
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnesium, calcium and zinc [N2N′] heteroscorpionate complexes†‡
Mariana
Luna Barros
,
Michael G.
Cushion
,
Andrew D.
Schwarz
,
Zoë R.
Turner
 * and
Philip
Mountford
*
* and
Philip
Mountford
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: zoe.turner@chem.ox.ac.uk; philip.mounford@chem.ox.ac.uk
First published on 8th January 2019
Abstract
A new family of sterically demanding N2N′ heteroscorpionate pro-ligands (HC(tBu2pz)2SiMe2N(H)R (R = iPr, tBu, Ph, Xyl)) has been prepared via a straightforward modular synthetic route. An extensive study into the synthesis and characterisation of lithium, magnesium, calcium and zinc complexes supported by both 3,5-tBu and 3,5-Me substituted N2N′ ligand families has been conducted. Attempted deprotonation of the pro-ligands with nBuLi afforded the corresponding lithium salts Li{HC(tBu2pz)2SiMe2NR} (R = iPr (1), tBu (2), Ph (3) and Xyl (4)) but air- and thermal-sensitivity limited the yields of these potentially useful precursors; only the sterically encumbered ligand system allowed clean reactivity. Magnesium methyl complexes Mg{HC(tBu2pz)2SiMe2NR}Me (R = iPr (5) and R = Ph (6)) were prepared using an excess of the Grignard reagent MeMgCl. Magnesium butyl complexes were synthesised in good yields using the dialkyl precursor MgnBu2 to afford Mg{HC(R′2pz)2SiMe2NR}nBu (R′ = Me; R = iPr (7), tBu (8), Ad (9), Ph (10). R′ = tBu; R = iPr (11), Ph (12)). Protonolylsis reactions were used to synthesise magnesium and calcium amide complexes Mg{HC(R′2pz)2SiMe2NR}{N(SiHMe2)2} (R′ = Me; R = iPr (13), tBu (14), Ph (15). R′ = tBu; R = Ph (16)) or Mg{HC(R′2pz)2SiMe2NR}{N(SiMe3)2} (R′ = Me; R = iPr (17), tBu (18), Ph (19). R′ = tBu; R = Ph (20)), and Ca{HC(R′2pz)2SiMe2NR}{N(SiMe2)2} (L) (R′ = Me; L = thf; R = iPr (21), tBu (22), Ph (23). R′ = tBu; L = none; R = Ph (24). Zinc methyl complexes Zn{HC(R′2pz)2SiMe2NR}Me (R′ = Me; R = iPr (25), tBu (26), Ph (27). R′ = tBu; R = Ph (28)) were prepared by reaction of the N2N′ heteroscorpionate pro-ligands with ZnMe2. In preliminary studies, magnesium amide complexes 16 and 20 were evaluated as initiators for the ring-opening polymerisation (ROP) of ε-caprolactone (ε-CL) and rac-lactide (rac-LA). Although the overall polymerisation control was poor, 16 and 20 were found to be active initiators.
Introduction
Heteroscorpionates, E(Rnpz)2(X),1 are a versatile class of tridentate poly(pyrazolyl) ligand where one of the pyrazolyl groups of a classical homoscorpionate, E(Rnpz)3,2 is replaced by a different C, N, O or S donor. Chiral classes of heteroscorpionate ligands have also been developed.3 They have demonstrated a wide range of uses in homogeneous catalysis4 and supramolecular3c,5 and bioinorganic chemistry,6 as well as a supporting ligand set for studying fundamental stoichiometric transformations.7Substitution of the 3-position of the pyrazolyl ring has been demonstrated to clearly influence complex nuclearity; incorporation of sterically demanding tert-butyl substituents is an efficient way to favour mononuclear systems (Chart 1).8 This is particularly important in the case of group 2 and related metal complexes which are prone to redistribute via the Schlenk equilibrium and makes the synthesis of well-defined heteroleptic compounds LnMX (L and X = a monoanionic ligand or ligand set) challenging.
 | ||
| Chart 1 Scorpionate complexes with 3-tBu substituents. | ||
Scorpionate ligands have found particular use in the ring-opening polymerisation (ROP) of cyclic esters; Chisholm et al. first reported bulky tris(pyrazolyl)hydroborate metal alkoxide complexes M{HB(tBu2pz)3}OR (M = Mg, Zn, Ca) as active polymerisation initiators where stereoselectivity was ascribed to chain-end control.9 Otero and co-workers have extensively developed heteroscorpionate complexes based on Mg8a,d,10 and Zn,10b,11 (κ3 coordination), demonstrating moderate to good levels of heterotacticity (Ps = 0.78–0.85) or isotacticity (Pi = 0.77), and Al11c,12 (κ2 coordination). Cui and co-workers have also reported zwitterionic, achiral N2N′ heteroscorpionate complexes to obtain highly isotactic-biased polylactide (Pi up to 0.85).13
Use of alkaline earth metals is particularly desirable in homogeneous catalysis as they are earth-abundant and possess low toxicity.14 Furthermore, Hill and co-workers have recently demonstrated that the organometallic calcium complexes [(BDI)CaR]2 are capable of nucleophilic alkylation of benzene, opening new possibilities for catalytic transformations with suitable ligand design.15
Our group has previously reported on the structures and reactivity of group 2 and group 12 scorpionate complexes.16 Of particular relevance to our current contribution is our previous communication on the preparation of the N2N′ heteroscorpionate rare earth complex Sc{HC(Me2pz)2SiMe2NiPr}(CH2SiMe3)2 and its ability to polymerise ethylene.17 Now we have developed a N2N′ heteroscorpionate ligand family with sterically demanding tert-butyl substituents in the 3-position to ensure that that only ligation of a single scorpionate ligand occurs, leaving one reactive M–X bond for further controlled reactivity. Herein, we report a study into the synthesis and structure of group 1, 2 and 12 complexes supported by this ligand type.
Results and discussion
Synthesis of a new family of sterically demanding N2N′ heteroscorpionate pro-ligands
We have previously reported the modular synthesis of the N2N′ heteroscorpionate pro-ligand HC(Me2pz)2SiMe2N(H)iPr based on the 3,5-Me pyrazolyl substitution motif.17 This family has now been extended to include HC(Me2pz)2SiMe2N(H)R (R = tBu, Ad, Ph). These new pro-ligands were prepared by the same methodology of nucleophilic attack of the desired amine at electropositive silicon in HC(Me2pz)2SiMe2Cl in 85–96% yield. For R = tBu and Ad, an excess of amine was used in order to neutralise the HCl produced by generating the ammonium salt, [RNH3]Cl. For R = Ph, NEt3 is added to act as a superior Brønsted base and quench the HCl produced; the use of an excess of NH2Ph is also unfavourable due to its low volatility (b.p. 184 °C at 1 atm).The 3,5-tBu and parent 3,5-H systems were initially targeted in order to investigate how modifying the stereoelectronic properties of the pyrazolyl rings would affect the subsequent properties of metal–ligand complexes. The ligand precursor HC(tBu2pz)2SiMe2Cl was prepared by lithiation of the appropriate bis(pyrazolyl)methane followed by in situ addition of Li{HC(tBu2pz)2} to an excess of SiMe2Cl2 (Scheme 1). The specific order of addition and non-stoichiometric conditions are used to prevent the double displacement of Cl−. The solid state structure was determined by a single crystal X-ray diffraction study and is depicted in Fig. S10 of the ESI.‡
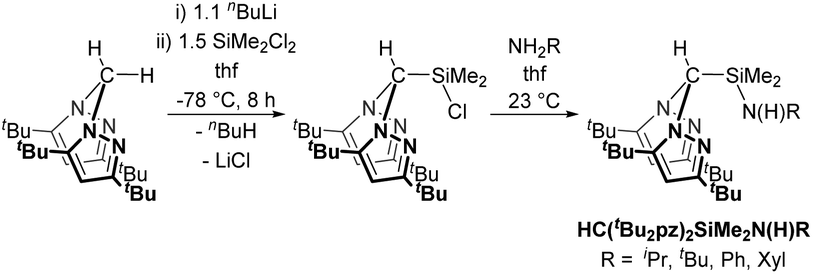 | ||
| Scheme 1 Synthesis of a new N2N′ heteroscorpionate pro-ligand family. See main text for further details of the second step. | ||
Attempts to prepare HC(pz)2SiMe2Cl using the same methodology resulted in an intractable mixture of products; deprotonation at the apical sp3 C is stabilised by the inductive effect of the two adjacent α-N atoms whereas the pyrazolyl ring C(5) is sp2-hybridised and is inductively stabilised by a single α-N atom. Use of a base such as nBuLi facilitates both pathways; coordination to N(1) favours C(5) deprotonation and coordination to N(2) favours apical carbon deprotonation. The mixture of lithiated species is trapped out by the hard SiMe2Cl2 electrophile to afford the mixture of substituted products where ring substitution is the major product.18
New pro-ligands HC(tBu2pz)2SiMe2N(H)R (R = iPr, tBu, Ph, Xyl (Xyl = 2,6-C6H3Me2)) were then prepared using procedures related to the 3,5-Me system in excellent yields of 94–99%. The influence of the sterically demanding 3,5-tBu groups becomes apparent with all but one reaction (that for R = iPr) taking place at a higher temperature and over a longer time period than for the analogous 3,5-Me substituted homologues. For R = Xyl, lithiation afforded LiN(H)Xyl which is a potent nucleophile and reacts with 1 equivalent of HC(tBu2pz)2SiMe2Cl in a salt elimination reaction.
The 1H and 13C{1H} NMR spectra of HC(tBu2pz)2SiMe2N(H)R (R = iPr, tBu, Ph, Xyl) all displayed the same type of diagnostic resonances. For example, where R = iPr, singlets at 6.99 and 6.01 ppm correspond to the apical ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) C(tBu2pz)2 and pyrazolyl N2C3tBu2 protons, respectively, and are in a 1
C(tBu2pz)2 and pyrazolyl N2C3tBu2 protons, respectively, and are in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, a consequence of Cs molecular symmetry. Singlets at 1.40 and 1.15 ppm are attributed to the tert-butyl groups in the 3- and 5-positions. This assignment is analogous to the 3,5-Me system and is supported by two dimensional 13C–1H correlation experiments. The SiMe singlet resonance at 0.37 ppm appears at a typically low frequency, as expected for methyl protons shielded by an electropositive Si atom. The molecular structure of HC(tBu2pz)2SiMe2N(H)Ph was also determined by a single crystal X-ray crystallographic study (see Fig. S11, Table S1 of the ESI‡ for further details). This structure shows an intramolecular interaction between the N(Ph)–H and pyrazolyl N atom, and contrasts that for HC(tBu2pz)2SiMe2Cl where the Si atom is in a pseudo trigonal bipyramidal environment, apparently as a result of electron donation from the lone pair of the pyrazolyl N(1) into the σ* orbital of the Si–Cl bond.
:2 ratio, a consequence of Cs molecular symmetry. Singlets at 1.40 and 1.15 ppm are attributed to the tert-butyl groups in the 3- and 5-positions. This assignment is analogous to the 3,5-Me system and is supported by two dimensional 13C–1H correlation experiments. The SiMe singlet resonance at 0.37 ppm appears at a typically low frequency, as expected for methyl protons shielded by an electropositive Si atom. The molecular structure of HC(tBu2pz)2SiMe2N(H)Ph was also determined by a single crystal X-ray crystallographic study (see Fig. S11, Table S1 of the ESI‡ for further details). This structure shows an intramolecular interaction between the N(Ph)–H and pyrazolyl N atom, and contrasts that for HC(tBu2pz)2SiMe2Cl where the Si atom is in a pseudo trigonal bipyramidal environment, apparently as a result of electron donation from the lone pair of the pyrazolyl N(1) into the σ* orbital of the Si–Cl bond.
Synthesis and characterisation of N2N′ heteroscorpionate lithium complexes
The preparation of alkali metal complexes of the N2N′ heteroscorpionate ligands was initially targeted in order to offer the potential of reactions via salt metathesis with appropriate metal salts as well as protonolysis routes. Several heteroscorpionate lithium salts from the NNO,19 NNS,8b,19a,20 NNN′11a,21 and NNCp22 heteroscorpionate classes have previously been reported; their syntheses all involve deprotonation at the methylene bridge of the appropriate bis(pyrazolyl)methane followed by nucleophilic attack upon a heterocumulene, α,β unsaturated system, aldehyde or imine to afford the desired lithium salt.Treatment of the N2N′ pro-ligands HC(tBu2pz)2SiMe2N(H)R with an excess of nBuLi at −78 °C afforded complexes formulated as [Li{HC(tBu2pz)2SiMe2NR}]n (R = iPr (1), tBu (2), Ph (3) and Xyl (4)) in poor to good isolated yields (5–70%) as colourless solids (eqn (1)). When thf is used as a reaction solvent, removal of the volatiles under reduced pressure served to leave no coordinated thf. These complexes are all air- and thermally-sensitive, showing degradation in both solution after a few hours and the solid state after 2 weeks when kept at −30 °C. This sensitivity was also noted in the reported thf-stabilised compound, Li{HC(tBu2pz)2CH2NXyl}(thf), which, on standing in the solid state, showed decomposition to the pro-ligand, HC(tBu2pz)2CH2N(H)Xyl.23 NNE heteroscorpionate lithium salts have most commonly been isolated as donor solvent-stabilised adducts. Examples of dimeric and oligomeric complexes have also been reported but are much rarer; Bassanetti et al. described the preparation and characterisation of [Li{HC(tBu2pz)2CS2}]3.20b Otero and co-workers reported dimeric (thio)acetamidinate NNN′ complexes [Li{HC(R′2pz)2CNR′′E}]2 (R′ = Me, Ph, tBu, R′′ = achiral or chiral moiety, E = O or S) complexes which provided a versatile route into the synthesis of enantiopure NNN′ ligands.
