
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme-catalysed enantioselective oxidation of alcohols by air exploiting fast electrochemical nicotinamide cycling in electrode nanopores†
Lei
Wan
 a,
Rachel S.
Heath
b,
Bhavin
Siritanaratkul
a,
Clare F.
Megarity
a,
Adam J.
Sills
a,
Matthew P.
Thompson‡
b,
Nicholas J.
Turner
*b and
Fraser A.
Armstrong
*a
a,
Rachel S.
Heath
b,
Bhavin
Siritanaratkul
a,
Clare F.
Megarity
a,
Adam J.
Sills
a,
Matthew P.
Thompson‡
b,
Nicholas J.
Turner
*b and
Fraser A.
Armstrong
*a
aDepartment of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: fraser.armstrong@chem.ox.ac.uk
bSchool of Chemistry, University of Manchester, Manchester Institute of Biotechnology, 131 Princess Street, Manchester M1 7DN, UK. E-mail: nicholas.turner@manchester.ac.uk
First published on 30th August 2019
Abstract
Enantioselective conversion of alcohols to ketones using air as the oxidant is achieved with high rates and efficiency using an indium tin oxide (ITO) electrode in which an alcohol dehydrogenase and a photosynthetic NADPH recycling enzyme are confined within nanopores. The massive catalytic enhancement arising from nanoconfinement is exploited in an air-driven electrochemical cell, which requires no complicating control features yet allows continuous monitoring of the reaction via the current that flows between anode (ITO: organic chemistry) and cathode (Pt: O2 from air).
As Biology's hydride carriers, nicotinamide cofactors NAD(P)(H) are involved in numerous enzyme-catalysed processes: an increasing number of these are being exploited by the pharmaceutical industry,1 and a wide range of options have emerged to deal with the challenges of rapid cofactor recycling.2–15 A potentially powerful new approach uses principles that govern rates and efficiency in living cells and exploits the observation that the photosynthetic enzyme ferredoxin-NADP+-reductase (FNR) and a NADP(H)-linked oxidoreductase (E2) are taken up collectively into the nanopores of an indium tin oxide (ITO) layer that is produced by depositing commercially available nanoparticles (diameter ca. 50 nm) onto a suitable conductive support, typically titanium foil or graphite.16 In the resulting electrode, abbreviated as (FNR + E2)@ITO/support, each of the two trapped enzyme partners are concentrated at a local level and the inter-site diffusion distance of NADP(H) is decreased to a very small length scale (e.g. <0.1 μm). The nanoconfinement of two interdependent catalysts massively enhances the rate and efficiency of biocatalytic transformations occurring as small reactants and products enter and leave the pores. Importantly, the whole process can be controlled electrochemically and monitored in real time through the electrical current that flows via FNR through the rapidly-recycling NADP(H).16–18 An attractive option for oxidation reactions is to connect (FNR + E2)@ITO/support to an O2-reducing Pt cathode that is supplied with a stream of air, thus rendering aerial oxidation continuously quantifiable. Here, we describe a simple, eco-friendly system for the enantioselective oxidation of an alcohol to a ketone. The current ‘green’ options for performing such reactions on an industrial scale are either chemical, using hydrogen peroxide, O2 or N-oxyl reagents, along with a transition-metal catalyst,19–21 or enzymatic, using O2 with laccases or alcohol oxidases.22,23 The nano-confined enzyme-electrochemical strategy could compete favourably with those options once details, including scalability, have been worked out.
Fig. 1 shows the two-compartment cell in schematic form along with a cartoon depicting the nanoconfinement effect: actual assembly of the cell is shown in ESI, Fig. S1.† The (FNR + E2)@ITO/Ti electrode is located in the main compartment. During operation, a gentle stream of compressed air is introduced to the smaller compartment, separated from the main compartment by a Nafion 115 proton-exchange membrane (PEM), which houses a Pt-electrode formed from platinised carbon paper (PCP: details given in ESI†). The reaction rate is monitored continuously by recording the current that flows, the accumulated charge reporting on the progress at any point in time.
