
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triel bonds within anion···anion complexes†
Mariusz
Michalczyk
 *a,
Wiktor
Zierkiewicz
*a,
Rafał
Wysokiński
a and
Steve
Scheiner
*b
*a,
Wiktor
Zierkiewicz
*a,
Rafał
Wysokiński
a and
Steve
Scheiner
*b
aFaculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: mariusz.michalczyk@pwr.edu.pl; wiktor.zierkiewicz@pwr.edu.pl
bDepartment of Chemistry and Biochemistry, Utah State University, Logan, Utah 84322-0300, USA. E-mail: steve.scheiner@usu.edu
First published on 1st November 2021
Abstract
The ability of two anions to interact with one another is tested in the context of pairs of TrX4− homodimers, where Tr represents any of the triel atoms B, Al, Ga, In, or Tl, and X refers to a halogen substituent F, Cl, or Br. None of these pairs engage in a stable complex in the gas phase, but the situation reverses in water where the two monomers are held together by Tr⋯X triel bonds, complemented by stabilizing interactions between X atoms. Some of these bonds are quite strong, notably those involving TrF4−, with interaction energies surpassing 30 kcal mol−1. Others are very much weaker, with scarcely exothermic binding energies. The highly repulsive electrostatic interactions are counteracted by large polarization energies.
Introduction
Interest in noncovalent bonds is exemplified by the longstanding and continuing level of scientific interest in hydrogen bonding.1,2 But noncovalent interactions are not limited only to this particular type, and the diversity and versatility of interactions other than hydrogen bonds represent an important ingredient in wide ranging and novel chemical phenomena,3 such as crystal engineering, catalysis, and biochemistry, just to name a few.4–17 The triel bond (TrB) is a new and intriguing member of this group, which has been the subject of ongoing study.18–23 Because the various Tr atoms are typically the center of a TrR3 planar trigonal molecule, they frequently have associated with them a positive region of molecular electrostatic potential (MEP) situated directly above the Tr atom. This so-called π-hole, connected with an empty Tr p-orbital, is amplified by electron-withdrawing R substituents23–31 and is able to attract a Lewis base so as to engage in a stable molecular complex.21,32–39 The resulting triel bond can be rather strong and has been shown to be important in interaction with N-heterocyclic carbenes,40 design of hydrogen storage materials,41 or play a catalytic role in organic and inorganic chemical reactions.42–44 Very recently, borane complexes have been screened for possible biomolecular recognition abilities.45Ideas concerning the σ-hole predate the earliest discussions of the π-hole.46–49 A σ-hole24,28,29 is generated along the extension of a covalent bond axis. A classic example is the σ-hole that develops on the halogen X atom of a R–X unit, which leads to the formation of a R–X⋯Nuc halogen bond with a nucleophile. The usual trigonal planar arrangement of a TrR3 molecule prevents the formation of a σ-hole within the molecular plane, which is why discussion of TrBs is usually limited to π-hole bonds. There are a few highly specialized circumstances in which a σ-hole, or something resembling it, can occur on a Tr atom,50–52 usually in either a highly strained cyclic unit or an aromatic structure where the hole does not lie opposite another atom, so does not fit the usual definition of a σ-hole. The issue of σ-hole triel bonding is thus in its infancy at this point.
The description of the magnitude of σ-hole or π-hole-based interactions are typically derived from calculation of the interaction or bond energies. Alternatively, the strength of such contacts may be estimated by the NBO or QTAIM approach and predicted in some degree by the measuring of molecular electrostatic potential extrema located at the interacting units. However, it was recently demonstrated that the strength of many noncovalent interaction types (as halogen, tetrel or pnictogen bonding) can be also quantitatively measured in terms of vibrational spectroscopy parameters obtained through the local mode analysis53–56 realized via LMODEA software.57
Another forefront area in current research is the idea that a pair of anions might be able to interact with one another in an attractive fashion. Despite the obvious strong Coulombic repulsive forces obstructing such an attraction, there are a growing number of works which document via both experimental and computational evidence that complexes can be formed between anions in certain situations.37,58–72 In the gas phase, these anion–anion complexes are usually metastable, in that their dissociation to a pair of anions is an exothermic process but one that is impeded by an energy barrier.37,58,59,61,66,71 Immersion of the system in a solvent, attenuates the electrostatic repulsions and typically allows a fully exothermic association process37,58,59,61,66,68 or a similar effect can occur if counterions are added to the system.67 Similar trends have been reported in the case of H-bonded complexes between ions of like charge.61–64,69 As an illustrative example, our own group recently showed that HgCl3− anions are stacked directly above one another in a crystal structure.67 This anion–anion stabilization is held together by a π-hole spodium bond, and facilitated by crystal packing forces and counterions. The electrostatic repulsions were counteracted by exchange, and dispersion components, and especially by polarization. Similar conclusions pertained to other model anion⋯anion (MX3−)2 dimers (M = Zn, Cd, Hg; X = Cl, Br, I)66 whose formation was exothermic when immersed in water or ethanol solvent. There still remains much to be learned as to the various forces involved in these anion-anion complexes, and what sorts of environments are needed to overcome the Coulombic repulsions.
The present work seeks to better understand this question of stabilizing anion–anion forces. There has as yet been no investigation of the ability of Tr atoms to participate in such anti-electrostatic complexes. In order to extend our understanding to triel bonds, a series of TrX4− units were examined (see Scheme 1), with regard to their possible interactions with one another within the context of homodimers. The full range of Tr atoms B, Al, Ga, In, and Tl were incorporated into these systems, as well as three different halogen X atoms F, Cl, and Br. As a particularly important aspect of this work, each TrX4− anion is tetrahedral, so the central Tr atom does not have the usual π-hole. Instead, there are four σ-holes, each along an extension of a Tr–X bond. This work thus encompasses the first study of σ-hole bonding by the triel family of atoms. A survey of the Cambridge Structural Database tells us how common such triel σ-hole anion–anion complexes might be, and provides some experimental contact to the calculations. Questions of particular interest include the sort of environment required for a pair of TrX4− anions to interact attractively with one another, and how strong might the triel bonds be. Can the interactions in fact be unambiguously attributed to triel bonds, and what might be the ancillary bonds that might emerge. How does the triel bond strength change as the central Tr atom is switched out for another, or as different X substituents are applied.