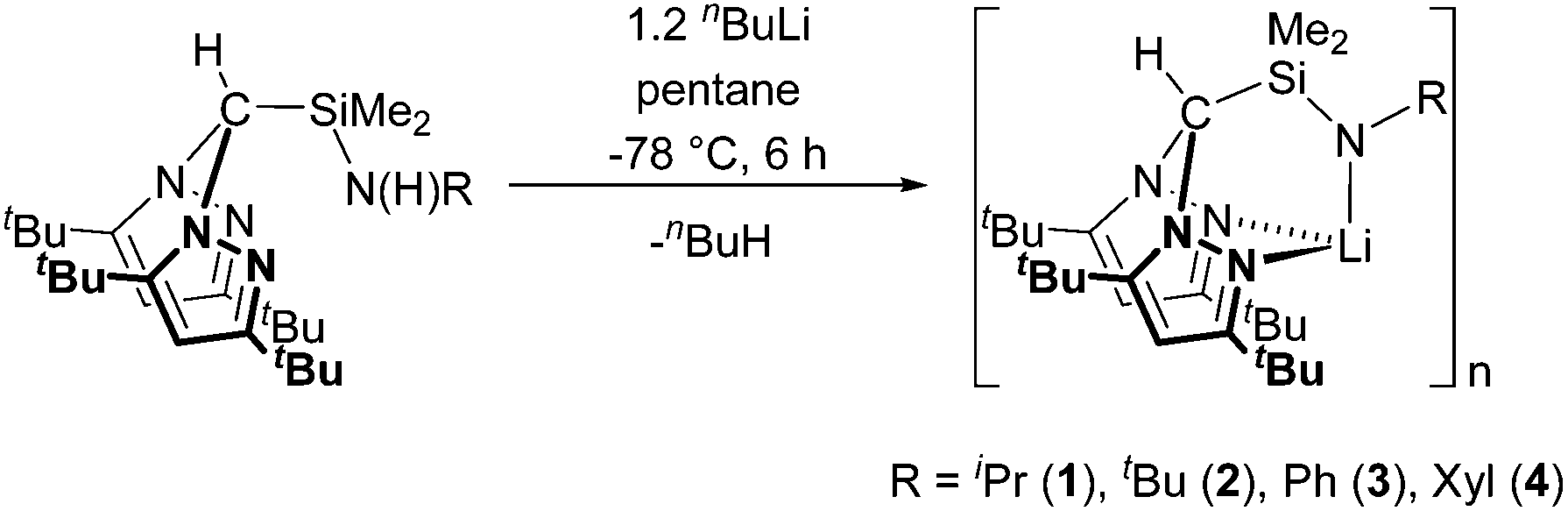 | (1) |
The 1H NMR spectra of complexes 1–4 all show resonances clearly shifted from those of the pro-ligand values, and the singlet attributed to the N proton is no longer present. In addition, 7Li NMR spectra were recorded at 23 °C and −80 °C for the most thermally robust complex, [Li{HC(tBu2pz)2SiMe2NXyl}]n (4), to investigate the possibility that the lithium salts existed as stabilised aggregates. However, at both high and low temperature a lone singlet was observed at 2.1 ppm. EI-MS data indicated a molecular ion peak at m/z = 555. The IR data were also diagnostic with the absorption attributed to ν(N–H) stretch in the pro-ligand being absent. All complexes were also characterised by elemental analysis. The steric demands of the 3,5-tBu ligand can stabilise complexes 1–4 with respect to sandwich complexes but it is most likely that these complexes exist as oligonuclear species. Analogous reactions with the corresponding 3,5-Me systems did not afford tractable, well-defined products.
Synthesis and characterisation of magnesium, calcium and zinc alkyl and amide complexes supported by N2N′ ligands
Treatment of HC(tBu2pz)2SiMe2N(H)R (R = iPr, Ph) with a two-fold excess of MeMgCl afforded Mg{HC(tBu2pz)2SiMe2NR}Me (R = iPr (5) and R = Ph (6)) in low isolated yields (24 and 7%, respectively) which is partially attributed to the high solubility of these complexes in aromatic and aliphatic solvents. EI-MS data of the crude reaction mixtures were also consistent with the presence of Mg{HC(tBu2pz)2SiMe2NR}Cl (e.g. for R = Ph, a molecular ion peak at m/z = 579 was observed). We have previously reported the reaction of HC(Me2pz)2(3,5-tBu2C6H3OH) with nBuMgCl which afforded Mg{HC(Me2pz)2(3,5-tBu2C6H3O)}2 alongside insoluble precipitates of MgCl2 and [Mg{HC(Me2pz)2(3,5-tBu2C6H3O)}Cl]n.16b Formation of similar homoleptic sandwich complexes has been reported;24 in our system, the bulky 3-tBu substituents act as ‘tetrahedral enforcers’ preventing sandwich formation.25 Parkin and co-workers reported a detailed study of the competitive alkyl and halide bond metathesis of a variety of Grignard reagents (RMgX) with tris(pyrazolyl)hydroborate (tpb) complexes M{HB(3-tBupz)3} (M = Tl, K) to afford Mg{HB(3-tBupz)3}R or Mg{HB(3-tBupz)3}X.26 The preference for alkyl or halide complexes was found to be dependent on the nature of Grignard reagent, steric profile of the tpb ligand, the Schlenk equilibrium and the molar ratio of M{HB(3-tBupz)3} to RMgX. Combination of Grignard reagents with alkali metal salts of ligands can be used to prepare metal alkyl complexes successfully.8d,10a,27MgR2 are therefore more appropriate starting materials for the preparation of the alkyl complexes Mg{HC(tBu2pz)2SiMe2NR′}R. The reaction of MgnBu2 with HC(R′2pz)2SiMe2N(H)R (R′ = Me, tBu) at 23 °C afforded the corresponding magnesium alkyl complexes Mg{HC(R′2pz)2SiMe2NR}nBu (R′ = Me; R = iPr (7), tBu (8), Ad (9), Ph (10). R′ = tBu; R = iPr (11), Ph (12)) in moderate yields (41–66%) (Scheme 2). Consistent with previous observations, the small scale reaction of HC(tBu2pz)2SiMe2N(H)iPr with nBuMgCl afforded complex 11 alongside MgCl2.
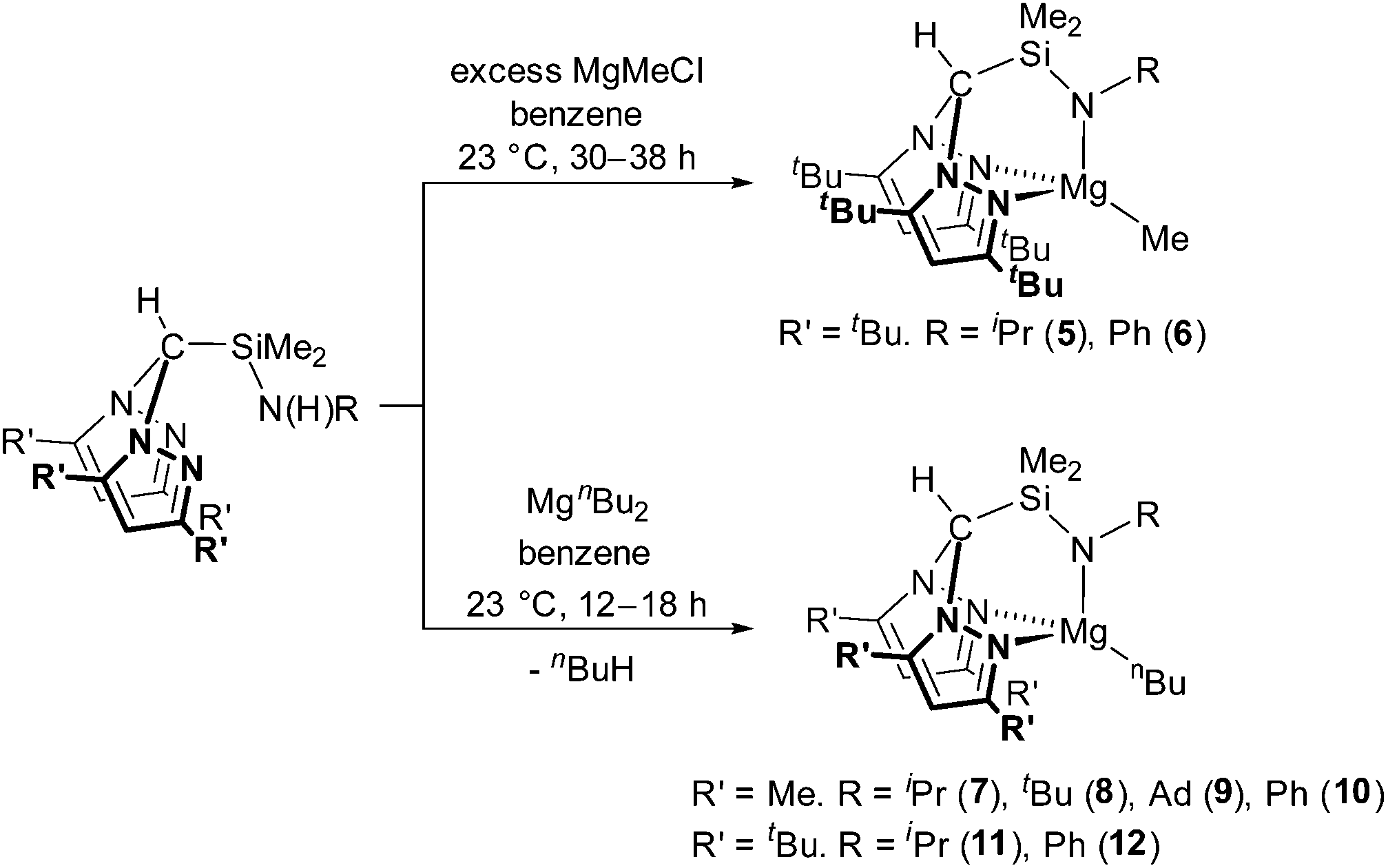 | ||
| Scheme 2 Synthesis of N2N′ supported magnesium alkyl complexes. | ||
In the 1H NMR spectra of complexes 7–12, the same diagnostic resonances were observed and demonstrated clearly shifted ligand resonances with respect to the pro-ligands. Four multiplets define the nBu protons at approximately 2.1, 1.8, 1.3 and 0.4 ppm in all complexes. Two dimensional COSY experiments show that these multiplets represent the 2-nBu, 3-nBu, 4-nBu and 1-nBu protons respectively.