 | ||
| Fig. 1 Sketches showing the setup of the two-compartment air-driven enzyme electrochemical cell and the basis for the nanoconfinement effect underpinning the action of the (FNR + E2)@ITO/support electrode for organic synthesis. A Nafion 115 proton exchange membrane (PEM) separates the main compartment (5 mL) from the cathode compartment (1 mL). The current flowing between the anode (ITO: organic chemistry) and cathode (Pt: O2 from air) yields the reaction rate. | ||
The scope of this system is demonstrated using, as E2, a recombinant alcohol dehydrogenase from Thermoanaerobacter ethanolicus 39E (TeSADH W110A), which has already been applied in hydrogen-borrowing cascades for synthesising amines, starting from either alcohols or alkanes.4,24 The enzyme favours oxidation of the (S)-enantiomer of secondary alcohols;25–27 some of which are highly valued as precursors or building blocks for pharmaceuticals.25,28 In a TeSADH-W110A-catalysed oxidation of a racemic mixture of chiral alcohol into the corresponding ketone, the (S)-enantiomer is transformed more rapidly, thereby concentrating the (R)-enantiomer, i.e. a kinetic resolution.28 Conversely, the reverse reaction favours production of the (S)-alcohol.
Two ways of forming the (FNR + E2)@ITO/support electrode were used for the experiments. In the first case, FNR is taken up alone by dropcasting a concentrated solution onto the electrode surface and incubating for approximately 30 s before rinsing with purified water (Milli-Q) or buffer and placing in the electrochemical cell. A small aliquot of a stock solution of TeSADH W110A (hereafter referred to as ADH) is then injected into the cell solution containing all other components: this initiates electrocatalysis, which develops with time as ADH binds and its ratio relative to FNR in the ITO pores increases. In the second case, both enzymes are dropcast as a concentrated mixture (various FNR/ADH ratios being used) onto the electrode surface and incubated for approximately 3 minutes: the electrode is then rinsed repeatedly with purified water (Milli-Q) or buffer, before being used in experiments. The second method allows rapid formation of a (FNR + ADH)@ITO/support electrode free of unbound enzyme and control over the FNR/ADH ratio to which ITO was originally exposed. In all cases the loading of FNR could be determined from the amplitude of non-turnover signals obtained in the absence of NADP(H).17
All experimental details are provided in ESI.†Fig. 2 shows the electrochemical characteristics of the anode and cathode of the enzyme electrochemical cell used for air-driven organic synthesis. The left side shows cyclic voltammograms (CVs) obtained with a stationary FNR@ITO/ITO-glass electrode ([FNR] = 1.25 mM, 3 μL, 30 seconds incubation) in an aqueous cell solution containing NADPH (50 μM) and a racemic mixture (10 mM) of 4-phenyl-2-butanol (4P2B), recorded at various times after introducing ADH. The initial cyclic voltammogram (scan 0, blue trace) corresponds to the quasi-reversible electrochemistry of NADPH. Upon introducing ADH to the cell solution (final concentration after initial stirring = 0.13 μM) the oxidation current increases steadily as ADH enters the ITO nanopores to form catalytically proficient nanozones with FNR.16 The right side of Fig. 2 shows catalytic reduction (negative current) of O2 in air, achieved at an electrochemically deposited PCP cathode. A useful driving potential of 0.7–0.8 V is clearly available for the aerial oxidation of alcohol (ΔE in Fig. 2).
