 101540 CrossRef.
- B. Wang, L. Yao, G. Xu, X. Zhang, D. Wang, X. Shu, J. Lv and Y.-C. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 20376–20382 CrossRef CAS PubMed.
- M. A. Mushtaq, A. Kumar, G. Yasin, M. Arif, M. Tabish, S. Ibraheem, X. Cai, W. Ye, X. Fang, A. Saad, J. Zhao, S. Ji and D. Yan, Appl. Catal., B, 2022, 317, 121711 CrossRef CAS.
- J. Gao, N. Shi, Y. Li, B. Jiang, T. Marhaba and W. Zhang, Environ. Sci. Technol., 2022, 56, 11602–11613 CrossRef CAS PubMed.
- T. Ren, Z. Duan, H. Wang, H. Yu, K. Deng, Z. Wang, H. Wang, L. Wang and Y. Xu, ACS Catal., 2023, 13, 10394–10404 CrossRef CAS.
- C. Lim, H. Roh, E. H. Kim, H. Kim, T. Park, D. Lee and K. Yong, Small, 2023, 19, 2304274 CrossRef CAS PubMed.
- A. Tayyebi, R. Mehrotra, M. A. Mubarok, J. Kim, M. Zafari, M. Tayebi, D. Oh, S.-H. Lee, J. E. Matthews, S.-W. Lee, T. J. Shin, G. Lee, T. F. Jaramillo, S.-Y. Jang and J.-W. Jang, Nat. Catal., 2024, 7, 510–521 CrossRef CAS.
- C. Lv, C. Lee, L. Zhong, H. Liu, J. Liu, L. Yang, C. Yan, W. Yu, H. H. Hng, Z. Qi, L. Song, S. Li, K. P. Loh, Q. Yan and G. Yu, ACS Nano, 2022, 16, 8213–8222 CrossRef CAS PubMed.
- X. Wei, Y. Liu, X. Zhu, S. Bo, L. Xiao, C. Chen, T. T. T. Nga, Y. He, M. Qiu, C. Xie, D. Wang, Q. Liu, F. Dong, C.-L. Dong, X.-Z. Fu and S. Wang, Adv. Mater., 2023, 35, 2300020 CrossRef CAS PubMed.
- Y. Zhao, Y. Ding, W. Li, C. Liu, Y. Li, Z. Zhao, Y. Shan, F. Li, L. Sun and F. Li, Nat. Commun., 2023, 14, 4491 CrossRef CAS PubMed.
- J. Geng, S. Ji, M. Jin, C. Zhang, M. Xu, G. Wang, C. Liang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202210958 CrossRef CAS PubMed.
- J. Shao, H. Jing, P. Wei, X. Fu, L. Pang, Y. Song, K. Ye, M. Li, L. Jiang, J. Ma, R. Li, R. Si, Z. Peng, G. Wang and J. Xiao, Nat. Energy, 2023, 8, 1273–1283 CrossRef CAS.
- Q. Lai, T. Cai, S. C. E. Tsang, X. Chen, R. Ye, Z. Xu, M. D. Argyle, D. Ding, Y. Chen, J. Wang, A. G. Russell, Y. Wu, J. Liu and M. Fan, Sci. Bull., 2022, 67, 2124–2138 CrossRef CAS PubMed.
- H. Li, F. Pan, C. Qin, T. Wang and K.-J. Chen, Adv. Energy Mater., 2023, 13, 2301378 CrossRef CAS.
- M. Appl, Ammonia, Ullmann's Encyclopedia of Industrial Chemistry, 2006 DOI:10.1002/14356007.a02_143.pub2.
- R. D. Milton, R. Cai, S. Abdellaoui, D. Leech, A. L. De Lacey, M. Pita and S. D. Minteer, Angew. Chem., Int. Ed., 2017, 56, 2680–2683 CrossRef CAS PubMed.
- J. Guo and P. Chen, Acc. Chem. Res., 2021, 54, 2434–2444 CrossRef CAS PubMed.
- O. Westhead, J. Barrio, A. Bagger, J. W. Murray, J. Rossmeisl, M.-M. Titirici, R. Jervis, A. Fantuzzi, A. Ashley and I. E. L. Stephens, Nat. Rev. Chem., 2023, 7, 184–201 CrossRef CAS PubMed.
- N. R. Dhar, E. V. Seshacharyulu and N. N. Biswas, Proc. Natl. Inst. Sci., 1941, 7, 115–131 CAS.
- G. N. Schrauzer, N. Strampach, L. N. Hui, M. R. Palmer and J. Salehi, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 3873–3876 CrossRef CAS PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- Q. Han, C. Wu, H. Jiao, R. Xu, Y. Wang, J. Xie, Q. Guo and J. Tang, Adv. Mater., 2021, 33, 2008180 CrossRef CAS PubMed.
- X. Gao, L. An, D. Qu, W. Jiang, Y. Chai, S. Sun, X. Liu and Z. Sun, Sci. Bull., 2019, 64, 918–925 CrossRef CAS PubMed.
- X. Chen, X. Zhang, Y.-H. Li, M.-Y. Qi, J.-Y. Li, Z.-R. Tang, Z. Zhou and Y.-J. Xu, Appl. Catal., B, 2021, 281, 119516 CrossRef CAS.
- P. Li, S. Gao, Q. Liu, P. Ding, Y. Wu, C. Wang, S. Yu, W. Liu, Q. Wang and S. Chen, Adv. Energy Sustainability Res., 2021, 2, 2000097 CrossRef CAS.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2020, 10, 1901973 CrossRef CAS.
- Y. Zhao, L. Zheng, R. Shi, S. Zhang, X. Bian, F. Wu, X. Cao, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2020, 10, 2002199 CrossRef CAS.
- W. Wang, H. Zhou, Y. Liu, S. Zhang, Y. Zhang, G. Wang, H. Zhang and H. Zhao, Small, 2020, 16, 1906880 CrossRef CAS PubMed.
- Y. Lu, Y. Yang, T. Zhang, Z. Ge, H. Chang, P. Xiao, Y. Xie, L. Hua, Q. Li, H. Li, B. Ma, N. Guan, Y. Ma and Y. Chen, ACS Nano, 2016, 10, 10507–10515 CrossRef CAS PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- Y. Guan, H. Wen, K. Cui, Q. Wang, W. Gao, Y. Cai, Z. Cheng, Q. Pei, Z. Li, H. Cao, T. He, J. Guo and P. Chen, Nat. Chem., 2024, 16, 373–379 CrossRef CAS PubMed.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706–709 CrossRef CAS.
- M. Ali, F. Zhou, K. Chen, C. Kotzur, C. Xiao, L. Bourgeois, X. Zhang and D. R. MacFarlane, Nat. Commun., 2016, 7, 11335 CrossRef CAS PubMed.
- H. E. Kim, J. Kim, E. C. Ra, H. Zhang, Y. J. Jang and J. S. Lee, Angew. Chem., Int. Ed., 2022, 61, e202204117 CrossRef CAS PubMed.
- M. A. Mushtaq, M. Arif, X. Fang, G. Yasin, W. Ye, M. Basharat, B. Zhou, S. Yang, S. Ji and D. Yan, J. Mater. Chem. A, 2021, 9, 2742–2753 RSC.
- M. S. Yu, S. C. Jesudass, S. Surendran, J. Y. Kim, U. Sim and M.-K. Han, ACS Appl. Mater. Interfaces, 2022, 14, 31889–31899 CrossRef CAS PubMed.
- C. Sun, Z. Shao, Y. Hu, Y. Peng and Q. Xie, ACS Appl. Mater. Interfaces, 2023, 15, 23085–23092 CrossRef CAS PubMed.