The molecular structures of complexes 5 and 10 were elucidated by single crystal X-ray diffraction studies and are depicted in Fig. 1 with selected metric parameters in Table 1. In both complexes, the magnesium is in a distorted tetrahedral environment with the N2N′ heteroscorpionate ligand coordinated facially in a κ3 binding mode. The increased steric demands of 3,5-tBu substituents in complex 5 with respect to 3,5-Me groups in complex 10 are observed through longer M–Npz bond lengths (5: 2.1799(16) and 2.1911(16) Å. 10: 2.1699(19) and 2.138(2) Å) and a more acute Namide–Mg–C bond angle (5: 122.09(8)° and 10: 138.18(9)°). The M–Npz and M–C bond lengths are comparable to previously reported four-coordinate tris(pyrazolyl)hydroborate (tpb) magnesium methyl complexes,28 the NNN′ heteroscorpionate complex Mg{HC(tBu2pz)2CNEtNtBu}Me reported by Otero and co-workers,8d and the NNO magnesium butyl complex Mg{HC(Me2pz)2(3,5-tBu2C6H3O)}nBu reported by our group.16b
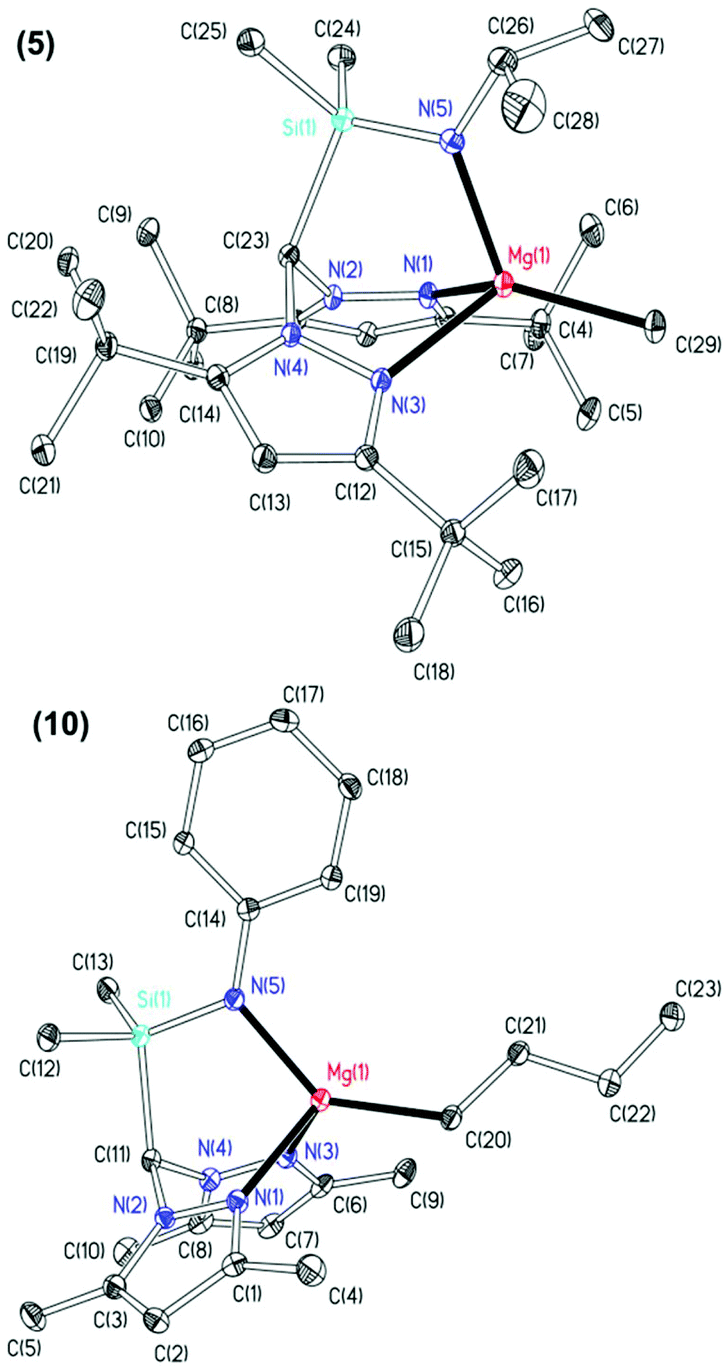 | ||
| Fig. 1 Thermal displacement ellipsoid drawings (20% probability ellipsoids) of Mg{HC(tBu2pz)2SiMe2NiPr}Me (5, top) and Mg{HC(Me2pz)2SiMe2NPh}nBu (10, bottom). All hydrogen atoms have been omitted for clarity. | ||
| 5 | 10 | |
|---|---|---|
| Mg(1)–N(1) | 2.1799(16) | 2.1699(19) |
| Mg(1)–N(3) | 2.1911(16) | 2.138(2) |
| Mg(1)–N(5) | 2.0083(17) | 2.0436(19) |
| Mg(1)–C(20)/(29) | 2.145(2) | 2.144(2) |
| N(1)–Mg(1)–C(20)/(29) | 123.01(7) | 112.98(9) |
| N(3)–Mg(1)–C(20)/(29) | 123.88(7) | 114.15(9) |
| N(5)–Mg(1)–C(20)/(29) | 122.09(8) | 138.18(9) |
Straightforward protonolysis reactions with 1 equivalent of the N2N′ heteroscorpionate ligands with 1 equivalent of Mg{N(SiHMe2)2}2 or Mg{N(SiMe3)2}2 yielded Mg{HC(R′2pz)2SiMe2NR}{N(SiHMe2)2} (R′ = Me; R = iPr (13), tBu (14), Ph (15). R′ = tBu; R = tBu (16)) or Mg{HC(R′2pz)2SiMe2NR}{N(SiMe3)2} (R′ = Me; R = iPr (17), tBu (18), Ph (19). R′ = tBu; R = Ph (20)), respectively, in moderate to good isolated yields (49–77%) (Scheme 3). Homoleptic sandwich complexes of the 3,5-Me substituted ligand can be isolated when a 2:1 ligand:metal stoichiometry is used, and the molecular structure of Mg{HC(Me2pz)2SiMe2NPh}2 has been determined by a single crystal X-ray diffraction study (see ESI Fig. S12 and Table S3‡). Using the more sterically demanding 3,5-tBu substituted ligand, the reactions proceeded cleanly at 60 °C and there was no evidence of sandwich complex formation; indeed, complexes 16 and 20 were the only products of 2:1 NMR scale reactions under identical conditions.
 | ||
| Scheme 3 Synthesis of N2N′ supported magnesium amide complexes. | ||
The NMR spectra of complexes 13–20 are consistent with Cs molecular symmetry on the NMR timescale, assuming fast rotation about the Mg–N(SiHMe2)2 and Mg–N(SiMe3)2 bonds. Complexes 16, 17, 19 and 20 have been structurally authenticated in the solid state by single crystal X-ray diffraction studies and are the first examples of N2N′ heteroscorpionate magnesium amide molecular structures. The molecular structures of complex 16 and 20 are depicted in Fig. 2 with selected metric parameters in Table 2 (see ESI Fig. S13 and S14 and Table S2‡ for complexes 17 and 19). The approximately tetrahedral magnesium centres are κ3 bound to the N2N′ heteroscorpionate ligand. The average Mg–Npz bond lengths (16: 2.192 Å. 20: 2.230 Å) are longer than those in Mg{C(Me2pz)3}{N(SiMe3)2} (2.115 Å) consistent with the changing steric profiles of the three compounds; the Mg–N(SiRMe2)2 distances are within the expected ranges. The largest N–Mg–N(SiRMe2)2 angle and longest Mg–Npz bond in both 16 and 20 are associated with N(5) due to steric repulsion between the methyl groups attached to Si(2) and the phenyl group attached to N(5). The Si(3)Me3 group has less steric impact as it is able to lodge into the cleft between the pyrazolyl rings. The smaller average Npz–Mg–N(SiRMe2)2 angle in 16 (114.2°) compared to that in 20 (120.4°) is consistent with the presence of the less bulky SiHMe2 group with respect to SiMe3 group.
 | ||
| Fig. 2 Thermal displacement ellipsoid drawings (20% probability ellipsoids) of Mg{HC(tBu2pz)2SiMe2NPh}{N(SiHMe2)2} (16, top) and Mg{HC(tBu2pz)2SiMe2NPh}{N(SiMe3)2} (20, bottom). C-Bound hydrogen atoms have been omitted for clarity. | ||
| 16 | 20 | |
|---|---|---|
| Mg(1)–N(1) | 2.167(4) | 2.254(2) |
| Mg(1)–N(3) | 2.217(4) | 2.206(2) |
| Mg(1)–N(5) | 2.027(4) | 2.023(2) |
| Mg(1)–N(6) | 2.002(4) | 2.023(2) |
| N(1)–Mg(1)–N(6) | 109.39(15) | 123.88(8) |
| N(3)–Mg(1)–N(6) | 119.10(15) | 116.93(8) |
| N(5)–Mg(1)–N(6) | 138.33(16) | 129.76(8) |
Treatment of HC(R′2pz)2SiMe2N(H)R (R′ = Me, tBu) with 1 equivalent of Ca{N(SiMe3)2}2(thf)2 afforded Ca{HC(Me2pz)2SiMe2NR}{N(SiMe2)2}(thf) (R = iPr (21), tBu (22), Ph (23)) or, in the case of the more sterically demanding ligand system, thf-free Ca{HC(tBu2pz)2SiMe2NPh}{N(SiMe2)2} (24) in good yields (60–74%), alongside 1 equivalent of HN(SiMe3)2 (eqn (2)). As previously observed, the reaction of the 3,5-Me pro-ligand proceeded at 23 °C whilst the bulkier 3,5-tBu pro-ligand required elevated temperatures of 60 °C to proceed.
 | (2) |
Due to the similar ionic radii of MgII and ZnII (MgII,4 C.N. = 0.570 Å, ZnII,4 C.N. = 0.600 Å), zinc alkyl complexes were targeted as a comparison to magnesium alkyl complexes 5–11. Protonolysis of the N2N′ heteroscorpionate pro-ligands with ZnMe2 afforded Zn{HC(R′2pz)2SiMe2NR}Me (R′ = Me; R = iPr (25), tBu (26), Ph (27). R′ = tBu; R = Ph (28)) in good yields (70–85%) (eqn (3)). The preparation of complex 28 required an elevated temperature of 60 °C, extended reaction time of 72 h and an excess of ZnMe2 in order to drive the reaction cleanly to completion.
 | (3) |
The molecular structures of complexes 25, 27 and 28 were elucidated by single crystal X-ray diffraction studies and are depicted in Fig. 3 with selected metric parameters in Table 3 (see ESI Fig. S15, Table S3‡ for data regarding complex 25). The zinc centre in complexes 27 and 28 is in a distorted tetrahedral coordination environment. The more acute N(5)–Zn(1)–CCH3 bond angle (27: 135.78(11)° and 28: 125.94(14)°), longer Zn(1)–Npz (27: 2.144(2) and 2.155(2) Å and 28: 2.171(4) and 2.202(4) Å) and Zn(1)–CCH3 bond distances (27: 1.981(3) and 28: 2.041(3) Å) of complex 28 with respect to 27 clearly reflect the differing steric demands of the 3,5-Me and 3,5-tBu pyrazolyl substituents. This trend is also observed in other examples of structurally characterised NNN′, NNO,8e,16b,29 NNCp4f heteroscorpionate and tris(pyrazolyl)hydroborate28b,30 zinc methyl complexes. Otero and co-workers reported the only other example of a structurally characterised NNN′ zinc methyl complex, Zn{HC(tBu2pz)2CN2iPr2}Me.11a Here some asymmetry in the Zn–Npz bond lengths is observed (2.236(3) and 2.118(3) Å). The M–Npz and M–CCH3 bond lengths in 28 (2.171(4), 2.202(4) and 1.976(3) Å, respectively) are shorter than those in Mg{HC(tBu2pz)2SiMe2NiPr}Me (1) (2.1799(16), 2.1911(16) and 2.145(2) Å, respectively) in accord with zinc being more polarising and covalent.16b
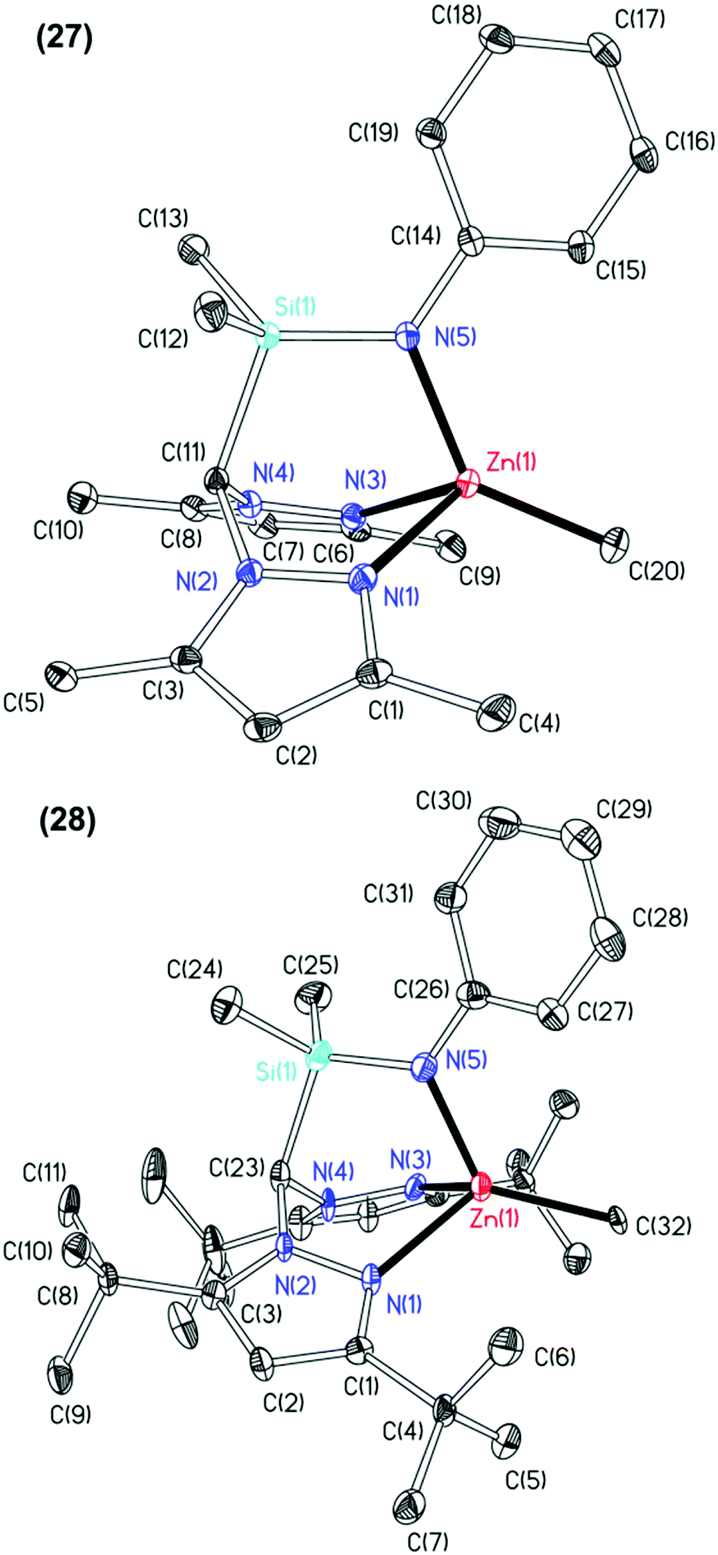 | ||
| Fig. 3 Thermal displacement ellipsoid drawings (20% probability ellipsoids) of (top) Zn{HC(Me2pz)2SiMe2NPh}Me (27, top) and Zn{HC(tBu2pz)2SiMe2NPh}Me (28, bottom). All hydrogen atoms have been omitted for clarity. | ||
| 27 | 28 | |
|---|---|---|
| Zn(1)–N(1) | 2.144(2) | 2.171(4) |
| Zn(1)–N(3) | 2.155(2) | 2.202(4) |
| Zn(1)–N(5) | 1.979(2) | 1.976(3) |
| Zn(1)–C(20) | 1.981(3) | — |
| Zn(1)–C(32) | 2.041(3) | |
| N(1)–Zn(1)–C(20) | 114.48(11) | — |
| N(1)–Zn(1)–C(32) | — | 122.59(14) |
| N(3)–Zn(1)–C(20) | 116.86(11) | — |
| N(3)–Zn(1)–C(32) | — | 120.12(14) |
| N(5)–Zn(1)–C(20) | 135.78(11) | — |
| N(5)–Zn(1)–C(32) | — | 125.94(14) |
Ring-opening polymerisation of ε-caprolactone and rac-lactide
One of the aims of this work was to assess the performance of the NNN′ heteroscorpionate amide complexes for the catalytic ring-opening polymerisation (ROP) of ε-caprolactone (ε-CL) and rac-lactide (rac-LA) monomers, in light of the success of related complexes and our previous studies into the ROP of cyclic esters.16b,31 An initial study was undertaken using ε-CL owing to the relative ease of its polymerisation with respect to LA due to the favourable release of 7-membered ring strain.Initial studies at room temperature in thf or toluene showed the amide complexes 16 and 20 to be highly active for the ROP of ε-CL, reaching full conversion of 100 equivalents of ε-CL in 20–30 minutes in either solvent. The results are summarised in Table 4. Both complexes afforded much higher than expected Mn values by gel permeation chromatography (GPC) (66620–89740 g mol−1), consistent with a much faster rate of propagation than rate of initiation in a coordination-insertion mechanism.32 The observed Mn values are in slightly better agreement with calculated Mn for polymerisations performed in thf. This is likely to be due to the effect of thf competing with the monomer for coordination to the metal centre, thus reducing the relative rate of propagation (compared to that in non-coordinating toluene solvent).