 | ||
| Fig. 2 Left. Cyclic voltammograms (CVs) for NADP+/NADPH interconversion coupled to the ADH-catalysed oxidation of 4-phenyl-2-butanol recorded at an FNR@ITO/ITO-glass electrode (1.0 cm2, [FNR] = 1.25 mM, 3 μL, 30 seconds incubation); cycle numbers are indicated, the ‘0’ cycle being recorded before introducing alcohol dehydrogenase. Right. A cyclic voltammogram for O2 reduction at a platinised carbon paper (PCP) electrode (2.4 cm2). The CVs were performed at scan rates of 1 mV s−1 for the FNR@ITO/ITO electrode and 5 mV s−1 for the PCP electrode. Experimental conditions: Cell volume = 8 ml, 50 mM TAPS buffer (pH 9.0), 25 °C, [ADH] = 0.13 μM, [NADPH] = 50 μM, [(rac)-4-phenyl-2-butanol ((rac)-4P2B)] = 10 mM. SHE = Standard Hydrogen Electrode, RHE = Reversible Hydrogen Electrode (corrected for pH). The air flow rate for PCP-catalysed O2 reduction was set at 30 scc min−1. | ||
Next, experiments were performed under hydrodynamic control to compare the system's overall activity and response towards product inhibition at different pH values. In each case, a (FNR + ADH)@ITO/PGE rotating disc electrode was prepared by dropcasting a concentrated mixture of FNR (705 μM) and ADH (42 μM) onto the ITO layer for approximately 3 min, taking care to ensure that the film did not dry. The electrode was then rinsed thoroughly with purified water (Milli-Q) before connecting and placing in the electrochemical cell. The upper cyclic voltammogram in Fig. 3A shows the oxidation of 5 mM (S)-4P2B at pH 9.0 carried out with 50 μM NADP(H) present in solution (a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of NADP+:NADPH was used in this case). Upon adding the product (4-phenyl-2-butanone) to a final concentration of 5 mM, the oxidation current dropped sharply and remained suppressed at a level of approximately 50% of that recorded before its introduction: continuing the cycle, a large current due to ketone reduction was observed. Fig. 3B shows the same experiment carried out at pH 8.0, the upper voltammogram again being recorded before adding ketone. Comparing the two sets of conditions, it is clear how the catalytic bias shifts in favour of alcohol oxidation as the pH is increased, and the effect of ketone as a product inhibitor is particularly marked at pH 8. The enantioselectivity for (S)- vs. (R)-4P2B was also measured under ADH-limited conditions using an electrode prepared by dropcasting a concentrated FNR-rich ([FNR]:[ADH] = 22:1) mixture: a >95% selectivity for the (S)-isomer was confirmed by comparing the S vs. R catalytic currents measured over the course of several hours. Results are shown in ESI (Fig. S2†). The enantiomeric ratio E of TeSADH W110A, defined as (kScat/KSm)/(kRcat/KRm), which reflects the enzyme's intrinsic kinetic enantioselectivity, was determined independently by solution kinetics (Fig. S3†) using fixed concentrations of NADP+ and a known concentration of ADH. The E value was calculated to be 45.8.
:1 mixture of NADP+:NADPH was used in this case). Upon adding the product (4-phenyl-2-butanone) to a final concentration of 5 mM, the oxidation current dropped sharply and remained suppressed at a level of approximately 50% of that recorded before its introduction: continuing the cycle, a large current due to ketone reduction was observed. Fig. 3B shows the same experiment carried out at pH 8.0, the upper voltammogram again being recorded before adding ketone. Comparing the two sets of conditions, it is clear how the catalytic bias shifts in favour of alcohol oxidation as the pH is increased, and the effect of ketone as a product inhibitor is particularly marked at pH 8. The enantioselectivity for (S)- vs. (R)-4P2B was also measured under ADH-limited conditions using an electrode prepared by dropcasting a concentrated FNR-rich ([FNR]:[ADH] = 22:1) mixture: a >95% selectivity for the (S)-isomer was confirmed by comparing the S vs. R catalytic currents measured over the course of several hours. Results are shown in ESI (Fig. S2†). The enantiomeric ratio E of TeSADH W110A, defined as (kScat/KSm)/(kRcat/KRm), which reflects the enzyme's intrinsic kinetic enantioselectivity, was determined independently by solution kinetics (Fig. S3†) using fixed concentrations of NADP+ and a known concentration of ADH. The E value was calculated to be 45.8.