- X. Chen, N. Li, Z. Kong, W.-J. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- B. Rytelewska, A. Chmielnicka, T. Chouki, M. Skunik-Nuckowska, S. Naghdi, D. Eder, A. Michalowska, T. Ratajczyk, E. Pavlica, S. Emin, Y. Fu, I. A. Rutkowska and P. J. Kulesza, Electrochim. Acta, 2023, 471, 143360 CrossRef CAS.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- H. Wan, A. Bagger and J. Rossmeisl, Angew. Chem., Int. Ed., 2021, 60, 21966–21972 CrossRef CAS PubMed.
- X. Lu, H. Song, J. Cai and S. Lu, Electrochem. Commun., 2021, 129, 107094 CrossRef CAS.
- H. Liu, L. Bai, A. Bergmann, B. R. Cuenya and J. Luo, Chem, 2024, 10, 2963–2986 CAS.
- T. Ouyang, S. Huang, X.-T. Wang and Z.-Q. Liu, Chem. – Eur. J., 2020, 26, 14024–14035 CrossRef CAS PubMed.
- H. Gleiter, Nanostruct. Mater., 1995, 6, 3–14 CrossRef CAS.
- V. V. Pokropivny and V. V. Skorokhod, Mater. Sci. Eng., C, 2007, 27, 990–993 CrossRef CAS.
- Z.-H. Xue, S.-N. Zhang, Y.-X. Lin, H. Su, G.-Y. Zhai, J.-T. Han, Q.-Y. Yu, X.-H. Li, M. Antonietti and J.-S. Chen, J. Am. Chem. Soc., 2019, 141, 14976–14980 CrossRef CAS PubMed.
- L. Meng, J. M. D. Lane, L. Baca, J. Tafoya, T. Ao, B. Stoltzfus, M. Knudson, D. Morgan, K. Austin, C. Park, P. Chow, Y. Xiao, R. Li, Y. Qin and H. Fan, J. Am. Chem. Soc., 2020, 142, 6505–6510 CrossRef CAS PubMed.
- B. Guan, X. Wang, Y. Xiao, Y. Liu and Q. Huo, Nanoscale, 2013, 5, 2469–2475 RSC.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. B. Na, J. Am. Chem. Soc., 2001, 123, 12798–12801 CrossRef CAS PubMed.
- J. Xu, S. Zhang, H. Liu, S. Liu, Y. Yuan, Y. Meng, M. Wang, C. Shen, Q. Peng, J. Chen, X. Wang, L. Song, K. Li and W. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308044 CrossRef CAS PubMed.
- G. Liu, M. Zavelani-Rossi, G. Han, H. Zhao and A. Vomiero, J. Mater. Chem. A, 2023, 11, 8950–8960 RSC.
- M. Parente, M. van Helvert, R. F. Hamans, R. Verbroekken, R. Sinha, A. Bieberle-Hütter and A. Baldi, Nano Lett., 2020, 20, 5759–5764 CrossRef CAS PubMed.
- J. E. Omoriyekomwan, A. Tahmasebi, J. Zhang and J. Yu, Energy Convers. Manage., 2019, 192, 88–99 CrossRef CAS.
- H. Du, H. Guo, K. Wang, X. Du, B. A. Beshiwork, S. Sun, Y. Luo, Q. Liu, T. Li and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202215782 CrossRef CAS PubMed.
- G. Liu, Z. Tang, X. Gu, N. Li, H. Lv, Y. Huang, Y. Zeng, M. Yuan, Q. Meng, Y. Zhou and C. Wang, Appl. Catal., B, 2022, 317, 121752 CrossRef CAS.
- Z. Chen, D. Chao, J. Liu, M. Copley, J. Lin, Z. Shen, G.-T. Kim and S. Passerini, J. Mater. Chem. A, 2017, 5, 15669–15675 RSC.
- R. Khare, D. B. Shinde, S. Bansode, M. A. More, M. Majumder, V. K. Pillai and D. J. Late, Appl. Phys. Lett., 2015, 106, 023111 CrossRef.
- D. J. Lee, S.-H. Yu, H. S. Lee, A. Jin, J. Lee, J. E. Lee, Y.-E. Sung and T. Hyeon, J. Mater. Chem. A, 2017, 5, 8744–8751 RSC.
- W. Bai, S. Li, J. Ma, W. Cao and J. Zheng, J. Mater. Chem. A, 2019, 7, 9086–9098 RSC.
- G. Xia, S. Wang, X. Zhao and L. Zhou, J. Mater. Chem. C, 2013, 1, 3291–3296 RSC.
- Y. Min, G. D. Moon, B. S. Kim, B. Lim, J.-S. Kim, C. Y. Kang and U. Jeong, J. Am. Chem. Soc., 2012, 134, 2872–2875 CrossRef CAS PubMed.
- S. Mahasivam, V. Bansal and M. Sastry, J. Phys. Chem. Lett., 2024, 15, 3923–3928 CrossRef CAS PubMed.
- Z. L. Shaw, S. Kuriakose, S. Cheeseman, M. D. Dickey, J. Genzer, A. J. Christofferson, R. J. Crawford, C. F. McConville, J. Chapman, V. K. Truong, A. Elbourne and S. Walia, Nat. Commun., 2021, 12, 3897 CrossRef CAS PubMed.
- J. N. Tiwari, R. N. Tiwari and K. S. Kim, Prog. Mater. Sci., 2012, 57, 724–803 CrossRef CAS.
- S. Sun, M. Lu, X. Gao, Z. Shi, X. Bai, W. W. Yu and Y. Zhang, Adv. Sci., 2021, 8, 2102689 CrossRef PubMed.
- P. Innocenzi and L. Stagi, Acc. Mater. Res., 2024, 5, 413–425 CrossRef CAS.
- Y. Zhang, F. Gao, H. You, Z. Li, B. Zou and Y. Du, Coord. Chem. Rev., 2022, 450, 214244 CrossRef CAS.
- Y. Lu, S. Du and R. Steinberger-Wilckens, Appl. Catal., B, 2016, 199, 292–314 CrossRef CAS.
- H. Hou, G. Shao, W. Yang and W.-Y. Wong, Prog. Mater. Sci., 2020, 113, 100671 CrossRef CAS.
- X. Lu, C. Wang and Y. Wei, Small, 2009, 5, 2349–2370 CrossRef CAS PubMed.
- L. Liang, X. Kang, Y. Sang and H. Liu, Adv. Sci., 2016, 3, 1500358 CrossRef PubMed.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- M. Byakodi, N. S. Shrikrishna, R. Sharma, S. Bhansali, Y. Mishra, A. Kaushik and S. Gandhi, Biosens. Bioelectron.: X, 2022, 12, 100284 CAS.
- T. A. Shifa, F. Wang, Y. Liu and J. He, Adv. Mater., 2019, 31, 1804828 CrossRef CAS PubMed.
- W. Huang, L. Hu, Y. Tang, Z. Xie and H. Zhang, Adv. Funct. Mater., 2020, 30, 2005223 CrossRef CAS.
- Z. He, J. Zhang, X. Li, S. Guan, M. Dai and S. Wang, Small, 2020, 16, 2005051 CrossRef CAS PubMed.
- P. Sabatier, La catalyse en chimie organique, Paris & Liége Ch. Béranger Editeur, 1920 Search PubMed.
- Y. Zhu, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, Small Methods, 2019, 3, 1800438 CrossRef.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- B. Bajorowicz, M. P. Kobylański, A. Gołąbiewska, J. Nadolna, A. Zaleska-Medynska and A. Malankowska, Adv. Colloid Interface Sci., 2018, 256, 352–372 CrossRef CAS PubMed.
