
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShifted-excitation Raman difference spectroscopy and charge-shifting detection coupled with spatially offset Raman spectroscopy for heritage science†
Alberto
Lux
 *abc,
Claudia
Conti
a,
Alessandra
Botteon
a,
Sara
Mosca
c and
Pavel
Matousek
*ac
*abc,
Claudia
Conti
a,
Alessandra
Botteon
a,
Sara
Mosca
c and
Pavel
Matousek
*ac
aInstitute of Heritage Science, National Research Council (CNR-ISPC), Via Cozzi 53, 20125, Milan, Italy. E-mail: alberto.lux@mail.polimi.it
bSapienza University of Rome, Faculty of Literature, Department of Classics, Piazzale Aldo Moro 5, 00185, Rome, Italy
cCentral Laser Facility, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, UK Research and Innovation (UKRI), Harwell Campus, OX11 0QX, UK
First published on 12th February 2025
Abstract
In situ measurements have great importance since in many scientific fields certain samples cannot be moved because of diverse reasons (excessive dimensions or weight, security, logistics etc.). In heritage science, this is a crucial requirement due to the high value of art objects, requiring non-invasive and in situ analyses. Therefore, it is important to have analytical methods capable of providing relevant information also outside laboratory environments. Such measurements face multiple challenges: for example, interference from ambient light or formation of artefacts due to undesired motions of the instruments. In Raman spectroscopy, a number of solutions have been demonstrated to mitigate these effects. For instance, Shifted Excitation Raman Difference Spectroscopy (SERDS) has proven efficient in removing the fluorescence of the sample and ambient light interference, and a charge-shifting detection approach was shown to be valuable in dealing with varying ambient light. In this study, we provide a comparison of conventional Raman spectroscopy, Shifted Excitation Raman Difference Spectroscopy (SERDS), charge-shifting detection technology and a combined SERDS and charge-shifting approach, in order to evaluate their effectiveness in mitigating fast evolving interfering backgrounds (e.g., varying ambient light). Further investigations were also carried out into the potential of coupling of these methods with Spatially Offset Raman Spectroscopy (SORS) to facilitate more effective non-invasive investigations of subsurface sample components (e.g. paint layers). The study was carried out using samples mimicking cultural heritage materials with different degrees of complexity and in the presence of fluorescence and ambient light interference. The results are, nevertheless, applicable more generally to other areas such as forensics or biomedical fields, where both dynamic and static interferences can hinder measurements.
Introduction
The removal of fluorescence and ambient light from Raman spectra is of great importance in many application areas including forensics,1–3 biomedical field,4,5 food6 or heritage science,7 especially in cases where in situ measurements are required, for example, due to security reasons, inability to move the sample or logistics issues. In this context, many solutions have been developed in the past few decades to deal with such scenarios. These include Shifted Excitation Raman Difference Spectroscopy8–11 (SERDS) and charge-shifting detection (CS). The specifics of the working principles of SERDS8,12–14 and charge-shifting15–17 are detailed in previous works: briefly, SERDS employs two excitation laser wavelengths that possess a very narrow wavelength difference, on the scale of bandwidths of Raman bands present, and since the fluorescence and ambient light background signals remain constant, whereas Raman signals shift with the corresponding amount, a subtraction of the pair of spectra obtained with each excitation wavelength effectively removes fluorescence and ambient light.15–17 However, difficulties may arise when the background interference is not static but varies in some manner (such as due to fluorescence bleaching,18 variation of ambient light intensity, the passing of a cloud in outdoor measurements or the shadow cast by the operator onto the instrument during acquisition) as SERDS is not able to deal with background intensity changes taking place between the two consecutive acquisitions at the two different excitation wavelengths. To address this issue, charge-shifting (CS) technology offers potential advantages: it employs an adapted CCD that receives signals illuminating only certain rows on its chip with alternating blocks of rows being unilluminated. In conjunction with rapidly alternating switching between the two excitation wavelengths, or between ON and OFF states where only one laser wavelength is used, the charges are shifted in synchrony up or down between the illuminated and obscured zones.15 After the acquisition, by subtracting the incoming signals of the two different sets of rows, it is possible to reject the dynamic interference of ambient light, since the frequency at which the charges are shifted can be much higher (1–10 kHz)17 than the typical frequency of the considered background variations. However, charge-shifting alone is not able to deal with the static component of sample fluorescence, which is the reason for its coupling with SERDS. Moreover, the employment of SORS19 with said technologies paves the way also for the investigations of subsurface layers of materials in a non-invasive manner, in addition to the rejection of both static and dynamic interference backgrounds.20–22 In this study, we evaluate the capabilities and limitations of these methods applied to cultural heritage samples. The samples were selected for their intrinsic complexity and challenges (e.g. Raman scattering efficiency and fluorescence). The experiments were further challenged by carrying out these measurements under dynamically varying ambient light to mimic in situ conditions, combining and comparing these techniques for the first time.Materials and methods
Set-up