 | ||
| Scheme 1 Monomers considered in this work. | ||
Computational methods
Full unconstrained optimization of isolated anions and complexes were performed at the MP2/aug-cc-pVDZ level of theory.73–75 The aug-cc-pVDZ-PP basis set including effective core potentials was used for heavy triel atoms (In and Tl) in order to incorporate relativistic effects.76,77 The B3LYP-D3/def2TZVPP78–80 level of theory was used to more fully model the full crystal geometry (two anions and surroundings) of YORZEK81 as found in the Cambridge Structural Database (CSD, ver. 5.42) archives.82 It has been shown in the literature that this functional yield structures and cohesive energies, for a representative set of systems, including those with triel atoms.83–88 Harmonic vibrational analysis verified true minima on the potential energy surfaces with all positive frequencies. Calculations were carried out in the gas phase and several other solvents including water, 1,2-ethane-diol, acetone, and dichlorethane using the PCM (polarizable continuum model) simulation.89 The interaction energy (Eint) of each complex was evaluated as the difference in total electronic energy between the fully optimized complex and its subunits in the geometries adopted within the complex; the binding energy (Eb) takes as its reference fully optimized isomers. The basis set superposition error (BSSE) was removed via the counterpoise procedure introduced by Boys and Bernardi.90 All calculations were carried out with the aid of the Gaussian 16, Rev. C.01 code.91 The MEP (molecular electrostatic potential) was evaluated on the 0.001 a.u. electronic isodensity contour via the MultiWFN program,92,93 and visualized by VMD software.94 The QTAIM (atoms-in-molecules)95 and NCI96,97 (noncovalent index) techniques were applied so as to display and quantify molecular diagrams regarding the topological properties by means of the AIMAll98 and MultiWFN programs, respectively. Decomposition of the interaction energies in water solvent was carried out through the LMOEDA protocol99 embedded in GAMESS-US (version 2020-R2) software100 at MP2/aug-cc-pVDZ level. NBO analysis was conducted using the DFT generated wavefunction via the NBO 7.0 program.101,102Results
The salient components of the system of interest were extracted from the crystal structure and are displayed in Fig. 1. The two TlCl4− anions are disposed in such a way that a Cl atom of one anionic unit lies nearly along the extension of a Cl–Tl axis of the other, with a R(Tl⋯Cl) distance of some 3.641 Å. There is a mirror-image second interaction with the same dimensions involving the Tl of the other anion. These arrangements are closely parallel to what would be expected were there a pair of Tl⋯Cl triel bonds present, utilizing the σ-hole on each Tl atom. To probe this situation in a bit more depth, AIM analysis of the TlCl4− dimer supported the presence of these two triel bonds, with bond critical point densities of 0.007 a.u., adding also three Cl⋯Cl bond paths with a very similar density. NCI analysis buttressed this conclusion, as illustrated in Fig. S1 (ESI†). The CSD82 archives contain 6 similar crystal structures (see Fig S2 in ESI†) of TrX42− dimer geometries. Among them, four contain an Al⋯Cl linkage, one Ga⋯Cl and one Tl⋯Br interaction. The triel bond distance varies between 3.562 and 3.727 Å.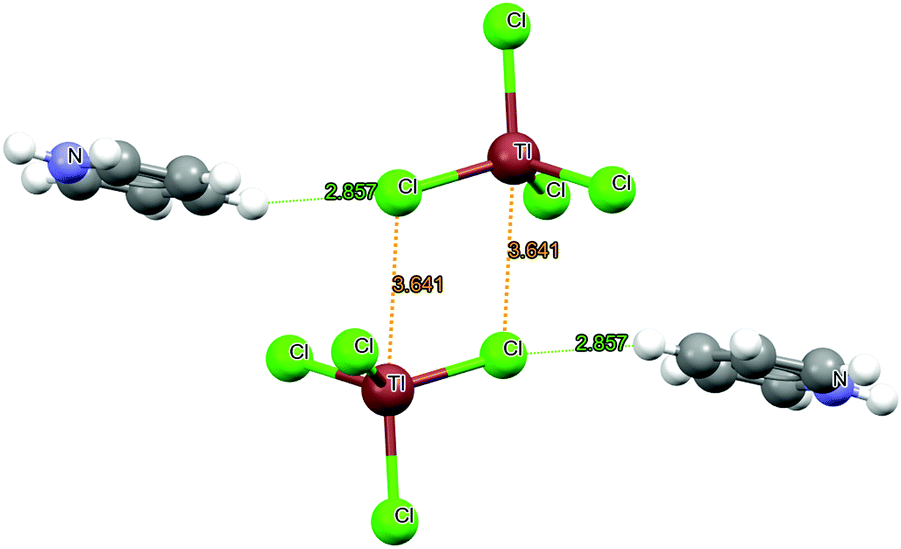 | ||
| Fig. 1 The original geometry of YORZEK crystalline solid.81 | ||
This idea that a pair of TrX4− anions can interact with one another in such a way as to form a pair of triel bonds was examined in a much broader sense, by considering five Tr atoms B, Al, Ga, In, and Tl. As halogen ligands, X = F, Cl, and Br were brought into the mix. The first step in the analysis is an examination of the molecular electrostatic potential (MEP) surrounding each anion monomer. This MEP was computed in both the gas phase and immersed in aqueous solvent. An example is displayed in Fig. 2 for TlCl4− in water. Since the moiety being probed is an anion, the MEP is of course negative throughout. But it is least negative along the extension of each Tr–X bond axis, as indicated by the red area in Fig. 2. Vs,max is defined as the maximal value of the MEP on the ρ = 0.001 a.u. isodensity surface, and is designated as a σ-hole, despite its negative sign.