 | ||
| Fig. 3 Cyclic voltammograms to measure the electrochemical oxidation of 5 mM (S)-4P2B achieved at a (FNR + ADH)@ITO/graphite rotating disc electrode. The experiments compare the reversibility, catalytic bias and product (ketone) inhibition obtained at pH 9 (A) and pH 8 (B). An aliquot of 4-phenyl-2-butanone was injected (giving 5 mM final concentration) in each case during the scan (shown in red and enlarged in the inset). The subsequent cycle following ketone injection is shown in blue. Experimental conditions: 50 mM TAPS + 50 mM MOPS + 50 mM CHES buffer (pH 9.0), [NADP+] = 25 μM, [NADPH] = 25 μM, 20 °C, scan rate = 3 mV s−1, electrode rotation speed = 500 rpm. | ||
Air-driven alcohol electrooxidation runs were carried out at pH 9.0 and 25 °C using a much larger electrode (7.2 cm2) to obtain products for analysis. Additional insight was also obtained by dropcasting FNR first then allowing ADH to enter the pores from dilute solution. Fig. 4 shows oxidations of (rac)-, (S)- and (R)-4P2B, monitored as a function of time after introducing ADH to the stirred electrochemical cell, a FNR@ITO/Ti electrode having been pre-prepared by dropcasting an aliquot of FNR solution ([FNR] = 425 μM, 24 μL, 30 seconds incubation) onto a freshly prepared ITO layer. Each experiment was initiated by injecting a 25 μL aliquot of ADH into the main compartment to give a final concentration of 0.44 μM,29 while the cell was previously charged with the reactant and NADP+ at the levels indicated. The alcohol concentration in these experiments was limited by its solubility in water (<20 mM). The traces contain four pieces of information: (1) the current at any particular time is proportional to an overall empirical catalytic turnover rate k′ (mole per s per unit area of electrode) via the relationship i = 2k′FA, where F is Faraday's constant and A is the surface area of the electrode. (2) The slope of the initial increase in current gives the rate at which optimised catalytic zones comprised of nanoconfined FNR and ADH become assembled in the ITO layer. (3) The total area (integral) under each trace gives the charge passed, which is related to the total chemical yield via the equation  (segments of the integral may be sampled to show the progress at any point in time). (4) The current eventually decreases, which can be due to several factors, i.e. the reaction approaches completion, becomes increasingly product-inhibited or there is a degradation in the number or performance of active nanozones. Results of experiments performed over a range of reactants and NADP+ concentrations, as analysed by coulometry, 1H NMR and GC-FID are provided in Tables 1, S1 and Fig. S4–S12.† Runs A–C were carried out with a racemic mixture, while D and E were carried out using S and R forms.
(segments of the integral may be sampled to show the progress at any point in time). (4) The current eventually decreases, which can be due to several factors, i.e. the reaction approaches completion, becomes increasingly product-inhibited or there is a degradation in the number or performance of active nanozones. Results of experiments performed over a range of reactants and NADP+ concentrations, as analysed by coulometry, 1H NMR and GC-FID are provided in Tables 1, S1 and Fig. S4–S12.† Runs A–C were carried out with a racemic mixture, while D and E were carried out using S and R forms.
 | ||
| Fig. 4 Timecourses (chronoamperograms, CA) showing oxidations of (rac)-, (S)- and (R)-4P2B in the air-driven electrochemical cell. Experimental conditions: Buffer = 0.2 M TAPS, pH = 9.0, 25 °C, [ADH] (after introduction to solution) = 0.44 μM, cell volume, 1 mL for the PCP-containing compartment and 5 mL for the main compartment, active surface of (FNR + ADH)@ITO/Ti electrode = 7.2 cm2 ([FNR] = 425 μM, 24 μL, 30 seconds incubation), air flow rate for the PCP-containing compartment = 2.6 scc min−1, [(rac)-4P2B] = 11.6 mM, [(R)-4P2B] = 9.9 mM, [(S)-4P2B] = 10.5 mM. The concentrations of NADP+ and types of enantiomers used for each plot are shown. Detailed results for these four experiments are shown in entries A, B, D, and E of Table 1. | ||