The experimental set up used in this study is based around a custom-built SORS system described in detail in previous publications.16,17 Its schematic diagram is shown in Fig. S1 of the ESI.† Briefly, the system is a SORS point-like excitation–collection system that utilizes a purpose-designed integrated SERDS laser module23 emitting at λ1 = 829.40 nm (L1) and λ2 = 828.85 nm (L2). The laser spot size and the collection area diameters are ∼500 μm and ∼1.5 mm, respectively. The SORS spatial offset is achieved by moving the entire collection path assembly with a motorized stage (MTS25-Z8 with a KDC101 controller, Thorlabs). The collected Raman signal is then sent to an imaging spectrometer (HoloSpec f/1.8i, Kaiser Optical Systems) coupled with a custom-made charge-shifting CCD (DU420A-BR-DD-9UW, Andor Technology). A custom made micro-machined metal mask, placed at the entrance slit of the spectrograph, made out of a tungsten foil (thickness = 100 μm, lateral dimensions: 10 mm × 5 mm, grid dimensions: height = 6 mm, width = 3 mm), was employed to shield (with a periodical pattern corresponding to CCD 8-pixels ON and OFF) the CCD sensor along the vertical axis, as required for the CS method.17 Raman measurements were performed both at 0 mm (i.e. imaged) and 2 mm spatial offsets. Laser intensity and acquisition times were adjusted to attain equivalent illumination conditions (the overall laser energy delivered to the sample, in multiple pulses and per measurement, was set to ∼2.1 J) in all the charge-shifting, SERDS and conventional acquisition modalities (a detailed schematic diagram is shown in Fig. S2 of the ESI†). An overview of experimental parameters for conventional, SERDS and CS read-out modalities is provided in Table 1. The wavelength calibration (pixel to relative Raman shift) was carried out using an aspirin tablet (using laser L1). To improve comparison between techniques, signal-to-noise ratios (S/N) have been added in the captions of the figures, calculated using the method described by Mosca et al.24 where the noise is assumed to be the largest artefact present in the spectra or photon shot noise amplitude, whichever is greater. In the case of SORS measurements, only ratios relative to the offset spectra have been computed assuming that the bottom layer signal is the signal of interest.| Modalities | Average power (mW) | Time (s) | |
|---|---|---|---|
| Conventional (L1) | 60 | 35 | |
| SERDS | L1 | 60 | 17.5 |
| L2 | 60 | 17.5 | |
| CS (L1 only) | 30 | 70 | |
| CS + SERDS | L1 | 60 | 35 |
| L2 | 60 | 35 | |
Conventional. Spectra were initially acquired in conventional mode using laser L1, with a laser power equivalent to 60 mW and acquisition time set to 35 s (which confers to the sample an overall energy of 2.1 J per measurement, as shown in the schematics in Fig. S2†).
SERDS. Similar to the conventional method, spectra with L1 and L2 were collected one after the other. The same laser power was employed for both lasers and acquisition time was halved to 17.5 s, so that the overall energy was equal to the conventional measurements. Difference spectra were obtained by subtracting the two Raman spectra obtained at the two respective excitation wavelengths. Additionally, reconstructed SERDS spectra were calculated using a reconstruction algorithm developed in Python, based on previous work.25 The routine is particularly basic, considering that our aim was to show how many artefacts might appear due to background interferences and how much effort would be required to design and implement a suitable routine to remove them all. Instead, employing a suitable combination of techniques and approaches might resolve the issue at its root. Moreover, removing narrow room light bands becomes even more complicated for in-field instrumentation, as the resolution of these spectrometers is comparable with the Raman bandwidths (as is typical of these devices, in order to maximise light throughput) and consequently the interfering emission lines have comparable widths with Raman bands: as such, their differentiation from Raman signals is not straightforward. Additional subtraction of polynomial backgrounds could further improve the outcomes in a number of cases. We have not performed this correction here to retain background distortions present as they enable the most direct comparison between the performances of individual techniques.
CS. The system was operated at 1 kHz. The timing and synchronization between the laser wavelengths and the charge-shifting CCD read-out were controlled by a digital delay generator (DG645, Stanford Research Systems) connected to the laser driver module and external CCD trigger (further details can be found in our previous study15–17). The trigger voltage value for each laser was adjusted to provide an average power of 30 mW at the sample in a 50% duty cycle mode. The equivalent acquisition time was set to 70 s (laser pulse width (Tw) × number of cycles × number of repetitions). It is worth underlining that we operated the charge shifting instrument at 50
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 numbers of cycles (maximum possible value for CS cycles – additional details are given in the ESI in Fig. S3 and 4†) to achieve the desired overall energy on the sample, equal to those from the other techniques. Thus, we accepted that some degree of charge leakage between alternating CCD rows could occur,17 adding some extra noise-like features to the spectrum.
000 numbers of cycles (maximum possible value for CS cycles – additional details are given in the ESI in Fig. S3 and 4†) to achieve the desired overall energy on the sample, equal to those from the other techniques. Thus, we accepted that some degree of charge leakage between alternating CCD rows could occur,17 adding some extra noise-like features to the spectrum.
Samples
The samples were chosen with increasing complexity in order to challenge the techniques at different levels. We started with a standard non-fluorescent sample and then moved on to more realistic, fluorescing ones.• S0 – a sample consisting of polytetrafluoroethylene (PTFE), about ∼8 mm thick and roughly 4 × 4 cm wide, used as a standard non-fluorescent sample.