- G. Zhang, Y. Li, C. He, X. Ren, P. Zhang and H. Mi, Adv. Energy Mater., 2021, 11, 2003294 CrossRef CAS.
- M. Li, H. Huang, J. Low, C. Gao, R. Long and Y. Xiong, Small Methods, 2019, 3, 1800388 CrossRef.
- J. Li, J. Wang, S. Shen, R. Chen, M. Liu and F. Dong, Environ. Sci. Technol., 2023, 57, 5445–5452 CrossRef CAS PubMed.
- H. Kominami, A. Furusho, S.-Y. Murakami, H. Inoue, Y. Kera and B. Ohtani, Catal. Lett., 2001, 76, 31–34 CrossRef CAS.
- M. Yamauchi, R. Abe, T. Tsukuda, K. Kato and M. Takata, J. Am. Chem. Soc., 2011, 133, 1150–1152 CrossRef CAS PubMed.
- G. Ren, J. Zhao, Z. Zhao, Z. Li, L. Wang, Z. Zhang, C. Li and X. Meng, Angew. Chem., Int. Ed., 2024, 63, e202314408 CrossRef CAS PubMed.
- G. Ren, M. Shi, Z. Li, Z. Zhang and X. Meng, Appl. Catal., B, 2023, 327, 122462 CrossRef CAS.
- J. Bao, H. Maimaiti, J. Sun, L. Feng and X. Zhao, Int. J. Hydrogen Energy, 2024, 95, 1–11 CrossRef CAS.
- X. Yu, P. Qiu, Y. Wang, B. He, X. Xu, H. Zhu, J. Ding, X. Liu, Z. Li and Y. Wang, J. Colloid Interface Sci., 2023, 640, 775–782 CrossRef CAS PubMed.
- P. Li, Z. Zhou, Q. Wang, M. Guo, S. Chen, J. Low, R. Long, W. Liu, P. Ding, Y. Wu and Y. Xiong, J. Am. Chem. Soc., 2020, 142, 12430–12439 CrossRef CAS PubMed.
- C. Liu, D. Hao, J. Ye, S. Ye, F. Zhou, H. Xie, G. Qin, J. Xu, J. Liu, S. Li and C. Sun, Adv. Energy Mater., 2023, 13, 2204126 CrossRef CAS.
- Y. Huang, Y. Zhu, S. Chen, X. Xie, Z. Wu and N. Zhang, Adv. Sci., 2021, 8, 2003626 CrossRef CAS PubMed.
- K. Li, W. Cai, Z. Zhang, H. Xie, Q. Zhong and H. Qu, Chem. Eng. J., 2022, 435, 135017 CrossRef CAS.
- Y. Fang, Y. Xue, L. Hui, H. Yu and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3170–3174 CrossRef CAS PubMed.
- K. Pournemati, A. Habibi-Yangjeh and A. Khataee, ACS Appl. Nano Mater., 2024, 7, 2200–2213 CrossRef CAS.
- D. Cui, S. Wang, X. Yang, L. Xu and F. Li, Small, 2024, 20, 2306229 CrossRef CAS PubMed.
- D. Hao, J. Ren, Y. Wang, H. Arandiyan, M. Garbrecht, X. Bai, H. K. Shon, W. Wei and B.-J. Ni, Energy Mater. Adv., 2021, 2021, 9761263 Search PubMed.
- R. Fu, Y. Wang, G. Wang, Q. Zhan, L. Zhang and L. Liu, Green Chem., 2023, 25, 8531–8538 RSC.
- Z. Zhao, R. Tan, Y. Kong, Z. Zhang, S. Qiu, X. Mu and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202303629 CrossRef CAS PubMed.
- J. Li, R. Chen, J. Wang, Y. Zhou, G. Yang and F. Dong, Nat. Commun., 2022, 13, 1098 CrossRef CAS PubMed.
- J. Li, R. Chen, J. Wang, K. Wang, Y. Zhou, M. Xing and F. Dong, Angew. Chem., Int. Ed., 2024, 63, e202317575 CrossRef CAS PubMed.
- P. Li, R. Wu, P. Li, S. Gao, Z. Qin, X. Song, W. Sun, Z. Hua, Q. Wang and S. Chen, Adv. Sci., 2024, 11, 2408829 CrossRef CAS PubMed.
- V. Jain, S. Tyagi, P. Roy and P. P. Pillai, J. Am. Chem. Soc., 2024, 146, 32356–32365 CrossRef CAS PubMed.
- C. Feng, F. Raziq, M. Hu, H. Huang, Z.-P. Wu, S. Zuo, J. Luo, Y. Ren, B. Chang, D. Cha, S. Ayirala, A. Al-Yousef, T. D. Dao and H. Zhang, Adv. Energy Mater., 2024, 14, 2303792 CrossRef CAS.
- P. Xia, X. Pan, S. Jiang, J. Yu, B. He, P. M. Ismail, W. Bai, J. Yang, L. Yang, H. Zhang, M. Cheng, H. Li, Q. Zhang, C. Xiao and Y. Xie, Adv. Mater., 2022, 34, 2200563 CrossRef CAS PubMed.
- R. Wu, S. Gao, C. Jones, M. Sun, M. Guo, R. Tai, S. Chen and Q. Wang, Adv. Funct. Mater., 2024, 34, 2314051 CrossRef CAS.
- L. Zhang, R. Li, L. Cui, Z. Sun, L. Guo, X. Zhang, Y. Wang, Y. Wang, Z. Yu, T. Lei, X. Jian, X. Gao, C. Fan and J. Liu, Chem. Eng. J., 2023, 461, 141892 CrossRef CAS.
- K. Wu, Z. Wang, X. Zhang, C. Sun, Q. Li, H. Zhang, X. Bai, A. Khosla and Z. Zhao, Langmuir, 2024, 40, 13603–13612 CrossRef CAS PubMed.
- Q. Zhang, Q. Li, H. Li, X. Shi, Y. Zhou, Q. Ye, R. Yang, D. Li and D. Jiang, Inorg. Chem., 2023, 62, 12138–12147 CrossRef CAS PubMed.
- Y. Liu, Y. Xue, L. Hui, H. Yu, Y. Fang, F. He and Y. Li, Nano Energy, 2021, 89, 106333 CrossRef CAS.
- S. Liu, M. Wang, H. Ji, L. Zhang, J. Ni, N. Li, T. Qian, C. Yan and J. Lu, Adv. Mater., 2023, 35, 2211730 CrossRef CAS PubMed.
- T. He, Z. Zhao, R. Liu, X. Liu, B. Ni, Y. Wei, Y. Wu, W. Yuan, H. Peng, Z. Jiang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 6057–6066 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, S. Feng, X. Gu, M. Zhou, H. Wang and J. Hua, Appl. Catal., B, 2024, 366, 125013 CrossRef.
- M. Yu, Y. Chen, M. Gao, G. Huang, Q. Chen and J. Bi, Small, 2023, 19, 2206407 CrossRef CAS PubMed.
- X. Wang, G. Fan, S. Guo, R. Gao, Y. Guo, C. Han, Y. Gao, J. Zhang, X. Gu and L. Wu, Angew. Chem., Int. Ed., 2024, 63, e202404258 CrossRef CAS PubMed.
- J. Tan, H. Ren, Z. Zhao, X. Xin, Y. Shi, D. Yang and Z. Jiang, Chem. Eng. J., 2023, 466, 143259 CrossRef CAS.
- Z. Shen, F. Li, L. Guo, X. Zhang, Y. Wang, Y. Wang, X. Jian, X. Gao, Z. Wang, R. Li, C. Fan and J. Liu, Appl. Catal., B, 2024, 346, 123732 CrossRef CAS.
