
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe trimorphism of 3-hydroxybenzoic acid: an experimental and computational study†
Doris E.
Braun
 *
*
Institute of Pharmacy, University of Innsbruck, Innrain 52c, 6020 Innsbruck, Austria. E-mail: doris.braun@uibk.ac.at
First published on 19th March 2021
Abstract
Following the computational prediction that 3-hydroxybenzoic acid (3HBA) could exist in more than the two literature polymorphs, an experimental investigation targeting computationally generated structures was performed. The third polymorph III, solved from powder X-ray diffraction data, and the literature form II share a common hydrogen-bonded ladder motif in contrast to the carboxylic acid dimer based form I. The two metastable polymorphs (II and III) are storage stable if phase pure and monotropically related to form I. Calorimetric measurements and (lattice) energy minimisations revealed that the three polymorphs are close in energy.
Depending on its chemical nature each compound may exist or can be designed in a number of solid-state forms (anhydrate, solvate/hydrate, cocrystal, amorphous form).1,2 The increasing awareness that the solid form may have tremendous impact on materials properties (solubility, stability, etc.)3–5 and consequently can profoundly influence industrial processes and the performance of a fine chemical (drug compound) triggered the research in the field of polymorphism. A hot class of solid-state forms are cocrystals, in particular since the 2018 US Food and Drug Administration (FDA) guidance on pharmaceutical cocrystals6 has been released. Cocrystallisation involves the formation of novel materials containing at least two neutral molecules in a crystalline array.7 Hydroxybenzoic acids are routinely investigated as coformers8–10 because they have the potential to form strong hydrogen-bonding interactions to other functional groups. The acids are chemically simple, yet important molecules (e.g. ref. 11) and many have been found to exhibit polymorphism12 and to form solvates.13,14
To extend previous studies on hydroxybenzoic acids (e.g. ref. 12, 13, 15 and 16), 3-hydroxybenzoic acid (3HBA, Fig. 1) was subjected to a complementary computational17 and experimental investigation, with the aim to unravel polymorphism, preferential packing arrangements and the stability of the experimental forms. Single crystal structures of two polymorphs (form I,18 monoclinic, CSD-refcode19 BIDLOP; form II, orthorhombic, BIDLOP01 (ref. 18)/02 (ref. 20)) and their monotropic relationship, with form I being the stable form,21 have already been reported.
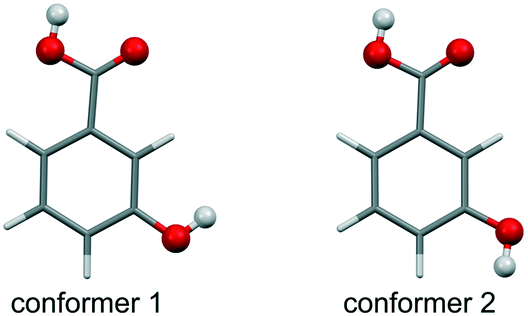 | ||
| Fig. 1 Molecular diagram of 3-hydroxybenzoic acid low-energy conformers 1 and 2, with conformer 1 being the global minimum and 3.25 kJ mol−1 more stable than conformer 2 (estimated at the PBE0/6-31G(d,p) level of theory). | ||