| Solvent | Yieldb (%) | Timec |
M
nd,e |
M w/Mn | |
|---|---|---|---|---|---|
|
a [ε-CL]:[catalyst] = 100:1, 3.4 mL solvent at 23 °C.
b Isolated yield at 100% conversion.
c Minutes.
d Molecular weights (g mol−1) determined from GPC using the appropriate Mark–Houwink corrections.
e Expected Mn (g mol−1) for 1 chain growing per metal centre at 100% conversion, ignoring end groups = 11414.
|
|||||
| 16 | thf | 76 | 25 | 71450 |
1.86 |
| 16 | Toluene | 79 | 25 | 89740 |
3.17 |
| 20 | thf | 81 | 30 | 66620 |
1.67 |
| 20 | Toluene | 73 | 20 | 84840 |
2.79 |
The PDIs (polydispersity indices, Mw/Mn) for polymerisations performed in thf are also narrower than corresponding polymerisations performed in toluene (although these are collectively all in the moderate to poor range, 1.67–3.17). This is also likely to be a direct consequence of the coordinative nature of thf compared to toluene. Broad PDIs are well-documented to be the result of intra- and inter-molecular transesterification reactions.32 It is reasonable to assume that the presence of a thf molecule bound to a metal coordination site would reduce the likelihood of ‘backbiting’ by a growing polymer chain at the metal centre. Due to the viscous nature of the polymer formed and the short times involved, it was not possible to acquire full kinetic data over the course of each polymerisation, nor was it possible to collect MALDI-ToF analysis of polymer samples due to the high observed Mn values. Coordination-insertion has been deduced to be the most likely mechanism of polymerisation with these complexes.
There are a relatively small number of metal complexes supported by heteroscorpionate ligands used as initiators for polymerisation of ε-CL. Examples include zinc amides and alkyls,11a,b,16b,33 rare earth dialkyls,34 aluminium alkyls and alkoxides,35 and magnesium alkyls and amides.8d,16b,24a We have previously reported the N2O heteroscorpionate complex Mg{HC(Me2pz)2(3,5-tBu2C6H3O)}{N(SiHMe2)2} which afforded complete conversion of 100 equivalents of ε-CL in toluene (but not thf) with high Mn (30870 g mol−1) and broad PDI (2.76). The related N2N′ magnesium alkyl complexes described by Otero and co-workers, Mg{HC(Me2pz)2CN2R2}R′, afforded rapid polymerisation (e.g. R = Et, R′ = CH2SiMe3, 97% conversion of 500 equivalents in 1 minute at 20 °C and R = iPr, R′ = CH2SiMe3, 62% at −60 °C) with narrow PDIs (1.41 and 1.12, respectively). The steric demands of the amidinate group were found to be important, with increased bulk resulting in slower initiators.
Polymerisation studies were next extended to the more challenging monomer, rac-LA. Due to the more encouraging results with ε-CL observed using thf as a solvent, it was decided to focus solely on thf-mediated polymerisations. The results are summarised in Table 5.
| Conv.b (%) | Time (h) |
M
nc |
M n (calcd)d | M w/Mn | k obs (min−1) | |
|---|---|---|---|---|---|---|
|
a Conditions: [rac-LA]:[catalyst] = 100:1, 6.0 mL thf at 70 °C.
b % conversion by 1H NMR spectroscopy.
c Molecular weights (g mol−1) determined from GPC using the appropriate Mark–Houwink corrections.36
d Expected Mn (g mol−1) for 1 chain growing per metal centre at the % conversion by 1H NMR spectroscopy.
|
||||||
| 16 | 90 | 1.5 | 55080 |
12972 |
1.48 | 0.03(2) |
| 20 | 89 | 0.75 | 51480 |
12799 |
1.45 | 0.04(3) |
| Ref 8a | 96 | 1.0 | 14000 |
13800 |
1.09 | — |
| Ref 8d | 91 | 0.03 | 12800 |
13100 |
1.09 | — |
One of the several interesting contrasts with the ε-CL polymerisation results is that the rac-LA polymerisations all required elevated temperatures of 70 °C and extended reaction times, largely due to the unfavourable release of the stable 6-membered ring strain in rac-LA when compared to the less thermodynamically stable 7-membered ring strain in ε-CL. The observed Mn values (51080–51480 g mol−1) are again large, and the relatively broad PDIs (1.45–1.48) are also evident in the rac-LA polymerisations, no doubt due in part to the increased tendency of transesterification side reactions at elevated temperatures. Disappointingly, no stereochemical enrichment in the PLA polymer produced was observed (Pr ≈ 0.5).
Despite the relatively poor polymerisation control demonstrated by the complexes tested, kinetic studies were carried out in some cases to investigate further the polymerisation process. All of the catalysts tested showed first order consumption of rac-LA after an initial induction period. Similar induction periods have also been observed in the ROP of cyclic esters using aluminium,37 yttrium,38 iron,39 tin alkoxides and amides. In these instances the induction period was thought to correspond to a period of ligand rearrangement around the metal centre, in order to allow the monomer access to the coordination sphere.40 An example semi-logarithmic plot is shown in Fig. 4 for the catalyst Mg{HC(tBu2pz)2SiMe2NPh}{N(SiHMe2)2} (16). The apparent rate constant for the polymerisation, kobs, was 0.03(2) min−1. This compares with a kobs value of 0.04(3) min−1 for the related catalyst Mg{HC(tBu2pz)2SiMe2NPh}{N(SiMe3)2} (20).
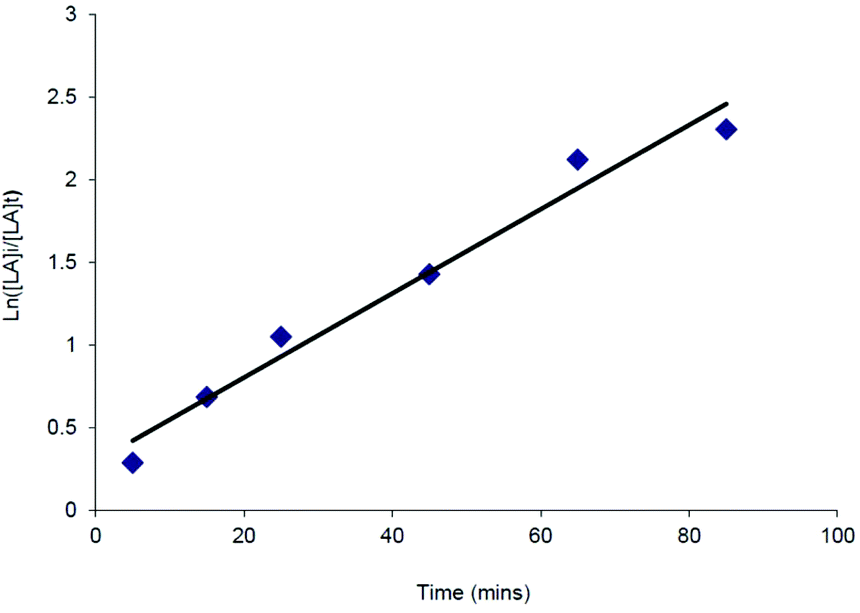 | ||
| Fig. 4 First order semi-logarithmic plot for rac-LA consumption using Mg{HC(tBu2pz)2SiMe2NPh}{N(SiHMe2)2} (16). Conditions [rac-LA0]:[16] = 100:1, 6 mL thf, 70 °C, 0.1 mL aliquots taken at the given intervals. | ||
Though there are no other specific examples of scorpionate magnesium amide complexes for lactide polymerisation, complexes 16 and 20 compare well with initiators with respect to the pioneering homoscorpionate tris(pyrazolyl)hydroborate (tpb) magnesium and zinc alkoxides for lactide polymerisation reported by Chisholm.9c Kinetic studies under pseudo first order conditions resulted in a value of kobs = 0.039 min−1 for Mg{HC(3-tBupz)3}OEt. When extended to calcium, initiators were found to be two orders of magnitude faster.9b Complexes 16 and 20 prove to be slow initiators with respect to some related heteroscorpionate systems.10a,29a,41 The N2N′ heteroscorpionate magnesium alkyl complex, Mg{HC(tBu2pz)2CNEtNnBu}CH2SiMe3, reported by Otero and co-workers was shown to polymerise 200 equivalents of rac-LA at 20 °C, reaching with 83% conversion in 2 minutes, to afford heterotactically-enriched PLAs (Pr = 0.70–0.79) with narrow PDIs (1.01–1.05) and no induction period (kapp = 2.0 min−1).8d These values compare well with the most active magnesium catalysts reported in the literature.42
Conclusions
A new family of sterically demanding N2N′ heteroscorpionate pro-ligands (HC(tBu2pz)2SiMe2N(H)R (R = iPr, tBu, Ph, Xyl)) has been developed and the synthesis and characterisation of lithium, magnesium, calcium and zinc complexes, M{HC(R′2pz)2SiMe2NR}(X) (M = Mg, Zn, Ca; R′ = Me or tBu, X = amide or alkyl), supported by both 3,5-tBu and 3,5-Me substituted N2N′ ligand families has been conducted.The 3,5-tBu substituents enabled isolation and characterisation of highly sensitive oligomeric lithium salts, [Li{HC(tBu2pz)2SiMe2NR}]n, which could not be identified with the 3,5-Me ligand system. Furthermore, they have also successfully ensured that ML2 bis(ligand) complexes do not form even when the M:L reaction stoichiometry is 2:1; retaining structural integrity and avoiding formation of such sandwich complexes is particularly desirable when considering further potential applications in homogeneous catalysis. Preliminary investigations into the polymerisation of ε-caprolactone and rac-lactide indicated poor control of polymerisation but nonetheless active catalysts.
In general, the N2N′ ligand set seems well suited to stabilising a variety of reactive group 2 and 12 complexes with alkyl and amide substituents and provides a platform for further reactivity or catalytic studies.