- Y. Xi, Y. Xiang, T. Bao, Z. Li, C. Zhang, L. Yuan, J. Li, Y. Bi, C. Yu and C. Liu, Angew. Chem., Int. Ed., 2024, 63, e202409163 CrossRef CAS PubMed.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 108, 73–78 CrossRef CAS.
- A. Pandikumar, S. Manonmani and R. Ramaraj, Catal. Sci. Technol., 2012, 2, 345–353 RSC.
- K. T. Ranjit, R. Krishnamoorthy, T. K. Varadarajan and B. Viswanathan, J. Photochem. Photobiol., A, 1995, 86, 185–189 CrossRef CAS.
- I. G. Zacharia and W. M. Deen, Ann. Biomed. Eng., 2005, 33, 214–222 CrossRef PubMed.
- R. Chen, J. Li, J. Wang, W. Yang, S. Shen and F. Dong, Environ. Sci. Technol., 2023, 57, 12127–12134 CrossRef CAS PubMed.
- P. Zhang, L. Chen, D.-H. Kuo, B. Wu, Z. Su, D. Lu, Q. Wu, J. Li, J. Lin and X. Chen, J. Mater. Chem. A, 2024, 12, 7163–7177 RSC.
- L. Chen, D. Shen, B. Li, Z. Xiao, W. Sun, X. Liu, J. Ma, C. Li and W. Wang, Ceram. Int., 2023, 49, 23129–23139 CrossRef CAS.
- J. Zhang, G. Zhang, H. Lan, H. Liu and J. Qu, Chem. Eng. J., 2022, 428, 130373 CrossRef CAS.
- G. Ren, M. Shi, S. Liu, Z. Li, Z. Zhang and X. Meng, Chem. Eng. J., 2023, 454, 140158 CrossRef CAS.
- X. Lv, W. Wei, F. Li, B. Huang and Y. Dai, Nano Lett., 2019, 19, 6391–6399 CrossRef CAS PubMed.
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, ACS Catal., 2017, 7, 3713–3720 CrossRef CAS.
- X. Huang, R. Du, J. Ren, X. Li, M. Fu, S. Fu, T. Ma, L. Guo, R. A. Soomro, C. Yang and D. Wang, ACS Catal., 2024, 14, 13542–13549 CrossRef CAS.
- A. Mallick, J. Kim and M. Pumera, Small, 2025, 21, 2407050 CrossRef CAS PubMed.
- H. Yu, F. Chen, X. Li, H. Huang, Q. Zhang, S. Su, K. Wang, E. Mao, B. Mei, G. Mul, T. Ma and Y. Zhang, Nat. Commun., 2021, 12, 4594 CrossRef CAS PubMed.
- H. Zheng, X. Li, K. Zhu, P. Liang, M. Wu, Y. Rao, R. Jian, F. Shi, J. Wang, K. Yan and J. Liu, Nano Energy, 2022, 93, 106831 CrossRef CAS.
- S. Gao, H. Ji, P. Yang, M. Guo, J. Tressel, S. Chen and Q. Wang, Small, 2023, 19, 2206114 CrossRef CAS PubMed.
- K.-C. Ma, H.-Y. Lin, Y.-C. Chen, C.-H. Tsai, K.-H. Zheng and J. M. Wu, J. Mater. Chem. A, 2024, 12, 26866–26876 RSC.
- Y. Fang, Y. Liu, L. Qi, Y. Xue and Y. Li, Chem. Soc. Rev., 2022, 51, 2681–2709 RSC.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- Z. Zhao, Y. Nian, J. Shi, X. Xin, X. Huang, Y. Shi, J. Tan, Y. Zhang, Y. Han, D. Yang and Z. Jiang, ACS Catal., 2024, 14, 11626–11634 CrossRef CAS.
- H. Zhao, D. Zhang, H. Li, W. Qi, X. Wu, Y. Han, W. Cai, Z. Wang, J. Lai and L. Wang, Adv. Energy Mater., 2020, 10, 2002131 CrossRef CAS.
- L. Tan, N. Yang, X. Huang, L. Peng, C. Tong, M. Deng, X. Tang, L. Li, Q. Liao and Z. Wei, Chem. Commun., 2019, 55, 14482–14485 RSC.
- Z. Liu, C. Wang, C. Chen, C. Li and C. Guo, Electrochem. Commun., 2021, 131, 107121 CrossRef CAS.
- X. Min and B. Liu, Small, 2023, 19, 2300794 CrossRef CAS PubMed.
- D. Yao, C. Tang, L. Li, B. Xia, A. Vasileff, H. Jin, Y. Zhang and S.-Z. Qiao, Adv. Energy Mater., 2020, 10, 2001289 CrossRef CAS.
- D. Kim, S. Surendran, G. Janani, Y. Lim, H. Choi, M.-K. Han, S. Yuvaraj, T.-H. Kim, J. K. Kim and U. Sim, Mater. Lett., 2022, 314, 131808 CrossRef CAS.
- W. P. Utomo, H. Wu and Y. H. Ng, Small, 2022, 18, 2200996 CrossRef CAS PubMed.
- L. Liu, T. Xiao, H. Fu, Z. Chen, X. Qu and S. Zheng, Appl. Catal., B, 2023, 323, 122181 CrossRef CAS.
- X. Wang, X. Wu, W. Ma, X. Zhou, S. Zhang, D. Huang, L. R. Winter, J.-H. Kim and M. Elimelech, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217703120 CrossRef CAS PubMed.
- Y. Xue, Q. Yu, Q. Ma, Y. Chen, C. Zhang, W. Teng, J. Fan and W.-X. Zhang, Environ. Sci. Technol., 2022, 56, 14797–14807 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, L. Han, S. Wang, T. Cui, Y. Yan, M. Xu, H. Duan, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202213711 CrossRef CAS PubMed.
- F.-Y. Chen, Z.-Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Rüscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Angew. Chem., Int. Ed., 2022, 61, e202202556 CrossRef CAS PubMed.
- S. Liu, S. Yin, S. Jiao, H. Zhang, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Mater. Today Energy, 2021, 21, 100828 CrossRef CAS.
- Y. Wang, Y. Xiong, M. Sun, J. Zhou, F. Hao, Q. Zhang, C. Ye, X. Wang, Z. Xu, Q. Wa, F. Liu, X. Meng, J. Wang, P. Lu, Y. Ma, J. Yin, Y. Zhu, S. Chu, B. Huang, L. Gu and Z. Fan, Angew. Chem., Int. Ed., 2024, 63, e202402841 CrossRef CAS PubMed.
- L. Ouyang, X. He, S. Sun, Y. Luo, D. Zheng, J. Chen, Y. Li, Y. Lin, Q. Liu, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2022, 10, 23494–23498 RSC.
- J. Liang, P. Liu, Q. Li, T. Li, L. Yue, Y. Luo, Q. Liu, N. Li, B. Tang, A. A. Alshehri, I. Shakir, P. O. Agboola, C. Sun and X. Sun, Angew. Chem., Int. Ed., 2022, 61, e202202087 CrossRef CAS PubMed.
- Q. Liu, L. Xie, J. Liang, Y. Ren, Y. Wang, L. Zhang, L. Yue, T. Li, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, I. Shakir, P. O. Agboola, Q. Kong, Q. Wang, D. Ma and X. Sun, Small, 2022, 18, 2106961 CrossRef CAS PubMed.
- D. Liu, L. Qiao, Y. Chen, P. Zhou, J. Feng, C. C. Leong, K. W. Ng, S. Peng, S. Wang, W. F. Ip and H. Pan, Appl. Catal., B, 2023, 324, 122293 CrossRef CAS.