The Z′ = 1 & 2 crystal energy landscape (Fig. 2) was generated starting from the two low-energy conformers of the acid (Fig. 1),22–24 allowing for conformational changes in the COOH function and the m-OH proton position25–29 and estimating room temperature Helmholtz free energies in the rigid-body harmonic approximation,30–32 as detailed in the ESI.†
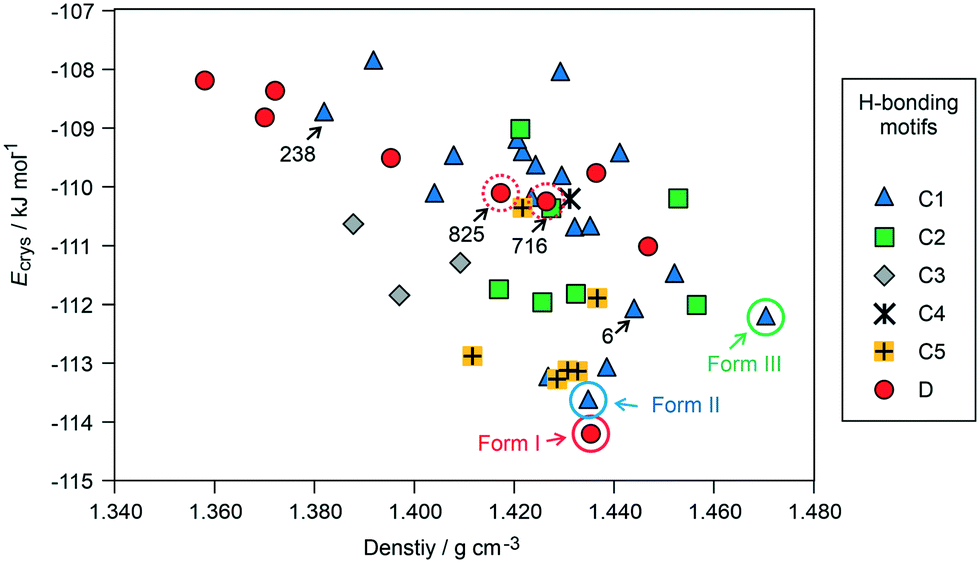 | ||
| Fig. 2 Lowest-energy crystal structures for 3HBA generated in CSP searches, with the experimental structures highlighted. The structures are classified according to hydrogen-bonding motifs (defined in Fig. 3). The experimental and selected hypothetical structures are labelled/encircled. | ||
The crystal energy landscape (Fig. 2) of 3HBA exhibits numerous structures, which are all in the energy range expected for polymorphism.33,34 The two known polymorphs were found as the global minimum structure (form I) and the second most stable structure (form II), differing only by 0.58 kJ mol−1 in energy. The form I structure contains the common carboxylic acid R(22)8 dimer motif35 D (Fig. 3), whereas form II features a chain motif C, with alternate COOH and OH-groups creating a ribbon motif, C1 hereafter, composed of two C chain motifs. Overall, all of the 45 low-energy structures can be classified into the two groups according to strong hydrogen-bonding interactions, i.e. motif C or D (Fig. 3). Only one fifth of the structures in the crystal energy landscape forms the dimer motif D. Thus, the majority of the hypothetical structures features the motif C, which is in contrast to previously computed (lattice) energy landscapes of dihydroxybenzoic acids.13,14Fig. 2 illustrates the competition of different hydrogen-bonding possibilities between carboxylic acid and hydroxyl functions, as already seen in the two literature polymorphs. A subclassification of the motif C structures revealed that five types of interactions of the C motif, two ribbon motifs (C1 and C2) and three cross-linking options (C3–5), all based on O–H⋯O (acid) hydrogen bonds, are feasible. Within three kJ mol−1 of the global minimum structure, apart from form I, exclusively motif C packings are found. The C5 structures are, based on crystal energy, very competitive with the experimentally observed motifs.
 | ||
| Fig. 3 Illustration of the hydrogen-bonding motifs observed in structures in the 3-hydroxybenzoic acid crystal energy landscape (Fig. 2). | ||
A packing similarity analysis of all low-energy structures, using the program CrystalCMP (Fig. 4),36 revealed isostructural packings for form I. The alternative form I structures (716 and 825, both Z′ = 2), four kJ mol−1 less stable than the experimental arrangement, differ in the position of the phenol hydroxyl proton and thus, the directionality of the strong O(phenol)–H⋯O(phenol) intermolecular interactions (Fig. S7 of the ESI†). The latter might indicate disorder over two sites of the polar phenol hydrogen atom as seen in related structures (e.g. phloroglucinol,37,38 gallic