Single layer measurements:
• S1 – phthalocyanine blue (C32H16N8Cu), a common pigment used in art, painted on a wooden support. This sample was collected from an atelier in Milan, which refused citation. It was effectively a single layer system since the Raman signal from the substrate was undetectable. In this case, phthalocyanine blue paint was highly absorbing and fluorescing. The blue layer was about 10 μm thick and roughly 5 × 5 cm wide.
• S2 – a gypsum (CaSO4·2H2O) layer, cut from a solidified mixture of gypsum powder and water, mimicking a plaster, stucco or preparation layer for paintings. Preliminary spectra showed some fluorescence signal from this sample. The sample was 0.5 mm thick and 2 × 5 cm wide, and was prepared in ISPC laboratory in Milan.
• S3 – a calcite (CaCO4) layer, often employed as plaster or a preparation layer for paintings, applied on paper (the latter did not provide any Raman signal). This sample was not prepared in our laboratory, but instead was acquired from the atelier in Milan mentioned above. Also, in this case only the calcite signal (top layer) from the preparation layer was detected, along with fluorescence possibly originating from the paper. The whole sample is about 1 mm thick and 5 × 5 cm wide.
The first four samples (S0, S1, S2, S3) were all analysed on the front and on the back sides. The only samples that showed any difference between the front and back signals were the S1 sample, which, as already mentioned, did not provide any signal from the wooden substrate, and the S3 sample, where the preparation layer was likely applied only on one side of the paper.
SORS measurements:
• S4 – a thin tape of PTFE (0.25 mm thick) placed on top of a marble support, which itself yields a strong calcite Raman signal. Both layers possessed appreciable Raman scattering efficiency and a very small degree of fluorescence.
• S5 – the sample was created using a block of PTFE as a substrate (8 mm thick) and S3 used as the top layer.
S4 and S5 samples mimic contemporary art and design materials (plastics) or collages.
All samples were analysed with room light with an additional incandescent light being present. Dynamic variation of these emission sources was achieved by a random pattern hand waving between the emission sources and the sample. More details are given in the following section.
Results and discussion
Ambient light interference
In order to be able to mimic both static and dynamic interferences, two ambient light sources were employed, namely a room ceiling lighting of the laboratory, which consisted of fluorescent lamps that had narrow bands at around 852, 912, 922 and 966 nm (equivalent to 320, 1093, 1215 and 1706 cm−1 and highlighted by asterisks in Fig. 1), and a broadband light source consisting of a 35 W halogen bulb (i.e. incandescent light) that emitted black body radiation to simulate sun type of radiation. Their impact on a standard Raman spectrum of PTFE is shown in Fig. 1.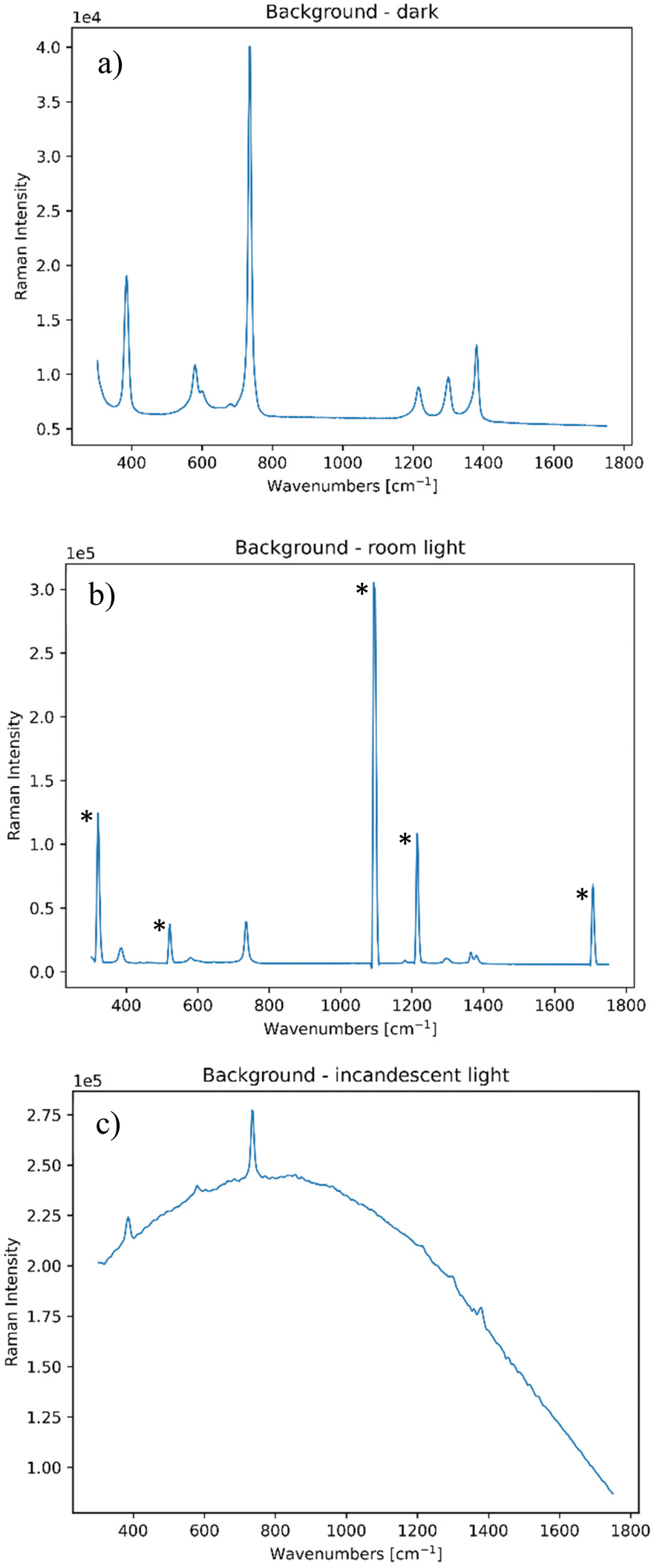 | ||
| Fig. 1 Standard PTFE spectrum collected (a) in the dark; (b) in the presence of a room ceiling light and (c) in the presence of an incandescent lamp. All these spectra have been collected at zero spatial offset. | ||
It is important to note that all these contributions are static, and so one needs to implement a dynamic contribution to these, in order to provide a random variation of the background light to mimic the above discussed non-static situations encountered in field measurements. We mimicked a common situation of passing of a cloud in front of the sun by randomly obstructing the incandescent light with different degrees of hand coverage. The random occurrence of shadows covering the room light was simulated by moving a hand in an irregular fashion, so to slightly obstruct its incoming ceiling room light radiation on the collection side.