- R. Zhang, S. Zhang, Y. Guo, C. Li, J. Liu, Z. Huang, Y. Zhao, Y. Li and C. Zhi, Energy Environ. Sci., 2022, 15, 3024–3032 RSC.
- N. J. Harmon, C. L. Rooney, Z. Tao, B. Shang, N. Raychaudhuri, C. Choi, H. Li and H. Wang, ACS Catal., 2022, 12, 9135–9142 CrossRef CAS.
- D. Dhanabal, S. S. Markandaraj and S. Shanmugam, ACS Catal., 2023, 13, 9136–9149 CrossRef CAS.
- L. Huang, L. Cheng, T. Ma, J.-J. Zhang, H. Wu, J. Su, Y. Song, H. Zhu, Q. Liu, M. Zhu, Z. Zeng, Q. He, M.-K. Tse, D.-T. Yang, B. I. Yakobson, B. Z. Tang, Y. Ren and R. Ye, Adv. Mater., 2023, 35, 2211856 CrossRef CAS PubMed.
- L. Cheng, T. Ma, B. Zhang, L. Huang, W. Guo, F. Hu, H. Zhu, Z. Wang, T. Zheng, D.-T. Yang, C.-K. Siu, Q. Liu, Y. Ren, C. Xia, B. Z. Tang and R. Ye, ACS Catal., 2022, 12, 11639–11650 CrossRef CAS.
- X.-F. Cheng, J.-H. He, H.-Q. Ji, H.-Y. Zhang, Q. Cao, W.-J. Sun, C.-L. Yan and J.-M. Lu, Adv. Mater., 2022, 34, 2205767 CrossRef CAS PubMed.
- L. Hui, Y. Xue, H. Yu, Y. Liu, Y. Fang, C. Xing, B. Huang and Y. Li, J. Am. Chem. Soc., 2019, 141, 10677–10683 CrossRef CAS PubMed.
- X. Feng, J. Liu, L. Chen, Y. Kong, Z. Zhang, Z. Zhang, D. Wang, W. Liu, S. Li, L. Tong and J. Zhang, J. Am. Chem. Soc., 2023, 145, 10259–10267 CrossRef CAS PubMed.
- Y. Wu, J. Lv, F. Xie, R. An, J. Zhang, H. Huang, Z. Shen, L. Jiang, M. Xu, Q. Yao and Y. Cao, J. Colloid Interface Sci., 2024, 656, 155–167 CrossRef CAS PubMed.
- X. Zheng, Y. Xue, C. Zhang and Y. Li, CCS Chem., 2023, 5, 1653–1662 CrossRef CAS.
- B. Chang, L. Li, D. Shi, H. Jiang, Z. Ai, S. Wang, Y. Shao, J. Shen, Y. Wu, Y. Li and X. Hao, Appl. Catal., B, 2021, 283, 119622 CrossRef CAS.
- W. Lin, H. Chen, G. Lin, S. Yao, Z. Zhang, J. Qi, M. Jing, W. Song, J. Li, X. Liu, J. Fu and S. Dai, Angew. Chem., Int. Ed., 2022, 61, e202207807 CrossRef CAS PubMed.
- Y. Li, C. Cheng, S. Han, Y. Huang, X. Du, B. Zhang and Y. Yu, ACS Energy Lett., 2022, 7, 1187–1194 CrossRef CAS.
- D. Wang, Z.-W. Chen, K. Gu, C. Chen, Y. Liu, X. Wei, C. V. Singh and S. Wang, J. Am. Chem. Soc., 2023, 145, 6899–6904 CrossRef CAS PubMed.
- J.-Y. Fang, Q.-Z. Zheng, Y.-Y. Lou, K.-M. Zhao, S.-N. Hu, G. Li, O. Akdim, X.-Y. Huang and S.-G. Sun, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed.
- Y. Wang, H. Li, W. Zhou, X. Zhang, B. Zhang and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202202604 CrossRef CAS PubMed.
- T. Hu, M. Wang, C. Guo and C. M. Li, J. Mater. Chem. A, 2022, 10, 8923–8931 RSC.
- J. Cai, J. Huang, A. Cao, Y. Wei, H. Wang, X. Li, Z. Jiang, G. I. N. Waterhouse, S. Lu and S.-Q. Zang, Appl. Catal., B, 2023, 328, 122473 CrossRef CAS.
- X. Luo, Y. Wu, H. Hu, T. Wei, B. Wu, J. Ding, Q. Liu, J. Luo and X. Liu, Small, 2024, 20, 2403399 CrossRef CAS PubMed.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS PubMed.
- J. Liang, S. Ma, J. Li, Y. Wang, J. Wu, Q. Zhang, Z. Liu, Z. Yang, K. Qu and W. Cai, J. Mater. Chem. A, 2020, 8, 10426–10432 RSC.
- M. Yu, H. Huang, J. Hu, S. Wang, J. Li and D. Wang, J. Mater. Chem. A, 2022, 10, 23990–23997 RSC.
- G. Lin, Q. Ju, X. Guo, W. Zhao, S. Adimi, J. Ye, Q. Bi, J. Wang, M. Yang and F. Huang, Adv. Mater., 2021, 33, 2007509 CrossRef CAS PubMed.
- R. Luo, B.-J. Li, Z.-P. Wang, M.-G. Chen, G.-L. Zhuang, Q. Li, J.-P. Tong, W.-T. Wang, Y.-H. Fan and F. Shao, JACS Au, 2024, 4, 3823–3832 CrossRef CAS PubMed.
- Y. Lv, S.-W. Ke, Y. Gu, B. Tian, L. Tang, P. Ran, Y. Zhao, J. Ma, J.-L. Zuo and M. Ding, Angew. Chem., Int. Ed., 2023, 62, e202305246 CrossRef CAS PubMed.
- J. Ma, Y. Zhang, B. Wang, Z. Jiang, Q. Zhang and S. Zhuo, ACS Nano, 2023, 17, 6687–6697 CrossRef CAS PubMed.
- A. K. K. Padinjareveetil, J. V. Perales-Rondon, D. Zaoralová, M. Otyepka, O. Alduhaish and M. Pumera, ACS Appl. Mater. Interfaces, 2023, 15, 47294–47306 CrossRef CAS PubMed.
- J. V. Perales-Rondon, D. Rojas, W. Gao and M. Pumera, ACS Sustainable Chem. Eng., 2023, 11, 6923–6931 CrossRef CAS.
- W. Gao, J. V. Perales-Rondon, J. Michalička and M. Pumera, Appl. Catal., B, 2023, 330, 122632 CrossRef CAS.
- W. Gao, J. Michalička and M. Pumera, J. Mater. Chem. A, 2024, 12, 32458–32470 RSC.
- Z. Huang, M. Rafiq, A. R. Woldu, Q.-X. Tong, D. Astruc and L. Hu, Coord. Chem. Rev., 2023, 478, 214981 CrossRef CAS.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- F. F. Tao, L. Nguyen and S. Zhang, Metal nanoparticles for catalysis: advances and applications, The Royal Society of Chemistry, 2014 Search PubMed.
- B. H. Ko, B. Hasa, H. Shin, Y. Zhao and F. Jiao, J. Am. Chem. Soc., 2022, 144, 1258–1266 CrossRef CAS PubMed.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- T. Hu, C. Wang, M. Wang, C. M. Li and C. Guo, ACS Catal., 2021, 11, 14417–14427 CrossRef CAS.
- J. Wang, Y. Wang, C. Cai, Y. Liu, D. Wu, M. Wang, M. Li, X. Wei, M. Shao and M. Gu, Nano Lett., 2023, 23, 1897–1903 CrossRef CAS PubMed.