acid17,39). In case of form II, no isostructural packings were found in Fig. 2. The structure showing the highest resemblance with form II, structure 6, shares identical stacks of the C1 motif with the experimental form. In structure 6 adjacent stacks of C1 are related by translation symmetry, whereas in form II a glide plane is present and adjacent C1 stacks are tilted with respect to one another (Fig. S8 of the ESI†). Structure 238 is another packing arrangement that exhibits a 2-dimensional relationship with form II, not involving the C1 motif but adjacent C layers. The packing comparisons highlight that the motifs can, despite of the same connectivity of strong hydrogen-bonding interactions, pack in different orientations (e.g., planar vs. tilted). The highest resemblance between different (hypothetical) structures was found between packings exhibiting the same hydrogen-bonding motifs, as expressed by lowest PSab (similarity) values in Fig. 4 and S6 of the ESI.†
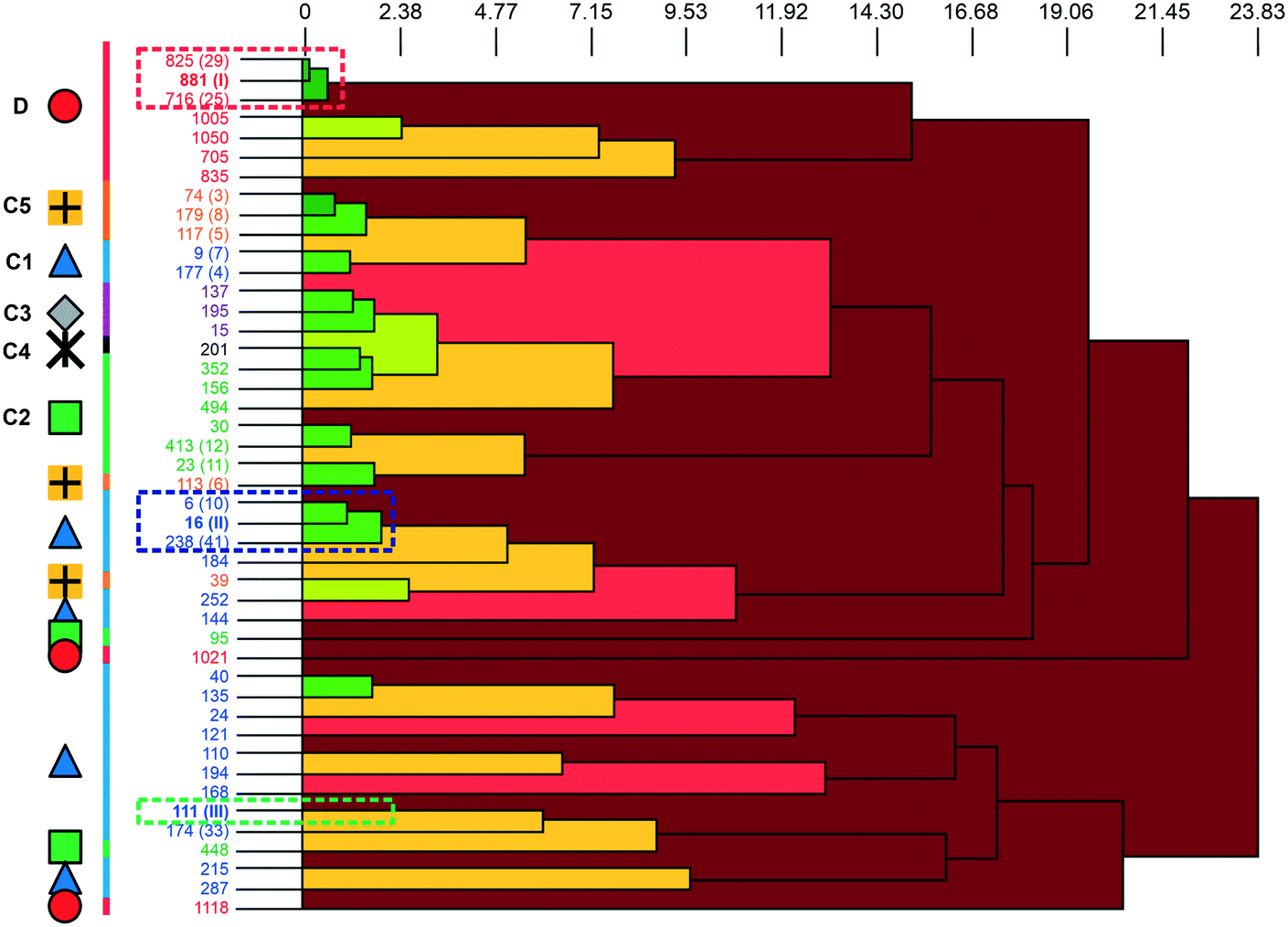 | ||
| Fig. 4 The dendrogram shows the packing similarity of experimental and computed 3HBA structures. In the dendrogram the horizontal axis corresponds to the PSab value (similarity). In this case, dark green indicates almost identical packing and dark red indicates dissimilar packing. PSab values are given in the ESI.† Symbols and colour coding of the vertical axis are according to hydrogen-bonding motifs (see Fig. 3). Numbers correspond to the CrystalPredictor rank and numbers in parenthesis to either the experimental forms (I, II, III) or to the rank in Fig. 2 (given for the lowest-energy structures). For an enlarged version see Fig. S5 of the ESI.† | ||
From the crystal energy landscape of 3HBA (Fig. 2) it can be derived that the majority of the computed structures are packed densely, with the density values spanning the range of 1.37 to 1.47 g cm−3. None of the low-energy structures features only the conformer 2. Finally, and most important, the computational search for 3HBA forms indicates that other polymorphs might be feasible, with the C5 motif and the highest-density structure being possible targets for further experimentation.
In the next step, the experimental forms I and II were reproduced according to literature procedures.21 Phase identity could be confirmed using IR spectroscopy (Fig. 5) and X-ray powder diffraction (Fig. S13, ESI†). Inspired by the computed low-energy structures, preliminarily investigations related to the successful preparation of the higher density 4-aminoquinaldine Hy1B° (ref. 40) were performed, using 1-octanol instead of water. The PXRD pattern of the obtained sample featured, in addition to the reflection positions of the known polymorphs I and II, additional peaks. The experiment was then repeated without solvent in DSC heating–cooling experiments,‡ and surprisingly the crystallisation product from the melt corresponded to a distinct solid-state form (Fig. S13, ESI†).