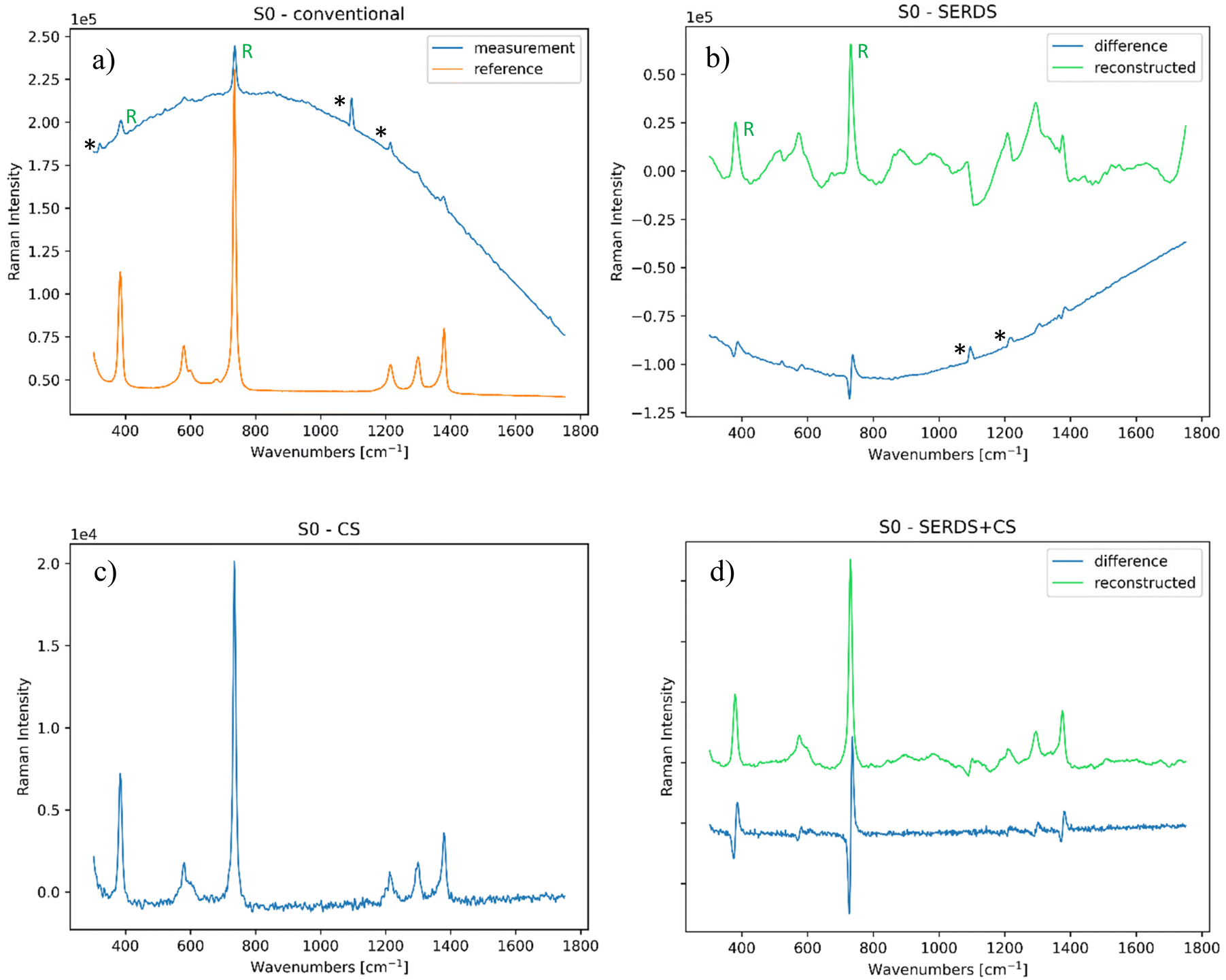 | ||
| Fig. 2 (a) Conventional Raman spectrum of the PTFE sample, compared with the reference PTFE spectrum (S/N = 1.9); (b) SERDS measurement of the same sample (S/N = 2.1); (c) CS measurement of the same sample (S/N = 89); (d) SERDS + CS measurement of the same sample (S/N = 82). The bands highlighted with (*) are ambient light artefacts, the ones with (R) in a and b are the most intense PTFE Raman bands. All these spectra were analysed at zero spatial offset. | ||
Single layers
In the first part of this study, we considered single layer samples before moving to more complex ones such as multilayer systems where SORS was additionally deployed. | ||
| Fig. 3 (a) Conventional Raman spectrum of the BW sample, compared to the reference (S/N = 0.1); (b) SERDS measurement of the same sample (S/N = 0.01). Bands highlighted with (R) are S1, more intense Raman bands; (c) CS measurement of the same sample (S/N = 14); and (d) SERDS + CS measurement of the same sample (S/N = 4). All bands highlighted with (*) are artefacts or background interferences. | ||
 | ||
| Fig. 4 (a) Conventional Raman spectrum of the gypsum sample, compared to the reference baseline (S/N = 0.24); (b) SERDS measurement of the same sample (S/N = 0.1); (c) CS measurement of the same sample (S/N = 19); and (d) SERDS + CS measurement of the same sample (S/N = 17). All bands highlighted with (*) are artefacts or background interferences. | ||
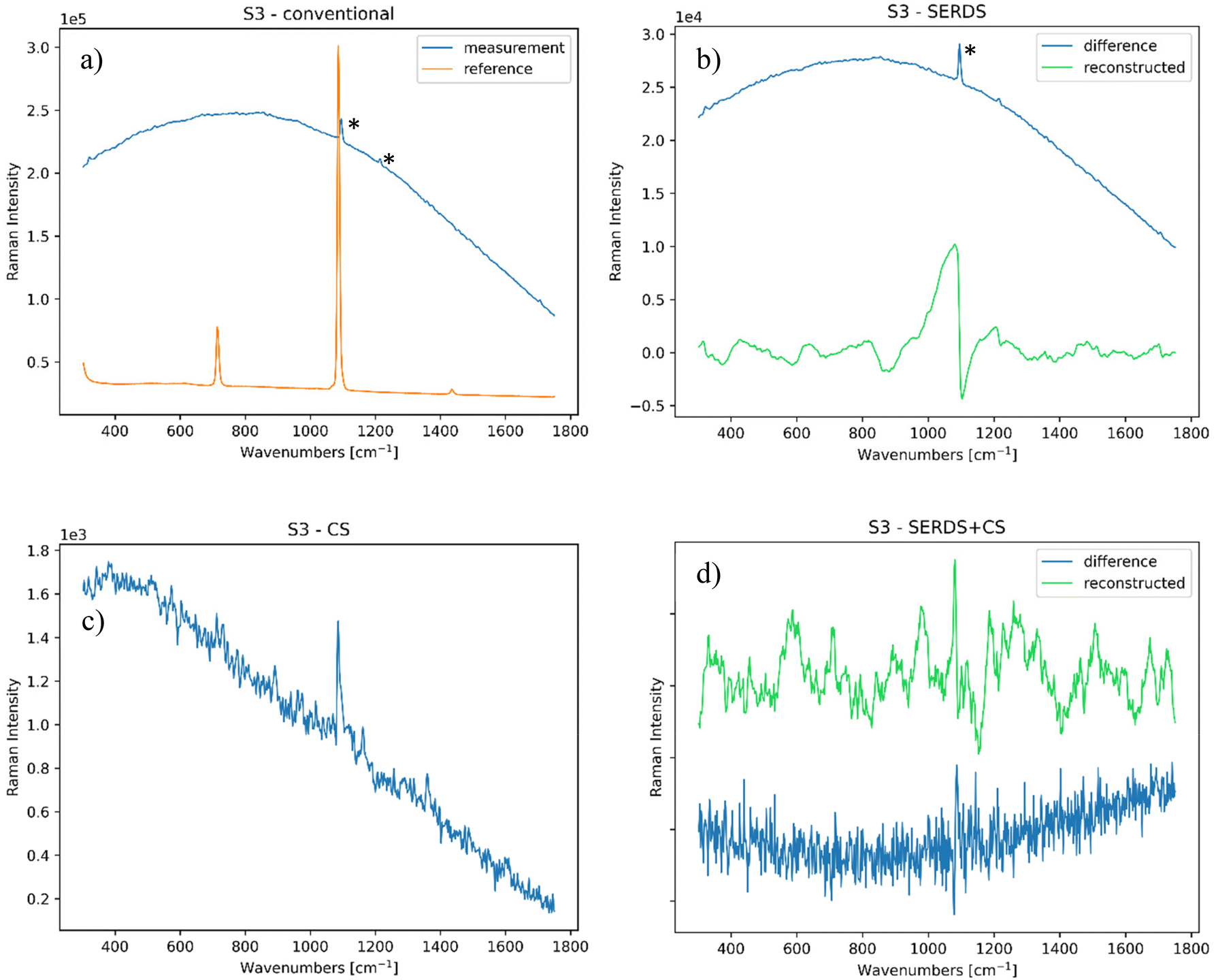 | ||