- W. Peng, M. Luo, X. Xu, K. Jiang, M. Peng, D. Chen, T.-S. Chan and Y. Tan, Adv. Energy Mater., 2020, 10, 2001364 CrossRef CAS.
- Y. Xu, K. Ren, T. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 306, 121094 CrossRef CAS.
- Y. Han, X. Zhang, W. Cai, H. Zhao, Y. Zhang, Y. Sun, Z. Hu, S. Li, J. Lai and L. Wang, J. Colloid Interface Sci., 2021, 600, 620–628 CrossRef CAS PubMed.
- L. Lv, Y. Shen, J. Liu, X. Meng, X. Gao, M. Zhou, Y. Zhang, D. Gong, Y. Zheng and Z. Zhou, J. Phys. Chem. Lett., 2021, 12, 11143–11150 CrossRef CAS PubMed.
- G. A. Cerrón-Calle, A. S. Fajardo, C. M. Sánchez-Sánchez and S. Garcia-Segura, Appl. Catal., B, 2022, 302, 120844 CrossRef.
- N. Zhou, Z. Wang, N. Zhang, D. Bao, H. Zhong and X. Zhang, ACS Catal., 2023, 13, 7529–7537 CrossRef CAS.
- M. Yang, T. Wei, J. He, Q. Liu, L. Feng, H. Li, J. Luo and X. Liu, Nano Res., 2024, 17, 1209–1216 CrossRef CAS.
- X. Qu, L. Shen, Y. Mao, J. Lin, Y. Li, G. Li, Y. Zhang, Y. Jiang and S. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 31869–31877 CrossRef CAS PubMed.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. T. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- W.-D. Zhang, H. Dong, L. Zhou, H. Xu, H.-R. Wang, X. Yan, Y. Jiang, J. Zhang and Z.-G. Gu, Appl. Catal., B, 2022, 317, 121750 CrossRef CAS.
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J.-F. Li, Z.-Q. Tian, W. Liu, A. Wang and T. Zhang, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed.
- J. Cheng, W. Sun, G. Dai, X. Yang, R. Xia, Y. Xu, X. Yang and W. Tu, Fuel, 2023, 332, 126106 CrossRef CAS.
- T. Xiang, Y. Liang, Y. Zeng, J. Deng, J. Yuan, W. Xiong, B. Song, C. Zhou and Y. Yang, Small, 2023, 19, 2303732 CrossRef CAS PubMed.
- P. Guo, D. Luan, H. Li, L. Li, S. Yang and J. Xiao, J. Am. Chem. Soc., 2024, 146, 13974–13982 CrossRef CAS PubMed.
- J. Deng, Y. Su, D. Liu, P. Yang, B. Liu and C. Liu, Chem. Rev., 2019, 119, 9221–9259 CrossRef CAS PubMed.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- H. Jiang, G.-F. Chen, O. Savateev, J. Xue, L.-X. Ding, Z. Liang, M. Antonietti and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202218717 CrossRef CAS PubMed.
- F. Gou, H. Wang, M. Fu, Y. Jiang, W. Shen, R. He and M. Li, Appl. Surf. Sci., 2023, 612, 155872 CrossRef CAS.
- Q. He, S. Qiao, Y. Zhou, R. Vajtai, D. Li, P. M. Ajayan, L. Ci and L. Song, Adv. Funct. Mater., 2022, 32, 2106684 CrossRef CAS.
- J. O. Olowoyo and R. J. Kriek, Small, 2022, 18, 2203125 CrossRef CAS PubMed.
- Z. Deng, C. Ma, Z. Li, Y. Luo, L. Zhang, S. Sun, Q. Liu, J. Du, Q. Lu, B. Zheng and X. Sun, ACS Appl. Mater. Interfaces, 2022, 14, 46595–46602 CrossRef CAS PubMed.
- J. Yuan, H. Yin, X. Jin, D. Zhao, Y. Liu, A. Du, X. Liu and A. P. O’Mullane, Appl. Catal., B, 2023, 325, 122353 CrossRef CAS.
- A. Ambrosi, C. K. Chua, N. M. Latiff, A. H. Loo, C. H. A. Wong, A. Y. S. Eng, A. Bonanni and M. Pumera, Chem. Soc. Rev., 2016, 45, 2458–2493 RSC.
- C. Huang, Y. Li, N. Wang, Y. Xue, Z. Zuo, H. Liu and Y. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS PubMed.
- H. Yu, Y. Xue, L. Hui, C. Zhang, Y. Fang, Y. Liu, X. Chen, D. Zhang, B. Huang and Y. Li, Natl. Sci. Rev., 2021, 8, nwaa213 CrossRef CAS PubMed.
- H. Zou, W. Rong, S. Wei, Y. Ji and L. Duan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29462–29468 CrossRef CAS PubMed.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- S. Wang, C. Song, Y. Cai, Y. Li, P. Jiang, H. Li, B. Yu and T. Ma, Adv. Energy Mater., 2023, 13, 2301136 CrossRef CAS.
- L. Verger, V. Natu, M. Carey and M. W. Barsoum, Trends Chem., 2019, 1, 656–669 CrossRef CAS.
- Y. Li, J. Ma, T. D. Waite, M. R. Hoffmann and Z. Wang, Environ. Sci. Technol., 2021, 55, 10695–10703 CrossRef CAS PubMed.
- Y. Xiao and C. Shen, Small, 2021, 17, 2100776 CrossRef CAS PubMed.
- L. Zhang, J. Liang, Y. Wang, T. Mou, Y. Lin, L. Yue, T. Li, Q. Liu, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, X. Guo, D. M. Ma and X. Sun, Angew. Chem., Int. Ed., 2021, 60, 25263–25268 CrossRef CAS PubMed.
- S. Zhang, M. Li, J. Li, Q. Song and X. Liu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2115504119 CrossRef CAS PubMed.
- D. Pan, M. Austeria P, S. Lee, H.-S. Bae, F. He, G. H. Gu and W. Choi, Nat. Commun., 2024, 15, 7243 CrossRef CAS PubMed.
- Y. Ying, M. P. Browne and M. Pumera, Sustainable Energy Fuels, 2020, 4, 3732–3738 RSC.
- K. P. A. Kumar, K. Ghosh, O. Alduhaish and M. Pumera, Electrochem. Commun., 2021, 122, 106890 CrossRef.
- E. Redondo and M. Pumera, Appl. Mater. Today, 2021, 25, 101253 CrossRef.
- C. Didier, A. Kundu and S. Rajaraman, Microsyst. Nanoeng., 2020, 6, 15 CrossRef CAS PubMed.
- C. Iffelsberger, S. Ng and M. Pumera, Appl. Mater. Today, 2020, 20, 100654 CrossRef.
- V. Urbanová, J. Plutnar and M. Pumera, Appl. Mater. Today, 2021, 24, 101131 CrossRef.
- G. Bharath, C. Liu, F. Banat, A. Kumar, A. Hai, A. K. Nadda, V. K. Gupta, M. A. Haija and J. Balamurugan, Chem. Eng. J., 2023, 465, 143040 CrossRef CAS.
- V. R. Silveira, R. Bericat-Vadell and J. Sá, J. Phys. Chem. C, 2023, 127, 5425–5431 CrossRef CAS.
- N. Gao, H. Yang, D. Dong, D. Dou, Y. Liu, W. Zhou, F. Gao, C. Nan, Z. Liang and D. Yang, J. Colloid Interface Sci., 2022, 611, 294–305 CrossRef CAS PubMed.
- C. H. Kim, J. Kim, F. Hollmann and C. B. Park, Appl. Catal., B, 2023, 336, 122925 CrossRef CAS.