| Reactant | Reactant amounta/mM | NADP+/μM | Conversionb/% | eec (R)/% | TTNd (approx.) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Charge passed | NMR | GC | Charge passed | NMR | GC | |||||
Experimental conditions: FNR@ITO/Ti (7.2 cm2), PCP electrode (2.4 cm2), temperature 25 °C, pH = 9.0, 0.2 M TAPS, [ADH] (after being introduced to solution) = 0.44 μM, solution volume in main compartment = 5 ml, solution volume in PCP-containing compartment = 1 mL, gas flow rate in PCP-containing compartment = 2.6 scc min−1.a Concentrations of reactants determined by 1H NMR.b Defined as [product]/[reactant] × 100%. The product concentration has been normalised for volume change (details shown in Table S1†).c Defined as  .d Defined as [product]/[NADP+]. .d Defined as [product]/[NADP+]. |
||||||||||
| A |

|
11.6 | 5 | 38.7 | 39.7 | 38.3 | 85 (R) | 898 | 922 | 889 |
| B | 11.6 | 20 | 48.4 | 48.5 | 48.5 | 99 (R) | 279 | 281 | 282 | |
| C | 11.6 | 50 | 50.4 | 55.4 | 53.0 | 96 (R) | 117 | 129 | 123 | |
| D |
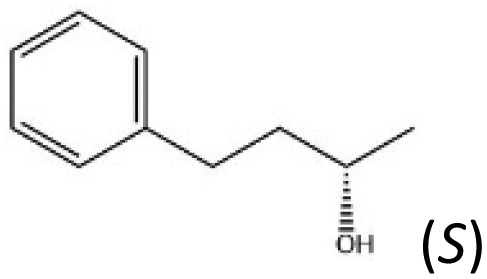
|
10.5 | 20 | 71.1 | 68.3 | 60.7 | 372 | 360 | 319 | |
| E |
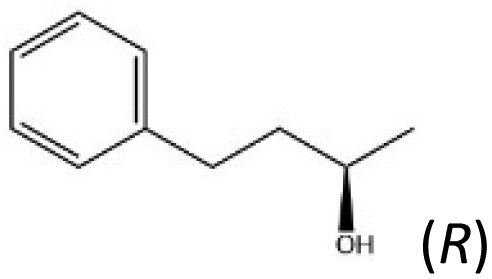
|
9.9 | 20 | 27.3 | 36.9 | 33.8 | 135 | 182 | 167 | |
Taking the experiment with ([NADP+] = 20 μM and [(rac)-4P2B)] = 11.6 (mM) (red trace in Fig. (4) as an exemplary case, the oxidation current increases for approximately 40 minutes after the injection of ADH, before peaking and eventually decreasing to a low level after 15 h. The charge passed after 15 hours was equivalent to 48.4% conversion, while product analyses by 1H NMR and GC-FID each gave values of 48.5% (Table 1). These values are encouraging considering that some losses of reactant and product occur due to crossover (leakage into the Pt-electrode compartment) and volatility (the cell not being tightly sealed). Upon lowering the concentration of NADP+ to 5 μM, the experiment with (rac)-4P2B still resulted in a peak current of 0.2 mA, i.e. approximately 70% of the value obtained using 20 μM NADP+. As expected, the total turnover number (TTN, defined as [ketone]/[NADP+], is highest for the lower NADP+ concentration, and must ultimately in these experiments be limited by the low amount of alcohol available for conversion. The (R) enantiomer is recovered at enantiomeric excess (ee) levels of 85, 99 and 96% respectively for the runs with 5, 20 and 50 μM NADP+. The results are as expected if S-4P2B is oxidised much more rapidly than R-4P2B, and the analogous experiment carried out with the favoured enantiomer (S)-4P2B, alone (Fig. 4, blue trace), revealed that a higher rate and greater yield are achieved. Unlike the solution assays which involve known, fixed concentrations of ADH, the electrochemical system involves unknown active amounts of ADH in the electrode pores. It is therefore not possible to calculate the kinetic constants needed to give a meaningful enantiomeric ratio. Accordingly, the experiment carried out with pure (R)-4P2B showed that significant conversion still occurred, albeit with a lower catalytic rate and decreased yield. Significantly, when (R)-4P2B is the sole substrate, a much longer time is taken to reach an optimum level, suggesting that the much lower natural activity for this isomer – as reflected in the enantiomeric ratio E determined from the solution kinetics, can be compensated for as a greater number of ADH molecules become incorporated into catalytic nanozones in the ITO layer that already contain FNR. Obvious comparison can be made with the experiment described above conducted using dropcasting with a fixed high FNR/ADH ratio (Fig. S2†) where a very high kinetic preference for S vs. R alcohol is maintained throughout.
