
A novel Cu(I)-based coordination polymer for efficient photocatalytic oxidation of C(sp3)–H bonds†
Jiangtao
Deng
,
Huilin
Huang
,
Zhentao
Li
,
Xu
Jing
 * and
Chunying
Duan
* and
Chunying
Duan
State Key Laboratory of Fine Chemicals, College of Chemistry, Dalian University of Technology, 116024, P. R. China. E-mail: xjing@dlut.edu.cn
First published on 3rd June 2024
Abstract
A novel coordination polymer CuCl-Pyhc was successfully synthesized, which can catalyze efficient and selective oxidation of C(sp3)–H bonds under mild conditions, exhibiting exceptional stability and remarkable recyclability. Furthermore, CuCl-Pyhc can mimic natural monooxygenases and activate oxygen into singlet oxygen (1O2).
The selective oxidation of C(sp3)–H bonds is extensively employed in the chemical industry for the synthesis of ketones and various high-quality chemicals.1 Traditional homogeneous processes typically rely on transition metal complexes, leading to significant environmental impacts and substantial costs for waste treatment procedures.2,3 Recently, photocatalysis has garnered attention as a promising approach for harnessing solar energy to generate photoexcited molecules. This process activates molecular oxygen, producing reactive oxygen species such as superoxide radicals (˙O2−) and singlet oxygen (1O2) through single electron transfer (SET) or energy transfer (EnT) pathways.4,5 These reactive species facilitate the gentle conversion of C(sp3)–H bonds. Meanwhile, molecular oxygen exhibits advantages such as a high content of reactive oxygen species, low cost, and no by-products generated during the reaction process, which are in line with the concept of green chemistry.6 And the high natural abundance and low toxicity of copper in nature make copper-based catalysts more economically and environmentally attractive.7 Recent studies have revealed that Cu(I) complexes exhibit excellent capability in activating molecular oxygen under visible light irradiation, displaying high catalytic activity and favorable substrate selectivity.8 Additionally, the extensive adjustability of ligand coordination modes provides advantages to cuprous complexes in optimizing redox properties and excited state lifetimes, facilitating the development of photocatalytic systems.9,10
As heterogeneous catalysts, coordination polymers have advantages such as controllable structure, structural modification, and thermal stability.11 Their unique structures can effectively hinder the aggregation and leaching of metal sites.12 Moreover, the tunable nature of coordination polymer materials allows the fixation of functional groups and photocatalysts into one specific structure to further prolong the excited-state lifetime and overcome restrictions of homogeneous catalytic systems.13 Therefore, they exhibit excellent photocatalytic oxidation properties.14 Based on these considerations, we aimed to construct a novel multiphase catalyst by combining the structural features of copper catalysts and coordination polymers, exploring its capability to catalyze the oxidation of C(sp3)–H bonds. Furthermore, the extended durability of copper in coordination polymers opens up the potential for substrate activation via photoinduced electron transfer (PET) or excited state energy transfer.15
In this study, we constructed a novel coordination polymer CuCl-Pyhc, with the cuprous ion as the metal center by introducing bidentate thiourea ligands. CuCl-Pyhc exhibited efficient activation of molecular oxygen, producing 1O2 reactive oxygen species through the EnT pathway. During the reaction process, the cocatalyst benzoquinone was introduced. The photoexcited CuCl-Pyhc can form Cu(II) through a PET process with benzoquinone. And according to previous reports, the excited state of benzoquinone exhibits excellent hydrogen atom transfer activity and can extract hydrogen atoms from substrates.16 Using this catalytic platform can effectively promote the oxidation of C(sp3)–H bonds. Additionally, as a multiphase catalyst, CuCl-Pyhc has exhibited excellent recyclability and stability.
The Pyhc ligand was synthesized according to a previously reported procedure (Fig. S1†). CuCl-Pyhc was obtained using Pyhc and CuCl by the layered diffusion method. The single crystal X-ray crystallographic study reflected that CuCl-Pyhc crystallizes in a monoclinic crystal system with the space group P21/c. Each central Cu atom coordinates with two bridging sulfur atoms, one N atom from different ligands, and a terminal chlorine atom to form a twisted tetrahedral coordination configuration (Fig. 1b). This coordination method allowed the ligands and copper atoms to connect end-to-end, forming a regular two-dimensional structural material (Fig. 1a). The adjacent sheets then assembled in an ABAB fashion via π⋯π stacking interactions between the ligands (Fig. 1c). Scanning electron microscopy revealed that the CuCl-Pyhc crystals were arranged in thin layers (Fig. S4†). And the elemental spectrum from Energy Dispersive X-ray Spectroscopy (EDS) showed a uniform distribution of elements throughout the structure (Fig. S5†). With its characteristics of exposed surface atoms and active sites, CuCl-Pyhc exhibits excellent catalytic potential.
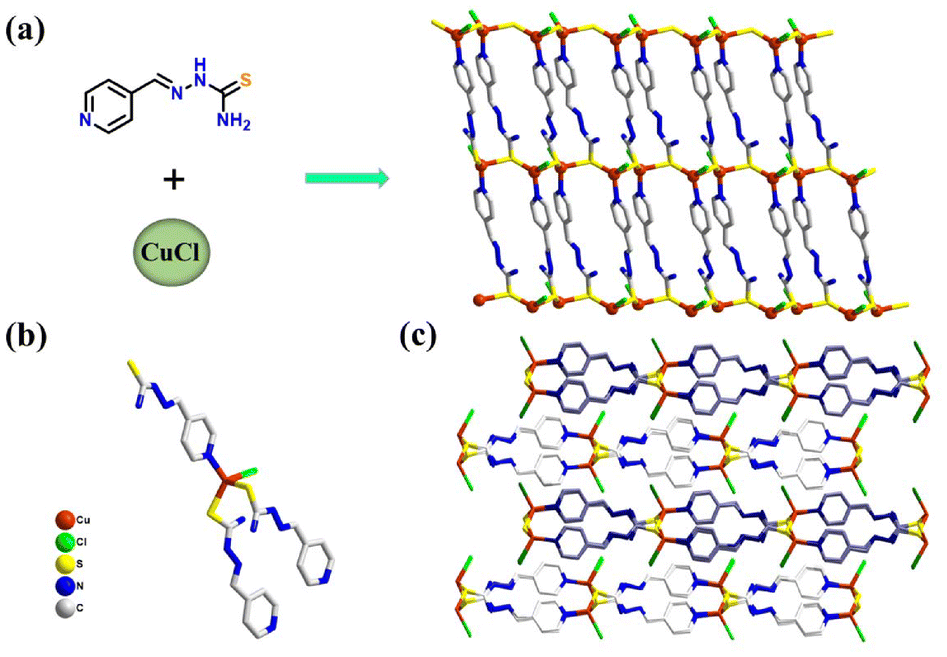 | ||
| Fig. 1 (a) The constituents and two-dimensional lamellar structure of CuCl-Pyhc. (b) Connecting configuration between the ligands and the Cu atom. (c) Stacking in an ABAB fashion of CuCl-Pyhc. | ||
The high phase purity of CuCl-Pyhc was revealed by powder X-ray diffraction (PXRD) (Fig. S6†). And X-ray photoelectron spectroscopy (XPS) spectra of CuCl-Pyhc showed that the copper is +1 valent (Fig. S7†). Additionally, the thermal analysis conducted through TGA on the freshly synthesized CuCl-Pyhc revealed that it started to decompose at approximately 200 °C, indicating its thermal stability under catalytic reaction conditions (Fig. S8†). The photocurrent–time curves of Pyhc and CuCl-Pyhc showed that the photocurrent density increased and returned to the initial value when the light was turned on and off (Fig. S9†). CuCl-Pyhc exhibited higher photocurrent density compared to Pyhc, indicating its superior charge separation efficiency. The results of electrochemical impedance spectroscopy (EIS) revealed that the semicircle of CuCl-Pyhc was smaller compared to that of Pyhc (Fig. S10†), indicating a lower charge transfer resistance.
The photoluminescence (PL) spectrum of CuCl-Pyhc showed a strong emission peak at 570 nm (λex = 455 nm) (Fig. S12†). The solid-state UV-vis absorption spectrum of CuCl-Pyhc exhibited a wider absorption band and a significant red shift in comparison to Pyhc, which was likely related to the effect of coordinated Cu(I) ions on the excited state of the ligand and the d–d transitions of Cu(I) ions (Fig S11†).17 The solid state cyclic voltammogram of CuCl-Pyhc exhibited redox peaks at 0.06 V (vs. Ag/AgCl). In addition to the free energy change (E0–0 = 2.33 eV) between the ground and excited states, the reduction potential of the excited state CuCl-Pyhc was calculated to be −2.36 V (Fig. S13 and S14†).18 This potential indicates that the excited state of CuCl-Pyhc can effectively activate O2.19 The oxygen quenching experiment conducted on the CuCl-Pyhc suspension showed a gradual decrease in emission intensity at 570 nm as the duration of oxygen exposure of the suspension increased. Moreover, the fluorescence lifetime of CuCl-Pyhc decreased from 3.38 ns in an Ar atmosphere to 2.71 ns in the presence of an oxygen atmosphere (Fig. 2a). The luminescence quenching process was attributed to photoinduced electron transfer or energy transfer from the excited state of CuCl-Pyhc to molecular oxygen, activating the oxygen to form active oxygen species. Afterwards, we conducted an experiment on the oxidation of 1,3-diphenylisobenzofuran (DPBF) by CuCl-Pyhc to elucidate its reactive oxygen species behavior. DPBF is a well-known 1O2 scavenger used to detect 1O2 produced in a system. When 1O2 oxidizes and degrades DPBF, the absorption intensity of the DPBF at λ = 410 nm decreases, serving as an indicator of the produced 1O2.20 We tested DPBF photooxidation with and without CuCl-Pyhc under visible light conditions. The experimental results showed that after 200 s of light irradiation with the addition of CuCl-Pyhc, the UV-vis absorption of DPBF at 410 nm decreased significantly (Fig. 2b). In contrast, the blank control group without CuCl-Pyhc only showed a slight decrease (Fig. 2c and S15†), which confirmed that CuCl-Pyhc can activate oxygen to form 1O2. Electron paramagnetic resonance (EPR) spectroscopy was further used to prove this point. In this analysis, 2,2,6,6-tetramethyl-4-piperidone (TEMP) was used as a radical trapping agent, specifically to identify the presence of 1O2. The EPR results of CuCl-Pyhc displayed the characteristic signal of TEMP–1O2, indicating the generation of singlet oxygen (Fig. 2d).
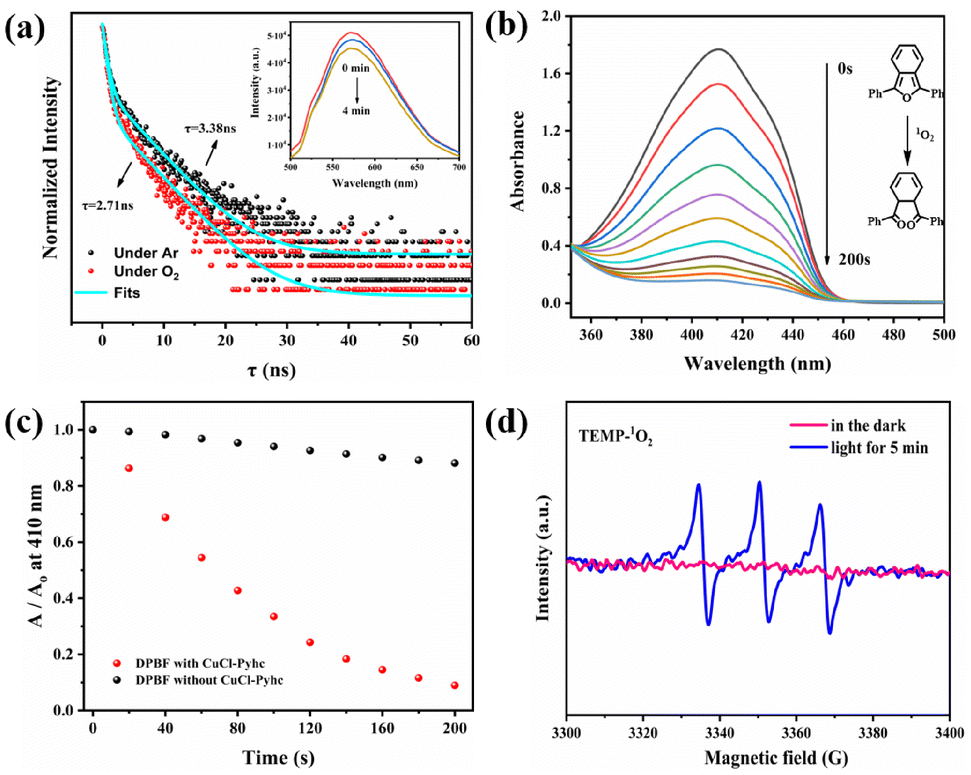 | ||
| Fig. 2 (a) Luminescence decays of CuCl-Pyhc in CH3CN suspensions with O2 (red) and without O2 (black) (the inset shows the time-dependent emission of CuCl-Pyhc in the CH3CN suspension under an O2 atmosphere; the intensity was recorded at 570 nm with excitation at 455 nm). (b) The change in the UV-vis absorption spectrum of DPBF with the duration of illumination in the presence of CuCl-Pyhc. (c) Absorbance decay of DPBF with (red) or without (black) CuCl-Pyhc at 410 nm. (d) EPR spectra of CuCl-Pyhc in CH3CN suspensions containing TEMP (0.1 mM) in the dark (red) and with 455 nm LED irradiation (blue). | ||
Based on the above experimental results, we found that CuCl-Pyhc exhibited the ability to activate oxygen. So we used it as a photocatalyst for selective C(sp3)–H activation and oxidation, wherein 4-ethylphenol was chosen as a model substrate. However, we found that when only CuCl-Pyhc was added as a photocatalyst, the reaction scarcely occurred (Table 1, entry 3). After adding the cocatalyst benzoquinone, the reaction could proceed efficiently. Next, we selected the optimal light source and reaction solvent (Table S2†). Under the optimized conditions, a loading of 5 mg of CuCl-Pyhc and benzoquinone (0.1 mmol) caused an 84% conversion of 4-ethylphenol (0.1 mmol) into 1-(4-hydroxyphenyl) ethan-1-one with a selectivity >99% under irradiation with a 455 nm LED for 8 h (Table 1, entry 1). Control experiments in the dark, or without O2, showed negligible amounts of products, confirming the photocatalytic nature and O2 were indispensable (Table 1, entries 4 and 5). And when a control experiment was performed without CuCl-Pyhc, the reaction scarcely occurred (Table 1, entry 2). When using the ligand Pyhc instead of CuCl-Pyhc or the copper salt CuCl with the same loading to participate in the reaction, the yields of the product were low, which also indicated the irreplaceability of CuCl-Pyhc (Table 1, entries 6 and 7).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
| a Reaction conditions: CuCl-Pyhc (5 mg), 4-ethylphenol (0.1 mmol), benzoquinone (0.1 mmol), CH3CN (4 mL), 455 nm LED, O2, 40 °C, 8 h. b The yields were determined using 1H NMR spectroscopy with an internal standard substance. N.R. = no reaction. | ||
| 1 | None | 84 |
| 2 | No CuCl-Pyhc | N.R. |
| 3 | No benzoquinone | N.R. |
| 4 | N2 instead of O2 | N.R. |
| 5 | No light | N.R. |
| 6 | Pyhc instead of CuCl-Pyhc | N.R. |
| 7 | CuCl instead of CuCl-Pyhc | 35 |
To demonstrate the broad applicability of this catalytic method in C–H bond catalytic oxidation, we conducted experiments using different substrates containing aryl R1 and R2 groups. The outcomes revealed that both electron-rich and electron-deficient substrates were successfully transformed into the desired ketones with impressive yields (Table S3†).
Afterwards, we conducted emission quenching experiments on CuCl-Pyhc after adding benzoquinone. The luminescence intensity of CuCl-Pyhc was significantly reduced when it adsorbed benzoquinone. A decrease in the fluorescence lifetime of benzoquinone-adsorbed CuCl-Pyhc of 1.02 ns (Fig. S16†) suggested the possibility of a PET process from the excited state of CuCl-Pyhc to the benzoquinone molecules.21 The presence of benzoquinone (0.067 M) in the acetonitrile suspension of CuCl-Pyhc (3.0 mg) effectively quenched the emission at 570 nm, and the titration curve obeyed a typical Stern–Volmer equation with the Volmer constant (KSV) calculated as 1959 M−1 (Fig. 3a). Subsequently, we compared the changes in Cu valence state of CuCl-Pyhc under 455 nm LED illumination under an oxygen atmosphere or with the addition of benzoquinone. XPS spectra revealed that the addition of benzoquinone resulted in a greater portion of Cu(I) being converted to Cu(II), which further demonstrated the PET process from CuCl-Pyhc to benzoquinone. And we conducted an investigation on the impact of the O2 atmosphere and benzoquinone on the photocurrent of CuCl-Pyhc. The results indicated that oxygen and benzoquinone can enhance the charge separation efficiency of CuCl-Pyhc (Fig. S17†).
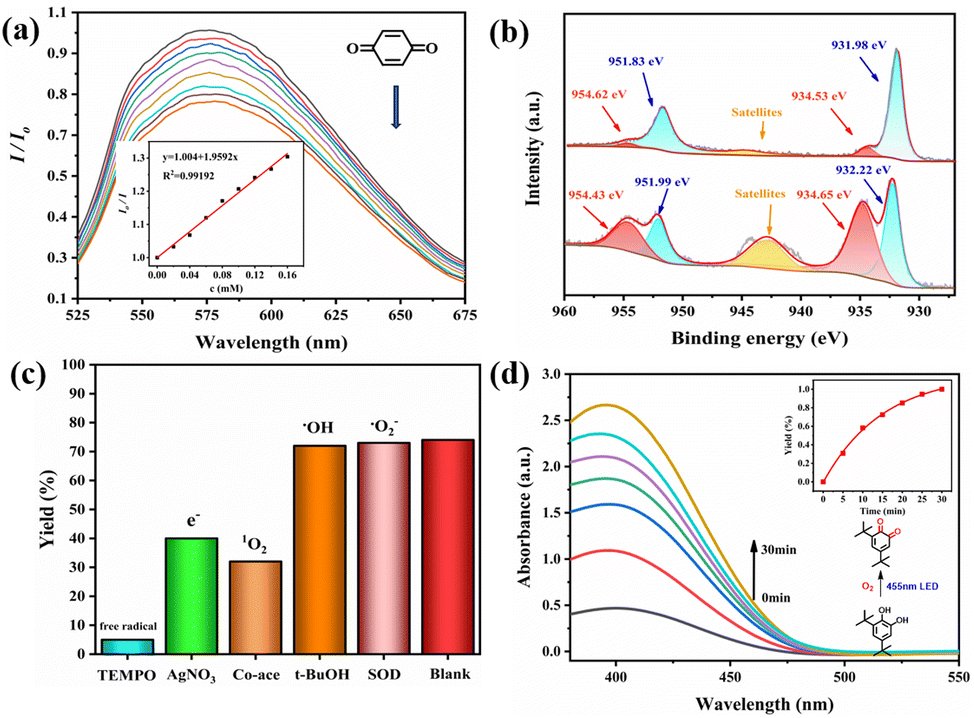 | ||
| Fig. 3 (a) Emission quenching of CuCl-Pyhc after the addition of BQ (inset shows the simulated Stern–Volmer curve; the intensity was recorded at 570 nm with excitation at 455 nm). (b) XPS spectra of CuCl-Pyhc for the Cu2p signal after 8 h illumination under an O2 atmosphere (upper panel) and with the addition of benzoquinone (lower panel). (c) Effect of scavengers on the photocatalytic oxidation of 4-ethylphenol over CuCl-Pyhc. (d) UV-vis spectra recording 3,5-DTBC oxidation by CuCl-Pyhc (inset shows the relationship between the yield of 3,5-DTBQ and the reaction time). | ||
Furthermore, we investigated the impact of incorporating various inhibitors of active species under the optimal reaction conditions on this catalytic system (Fig. 3c). When the 1O2 scavenger cobalt acetylacetonate was added, a significant decrease in yield to 32% was observed, indicating that 1O2 played a more dominant role in the reaction. In contrast, the addition of the ˙O2− scavenger SOD and the hydroxyl radical scavenger t-BuOH resulted in only minor decreases in conversion, suggesting that ˙O2− and ˙OH are not the primary reactive oxygen species controlling the monooxygenation process. The introduction of the electron scavenger AgNO3 into the reaction system led to a considerable reduction in the yield by 38%, underscoring the critical role of photogenerated electrons in the catalytic process.22 And after the addition of the radical capturing agent TEMPO, the yield was only 5%, which proved that the reaction possibly underwent a free radical process.23 To verify the similarity between the catalyst and natural enzymes, we conducted a typical experiment to oxidize ortho diphenol to the corresponding quinone, simulating a natural copper based oxidase, which is of great significance for the biosynthesis of melanin and other polyphenolic natural products.24 When 3,5-di-tert-butylcatechol (3,5-DTBC) was selected as the substrate, the photocatalysis by CuCl-Pyhc demonstrated that it simulated the active center of natural catechol oxidase for the oxidation of 3,5-DTBC to 3,5-di-tert-butyl-o-benzoquinone (3,5-DTBQ), and it exhibited excellent catalytic potential (Fig. 3d).
And we evaluated the adsorption capability of CuCl-Pyhc for the substrate. The 1H NMR spectrum of the impregnated crystals (denoted CuCl-Pyhc@4-ethylphenol) showed that the CuCl-Pyhc adsorbed about 0.48 moles of 4-ethylphenol per copper node (Fig. 4b). And the IR spectrum displayed a new C–H stretching vibration at 2963.62 cm−1, while the peak for the C–H bond of free 4-ethylphenol appeared at 2962.17 cm−1 (Fig. 4a). These findings demonstrated that the substrate could be adsorbed by CuCl-Pyhc. Afterwards, we examined the reaction yield over time, finding the highest yield was achieved when the reaction time reached 8 hours. In addition, we conducted two sets of experiments to remove CuCl-Pyhc through filtration at 2 and 4 hours, and the yields only showed a marginal improvement (Fig. 4c), confirming its significant role as a heterogeneous catalyst. CuCl-Pyhc also proved to be highly recyclable, maintaining its activity after five reuse cycles (Fig. 4d). The PXRD and IR spectra of CuCl-Pyhc obtained simultaneously indicated good structural stability (Fig. S18 and S19†).
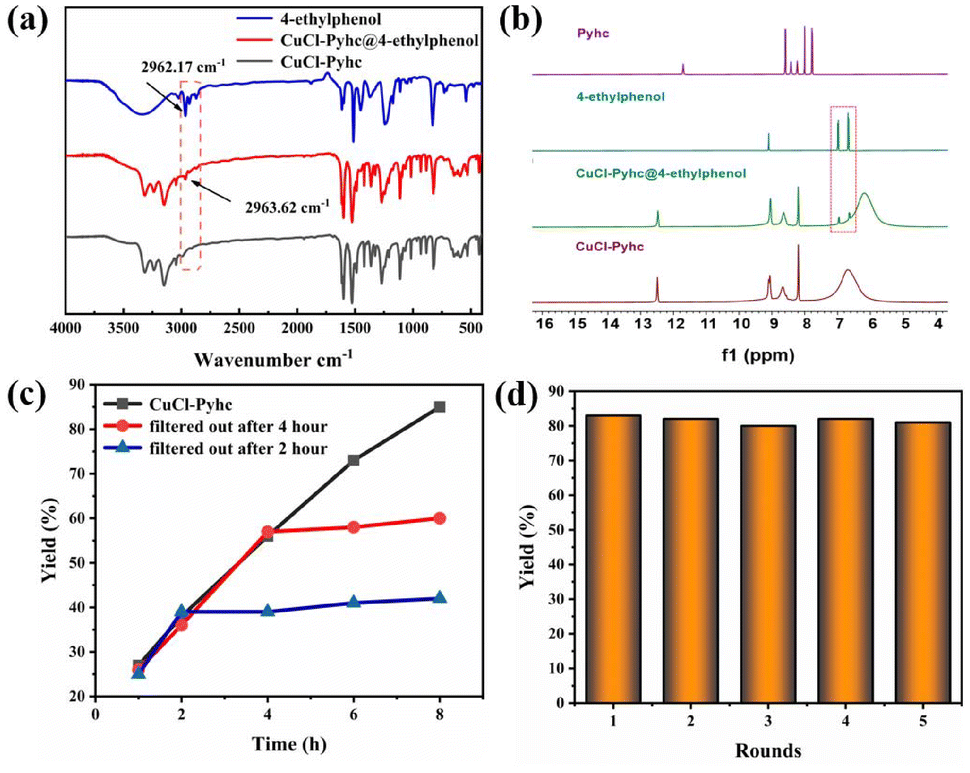 | ||
| Fig. 4 (a) IR spectra of CuCl-Pyhc impregnated with 4-ethylphenol (red); free CuCl-Pyhc (black); and free 4-ethylphenol (blue). (b) 1H NMR spectra of the crystals of Pyhc, 4-ethylphenol, CuCl-Pyhc, and CuCl-Pyhc@4-ethylphenol (in DMSO-d6 and sulfuric acid-d2). (c) Catalytic traces of oxidation of 4-ethylphenol under the optimal conditions with the catalyst CuCl-Pyhc filtered after 2 h and 4 h. (d) Recycling experiments of oxidation of 4-ethylphenol with CuCl-Pyhc under optimal conditions. | ||
A possible reaction mechanism was proposed based on our experimental observations and literature review. Firstly, the light irradiation promoted the formation of excited states of CuCl-Pyhc, and a PET process and an EnT process possibly resulted in the production of benzoquinone radicals and superoxide species Cu(II)–O2˙−. The in situ generated benzoquinone radicals acted as hydrogen transfer reagents to capture hydrogen atoms from the substrates and generated the corresponding alkyl radicals,25 then exchanged hydrogen atoms to obtain Cu(II)–O–OH. The alkyl radicals underwent an oxygen transfer step with Cu(II)–O–OH to obtain the final oxidation products (Fig. S20†).26,27
In summary, we reported a new platform for the oxidation of C(sp3)–H bonds with high efficiency and selectivity by merging of the energy transfer and photoinduced electron transfer process into one cuprous coordination polymer. The CuCl-Pyhc has similar catalytic activity to natural enzymes, which can efficiently activate O2 to form the active metallic oxygen active species for the oxidation of C(sp3)–H bonds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21971030).References
- S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- N. A. Hirscher, N. Ohri, Q. Yang, J. Zhou, J. M. Anna, E. J. Schelter and K. I. Goldberg, J. Am. Chem. Soc., 2021, 143, 19262–19267 CrossRef CAS PubMed.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed.
- T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, RSC Adv., 2020, 10, 34429–34458 RSC.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- C. Minozzi, A. Caron, J. C. Grenier-Petel, J. Santandrea and S. K. Collins, Angew. Chem., Int. Ed., 2018, 57, 5477–5481 CrossRef CAS PubMed.
- X.-Q. Liang, R. K. Gupta, Y.-W. Li, H.-Y. Ma, L.-N. Gao, C.-H. Tung and D. Sun, Inorg. Chem., 2020, 59, 2680–2688 CrossRef CAS PubMed.
- H. M. Tay, N. Kyratzis, S. Thoonen, S. A. Boer, D. R. Turner and C. Hua, Coord. Chem. Rev., 2021, 435, 213763 CrossRef CAS.
- Z. Hu, E. M. Mahdi, Y. Peng, Y. Qian, B. Zhang, N. Yan, D. Yuan, J.-C. Tan and D. Zhao, J. Mater. Chem. A, 2017, 5, 8954–8963 RSC.
- B. Zhong, H. Huang, X. Jing and C. Duan, Chem. Commun., 2022, 58, 3961–3964 RSC.
- K. Wu, X.-Y. Liu, P.-W. Cheng, M. Xie, W. Lu and D. Li, Sci. China: Chem., 2023, 66, 1634–1653 CrossRef CAS.
- S. Garakyaraghi, C. E. McCusker, S. Khan, P. Koutnik, A. T. Bui and F. N. Castellano, Inorg. Chem., 2018, 57, 2296–2307 CrossRef CAS PubMed.
- L. C. Finney, L. J. Mitchell and C. J. Moody, Green Chem., 2018, 20, 2242–2249 RSC.
- D. Shi, R. Zheng, M. J. Sun, X. Cao, C. X. Sun, C. J. Cui, C. S. Liu, J. Zhao and M. Du, Angew. Chem., Int. Ed., 2017, 56, 14637–14641 CrossRef CAS PubMed.
- Y. Shi, T. Zhang, X.-M. Jiang, G. Xu, C. He and C. Duan, Nat. Commun., 2020, 11, 5384 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- J. Park, D. Feng, S. Yuan and H. C. Zhou, Angew. Chem., Int. Ed., 2014, 54, 430–435 CrossRef PubMed.
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS.
- M.-H. Li, Z. Yang, Z. Li, J.-R. Wu, B. Yang and Y.-W. Yang, Chem. Mater., 2022, 34, 5726–5739 CrossRef CAS.
- W. Lee, S. Jung, M. Kim and S. Hong, J. Am. Chem. Soc., 2021, 143, 3003–3012 CrossRef CAS PubMed.
- M. Li, J. Chen, W. Wu, Y. Fang and S. Dong, J. Am. Chem. Soc., 2020, 142, 15569–15574 CrossRef CAS PubMed.
- C. Yang, J. Yang, X. Gong, Y. Wei and X. Xu, Phys. Chem. Chem. Phys., 2022, 24, 14947–14952 RSC.
- L. Kimberley, A. M. Sheveleva, J. Li, J. H. Carter, X. Kang, G. L. Smith, X. Han, S. J. Day, C. C. Tang, F. Tuna, E. J. L. McInnes, S. Yang and M. Schröder, Angew. Chem., Int. Ed., 2021, 60, 15243–15247 CrossRef CAS PubMed.
- H. Huang, X. Jing, J. Deng, C. Meng and C. Duan, J. Am. Chem. Soc., 2023, 145, 2170–2182 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2329162. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01257g |
| This journal is © The Royal Society of Chemistry 2024 |