- F. Xu, F. Wu, K. Zhu, Z. Fang, D. Jia, Y. Wang, G. Jia, J. Low, W. Ye, Z. Sun, P. Gao and Y. Xiong, Appl. Catal., B, 2021, 284, 119689 CrossRef CAS.
- S. Zhou, K. Sun, C. Y. Toe, J. Yin, J. Huang, Y. Zeng, D. Zhang, W. Chen, O. F. Mohammed, X. Hao and R. Amal, Adv. Mater., 2022, 34, 2201670 CrossRef CAS PubMed.
- L. Paramanik, S. Sultana and K. M. Parida, J. Colloid Interface Sci., 2022, 625, 83–99 CrossRef CAS PubMed.
- X. Li, W. Fan, Y. Bai, Y. Liu, F. Wang, H. Bai and W. Shi, Chem. Eng. J., 2022, 433, 133225 CrossRef CAS.
- D. Liu, J. Wang, S. Bian, Q. Liu, Y. Gao, X. Wang, P. K. Chu and X.-F. Yu, Adv. Funct. Mater., 2020, 30, 2002731 CrossRef CAS.
- Y. Bai, S. Gao, W. Xie, Z. Fang, H. Bai and W. Fan, Int. J. Hydrogen Energy, 2023, 48, 10882–10890 CrossRef CAS.
- P. Li, Y. Liu, M. A. Mushtaq and D. Yan, Inorg. Chem. Front., 2023, 10, 4650–4667 RSC.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef.
- U. S. Environmental Protection Agency, Drinking Water Standards and Health Advisories, EPA 822-F-18-001, 2018, https://www.epa.gov/system/files/documents/2022-2001/dwtable2018.pdf.
- L. Su, D. Han, G. Zhu, H. Xu, W. Luo, L. Wang, W. Jiang, A. Dong and J. Yang, Nano Lett., 2019, 19, 5423–5430 CrossRef CAS PubMed.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- S. Lee, S. Kim, C. Park, W. Kim, S. Ryu and W. Choi, Energy Environ. Sci., 2021, 14, 4437–4450 RSC.
- C. Park, H. Kwak, G.-H. Moon and W. Kim, J. Mater. Chem. A, 2021, 9, 19179–19205 RSC.
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, ACS Sustainable Chem. Eng., 2020, 8, 2672–2681 CrossRef CAS.
- W. Zheng, L. Zhu, Z. Yan, Z. Lin, Z. Lei, Y. Zhang, H. Xu, Z. Dang, C. Wei and C. Feng, Environ. Sci. Technol., 2021, 55, 13231–13243 CAS.
- N. C. Kani, J. A. Gauthier, A. Prajapati, J. Edgington, I. Bordawekar, W. Shields, M. Shields, L. C. Seitz, A. R. Singh and M. R. Singh, Energy Environ. Sci., 2021, 14, 6349–6359 RSC.
- X. Wu, Z. Song, Z. Liu, X. Tang, F. Yao, F. Zhao, X. Min and C.-J. Tang, Appl. Catal., B, 2024, 359, 124467 CrossRef CAS.
- F.-Y. Chen, A. Elgazzar, S. Pecaut, C. Qiu, Y. Feng, S. Ashokkumar, Z. Yu, C. Sellers, S. Hao, P. Zhu and H. Wang, Nat. Catal., 2024, 7, 1032–1043 CrossRef CAS.
- Q. Zhang, Y. Li, M. Geng, J. Zhu, H. Sun and B. Jiang, Appl. Catal., B, 2023, 330, 122658 CrossRef CAS.
- G. Zhang, B. Li, Y. Shi, Q. Zhou, W.-J. Fu, G. Zhou, J. Ma, S. Yin, W. Yuan, S. Miao, Q. Ji, J. Qu and H. Liu, Nat. Sustainable, 2024, 7, 1251–1263 CrossRef.
- M. Jiang, M. Zhu, M. Wang, Y. He, X. Luo, C. Wu, L. Zhang and Z. Jin, ACS Nano, 2023, 17, 3209–3224 CrossRef CAS PubMed.
- A. Khan, A. Abbas and R. Dickson, Green Chem., 2024, 26, 1551–1565 RSC.
- Y. Wan, M. Zheng, W. Yan, J. Zhang and R. Lv, Adv. Energy Mater., 2024, 14, 2303588 CrossRef CAS.
- J. Leverett, T. Tran-Phu, J. A. Yuwono, P. Kumar, C. Kim, Q. Zhai, C. Han, J. Qu, J. Cairney, A. N. Simonov, R. K. Hocking, L. Dai, R. Daiyan and R. Amal, Adv. Energy Mater., 2022, 12, 2201500 CrossRef CAS.
- J. Zheng, S. Xu, J. Sun, J. Zhang, L. Sun, X. Pan, L. Li and G. Zhao, Appl. Catal., B, 2023, 338, 123056 CrossRef CAS.
- Y. Luo, K. Xie, P. Ou, C. Lavallais, T. Peng, Z. Chen, Z. Zhang, N. Wang, X.-Y. Li, I. Grigioni, B. Liu, D. Sinton, J. B. Dunn and E. H. Sargent, Nat. Catal., 2023, 6, 939–948 CrossRef CAS.
- D.-S. Huang, X.-F. Qiu, J.-R. Huang, M. Mao, L. Liu, Y. Han, Z.-H. Zhao, P.-Q. Liao and X.-M. Chen, Nat. Synth., 2024, 3, 1404–1413 CrossRef CAS.
- H. Xiong, P. Yu, K. Chen, S. Lu, Q. Hu, T. Cheng, B. Xu and Q. Lu, Nat. Catal., 2024, 7, 785–795 CrossRef CAS.
- N. Meng, X. Ma, C. Wang, Y. Wang, R. Yang, J. Shao, Y. Huang, Y. Xu, B. Zhang and Y. Yu, ACS Nano, 2022, 16, 9095–9104 CrossRef CAS PubMed.
- X. Liu, P. V. Kumar, Q. Chen, L. Zhao, F. Ye, X. Ma, D. Liu, X. Chen, L. Dai and C. Hu, Appl. Catal., B, 2022, 316, 121618 CrossRef CAS.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustainable, 2021, 4, 868–876 CrossRef.
- X. Wei, X. Wen, Y. Liu, C. Chen, C. Xie, D. Wang, M. Qiu, N. He, P. Zhou, W. Chen, J. Cheng, H. Lin, J. Jia, X.-Z. Fu and S. Wang, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- Y. Wang, S. Xia, R. Cai, J. Zhang, C. Yu, J. Cui, Y. Zhang, J. Wu and Y. Wu, Angew. Chem., Int. Ed., 2024, 63, e202318589 CrossRef CAS PubMed.
- Y. Yang, J. Peng, Z. Shi, P. Zhang, A. Arramel and N. Li, J. Mater. Chem. A, 2023, 11, 6428–6439 RSC.
- Y. Zhang, L. Lu, T. Zhao, J. Zhao, Q. Cai and Z. Chen, J. Mater. Chem. A, 2024, 12, 16704–16715 RSC.
- B. Govindan, K. Annamalai, A. Kumar, S. Palanisamy, M. Abu Haija and F. Banat, ACS Sustainable Chem. Eng., 2024, 12, 8174–8187 CrossRef CAS.
- X. Zhu, X. Zhou, Y. Jing and Y. Li, Nat. Commun., 2021, 12, 4080 CrossRef CAS PubMed.
- X. Cao, D. Zhang, Y. Gao, O. V. Prezhdo and L. Xu, J. Am. Chem. Soc., 2024, 146, 1042–1052 CrossRef CAS PubMed.