Experimental details
General considerations
All manipulations were carried out under an inert atmosphere of argon or dinitrogen using standard Schlenk-line or drybox procedures. Solvents were pre-dried over activated 4 Å molecular sieves and refluxed over sodium (toluene), sodium/potassium (pentane, diethyl ether), or calcium hydride (dichloromethane) under a dinitrogen atmosphere and collected by distillation. Alternatively, solvents were degassed by sparging with dinitrogen and dried by passing through a column of activated alumina. Deuterated solvents were dried over potassium (C6D6) or P2O5 (CD2Cl2), distilled under reduced pressure, and stored under dinitrogen in J. Young Teflon valve ampoules. Solution NMR samples were prepared under a dinitrogen atmosphere in a drybox, in 5 mm Wilmad NMR tubes possessing Young's Teflon valves. 1H and 13C NMR spectra were recorded on a Varian Mercury 300 spectrometer or a Bruker AVII 500 spectrometer. 1H and 13C{1H} NMR spectra were referenced internally to residual protio-solvent (1H) or solvent (13C) resonances, and are reported relative to tetramethylsilane (δ = 0 ppm). 7Li NMR spectra are referenced relative to LiCl. Chemical shifts are quoted in δ (ppm) and coupling constants in Hz. Where necessary, 1H and 13C assignments were assisted by the use of two-dimensional 1H–1H and 1H–13C correlation experiments. IR spectra were recorded on a PerkinElmer 1710 or Nicolet magna 560 FTIR spectrometer. Samples were prepared in a drybox as Nujol mulls between NaCl plates. IR data are quoted as wavenumbers (cm−1) within the range 4000–400 cm−1. Mass spectra were recorded by the mass spectrometry service of the University of Oxford Inorganic Chemistry Laboratory. Elemental analyses were carried out by the Elemental Analysis Service at the London Metropolitan University. The compounds H2C(tBu2pz)2,8f HC(Me2pz)2SiMe2N(H)iPr,17 Ca{N(SiMe3)2}2(thf)2,43 Mg{N(SiMe3)2}243 and Mg{N(SiHMe2)2}243 were prepared according to literature procedures. ε-Caprolactone was dried over freshly ground CaH2, stored over molecular sieves (4 Å) at 4 °C and distilled before use. rac-Lactide was recrystallized twice from toluene and subsequently sublimed twice prior to use. All other reagents were purchased and used without further purification. Polymer molecular weights (Mn, Mw) were determined by GPC using a Polymer Laboratories Plgel Mixed-D column (300 mm length, 7.5 mm diameter) and a Polymer Laboratories PL-GPC50 Plus instrument equipped with a refractive index detector. Tetrahydrofuran (HPLC grade) was used as an eluent at 30 °C with a rate of 1 mL min−1. Linear polystyrenes were used as primary calibration standards, and Mark–Houwink corrections for poly(ε-CL) or poly(rac-LA) in thf were applied for the experimental samples.36
X-ray crystallography
Crystal data collection and processing parameters for HC(tBu2pz)2SiMe2Cl, HC(tBu2pz)2SiMe2N(H)Ph, Mg{HC(Me2pz)2SiMe2NPh}2, Mg{HC(tBu2pz)2SiMe2NiPr}Me (5), Mg{HC(Me2pz)2SiMe2NPh}nBu (10), Mg{HC(tBu2pz)2SiMe2NPh}-{N(SiHMe2)2} (16), Mg{HC(Me2pz)2SiMe2NiPr}{N(SiMe3)2} (17), Mg{HC(Me2pz)2SiMe2NPh}{N(SiMe3)2} (19), Mg{HC(tBu2pz)2-SiMe2NPh}{N(SiMe3)2} (20), Zn{HC(Me2pz)2SiMe2NiPr}Me (25), Zn{HC(Me2pz)2SiMe2NPh}Me (27), Zn{HC(tBu2pz)2SiMe2NPh}Me (28) are given in Tables S1–3 and Fig. S10–15 of the ESI.‡ The structures have been deposited in the Cambridge Structural Database as CCDC 1885899–1885910.‡ Crystals were mounted on glass fibres using perfluoropolyether oil and cooled rapidly under a stream of cold N2 using an Oxford Cryosystems Cryostream unit. Diffraction data were measured using a Enraf-Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å). As appropriate, absorption and decay corrections were applied to the data and equivalent reflections merged.44 The structures were solved with SIR9245 or Superflip,46 and further refinements and all other crystallographic calculations were performed using the CRYSTALS program suite.47 Other details of the structure solution and refinements are given in the ESI.‡
Synthetic details and characterising data
C(Me2pz)2), 5.67 (2 H, s, N2C3Me2), 2.21 (6 H, s, 3-N2C3H![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) 2), 1.94 (6 H, s, 5-N2C3H2), 1.90 (3 H, m, Ad), 1.67 (6 H, app. d, app. 3JHH = 3 Hz, Ad), 1.51 (6 H, m, Ad), 0.54 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 146.5 (3-N2
2), 1.94 (6 H, s, 5-N2C3H2), 1.90 (3 H, m, Ad), 1.67 (6 H, app. d, app. 3JHH = 3 Hz, Ad), 1.51 (6 H, m, Ad), 0.54 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 146.5 (3-N2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) 3HMe2), 140.2 (5-N23HMe2), 106.4 (4-N23HMe2), 69.9 (H(Me2pz)2), 50.0 (1-Ad), 47.6 36.6 (2,4,6,8,9,10-Ad), 30.4 (3,5,7-Ad), 13.8 (3-N2C3H2), 11.0 (5-N2C3H2), 2.2 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3342 (m) 2951 (s), 2853 (m), 1560 (s), 1461 (w), 1376 (m), 1258 (m), 1096 (m), 1018 (w), 967 (m), 822 (w), 799 (m), 721 (w). EI-MS: m/z = 411 (100%) [M]+, 316 (10%) [M − Me2pz]+. Anal. found (calcd. for C23H37N5Si): C, 67.0 (67.1); H, 9.1 (9.1); N, 17.0 (17.0)%.
C(Me2pz)2), 5.61 (2 H, s, N2C3Me2), 4.34 (1 H, s, NH), 2.16 (6 H, s, 3-N2C3H2), 1.73 (6 H, s, 5-N2C3H2), 0.60 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 147.7 (3-N23HMe2), 142.9 (1-C6H5), 139.9 (5-N23HMe2), 129.8 (3-C6H5), 118.7 (4-C6H5), 117.5 (2-C6H5), 106.7 (4-N23HMe2), 67.0 (H(Me2pz)2), 13.9 (3-N2C3H2), 10.6 (5-N2C3H2), −0.2 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3345 (m), 2930 (s), 2731 (m), 1602 (w), 1552 (s), 1463 (m), 1376 (w), 1296 (m), 1246 (m), 1145 (w), 1110 (w), 1025 (s), 972 (m), 907 (m), 807 (s), 787 (s), 747 (m), 691 (m). EI-MS: m/z = 353 (100%) [M]+. Anal. found (calcd for C19H27N5Si): C, 64.5 (64.5); H, 7.7 (7.7); N, 19.7 (19.7)%.
C(tBu2pz)2), 6.01 (2 H, s, N2C3tBu2), 1.27 (18 H, s, 3-N2C3H
3HMe2), 140.2 (5-N23HMe2), 106.4 (4-N23HMe2), 69.9 (H(Me2pz)2), 50.0 (1-Ad), 47.6 36.6 (2,4,6,8,9,10-Ad), 30.4 (3,5,7-Ad), 13.8 (3-N2C3H2), 11.0 (5-N2C3H2), 2.2 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3342 (m) 2951 (s), 2853 (m), 1560 (s), 1461 (w), 1376 (m), 1258 (m), 1096 (m), 1018 (w), 967 (m), 822 (w), 799 (m), 721 (w). EI-MS: m/z = 411 (100%) [M]+, 316 (10%) [M − Me2pz]+. Anal. found (calcd. for C23H37N5Si): C, 67.0 (67.1); H, 9.1 (9.1); N, 17.0 (17.0)%.
C(Me2pz)2), 5.61 (2 H, s, N2C3Me2), 4.34 (1 H, s, NH), 2.16 (6 H, s, 3-N2C3H2), 1.73 (6 H, s, 5-N2C3H2), 0.60 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 147.7 (3-N23HMe2), 142.9 (1-C6H5), 139.9 (5-N23HMe2), 129.8 (3-C6H5), 118.7 (4-C6H5), 117.5 (2-C6H5), 106.7 (4-N23HMe2), 67.0 (H(Me2pz)2), 13.9 (3-N2C3H2), 10.6 (5-N2C3H2), −0.2 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3345 (m), 2930 (s), 2731 (m), 1602 (w), 1552 (s), 1463 (m), 1376 (w), 1296 (m), 1246 (m), 1145 (w), 1110 (w), 1025 (s), 972 (m), 907 (m), 807 (s), 787 (s), 747 (m), 691 (m). EI-MS: m/z = 353 (100%) [M]+. Anal. found (calcd for C19H27N5Si): C, 64.5 (64.5); H, 7.7 (7.7); N, 19.7 (19.7)%.
C(tBu2pz)2), 6.01 (2 H, s, N2C3tBu2), 1.27 (18 H, s, 3-N2C3H![[t with combining low line]](https://www.rsc.org/images/entities/i_char_0074_0332.gif)
![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif)
![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) 2), 1.25 (18 H, s, 5-N2C3H2), 0.99 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.8 (3-N23HtBu2), 152.3 (5-N23HtBu2), 102.7 (4-N23HtBu2), 74.0 (H(tBu2pz)2), 32.9 (3-N2C3H(Me3)2), 32.1 (5-N2C3H(Me3)2), 31.4 (3-N2C3H(C3)2), 30.7 (5-N2C3H(C3)2), 6.7 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 1541 (m), 1531 (m), 1361 (s), 1334 (m), 1319 (w), 1249 (s), 1230 (m), 1203 (w), 1125 (w), 1084 (w), 1059 (w), 1012 (w), 999 (m), 867 (w), 844 (w), 652 (w). EI-MS: m/z = 465 (6%) [M]+, 429 (100%) [M − Cl]+, 372 (82%) [M − SiMe2Cl]+. Anal. found (calcd for C25H45ClN4Si): C, 64.5 (64.6); H, 9.7 (9.8); N, 11.9 (12.0)%.
C(tBu2pz)2), 6.01 (2 H, s, N2C3tBu2), 3.09 (1 H, sept, 3JHH = 6.2 Hz, CMe2), 1.40 (18 H, s, 3-N2C3H2), 1.15 (18 H, s, 5-N2C3H2), 0.96 (6 H, d, 3JHH = 6.2 Hz, HC2), 0.37 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.1 (3-N23HtBu2), 153.9 (5-N23HtBu2), 102.5 (4-N23HtBu2), 76.5 (H(tBu2pz)2), 43.3 (HMe2), 32.4 (3-N2C3H(Me3)2), 32.2 (5-N2C3H(Me3)2), 30.8 (3-N2C3H(C3)2), 30.5 (5-N2C3H(C3)2), 28.3 (HC2), 0.7 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3363 (w), 2712 (w), 1536 (s), 1394 (s), 1362 (s), 1343 (s), 1316 (s), 1276 (m), 1213 (s), 1168 (s), 1131 (s), 1067 (m), 1021 (s), 1003 (s), 931 (w), 880 (m), 828 (m), 685 (w). EI-MS: m/z = 487 (75%) [M]+, 430 (100%) [M − N(H)iPr]+. Anal. found (calcd for C28H53N5Si): C, 69.0 (68.9); H, 11.1 (11.0); N, 14.2 (14.4)%.
C(tBu2pz)2), 7.00 (2 H, d, 3JHH = 7.0 Hz, 3-C6H3Me2), 6.87 (1 H, t, 3JHH = 7.0 Hz, 4-C6H3Me2), 6.02 (2 H, s, N2C3tBu2), 3.55 (1 H, s, NH), 2.12 (6 H, s, C6H32), 1.35 (18 H, s, 3-N2C3H2), 1.12 (18 H, s, 5-N2C3H2), 0.42 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.3 (3-N23HtBu2), 154.2 (5-N23HtBu2), 143.2 (1-C6H3Me2), 133.0 (2-C6H3Me2), 128.7 (3-C6H3Me2), 122.7 (4-C6H3Me2), 102.9 (4-N23HtBu2), 75.4 (H(tBu2pz)2), 32.4 (3-N2C3H(Me3)2), 32.2 (5-N2C3H(Me3)2), 30.8 (3-N2C3H(C3)2), 30.4 (5-N2C3H(C3)2), 19.7 (C6H32), 1.3 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3319 (w), 1593 (w), 1537 (m), 1392 (w), 1363 (m), 1341 (m), 1316 (m), 1276 (w), 1237 (m), 1215 (m), 1176 (w), 1159 (w), 1129 (w), 1094 (w), 1070 (w), 1001 (m), 902 (m), 877 (m), 828 (m), 792 (s), 759 (s). EI-MS: m/z = 429 (100%) [M − N(H)C6H3Me2]+. Anal. found (calcd for C33H55N5Si): C, 72.2 (72.1); H, 10.2 (10.1); N, 12.7 (12.6)%.
C(tBu2pz)2), 5.91 (2 H, s, N2C3tBu2), 2.39 (6 H, s, C6H32), 1.23 (18 H, s, 5-N2C3H2), 1.16 (18 H, s, 3-N2C3H2), 0.13 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): 159.7 (3-N23HtBu2), 157.2 (1-6H3Me2), 153.2 (5-N23HtBu2), 131.8 (2,6-6H3Me2), 127.9 (3-6H3Me2), 115.4 (4-C6H3Me2), 101.3 (4-N23HtBu2), 71.5 (H(tBu2pz)2), 32.8 (5-N2C3H(Me3)2), 32.0 (3-N2C3H(Me3)2), 31.6 (5-N2C3H(C3)2), 30.3 (3-N2C3H(C3)2), 21.01 (C6H32), 3.24 (SiMe) ppm. 7Li NMR (toluene-d8, 194.3 MHz, 218 and 293 K): 2.1 (N
2), 1.25 (18 H, s, 5-N2C3H2), 0.99 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.8 (3-N23HtBu2), 152.3 (5-N23HtBu2), 102.7 (4-N23HtBu2), 74.0 (H(tBu2pz)2), 32.9 (3-N2C3H(Me3)2), 32.1 (5-N2C3H(Me3)2), 31.4 (3-N2C3H(C3)2), 30.7 (5-N2C3H(C3)2), 6.7 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 1541 (m), 1531 (m), 1361 (s), 1334 (m), 1319 (w), 1249 (s), 1230 (m), 1203 (w), 1125 (w), 1084 (w), 1059 (w), 1012 (w), 999 (m), 867 (w), 844 (w), 652 (w). EI-MS: m/z = 465 (6%) [M]+, 429 (100%) [M − Cl]+, 372 (82%) [M − SiMe2Cl]+. Anal. found (calcd for C25H45ClN4Si): C, 64.5 (64.6); H, 9.7 (9.8); N, 11.9 (12.0)%.
C(tBu2pz)2), 6.01 (2 H, s, N2C3tBu2), 3.09 (1 H, sept, 3JHH = 6.2 Hz, CMe2), 1.40 (18 H, s, 3-N2C3H2), 1.15 (18 H, s, 5-N2C3H2), 0.96 (6 H, d, 3JHH = 6.2 Hz, HC2), 0.37 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.1 (3-N23HtBu2), 153.9 (5-N23HtBu2), 102.5 (4-N23HtBu2), 76.5 (H(tBu2pz)2), 43.3 (HMe2), 32.4 (3-N2C3H(Me3)2), 32.2 (5-N2C3H(Me3)2), 30.8 (3-N2C3H(C3)2), 30.5 (5-N2C3H(C3)2), 28.3 (HC2), 0.7 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3363 (w), 2712 (w), 1536 (s), 1394 (s), 1362 (s), 1343 (s), 1316 (s), 1276 (m), 1213 (s), 1168 (s), 1131 (s), 1067 (m), 1021 (s), 1003 (s), 931 (w), 880 (m), 828 (m), 685 (w). EI-MS: m/z = 487 (75%) [M]+, 430 (100%) [M − N(H)iPr]+. Anal. found (calcd for C28H53N5Si): C, 69.0 (68.9); H, 11.1 (11.0); N, 14.2 (14.4)%.