| Fig. 5 (a) Conventional Raman spectrum of the paper sample, compared to the reference (S/N = 0.05); (b) SERDS measurement of the same sample (S/N = 0.03); (c) CS measurement of the same sample (S/N = 6.9); and (d) SERDS + CS measurement of the same sample (S/N = 3). All bands highlighted with (*) are artefacts or background interferences. | ||
SORS measurements
In the second part of the study, we selected layered samples with increasing complexity (considering their scattering efficiency) to evaluate the performance of the combined use of SORS with SERDS and charge-shifting approaches. As mentioned above, these samples are also relevant to the field of cultural heritage, mimicking contemporary art and design materials (plastics) or collages.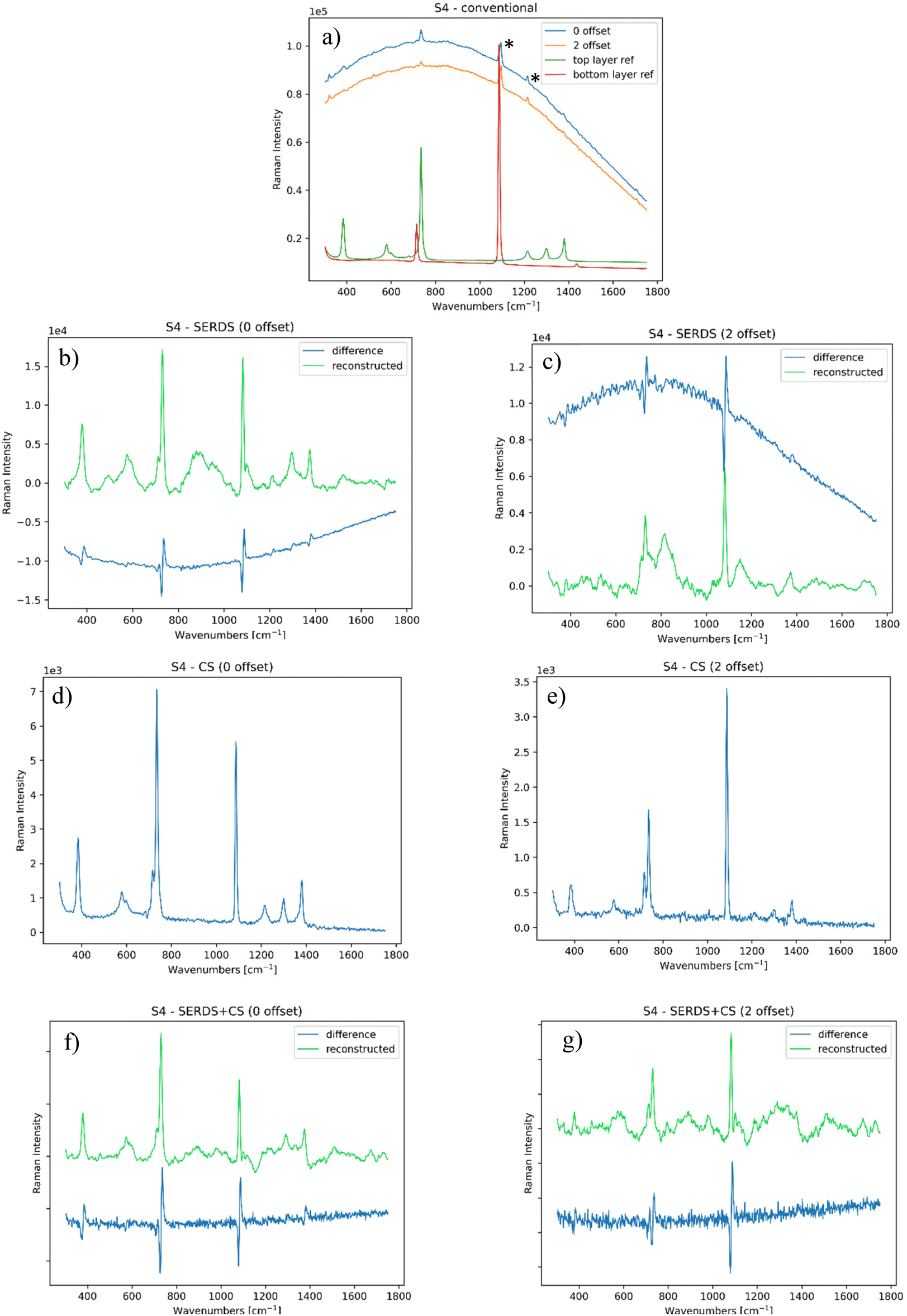 | ||
| Fig. 6 (a) Conventional SORS Raman spectrum of PTFE over marble, compared to the reference (S/N = 0.38); SERDS measurement of the same sample at (b) 0 and (c) 2 mm offset (S/N = 17); charge-shifting measurement of PTFE over marble at (d) 0 and (e) 2 mm offset (S/N = 62); SERDS + CS analyses of the same sample at (f) 0 and (g) 2 mm offset (S/N = 30). | ||
CS measurements (shown in Fig. 6d and e) removed most of the dynamic ambient light contributions and given the nature of the sample there is no significant fluorescence contribution present. The spectra also showed an appreciable SORS effect. In comparison, SERDS + CS measurements (see Fig. 6f and g) yielded poorer results. This is attributed to the fact that the contribution of fluorescence from the sample was relatively low, making SERDS redundant and consequently the deployment of SERDS + CS only added an extra noise under our specific acquisition conditions, as noted earlier.17