To elucidate further, the factors controlling the catalytic activity, experiments were also carried out to determine the response to specific interventions such as injections of further reactant, product, cofactor and enzymes. A timecourse of the reaction monitored over 18 h is shown in Fig. 5, electrocatalysis at the FNR@ITO/Ti electrode having been initiated by introducing 0.19 μM ADH to the cell solution. When the current had decreased to approximately 25% of its starting value after 6 h, a further amount of the reactant (rac)-4P2B was introduced (the extra amounting to 2.5 mM in concentration), resulting in an immediate increase in current. The next intervention was the introduction of the ketone product (again, giving a 2.5 mM final concentration increase) which caused the current to drop immediately by a third. Subsequent interventions involved additions of TeSADH W110A, FNR, NADP+, and second injections of alcohol and ketone. Whereas addition of more alcohol dehydrogenase to augment the original 0.19 μM level resulted in an increase in current, addition of FNR caused no change and the injection of NADP+ to give a total concentration of 100 μM, doubling the original amount, increased the current by less than 10%. Subsequent additions of ketone and alcohol gave a decrease and increase in current, respectively, proportional to those observed earlier. The results show that the decrease in activity arises mainly from product inhibition, which greatly exceeds losses due to deterioration of the (FNR + ADH)@ITO/Ti electrode system or any instability of NADP+. The fact that the ultimate position of equilibrium with the air-driven experiments lies very much in favour of the ketone product rules out the possibility that the reaction is approaching its thermodynamic end point.
 | ||
| Fig. 5 Time course showing how the system responds to different interventions. Buffer = 0.2 M TAPS, pH = 9.0, 25 °C, initial [(rac)-4P2B] = 2.5 mM in the main compartment, initial [NADP+] = 50 μM, [ADH] (after introduction to solution) = 0.19 μM, air flow rate = 10.0 scc min−1, cell solution = 9 mL for main compartment and 1.2 mL for PCP-containing compartment, active surface area of (FNR + ADH)@ITO/Ti anode = 3.6 cm2 ([FNR] = 425 μM, 12 μL, 30 seconds incubation), active surface area of PCP cathode = 2.4 cm2. Argon was bubbled through the main compartment throughout. The inset shows an enlarged view of the time period during which several interventions were made. Inside this figure, each injection of (rac)-4P2B and 4-phenyl-2-butanone raises the cell concentration by 2.5 mM, injection of [NADP+] raises its concentration by 50 μM, the concentration of ADH is raised by 0.1 μM, the concentration of FNR is raised by 0.57 μM. | ||
Conclusions
The experiments and results we have described suggest a new and simple approach to conduct enzyme-catalysed selective oxidations of organic compounds – one that provides real-time control and monitoring as well as significant scope for scaling up to meet levels for special chemicals. The high catalytic rate due to the intense local concentration of two NADP(H) enzyme partners within electrode nanopores is a key factor. Although the catalysis is electrochemical in kind, the cell requires only anode and cathode, and reactor design is simplified. The reduction of O2 in air to H2O at a platinum cathode is clean and well established, and provides a large, steady overpotential to drive the efficient, near-reversible organic oxidation at the anode. No non-aqueous solvents are needed apart from the acetone that is used in rapid electrophoretic preparation of the ITO layer. Scalability should be feasible as ITO is inexpensive and non-toxic and nanoparticle powders are already produced commercially for manufacturing electronic displays. The enzymes are required in only small amounts, recombinant FNR being easy to produce and stable. The enzyme-ITO layer (<3 μm) is easily removed and replaced between runs, while the titanium and platinum electrodes are re-used continually. The system uses a low concentration of NADP+ (<20 μM, vs. 0.5 mM for typical analytical scale biotransformations). The results suggest that enantioselectivity will be optimised by ensuring that the FNR/ADH ratio is high. Obviously, the cell is easily adapted to driving reversible reactions in either direction, as all that is required to reduce ketones is to replace air by H2.18 Both TTN and yields are expected to increase greatly by implementing a two-phase system in which a much higher amount of alcohol is presented as an organic phase into which the ketone can also partition, thus also limiting product inhibition.Real-time monitoring (via the current response) allows continuous probing of the system with additions of reactants, products or enzymes to determine their effects on the reaction. One could envisage changing other factors such as temperature and pH during the timecourse of the reaction and monitoring their effects. Overall therefore, a single timecourse/experiment with interventions could quickly yield all the necessary information regarding the conditions required to optimise a particular reaction, and this capability could be implemented in batch processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported in Oxford by the Biological and Biotechnological Research Council (Follow-on Fund, Grant BB/P023797/1) and SCG Chemicals (Thailand), and in Manchester by an ERC Advanced Grant awarded to N. J. T (Grant number 742987) and the CoEBio3 Affiliates Program. Lei Wan is grateful to the China Scholarship Council for funding.Notes and references
- C. Wandrey, A. Liese and D. Kihumbu, Org. Process Res. Dev., 2000, 4, 286–290 CrossRef CAS.