C(tBu2pz)2), 7.00 (2 H, d, 3JHH = 7.0 Hz, 3-C6H3Me2), 6.87 (1 H, t, 3JHH = 7.0 Hz, 4-C6H3Me2), 6.02 (2 H, s, N2C3tBu2), 3.55 (1 H, s, NH), 2.12 (6 H, s, C6H32), 1.35 (18 H, s, 3-N2C3H2), 1.12 (18 H, s, 5-N2C3H2), 0.42 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.3 (3-N23HtBu2), 154.2 (5-N23HtBu2), 143.2 (1-C6H3Me2), 133.0 (2-C6H3Me2), 128.7 (3-C6H3Me2), 122.7 (4-C6H3Me2), 102.9 (4-N23HtBu2), 75.4 (H(tBu2pz)2), 32.4 (3-N2C3H(Me3)2), 32.2 (5-N2C3H(Me3)2), 30.8 (3-N2C3H(C3)2), 30.4 (5-N2C3H(C3)2), 19.7 (C6H32), 1.3 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 3319 (w), 1593 (w), 1537 (m), 1392 (w), 1363 (m), 1341 (m), 1316 (m), 1276 (w), 1237 (m), 1215 (m), 1176 (w), 1159 (w), 1129 (w), 1094 (w), 1070 (w), 1001 (m), 902 (m), 877 (m), 828 (m), 792 (s), 759 (s). EI-MS: m/z = 429 (100%) [M − N(H)C6H3Me2]+. Anal. found (calcd for C33H55N5Si): C, 72.2 (72.1); H, 10.2 (10.1); N, 12.7 (12.6)%.
C(tBu2pz)2), 5.91 (2 H, s, N2C3tBu2), 2.39 (6 H, s, C6H32), 1.23 (18 H, s, 5-N2C3H2), 1.16 (18 H, s, 3-N2C3H2), 0.13 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): 159.7 (3-N23HtBu2), 157.2 (1-6H3Me2), 153.2 (5-N23HtBu2), 131.8 (2,6-6H3Me2), 127.9 (3-6H3Me2), 115.4 (4-C6H3Me2), 101.3 (4-N23HtBu2), 71.5 (H(tBu2pz)2), 32.8 (5-N2C3H(Me3)2), 32.0 (3-N2C3H(Me3)2), 31.6 (5-N2C3H(C3)2), 30.3 (3-N2C3H(C3)2), 21.01 (C6H32), 3.24 (SiMe) ppm. 7Li NMR (toluene-d8, 194.3 MHz, 218 and 293 K): 2.1 (N![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) ) ppm. IR (NaCl plates, Nujol): 1587 (w), 1544 (m), 1418 (s), 1359 (m), 1297 (s), 1235 (m), 1207 (m), 1097 (w), 1064 (w), 1023 (w), 994 (m), 982 (m), 829 (m), 821 (m), 776 (m), 755 (s), 709 (w), 693 (w), 615 (w), 530 (w) cm−1. EI-MS: m/z: 555 [M]+ (8%), 429 [M − N(Li)Xyl]+ (100%), 371 [M − SiMe2N(Li)Xyl]+ (8%), Anal. Found (calcd for C33H54LiN5Si): C, 71.4 (71.3); H, 9.7 (9.8); N, 12.7 (12.6)%.
C(tBu2pz)2), 5.96 (2 H, s, N2C3tBu2), 3.61 (1 H, sept, 3JHH = 6.3 Hz, CMe2), 1.54 (6 H, d, 3JHH = 6.4 Hz, HC2), 1.52 (18 H, s, 3-N2C3H2), 1.16 (18 H, s, 5-N2C3H2), 0.05 (6 H, s, SiMe), −0.20 (3 H, s, MgMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 163.2 (3-N23HtBu2), 153.1 (5-N23HtBu2), 102.9 (4-N23HtBu2), 69.8 (H(tBu2pz)2), 46.8 (HMe2), 33.0 (HC2), 32.3 (3-N2C3H(Me3)2), 32.1 (5-N2C3H(Me3)2), 31.6 (3-N2C3H(C3)2), 30.7 (5-N2C3H(C3)2), 1.2 (SiMe), −3.6 (MgMe). IR (NaCl plates, Nujol mull, cm−1): 2795 (w), 2599 (w), 1615 (w), 1526 (m), 1423 (m), 1348 (m), 1305 (m), 1244 (m), 1220 (m), 1209 (m), 1139 (m), 1111 (m), 1061 (s), 1029 (m), 899 (m), 829 (m), 755 (s), 733 (w), 710 (w). EI-MS: m/z = 429 (100%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C29H55MgN5Si): C, 65.9 (66.2); H, 10.4 (10.5); N, 13.1 (13.3)%.
Me2), 5.04 (1 H, s, C(Me2pz)2), 2.27 (2 H, m, 2-MgnBu), 2.09 (6 H, s, 3-N2C3H2), 1.90 (2 H, m, 3JHH = 7.2 Hz, 3-MgnBu), 1.57 (6 H, s, 5-N2C3H2), 1.30 (3 H, t, 3JHH = 7.3 Hz, 4-MgnBu), 0.49 (2 H, m, 1-MgnBu), 0.05 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 157.7 (1-C6H5), 150.4 (3-N23HMe2), 139.5 (5-N23HMe2), 129.5 (3-C6H5), 121.7 (2-C6H5), 115.0 (4-C6H5), 105.9 (4-N23HMe2), 63.3 (H(Me2pz)2), 33.4 (2-MgnBu), 32.7 (3-MgnBu), 14.7 (1-MgnBu), 12.9 (3-N2C3H2), 10.3 (5-N2C3H2), 8.8 (4-MgnBu), −1.4 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2925 (s), 2361 (m), 1587 (s), 1553 (s), 1489 (w), 1457 (w), 1377 (m), 1291 (m), 1183 (m), 1078 (s), 1047 (s), 990 (s), 942 (s), 827 (m), 804 (w), 768 (s), 738 (m), 694 (m). EI-MS: m/z = 376 (100%) [M − nBu]+. Anal. found (calcd for C23H35MgN5Si): C, 63.7 (63.7); H, 8.1 (8.1); N, 16.2 (16.1)%.
C(tBu2pz)2), 5.96 (2 H, s, N2C3tBu2), 2.08 (2 H, m, 2-MgnBu), 1.83 (2 H, app. sext, app. 3JHH = 7.2 Hz, 3-MgnBu), 1.47 (18 H, s, 3-N2C3H2), 1.27 (3 H, m, 4-MgnBu), 1.10 (18 H, s, 5-N2C3H2), 0.49 (2 H, m, 1-MgnBu), 0.15 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 164.0 (3-N23HtBu2), 157.1 (1-C6H5), 153.8 (5-N23HtBu2), 129.0 (3-C6H5), 124.4 (2-C6H5), 116.2 (4-C6H5), 103.2 (4-N23HtBu2), 69.2 (H(tBu2pz)2), 33.0 (3-N2C3H(Me3)2), 32.8 (2-MgnBu), 32.6 (3-MgnBu), 32.2 (5-N2C3H(Me3)2), 31.5 (3-N2C3H(C3)2), 30.7 (5-N2C3H(C3)2), 15.8 (1-MgnBu), 14.6 (4-MgnBu), 0.33 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 1587 (s), 1534 (w), 1486 (s), 1425 (w), 1367 (m), 1284 (s), 1222 (w), 1209 (m), 1173 (w), 1148 (w), 1059 (m), 1027 (m), 995 (m), 957 (s), 871 (w), 825 (m), 800 (s), 791 (m), 764 (m). EI-MS: m/z = 429 (60%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C35H59MgN5Si): C, 69.7 (69.8); H, 9.9 (9.9); N, 11.7 (11.6)%.
Me2)2), 5.34 (2 H, s, N2C3Me2), 5.00 (1 H, s, C(Me2pz)2), 3.85 (1 H, sept, 3JHH = 6.3 Hz, CMe2), 2.36 (6 H, s, 3-N2C3H2), 1.55 (6 H, s, 5-N2C3H2), 1.49 (6 H, d, 3JHH = 6.3 Hz, HC2), 0.55 (12 H, d, 3JHH = 3.4 Hz, N(SiH2)2), −0.90 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 149.9 (3-N23HMe2), 139.4 (5-N23HMe2), 105.8 (4-N23HMe2), 63.8 (H(Me2pz)2), 47.5 (HMe2), 31.5 (HC2), 13.8 (3-N2C3H2), 10.4 (5-N2C3H2), 4.6 (N(SiHMe2)2), 1.3 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2925 (s), 2361 (m), 2067 (w), 1553 (m), 1506 (w), 1459 (s), 1377 (s), 1315 (s), 1280 (m), 1242 (s), 1164 (s), 1132 (m), 1045 (m), 945 (m), 892 (s), 826 (m), 800 (m), 756 (m), 729 (m). EI-MS: m/z = 474 (100%) [M]+, 342 (20%) [M − N(SiHMe2)2]+. Anal. found (calcd for C20H42MgN6Si3): C, 50.2 (50.5); H, 8.7 (8.9); N, 17.6 (17.7)%.
C(tBu2pz)2), 6.03 (2 H, s, N2C3tBu2), 5.24 (2 H, sept, 3JHH = 2.7 Hz, SiMe2), 1.59 (18 H, s, 3-N2C3H2), 1.11 (18 H, s, 5-N2C3H2), 0.38 (12 H, 3JHH = 2.7 Hz, SiH2), −0.05 (6 H, s, Si2). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): 165.5 (3-N23HtBu2), 156.8 (1-C6H5), 155.9 (5-N23HtBu2), 128.8 (3-C6H5), 126.4 (2-C6H5), 117.5 (4-C6H5), 104.7 (4-N23HtBu2), 69.0 (H(tBu2pz)2), 33.1 (5-N2C3H(Me3)2), 33.0 (3-N2C3H(Me3)2), 31.6 (5-N2C3H(Me3)2), 31.4 (3-N2C3H(Me3)2), 3.7 (SiH2), 0.4 (Si2). IR (NaCl plates, Nujol, cm−1): 2854 (s), 2058 (m), 1586 (w), 1261 (w), 1093 (s), 976 (w), 886 (m), 759 (w). EI-MS: m/z 676 [M − Mg{N(SiHMe2)2} − NPh]+ (20%). Anal. found (calcd for C35H64MgN6Si3): C, 62.0 (62.1); H, 9.6 (9.5); N, 12.4 (12.4)%.
Me2), 4.99 (1 H, s, C(Me2pz)2), 2.42 (6 H, s, 3-N2C3H2), 1.49 (6 H, s, 5-N2C3H2), 0.41 (18 H, s, N(SiMe3)2), −0.13 (SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 157.0 (1-C6H5), 150.6 (3-N23HMe2), 140.0 (5-N23HMe2), 129.2 (3-C6H5), 125.3 (2-C6H5), 117.0 (4-C6H5), 106.2 (4-N23HMe2), 62.9 (H(Me2pz)2), 14.1 (3-N2C3H2), 10.4 (5-N2C3H2), 6.3 (N(SiMe3)2), −1.1 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2972 (w), 2728 (m), 1581 (s), 1521 (s), 1419 (m), 1377 (s), 1312 (s), 1243 (s), 1172 (m), 1096 (s), 1005 (s), 959 (m), 882 (m), 826 (s), 773 (m). EI-MS: m/z = 536 (40%) [M]+, 376 (85%) [M − N(SiMe3)2]+. Anal. found (calcd for C25H44MgN6Si3): C, 55.8 (55.9); H, 8.3 (8.3); N, 15.6 (15.7)%.
C(tBu2pz)2), 6.05 (2 H, s, N2C3tBu2), 1.61 (18 H, s, 3-N2C3H2), 1.07 (18 H, s, 5-N2C3H2), 0.36 (18 H, Si3), −0.08 (6 H, s, Si2). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): 165.4 (3-N23HtBu2), 156.8 (1-C6H5), 156.3 (5-N23HtBu2), 128.4 (3-C6H5), 126.9 (2-C6H5), 118.0 (4-C6H5), 104.8 (4-N23HtBu2), 68.5 (H(tBu2pz)2), 32.8 (5-N2C3H(Me3)2), 32.6 (3-N2C3H(Me3)2), 31.4 (5-N2C3H(Me3)2), 31.2 (3-N2C3H(Me3)2), 6.1 (Si3), 0.2 (Si2). IR (NaCl plates, Nujol, cm−1): 2726 (s), 2670 (m), 1589 (w), 1549 (w), 1196 (w), 1167 (w), 1067 (s), 951 (w), 837 (m). Anal. found (calcd for C37H68MgN6Si3): C, 59.1 (63.0); H, 9.2 (9.7); N, 11.2 (11.9)%. This was the best of several attempts using recrystallised material.