 | ||
| Fig. 7 (a) Conventional SORS Raman spectrum of paper over PTFE, compared to the reference (S/N = 0.05); SERDS measurement of the same sample at (b) 0 and (c) 2 mm offset (S/N = 0.22); charge-shifting measurement of paper over PTFE at (d) 0 and (e) 2 mm offset (S/N = 6); SERDS + CS analyses of the same sample at (f) 0 and (g) 2 mm offset (S/N = 1.3). | ||
Similarly, the 0 mm offset CS and SERDS + CS measurements (Fig. 7d and f) were able to detect the Raman bands of the compounds involved, with the former yielding a higher quality spectrum. The outcome with the 2 mm offset was however different as in this case the charge-shifting method alone (Fig. 7e) was not able to retrieve any signal from the top or bottom layer, possibly compounded by photon absorption within the paper. In contrast, the SERDS + CS approach detected the PTFE signal of the subsurface, removing effectively the calcite band coming from the top layer (Fig. 7g). In this scenario, the full capability of the combined SERDS and charge-shifting method was essential to render a satisfactory result. The combined use of SORS, SERDS and CS readout approaches was capable of non-invasive probing of the inner layers of a sample while rejecting any static and dynamic interfering contributions. As previously mentioned, the amount of noise-like features in these spectra are related to the necessity of setting of the charge-shifting measurements out of its optimal regime, in order to obtain results comparable to those of other techniques.