 | ||
| Fig. 2 MEP of TlCl4− in water solvent. Color scale is −0.14 a.u. (blue) to −0.11 a.u. (red). | ||
The maxima on these surfaces are listed in Table 1 for the various anions in both the gas phase and in aqueous solution. There seems to be little influence of solvent on the depth of these σ-holes. There are several principal trends in the data. For the lighter triel atoms, Vs,max becomes less negative as the X ligand changes from F to Cl to Br, but this trend reverses as the heavier Tr atoms display less sensitivity to the nature of X. For any given ligand, the σ-hole becomes less negative as the Tr atom grows in size, especially noticeable for X = F. Overall, the least negative σ-hole is associated with InCl4−, and BF4− has the most negative.
| Anion | Gas | Water |
|---|---|---|
| BF4− | −116.2 | −116.3 |
| AlF4− | −85.4 | −85.6 |
| GaF4− | −78.8 | −78.5 |
| InF4− | −57.9 | −55.5 |
| TlF4− | −62.1 | −59.4 |
| BCl4− | −82.7 | −81.7 |
| AlCl4− | −80.4 | −81.0 |
| GaCl4− | −79.0 | −79.8 |
| InCl4− | −61.6 | −62.2 |
| TlCl4− | −63.2 | −63.4 |
| BBr4− | −73.7 | −72.3 |
| AlBr4− | −73.7 | −72.7 |
| GaBr4− | −72.6 | −71.5 |
| InBr4− | −63.0 | −64.7 |
| TlBr4− | −64.2 | −65.7 |
Dimers
In the gas phase, only three of the anions are able to form a homodimer that represents a minimum on the potential energy surface. Three fluorinated systems (TrF4)−2 where Tr is one of the three heaviest atoms Ga, In, and Tl, are metastable minima with positive interaction energies of 37, 15, and 22 kcal mol−1, respectively. It is only when immersed in water that all of the anions engage in stable homodimers. Most of these structures are symmetric with two equivalent (TrX4)− units and both intermolecular R(Tr⋯X) distances equal to one another. These bond lengths are contained in Table 2 where they are compared with the sum of their vdW radii in the last column. There is a good deal of variety in this comparison. With the exception of BF4−, the fluorinated dimers have very short distances, just barely longer than 2 Å. This distance is much shorter than the vdW sum, and only slightly longer than the sum of covalent radii. Those systems containing Cl and Br are quite different. Whereas the R(Tr⋯X) distance exceeds the vdW sum for the lighter Tr atoms, it is considerably shorter than this point of comparison for In and Tl.| Constituents | R(Tr⋯X) | vdW radii sum136 |
|---|---|---|
| a Dimers with R(Tr⋯X) longer than sum of corresponding vdW radii. | ||
| BF4− | 4.047/4.060a | 3.37 |
| AlF4− | 2.026 | 3.71 (cov: 1.78) |
| GaF4− | 2.093 | 3.78 (cov: 1.79) |
| InF4− | 2.187 | 3.89 (cov: 1.99) |
| TlF4− | 2.259 | 3.93 (cov: 2.02) |
| BCl4− | 4.481/4.500a | 3.73 |
| AlCl4− | 4.414/4.421a | 4.07 |
| GaCl4− | 4.350/4.377a | 4.14 |
| InCl4− | 3.033/3.036 | 4.25 |
| TlCl4− | 3.249/3.234 | 4.29 |
| BBr4− | 4.274a | 3.77 |
| AlBr4− | 4.355/4.358a | 4.11 |
| GaBr4− | 4.312/4.349a | 4.18 |
| InBr4− | 3.934/3.941 | 4.29 |
| TlBr4− | 3.722/3.718 | 4.33 |
The energetics of these complexes reflect many of the trends observed in the distances. As reported in Table 3, the interaction energies of the fluorinated dimers (again with the exception of BF4) are quite negative, commensurate with their short bond lengths that approach covalent values. This exothermicity is tempered when the monomer deformation energies are factored in, so that the binding energies in the last column are somewhat less negative, some even slightly positive after counterpoise correction. The interaction energies of the chlorinated and brominated dimers are consistent with bond lengths: these quantities are more negative for the heavier Tr atoms. Note however, that negative interaction and binding energies are achieved even for the smaller Tr atoms for which the intermolecular distance exceeded the vdW sum. With respect to comparison between different halogen ligands, the brominated dimers are more stable than X = Cl for the lighter Tr, but this pattern reverses for In and Tl. Of course, correlations between bond length and strength are not perfect103 but the former does serve as a useful indicator of the latter in most cases.
| Constituents | E int | E b |
|---|---|---|
| a Energies corrected by BSSE in parentheses. | ||
| BF4− | −0.22 (0.66)a | −0.23 (0.65) |
| AlF4− | −27.66 (−23.22) | −0.64 (3.80) |
| GaF4− | −28.79 (−21.20) | −7.24 (0.34) |
| InF4− | −39.66 (−33.65) | −22.04 (−16.03) |
| TlF4− | −34.68 (−26.57) | −20.48 (−12.37) |
| BCl4− | −1.96 (−0.77) | −1.96 (−0.77) |
| AlCl4− | −2.58 (−0.83) | −2.55 (−0.80) |
| GaCl4− | −2.87 (−0.89) | −2.83 (−0.85) |
| InCl4− | −11.31 (−5.28) | −4.41 (1.62) |
| TlCl4− | −9.08 (−3.03) | −5.80 (0.25) |
| BBr4− | −2.63 (−0.50) | −2.61 (−0.48) |
| AlBr4− | −4.43 (−0.89) | −4.34 (−0.80) |
| GaBr4− | −4.84 (−1.04) | −4.73 (−0.93) |
| InBr4− | −6.06 (−2.52) | −5.37 (−0.64) |
| TlBr4− | −7.79 (−1.69) | −6.47 (−0.37) |
Nature of bonding
AIM analysis of these various dimers leads to some intriguing findings. As may be seen in Fig. S3 (ESI†), the molecular diagrams suggest not only Tr⋯X bond paths, but also X⋯X paths between atoms on different subunits. In fact, in several of these dimers, particularly the weak ones, it is only the latter sort of bond path that appears. More specifically, AIM shows only X⋯X bond paths in the dimers where Tr = B, combinations of Al or Ga with Cl and Br, and In combined with Br. In short, there are triel bond paths only for the more strongly bound dimers, with interaction energy greater than 6 kcal mol−1. Two of these molecular diagrams are presented in Fig. 3 for (GaCl4−)2 and (InCl4−)2 as an illustration of the different sorts of noncovalent interactions. | ||
| Fig. 3 AIM molecular diagrams of (a) (GaCl4−)2 and (b) (InCl4−)2. Numbers on bond paths refer to the density at the bond critical point. | ||
 | ||
| Fig. 4 Three views of NCI diagram of (a) (GaCl4−)2 and (b) (InCl4−)2. Colors represent various type of interaction: blue – strong noncovalent interactions, green – interaction of average strength, brown – very weak interactions, red – repulsion. Black arrows point to the green regions corresponding to the attractive interaction between the triel atom and the chlorine from the other subsystem. The interactions between the chlorine atoms correspond to the orange arrows. | ||
The salient characteristics of the bond paths are collected in Table 4, for those complexes in which there is a Tr⋯X bond path present. (Of course, there are actually two of these bonds present due to the molecular symmetry). The other sort of bond path reported in Table 4 is the strongest of the X⋯X bonds, that between the two X atoms that are involved in the triel bond. According to the AIM analysis, the latter X⋯X bond cannot be ignored as some of its properties are comparable to those of the dominant Tr⋯X triel bonds. Focusing on the triel bonds, the density tends to grow as the Tr bond becomes stronger. Its Laplacian is positive, and also shows larger values for the triel bonds than for the X⋯X interactions. Coupled with the very small values of the energy density H, these bonds can be comfortably characterized as noncovalent bonds. The reader should be aware that there are instances in the literature wherein AIM interpretation of noncovalent bonding can be problematic.104,105
| Bond | ρ | ∇2ρ | H |
|---|---|---|---|
| AlF4− | |||
| Al⋯F | 0.033 | 0.258 | 0.006 |
| F⋯F | 0.039 | 0.161 | −0.005 |
| GaF4− | |||
| Ga⋯F | 0.049 | 0.245 | −0.009 |
| F⋯F | 0.032 | 0.132 | −0.004 |
| InF4− | |||
| In⋯F | 0.056 | 0.322 | −0.001 |
| F⋯F | 0.025 | 0.099 | −0.002 |
| TlF4− | |||
| Tl⋯F | 0.058 | 0.308 | −0.002 |
| InCl4− | |||
| In⋯Cl | 0.017 | 0.056 | 0.001 |
| Cl⋯Cl | 0.013 | 0.038 | 0.001 |
| TlCl4− | |||
| Tl⋯Cl | 0.014 | 0.044 | 0.001 |
| Cl⋯Cl | 0.010 | 0.029 | 0.001 |
| TlBr4− | |||
| Tl⋯Br | 0.007 | 0.021 | 0.001 |
| Br⋯Br | 0.007 | 0.019 | 0.000 |
NCI represents an alternate pictorial means of analyzing the topology of the electron density. Specific noncovalent interactions between subsystems are indicated by arrows as indicated in Fig. 4. Black arrows point to green regions corresponding to the attractive interaction between the triel atom and the Cl from the partner subsystem. Interactions between Cl atoms are indicated by the orange arrows. There are arrows of both colors present in both (GaCl4−)2 and (InCl4−)2, suggesting both systems contain noncovalent bonds of both types. A point of disagreement with AIM arises in that AIM did not suggest a triel bond between the Tr and Cl atoms in the (GaCl4−)2 complex, NCI does predict such an attraction. As a secondary issue, NCI detects more Cl⋯Cl interactions than does AIM in the case of (InCl4−)2.
NBO offers a somewhat different perspective on the binding in these complexes. The (GaCl4−)2 and (InCl4−)2 dimers again offer an illuminating view of these two viewpoints. According to AIM, the only interactions within the former structure are six bonds between Cl atoms on the two subunits, albeit only weak ones with ρBCP equal to 0.003 a.u. or less. (InCl4−)2, on the other hand, is viewed by AIM as containing a pair of In⋯Cl interactions with density 0.017 a.u., with a supplement arising from one Cl⋯Cl pair of 0.013 a.u. The results of NBO analysis of (GaCl4−)2 are presented in Fig. 5a and b in the form of the orbitals that interact with one another with appreciable values of second-order perturbation energy E(2). The primary interaction, with E(2) = 0.6 kcal mol−1, involves the lone pair of one Cl atom of the upper molecule and the antibonding σ*(GaCl) of its partner in Fig. 5a. It is the Cl atom that lies directly opposite the donor Cl lone pair which is involved in this transfer. This sort of diagram is typical of triel and related noncovalent bonds so offers evidence of a triel bond in this dimer, contrary to the AIM view. The only other interactions in this complex transfer charge from the same Cl lone pair to each of the three other σ*(GaCl) orbitals, one of which is illustrated in Fig. 5b, but E(2) for these transfers are much smaller, only 0.10–0.13 kcal mol−1. (It should be mentioned here that the symmetry of these complexes leads to a mirror image picture of the interactions where it is the lone pair of the lower molecule transferring charge to the antibonding orbitals of the upper one.).
 | ||
| Fig. 5 View of interacting NBO localized orbitals in (a) and (b) (GaCl4−)2 and (c) and (d) (InCl4−)2. | ||
Turning next to the (InCl4−)2 complex, there are much stronger inter-orbital interactions as pictured in Fig. 5c and d. As in the (InCl4−)2 case, the Cl lone pair of the upper molecule is the source of the transfer in all these pairs. The acceptor orbitals are again σ*(InCl), with the highest degree of transfer involving that Cl which lies opposite the Cl donor in Fig. 5c, with E(2) amounting to 11.2 kcal mol−1. E(2) for the other three σ*(InCl) orbitals, one of which is pictured in Fig. 5d, range between 6.5 and 7.7 kcal mol−1. This entire picture of NBO orbitals involved in these transfers in either the Ga or In systems is entirely consistent with a typical triel or other related noncovalent bond, so the NBO interpretation of these complexes, both weak and strong, would be that of a triel bond.
Another window into the nature of the bonding in these dimers is derived from electron density shifts that accompany the complexation. Maps of these shifts in three dimensions are computed by subtracting the sum of the densities of the two unperturbed monomers from that of the full dimer, with all atoms fixed in their dimer positions. The purple regions in Fig. 6 denote increases in density that arise as the two monomers interact with one another, and losses are shown in green. Due to the much closer contact within the (InCl4−)2 dimer, the shifts are of greater intensity than they are in (GaCl4−)2. In order then to compare the density shifts on a more equal footing, the contours for (GaCl4−)2 in Fig. 6a represent a density change of 0.0006 a.u. as compared to the larger 0.0015 a.u. contours in Fig. 6b.
 | ||
| Fig. 6 Electron density difference maps caused by dimerization of (a) (GaCl4−)2 and (b) (InCl4−)2. Purple and green regions respectively denote gain and loss of density. Contour shown in (a) is 0.0006 a.u. and that in (b) is 0.0015 a.u. | ||
There are fundamental differences between the patterns for the two dimers. The changes within (GaCl4−)2 indicate nearly equal polarizations of each of the three Cl atoms on each monomer that are oriented toward the partner. These polarizations from green to purple shift density away from the partner. The fourth Cl atom accrues a bit of additional density, indicated by the surrounding purple regions. There is very little charge motion that involves the Ga atoms, nor the region between them and the Cl atoms on the neighboring subunit. This pattern is consistent with the AIM diagnosis of weak interactions between neighboring Cl atoms, with no Tr atom involvement.
The shifts within (InCl4−)2 in Fig. 6b are quite different. There are both gains and losses in the intermolecular region. There is a clear loss/gain pattern along the In⋯Cl path which is characteristic of triel and other noncovalent bonds. There is also a sizable green region of density loss at the center of the system, coupled with gains along the Cl⋯Cl axis, which may be symptomatic of the Cl⋯Cl bonding interaction that is flagged by the AIM diagram. All in all, these density shift patterns are consistent with the AIM version of the bonding in these two complexes.
The individual components of decomposition of the total interaction energy of the chloride cases are collected in Table 5. The trends appear to break down into two overarching categories. The first three dimers listed are the weakest of the five. A second distinguishing feature is that there is no AIM Tr⋯Cl triel bond path in these weaker complexes, while such a path is present in the other two. The interaction energy in the three weakly bound dimers are completely dominated by polarization which accounts for 95% of the total attractive energy. While polarization is also important for the two stronger complexes, its contribution drops a bit to 74–78%, as exchange vastly raises its contribution from 4 to 35–50 kcal mol−1. On a percentage basis, this enhancement brings the exchange component up to roughly 20%, and dispersion ramps up to about 5%. It might also be noted that the electrostatic component is highly repulsive in all cases, and must be overcome by the other factors. While factors such as geometry relaxation and electron density reorganization can at times complicate the interpretation of individual components of the total interaction energy,104–107 they nonetheless offer a valuable window into the sources of stability. Similar caveats apply to the MEP since it is most apt when species are some distance apart, and interactions between potentials do not include the kinetic aspects of the interaction.
| E es | E ex | % | E rep | E pol | % | E disp | % | E int | |
|---|---|---|---|---|---|---|---|---|---|
| BCl4−⋯BCl4− | 157.5 | −3.24 | 2 | 4.60 | −156.6 | 96 | −3.01 | 2 | −0.75 |
| AlCl4−⋯AlCl4− | 147.2 | −3.52 | 2 | 5.34 | −147.4 | 95 | −4.20 | 3 | −2.58 |
| GaCl4−⋯GaCl4− | 146.5 | −4.31 | 3 | 6.29 | −146.1 | 95 | −3.45 | 2 | −1.07 |
| InCl4−⋯InCl4− | 125.6 | −49.2 | 22 | 86.6 | −161.0 | 74 | −8.50 | 4 | −6.5 |
| TlCl4−⋯TlCl4− | 133.1 | −34.6 | 18 | 59.3 | −153.7 | 78 | −9.40 | 5 | −5.3 |
Environmental effects
The forgoing data were focused on either the in vacuo situation, or when the systems are placed in the very high dielectric solvent water. These two scenarios represent two extremes on a continuum. Many environments in which these systems might find themselves, especially in a crystal, represent some intermediate situation. In order to examine how other environments might affect these systems, (TlCl4−)2 was taken as a prototype dimer, and its properties calculated in the situations listed in Table 6. When immersed in water, the optimized distance is 3.24 Å, at which point the interaction energy is −3.03 kcal mol−1. When the monomer deformations are added, the binding energy becomes slightly positive. As the solvent becomes less polar, i.e. as ε drops down to 40 and then 20, the interaction energy rises in magnitude to −10 kcal mol−1, although Eb remains slightly endothermic. For less polar solvent, in this case dichloroethane with ε = 10, the dimer is no longer able to form a stable complex. A single point calculation of the dimer in the geometry it adopts within the crystal is highly endothermic, and does not even represent a true minimum. The last row of Table 6 considers the environment not as a surrounding polarizable continuum, but rather as a pair of C5H6N+ pyridinium cations, positioned just as they are in the crystal. With these two counterions present, the interaction energy between the pair of (TlCl4−)⋯pyr+ units becomes negative, albeit only slightly so. This result suggests that the presence of the counterions are a crucial factor in the ability of the two anions to engage with one another.| Environment | ε | R(Tr⋯Cl), Å | E int | E b |
|---|---|---|---|---|
| a System is not converged. b Not optimized. | ||||
| Water | 78 | 3.234/3.249 | −3.03 | 0.25 |
| 1,2-Ethane-diol | 40 | 3.257/3.230 | −10.20 | 0.92 |
| Acetone | 20 | 3.275/3.263 | −9.20 | 1.97 |
| Dichloroethane | 10 | —a | — | — |
| Gas phase (in geometry of crystal) | 1 | 3.641b | +48.00 | — |
Gas phase (in geometry of crystal) with two counterions |
1 | 3.641b | −0.58 | — |
Summary and discussion
The Coulombic repulsion between the two anions overwhelms any attractive force in the gas phase. However, the attenuation of these repulsions by placement of the system within a dielectric continuum enables the noncovalent bonds to form, leading to stable dimers. The most strongly bound involve TrF4− dimers where Tr is Al, Ga, In, or Tl, and interaction energies are greater than 20 kcal mol−1. Binding energies which include monomer deformations are less exothermic, some even slightly positive. BF4− dimer, in contrast, is barely bound at all. Within the group of chlorinated systems InCl4− and TlCl4− are more strongly bound 3–5 (kcal mol−1) than are those of the lighter Tr atoms, less than 1 kcal mol−1. With respect to X = Br, the interaction energies of the TrBr4− dimers, range between 0.5 and 2.5 kcal mol−1.As for the sorts of noncovalent bonds that are present, there is some disagreement amongst the analyses that rest on electron density topology and the NBO treatment of interorbital transfers. AIM bond paths are not present between the Tr of one monomer and X on its partner in all cases. Triel bond paths are present only for the more strongly bound dimers, with interaction energy greater than 6 kcal mol−1. Other AIM bond paths suggest intermolecular X⋯X bonds, for most but not all of the dimers. NCI analysis indicates both Tr⋯X and X⋯X bonds are present. The NBO interorbital second-order perturbation energies are consistent with the presence of a triel bond in these dimers, whereas maps of electron density shifts are more consistent with the AIM interpretation.
The observed patterns of anion⋯anion interactions observed here are consistent with prior work involving related systems. The overall conclusion from many of these studies is that formation of a complex may be possible in the gas phase, but the dimer is typically only metastable with respect to dissociation to a pair of anions. The situation changes in solution where the dimer represents a thermodynamically stable minimum. H-bonded complexes constitute the largest subset of these sorts of systems63,108–122 but the categories continue to grow. The number of halogen-bonded dimers between pairs of anions is rising,72,123–129 augmented recently by pnicogen bonds.130 Another category places a metal atom at the center of each anion,131–133 whether alkaline earth metal or the Zn family. Inert gas atoms too can involve themselves in anion⋯anion interactions as when AeX5− (Ae = Kr, Xe) binds to a small anion.134
In terms of overall stability, some of these complexes are fairly tightly bound. In aqueous solution the binding energy of CN− with the various MCl3− anions, where M is a group 2A atom ranges up to 20 kcal mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 131 and lies in a similar range for 2B atoms Zn, Cd, or Hg.132 Pnicogen bonds between anions are even stronger, more than 20 kcal mol−1 for the ZCl4− series, with Z = P, As or Sb.130 Interaction energies are quite a bit smaller, less than 2 kcal mol−1, when any of several small anions engages in an aerogen bond with AeX5−.134 Of particular relevance to the systems examined here, when TrCl4− interacts with CN− in water, the interaction energy approaches 50 kcal mol−1.135
131 and lies in a similar range for 2B atoms Zn, Cd, or Hg.132 Pnicogen bonds between anions are even stronger, more than 20 kcal mol−1 for the ZCl4− series, with Z = P, As or Sb.130 Interaction energies are quite a bit smaller, less than 2 kcal mol−1, when any of several small anions engages in an aerogen bond with AeX5−.134 Of particular relevance to the systems examined here, when TrCl4− interacts with CN− in water, the interaction energy approaches 50 kcal mol−1.135
Turning away from highly compact anions like CN− anion, to a pair of MX3− units where M is a member of the Zn family, interaction energies lie in the 2–8 kcal mol−1 range, again in water.66 These quantities are similar to those computed here for the TrX4− anions, with the exceptions of some of the more strongly bound TrF4− homodimers. With respect to the electrostatic component of these dimerizations, Ees is large and positive, highly repulsive, much like the aforementioned ZnX3− spodium-bonded dimers.66
Evidence of anion⋯anion interactions based on the σ-hole which is considered in the current work may stimulate further studies of new supramolecular dianionic synthons of similar architecture and stabilization scheme. It is anticipated that these ideas may find application in crystal engineering or materials chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed in part by a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology and by the US National Science Foundation under Grant No. 1954310. A generous allotment of computer time from the Wroclaw Supercomputer and Networking Center is acknowledged.References
- S. Scheiner, Struct. Chem., 2019, 30, 1119–1128 CrossRef CAS.
- S. Scheiner, J. Indian Inst. Sci., 2020, 100, 61–76 CrossRef.
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- C. Zhou, Y. Tian, M. Wang, A. Rose, T. Besara, N. K. Doyle, Z. Yuan, J. C. Wang, R. Clark, Y. Hu, T. Siegrist, S. Lin and B. Ma, Angew. Chem., Int. Ed., 2017, 56, 9018–9022 CrossRef CAS PubMed.
- M. Brynda, M. Geoffroy and G. Bernardinelli, Chem. Commun., 1999, 961–962, 10.1039/a901083a.
- P. Vermeeren, T. A. Hamlin, F. M. Bickelhaupt and I. Fernandez, Chem. – Eur. J., 2021, 27, 5180–5190 CrossRef CAS PubMed.
- T. A. Hamlin, F. M. Bickelhaupt and I. Fernandez, Acc. Chem. Res., 2021, 54, 1972–1981 CrossRef CAS PubMed.
- R. M. Gomila and A. Frontera, Front. Chem., 2020, 8, 395 CrossRef CAS PubMed.
- A. Gini, M. Paraja, B. Galmes, C. Besnard, A. I. Poblador-Bahamonde, N. Sakai, A. Frontera and S. Matile, Chem. Sci., 2020, 11, 7086–7091 RSC.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- A. Frontera and A. Bauzá, Org. Biomol. Chem., 2021, 19, 6858–6864 RSC.
- J. A. Fernández Riveras, A. Frontera and A. Bauzá, Phys. Chem. Chem. Phys., 2021, 23, 17656–17662 RSC.
- M. D. N. Pina, A. Frontera and A. Bauza, ACS Chem. Biol., 2020, 15, 1942–1948 CrossRef CAS PubMed.
- M. D. Pina, A. Frontera and A. Bauz, J. Phys. Chem. Lett., 2020, 11, 8259–8263 CrossRef CAS PubMed.
- M. K. Bhattacharyya, U. Saha, D. Dutta, A. Frontera, A. K. Verma, P. Sharma and A. Das, New J. Chem., 2020, 44, 4504–4518 RSC.
- P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CrossRef CAS PubMed.
- S. J. Grabowski, ChemPhysChem, 2014, 15, 2985–2993 CrossRef CAS PubMed.
- S. J. Grabowski, J. Comput. Chem., 2018, 39, 472–480 CrossRef CAS PubMed.
- S. J. Grabowski, Struct. Chem., 2017, 28, 1163–1171 CrossRef CAS.
- S. J. Grabowski, ChemPhysChem, 2015, 16, 1470–1479 CrossRef CAS PubMed.
- S. J. Grabowski, Molecules, 2015, 20, 11297–11316 CrossRef CAS PubMed.
- S. J. Grabowski, ChemPhysChem, 2015, 16, 1470–1479 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, J. Comput. Chem., 2018, 39, 464–471 CrossRef CAS PubMed.
- A. Bauza, A. Frontera and T. J. Mooibroek, Cryst. Growth Des., 2016, 16, 5520–5524 CrossRef CAS.
- A. Bauza and A. Frontera, Phys. Chem. Chem. Phys., 2015, 17, 24748–24753 RSC.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2021, 23, 16458–16468 RSC.
- A. Bauza, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef CAS PubMed.
- A. Bauza, T. J. Mooibroek and A. Frontera, Chem. Commun., 2015, 51, 1491–1493 RSC.
- P. Politzer and J. S. Murray, Theor. Chem. Acc., 2021, 140, 7 Search PubMed.
- W. Zierkiewicz, M. Michalczyk and S. Scheiner, Molecules, 2020, 25, 635 Search PubMed.
- S. J. Grabowski, Coord. Chem. Rev., 2020, 407, 213171 CrossRef CAS.
- T. A. Ford, J. Mol. Struct., 2020, 1210, 128020 CrossRef CAS.
- M. D. Esrafili and P. Mousavian, Mol. Phys., 2018, 116, 388–398 CrossRef CAS.
- Q. Yang, Q. Li and S. Scheiner, ChemPhysChem, 2021, 22, 1461–1469 CrossRef CAS PubMed.
- R. Wysokinski, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 4818–4828 RSC.
- R. Wang, C. Luo, Q. Li and S. Scheiner, Int. J. Quantum Chem., 2021, 121, e26526 CAS.
- A. Bauza and A. Frontera, ChemPhysChem, 2016, 17, 3181–3186 CrossRef CAS PubMed.
- Z. Chi, W. Dong, Q. Li, X. Yang, S. Scheiner and S. Liu, Int. J. Quantum Chem., 2019, 119, e25867 CrossRef.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- S. Bhunya, T. Malakar, G. Ganguly and A. Paul, ACS Catal., 2016, 6, 7907–7934 CrossRef CAS.
- J. P. Mancinelli and S. M. Wilkerson-Hill, ACS Catal., 2020, 10, 11171–11176 CrossRef CAS.
- K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. C. G. da Silva, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS.
- D. B. Diaz and A. K. Yudin, Nat. Chem., 2017, 9, 731–742 CrossRef CAS PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. G. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- J. S. Murray, P. Lane and P. Politzer, Int. J. Quantum Chem., 2007, 107, 2286–2292 CrossRef CAS.
- J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033–1038 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- M. D. Esrafili and P. Mousavian, Chem. Phys. Lett., 2017, 678, 275–282 CrossRef CAS.
- L. Gao, Y. Zeng, X. Zhang and L. Meng, J. Comput. Chem., 2016, 37, 1321–1327 CrossRef CAS PubMed.
- Q. Q. Yang, B. H. Zhou, Q. Z. Li and S. Scheiner, ChemPhysChem, 2021, 22, 481–487 CrossRef CAS PubMed.
- D. Setiawan, E. Kraka and D. Cremer, J. Phys. Chem. A, 2015, 119, 1642–1656 CrossRef CAS PubMed.
- V. Oliveira and E. Kraka, J. Phys. Chem. A, 2017, 121, 9544–9556 CrossRef CAS PubMed.
- V. Oliveira, E. Kraka and D. Cremer, Inorg. Chem., 2017, 56, 488–502 CrossRef CAS PubMed.
- D. Sethio, V. Oliveira and E. Kraka, Molecules, 2018, 23, 2763 CrossRef PubMed.
- E. Kraka, W. Zou and Y. Tao, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1480 CAS.
- W. Zierkiewicz, R. Wysokinski, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 870–877 CrossRef CAS PubMed.
- R. Wysokinski, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 1119–1125 CrossRef CAS PubMed.
- O. Mo, M. M. Montero-Campillo, M. Yanez, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2020, 124, 1515–1521 CrossRef CAS PubMed.
- M. O. Miranda, D. J. R. Duarte and I. Alkorta, ChemPhysChem, 2020, 21, 1052–1059 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, J. Phys. Chem. A, 2020, 124, 2207–2214 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, Phys. Chem. Chem. Phys., 2020, 22, 11348–11353 RSC.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2015, 119, 183–194 CrossRef CAS PubMed.
- G. Blanger-Chabot and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 14270–14274 CrossRef PubMed.
- R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 13853–13861 RSC.
- R. Wysokinski, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2021, 22, 818–821 CrossRef CAS PubMed.
- A. Grabarz, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Molecules, 2021, 26, 2116 CrossRef CAS PubMed.
- A. S. R. Chesman, J. L. Hodgson, E. I. Izgorodina, A. Urbatsch, D. R. Turner, G. B. Deacon and S. R. Batten, Cryst. Growth Des., 2014, 14, 1922–1932 CrossRef CAS.
- R. Prohens, A. Portell, M. Font-Bardia, A. Bauza and A. Frontera, Chem. Commun., 2018, 54, 1841–1844 RSC.
- D. Quinonero, I. Alkorta and J. Elguero, ChemPhysChem, 2020, 21, 1597–1607 CrossRef CAS PubMed.
- D. Quinonero, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2016, 18, 27939–27950 RSC.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- C. Moller and M. S. Plesset, Phys. Rev., 1934, 46, 0618–0622 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 Search PubMed.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- B. D. James, M. B. Millikan, B. W. Skelton and A. H. White, Main Group Met. Chem., 1993, 16, 335 CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- A. N. Alexandrova, K. A. Birch and A. I. Boldyrev, J. Am. Chem. Soc., 2003, 125, 10786–10787 CrossRef CAS PubMed.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- T. R. Galeev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2011, 13, 21655–21656 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- A. P. Sergeeva, Z. A. Piazza, C. Romanescu, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 18065–18073 CrossRef CAS PubMed.
- D. Y. Zhao, Y. S. Li, M. Y. Xu, Z. M. Li, H. Zhang and L. Yu, J. Mol. Liq., 2021, 343, 117528 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- R. Bader, Atoms In Molecules. A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- P. de Silva and C. Corminboeuf, J. Chem. Theory Comput., 2014, 10, 3745–3756 CrossRef CAS PubMed.
- A. T. Keith, AIMAll (Version 14.11.23), TK Gristmill Software, Overland Park KS, USA, 2014 Search PubMed.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina, E. Guidez, T. Harville, S. Irle, J. Ivanic, K. Kowalski, S. S. Leang, H. Li, W. Li, J. J. Lutz, I. Magoulas, J. Mato, V. Mironov, H. Nakata, B. Q. Pham, P. Piecuch, D. Poole, S. R. Pruitt, A. P. Rendell, L. B. Roskop, K. Ruedenberg, T. Sattasathuchana, M. W. Schmidt, J. Shen, L. Slipchenko, M. Sosonkina, V. Sundriyal, A. Tiwari, J. L. Galvez Vallejo, B. Westheimer, M. Włoch, P. Xu, F. Zahariev and M. S. Gordon, J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- M. Kaupp, B. Metz and H. Stoll, Angew. Chem., Int. Ed., 2000, 39, 4607–4609 CrossRef CAS PubMed.
- M. Jabłoński, J. Comput. Chem., 2018, 39, 2183–2195 CrossRef PubMed.
- C. R. Wick and T. Clark, J. Mol. Model., 2018, 24, 142 CrossRef PubMed.
- C. Dieter and K. Elfi, Curr. Org. Chem., 2010, 14, 1524–1560 CrossRef.
- J. Andrés, P. W. Ayers, R. A. Boto, R. Carbó-Dorca, H. Chermette, J. Cioslowski, J. Contreras-García, D. L. Cooper, G. Frenking, C. Gatti, F. Heidar-Zadeh, L. Joubert, Á. Martín Pendás, E. Matito, I. Mayer, A. J. Misquitta, Y. Mo, J. Pilmé, P. L. A. Popelier, M. Rahm, E. Ramos-Cordoba, P. Salvador, W. H. E. Schwarz, S. Shahbazian, B. Silvi, M. Solà, K. Szalewicz, V. Tognetti, F. Weinhold and É.-L. Zins, J. Comput. Chem., 2019, 40, 2248–2283 CrossRef PubMed.
- S. R. Kass, J. Am. Chem. Soc., 2005, 127, 13098–13099 CrossRef CAS PubMed.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, ChemPhysChem, 2012, 13, 1421–1424 CrossRef CAS PubMed.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. Phys. Lett., 2013, 555, 106–109 CrossRef CAS.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2014, 119, 183–194 CrossRef PubMed.
- F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2014, 53, 11214–11217 CrossRef CAS PubMed.
- G. Frenking and G. F. Caramori, Angew. Chem., Int. Ed., 2015, 54, 2596–2599 CrossRef CAS PubMed.
- I. Alkorta, I. Mata, E. Molins and E. Espinosa, Chem. – Eur. J., 2016, 22, 9226–9234 CrossRef CAS PubMed.
- E. M. Fatila, E. B. Twum, A. Sengupta, M. Pink, J. A. Karty, K. Raghavachari and A. H. Flood, Angew. Chem., Int. Ed., 2016, 55, 14057–14062 CrossRef CAS PubMed.
- P. R. Horn, Y. Mao and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067–23079 RSC.
- F. Weinhold, Inorg. Chem., 2018, 57, 2035–2044 CrossRef CAS PubMed.
- R. Barbas, R. Prohens, A. Bauzá, A. Franconetti and A. Frontera, Chem. Commun., 2019, 55, 115–118 RSC.
- D. A. Cullen, M. G. Gardiner and N. G. White, Chem. Commun., 2019, 55, 12020–12023 RSC.
- N. G. White, CrystEngComm, 2019, 21, 4855–4858 RSC.
- W. Zhao, B. Qiao, J. Tropp, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2019, 141, 4980–4989 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, J. Phys. Chem. A, 2020, 124, 2207–2214 CrossRef CAS PubMed.
- G. Wang, Z. Chen, Z. Xu, J. Wang, Y. Yang, T. Cai, J. Shi and W. Zhu, J. Phys. Chem. B, 2016, 120, 610–620 CrossRef CAS PubMed.
- C. Wang, Y. Fu, L. Zhang, D. Danovich, S. Shaik and Y. Mo, J. Comput. Chem., 2018, 39, 481–487 CrossRef CAS PubMed.
- Z. Zhu, G. Wang, Z. Xu, Z. Chen, J. Wang, J. Shi and W. Zhu, Phys. Chem. Chem. Phys., 2019, 21, 15106–15119 RSC.
- J. M. Holthoff, E. Engelage, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 11150–11157 CrossRef CAS PubMed.
- T. Maxson, A. S. Jalilov, M. Zeller and S. V. Rosokha, Angew. Chem., Int. Ed., 2020, 59, 17197–17201 CrossRef CAS PubMed.
- K. Ghosh, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 1429–1438 RSC.
- Y. Li, L. Meng and Y. Zeng, ChemPlusChem, 2021, 86, 232–240 CrossRef CAS PubMed.
- S. Scheiner, R. Wysokiński, M. Michalczyk and W. Zierkiewicz, J. Phys. Chem. A, 2020, 124, 4998–5006 CrossRef CAS PubMed.
- W. Zierkiewicz, R. Wysokiński, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 870–877 CrossRef CAS PubMed.
- R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 1119–1125 CrossRef PubMed.
- R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2021, 22, 818–821 CrossRef PubMed.
- A. Grabarz, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Molecules, 2021, 26, 2116 CrossRef CAS PubMed.
- R. Wysokiński, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 4818–4828 RSC.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp04296c |
| This journal is © the Owner Societies 2021 |