Me2), 5.20 (1 H, s, C(Me2pz)2), 3.74 (4 H, br s, OC2CH2), 2.38 (6 H, s, 3-N2C3H2), 1.72 (6 H, s, 5-N2C3H2), 1.34 (4 H, br s, OCH2C2), 0.45 (18 H, s, N(SiMe3)2), 0.07 (SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.8 (1-C6H5), 150.2 (3-N23HMe2), 139.9 (5-N23HMe2), 129.2 (3-C6H5), 121.7 (2-C6H5), 113.9 (4-C6H5), 106.4 (4-N23HMe2), 68.7 (OH2CH2), 64.7 (H(Me2pz)2), 25.4 (OCH2H2), 14.4 (3-N2C3H2), 11.0 (5-N2C3H2), 6.0 (N(SiMe3)2), 2.9 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2854 (w), 2361 (m), 1576 (s), 1521 (s), 1496 (w), 1458 (s), 1377 (s), 1259 (m), 1019 (m), 975 (m), 881 (m), 837 (w), 722 (s), 667 (m). EI-MS: m/z = 460 (25%) [M − NPh(thf)]+. Anal. found (calcd for C29H51CaN6OSi3): C, 55.7 (55.8); H, 8.2 (8.2); N, 13.4 (13.5)%.
C(tBu2pz)2), 5.99 (2 H, s, N2C3tBu2), 1.44 (18 H, s, 3-N2C3H2), 1.17 (18 H, s, 5-N2C3H2), 0.28 (18 H, s, N(SiMe3)2), 0.03 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 164.1 (3-N23HtBu2), 157.1 (1-C6H5), 156.6 (5-N23HtBu2), 129.6 (3-C6H5), 125.3 (2-C6H5), 116.7 (4-C6H5), 103.9 (4-N23HtBu2), 71.1 (H(tBu2pz)2), 33.3 (3-N2C3H(Me3)2), 32.8 (5-N2C3H(Me3)2), 31.8 (3-N2C3H(C3)2), 30.9 (5-N2C3H(C3)2), 5.8 (N(SiMe3)2), 1.4 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 1584 (w), 1549 (w), 1533 (w), 1364 (m), 1197 (w), 1171 (w), 1046 (s), 995 (w), 955 (m), 879 (m), 703 (w), 589 (w). EI-MS: m/z = 429 (40%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C37H68CaN6Si3): C, 61.6 (61.6); H, 9.4 (9.5); N, 11.7 (11.7)%.
Me2), 5.00 (1 H, s, C(Me2pz)2), 1.97 (6 H, s, 3-N2C3H2), 1.51 (6 H, s, 5-N2C3H2), 0.13 (3 H, s, ZnMe), 0.01 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 163.6 (1-C6H5), 149.4 (3-N23HMe2), 138.5 (5-N23HMe2), 129.4 (3-C6H5), 121.1 (2-C6H5), 115.1 (4-C6H5), 105.7 (4-N23HMe2), 62.8 (H(Me2pz)2), 12.8 (3-N2C3H2), 10.3 (5-N2C3H2), −1.5 (SiMe), −9.8 (ZnMe). IR (NaCl plates, Nujol mull, cm−1): 2923 (w), 2360 (m), 1576 (s), 1521 (w), 1489 (w), 1457 (s), 1376 (s), 1313 (w), 1290 (s), 1248 (m), 1141 (w), 991 (w), 949 (s), 853 (w), 835 (m), 771 (m), 722 (w). EI-MS: m/z = 431 (100%) [M]+, 416 (20%) [M − Me]+. Anal. found (calcd for C20H29N5SiZn): C, 55.4 (55.5); H, 6.8 (6.8); N, 16.1 (16.2)%.
C(tBu2pz)2), 6.01 (2 H, s, N2C3tBu2), 1.45 (18 H, s, 3-N2C3H2), 1.12 (18 H, s, 5-N2C3H2), 0.41 (3 H, s, ZnMe), 0.18 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 162.9 (3-N23HtBu2), 157.0 (1-C6H5), 153.1 (5-N23HtBu2), 129.1 (3-C6H5), 123.0 (2-C6H5), 115.8 (4-C6H5), 103.2 (4-N23HtBu2), 68.5 (H(tBu2pz)2), 32.9 (3-N2C3H(Me3)2), 32.3 (5-N2C3H(Me3)2), 31.6 (3-N2C3H(C3)2), 30.6 (5-N2C3H(C3)2), 0.0 (SiMe), −3,4 (ZnMe). IR (NaCl plates, Nujol mull, cm−1): 3143 (w), 3060 (w), 1588 (s), 1557 (m), 1542 (m), 1528 (s), 1489 (s), 1396 (w), 1364 (m), 1357 (m), 1291 (s), 1211 (m), 1178 (w), 1169 (m), 1152 (w), 1130 (w), 1075 (w), 1060 (m), 1023 (m), 993 (m), 961 (s), 867 (w), 843 (s), 793 (m), 765 (m), 751 (m), 709 (w), 692 (m). EI-MS: m/z = 429 (100%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C32H53N5SiZn): C, 63.8 (63.9); H, 8.8 (8.9); N, 11.6 (11.7)%.
) ppm. IR (NaCl plates, Nujol): 1587 (w), 1544 (m), 1418 (s), 1359 (m), 1297 (s), 1235 (m), 1207 (m), 1097 (w), 1064 (w), 1023 (w), 994 (m), 982 (m), 829 (m), 821 (m), 776 (m), 755 (s), 709 (w), 693 (w), 615 (w), 530 (w) cm−1. EI-MS: m/z: 555 [M]+ (8%), 429 [M − N(Li)Xyl]+ (100%), 371 [M − SiMe2N(Li)Xyl]+ (8%), Anal. Found (calcd for C33H54LiN5Si): C, 71.4 (71.3); H, 9.7 (9.8); N, 12.7 (12.6)%.
C(tBu2pz)2), 5.96 (2 H, s, N2C3tBu2), 3.61 (1 H, sept, 3JHH = 6.3 Hz, CMe2), 1.54 (6 H, d, 3JHH = 6.4 Hz, HC2), 1.52 (18 H, s, 3-N2C3H2), 1.16 (18 H, s, 5-N2C3H2), 0.05 (6 H, s, SiMe), −0.20 (3 H, s, MgMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 163.2 (3-N23HtBu2), 153.1 (5-N23HtBu2), 102.9 (4-N23HtBu2), 69.8 (H(tBu2pz)2), 46.8 (HMe2), 33.0 (HC2), 32.3 (3-N2C3H(Me3)2), 32.1 (5-N2C3H(Me3)2), 31.6 (3-N2C3H(C3)2), 30.7 (5-N2C3H(C3)2), 1.2 (SiMe), −3.6 (MgMe). IR (NaCl plates, Nujol mull, cm−1): 2795 (w), 2599 (w), 1615 (w), 1526 (m), 1423 (m), 1348 (m), 1305 (m), 1244 (m), 1220 (m), 1209 (m), 1139 (m), 1111 (m), 1061 (s), 1029 (m), 899 (m), 829 (m), 755 (s), 733 (w), 710 (w). EI-MS: m/z = 429 (100%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C29H55MgN5Si): C, 65.9 (66.2); H, 10.4 (10.5); N, 13.1 (13.3)%.
Me2), 5.04 (1 H, s, C(Me2pz)2), 2.27 (2 H, m, 2-MgnBu), 2.09 (6 H, s, 3-N2C3H2), 1.90 (2 H, m, 3JHH = 7.2 Hz, 3-MgnBu), 1.57 (6 H, s, 5-N2C3H2), 1.30 (3 H, t, 3JHH = 7.3 Hz, 4-MgnBu), 0.49 (2 H, m, 1-MgnBu), 0.05 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 157.7 (1-C6H5), 150.4 (3-N23HMe2), 139.5 (5-N23HMe2), 129.5 (3-C6H5), 121.7 (2-C6H5), 115.0 (4-C6H5), 105.9 (4-N23HMe2), 63.3 (H(Me2pz)2), 33.4 (2-MgnBu), 32.7 (3-MgnBu), 14.7 (1-MgnBu), 12.9 (3-N2C3H2), 10.3 (5-N2C3H2), 8.8 (4-MgnBu), −1.4 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2925 (s), 2361 (m), 1587 (s), 1553 (s), 1489 (w), 1457 (w), 1377 (m), 1291 (m), 1183 (m), 1078 (s), 1047 (s), 990 (s), 942 (s), 827 (m), 804 (w), 768 (s), 738 (m), 694 (m). EI-MS: m/z = 376 (100%) [M − nBu]+. Anal. found (calcd for C23H35MgN5Si): C, 63.7 (63.7); H, 8.1 (8.1); N, 16.2 (16.1)%.
C(tBu2pz)2), 5.96 (2 H, s, N2C3tBu2), 2.08 (2 H, m, 2-MgnBu), 1.83 (2 H, app. sext, app. 3JHH = 7.2 Hz, 3-MgnBu), 1.47 (18 H, s, 3-N2C3H2), 1.27 (3 H, m, 4-MgnBu), 1.10 (18 H, s, 5-N2C3H2), 0.49 (2 H, m, 1-MgnBu), 0.15 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 164.0 (3-N23HtBu2), 157.1 (1-C6H5), 153.8 (5-N23HtBu2), 129.0 (3-C6H5), 124.4 (2-C6H5), 116.2 (4-C6H5), 103.2 (4-N23HtBu2), 69.2 (H(tBu2pz)2), 33.0 (3-N2C3H(Me3)2), 32.8 (2-MgnBu), 32.6 (3-MgnBu), 32.2 (5-N2C3H(Me3)2), 31.5 (3-N2C3H(C3)2), 30.7 (5-N2C3H(C3)2), 15.8 (1-MgnBu), 14.6 (4-MgnBu), 0.33 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 1587 (s), 1534 (w), 1486 (s), 1425 (w), 1367 (m), 1284 (s), 1222 (w), 1209 (m), 1173 (w), 1148 (w), 1059 (m), 1027 (m), 995 (m), 957 (s), 871 (w), 825 (m), 800 (s), 791 (m), 764 (m). EI-MS: m/z = 429 (60%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C35H59MgN5Si): C, 69.7 (69.8); H, 9.9 (9.9); N, 11.7 (11.6)%.
Me2)2), 5.34 (2 H, s, N2C3Me2), 5.00 (1 H, s, C(Me2pz)2), 3.85 (1 H, sept, 3JHH = 6.3 Hz, CMe2), 2.36 (6 H, s, 3-N2C3H2), 1.55 (6 H, s, 5-N2C3H2), 1.49 (6 H, d, 3JHH = 6.3 Hz, HC2), 0.55 (12 H, d, 3JHH = 3.4 Hz, N(SiH2)2), −0.90 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 149.9 (3-N23HMe2), 139.4 (5-N23HMe2), 105.8 (4-N23HMe2), 63.8 (H(Me2pz)2), 47.5 (HMe2), 31.5 (HC2), 13.8 (3-N2C3H2), 10.4 (5-N2C3H2), 4.6 (N(SiHMe2)2), 1.3 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2925 (s), 2361 (m), 2067 (w), 1553 (m), 1506 (w), 1459 (s), 1377 (s), 1315 (s), 1280 (m), 1242 (s), 1164 (s), 1132 (m), 1045 (m), 945 (m), 892 (s), 826 (m), 800 (m), 756 (m), 729 (m). EI-MS: m/z = 474 (100%) [M]+, 342 (20%) [M − N(SiHMe2)2]+. Anal. found (calcd for C20H42MgN6Si3): C, 50.2 (50.5); H, 8.7 (8.9); N, 17.6 (17.7)%.
C(tBu2pz)2), 6.03 (2 H, s, N2C3tBu2), 5.24 (2 H, sept, 3JHH = 2.7 Hz, SiMe2), 1.59 (18 H, s, 3-N2C3H2), 1.11 (18 H, s, 5-N2C3H2), 0.38 (12 H, 3JHH = 2.7 Hz, SiH2), −0.05 (6 H, s, Si2). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): 165.5 (3-N23HtBu2), 156.8 (1-C6H5), 155.9 (5-N23HtBu2), 128.8 (3-C6H5), 126.4 (2-C6H5), 117.5 (4-C6H5), 104.7 (4-N23HtBu2), 69.0 (H(tBu2pz)2), 33.1 (5-N2C3H(Me3)2), 33.0 (3-N2C3H(Me3)2), 31.6 (5-N2C3H(Me3)2), 31.4 (3-N2C3H(Me3)2), 3.7 (SiH2), 0.4 (Si2). IR (NaCl plates, Nujol, cm−1): 2854 (s), 2058 (m), 1586 (w), 1261 (w), 1093 (s), 976 (w), 886 (m), 759 (w). EI-MS: m/z 676 [M − Mg{N(SiHMe2)2} − NPh]+ (20%). Anal. found (calcd for C35H64MgN6Si3): C, 62.0 (62.1); H, 9.6 (9.5); N, 12.4 (12.4)%.
Me2), 4.99 (1 H, s, C(Me2pz)2), 2.42 (6 H, s, 3-N2C3H2), 1.49 (6 H, s, 5-N2C3H2), 0.41 (18 H, s, N(SiMe3)2), −0.13 (SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 157.0 (1-C6H5), 150.6 (3-N23HMe2), 140.0 (5-N23HMe2), 129.2 (3-C6H5), 125.3 (2-C6H5), 117.0 (4-C6H5), 106.2 (4-N23HMe2), 62.9 (H(Me2pz)2), 14.1 (3-N2C3H2), 10.4 (5-N2C3H2), 6.3 (N(SiMe3)2), −1.1 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2972 (w), 2728 (m), 1581 (s), 1521 (s), 1419 (m), 1377 (s), 1312 (s), 1243 (s), 1172 (m), 1096 (s), 1005 (s), 959 (m), 882 (m), 826 (s), 773 (m). EI-MS: m/z = 536 (40%) [M]+, 376 (85%) [M − N(SiMe3)2]+. Anal. found (calcd for C25H44MgN6Si3): C, 55.8 (55.9); H, 8.3 (8.3); N, 15.6 (15.7)%.