- M. Thompson and N. J. Turner, ChemCatChem, 2017, 9, 3833–3866 CrossRef CAS.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- S. Montgomery, J. Mangas-Sanchez, M. Thompson, G. Aleku, B. Dominguez and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 10491–10494 CrossRef CAS PubMed.
- A. Angelastro, W. M. Dawson, L. Y. Luk and R. K. Allemann, ACS Catal., 2017, 7, 1025–1029 CrossRef CAS.
- G. T. R. Palmore, H. Bertschy, S. H. Bergens and G. M. Whitesides, J. Electroanal. Chem., 1998, 443, 155–161 CrossRef CAS.
- J. Komoschinski and E. Steckhan, Tetrahedron Lett., 1988, 29, 3299–3300 CrossRef CAS.
- I. Schröder, E. Steckhan and A. Liese, J. Electroanal. Chem., 2003, 541, 109–115 CrossRef.
- V. Höllrigl, K. Otto and A. Schmid, Adv. Synth. Catal., 2007, 349, 1337–1340 CrossRef.
- K. A. Brown, M. B. Wilker, M. Boehm, H. Hamby, G. Dukovic and P. W. King, ACS Catal., 2016, 6, 2201–2204 CrossRef CAS.
- S. Choudhury, J.-O. Baeg, N.-J. Park and R. K. Yadav, Green Chem., 2014, 16, 4389–4400 RSC.
- F. Hollmann, I. W. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- C. Zor, H. A. Reeve, J. Quinson, L. A. Thompson, T. H. Lonsdale, F. Dillon, N. Grobert and K. A. Vincent, Chem. Commun., 2017, 53, 9839–9841 RSC.
- R. Ruppert, S. Herrmann and E. Steckhan, Tetrahedron Lett., 1987, 28, 6583–6586 CrossRef CAS.
- J. Van Esch, M. Hoffmann and R. Nolte, J. Org. Chem., 1995, 60, 1599–1610 CrossRef CAS.
- C. F. Megarity, B. Siritanaratkul, R. S. Heath, L. Wan, G. Morello, S. R. Fitzpatrick, R. L. Booth, A. J. Sills, A. W. Robertson, J. H. Warner, N. J. Turner and F. A. Armstrong, Angew. Chem., Int. Ed., 2019, 58, 4948–4952 CrossRef CAS PubMed.
- B. Siritanaratkul, C. F. Megarity, T. G. Roberts, T. O. Samuels, M. Winkler, J. H. Warner, T. Happe and F. A. Armstrong, Chem. Sci., 2017, 8, 4579–4586 RSC.
- L. Wan, C. F. Megarity, B. Siritanaratkul and F. A. Armstrong, Chem. Commun., 2018, 54, 972–975 RSC.
- R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef CAS.
- G. Chen, Y. Zhou, Z. Long, X. Wang, J. Li and J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 4438–4446 CrossRef CAS PubMed.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- M. Pickl, M. Fuchs, S. M. Glueck and K. Faber, Appl. Microbiol. Biotechnol., 2015, 99, 6617–6642 CrossRef CAS PubMed.
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Green Chem., 2017, 19, 474–480 RSC.
- M. Tavanti, J. Mangas-Sanchez, S. L. Montgomery, M. P. Thompson and N. J. Turner, Org. Biomol. Chem., 2017, 15, 9790–9793 RSC.
- K. I. Ziegelmann-Fjeld, M. M. Musa, R. S. Phillips, J. G. Zeikus and C. Vieille, Protein Eng., Des. Sel., 2007, 20, 47–55 CrossRef CAS PubMed.
- D. Burdette and J. Zeikus, Biochem. J., 1994, 302, 163–170 CrossRef CAS PubMed.
- M. M. Musa, R. S. Phillips, M. Laivenieks, C. Vieille, M. Takahashi and S. M. Hamdan, Org. Biomol. Chem., 2013, 11, 2911–2915 RSC.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, J. Org. Chem., 2007, 72, 30–34 CrossRef CAS PubMed.
- F. O. Bryant, J. Wiegel and L. G. Ljungdahl, Appl. Environ. Microbiol., 1988, 54, 460–465 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc01534e |
| ‡ Current address: EnginZyme AB, Teknikringen 38 A, 114 28 Stockholm, Sweden. |
| This journal is © The Royal Society of Chemistry 2019 |