Conclusions
The challenges presented by in situ field measurements in heritage science are numerous, stemming from both ambient light contributions and sample fluorescence, and these are often combined creating specific, highly challenging scenarios. Given the necessity to perform accurate in situ measurements while rejecting any signals that differ from Raman signals of interest, we demonstrated that the synergy between SERDS and charge-shifting technologies, where the former is able to mitigate fluorescence interference and the latter allows us to deal with varying ambient light, is highly beneficial in dealing with such scenarios.When the sample is highly fluorescent and there is no dynamic variation of the ambient light, SERDS proved to be the most effective approach. In this situation, the charge-shifting approach was not able to deal with sample fluorescence as SERDS. However, when the background evolved dynamically, the employment of charge-shifting was therefore fundamentally important. We proved that through this combination of techniques it is possible to obtain promising results when simulating highly challenging in situ conditions, such as varying cloud coverage or shadows projected onto the collection side of the instrument or the variation of artificial room light. Quality of the spectra can be further improved, especially considering that the charge-shifting instrument worked outside its optimum regime. We have also shown that these techniques can be combined with SORS paving a way for the non-invasive investigations of the subsurface components of materials in situ while simultaneously rejecting ambient light and fluorescence interfering contributions.
Data availability
Data are openly available in a Mendeley Data repository.Lux, Alberto; Conti, Claudia; Botteon, Alessandra; Mosca, Sara; Matousek, Pavel (2024), “CS paper”, Mendeley Data, V1, https://doi.org/10.17632/6fdxbc8h83.1.
Conflicts of interest
There are no conflicts of interest to declare.References
- K. Shin and H. Chung, Wide area coverage Raman spectroscopy for reliable quantitative analysis and its applications, Analyst, 2013, 138(12), 3335, 10.1039/c3an36843b.
- W. J. Olds, E. Jaatinen, P. Fredericks, B. Cletus, H. Panayiotou and E. L. Izake, Spatially offset Raman spectroscopy (SORS) for the analysis and detection of packaged pharmaceuticals and concealed drugs, Forensic Sci. Int., 2011, 212, 69–77 CrossRef CAS PubMed.
- P. W. Loeffen, G. Maskall, S. Bonthron, M. Bloomfield, C. Tombling and P. Matousek, Chemical and explosives point detection through opaque containers using spatially offset Raman spectroscopy (SORS), Chem. Biol. Radiol. Nucl. Explos. Sens. XII, 2011, 8018: 80181E. DOI:10.1117/12.882126.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Raman Spectroscopy of Biological Tissues, Appl. Spectrosc. Rev., 2007, 42(5), 493–541, DOI:10.1080/05704920701551530.
- F. Korinth, T. A. Shaik, J. Popp and C. Krafft, Assessment of shifted excitation Raman difference spectroscopy in highly fluorescent biological samples, Analyst, 2021, 146(22), 6760–6767, 10.1039/d1an01376a.
- D. I. Ellis, R. Eccles, Y. Xu, J. Griffen, H. Muhamadali and P. Matousek, et al., Through-container, extremely low concentration detection of multiple chemical markers of counterfeit alcohol using a handheld SORS device, Sci. Rep., 2017, 7(1), 12082, DOI:10.1038/s41598-017-12263-0.
- C. Conti, A. Botteon, C. Colombo, M. Realini and P. Matousek, Fluorescence suppression using micro-scale spatially offset Raman spectroscopy, Analyst, 2016, 141, 5374–5381, 10.1039/c6an00852f.
- K. Sowoidnich and H.-D. Kronfeldt, Fluorescence Rejection by Shifted Excitation Raman Difference Spectroscopy at Multiple Wavelengths for the Investigation of Biological Samples, ISRN Spectrosc., 2012, 2012, 1–11, DOI:10.5402/2012/256326.
- K. Sowoidnich, M. Oster, K. Wimmers, M. Maiwald and B. Sumpf, Shifted excitation Raman difference spectroscopy as enabling technique for the analysis of animal feedstuff, J. Raman Spectrosc., 2021, 52(8), 1418–1427, DOI:10.1002/jrs.6140.
- J. Zhao, M. M. Carrabba and F. S. Allen, Automated Fluorescence Rejection Using Shifted Excitation Raman Difference Spectroscopy, Appl. Spectrosc., 2002, 56(7), 834–845, DOI:10.1366/000370202760171491.
- A. P. Shreve, N. J. Cherepy and R. A. Mathies, Effective Rejection of Fluorescence Interference in Raman Spectroscopy Using a Shifted Excitation Difference Technique, Appl. Spectrosc., 1992, 46(4), 707–711, DOI:10.1366/0003702924125122.
- B. Sumpf, M. Maiwald, A. Müller, J. Fricke, P. Ressel and F. Bugge, et al., Comparison of two concepts for dual-wavelength DBR ridge waveguide diode lasers at 785 nm suitable for shifted excitation Raman difference spectroscopy, Appl. Phys. B: Lasers Opt., 2015, 120(2), 261–269, DOI:10.1007/s00340-015-6133-x.
- M. Maiwald, B. Eppich, J. Fricke, A. Ginolas, F. Bugge and B. Sumpf, et al., Dual-Wavelength Y-Branch Distributed Bragg Reflector Diode Laser at 785 Nanometers for Shifted Excitation Raman Difference Spectroscopy, Appl. Spectrosc., 2014, 68(8), 838–843, DOI:10.1366/13-07331.
- P. Strobbia, V. Cupil-Garcia, B. M. Crawford, A. M. Fales, T. J. Pfefer and Y. Liu, et al., Accurate in vivo tumor detection using plasmonic-enhanced shifted-excitation Raman difference spectroscopy (SERDS), Theranostics, 2021, 11(9), 4090–4102, DOI:10.7150/thno.53101.
- K. Sowoidnich, M. Towrie and P. Matousek, Lock-in detection in Raman spectroscopy with charge-shifting CCD for suppression of fast varying backgrounds, J. Raman Spectrosc., 2019, 50(7), 983–995, DOI:10.1002/jrs.5597.
- K. Sowoidnich, M. Towrie, M. Maiwald, B. Sumpf and P. Matousek, Shifted Excitation Raman Difference Spectroscopy with Charge-Shifting Charge-Coupled Device (CCD) Lock-In Detection, Appl. Spectrosc., 2019, 73(11), 1265–1276, DOI:10.1177/0003702819859352.
- S. Mosca, K. Sowoidnich, M. Mehta, W. H. Skinner, B. Gardner and F. Palombo, et al., 10 kHz Shifted-Excitation Raman Difference Spectroscopy with Charge-Shifting Charge-Coupled Device Read-Out for Effective Mitigation of Dynamic Interfering Backgrounds, Appl. Spectrosc., 2023, 77(6), 569–582, DOI:10.1177/00037028231167441.
- D. Cebeci-Maltaş, P. Wang, M. A. Alam, R. Pinal and D. Ben-Amotz, Photobleaching profile of Raman peaks and fluorescence background, Eur. Pharm. Rev., 2017, 22(6), 18–21 Search PubMed.
- P. Matousek, I. P. Clark, E. R. C. Draper, M. D. Morris, A. E. Goodship and N. Everall, et al., Subsurface probing in diffusely scattering media using spatially offset Raman spectroscopy, Appl. Spectrosc., 2005, 59(4), 393–400, DOI:10.1366/0003702053641450.
- S. Mosca, C. Conti, N. Stone and P. Matousek, Spatially offset Raman spectroscopy, Nat. Rev. Methods Primers, 2021, 1(1) DOI:10.1038/s43586-021-00019-0.
- S. Mosca, P. Dey, M. Salimi, B. Gardner, F. Palombo and N. Stone, et al., Spatially Offset Raman Spectroscopy - How Deep?, Anal. Chem., 2021, 93(17), 6755–6762, DOI:10.1021/acs.analchem.1c00490.
- A. Lux, M. Realini, A. Botteon, M. Maiwald, A. Müller and B. Sumpf, et al., Advanced portable micro-SORS prototype coupled with SERDS for heritage science, Analyst, 2024, 149(8), 2317–2327, 10.1039/d3an02215c.
- B. Sumpf, A. Müller and M. Maiwald, Tailored diode lasers: enabling Raman spectroscopy in the presence of disturbing fluorescence and background light, in Proc. SPIE 10894, Plasmonics in Biology and Medicine XVI, 2019, 1089411, DOI:10.1117/12.2507425.
- S. Mosca, M. Mehta, W. H. Skinner, B. Gardner, F. Palombo and N. Stone, et al., Active Surface-Enhanced Raman Spectroscopy (SERS): A Novel Concept for Enhancing Signal Contrast in Complex Matrices Using External Perturbation, Appl. Spectrosc., 2024, 1–8, DOI:10.1177/00037028241267898.
- P. Matousek, M. Towrie and A. W. Parker, Simple reconstruction algorithm for shifted excitation Raman difference spectroscopy, Appl. Spectrosc., 2005, 59(6), 848–851, DOI:10.1366/0003702054280757.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01280a |
| This journal is © The Royal Society of Chemistry 2025 |