 | ||
| Fig. 5 Comparison of the IR spectra of the three polymorphs of 3HBA recorded at room temperature. | ||
The IR spectrum of the polymorph obtained from the quench cooled melt (form III hereafter) shows high resemblance with the IR spectrum of 3HBA form II, with the ν(O–H) and ν(C–O) vibrational regions being nearly superimposable. The latter implies that form III features a C hydrogen-bonded motif (Fig. 3). A comparison of the experimental form III PXRD pattern to the from the computed structures simulated patterns indicates a match with the rank 9 (highest density) structure in Fig. 2. Furthermore, comparing the lattice parameters of the rank 9 structure to the experimental values (form III was successfully indexed41,42 to a monoclinic Z′ = 1 P21/c cell) revealed a good agreement. To further confirm that the rank 9 structure corresponds to the experimental form a rigid-body Rietveld refinement43,44 starting from a PBE-TS optimised model of the structure (see section 1.5 of the ESI†) and a structure model derived from simulated annealing45 was carried out in tandem (Fig. S17, ESI†).§
The 3HBA molecule in the form III structure adopts, similar to the other two polymorphs, a conformation related to the global minimum conformer 1 (Fig. 1). As already indicated by the IR spectrum the acid forms the C(11)7 chain motif (Fig. 3) propagating parallel to the b crystallographic axis. Each two C(11)7 chains interlink through strong O–H⋯O (acid) hydrogen-bonding interactions forming the form II C1 ladder motif. The two polymorphs, form II and III, differ in the stacking of the C1 ladder and the orientation of adjacent stacks of ladders (Fig. 6). In form II the C1 motifs are stacked by translation symmetry along b, whereas in form III an inversion centre relates the C1 ladder motif leading to an approximate alternation of the positions of the COOH and OH functions along the c crystallographic axis (Fig. S20, ESI†). Adjacent stacks of ladder C1 motifs are roughly arranged in plane in III and tilted by approx. 133° in form II. Overall, the two structures exhibit only a 1-dimensional packing similarity, the C1 motif.
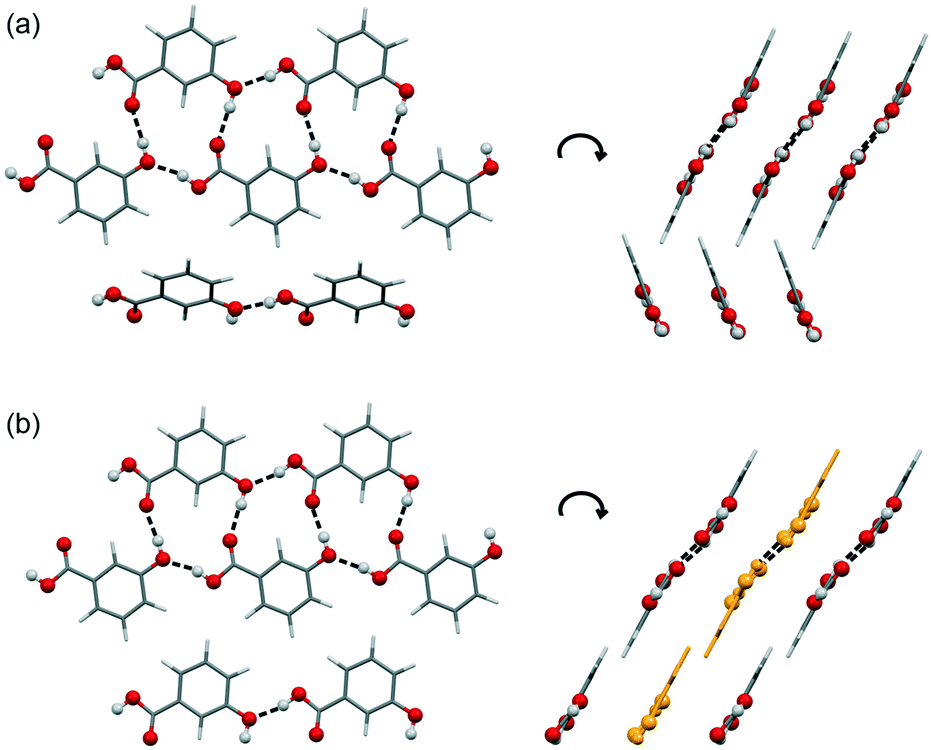 | ||
| Fig. 6 Comparison of hydrogen-bonding motifs and packings of 3HBA forms II (a) and III (b). Strong H-bonding interactions are denoted with dashed lines. Ladder motifs color-coded in yellow in (b) indicate the alternative orientation of the motif (related by inversion symmetry with respect to the neighbouring ladders). | ||
The stability of the experimental C1 and D motifs was estimated based on pairwise intermolecular interaction energy calculations (CrystalExplorer V17,46–48 section 1.4 of the ESI†). The carboxylic acid dimer (form I) was calculated to account for −74.9 kJ mol−1 in pairwise energy and the additional O–H⋯O hydrogen-bonding interaction for twice −29.0 kJ mol−1, leading to a motif D+ energy of 66.45 kJ mol−1. In contrast, the C1 ladder motif was estimated to be −71.7 kJ mol−1 and −71.4 kJ mol−1 for form II and III, respectively. Thus, the ladder motif contributes more to the lattice energy than the hydrogen-bonding interactions seen in the motif D based structure I. Nevertheless, the slightly lower contribution from strong hydrogen-bonding interactions in form I is compensated for by stronger aromatic and close contact interactions, leading to a more stable packing arrangement than the C1 structures.