- H. Cai, Z. Wang, G. Meng, T. Wei, Y. Liu, J. Luo, Q. Liu, G. Hu and X. Liu, Inorg. Chem., 2024, 63, 20935–20939 CrossRef CAS PubMed.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- Z. Fang, Z. Jin, S. Tang, P. Li, P. Wu and G. Yu, ACS Nano, 2022, 16, 1072–1081 CrossRef CAS PubMed.
- J.-Y. Zhu, Q. Xue, Y.-Y. Xue, Y. Ding, F.-M. Li, P. Jin, P. Chen and Y. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 14064–14070 CrossRef CAS PubMed.
- X. Deng, Y. Yang, L. Wang, X.-Z. Fu and J.-L. Luo, Adv. Sci., 2021, 8, 2004523 CrossRef CAS PubMed.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 13, 7958 CrossRef CAS PubMed.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 10804–10811 CrossRef CAS PubMed.
- E. A. Weiss, ACS Energy Lett., 2017, 2, 1005–1013 CrossRef CAS.
- S.-L. Meng, J.-H. Li, C. Ye, Y.-L. Yin, X.-L. Zhang, C. Zhang, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2024, 36, 2311982 CrossRef CAS PubMed.
- Z. Niu, S. Fan, X. Li, P. Wang, Z. Liu, J. Wang, C. Bai and D. Zhang, Chem. Eng. J., 2022, 450, 138343 CrossRef CAS.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- Á. Balog, E. Kecsenovity, G. F. Samu, J. He, D. Fekete and C. Janáky, Nat. Catal., 2024, 7, 522–535 CrossRef.
- J. Kim, J.-A. Lin, J. Kim, I. Roh, S. Lee and P. Yang, Nat. Catal., 2024, 7, 977–986 CrossRef CAS.
- W. Zhu, X. Zhang, F. Yao, R. Huang, Y. Chen, C. Chen, J. Fei, Y. Chen, Z. Wang and H. Liang, Angew. Chem., Int. Ed., 2023, 62, e202300390 CrossRef CAS PubMed.
- S. Li, P. Ma, C. Gao, L. Liu, X. Wang, M. Shakouri, R. Chernikov, K. Wang, D. Liu, R. Ma and J. Wang, Energy Environ. Sci., 2022, 15, 3004–3014 RSC.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- M. Yang, T. Wei, C. Zeng, J. Zhang, Y. Liu, J. Luo, G. Hu and X. Liu, Chem. Eng. J., 2024, 498, 155799 CrossRef CAS.
- R. Zaffaroni, D. Ripepi, J. Middelkoop and F. M. Mulder, ACS Energy Lett., 2020, 5, 3773–3777 CrossRef CAS.
- C. Zhang, J. Li, R. Chen, S. Shen, J. Wang, Y. Sun and F. Dong, ACS Catal., 2024, 14, 15721–15742 CrossRef CAS.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- N. Amornthammarong, J.-Z. Zhang and P. B. Ortner, Anal. Methods, 2011, 3, 1501–1506 RSC.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- J. B. Hansen and P. Han, Green Ammonia by Haldor Topsoe, Department of Energy, 2021, https://www.energy.gov/sites/default/files/2021-2008/2024-green-ammonia-haldor-topsoe.pdf Search PubMed.
- Small-Scale Green Ammonia Plants Open up New Storage Possibilities for Wind and Solar Power, ThyssenKrupp Industrial Solutions, 2022, https://insights.thyssenkrupp-industrial-solutions.com/story/small-scale-green-ammonia-plants-open-up-new-storage-possibilities-for-wind-and-solar-power/.
- Yara Selects Linde Engineering to Build Electrolysis Plant at Porsgrunn, Ammonia Energy Association, 2022, https://www.ammoniaenergy.org/articles/yara-selects-linde-engineering-to-build-electrolysis-plant-at-porsgrunn/.
- S. Wu, N. Salmon, M. M.-J. Li, R. Bañares-Alcántara and S. C. E. Tsang, ACS Energy Lett., 2022, 7, 1021–1033 CrossRef CAS.
- B. Bonnet-Cantalloube, M. Espitalier-Noël, P. F. de Carvalho, J. Fonseca and G. Pawelec, Clean ammonia in the future energy system, Hydrogen Europe, 2023, https://hydrogeneurope.eu/wp-content/uploads/2023/2003/2023.<?pdb_no 2003?>2003<?pdb<?db_id PDB?> END?>_H2022Europe_Clean_Ammonia_Report_DIGITAL_FINAL.pdf.
- ENGIE-YARA, Renewable Hydrogen and Ammonia Deployment in Pilbara, YURI Phase 0: Feasibility study public report, 2020, https://arena.gov.au/assets/2020/2011/engie-yara-renewable-hydrogen-and-ammonia-deployment-in-pilbara.pdf.
- Kapsom, The World's First Green Ammonia Plant Was Made By Kapsom, Green ammonia produced from renewable,to a renewable energy drive, 2022, https://www.kapsom.com/the-worlds-first-green-ammonia-plant-was-made-by-kapsom/.
- W. Bi, E. Gyenge and D. P. Wilkinson, Chem. Eng. J., 2023, 478, 147359 CrossRef CAS.
- H2F project, Green hydrogen, green ammonia and green fertiliser plant, https://www.fertiberia.com/en/greenammonia/h2f-project/.
- P. Mayer, A. Ramirez, G. Pezzella, B. Winter, S. M. Sarathy, J. Gascon and A. Bardow, iScience, 2023, 26, 107389 CrossRef CAS PubMed.
- M.-H. Yuan, Y.-H. Chen, J.-Y. Tsai and C.-Y. Chang, Process Saf. Environ. Prot., 2016, 102, 777–785 CrossRef CAS.
- L. Zeng, C. Mangan and X. Li, Water Sci. Technol., 2006, 54, 137–145 CrossRef CAS PubMed.
- J. Gao, Q. Ma, Z. Wang, B. E. Rittmann and W. Zhang, Nat. Commun., 2024, 15, 8455 CrossRef CAS PubMed.
- L. M. Wilder, K. Wyatt, C. A. Skangos, W. E. Klein, M. R. Parimuha, J. L. Katsirubas, J. L. Young and E. M. Miller, ACS Appl. Energy Mater., 2024, 7, 536–545 CrossRef CAS PubMed.
- K. Kim, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F1523 CrossRef CAS.
- S. Li, D. Han, G. Jiang, Z. Han, H. Lu, J. Gao, X. Wang, Y. Wang, C. Geng, Z. Weng and Q.-H. Yang, ACS Appl. Energy Mater., 2023, 6, 5067–5073 CrossRef CAS.
- W. Liao, J. Wang, G. Ni, K. Liu, C. Liu, S. Chen, Q. Wang, Y. Chen, T. Luo, X. Wang, Y. Wang, W. Li, T.-S. Chan, C. Ma, H. Li, Y. Liang, W. Liu, J. Fu, B. Xi and M. Liu, Nat. Commun., 2024, 15, 1264 CrossRef CAS PubMed.
- S.-N. Zhang, P. Gao, Q.-Y. Liu, Z. Zhang, B.-L. Leng, J.-S. Chen and X.-H. Li, Nat. Commun., 2024, 15, 10877 CrossRef PubMed.
- Z. Xu, L. Wan, Y. Liao, M. Pang, Q. Xu, P. Wang and B. Wang, Nat. Commun., 2023, 14, 1619 CrossRef CAS PubMed.
- J. Lan, Z. Wang, C.-W. Kao, Y.-R. Lu, F. Xie and Y. Tan, Nat. Commun., 2024, 15, 10173 CrossRef CAS PubMed.
- L. Mi, Q. Huo, J. Cao, X. Chen, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2202247 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |