
Leveraging peptide–cellulose interactions to tailor the hierarchy and mechanics of peptide–polymer hybrids†
Daseul
Jang
 a,
Laura E.
Beckett
a,
Jong
Keum
b and
LaShanda T.J.
Korley
*ac
a,
Laura E.
Beckett
a,
Jong
Keum
b and
LaShanda T.J.
Korley
*ac
aDepartment of Materials Science and Engineering, University of Delaware, 127 The Green, 201 Dupont Hall, Newark, Delaware 19716, USA. E-mail: lkorley@udel.edu
bCenter for Nanophase Materials Sciences and Neutron Scattering Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, 37830, Tennessee, USA
cDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, Delaware 19716, USA
First published on 31st May 2023
Abstract
Inspired by spider silk's hierarchical diversity, we leveraged peptide motifs with the capability to tune structural arrangement for controlling the mechanical properties of a conventional polymer framework. The addition of nanofiller with hydrogen bonding sites was used as another pathway towards hierarchical tuning via matrix–filler interactions. Specifically, peptide–polyurea hybrids (PPUs) were combined with cellulose nanocrystals (CNCs) to develop mechanically-tunable nanocomposites via tailored matrix–filler interactions (or peptide–cellulose interactions). In this material platform, we explored the effect of these matrix–filler interactions on the secondary structure, hierarchical ordering, and mechanical properties of the peptide hybrid nanocomposites. Interactions between the peptide matrix and CNCs occur in all of the PPU/CNC nanocomposites, preventing α-helical ordering, but promoting inter-molecular hydrogen bonded β-sheet formation. Depending on peptide and CNC content, the Young's modulus varies from 10 to 150 MPa. Unlike conventional cellulose-reinforced polymer nanocomposites, the mechanical properties of these composite materials are dictated by a balance of CNC reinforcement, peptidic ordering, and microphase-separated morphology. This research highlights that leveraging peptide–cellulose interactions is a strategy to create materials with targeted mechanical properties for a specific application using a limited selection of building blocks.
10th Anniversary StatementEarly in my career, the Journal of Materials Chemistry B provided a platform for disseminating my research group's investigations into bio-inspired strategies for materials chemistry utilizing peptide building blocks as a handle for hierarchical design. The interdisciplinary nature of this journal enabled a framework to highlight the link between synthetic design, architectural features, and functional behavior unique to our work, including a feature in the 2014 Emerging Investigators Themed Issue. Serving on the Editorial Advisory Board from 2014–2017 also provided an engagement opportunity to explore ways to promote and encourage interdisciplinary materials chemistry approaches relevant to biomaterials applications. On the occasion of the 10th Anniversary of the Journal of Materials Chemistry B, I am delighted to showcase our most recent pathway to nature-inspired materials, examining the interplay of peptide motifs and cellulose nanocrystals in the design of hybrid nanocomposites with highly tunable mechanics and hierarchical assembly. |
Introduction
The mechanical performance of materials is a crucial factor to consider when designing and engineering specific functionality. In tissue engineering, the mechanical microenvironments of biomaterials dictate cell attachment and proliferation, allowing for the regeneration of damaged tissues.1 Specifically, bone tissue requires scaffolds with relatively high stiffness (around 15–20 MPa) compared with other soft tissues (e.g., cartilage) to bear the weight of the body and resist bending and twisting forces.2 Thus, varying tissue types require different mechanical properties to invoke specific function, covering more than 10 orders of magnitude in Young's modulus.3 To address the need to design tunable materials with potential in biological applications, it is essential to explore and develop a suite of molecular design and composite strategies for mechanical modularity.4,5The incorporation of nanofillers in a polymer matrix often is used to achieve superior or desired mechanical properties, such as modulus and strength compared with the polymer alone.6,7 These polymer nanocomposites (PNCs) are multiphase materials comprised of a continuous polymer phase (matrix) and nanometer-sized additives (nanofiller). Interfacial interactions between the matrix and the filler play an important role in determining the properties of the final material.8,9 Specifically, favorable matrix–filler interactions generally improve mechanical performance. One approach toward modulating matrix–filler interfacial interactions is to utilize nanofillers that possess non-covalent bonding sites, such as nanocellulose. CNCs are are highly crystalline, rod-like nanomaterials extracted from cellulose fibers by chemical treatment, such as acid hydrolysis. CNCs have garnered significant attention as a reinforcing agent in PNCs because of their anisotropic morphology (i.e., high aspect ratio) and inherently high stiffness, which are directly related to the mechanical properties (e.g., strength, stiffness, and elongation) of PNCs.10,11 CNCs have been introduced into various polymers, such as poly(vinyl alcohol),12 poly(vinyl acetate),13 poly(ethylene oxide),12 chitosan,14 poly(butyl methacrylate),15 polybutadiene,16 poly[styrene-(ethylene-co-butylene)-styrene] triblock copolymer,17 polyurethane,18–21 and natural silks,22–27 to enhance mechanical behavior. For example, the mechanical properties (e.g., storage modulus) of polymer nanocomposites containing natural rubber (NR) latex (matrix) and nanocellulose (filler) increased through the surface modification of matrix, which is related to the quality of cellulose nanocrystal (CNC) dispersion and final morphology.28 The introduction of epoxy groups into the NR led to increased hydrogen bonding interactions between the polymer matrix and filler, and the epoxidized NR/CNC nanocomposites exhibited significant enhancement in storage modulus compared with pristine NR/CNC nanocomposites. Thus, the precise control of these matrix–filler physical associations can be a facile strategy to tailor material properties.29 Furthermore, systematic understanding of structure–property relationships facilitates the design of nanocomposites with desired mechanical properties for a specific application.30,31
Polypeptide-hybrids are an emerging class of nanocomposite matrix materials due to their nanoscale structure, tunable mechanical properties, and inherent biocompatibility.22–27 The abundant functional groups present in polypeptide-based materials facilitates integration with inorganic or organic nanomaterials through covalent and non-covalent interactions, such as hydrogen bonding and electrostatic interactions.32,33 Controlling weak physical associations within polypeptide-based nanocomposites can facilitate their hierarchical organization and promote desired mechanical function. For example, the mechanical properties of silk fibroins, which are comprised of natural polypeptide sequences, were enhanced via peptide–CNC interactions (i.e., hydrogen bonding).24 Their favorable interfacial associations induced the formation of a unique self-assembled “shish kebab” morphology, leading to increased Young's modulus (30 GPa) and strength (260 MPa). This study demonstrates that peptide–CNC interactions can be used to leverage structural hierarchy as a pathway towards enhanced mechanical response. However, recent polypeptide-based nanocomposites have been fabricated from natural biopolymers such as silk fibroin, amyloid fibril, keratin, elastin, and collagen, which are limited in scalability, processability, and durability.32,33
A promising approach toward overcoming the limitations of natural polypeptide-based nanocomposites is to harness peptide–polymer hybrids as matrix materials. These hybrids are silk-inspired, functional block copolymers that combine the structural and functional control of peptides and the versatility, processability, and scalability of traditional synthetic polymers, which have been applied to wide range of applications, such as drug delivery, tissue engineering, adhesives, electronics, actuators, and sensors.34–36 In these hybrids, secondary structures (e.g., α-helices and β-sheets) govern the development of unique microstructures (e.g., micellar aggregates, nanotubes, or fibrils) that significantly influence their thermal and mechanical properties, and stimuli-responsive behavior.37,38 For example, Johnson et al. utilized secondary structure to tune the mechanical performance of peptide–polyurea hybrids, comprised of poly(β-benzyl-L-aspartate)-b-poly(dimethylsiloxane)-b-poly(β-benzyl-L-aspartate) (PBLA-b-PDMS-b-PBLA) as a building block.32 In these PBLA-polyurea hybrids, β-sheet ordering exhibited superior strength and toughness compared to α-helical ordering due to inter-molecular hydrogen bonding at the same PBLA content. To date, peptide-polymer hybrids have limited exploration as nanocomposite matrices. Lei et al. explored mechanical modulation of polymer–peptide hybrids by designing nanocomposites comprised of polypeptide-functionalized graphene oxide (GO) dispersed in a poly(γ-benzyl-L-glutamate)-b-poly(dimethylsiloxane)-b-poly(γ-benzyl-L-glutamate) (PBLG-b-PDMS-b-PBLG) matrix.39 These GO/peptide copolymer composite gels formed a nanofibrous network morphology with well-dispersed GO sheets, resulting in increased modulus and fracture stress. However, fundamental understanding of the interrelated influence of matrix morphology and matrix–filler interactions on the mechanical behavior and hierarchical architecture has yet to be elucidated in peptide-polymer hybrid nanocomposites.
To bridge the knowledge gap in peptide hybrid nanocomposites, we incorporated CNCs as the functional filler in a series of peptide-polymer hybrids as the matrix. Specifically, peptidic polyureas (PPUs) comprised of PBLA-b-PDMS-b-PBLA and 1,6-hexamethylene diisocyanate (HDI), which have been previously investigated,37,38 were utilized due to the ability to tailor secondary conformation, hierarchical ordering, and mechanical properties. To assess the role of the structural hierarchy of the matrix on mechanical response, the peptidic ordering and morphology were varied by changing peptide composition. Using this platform, we examined the impact of matrix–filler interactions on the secondary structure, hydrogen bonding arrangement, microphase-separated morphology, and mechanical properties of these peptide hybrid nanocomposites. This investigation provides a facile approach to dictate secondary structure conformation, microstructure, and mechanical behavior via peptide–CNC interactions in polypeptide hybrid nanocomposites.
Results and discussion
We explored the influence of CNC incorporation on the secondary structure, microphase-separated morphology, and mechanical properties of PPUs with PBLA-b-PDMS-b-PBLA as the soft block and HDI as the hard segment. In this investigation, a non-chain extended peptidic polyurea (PPU) matrix was chosen (Fig. 1A) to limit the influence of the hard domain on matrix–filler interactions,37 focusing primarily on the peptidic (PBLA) segments with the ordered soft domain. PDMS, which has a low hydrogen bonding interaction energy (7.5 kJ mol−1),28 was utilized as the central block of the soft segment to: (1) minimize interactions within the soft segment (PDMS–PBLA interactions) and (2) allow CNCs to interact selectively with PBLA among the soft segment. Furthermore, the utilization of a peptide block (PBLA, repeat length ∼20) as the soft segment enhances the miscibility between the matrix (PPU) and nanofiller (CNC). The PBLA content was varied to examine the impact of matrix morphology or hierarchical ordering on the mechanical properties. The following nomenclature was used for neat PPUs and PPU/CNC nanocomposites (Fig. 1B): An–X/CNCY, where A refers to the PPUs consisting of PBLA-b-PDMS-b-PBLA and HDI, n is the PBLA block length, X is the peptide content, and Y is the CNC weight fraction in the PPUs. An-X/CNC0 represents the control PPUs. Table 1 details the molecular weight and dispersity of a series of PPUs with 10 (A20-10/CNC0) or 20 wt% (A20-20/CNC0) of PBLA. PPU/CNC nanocomposites were fabricated via solution casting, which is a scalable composite processing approach. Employing this nanocomposite platform, we correlated PPU–CNC interactions with the hierarchical structure and mechanical properties of the peptide hybrid/CNC composites.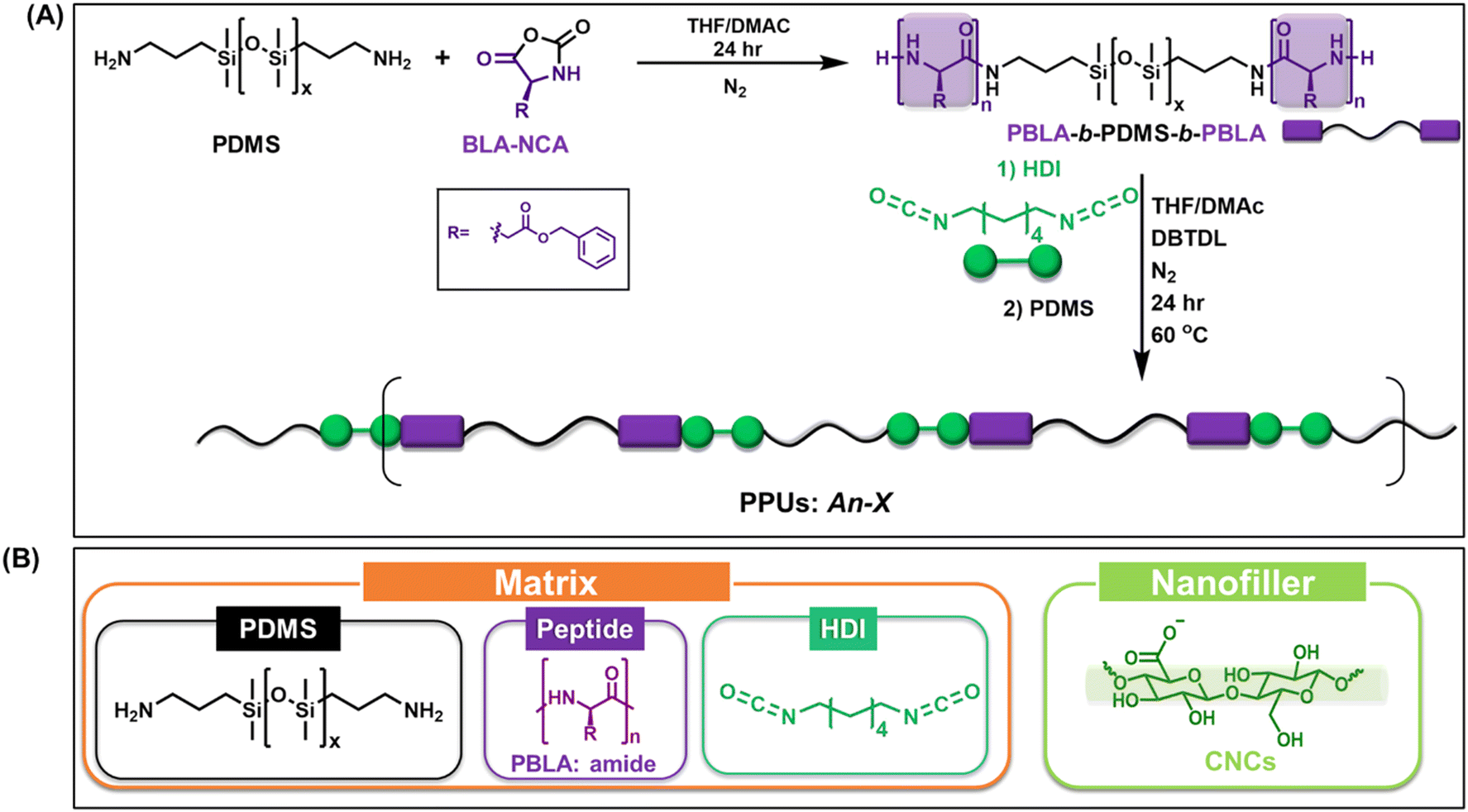 | ||
| Fig. 1 (A) Synthetic scheme of PPUs (matrix) and (B) building blocks (i.e., matrix and nanofiller) used to design PPU/CNC nanocomposites. | ||
Characterization of secondary conformation and interactions present in PPU/CNC composites: the effect of CNC incorporation on the peptidic ordering of PPUs
Analysis of the secondary structure and hydrogen bonding arrangement is essential to the evaluation of peptide-based/CNC nanocomposites due to the significant influence on the material performance, such as mechanical or stimuli-responsive properties.37,38 Attenuated total reflection – Fourier transform infrared spectroscopy (ATR-FTIR) can be utilized to determine secondary conformation, such as α-helices and β-sheets, in the solid state and to identify specific interactions within the PPU/CNC nanocomposites. In Fig. 2, the amide I carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching band was examined to confirm variations in secondary structure or PBLA ordering present in each sample. Specifically, the second derivative (Fig. S1, ESI†) was employed to define peaks and uncover any hidden peaks related to peptide structural analysis. A signal between 1620 and 1645 cm−1 is associated with β-sheet formation, while a peak between 1650 and 1665 cm−1 is indicative of α-helical structures.41,42 It is important to note that the CO stretching band of hydrogen-bonded and ordered urea groups appears at 1600–1625 cm−1.38 The neat PPUs (A20-10/CNC0; A20-20/CNC0) exhibit a mixture of α-helices and β-sheets. Comparing these two PPUs reveals that the intensity of the α-helix band increases with increasing PBLA weight fraction (A20-20), indicating that the higher PBLA content promotes an α-helical arrangement. A similar observation was identified in our previous investigations where this trend was attributed to the reduced mobility of PDMS segments with increasing PBLA content.37,38 For the A20-10/CNC nanocomposites, the peak position at ∼1624 cm−1 relatively remains constant, while the peak at ∼1662 cm−1 disappears (Fig. S1, ESI†). Furthermore, the peaks broaden with increasing CNC content. These observations imply that β-sheet formation is dominant in the composites and that the CNCs interact extensively with the PBLA blocks and urea groups (hard segments). In contrast, upon CNC incorporation in the A20-20 series, the peak assigned to α-helices disappears and the band associated with β-sheets shifts toward a lower wavenumber. These variations in amide I band suggest that CNC incorporation hinders the intramolecular hydrogen bonding required for α-helix formation, but facilitates intermolecular hydrogen bonding, leading to increased β-sheet content. The shifts in peptidic ordering provide evidence of extensive hydrogen bonding between CNCs and the PBLA segments. The modulation of polypeptide secondary structures through hydrogen bonding with additives has been reported.43–45 For example, a phenolic resin with hydrogen bonding sites was blended with poly(glutamate)s to control the secondary structure.43 In this blend, the stabilization of α-helical conformations was dependent on the rigidity of the protecting or side chain groups and the content of phenolic resin, which impacted the hydrogen bonding interactions between the polypeptide and phenolic resin.
O) stretching band was examined to confirm variations in secondary structure or PBLA ordering present in each sample. Specifically, the second derivative (Fig. S1, ESI†) was employed to define peaks and uncover any hidden peaks related to peptide structural analysis. A signal between 1620 and 1645 cm−1 is associated with β-sheet formation, while a peak between 1650 and 1665 cm−1 is indicative of α-helical structures.41,42 It is important to note that the CO stretching band of hydrogen-bonded and ordered urea groups appears at 1600–1625 cm−1.38 The neat PPUs (A20-10/CNC0; A20-20/CNC0) exhibit a mixture of α-helices and β-sheets. Comparing these two PPUs reveals that the intensity of the α-helix band increases with increasing PBLA weight fraction (A20-20), indicating that the higher PBLA content promotes an α-helical arrangement. A similar observation was identified in our previous investigations where this trend was attributed to the reduced mobility of PDMS segments with increasing PBLA content.37,38 For the A20-10/CNC nanocomposites, the peak position at ∼1624 cm−1 relatively remains constant, while the peak at ∼1662 cm−1 disappears (Fig. S1, ESI†). Furthermore, the peaks broaden with increasing CNC content. These observations imply that β-sheet formation is dominant in the composites and that the CNCs interact extensively with the PBLA blocks and urea groups (hard segments). In contrast, upon CNC incorporation in the A20-20 series, the peak assigned to α-helices disappears and the band associated with β-sheets shifts toward a lower wavenumber. These variations in amide I band suggest that CNC incorporation hinders the intramolecular hydrogen bonding required for α-helix formation, but facilitates intermolecular hydrogen bonding, leading to increased β-sheet content. The shifts in peptidic ordering provide evidence of extensive hydrogen bonding between CNCs and the PBLA segments. The modulation of polypeptide secondary structures through hydrogen bonding with additives has been reported.43–45 For example, a phenolic resin with hydrogen bonding sites was blended with poly(glutamate)s to control the secondary structure.43 In this blend, the stabilization of α-helical conformations was dependent on the rigidity of the protecting or side chain groups and the content of phenolic resin, which impacted the hydrogen bonding interactions between the polypeptide and phenolic resin.
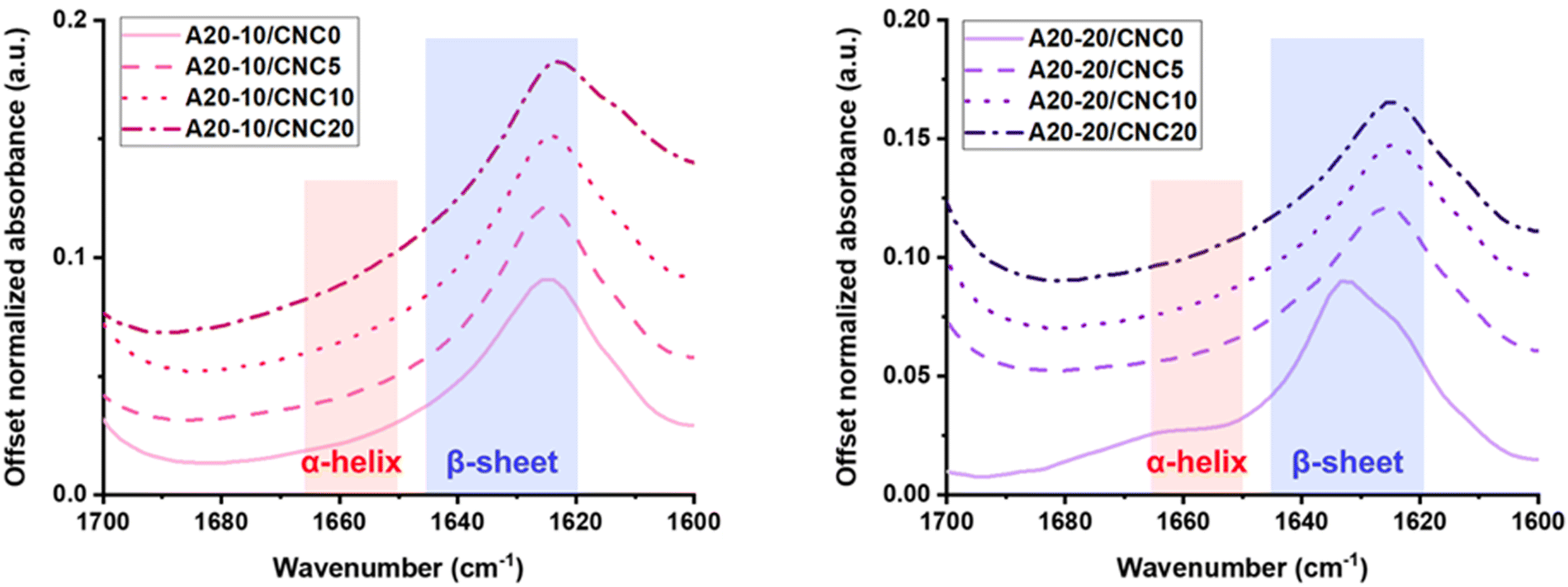 | ||
| Fig. 2 ATR-FTIR spectra of PPU/CNC nanocomposites as a function of the CNC weight fraction. Peptide secondary structure was identified based on the second derivative in the amide I stretching region (1700–1600 cm−1). CNC incorporation into PPUs leads to variations in secondary conformation, disrupting α-helical ordering but promoting intermolecular hydrogen bonding. This finding provides evidence of preferential interactions between the PBLA blocks and CNCs. | ||
To further investigate matrix–filler (PPU–CNC) interactions, the O–H stretching region (3100–3600 cm−1) and the N–H stretching absorption region (3200–3450 cm−1; hydrogen-bonded urea N–H) were monitored.46–49 We anticipated that competitive hydrogen bonding interactions would occur in the PPU/CNC composites because the N–H groups in the PPU and the hydroxyl groups in the CNCs can act as proton donors, while the carbonyl groups in PPUs can serve as proton acceptors.50 Fig. S2 (ESI†) shows variations in the peak positions and widths upon incorporation of CNCs into PPUs. Shifting to a higher or lower wavenumber indicates changes in the surrounding environment of the N–H groups.49 Both the A20-10 and A20-20 nanocomposites shifted toward a higher wavenumber (A20-10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3334 cm−1 → 3336 cm−1, A20-20:3322 cm−1 → 3336 cm−1), most likely due to the formation of intermolecular hydrogen bonding between the hard segments (urea groups) and the CNCs. Peak broadening occurs in all PPU/CNC nanocomposites, signifying the presence of differently hydrogen-bonded species with a wide range of proton donor–acceptor distances or increased phase mixing.51 Thus, the ATR-FTIR results suggest that: (1) the CNCs interact favorably with the peptidic polyurea matrices through PBLA–CNC or urea–CNC associations, leading to variations in hydrogen bonding arrangement, and (2) CNC–PPU interactions can be harnessed to tune secondary structures.
:3334 cm−1 → 3336 cm−1, A20-20:3322 cm−1 → 3336 cm−1), most likely due to the formation of intermolecular hydrogen bonding between the hard segments (urea groups) and the CNCs. Peak broadening occurs in all PPU/CNC nanocomposites, signifying the presence of differently hydrogen-bonded species with a wide range of proton donor–acceptor distances or increased phase mixing.51 Thus, the ATR-FTIR results suggest that: (1) the CNCs interact favorably with the peptidic polyurea matrices through PBLA–CNC or urea–CNC associations, leading to variations in hydrogen bonding arrangement, and (2) CNC–PPU interactions can be harnessed to tune secondary structures.
The impact of cellulose incorporation on the phase separation behavior of PPUs
With knowledge of the secondary structural arrangement and hydrogen bonding organization, the phase behavior and hierarchical structure of the PPU/CNC nanocomposites were explored via atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS). These investigations will enable morphological understanding of these PPU/CNC composites, which has been shown to play a critical role in mechanical performance in conventional polyureas and peptidic polyureas as well as polyurea/urethane nanocomposites.37,52,53AFM was utilized to visualize the microphase-separated morphology of the PPU/CNC. Fig. 3 represents the phase images, where the PDMS phase appears dark and the bright areas correspond to either the hard phase (PBLA and urea hard segments) or CNCs. All neat PPU films display randomly dispersed rod-like structures that are formed by the self-assembly of PBLA and hard segments. These fibrillar morphologies also have been observed in conventional segmented polyurethanes and peptide–polymer hybrids as a result of microphase segregation and peptidic segment ordering.19,37 Upon 5 wt% CNC loading in the A20-10 matrix, a globular-like morphology appears. The morphology of segmented polyurethanes is generally determined by hydrogen bonding arrangement and the degree of phase mixing.50 As shown in the ATR-FTIR spectra, a fraction of the PBLA–PBLA and urea–urea (hard segment-hard segment) hydrogen bonds are replaced by PBLA–CNC and urea–CNC interactions, which may lead to a shift in the microphase-separated morphology from rod-like to globular-like morphologies. A similar morphological transition was reported when the peptide weight fraction in PPUs is low (∼5 wt%) or peptide-driven long-range ordering is less pronounced.37 At a higher CNC content (10–20 wt%), CNC nanorods (∼10 nm in width) are observed in the A20-10/CNC nanocomposites, which are brighter than PPUs due to their higher stiffness. The CNC weight fraction in A20-10 highly influences the self-assembled morphology of A20-10 in the presence of CNC nanorods. For A20-10/CNC10, CNCs are surrounded by globular aggregates. In contrast, for A20-10/CNC20, CNC rods are entangled with a nanofibrous matrix. Similarly, adding 5 wt% of CNCs to A20-20 results in the lack of well-defined nanofibrils in comparison to the neat A20-20 matrix. Upon introduction of 10 and 20 wt% CNCs to A20-20, inter-connected fiber networks consisting of the CNC nanorods and PPU nanofibrils are observed. Thus, this AFM investigation highlights that CNC incorporation into the PPUs results in a hierarchical structural shift, which is dictated by hydrogen bonding organization and phase separation. The influence of morphology on mechanical properties will be explored in the following section: Mechanical response of the PPU/CNC nanocomposites.
 | ||
| Fig. 3 Tapping-mode AFM phase images of the A20-10/CNC and A20-20/CNC nanocomposites (image size: 1 × 1 μm, scan rate: 1 Hz). These images show the rod-like structures of A20-10 and A20-20 disappear upon CNC loading in PPUs, indicating that a change of hydrogen bonding arrangements from PBLA–PBLA to PBLA–CNC results in phase separation behavior. | ||
To complement AFM results and further probe the effect of CNCs on the degree of phase separation, SAXS experiments were conducted. SAXS is a useful tool for elucidating nanoscale structures of polyurethane and their nanocomposites because the shape, size, and distribution of the structural arrangement generally determine the scattering patterns and intensity distribution.54–56
Model fitting for the neat PPUs was conducted to determine the inter-domain spacing (Fig. S3, ESI†). The experimental scattering intensity I(Q) measured from the A20-20 peptidic polyurea was model fit using the following eqn (1) under the assumption of a stacked two-phase lamellar morphology,
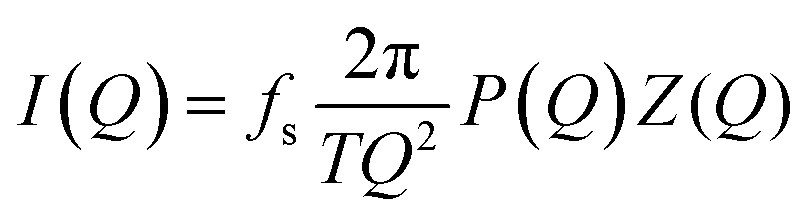 | (1) |
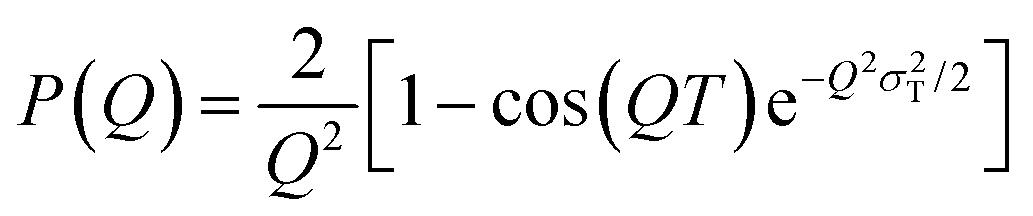 | (2) |
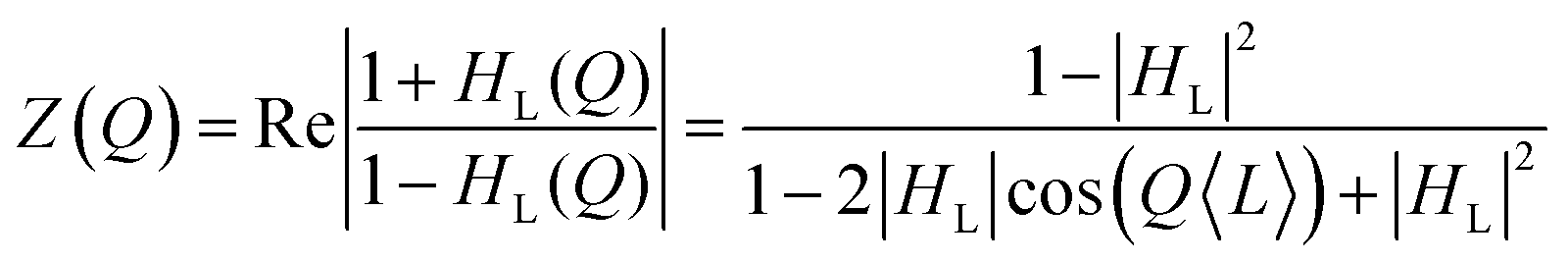 | (3) |
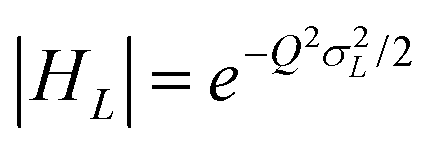 .
.
In the case of the A20-10 peptidic polyurea, the scattering function derived for a regularly or roughly ordered lamellar morphology did not adequately describe the experimental SAXS curves. To better understand the lamellar morphology of the A20-10 peptidic polyurea, an additional function, the broad peak model,61 was included in eqn (1) as follows,
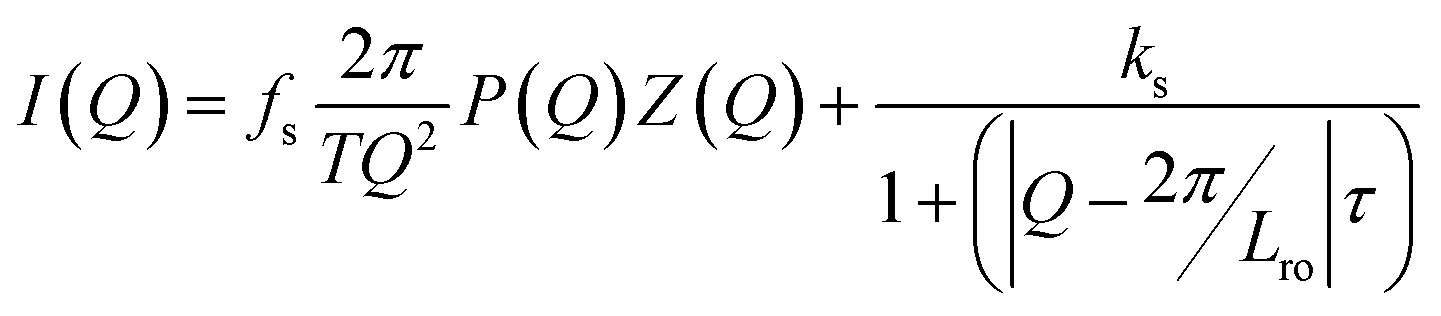 | (4) |
As shown in Fig. 4, all PPU controls exhibit a scattering peak as a result of their local heterogeneities in electron density, which is indicative of microphase separation between the ordered “pseudo” hard (PBLA + HDI) regions and disordered soft domains. Based on the model fitting results (Fig. S3, ESI†), an increase in PBLA content leads to an increase in the long spacing (L), shifting from 22 nm (A20-10/CNC0) to 26 nm (A20-20/CNC0). Additionally, Fig. 4 shows that A20-20/CNC0 possesses two reflections at ∼0.02 and 0.04 Å−1 (with spacing ratio of 2:1), indicative of either a lamellar organization or the presence of different inter-domain spacings.40 These findings demonstrate that an increased PBLA amount or peptide packing leads to a long-range, ordered structure. Upon CNC loading, the scattering peaks of all nanocomposites appear broader and indistinguishable, limiting model fitting for these samples. These broader peaks indicated electronic density variations, which are likely a result of increased phase mixing and irregular structural organization.62 Additionally, this peak broadening in all PPU/CNC nanocomposites indicates a change in the packing of the PPU domain, supporting the assertion that PPU–CNC hydrogen bonding (Fig. 2) promotes phase mixing or a disruption of the long-range connectivity between “pseudo” hard domains (PBLA and hard segments).63 To further probe the phase segregation behavior, the Lorentz correction (a plot of Q vs. I(Q)Q2) can be applied to the SAXS data.62–65 As illustrated in Fig. S4 (ESI†), this plot accentuates small scattering peaks. For all nanocomposite SAXS spectra, the scattering peaks are shifted to smaller q values (i.e., increase in L), which may result from the co-organization of PPUs and CNCs as seen in Fig. 3.
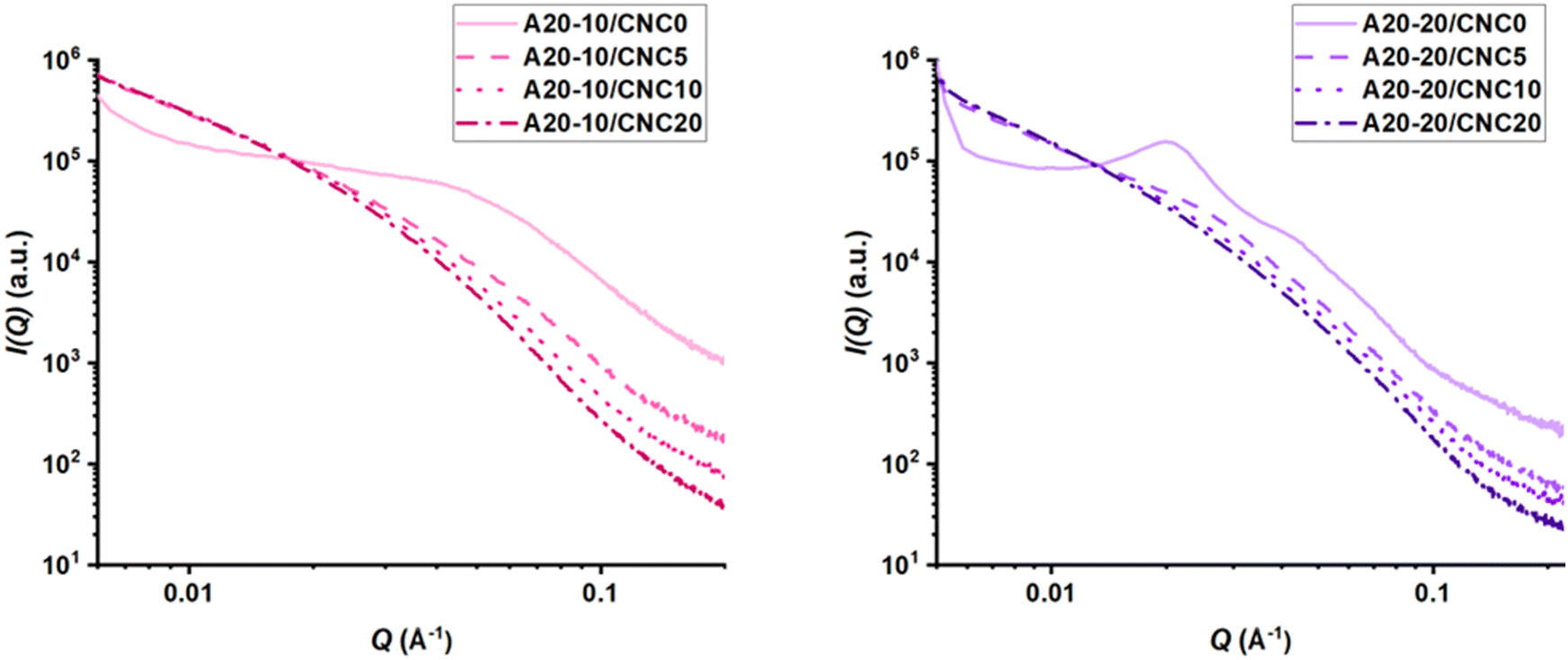 | ||
| Fig. 4 1D-SAXS profiles of the PPU/CNC nanocomposites with varying PBLA and CNC content collected at room temperature for 30 minutes. The neat PPU matrices (i.e., A20-10/CNC0 and A20-20/CNC0) exhibit a defined scattering peak, whereas the addition of CNC to the matrices results in a reduction or loss of the distinct peak. These results indicate that the introduction of CNCs into PPU matrices modulates long-range ordering and structural hierarchy. | ||
Mechanical response of the PPU/CNC nanocomposites
To probe the influence of the hydrogen bonding arrangement and morphology on the storage modulus and tanδ (molecular motion) in PPU/CNC nanocomposites, dynamic mechanical analysis (DMA) was utilized (Fig. 5). DMA studies of the peptidic polyurea/CNC nanocomposites were limited to CNC contents of 5 wt% and 10 wt% due to the brittle nature of A20-20/CNC20. For the control PPUs (A20-10/CNC0; A20-20/CNC0), increasing the PBLA content leads to a higher storage modulus in the glassy state (below the PBLA glass transition temperature (Tg)). At −50 °C, the storage modulus of A20-20/CNC0 (∼112 MPa) is 14 times higher than that of A20-10/CNC0 (∼8 MPa). A similar trend was observed in prior studies.37 The higher storage modulus of A20-20/CNC0 is likely a result of the rigid or “pseudo” hard segment character of the PBLA blocks.40 Furthermore, based on the AFM and SAXS findings, A20-20/CNC0 exhibits more elongated and larger fibrillar structures, which may allow for efficient energy dissipation and improved mechanical response. Upon CNC incorporation, significant differences are observed in the A20-10/CNC and A20-20/CNC series. For the A20-10/CNC nanocomposites, the plateau modulus below the Tg of PBLA increases from 23 MPa to 62 MPa as the CNC content shifts from 5 wt% to 10 wt%. Similar to traditional polyurethane/CNC nanocomposites, the storage modulus increases with increasing CNC content due to the reinforcement effect of crystalline nanocelluloses.20 However, in the A20-20/CNC5 nanocomposite, a reduction in the plateau modulus is observed compared to the control A20-20/CNC0, which may be ascribed to reduced α-helix content (Fig. 2) and/or disrupted rod-like morphology (Fig. 3). Unlike conventional CNC-reinforced polyurethane nanocomposites, this unusual storage modulus reduction suggests that the PBLA secondary structures and/or peptidic ordering-driven morphology may have a stronger influence on the storage modulus at the higher PBLA content. However, the storage modulus also increases at 10 wt% of CNC, which may be attributed to synergistic effects of increased PPU-CNC interactions and/or a morphological shift from globular-like to an interconnected, nanofibrous structure (Fig. 3). Above the Tg of PBLA, the storage moduli of all PPU/CNC nanocomposites are significantly higher than the control PPUs. For example, at 90 °C, the storage modulus of A20-10/CNC10 (∼33 MPa) is about 14-fold larger than the value of the neat A20-10 (∼2.3 MPa), which exhibits an abrupt drop above the PBLA Tg. The substantial reinforcement of the PPU/CNC nanocomposites above the glass transition temperature can be attributed to the efficient dispersion of the CNC nanofiller in the PPU matrix.19 These findings reveal that thermomechanical properties of A20-10, containing a lower α-helical content compared to A20-20, are linearly related to CNC weight fraction. In contrast, the thermomechanical behavior of A20-20, which contains a higher α-helical fraction, is dictated by a balance of PPU–CNC interactions and a hierarchical microstructure. Furthermore, the DMA results highlight that peptide–cellulose interactions can be a handle to tailor not only peptidic ordering and morphology, but also mechanical response.
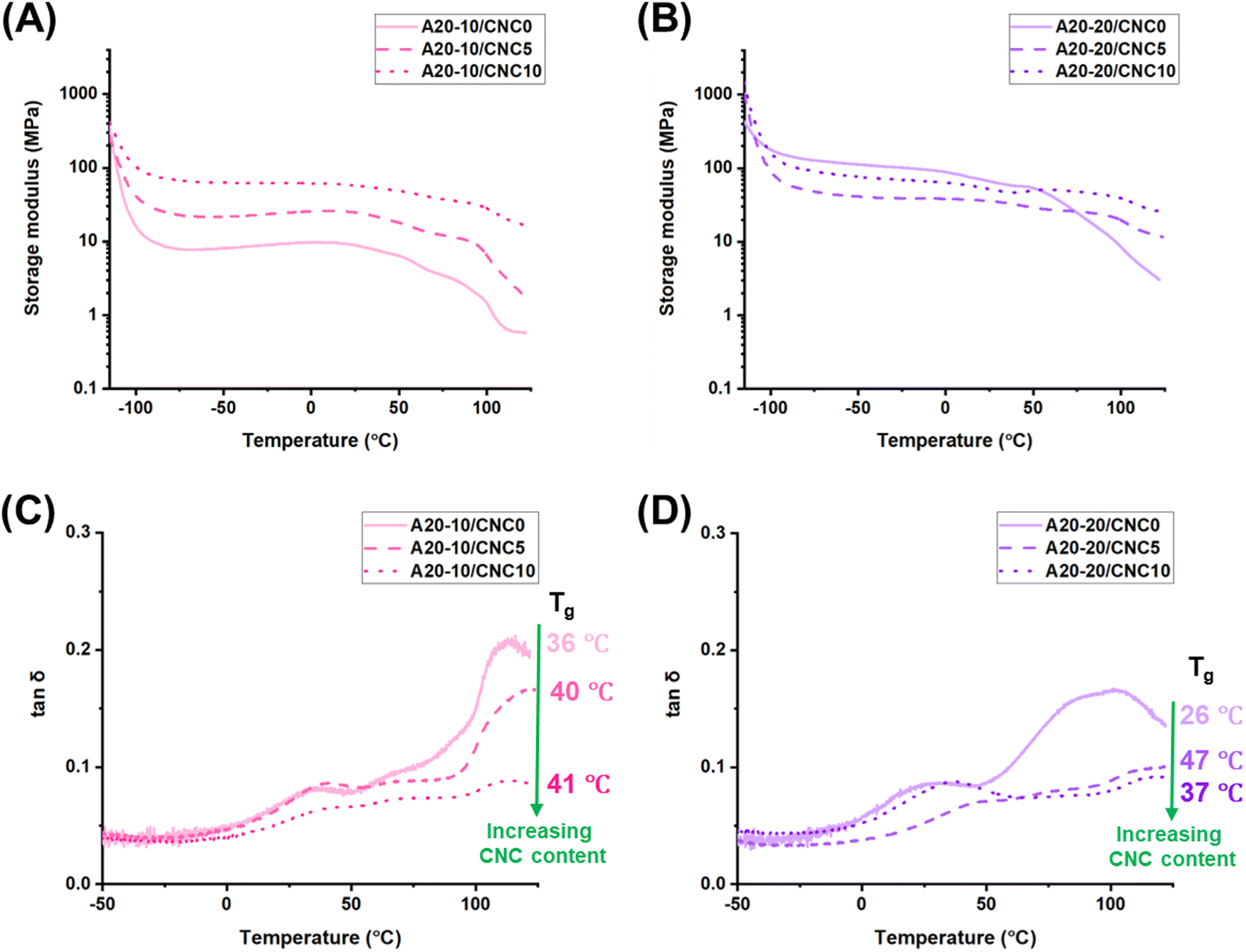 | ||
| Fig. 5 DMA curves of neat PPUs and CNC-reinforced PPUs with varying CNC weight fractions recorded in oscillatory film tension mode at a heating rate of 5 °C min−1 and a frequency of 1 Hz. Top: Storage modulus; Bottom: tanδ (the ratio of loss modulus over storage modulus) with summarized PBLA glass transition temperatures (Tgs – peak in tanδ). (A) and (C) A20-10/CNC series; (B) and (D) A20-20/CNC series. Increasing CNC content in A20-10 yields a higher storage modulus, whereas incorporating CNC into A20-20 reduces a storage modulus. In both A20-10 and A20-20, Tg increases when CNCs are added, suggesting that the chain mobility is restricted. These data support that the peptide content and hierarchical structures of PPUs have a significant impact on the thermomechanical properties of PPU/CNC nanocomposites. This finding demonstrates the importance of not only matrix–filler interactions, but also the hierarchical arrangement of the matrix material in tuning mechanical performance. | ||
Examination of the tanδ peaks (Fig. 5C and D) provides information on the impact of CNC incorporation on the mobility of the peptidic soft segments. With increasing CNC content, the tanδ peak of the A20-10 nanocomposites shifts to higher temperatures and broadens, suggesting a restriction on soft segment mobility as a result of specific PPU–CNC interactions.66 Similarly, the tanδ peak of the A20-20 nanocomposites occurs at higher temperatures (37–47 °C) compared to A20-20/CNC0 (26 °C) as indicated in Fig. 5D. It is important to note that both A20-10/CNC10 and A20-20/CNC5 exhibit the broadest tanδ peak among the PPU/CNC series. These observations suggests that PBLA–CNC interactions are the most favorable in A20-10/CNC10 and A20-20/CNC5.66 Furthermore, these observations can be corroborated by comparing the tanδ peak intensity, which is related to the extent of matrix–CNC interactions and phase mixing.67 The intensity reduction in the temperature range of 10–50 °C is apparent in A20-10/CNC10 and A20-20/CNC5 as a consequence of the extensive matrix–CNC interactions and possibly the existence of an interphase region with reduced mobility.67
The impact of the peptidic ordering and nanocomposite morphology on the mechanical properties also was explored via tensile testing at room temperature, which is below the Tg of all PPU/CNC samples. Based on prior literature related to cellulose-containing polyurethanes20 and peptide-containing polyureas,37,41 we anticipate that the main factors that impact mechanical properties in the cellulose-reinforced peptidic polyureas are: (1) peptide weight fraction, which modulates the degree of soft segment ordering, (2) CNC content that directly relates to mechanical reinforcement via matrix–filler and/or filler-filler interactions, and (3) the microphase-separated morphology driven by not only peptide secondary structure, but also matrix–CNC physical associations.
Fig. 6A and B depicts the stress-strain curves of the PPU/CNC samples and shows two distinct regimes. In the first regime, the stress is linearly proportional to the strain, suggesting elastic deformation dictated by “pseudo” hard segment ordering. The second regime reveals a yield point, which is correlated to the fracture of the hard domains and indicates the beginning of plastic deformation.19 For the control PPUs (A20-10/CNC0; A20-20/CNC0), an increase in PBLA weight fraction leads to a higher Young's modulus (59 MPa), tensile strength (4 MPa), elongation-at-break (263%), and toughness (87 MJ m−3), which demonstrates the importance of peptidic ordering and hierarchical structure on the mechanical properties. On the basis of AFM, SAXS, and DMA investigations, an increase in PBLA content induces long-range ordering, resulting in a material with high stiffness and toughness. For the A20-10/CNC composites, increased CNC content and matrix–filler interactions enhance stiffness and tensile strength (Fig. 6C). Specifically, the Young's moduli of A20-10/CNC nanocomposites increased from 9 MPa for the neat A20-10 to ∼24 MPa for the nanocomposites with 5 and 10 wt% of CNCs. A dramatic increase in Young's modulus to 95 MPa (∼10-fold higher than A20-10 control) is observed for A20-10/CNC20. This significant reinforcement effect may be due in part to the morphological shift shown in Fig. 3. A similar trend is observed for tensile strength. In contrast, a CNC loading of 5 wt% to A20-20 (Fig. 6D) results in a decrease in the Young's modulus (41 MPa), which is not observed in conventional cellulose/polyurethane nanocomposites. This reduction can be attributed to variations in the hierarchical structure: (1) the disruption of α-helical ordering (secondary structure) (Fig. 2) and 2) a lack of long-range ordering (Fig. 3 and 4). As reported in prior studies,37 densely-packed, connected fibrillar structures in PPUs yielded a significant improvement in the tensile modulus. It is important to highlight that the addition of 5 wt% CNCs to A20-10 and A20-20 shifts the microphase-separated morphology from highly-ordered nanofibrils to globular-like or short fibre structures, but results in differences in the mechanical response. The Young's modulus of A20-10 increases, but that of A20-20 decreases, when the CNC content is varied from 0 to 5 wt%. This finding supports the assertion that peptidic ordering in addition to a hierarchically-ordered morphology significantly influences mechanical deformation behavior. The higher CNC content (10–20 wt%) in the A20-20 series improves the stiffness (56 and 147 MPa at 10 and 20 wt%, respectively) due to: (1) the dominant influence of matrix–filler interactions over α-helical arrangement and (2) the formation of inter-connected nanofibrous morphologies (Fig. 3). To probe the prevalence of matrix–filler versus filler-filler interactions, both the Halpin-Tsai model and the percolation model were used to evaluate the tensile behavior of the PPU/CNC nanocomposites. As highlighted in Fig. S5 (ESI†), the moduli of our PPU/CNC composite systems more closely follow the Halpin–Tsai model than the percolation model, suggesting that: (1) the CNC nanofillers are homogeneously dispersed in the PPU matrix and (2) filler–filler interactions or the formation of a CNC percolating network are hindered.68–70 In both the A20-10 and A20-20 nanocomposites, CNC addition hinders extensibility (Fig. 6A and B) due to restricted chain mobility (Fig. 5B and C), which is generally driven by strong matrix–filler interactions.
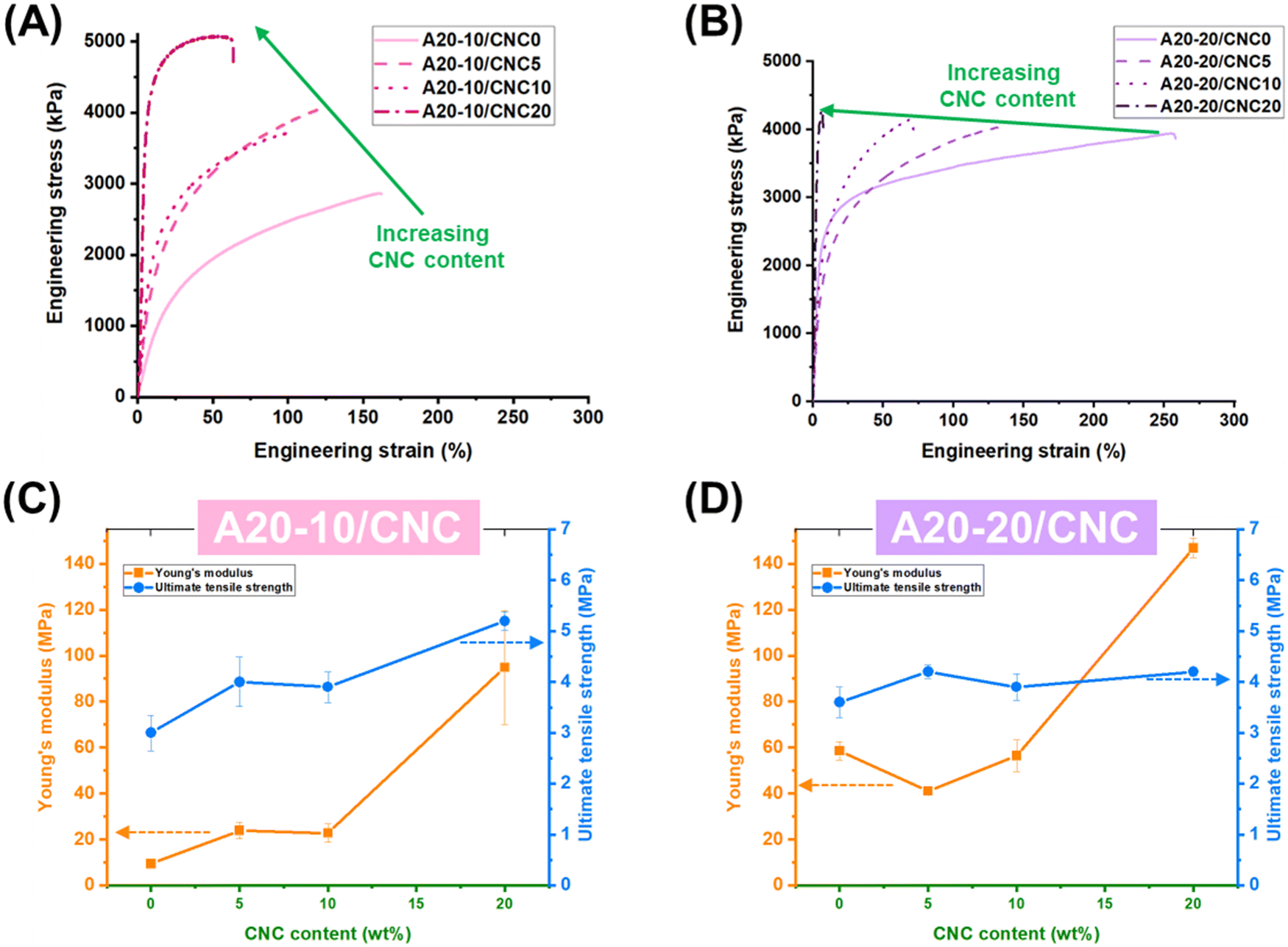 | ||
| Fig. 6 Representative stress-strain curves of (A) A20-10/CNC and (B) A20-20/CNC measured using a Zwick Roell mechanical testing machine (100 N load cell) in tension mode at room temperature under a constant strain rate of 100% of the initial gauge length/min. At least three samples with dimensions of ∼3 × 15 mm were tested for each material. Young's modulus (E) and ultimate tensile strength of (C) A20-10/CNC and (D) A20-20/CNC obtained by averaging the results of three samples, which highlight that the modulus is highly influenced by the PBLA content and CNC weight fraction compared with tensile strength. Young's modulus of A20-10/CNC nanocomposites increases from 9 MPa to 95 MPa with increasing CNC weight fractions from 0 wt% to 20 wt%, which contrasts with A20-20/CNC nanocomposites. Particularly, A20-20/CNC5 (E = 41 MPa) exhibits a lower Young's modulus compared to neat A20-20 (E = 59 MPa). Increasing the amount of CNCs in A20-20 to 20 wt% enables obtaining a higher Young's modulus up to 147 MPa. This tensile behavior reveals that a balance of peptidic ordering and PBLA–CNC interactions dictates the mechanical response of PPU/CNC nanocomposites. | ||
To understand the evolution of the phase behavior, in situ tensile deformation studies were conducted using SAXS (Fig. S6, ESI†). A20-20/CNC0 and A20-20/CNC5 samples were tested for this investigation due to their unique mechanical behavior compared with conventional polyurethane/CNC nanocomposites; the storage modulus and Young's modulus of A20-20 decrease upon the incorporation of CNCs. Fig. S6A and B (ESI†) represents that the 2D SAXS patterns for both A20-20/CNC0 and A20-20/CNC5 samples become more ellipsoidal, and the radii along the machine direction become smaller during elongation, indicating the occurrence of film deformation. In detail, during the deformation of A20-20/CNC0, the lamellar thickness remains relatively constant (Fig. S3, ESI†). However, the long spacing parallel to the stretching direction increases and the long spacing normal to the machine direction decreases (Fig. S3, ESI†), indicating that the interstitial amorphous chains between “pseudo” hard domains are stretched during this strain regime.71 Due to difficulty in fitting the weak scattering peak for A20-20/CNC5, azimuthal profiles were used to identify structural variations as a function of deformation. Fig. S6C (ESI†) reveals a four-point scattering pattern for the control A20-20/CNC0 at a strain of 100%, which is indicative of the orientation of pseudo hard domains or rod-like structures with respect to the stretching direction.72–74 However, upon the incorporation of 5 wt% CNCs into A20-20 (Fig. S6D, ESI†), the four-point scattering weakens, denoting that the orientation of pseudo hard domains is hindered. This limited rotation can be a result of disrupted peptidic ordering and a rod-like morphology via PBLA-CNC interactions. Overall, the mechanical testing results reveal that a balance of secondary structure, PPU-CNC interactions, and microphase-separated morphology define the mechanical response and deformation behavior of PPU/CNC composite materials.
Experimental
Materials
Tetrahydrofuran (THF, Optima grade) and anhydrous N,N-dimethylacetamide (DMAc, anhydrous, 99.8%)) were purchased from Fisher Scientific. While anhydrous DMAc was used as-received, THF was purified using a solvent purification system (Vacuum Atmosphere Company). Reagents, including β-benzyl-L-aspartate (BLA), triphosgene, 1,6-hexamethylene diisocyanate (HDI), dibutyltin dilaurate (DBTDL), and α,ω-Bis(3-aminopropyl)poly(dimethylsiloxane) (PDMS, 2500 g mol−1), were obtained from Sigma-Aldrich. PDMS was dried at 95 °C under vacuum for 18 hours prior to use to remove any residual water. BLA N-carboxyanhydride (NCA), poly(β-benzyl-L-aspartate)-b-poly(dimethylsiloxane)-b-poly(β-benzyl-L-aspartate) (PBLA-b-PDMS-b-PBLA), and non-chain extended PBLA-based polyurea hybrids were synthesized via established literature procedures.11 TEMPO-cellulose nanocrystals (CNCs) with carboxyl group content of 2.0 mmol g−1 were obtained from Cellulose Lab (Canada). These CNCs are 5–20 nm in width and 140–200 nm in length.Synthesis of non-chain extended PBLA-based polyurea hybrids
As reported previously,38 non-chain extended PBLA polyureas were synthesized using PBLA-b-PDMS-b-PBLA triblocks as the soft segment, which were prepared via ring-opening polymerization of BLA-NCA using diamine-terminated PDMS as the initiator. For all samples, an isocyanate/amine ([NCO]:[NH2]) ratio of 1 was used, and the ratio of PBLA-b-PDMS-b-PBLA to PDMS was modulated to achieve the target PBLA content. Specifically, the PBLA weight fraction was calculated using the following eqn (5):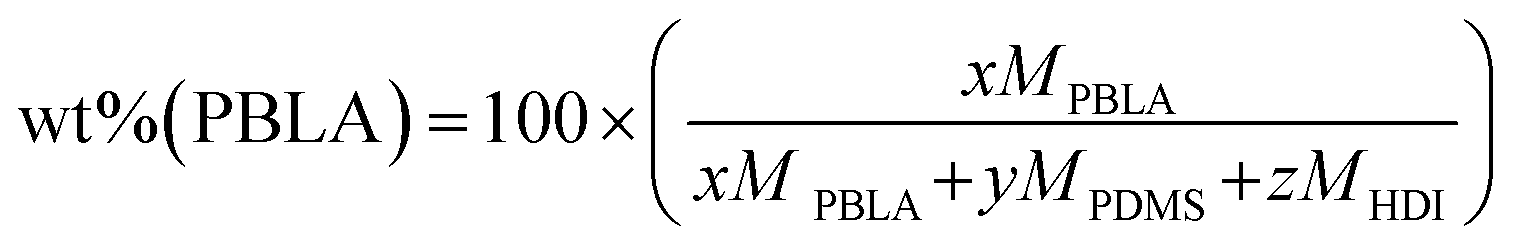 | (5) |
All PPUs were polymerized in glovebox under a nitrogen atmosphere. As an example, A20-20 was synthesized by adding HDI (0.4 g, 2.3 mmol) and 23 mL of 3:1 THF:DMAc to an oven dried 100 mL round bottom flask equipped with a magnetic stirrer and a Virgreux condenser. To this solution, the triblock (2.0 g, 0.2 mmol), predissolved in 12 mL of 3:1 THF:DMAc with 5 drops of DBTDL, was added dropwise using a dropping funnel over ∼20 minutes. This solution was heated to 60 °C and stirred for 16 hours before adding PDMS (5.3 g, 2.1 mmol) predissolved in 12 mL of 3:1 THF:DMAc. The reaction was allowed to proceed for an additional 24 hours. The reaction mixture was precipitated in deionized water and filtered. The filtrate was washed with water and methanol to purify. The purified precipitate (i.e., white rubbery solid) was dried under vacuum until a constant weight was achieved (∼2 days).
Fabrication of PPU/CNC nanocomposites
Neat PPU and PPU/CNC nanocomposite films with varying CNC content were fabricated by solvent casting. CNCs were dispersed in DMAc (6 mg mL−1) using a solvent exchange method followed by ultrasonication. PPU solutions in 1:1 THF:DMAc ratio (0.1 g mL−1) were mixed with different amounts of CNCs (5, 10, and 20 wt%), and the mixtures were stirred overnight. Each nanocomposite solution containing a PPU matrix and CNCs was poured into a Teflon mold and then vacuum dried at 60 °C for 4 days. Film thicknesses were approximately 0.2 mm. The nomenclature for all samples is as follows: An-X/CNCY, where A refers to non-chain extended PBLA-based polyurea hybrids, n is the PBLA block length fixed to 20, X is the peptide weight percentage, and Y is the CNC weight fraction in the PPUs. We observed that a PDMS-based polyurea without PBLA was precipitated upon CNC addition in the solution, suggesting that the PBLA blocks enhance the miscibility with CNCs. A series of A20-10/CNC and A20-20/CNC nanocomposites were prepared using PPUs matrices that were synthesized from the same batch. While three replicates were performed for each solvent-cast film sample during tensile testing, all the films were subjected to single tests for all other characterizations below.
Molecular weight characterization
1H nuclear magnetic resonance (1H-NMR) spectra (Fig. S7, ESI†) were recorded on Bruker 600 MHz (in CDCl3), and the block length of PBLA in the PBLA-b-PDMS-b-PBLA was calculated using end-group analysis. The number-average molecular weight (Mn) of pure PPU samples was measured by gel permeation chromatography (GPC) (a TOSOH Bioscience GPC equipped with refractive index and variable wavelength detectors) (Fig. S8, ESI†). Calibration was obtained using nine polystyrene standards (589–2110000 g mol−1) in THF at 40 °C.
Attenuated total reflection – Fourier transform infrared spectroscopy (ATR-FTIR)
The ATR-FTIR spectra were recorded using a Thermo Nicolet NEXUS 470 FTIR spectrometer with diamond crystal. All spectra of the solvent-cast films were collected averaging 128 scans with a resolution of 4 cm−1 in the range of 400–4000 cm−1.Atomic force microscopy (AFM)
AFM of the solvent-cast film was conducted on a Bruker Multimode in tapping mode using Bruker antimony doped silicon tips (320 kHz, 125 μm). 1 μm × 1 μm images were collected at a frequency of 1 Hz. All images were processed using the Bruker Nanoscope Analysis 1.5 software.Small-angle X-ray scattering (SAXS)
SAXS data were collected using a Xenocs Xeuss 2.0. X-rays were generated at 50 kV/0.6 mA at a beam wavelength of 1.542 Å (Cu Kα radiation) and a sample-to-detector distance of 1200 mm. The scattered beam was recorded on a CCD detector with a pixel resolution of 172 × 172 μm. The scattering patterns of solvent-cast films were recorded over 30 minutes of exposure time at room temperature. 2D patterns were azimuthally integrated to obtain the scattering intensity as a function of the absolute value of the scattering vector,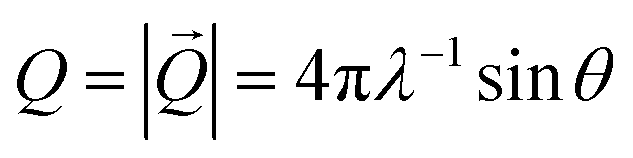 with λ and θ describing the wavelength of the X-ray beam and the half of the scattering angle, respectively. A stretcher was used to elongate samples and collect SAXS data at various strains. The data were reduced from the 2D patterns to 1D scattering profiles using SAXSGUI.
with λ and θ describing the wavelength of the X-ray beam and the half of the scattering angle, respectively. A stretcher was used to elongate samples and collect SAXS data at various strains. The data were reduced from the 2D patterns to 1D scattering profiles using SAXSGUI.
Dynamic mechanical analysis (DMA)
DMA studies were performed on a TA Instruments Q800 dynamic mechanical analyzer operating under tensile mode at a temperature range of −120 °C to 120 °C and a heating rate of 5 °C min−1. Films were cut into rectangular dimensions of approximately 15 × 3 mm for DMA measurement.Tensile testing
Tensile testing was carried out using a Zwick/Roell mechanical testing instrument equipped with a 100 N load cell. Solvent-cast films were cut into rectangles with dimensions of approximately 3 (width) mm × 15 (length) mm. All samples were elongated to failure at room temperature under a constant strain rate of 100% of the initial gauge length per minute. The reported mechanical properties were an average of three samples.Conclusions
We designed PPU/CNC nanocomposites to leverage peptide–cellulose interactions as an additional pathway to tailor phase behavior and mechanical response in peptide hybrid materials. It was demonstrated that matrix–filler (PPU-CNC) interactions influence the peptidic ordering, hydrogen bonding arrangement, microphase-separated morphology, and mechanical properties of the PPU/CNC nanocomposites.At a lower peptide content (10 wt% of PBLA, A20-10/CNC0), PPUs prefer a β-sheet conformation. In contrast, at a higher peptide content (20 wt% of PBLA, A20-20/CNC0), PPUs exhibit an increased α-helical arrangement and a larger inter-domain spacing than PPUs with 10 wt% of PBLA. The long-range, ordered structure of A20-20 leads to a high Young's modulus (59 MPa), tensile strength (4 MPa), strain-at-break (263%), and toughness (87 MJ m−3). CNC incorporation in A20-10 and A20-20 matrices not only induces PPU–CNC interactions, but also varies the self-assembled morphology of the final composite materials (from nanofibrillar to globular-like and inter-connected nanofibrous structures) as evidenced by ATR-FTIR, AFM, and SAXS. However, the mechanical response is highly dependent upon the PBLA weight fraction.
In the PPU/CNC composites, PPU–CNC interactions dominate over the influence of CNC–CNC interactions. Across all PPU/CNC nanocomposites, PPU–CNC interactions reduce the mobility of soft segments, resulting in a decrease in strain-at-break compared with neat PPUs. However, the storage modulus, Young's modulus, and ultimate tensile strength of the A20-10/CNC composite series are enhanced with varying CNC weight fractions from 5 to 20 wt%. In contrast, a low CNC loading (5 wt%) in A20-20 reduces the storage modulus and Young's modulus, while the tensile strength remains relatively constant. Upon increasing the CNC content (10–20 wt% of CNCs), the storage moduli and Young's moduli of the A20-20/CNC composite increase. These findings suggest that PPU–CNC interactions (i.e., inter-molecular hydrogen bonding between β-sheets, hard blocks, and CNCs) dictate the mechanical response of the A20-10/CNC series, whereas the tensile properties of the A20-20/CNC nanocomposites are governed by the balance of PPU–CNC interactions and hierarchically-ordered morphology. Thus, in these PPU/CNC composites, peptidic ordering, PPU–CNC interactions, and microphase-separated morphology define their mechanical behavior. This research highlights that leveraging peptide–cellulose interactions is a strategic pathway to tailor the secondary structure, hierarchical structure, and mechanical properties of nanocomposite materials. This design approach can enable new pathways toward functional and mechanically-robust peptide hybrid materials with potential applications relevant to health care technology, such as scaffolds and sutures. Future expansion of this platform targets the utilization of functionalized nanofillers to induce specific interactions between matrix components.
Author contributions
Conceptualization: D. J., L. T. J. K.; methodology: D. J., L. T. J. K.; investigation and analysis: D. J., L. B., J. K.; writing – original draft: D. J., L. B., L. T. J. K.; writing – review and editing: D. J., L. B., J. K., L. T. J. K.; supervision and funding acquisition: L. T. J. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research was provided by the National Science Foundation (NSF) PIRE: Bio-inspired Materials and Systems OISE 1844463]. AFM access was supported by the Delaware INBRE program, with grants from the NIH-NIGMS (#P20 GM103446) and the State of Delaware, and it was provided by the BioImaging Center at the University of Delaware. Access to the ATR-FTIR, SAXS, and DMA was provided by the Advanced Materials Characterization Laboratory (AMCL) at the University of Delaware. This research used the resources of the Center for Nanophase Materials Sciences (CNMS) under the CNMS user program and Spallation Neutron Source (SNS), which are DOE Office of Science User Facilities.Notes and references
- Y. Tao, J. Shao, P. Li and S. Q. Shi, Mater. Technol., 2019, 53, 71–76 CAS.
- S. S. Sheiko and A. V. Dobrynin, Macromolecules, 2019, 52, 7531 Search PubMed.
- A. M. Handorf, Y. Zhou, M. A. Halanski and W. J. Li, Organogenesis, 2015, 11, 1 CrossRef PubMed.
- Q. Xia, L. Chen, Y. Zhu, Z. Shao and M. Guo, J. Mater. Chem. B, 2019, 7, 1734 RSC.
- J. Liu, H. Zheng, P. S. P. Poh, H. G. Machens and A. F. Schilling, Int. J. Mol. Sci., 2015, 16, 15997 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14 CrossRef CAS PubMed.
- Y. Shi and Z. Chen, J. Mater. Chem. C, 2018, 6, 11817 RSC.
- V. Khoshkava and M. R. Kamal, Biomacromolecules, 2013, 14, 3155 CrossRef CAS PubMed.
- W. Meesorn, A. Shirole, D. Vanhecke, L. M. De Espinosa and C. Weder, Macromolecules, 2017, 50, 2364 CrossRef CAS.
- N. Lin and A. Dufresne, Eur. Polym. J., 2014, 59, 302–325 CrossRef CAS.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- J. W. Kim, H. Park, G. Lee, Y. R. Jeong, S. Y. Hong, K. Keum, J. Yoon and M. S. Kim, Adv. Funct. Mater., 2019, 29, 1905968 CrossRef CAS.
- K. Shanmuganathan, J. R. Capadona, S. J. Rowan and C. Weder, Prog. Polym. Sci., 2010, 35, 212 CrossRef CAS.
- P. Tyagi, R. Mathew, C. Opperman, H. Jameel, R. Gonzalez, L. Lucia, M. Hubbe and L. Pal, Langmuir, 2019, 35, 104 CrossRef CAS PubMed.
- L. Hsu, C. Weder and S. J. Rowan, J. Mater. Chem., 2011, 21, 2812 RSC.
- K. L. Dagnon, K. Shanmuganathan, C. Weder and S. J. Rowan, Macromolecules, 2012, 45, 4707 CrossRef CAS.
- C. Yadav, A. Saini and P. K. Maji, Cellulose, 2018, 25, 449 CrossRef CAS.
- J. Mendez, P. K. Annamalai, S. J. Eichhorn, R. Rusli, S. J. Rowan, E. J. Foster and C. Weder, Macromolecules, 2011, 44, 6827 CrossRef CAS.
- L. Rueda, A. Saralegui, B. Fernández, Q. Zhou, L. A. Berglund, M. A. Corcuera, I. Mondragon and A. Eceiza, Carbohydr. Polym., 2013, 92, 751 CrossRef CAS PubMed.
- A. Saralegi, L. Rueda, L. Martin, A. Arbelaiz, A. Eceiza and M. A. Corcuera, Compos. Sci. Technol., 2013, 88, 39 CrossRef CAS.
- M. L. Auad, M. A. Mosiewicki, T. Richardson, M. I. Aranguren and N. E. Marcovich, J. Appl. Polym. Sci., 2010, 115, 1215 Search PubMed.
- H. Cho, A. Shakil, A. A. Polycarpou and S. Kim, ACS Nano, 2021, 15, 19546–19558 CrossRef CAS PubMed.
- D. Yan, L. Qiu, K. J. Shea, Z. Meng and M. Xue, ACS Appl. Mater. Interfaces, 2019, 11, 39163–39170 Search PubMed.
- R. Xiong, H. S. Kim, S. Zhang, S. Kim, V. F. Korolovych, R. Ma, Y. G. Yingling, C. Lu and V. V. Tsukruk, ACS Nano, 2017, 11, 12008–12019 CrossRef CAS PubMed.
- A. Shakil, S. Kim and A. A. Polycarpou, Adv. Mater. Interfaces, 2022, 9, 2101640 CrossRef CAS.
- G. Guidetti, H. Sun, A. Ivanova, B. Marelli and B. Frka-petesic, Adv. Sustainable Syst., 2021, 5, 2000272 CrossRef CAS.
- P. Mohammadi, A. S. Aranko, C. P. Landowski, O. Ikkala, K. Jaudzems, W. Wagermaier and M. B. Linder, Sci. Adv., 2019, 5, 1 Search PubMed.
- E. Ojogbo, C. Tzoganakis and T. H. Mekonnen, ACS Sustainable Chem. Eng., 2022, 10, 8743 CrossRef CAS.
- N. D. Wanasekara and L. T. J. Korley, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 463 CrossRef CAS.
- R. A. Vaia and J. F. Maguire, Chem. Mater., 2007, 19, 2736 CrossRef CAS.
- S. Lu, Z. Wu and A. Jayaraman, J. Phys. Chem. B, 2021, 125, 2435 CrossRef CAS PubMed.
- Z. Wang, S. Kang, S. Cao, M. Krecker, V. V. Tsukruk and S. Singamaneni, MRS Bull., 2020, 45, 1017 CrossRef.
- M. K. Gupta, D. T. Wagner and M. C. Jewett, MRS Bull., 2020, 45, 999 CrossRef CAS.
- J. Y. Shu, B. Panganiban and T. Xu, Annu. Rev. Phys. Chem., 2013, 64, 631 CrossRef CAS PubMed.
- D. W. Lowik, L. Ayres, J. M. Smeenk and J. C. Van Hest, Adv. Polym. Sci., 2006, 202, 19 CrossRef.
- A. Maslovskis, I. Grillo, N. Hodson, A. F. Miller and A. Saiani, Langmuir, 2014, 30, 10471–10480 CrossRef CAS PubMed.
- J. C. Johnson, N. D. Wanasekara and L. T. J. Korley, J. Mater. Chem. B, 2014, 2, 2554 RSC.
- D. Jang, C. B. Thompson, S. Chatterjee and L. T. J. Korley, Mol. Syst. Des. Eng., 2021, 6, 1003 RSC.
- W. W. Lei, L. Y. Shi, H. Li, C. X. Li, Y. F. Diao, Y. L. Zhang and R. Ran, RSC Adv., 2017, 7, 1471 RSC.
- E. Yilgör, E. Burgaz, E. Yurtsever and I. Yilgör, Polymer, 2000, 41, 849 CrossRef.
- L. Matolyak, J. Keum and L. T. J. Korley, Biomacromolecules, 2016, 17, 3931 CrossRef CAS PubMed.
- S. A. Riou, S. L. Hsu and H. D. Stidham, Biophys. J., 1998, 75, 2451 CrossRef CAS PubMed.
- S.-W. Kuo and C.-J. Chen, Macromolecules, 2011, 44, 7315 CrossRef CAS.
- M. G. Mohamed, J.-H. Tu, S.-H. Huang, Y.-W. Chiang and S.-W. Kuo, RSC Adv., 2016, 6, 51456 RSC.
- C.-C. Tsai, Z. Gan, T. Chen and S.-W. Kuo, Macromolecules, 2018, 51, 3017 CrossRef CAS.
- A. Santamaria-Echart, L. Ugarte, A. Arbelaiz, N. Gabilondo, M. A. Corcuera and A. Eceiza, Eur. Polym. J., 2016, 76, 99 Search PubMed.
- C. Miao, D. Mauran and W. Y. Hamad, Soft Matter, 2022, 4572 RSC.
- M. C. Silva and G. G. Silva, J. Appl. Polym. Sci., 2005, 98, 336 CrossRef CAS.
- H. Bi, Z. Ren, G. Ye, H. Sun, R. Guo, X. Jia and M. Xu, Cellulose, 2020, 27, 8011 CrossRef CAS.
- P. S. De Oliveira Patricio, I. M. Pereira, N. C. F. Da Silva, E. Ayres, F. V. Pereira and R. L. Oréfice, Eur. Polym. J., 2013, 49, 3761 CrossRef.
- C. M. Brunette, S. L. Hsu and W. J. MacKnight, Macromolecules, 1982, 15, 71 CrossRef CAS.
- J. C. Johnson, N. D. Wanasekara and L. T. J. Korley, Biomacromolecules, 2012, 13, 1279 CrossRef CAS PubMed.
- L. E. Matolyak, J. K. Keum, K. M. Van De Voorde and L. T. J. Korley, Org. Biomol. Chem., 2017, 15, 7607 RSC.
- S. Pongkitwitoon, R. Hernández, J. Weksler, A. Padsalgikar, T. Choi and J. Runt, Polymer, 2009, 50, 6305 Search PubMed.
- M. Song, H. S. Xia, K. J. Yao and D. J. Hourston, Eur. Polym. J., 2005, 41, 259 CrossRef CAS.
- Q. Tian, I. Krakovský, G. Yan, L. Bai, J. Liu, G. Sun, L. Rosta, B. Chen and L. Almásy, Polymers, 2016, 8, 197 CrossRef PubMed.
- F. Nallet, R. Laversanne and D. Roux, J. Phys. II, 1993, 3, 487 CrossRef CAS.
- J. Berghausen, J. Zipfel, P. Lindner and W. Richtering, J. Phys. Chem. B, 2001, 105, 11081 CrossRef CAS.
- M. Bergström and J. S. Pedersen, J. Phys. Chem. B, 1999, 103, 8502 CrossRef.
- I. Dékány, F. Szántó and L. G. Nagy, Progress in Colloid & Polymer Science, 1978 Search PubMed.
- F. Horkay and B. Hammouda, Colloid Polym. Sci., 2008, 286, 611 CrossRef CAS.
- I. Erukhimovich and M. O. de la Cruz, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2927–3009 CrossRef.
- F. De Luca Bossa, C. Santillo, L. Verdolotti, P. Campaner, A. Minigher, L. Boggioni, S. Losio, F. Coccia, S. Iannace and G. C. Lama, Materials, 2020, 13, 211 CrossRef PubMed.
- Y. H. Zhan, R. Patel, M. Lavorgna, F. Piscitelli, A. Khan, H. S. Xia, H. Benkreira and P. Coates, Plast., Rubber Compos., 2010, 39, 400 CrossRef CAS.
- F. Cser, J. Appl. Polym. Sci., 2001, 80, 2300 CrossRef CAS.
- G. Tsagaropoulos and A. Eisenberg, Macromolecules, 1995, 28, 6067–6077 Search PubMed.
- Q. Wu, M. Henriksson, X. Liu and L. A. Berglund, Biomacromolecules, 2007, 8, 3687–3692 CrossRef CAS PubMed.
- A. Peterson, A. Y. Mehandzhiyski, L. Svenningsson, A. Ziolkowska, K. Roland, A. Lund, L. Sandblad, L. Even, G. Lo Re, I. Zozoulenko and C. Mu, Macromolecules, 2021, 54, 3507–3516 CrossRef CAS.
- K. Shanmuganathan, J. R. Capadona, S. J. Rowan and C. Weder, ACS Appl. Mater. Interfaces, 2010, 2, 165 CrossRef CAS PubMed.
- A. Redondo, N. Mortensen, K. Djeghdi, D. Jang, R. D. Ortuso, C. Weder, L. T. J. Korley, U. Steiner and I. Gunkel, ACS Appl. Mater. Interfaces, 2022, 14, 7270 CrossRef CAS PubMed.
- S. Lin, J. Liu, X. Liu and X. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10244 CrossRef CAS PubMed.
- C. R. Desper, N. S. Schneider, J. P. Jasinski and J. S. Lin, Macromolecules, 1985, 18, 2757 CrossRef.
- D. J. Blundell, G. Eeckhaut, W. Fuller, A. Mahendrasingam and C. Martin, Polymer, 2002, 43, 5197 CrossRef CAS.
- F. Yeh, B. S. Hsiao, B. B. Sauer, S. Michel and H. W. Siesler, Macromolecules, 2003, 36, 1940–1954 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ATR-FTIR, SAXS, Young's moduli according to the Halpin–Tsai model and percolation model, in situ SAXS, NMR, and GPC data. See DOI: https://doi.org/10.1039/d3tb00079f |
| This journal is © The Royal Society of Chemistry 2023 |