The thermal stability of the form III polymorph was investigated using hot-stage microscopy (HSM) and differential scanning calorimetry (DSC). By embedding 3HBA in high-viscosity silicon oil and then producing form IIIvia quench cooling the melt it was possible to determine the form III melting point at 195.5–196.5 °C. In case of a dry preparation (without high-viscosity silicon oil) it was not possible to avoid contamination with form I during the procedure. In the latter experiment seed crystals of form I always occurred due to sublimation of the compound upon heating and induced the form III to I phase transformation (Fig. 7). Sublimation of form I starts at 150 °C and the transformation of form I to III at approx. 165 °C. The form I sublimates occur in form of stems and rectangular plates and melt at 202 °C. The HSM behaviour of 3HBA has already been described in 1954,49 one polymorph was reported to melt at 198 °C and the other (form I) at 202 °C. Based on the microscopic description provided it may be concluded that the second polymorph from 1954 corresponds to form III.
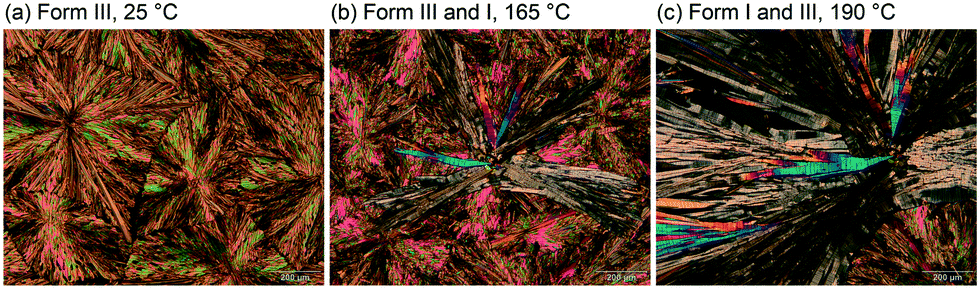 | ||
| Fig. 7 Hot-stage microscopic investigations showing the 3HBA form III to form I transformation upon heating from 25 °C to 190 °C. | ||
The three polymorphs were then subjected to DSC investigations. The DSC heating curve of form I (Fig. 8, closed DSC pan) shows only one thermal event, the melting of the polymorph at 202 °C, with a heat of fusion (ΔfusH) of 36.66 kJ mol−1. The latter values are in good agreement with the data determined by Nordström and Rasmuson50 and Perlovich et al.51 Upon cooling the melt, at approx. 190 °C, recrystallisation of form III occurs (confirmed with PXRD). Reheating form III in a closed DSC crucible allowed the determination of its melting point at 196 °C.
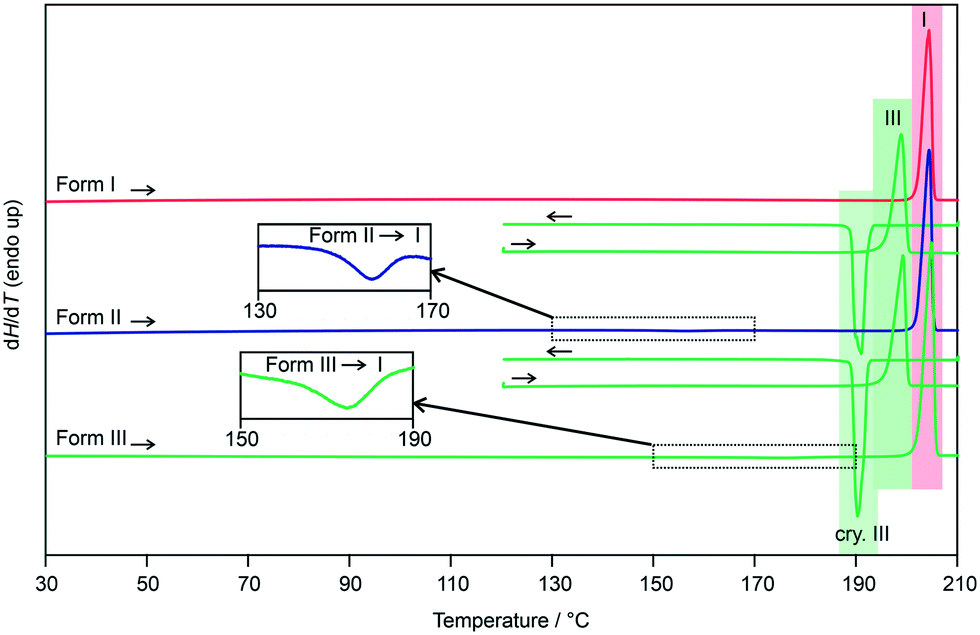 | ||
| Fig. 8 DSC heating and cooling curves of 3HBA polymorphs I–III. The insets show the form II to I and form III to I phase transformations; I, III – melting points; cry – crystallisation. | ||
Repeating the DSC experiments with either a form II or a form III sample, with form III being prepared by quench cooling the melt on the hot-bench (not embedded in silicon oil), resulted at a first glance in identical DSC curves. Upon carefully examining the heating curves, exothermic phase transformations are detectable for forms II and III. In case of form II the transition starts at approx. 150 °C and the enthalpy value corresponds to −0.53 kJ mol−1 (Table 1). The transformation product is form I, as confirmed with variable temperature IR spectroscopy (Fig. S26, ESI†) and PXRD. The small energy difference between the two polymorphs has already been reported in literature.50 According to the rules by Burger and Ramberger52 it may be assumed that the polymorphic pair II/I is monotropically related, with form I being the stable polymorph.
| Solid forma | Form I | Form II | Form III |
|---|---|---|---|
| a T fus – melting point, ΔfusH – heat of fusion, Ttrs – transition temperature, ΔtrsHX–I – heat of transition from form II or III to I, ΔsolH(DMSO) – heat of solution in DMSO, Elatt – lattice energy, DMA – DMACRYS, CO – CrystalOptimizer, Ecrys (RT) – crystal energy estimated at 25 °C. For details see section 1.2 of the ESI.† b The acid is very volatile at the melting point. Therefore the ΔfusH values, in particular for form III (produced from the melt of I), are too low and should not be used to calculate ΔtrsHIII–I. | |||
| Differential scanning calorimetry | |||
| T fus/°C | 201.9 ± 0.1 | — | 195.9 ± 0.1 |
| ΔfusH/kJ mol−1 | 36.33 ± 0.18 | — | 33.85 ± 0.09b |
| T trs/°C | — | 150 | 165 |
| ΔtrsHX–I/kJ mol−1 | — | −0.53 ± 0.02 | −1.26 ± 0.13 |
| Stability order (0 K) | a (most stable) | b | c |
| Solution calorimetry | |||
| ΔsolH(DMSO)/kJ mol−1 | −6.76 ± 0.06 | −7.39 ± 0.08 | −7.79 ± 0.09 |
| ΔtrsHX–I/kJ mol−1 | — | −0.64 ± 0.10 | −1.03 ± 0.11 |
| Stability order (0 K) | a (most stable) | b | c |
| Calculations | |||
| E latt (DMA)/kJ mol−1 | −97.65 | −96.87 | −96.90 |
| –ΔtrsUX–I/kJ mol−1 | — | −0.78 | −0.75 |
| Stability order (0 K) | a (most stable) | c | b |
| E latt (CO)/kJ mol−1 | −100.25 | −97.56 | −98.48 |
| –ΔtrsUX–I/kJ mol−1 | — | −2.69 | −1.77 |
| Stability order (0 K) | a (most stable) | c | b |
| E crys (RT)/kJ mol−1 | −114.20 | −113.61 | −112.19 |
| –ΔtrsUX–I/kJ mol−1 | — | −0.58 | −2.01 |
| Stability order (RT) | a (most stable) | b | c |
Similar to form II, the form III DSC trace shows at a temperature of approx. 165 °C an exothermic transformation to form I, with a transition enthalpy of −1.26 kJ mol−1 (Table 1). The formation of form I was confirmed using variable temperature IR spectroscopy (Fig. S27, ESI†) and PXRD. Due to the fact that form III shows a lower melting point and a lower heat of fusion than form I, in addition to the measured exothermic transformation, it can be concluded that the polymorphic pair III/I is monotropically related. The heat of fusion difference of the two polymorphs (I and III) of −2.48 kJ mol−1 indicates that the transition enthalpy is overestimated based on the latter calculation. The reason therefore can be seen in the fact that the compound is very volatile at high temperatures. A loss of approx. 3% of the compound was estimated if 3HBA is heated to its melting point, which was essential for preparing the form III sample in DSC runs.
To verify the 0 K stability order derived from DSC (Table 1), the heats of solution of the three polymorphs were recorded in DMSO (see section 2.5 of the ESI†). The heat of solution was highest for form I and lowest for form III (Table 1), resulting in an enthalpy difference of −0.64 kJ mol−1 and −1.03 kJ mol−1 for the polymorphic pairs II/I and III/I, respectively. The solution calorimetry and DSC results consistently indicate that form I is the most stable form, followed by form II and form III (Fig. S25 of the ESI†) and that the energy differences between the polymorphs are <1.3 kJ mol−1 and therefore a challenge for (lattice) energy calculations.
Based on the crystal density it can be possible to derive the 0 K stability order (‘density rule’52). Therefore, the cell volumes of the structures determined at RT were compared. Form III adopts the smallest volume (611.44 Å3) of the three polymorphs (I: 624.54 Å3; II: 624.32–626.16 Å3). Thus, 3HBA is an exception to the density rule, which may be due to the different hydrogen-bonding motifs seen in the experimental forms.
A comparison of the experimental and computational results shows that the method applied for the generation of the crystal energy landscape (Ecrys, Table 1 and Fig. 2) found the experimental structures and correctly reproduced the stability order. Form I is calculated to be the most stable structure at all stages of the process (Table S1, ESI†). In contrast, the stability order between II and III swaps depending on whether free energy contributions are added or not. The applied method reproduced the experimental results reasonably well, but for more complex and/or more flexible molecules computationally more accurate methods should to be engaged (e.g., ref. 53).
The storage stability of the metastable forms II and III was investigated at room conditions using phase pure samples and form II or III samples with form I impurities (1–2%). All samples were periodically analysed using PXRD. For pure form II and III samples no transition to any of the other polymorphs was observed within the investigation time of 15 weeks, whereas for the “impure” samples a very slow transformation to form I was already detectable after the first week (exemplarily shown in section 2.2 of the ESI† for form III spiked with form I). Thus, the metastable forms of 3HBA are only storage stable if phase pure. It was not possible to induce a form III to form II transformation in the solid-state.
Conclusions
3-Hydroxybenzoic acid is used as a coformer for modifying physico-chemical properties of (drug) molecules. Therefore, it is essential to have a thorough knowledge of its solid form landscape. As demonstrated in this study, understanding the solid-state, even for a small molecule, is still much a work in progress. This work highlights the potential of computational chemistry in finding and characterising solid-state forms. The fact that numerous structures in the crystal energy landscape are highly competitive in energy, despite showing different hydrogen-bonding motifs and distinct packing arrangements, explains why 3-hydroxybenzoic acid is prone for polymorphism. Based on the computational results it can be assumed that other polymorphs of the compound can exist. Furthermore, this carefully conducted study allowed to measure transition enthalpies for polymorphs which are close in energy, and thus, provides reference data for improving computational algorithms.The author would like to thank Prof. Price (University College London) for the use of DMACRYS, Profs. Pantelides and Adjiman (Imperial College London) for the use of CrystalPredictor and CrystalOptimizer, and Prof. Griesser (University of Innsbruck) for access to instrumentation. The computational results presented have been achieved using the HPC infrastructure LEO of the University of Innsbruck.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. von Raumer and R. Hilfiker, in Polymorphism in the Pharmaceutical Industry, ed. R. Hilfiker and M. von Raumer, Wiley-VCH, Weinheim, Germany, 2nd edn, 2019, pp. 1–30 Search PubMed.
- N. K. Duggirala, M. L. Perry, O. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- A. Y. Lee, D. Erdemir and M. S. Myerson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef CAS PubMed.
- H. G. Brittain, D. J. R. Grant and P. B. Myrdal, Drugs Pharm. Sci., 2009, 192, 436–480 CAS.
- R. Hilfiker, F. Blatter and M. von Raumer, in Polymorphism in the Pharmaceutical Industry, ed. R. Hilfiker, 2006, pp. 1–19 Search PubMed.
- Food and Drug Administration/Center for Drug Evaluation and Research (CDER), Regulatory Classification of Pharmaceutical Co-Crystals Guidance for Industry, 2018.
- C. Zhang, Y. Xiong, F. Jiao, M. Wang and H. Li, Cryst. Growth Des., 2019, 19, 1471–1478 CrossRef CAS.
- D.-K. Bucar, R. F. Henry, G. G. Z. Zhang and L. R. MacGillivray, Cryst. Growth Des., 2014, 14, 5318–5328 CrossRef CAS.
- M. Goldyn, D. Larowska, W. Nowak and E. Bartoszak-Adamska, CrystEngComm, 2019, 21, 7373–7388 RSC.
- R. Kaur, R. Gautam, S. Cherukuvada and T. N. Guru Row, IUCrJ, 2015, 2, 341–351 CrossRef CAS PubMed.
- C. Liu, C. Kuei, J. Zhu, J. Yu, L. Zhang, A. Shih, T. Mirzadegan, J. Shelton, S. Sutton, M. A. Connelly, G. Lee, N. Carruthers, J. Wu and T. W. Lovenberg, J. Pharmacol. Exp. Ther., 2012, 341, 794–801 CrossRef CAS PubMed.
- B. Sarma, P. Sanphui and A. Nangia, Cryst. Growth Des., 2010, 10, 2388–2399 CrossRef CAS.
- D. E. Braun, P. G. Karamertzanis, J.-B. Arlin, A. J. Florence, V. Kahlenberg, D. A. Tocher, U. J. Griesser and S. L. Price, Cryst. Growth Des., 2011, 11, 210–220 CrossRef CAS PubMed.
- D. E. Braun, P. G. Karamertzanis and S. L. Price, Chem. Commun., 2011, 47, 5443–5445 RSC.
- R. A. Haak and B. W. Benson, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1973, 23, 157–166 CrossRef CAS PubMed.
- F. L. Nordstroem and A. C. Rasmuson, Eur. J. Pharm. Sci., 2006, 28, 377–384 CrossRef CAS PubMed.
- D. E. Braun, R. M. Bhardwaj, A. J. Florence, D. A. Tocher and S. L. Price, Cryst. Growth Des., 2013, 13, 19–23 CrossRef CAS PubMed.
- G. V. Gridunova, N. G. Furmanova, Y. T. Struchkov, Z. I. Ezhkova, L. P. Grigoryeva and B. A. Chayanov, Kristallografiya, 1982, 27, 267–272 CAS.
- R. Taylor and P. A. Wood, Chem. Rev., 2019, 119, 9427–9477 CrossRef CAS PubMed.
- S. Clevers, F. Simon, M. Sanselme, V. Dupray and G. Coquerel, Cryst. Growth Des., 2013, 13, 3697–3704 CrossRef CAS.
- M. Svaerd and A. C. Rasmuson, Cryst. Growth Des., 2013, 13, 1140–1152 CrossRef CAS.
- P. G. Karamertzanis and C. C. Pantelides, J. Comput. Chem., 2005, 26, 304–324 CrossRef CAS PubMed.
- P. G. Karamertzanis and C. C. Pantelides, Mol. Phys., 2007, 105, 273–291 CrossRef CAS.
- M. Habgood, I. J. Sugden, A. V. Kazantsev, C. S. Adjiman and C. C. Pantelides, J. Chem. Theory Comput., 2015, 11, 1957–1969 CrossRef CAS PubMed.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478–8490 RSC.
- A. J. Stone, J. Chem. Theory Comput., 2005, 1, 1128–1132 CrossRef CAS PubMed.
- A. J. Stone, GDMA: A Program for Performing Distributed Multipole Analysis of Wave Functions Calculated Using the Gaussian Program System [2.2], University of Cambridge, Cambridge, United Kingdom, 2010 Search PubMed.
- D. S. Coombes, S. L. Price, D. J. Willock and M. Leslie, J. Phys. Chem., 1996, 100, 7352–7360 CrossRef CAS.
- A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman and C. C. Pantelides, J. Chem. Theory Comput., 2011, 7, 1998–2016 CrossRef CAS PubMed.
- A. T. Anghel, G. M. Day and S. L. Price, CrystEngComm, 2002, 4, 348–355 RSC.
- G. M. Day, S. L. Price and M. Leslie, Cryst. Growth Des., 2001, 1, 13–27 CrossRef CAS.
- G. M. Day, S. L. Price and M. Leslie, J. Phys. Chem. B, 2003, 107, 10919–10933 CrossRef CAS.
- S. L. Price and S. M. Reutzel-Edens, Drug Discovery Today, 2016, 21, 912–923 CrossRef PubMed.
- S. L. Price, D. E. Braun and S. M. Reutzel-Edens, Chem. Commun., 2016, 52, 7065–7077 RSC.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- J. Rohlicek, E. Skorepova, M. Babor and J. Cejka, J. Appl. Crystallogr., 2016, 49, 2172–2183 CrossRef CAS.
- L. H. Thomas, G. A. Craig, C. A. Morrison, A. M. Reilly and C. C. Wilson, Cryst. Growth Des., 2011, 11, 2045–2049 CrossRef CAS.
- D. E. Braun, D. A. Tocher, S. L. Price and U. J. Griesser, J. Phys. Chem. B, 2012, 116, 3961–3972 CrossRef CAS PubMed.
- A. A. Hoser, I. Sovago, A. Lanza and A. O. Madsen, Chem. Commun., 2017, 53, 925–928 RSC.
- D. E. Braun, H. Oberacher, K. Arnhard, M. Orlova and U. J. Griesser, CrystEngComm, 2016, 18, 4053–4067 RSC.
- A. J. Markvardsen, W. I. F. David, J. C. Johnson and K. Shankland, Acta Crystallogr., Sect. A: Found. Crystallogr., 2001, 57, 47–54 CrossRef CAS PubMed.
- W. I. F. David, K. Shankland, J. van de Streek, E. Pidcock, W. D. S. Motherwell and J. C. Cole, J. Appl. Crystallogr., 2006, 39, 910–915 CrossRef CAS.
- A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249–4255 CrossRef CAS PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- L. Kofler and A. Kofler, Thermo-Mikro-Methoden zur Kennzeichnung organischer Stoffe und Stoffgemische, Verlag Chemie GmbH, Weinheim/Bergstrasse, 1954 Search PubMed.
- F. L. Nordström and A. C. Rasmuson, J. Pharm. Sci., 2006, 95, 748–760 CrossRef PubMed.
- G. L. Perlovich, T. V. Volkova and A. Bauer-Brandl, J. Pharm. Sci., 2006, 95, 1448–1458 CrossRef CAS PubMed.
- A. Burger and R. Ramberger, Microchim. Acta, 1979, 2, 273–316 CrossRef CAS.
- J. Hoja, H.-Y. Ko, M. A. Neumann, R. Car, R. A. DiStasio and A. Tkatchenko, Sci. Adv., 2019, 5, eaau3338 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Low-energy conformers, computational generation of the crystal energy landscape, packing comparisons, pairwise intermolecular energy calculations, powder X-ray diffraction, Rietveld refinement of form III, solution calorimetry, semi-schematic energy temperature diagram, and variable temperature IR spectroscopy. See DOI: 10.1039/d1ce00159k |
| ‡ 3HBA form I or II was heated in a closed DSC crucible above its melting point and then cooled to RT. |
| § Crystal data of form III: C7H6O3, Mr = 138.12, monoclinic, P21/c, T = 25 °C, sample formulation: powder, wavelength: 1.54184 Å, a = 9.6308(3) Å, b = 8.2989(2) Å, c = 7.7396(2) Å, β = 98.716(3), Z = 4, density = 1.500 g cm−3, 2 theta range for data collection: 2 to 70°, background treatment: Chebyshev polynomial, No. of measured reflections: 265, refinement method: rigid-body (PBE-TS), data/parameter: 265/50, Rwp = 10.40%, Rexp = 1.76%, Rp = 8.17%. |
| This journal is © The Royal Society of Chemistry 2021 |