
A proof-of-concept study on a fully biobased and degradable polymer network based on vanillin and myrcene†
Pia S.
Löser
a,
Arthur
Lamouroux
b,
Michael A. R.
Meier
ac and
Audrey
Llevot
 *b
*b
aLaboratory of Applied Chemistry, Institute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Straße am Forum 7, 76131 Karlsruhe, Germany
bUniv. Bordeaux, CNRS, Bordeaux INP, LCPO, UMR 5629, F-33600, Pessac, France. E-mail: audrey.llevot@enscbp.fr
cLaboratory of Applied Chemistry, Institute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 15th May 2024
Abstract
Despite the widespread use of polymer networks, their development has tended to disregard crucial sustainability considerations. In particular, their production and their management at the end-of-life can generate high environmental impacts. Therefore, the design of fully biobased and degradable polymer networks is of great importance. In this work, vanillin and myrcene were selected as renewable resources and a carbonate unit as cleavable linkage to induce a possible network disassembly. Vanillin was converted into a symmetric α,ω-diene featuring a carbonate unit in its center. This monomer was copolymerized by thiol–ene reaction with a myrcene derived trithiol to form a polymer network. An elastic transparent film exhibiting a glass transition temperature of 14 °C, a Young's modulus of 6.87 MPa, an ultimate tensile strength of 1.48 MPa and an elongation at break of 148% was obtained. Owing to the inherent carbonate group, the polymer network could be degraded under acidic conditions into a polyol with an OH content of 2.23 mmol g−1, as determined by 13P NMR. The polyol obtained by degradation was effectively repolymerized to produce a new polyurethane. These proof-of-concept findings provide a potential strategy for circularization of biobased polymer networks.
Introduction
Reducing the environmental impact of cross-linked polymers is a crucial matter for our society. Over the last decades, this topic has become the focus of many researchers, both in academia and industry, as cross-linked polymers are extensively employed. Initial efforts were mainly devoted to the transition from fossil to renewable feedstocks. Terpenes, vegetable oils, lignin, carbohydrates and thereof derived platform chemicals have been used successfully for the synthesis of polymer networks, including epoxy, polyurethane and polyester resins.1–5 These first steps towards more sustainable polymer networks are important, but problems arising at their end-of-life should also be addressed. The permanently cross-linked nature of these materials provides them with outstanding thermo-mechanical properties and chemical resistance. These characteristics are the driving factors behind their applications in the field of automotive, aerospace, electronics, construction etc. However, this structure hinders their recyclability, traditionally limiting their end-of-life options to either incineration or grinding for filler applications.6 To tackle this issue, the introduction of degradable and/or dynamic units as cross-links or into the polymer backbone has been proposed. Different concepts, such as covalent adaptable networks, vitrimers, structurally tailored and engineered macromolecular networks, living additive manufacturing or macromolecular metamorphosis have emerged to ensure the post-modification or recycling of polymer networks.7–12 In order to decrease the environmental impact of polymer networks, combining recyclability with the use of bioresources is crucial.13–19Among all the biobased monomers used for the preparation of polymer networks, vanillin is an interesting precursor due to its aromatic structure and its aldehyde and phenolic functional groups. Vanillin, which is historically extracted from vanilla beans, can also be industrially produced from lignosulfonates, a side-product of the sulfite pulping industry. A large number of studies report the synthesis of polymer networks from vanillin, which are also summarized in a few reviews.20–24 Contrary, the literature about biobased recyclable vanillin-based polymer networks is less abundant. This field is dominated by the preparation of vitrimers featuring dynamic imine motives.25–32 Moreover, different synthetic approaches have been described to synthesize polyimine networks,33–35 imine/epoxy networks,36–39 imine/urethane networks40–42 or polymer networks by radical polymerization.43 Dihydrazone-based dynamic covalent epoxy networks,44 vinylogous urethane vitrimers45 and disulfide-based epoxy vitrimers46 were also produced.
Alternatively, in line with the Green Chemistry concept “design to degrade”, the presence of cleavable units enables the degradation of polymer networks into monomers or oligomers that can potentially be repolymerized or used as value-added chemicals for other applications.47 In 1996, Buchwalter and coworkers reported the design of cycloaliphatic diepoxides featuring a cleavable linkage (ketal, acetal, formal or ester) for the preparation of thermosets capable of being disassembled in acid-containing solvent mixtures.48 Vanillin constitutes a relevant building block to apply this strategy to biobased polymer networks. For instance, the ester moiety introduced during the synthesis of doubly glycidylated vanillic acid (at the hydroxyl and the carboxylic acid group), in combination with the presence of ester moieties in epoxidized soybean oil, was used as disassembly points for the degradation of fully biobased epoxy resins in acidic media.49,50 Vanillin was also converted into an α,ω-diene monomer with degradable ester groups in its center via Williamson and Tishchenko reactions and reacted with multifunctional thiols to form degradable thermosets.51 Vanillin-based degradable epoxy resins were synthesized using monomers incorporating Schiff base linkages.52,53 Several monomers exhibiting acetal groups in their center were also prepared from vanillin by exploiting its aldehyde group. Wu and coworkers synthesized an α,ω-diene monomer by Williamson reaction followed by acetalization in the presence of pentaerythritol.54 This monomer was converted to a degradable polymer network by thiol–ene reaction, with potential applications in flexible devices. A spirodiacetal building block was synthesized by Lin and coworkers by reacting vanillin with erythritol.55 This cleavable monomer was copolymerized with epoxidized soybean oil to yield a fully biobased polymer network. Owing to the dynamic and degradable features of acetal motive, the obtained materials exhibited both good recyclability (covalent adaptable network) and acid degradability, and the degraded products could be reused for the preparation of new polymer networks.
Carbonates represent an interesting class of dynamic and acid-degradable linkages that have not been deeply investigated for the preparation of recyclable polymer networks up to date. In 2000, Wong and coworkers synthesized cycloaliphatic epoxides containing thermally cleavable carbonate linkages. These monomers underwent curing reactions with cyclic anhydrides, similarly to a commercial cycloaliphatic diepoxide and the resulting polymer networks could be disassembled at 220 °C.56 Recently, transcarbonation reactions in the presence of free hydroxyl groups were exploited to prepare acid-degradable polycarbonate vitrimers.57 To the best of our knowledge, there is no example of degradable and fully biobased polymer network based on cleavable carbonate units.
In this work, a symmetric α,ω-diene monomer featuring a cleavable carbonate unit in its center was designed from vanillyl alcohol. A myrcene-based trithiol was further synthesized as biobased comonomer for thiol–ene polymerization. The photopolymerization of these monomers and the thermomechanical properties of the obtained polymer network were studied. Subsequently, the cleavage of the symmetric monomer was investigated as a model, before transferring the most promising conditions to the polymer networks. Finally, the polyols obtained as degradation products were thoroughly characterized and repolymerized into polyurethanes to demonstrate the feasibility of reusing the degraded polymer network (Fig. 1).
 | ||
| Fig. 1 General strategy implemented in this study. | ||
Results and discussion
A cleavable α,ω-diene (AVACarb) was synthesized from vanillyl alcohol in a 2-step approach. The difference of reactivity between the phenolic and benzylic hydroxyl groups of vanillyl alcohol was exploited to selectively introduce the two desired functionalities: first allyl groups for later thiol–ene polymerization and subsequently a cleavable carbonate linker (Scheme 1). | ||
| Scheme 1 Reaction scheme of the synthesis of the cleavable α,ω-diene: allylated vanillyl alcohol carbonate (AVACarb). | ||
The allylation of vanillyl alcohol (VA) was carried out via Tsuji–Trost reaction in water employing palladium nanoparticles stabilized on poly(vinyl pyrrolidone) (PVP) as catalyst, triphenyl phosphine as additive and allyl methyl carbonate as allylating agent.58 This procedure enabled a selective allylation of the phenolic hydroxyl group, as evidenced by the unmodified peak of the benzylic OH group in the 1H NMR spectrum at 5.08 ppm (signal 12 Fig. 2a). After extraction, AVA was obtained in a yield of 75%. This methodology constitutes a sustainable alternative to classic allylation reactions using reactants such as allyl halides. A symmetric cleavable α,ω-diene (AVACarb) was then synthesized from AVA using potentially renewable and non-toxic dimethyl carbonate (DMC) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as organocatalyst.59 The carbonate moiety was introduced in a 2-step procedure. AVA was reacted with 10.0 eq. of DMC and 5.00 mol% TBD at 80 °C. After 6 h reaction time, AVA was almost completely converted into vanillyl methyl carbonate. Subsequently, the excess of DMC was removed under reduced pressure and 1.05 eq. AVA were added. The mixture was heated to 100 °C for 24 h without additional solvent, as the higher temperature enabled the reaction to proceed in a molten state. After purification, the symmetric allylated vanillyl alcohol carbonate (AVACarb) was obtained in a 67% yield. The structure of this monomer was confirmed by 1H NMR (Fig. 2b). The disappearance of the peak at 5.08 ppm (signal 12), characteristic of the proton of the benzylic OH group and the shift of the peak characteristic of the benzylic CH2 from 4.43 ppm to 5.06 ppm (signal 11) due to the incorporation of the adjacent electron-withdrawing carbonate group were observed. This monomer was also characterized by 13C NMR, mass spectrometry and IR spectroscopy (see ESI†).
 | ||
| Fig. 2 1H-NMR spectra of allylated vanillyl alcohol AVA (a) and the cleavable α,ω-diene AVACarb (b) in DMSO-d6. | ||
A biobased trithiol was synthesized from myrcene, following a 2-step procedure described before for the preparation of limonene dithiol (Scheme 2).60
 | ||
| Scheme 2 Reaction scheme of the synthesis of myrcene trithiol monomer. | ||
In a first step, myrcene was reacted with thioacetic acid by stirring the mixture at room temperature for 23 h. The reaction was monitored by gas chromatography (GC) (Fig. 3a). The disappearance of the signal at 2.4 min, attributed to myrcene, and the appearance of a new signals at 16 min indicated the completion of the reaction. Subsequently, unreacted thioacetic acid was removed under reduced pressure. Then, a transesterification was performed in methanol in the presence of TBD as catalyst. The solution was stirred at 75 °C for 25 h to cleave the thioester groups. The GC chromatogram (crude product) showed the disappearance of the signals at 16 min and the appearance of three main signals at 9.8, 10.5 and 11.2 min. The separation of the products was attempted by column chromatography. The 1st collected fraction mostly contained by-products. Fractions 2 and 3 both contained a mixture of the main products, which consist of stereo- and regio-isomeric structures, explaining the multiple peaks observed by GC analysis. These two fractions, which represented a yield of 58%, were gathered and further analyzed. Compared to myrcene, the 1H NMR of the product confirmed the complete conversion of the double bonds (Fig. 3b). The appearance of peaks between 2.5 and 3.5 ppm, characteristics of protons adjacent to the thiol moiety is observed. The 13C NMR showed a multitude of peaks which were attributed to CH, CH2 and CH3 with the help of DEPT and HSQC analyses (Fig. S4 and S5†). However, due to the presence of several isomers (potentially regio and stereo isomers), peaks could not be assigned precisely. Therefore, the structure of the trithiol was assessed by elemental analysis (C10H22S3, calculated: C 50.37%, H 9.30%, S 40.33%; found: C 50.67%, H 8.61%, S 38.67%) and mass spectrometry (EI-HRMS of C10H22S3 [M]+, calculated: 238.0878; found: 238.0882). In the literature, the synthesis of this trithiol has been reported in a patent.61 In this case, thiol formation was achieved by radical and acid-catalyzed sulfhydration of myrcene using H2S as sulfur source. After distillation, the product was also obtained as a mixture of regioisomers in a purity of 98%, but no characterization was provided.
 | ||
| Fig. 3 (a) GC-MS monitoring of the synthesis of myrcene trithiol, from the top to bottom: starting material myrcene, reaction mixture after reaction with thioacetic acid for 23 h, crude product after thioester hydrolysis, fraction 1, fraction 2 and fraction 3 collected after column chromatography, (b) 1H NMR spectra of myrcene (top) and fraction 2 (bottom) in CDCl3. | ||
Having both monomers in hand, thiol–ene polymerization was investigated (Fig. 4). The monomers were mixed in a double bond![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thiol ratio of 1:1 in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) as initiator. In order to prepare films, the mixture was gently heated to ensure melting of the monomers and was poured onto a glass plate. The samples were placed in a UV-oven and irradiated at 365 nm for 10 min. After polymerization, the resulting transparent free-standing film could be peeled off the glass plate. FTIR was used to verify the completion of the thiol–ene photopolymerization (Fig. 5a).62 Full conversion of the thiol was observed through disappearance of the peak located at 2543 cm−1 (S–H stretching). The signals associated to the allyl groups at 1647 cm−1 (C
:thiol ratio of 1:1 in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) as initiator. In order to prepare films, the mixture was gently heated to ensure melting of the monomers and was poured onto a glass plate. The samples were placed in a UV-oven and irradiated at 365 nm for 10 min. After polymerization, the resulting transparent free-standing film could be peeled off the glass plate. FTIR was used to verify the completion of the thiol–ene photopolymerization (Fig. 5a).62 Full conversion of the thiol was observed through disappearance of the peak located at 2543 cm−1 (S–H stretching). The signals associated to the allyl groups at 1647 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching), and 3075 cm−1 (olefinic C–H stretching) showed very low intensity and a significant decrease compared to the AVACarb. FTIR analyses of the polymer film also highlighted the presence of CO vibration at 1738 cm−1, indicating that the carbonate remained unaffected during the cross-linking reaction (Fig. 5a). Moreover, this fact was confirmed by the synthesis and characterization of a model thiol–ene adduct from AVACarb and 2 equivalents of 2-phenylethanethiol (see ESI pp. 4–9†). The polymer film exhibited a relatively low soluble fraction in THF (14 wt% soluble fraction), a glass transition temperature (Tg) of 14 °C and a temperature at 5% weight loss (Td5%) of 202 °C (Fig. 5b and c).
C stretching), and 3075 cm−1 (olefinic C–H stretching) showed very low intensity and a significant decrease compared to the AVACarb. FTIR analyses of the polymer film also highlighted the presence of CO vibration at 1738 cm−1, indicating that the carbonate remained unaffected during the cross-linking reaction (Fig. 5a). Moreover, this fact was confirmed by the synthesis and characterization of a model thiol–ene adduct from AVACarb and 2 equivalents of 2-phenylethanethiol (see ESI pp. 4–9†). The polymer film exhibited a relatively low soluble fraction in THF (14 wt% soluble fraction), a glass transition temperature (Tg) of 14 °C and a temperature at 5% weight loss (Td5%) of 202 °C (Fig. 5b and c).
 | ||
| Fig. 4 Thiol–ene coupling between AVACarb and myrcene-based trithiol resulting in a cross-linked polymer film (picture). | ||
 | ||
| Fig. 5 (a) FTIR spectra of myrcene trithiol (bottom), AVACarb (top) and resulting film (middle); (b) TGA thermogram, (c) DSC thermogram, (d) tensile tests (5 replicates), of polymer films obtained by thiol–ene polymerization of myrcene trithiol and AVACarb. | ||
Tensile test measurements were performed on 5 replicates to determine the mechanical properties and show the reproducibility of the synthesis. The film exhibited a Young's modulus of 6.87 ± 1.29 MPa, an ultimate tensile strength of 1.48 ± 0.194 MPa and an elongation at break of 148 ± 10.7% (Fig. 5d). These mechanical properties fall in the same range as similar systems based on vanillin or lignin derivatives thiol–ene networks.51,54,62,63 Finally, the crosslinking density (νe) was calculated from the classical rubbery elasticity theory by DMA analysis (Fig. S12†). A value of 50 mmol dm−3 lying in the low range of what has been reported in the literature for similar systems, i.e. polymer networks synthesized by thiol–ene reaction, was obtained.54,64 The polymer film obtained in this study constituted a good base to investigate the degradability of the carbonate linker in a polymer network. A deeper screening of the polymerization conditions could lead to a decrease of the soluble fraction and an increase of the cross-linking density and thus, polymer thermomechanical properties.
With the objective to develop a solvent-assisted depolymerization method for the polymer networks, the cleavage conditions of the carbonate units were first screened using the AVACarb monomer (Fig. 6a). The cleavage of AVACarb into AVA was monitored by size exclusion chromatography (SEC). We normalized the peak characteristic of AVACarb (retention time: 34 min) and observed the appearance and further increase of a peak at 36 min characteristic to AVA (Fig. 6b). The structure of the obtained AVA was also confirmed by GC-MS (Fig. S14†). The integration of these peaks enabled the estimation of a degradation percentage when screening different reaction conditions for the degradation (Fig. 6c). The use of DMF as organic solvent proved to be beneficial to solubilize both the starting material and degradation product. The increase of the acid concentration (from 0.5 M to 1 M) led to a faster and more complete degradation. Under optimized conditions, 92% of the AVACarb was degraded into AVA within 46 h.
 | ||
| Fig. 6 (a) Molecular model: cleavage of AVACarb into AVA under acidic conditions, (b) monitoring of the degradation in DMF/HCl 1 M at 100 °C via SEC, (c) establishment of degradation conditions for AVACarb: test of different reaction conditions. | ||
Following the promising degradation results of AVACarb, the suitability of the established procedure was evaluated on the polymer network. Initially, the sample was insoluble in the DMF/HCl mixture. After 24 h however, complete solubilization was observed (Fig. 7a) and the degradation of the solubilized polymer could be monitored by SEC (Fig. 6b). From 24 h to 73 h, the SEC trace of the degradation product showed a shift to lower molar masses and a decrease of the dispersity. The reaction time was prolonged to 120 h, but no further polymer network disassembly could be observed. The solvent was removed under reduced pressure and the resulting powder was analyzed by ESI-MS. The most abundant peak (855.3418 g mol−1) could be assigned to the expected degradation product (Fig. 6c and d), consisting of the myrcene trithiol linked to three AVA moieties. The good agreement between the measured and the calculated isotopic patterns (Fig. 7e) of this product confirmed its structure. This structure identification confirms that (i) the network can be degraded as expected and moreover that (ii) the cross-links within the network were formed as anticipated.
 | ||
| Fig. 7 (a) Visual observation of the cross-linked polymer solubilization under degradation conditions, (b) SEC monitoring of the solubilized polymer, (c) ESI-MS spectrum of degradation product, (d) chemical structure of the expected main degradation product, (e) calculated isotopic pattern (top), measured isotopic pattern of the expected degradation product. | ||
Further characterization was performed to confirm the chemical structure of the degradation product. The hydrolysis of the carbonate group was confirmed by FTIR with the disappearance of the band at 1738 cm−1 (Fig. 8a) and by 13C NMR with the absence of peak between 155 and 157 ppm (Fig. 8b). In both analyses, a new signal characteristic of another carbonyl function at 1718 cm−1 in the FTIR spectrum and at 161 ppm on the 13C NMR spectrum was detected. We suggest that these signals belong to a carbonic acid monobenzylester, which is an intermediate in the hydrolysis reaction (Scheme S2†). Monoesters of carbonic acid are known to be unstable and tend to decompose into alcohol and carbon dioxide, but some studies reported their isolations, especially in acidic solutions.65–68 Additionally, the FTIR spectrum of the degradation products revealed the appearance of a band attributed to OH vibrations at 3360 cm−1. The identification of the nature of hydroxyl groups and their quantitative determination has been carried out by 31P NMR spectroscopy. For this analysis, the degradation product was phosphorylated using 2-chloro-4,4,5,5-tetramthyl-1,3,2-diaxophospholane and the proportions of each type of hydroxyl group were evaluated against an internal standard (N-hydroxy-5-norbornene-2,3-dicarboximide).69 The 31P NMR spectrum of the degradation product exhibited one main peak at around 147 ppm, the area of aliphatic hydroxyl groups (Fig. 8c). By relative integration to the internal standard, an OH content of 2.23 mmol g−1, corresponding to 124 mg KOH per g, was calculated for the polyol received by network degradation. This value is in the range of commercially available polyols prepared from vegetable oils constituting classic building blocks for polyurethane synthesis (Table S1†).70
 | ||
| Fig. 8 (a) FTIR spectra of the polymer network (top) and degradation product (bottom), (b) zoom on the 13C NMR spectra of the AVACarb (top) and degradation product (bottom), (c) 31P NMR spectrum of the phosphorylated degradation product. | ||
As a preliminary test of the degradation product's reactivity in polyurethane synthesis, the obtained polyol was mixed with 4,4′-methylene-bis(cyclohexyl isocyanate) (HMDI) in a DSC pan in the presence of dibutyltin dilaurate catalyst. During the first heating ramp, an exothermic peak characteristic of the polymerization was observed (Fig. S15†). Afterwards, the resulting polymer exhibited a Tg of 9 °C (Fig. S16†). The success of the repolymerization was also confirmed by FTIR with the disappearance of the isocyanate stretching vibration at 2246 cm−1, the decrease of the alcohol stretching vibration at 3360 cm−1 and the appearance of the stretching vibration of the urethane CO bond at 1710 cm−1 and of the bending vibration of the N–H bond at 1560 cm−1 (Fig. S17†). Acknowledging the extensive range of applications of polyurethanes, the valorization of degradation products for their synthesis is a promising approach for the chemical recycling of materials and especially cross-linked polymers.
Conclusions
A vanillin-derived α,ω-diene monomer featuring a carbonate linkage in its center was synthesized and copolymerized with a biobased myrcene-based trithiol. The obtained fully biobased polymer network exhibited a relatively low soluble fraction, elastic properties and good thermal properties. The presence of the cleavable carbonate unit endows the polymer network with good degradation properties. In a DMF/HCl mixture at 100 °C, the polymer film became soluble after 17 h and the degradation continued in solution until 73 h. The disappearance of the carbonate unit and formation of OH groups was confirmed. The obtained polyol exhibited an OH content of 2.23 mmol g−1, a value that is in the range of commercially available polyols prepared from vegetable oils. This work showed the suitability of carbonate groups for the preparation of degradable polymer networks by thiol–ene reaction. The degradation products being polyols, we showed a proof of concept of their upcycling in the synthesis of polyurethane. By using simple chemistry, greener synthetic pathways and bioresources, this example opens a way for the development of more sustainable polymer networks that can reduce reliance on virgin feedstocks, minimize waste generation, and enhance overall resource efficiency.Experimental section
Materials
All solvents were used without further purification. Water, when used in the synthesis, was deionised.The following chemicals were used as received: chloroform-d3 (CDCl3, 99.8 atom% D, Euriso-Top), dimethyl sulfoxide-d6 (99.8 atom% D, Euriso-Top), silica gel 60 (0.040–0.063, Sigma-Aldrich), sodium sulfate (99%, Bernd Kraft), 2,2-dimethoxy-2-phenylacetophenone (DMPA, 99%, Sigma-Aldrich), sodium chloride (99.5%, Fisher chemical), allyl alcohol (99%, Acros Organics), dimethyl carbonate (>99%, Sigma-Aldrich), 1,5,7-triazabicyclo(4.4.0)dec-5-ene (TBD, 98%, Sigma-Aldrich), vanillyl alcohol (99%, Acros Organics), palladium nanoparticles (PdNP@PVP, particles were synthesized by Dr Silke Behrens (IKFT) and analyzed by TEM, DLS and AES-ICP,58 palladium content: 0.0404 mmol mL−1), methanol (HPLC grade, VWR chemicals), tetrahydrofuran (>99.7%, VWR chemicals), thioacetic acid (96%, Sigma-Aldrich), myrcene (90%, Acros Organics), N,N-dimethylformamide (99.8%, Sigma-Aldrich), hydrochloric acid (12 M, 37%, VWR chemicals), 2-chloro-4,4,5,5-tetramethyl-1,3,2-diaxophospholane (95%, Sigma-Aldrich), chromium(III) acetylacetonate (>99% trace metals basis, Sigma-Aldrich) and N-hydroxy-5-norbornene-2,3-dicarboximide (97%, Sigma-Aldrich), dibutyltin dilaurate (95%, Sigma-Aldrich) and 4,4′-methylene-bis(cyclohexyl isocyanate) (90%, Fisher chemical).
Methods
:1) using helium as carrier gas (flow rate 1.0 mL min−1).
| Soluble part/% = 100 × (1 − mD/m0) |
A temperature sweep was performed on the same instrument. This experiment was carried out on the interval from −10 to 80 °C with a heating rate of 3 °C min−1 at a frequency of 1.0 Hz and a strain of 0.2%. Crosslinking density (νe) was calculated from classical rubbery elasticity theory: E′ = 3νeRT with E′ the storage modulus of the polymer network in the rubbery plateau region at Tα + 50 °C, R the universal constant (R = 8.314 J mol−1 K−1), and T is the absolute temperature corresponding to E′.
000 Da). This set is preceded by a GF-1G 7B (7.5 × 50) guard column. The flow rate was kept stable at 0.5 ml min−1 and the column temperature was maintained at 50 °C. Easivial kit of Polystyrene from Agilent was used as the standard (Mn from 162 to 364000 Da).
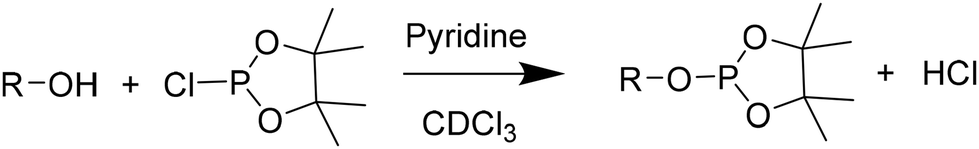 | ||
| Scheme 3 Phosphorylation reaction of OH groups. | ||
In a dry Schlenk flask, dried polyols (15.7 mg), internal standard (4.5 mg) and relaxation agent (1.5 mg) chromium(III) acetylacetonate were precisely weighted. Under an argon flow, a mixture of dry pyridine/CDCl3 (0.5 mL; 1.6:1 vol/vol) was added. If the mixture was not homogeneous, dry DMF (0.5 mL) was added. Finally, phosphorylating reagent (0.1 mL) was added. The sample was stirred for 1 hour under argon flow. Afterwards, 0.4 mL of the mixture were transferred into an NMR tube in which 0.2 mL of dry CDCl3 were added. The NMR analysis was immediately performed to prevent hydrolysis. The analysis was performed on BRUKER Advance DPX spectrometers (Billerica, MA) 400 MHz spectrometer with 512 scans, a relaxation time d1 of 5 seconds.
The chemical shifts are reported relative to the reaction product of 2-chloro-4,4,5,5-tetramethyl1,3,2-dioxaphospholane with water at 132.2 ppm. Integrals are assigned to the functional groups as followed: δ = 150.0–145.5 ppm for aliphatic hydroxyl groups, 145.5–144.7 ppm for cyclohexanol, 144.7–136.6 ppm for phenolic hydroxyl groups.
Experimental procedures
 | ||
| Scheme 4 Synthesis of allyl methyl carbonate from DMC and allyl alcohol. | ||
In a round bottom flask, a mixture of allyl alcohol (1 mol, 68.3 mL) and dimethyl carbonate (631 mL, 7.5 mol, 7.50 eq.) was stirred at 80 °C for five min. Subsequently, TBD (10 mmol, 1.39 g, 0.01 eq.) was added and the reaction mixture was run for 1 hour. A mixture of dimethyl carbonate, allyl methyl carbonate and diallyl carbonate was obtained and separated by distillation. The product was distilled at 100 mbar and 70 °C yielding to allyl methyl carbonate as an oily compound.
Yield: 28%.
1 H-NMR (400 MHz, CDCl3) δ/ppm = 5.96–5.87 (m, 1H), 5.37–5.23 (m, 2H), 4.62–4.60 (m, 2H), 3.77 (s, 3H).
13 C-NMR (101 MHz, CDCl3) δ/ppm = 155.70, 131.70, 118.91, 68.56, 54.88.
IR (ATR platinum diamond): ν/cm−1 = 2959.3, 1745.5, 1650.6, 1444.1, 1365.1, 1249.1, 965.0, 927.5, 790.8, 555.2.
In a round bottom flask, vanillyl alcohol (64.9 mmol, 10 g) was mixed with allyl methyl carbonate (162 mmol, 18.8 g, 2.5 eq.) and triphenylphosphine (3.25 mmol, 851 mg, 0.05 eq.) in water (90 mL). Palladium nanoparticles stabilized in water (PdNP@PVP, 0.10 mol%, 64.9 μmol, 1.60 mL) were added to the mixture and the reaction was stirred at 90 °C for 16 hours. The reaction mixture was poured in a separation funnel and extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and solvent was removed under reduced pressure. The crude mixture was purified by column chromatography (eluent cyclohexane/EtOAc; 7/3, vol/vol) to give AVA.
Yield: 75%.
R f = 0.45 (cyclohexane/EtOAc; 5/5, vol/vol).
1 H-NMR (300 MHz, DMSO-d6) δ/ppm = 6.93–6.63 (m, 3H), 6.09–5.96 (m, 1H), 5.40–5.21 (m, 2H), 5.07 (t, J = 5.7 Hz, 1H), 4.51 (d, J = 3.9 Hz, 2H), 4.41 (d, J = 5.5 Hz, 2H), 3.75 (s, 3H).
13 C-NMR (75 MHz, DMSO-d6) δ/ppm = 148.93, 146.38, 135.51, 134.06, 118.49, 117.33, 113.44, 110.80, 69.09, 62.78, 55.42.
IR (ATR platinum diamond): ν/cm−1 = 3330.9, 2993.4, 2906.2, 2856.2, 2354.3, 2157.7, 2033.5, 1860.9, 1646.6, 1591.6, 1512.7, 1454.9, 1417.9, 1365.1, 1319.3, 1294.0, 1252.2, 1232.6, 1136.9, 1059.6, 1022.8, 1005.0, 989.0, 925.7, 916.5, 853.5, 820.2, 802.6, 739.7, 642.2, 606.0, 592.4, 575.2, 522.1, 462.538, 409.7.
EI-HRMS of C11H14O3 [M]+ calculated: 194.0937, found: 194.0937.
:5 to 7:3, vol/vol) to obtain AVAcarb as a white solid.
Yield: 67%.
R f : 0.56 (cyclohexane/EtOAc; 6/4, vol/vol).
1 H-NMR (400 MHz, DMSO-d6) δ/ppm: 7.00–6.89 (m, 6H), 6.07–5.98 (m, 2H), 5.40–5.22 (m, 4H, H10), 5.06 (s, 4H), 4.54 (dt, J = 5.3, 1.4 Hz, 4H), 3.74 (s, 6H).
13 C-NMR (101 MHz, DMSO-d6) δ/ppm: 154.46, 148.91, 147.77, 133.75, 128.00, 121.15, 117.55, 113.18, 112.60, 69.10, 68.90, 55.50.
IR (ATR platinum diamond): ν/cm−1: 2963.1, 2835.8, 1742.3, 1649.6, 1604.8, 1590.6, 1513.4, 1453.5, 1421.5, 1376.3, 1328.9, 1253.0, 1223.6, 1195.1, 1165.2, 1134.0, 1021.5, 917.4, 905.1, 886.3, 852.6, 801.3, 787.2, 738.2, 663.0, 633.3, 617.0, 550.7, 518.8, 468.1, 420.2.
FAB-HRMS of C23H26O7 [M]+ calculated: 414.1673, found: 414.1674.
In a round bottom flask, myrcene compound (36.7 mmol, 5 g) was mixed with thioacetic acid (132 mmol, 9.44 mL, 3.6 eq.) under ice cooling. The mixture was stirred at room temperature for 23 hours. The thioacetic acid was partly removed under reduced pressure. Subsequently, TBD (5.51 mmol, 766 mg, 0.15 eq.) and MeOH (45 mL, 1.1 mol, 30 eq.) were added and the solution was stirred at 75 °C for 25 hours. The solvent was removed under reduced pressure and the product was isolated by column chromatography (eluent from 0.5% to 1% of EtOAc in cyclohexane, vol/vol). The desired product was obtained as a mixture of isomers. Because of the indistinct NMR spectra (Fig. S4†), the product was identified via mass spectrometry and elemental analysis.
Yield: 59%.
R
f
= 0.61 (cyclohexane/EtOAc; 98:2, vol/vol).
IR (ATR platinum diamond): ν (cm−1): 2958.7, 2924.9, 2867.9, 2548.7, 1452.2, 1382.7, 1366.3, 1329.2, 1247.7, 1080.0, 921.6, 873.1, 809.4, 765.9, 713.5, 660.8, 468.9.
EI-HRMS of C10H22S3 [M]+ calculated: 238.0878, found: 238.0882.
Elemental analysis: calculated: C 50.37%, H 9.30%, S 40.33; found: C 50.67%, H 8.61%, S 38.67%.
:thiol ratio of 1:1. Then, DMPA (5 mol% per double bond, 3.30 mg) was added. After thorough mixing, the mixture was transferred to a glass plate with tape boundaries (1 cm × 2.5 cm). Solid samples were gently melted using a heatgun to ensure homogeneous film formation. The hot samples were placed into the UV oven and the curing was carried out under UV irradiation (365 nm, 50% power) for 10 min.
IR (ATR platinum diamond): ν/cm−1 = 2931.7, 1738.9, 1592.2, 1513.3, 1462.6, 1421.1, 1386.2, 1366.0, 1334.6, 1224.3, 1162.1, 1138.0, 1026.5, 908.7, 852.0, 789.5, 650.0, 552.3, 461.2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr A. Llevot acknowledges the Ministry of Science, Research and Arts of Baden-Wuerttemberg, MWK, for the Research Seed Capital (RiSC) funding. The authors would like to thank Emmanuel Ibarboure, Frédérique Ham Pichavant and Maëva Peloille for their respective help with the tensile test measurements, mass spectrometry and 13P NMR.References
- J. Liu, L. Zhang, W. Shun, J. Dai, Y. Peng and X. Liu, J. Polym. Sci., 2021, 59, 1474–1490 CrossRef CAS.
- R. L. Quirino, K. Monroe, C. H. Fleischer III, E. Biswas and M. R. Kessler, Polym. Int., 2021, 70, 167–180 CrossRef CAS.
- E. Feghali, K. M. Torr, D. J. van de Pas, P. Ortiz, K. Vanbroekhoven, W. Eevers and R. Vendamme, Top. Curr. Chem., 2018, 376, 32 CrossRef PubMed.
- A. Llevot, J. Am. Oil Chem. Soc., 2017, 94, 169–186 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817–823 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- M. Chen, Y. Gu, A. Singh, M. Zhong, A. M. Jordan, S. Biswas, L. T. J. Korley, A. C. Balazs and J. A. Johnson, ACS Cent. Sci., 2017, 3, 124–134 CrossRef CAS PubMed.
- A. Beziau, A. Fortney, L. Fu, C. Nishiura, H. Wang, J. Cuthbert, E. Gottlieb, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Polymer, 2017, 126, 224–230 CrossRef CAS.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- T. Vidil and A. Llevot, Macromol. Chem. Phys., 2022, 223, 2100494 CrossRef CAS.
- J. Liu, S. Wang, Y. Peng, J. Zhu, W. Zhao and X. Liu, Prog. Polym. Sci., 2021, 113, 101353 CrossRef CAS.
- X.-L. Zhao, P.-X. Tian, Y.-D. Li and J.-B. Zeng, Green Chem., 2022, 24, 4363–4387 RSC.
- A. Liguori and M. Hakkarainen, Macromol. Rapid Commun., 2022, 43, 2100816 CrossRef CAS PubMed.
- X.-L. Zhao, Y.-D. Li and J.-B. Zeng, Polym. Chem., 2022, 13, 6573–6588 RSC.
- A. Kumar and L. A. Connal, Macromol. Rapid Commun., 2023, 44, 2200892 CrossRef CAS PubMed.
- M. A. Lucherelli, A. Duval and L. Avérous, Prog. Polym. Sci., 2022, 127, 101515 CrossRef CAS.
- J. Wan, J. Zhao, X. Zhang, H. Fan, J. Zhang, D. Hu, P. Jin and D.-Y. Wang, Prog. Polym. Sci., 2020, 108, 101287 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- F. Ng, G. Couture, C. Philippe, B. Boutevin and S. Caillol, Molecules, 2017, 22, 149 CrossRef PubMed.
- E. A. Baroncini, S. Kumar Yadav, G. R. Palmese and J. F. Stanzione III, J. Appl. Polym. Sci., 2016, 133, 44103 CrossRef.
- Z. Zhou, X. Su, J. Liu and R. Liu, ACS Appl. Polym. Mater., 2020, 2, 5716–5725 CrossRef CAS.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2020, 8, 17272–17279 CrossRef CAS.
- V.-D. Mai, S.-R. Shin, D.-S. Lee and I. Kang, Polymers, 2019, 11, 293 CrossRef PubMed.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, W. Yuan, S. Zhou, S. You and J. Zhu, Green Chem., 2019, 21, 1484–1497 RSC.
- Q. Yu, X. Peng, Y. Wang, H. Geng, A. Xu, X. Zhang, W. Xu and D. Ye, Eur. Polym. J., 2019, 117, 55–63 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, W. Yuan, B. Wang and J. Zhu, Macromolecules, 2018, 51, 8001–8012 CrossRef CAS.
- H. Geng, Y. Wang, Q. Yu, S. Gu, Y. Zhou, W. Xu, X. Zhang and D. Ye, ACS Sustainable Chem. Eng., 2018, 6, 15463–15470 CrossRef CAS.
- M. A. Rashid, M. N. Hasan, M. A. R. Dayan, M. S. Ibna Jamal and M. K. Patoary, Reactions, 2023, 4, 66–91 CrossRef CAS.
- L. Jiang, Y. Tian, J. Cheng and J. Zhang, Polym. Chem., 2021, 12, 6527–6537 RSC.
- K. Hong, Q. Sun, X. Zhang, L. Fan, T. Wu, J. Du and Y. Zhu, ACS Sustainable Chem. Eng., 2022, 10, 1036–1046 CrossRef CAS.
- L. Jiang, Y. Tian, X. Wang, J. Zhang, J. Cheng and F. Gao, Polym. Chem., 2023, 14, 862–871 RSC.
- L. Xiaohong, L. Liyan, L. Maoping, S. Xuan, L. Hehua and C. Guoyang, Polymer, 2020, 210, 123030 CrossRef.
- H. Memon, Y. Wei and C. Zhu, Mater. Today Commun., 2021, 29, 102814 CrossRef CAS.
- A. Roig, P. Hidalgo, X. Ramis, S. De la Flor and À. Serra, ACS Appl. Polym. Mater., 2022, 4, 9341–9350 CrossRef CAS.
- M. A. Rashid, M. M. Mian, Y. Wei and W. Liu, Mater. Today Commun., 2023, 35, 106178 CrossRef CAS.
- Y. Sun, D. Sheng, H. Wu, X. Tian, H. Xie, B. Shi, X. Liu and Y. Yang, Polymer, 2021, 233, 124208 CrossRef CAS.
- D.-M. Xie, D.-X. Lu, X.-L. Zhao, Y.-D. Li and J.-B. Zeng, Ind. Crops Prod., 2021, 174, 114198 CrossRef CAS.
- H. Wang, Y. Fu, Y. Liu, J. Li, X. Sun and T. Liu, Eur. Polym. J., 2023, 196, 112309 CrossRef CAS.
- S. Grauzeliene, M. Kastanauskas, V. Talacka and J. Ostrauskaite, ACS Appl. Polym. Mater., 2022, 4, 6103–6110 CrossRef PubMed.
- X. Xu, S. Ma, S. Wang, J. Wu, Q. Li, N. Lu, Y. Liu, J. Yang, J. Feng and J. Zhu, J. Mater. Chem. A, 2020, 8, 11261–11274 RSC.
- S. Engelen, A. A. Wróblewska, K. De Bruycker, R. Aksakal, V. Ladmiral, S. Caillol and F. E. Du Prez, Polym. Chem., 2022, 13, 2665–2673 RSC.
- S. Guggari, F. Magliozzi, S. Malburet, A. Graillot, M. Destarac and M. Guerre, ACS Sustainable Chem. Eng., 2023, 11, 6021–6031 CrossRef CAS PubMed.
- S. Ma and D. C. Webster, Prog. Polym. Sci., 2018, 76, 65–110 CrossRef CAS.
- S. L. Buchwalter and L. L. Kosbar, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 249–260 CrossRef CAS.
- M. Shen, R. Almallahi, Z. Rizvi, E. Gonzalez-Martinez, G. Yang and M. L. Robertson, Polym. Chem., 2019, 10, 3217–3229 RSC.
- M. Shen and M. L. Robertson, ACS Sustainable Chem. Eng., 2021, 9, 438–447 CrossRef CAS.
- Z. Gao, Y. You, Q. Chen, M. North and H. Xie, Green Chem., 2023, 25, 172–182 RSC.
- S. Wenjie, Z. Lei, X. Jiazhu, C. Binlong and C. Yonghong, Mater. Lett., 2022, 323, 132589 CrossRef.
- M. Jinpeng, L. Guanxi, H. Xueni, L. Ning, L. Zhe, Z. Fan, Y. Liangliang, C. Xue, S. Lei and A. Yuhui, Polym. Degrad. Stab., 2022, 201, 109989 CrossRef.
- M. Ge, J.-T. Miao, K. Zhang, Y. Wu, L. Zheng and L. Wu, Polym. Chem., 2021, 12, 564–571 RSC.
- W. Zhang, F. Gao, X. Chen, L. Shen, Y. Chen and Y. Lin, ACS Sustainable Chem. Eng., 2023, 11, 3065–3073 CrossRef CAS.
- L. Wang, H. Li and C. P. Wong, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3771–3782 CrossRef CAS.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- A. Llevot, B. Monney, A. Sehlinger, S. Behrens and M. A. R. Meier, Chem. Commun., 2017, 53, 5175–5178 RSC.
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC.
- M. Firdaus, M. A. R. Meier, U. Biermann and J. O. Metzger, Eur. J. Lipid Sci. Technol., 2014, 116, 31–36 CrossRef CAS.
- P. Saint-louis-Augustin, G. Fremy, B. Monguillon and L. Corbel, US Pat., US20200010412A1, 2020 Search PubMed.
- G. Yang, S. L. Kristufek, L. A. Link, K. L. Wooley and M. L. Robertson, Macromolecules, 2016, 49, 7737–7748 CrossRef CAS.
- J. Xue, X. Yang, Y. Ke, Z. Yan, X. Dong, Y. Luo and C. Zhang, Ind. Crops Prod., 2021, 171, 113956 CrossRef CAS.
- L. Pezzana, G. Melilli, P. Delliere, D. Moraru, N. Guigo, N. Sbirrazzuoli and M. Sangermano, Prog. Org. Coat., 2022, 173, 107203 CrossRef CAS.
- J. Bernard, E.-M. Köck, R. G. Huber, K. R. Liedl, L. Call, R. Schlögl, H. Grothe and T. Loerting, RSC Adv., 2017, 7, 22222–22233 RSC.
- A. Dibenedetto, M. Aresta, P. Giannoccaro, C. Pastore, I. Pápai and G. Schubert, Eur. J. Inorg. Chem., 2006, 2006, 908–913 CrossRef.
- C. M. Comisar, S. E. Hunter, A. Walton and P. E. Savage, Ind. Eng. Chem. Res., 2008, 47, 577–584 CrossRef CAS.
- J. Østergaard and C. Larsen, Molecules, 2007, 12, 2396–2412 CrossRef PubMed.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- H. Sardon, D. Mecerreyes, A. Basterretxea, L. Avérous and C. Jehanno, ACS Sustainable Chem. Eng., 2021, 9, 10664–10677 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data: 1H NMR and 13C NMR spectra, DSC analyses and FTIR spectrum of the polyurethane. See DOI: https://doi.org/10.1039/d4py00269e |
| This journal is © The Royal Society of Chemistry 2024 |