C(tBu2pz)2), 6.05 (2 H, s, N2C3tBu2), 1.61 (18 H, s, 3-N2C3H2), 1.07 (18 H, s, 5-N2C3H2), 0.36 (18 H, Si3), −0.08 (6 H, s, Si2). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): 165.4 (3-N23HtBu2), 156.8 (1-C6H5), 156.3 (5-N23HtBu2), 128.4 (3-C6H5), 126.9 (2-C6H5), 118.0 (4-C6H5), 104.8 (4-N23HtBu2), 68.5 (H(tBu2pz)2), 32.8 (5-N2C3H(Me3)2), 32.6 (3-N2C3H(Me3)2), 31.4 (5-N2C3H(Me3)2), 31.2 (3-N2C3H(Me3)2), 6.1 (Si3), 0.2 (Si2). IR (NaCl plates, Nujol, cm−1): 2726 (s), 2670 (m), 1589 (w), 1549 (w), 1196 (w), 1167 (w), 1067 (s), 951 (w), 837 (m). Anal. found (calcd for C37H68MgN6Si3): C, 59.1 (63.0); H, 9.2 (9.7); N, 11.2 (11.9)%. This was the best of several attempts using recrystallised material.
Me2), 5.20 (1 H, s, C(Me2pz)2), 3.74 (4 H, br s, OC2CH2), 2.38 (6 H, s, 3-N2C3H2), 1.72 (6 H, s, 5-N2C3H2), 1.34 (4 H, br s, OCH2C2), 0.45 (18 H, s, N(SiMe3)2), 0.07 (SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 158.8 (1-C6H5), 150.2 (3-N23HMe2), 139.9 (5-N23HMe2), 129.2 (3-C6H5), 121.7 (2-C6H5), 113.9 (4-C6H5), 106.4 (4-N23HMe2), 68.7 (OH2CH2), 64.7 (H(Me2pz)2), 25.4 (OCH2H2), 14.4 (3-N2C3H2), 11.0 (5-N2C3H2), 6.0 (N(SiMe3)2), 2.9 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 2854 (w), 2361 (m), 1576 (s), 1521 (s), 1496 (w), 1458 (s), 1377 (s), 1259 (m), 1019 (m), 975 (m), 881 (m), 837 (w), 722 (s), 667 (m). EI-MS: m/z = 460 (25%) [M − NPh(thf)]+. Anal. found (calcd for C29H51CaN6OSi3): C, 55.7 (55.8); H, 8.2 (8.2); N, 13.4 (13.5)%.
C(tBu2pz)2), 5.99 (2 H, s, N2C3tBu2), 1.44 (18 H, s, 3-N2C3H2), 1.17 (18 H, s, 5-N2C3H2), 0.28 (18 H, s, N(SiMe3)2), 0.03 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 164.1 (3-N23HtBu2), 157.1 (1-C6H5), 156.6 (5-N23HtBu2), 129.6 (3-C6H5), 125.3 (2-C6H5), 116.7 (4-C6H5), 103.9 (4-N23HtBu2), 71.1 (H(tBu2pz)2), 33.3 (3-N2C3H(Me3)2), 32.8 (5-N2C3H(Me3)2), 31.8 (3-N2C3H(C3)2), 30.9 (5-N2C3H(C3)2), 5.8 (N(SiMe3)2), 1.4 (SiMe). IR (NaCl plates, Nujol mull, cm−1): 1584 (w), 1549 (w), 1533 (w), 1364 (m), 1197 (w), 1171 (w), 1046 (s), 995 (w), 955 (m), 879 (m), 703 (w), 589 (w). EI-MS: m/z = 429 (40%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C37H68CaN6Si3): C, 61.6 (61.6); H, 9.4 (9.5); N, 11.7 (11.7)%.
Me2), 5.00 (1 H, s, C(Me2pz)2), 1.97 (6 H, s, 3-N2C3H2), 1.51 (6 H, s, 5-N2C3H2), 0.13 (3 H, s, ZnMe), 0.01 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 163.6 (1-C6H5), 149.4 (3-N23HMe2), 138.5 (5-N23HMe2), 129.4 (3-C6H5), 121.1 (2-C6H5), 115.1 (4-C6H5), 105.7 (4-N23HMe2), 62.8 (H(Me2pz)2), 12.8 (3-N2C3H2), 10.3 (5-N2C3H2), −1.5 (SiMe), −9.8 (ZnMe). IR (NaCl plates, Nujol mull, cm−1): 2923 (w), 2360 (m), 1576 (s), 1521 (w), 1489 (w), 1457 (s), 1376 (s), 1313 (w), 1290 (s), 1248 (m), 1141 (w), 991 (w), 949 (s), 853 (w), 835 (m), 771 (m), 722 (w). EI-MS: m/z = 431 (100%) [M]+, 416 (20%) [M − Me]+. Anal. found (calcd for C20H29N5SiZn): C, 55.4 (55.5); H, 6.8 (6.8); N, 16.1 (16.2)%.
C(tBu2pz)2), 6.01 (2 H, s, N2C3tBu2), 1.45 (18 H, s, 3-N2C3H2), 1.12 (18 H, s, 5-N2C3H2), 0.41 (3 H, s, ZnMe), 0.18 (6 H, s, SiMe). 13C{1H} NMR (C6D6, 75.4 MHz, 293 K): δ 162.9 (3-N23HtBu2), 157.0 (1-C6H5), 153.1 (5-N23HtBu2), 129.1 (3-C6H5), 123.0 (2-C6H5), 115.8 (4-C6H5), 103.2 (4-N23HtBu2), 68.5 (H(tBu2pz)2), 32.9 (3-N2C3H(Me3)2), 32.3 (5-N2C3H(Me3)2), 31.6 (3-N2C3H(C3)2), 30.6 (5-N2C3H(C3)2), 0.0 (SiMe), −3,4 (ZnMe). IR (NaCl plates, Nujol mull, cm−1): 3143 (w), 3060 (w), 1588 (s), 1557 (m), 1542 (m), 1528 (s), 1489 (s), 1396 (w), 1364 (m), 1357 (m), 1291 (s), 1211 (m), 1178 (w), 1169 (m), 1152 (w), 1130 (w), 1075 (w), 1060 (m), 1023 (m), 993 (m), 961 (s), 867 (w), 843 (s), 793 (m), 765 (m), 751 (m), 709 (w), 692 (m). EI-MS: m/z = 429 (100%) [HC(tBu2pz)2SiMe2]+. Anal. found (calcd for C32H53N5SiZn): C, 63.8 (63.9); H, 8.8 (8.9); N, 11.6 (11.7)%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. D. S and M. G. C. thank the EPSRC (Grant EP/P502667/1 and EP/P503876/1 respectively) and M. L. B. thanks the University of Oxford and the European Union Alβan Programme for financial support (Grant E05D059950MX). The authors also thank Chemical Crystallography for use of the diffractometers and help with structure solutions.Notes and references
- (a) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda and A. Lara-Sánchez, Dalton Trans., 2004, 1499 RSC; (b) C. Pettinari and R. Pettinari, Coord. Chem. Rev., 2005, 249, 663 CrossRef CAS; (c) A. Otero, J. Fernández-Baeza, A. Lara-Sánchez and L. F. Sánchez-Barba, Coord. Chem. Rev., 2013, 257, 1806 CrossRef CAS.
- (a) S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 3170 CrossRef CAS; (b) S. Trofimenko, Chem. Rev., 1993, 93, 943 CrossRef CAS; (c) S. Trofimenko, Scorpionates. The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 CrossRef; (d) S. Trofimenko, Polyhedron, 2004, 23, 197 CrossRef CAS; (e) S. Trofimenko, in Prog. Inorg. Chem, John Wiley & Sons, Inc., 2007, p. 115 Search PubMed.
- (a) A. Otero, J. Fernández-Baeza, A. Lara-Sánchez, J. Tejeda and L. F. Sánchez-Barba, Eur. J. Inorg. Chem., 2008, 2008, 5309 CrossRef; (b) M. Honrado, A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez, J. Tejeda, M. P. Carrion, J. Martínez-Ferrer, A. Garcés and A. M. Rodríguez, Organometallics, 2013, 32, 3437 CrossRef CAS; (c) J. A. Castro-Osma, C. Alonso-Moreno, M. V. Gómez, I. Marquez-Segovia, A. Otero, A. Lara-Sánchez, J. Fernández-Baeza, L. F. Sánchez-Barba and A. M. Rodríguez, Dalton Trans., 2013, 42, 14240 RSC; (d) A. Otero, J. Fernández-Baeza, J. Tejeda, A. Lara-Sánchez, S. Franco, J. Martínez-Ferrer, M. P. Carrion, I. López-Solera, A. M. Rodríguez and L. F. Sánchez-Barba, Inorg. Chem., 2011, 50, 1826 CrossRef CAS PubMed; (e) T. Godau, S. M. Bleifuss, A. L. Mueller, T. Roth, S. Hoffmann, F. W. Heinemann and N. Burzlaff, Dalton Trans., 2011, 40, 6547 RSC; (f) A. Otero, J. Fernández-Baeza, J. Tejeda, A. Lara-Sánchez, M. Sánchez-Molina, S. Franco, I. López-Solera, A. M. Rodríguez, L. F. Sánchez-Barba, S. Morante-Zarcero and A. Garcés, Inorg. Chem., 2009, 48, 5540 CrossRef CAS PubMed.
- (a) M. Abubekerov, J. Wei, K. R. Swartz, Z. Xie, Q. Pei and P. L. Diaconescu, Chem. Sci., 2018, 9, 2168 RSC; (b) F. de la Cruz-Martínez, J. Martínez, M. A. Gaona, J. Fernández-Baeza, L. F. Sánchez-Barba, A. M. Rodríguez, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2018, 6, 5322 CrossRef; (c) G. Türkoglu, S. Tampier, F. Strinitz, F. W. Heinemann, E. Hübner and N. Burzlaff, Organometallics, 2012, 31, 2166 CrossRef; (d) A. Otero, A. Lara-Sánchez, C. Nájera, J. Fernández-Baeza, I. Márquez-Segovia, J. A. Castro-Osma, J. Martínez, L. F. Sánchez-Barba and A. M. Rodríguez, Organometallics, 2012, 31, 2244 CrossRef CAS; (e) A. F. R. Kilpatrick, S. V. Kulangara, M. G. Cushion, R. Duchateau and P. Mountford, Dalton Trans., 2010, 39, 3653 RSC; (f) A. Garcés, L. F. Sánchez-Barba, C. Alonso-Moreno, M. Fajardo, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez, I. López-Solera and A. M. Rodríguez, Inorg. Chem., 2010, 49, 2859 CrossRef PubMed; (g) H. Kopf, B. Holzberger, C. Pietraszuk, E. Hübner and N. Burzlaff, Organometallics, 2008, 27, 5894 CrossRef CAS; (h) A. Otero, J. Fernández-Baeza, A. Lara-Sánchez, A. Antiñolo, J. Tejeda, E. Martínez-Caballero, I. Márquez-Segovia, I. López-Solera, L. F. Sánchez-Barba and C. Alonso-Moreno, Inorg. Chem., 2008, 47, 4996 CrossRef CAS PubMed; (i) S. Milione, V. Bertolasi, T. Cuenca and A. Grassi, Organometallics, 2005, 24, 4915 CrossRef CAS; (j) Z. Mou, H. Xie, M. Wang, N. Liu, C. Yao, L. Li, J. Liu, S. Li and D. Cui, Organometallics, 2015, 34, 3944 CrossRef CAS.
- (a) J. R. Gardinier, J. S. Hewage, J. Hoffman, S. V. Lindeman, D. E. Williams and N. B. Shustova, Eur. J. Inorg. Chem., 2016, 2016, 2615 CrossRef CAS; (b) G. Durá, M. C. Carrión, F. A. Jalón, B. R. Manzano and A. M. Rodríguez, Cryst. Growth Des., 2015, 15, 5174 CrossRef; (c) I. Bassanetti and L. Marchiò, Inorg. Chem., 2011, 50, 10786 CrossRef CAS PubMed; (d) D. L. Reger, E. A. Foley and M. D. Smith, Inorg. Chem., 2009, 49, 234 CrossRef PubMed.
- (a) B. S. Hammes and C. J. Carrano, Inorg. Chem., 2001, 40, 919 CrossRef CAS PubMed; (b) B. S. Hammes, B. S. Chohan, J. T. Hoffman, S. Einwächter and C. J. Carrano, Inorg. Chem., 2004, 43, 7800 CrossRef CAS PubMed; (c) J. S. Fell, D. M. Steele, T. C. Hatcher and B. F. Gherman, Theor. Chem. Acc., 2015, 134, 71 Search PubMed; (d) C. J. Carrano, Eur. J. Inorg. Chem., 2016, 2016, 2377 CrossRef CAS; (e) M. Pellei, V. Gandin, C. Cimarelli, W. Quaglia, N. Mosca, L. Bagnarelli, C. Marzano and C. Santini, J. Inorg. Biochem., 2018, 187, 33 CrossRef CAS PubMed.
- D. J. Wasylenko, H. M. Tatlock, L. S. Bhandari, J. R. Gardinier and C. P. Berlinguette, Chem. Sci., 2013, 4, 734 RSC.
- (a) A. Garcés, L. F. Sánchez-Barba, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2017, 36, 884 CrossRef; (b) S. Tampier and N. Burzlaff, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 981 CrossRef CAS PubMed; (c) N. G. Spiropulos, G. C. Chingas, M. Sullivan, J. T. York and E. C. Brown, Inorg. Chim. Acta, 2011, 376, 562 CrossRef CAS; (d) L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, A. Otero, C. Alonso-Moreno, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2011, 30, 2775 CrossRef; (e) I. Hegelmann, A. Beck, C. Eichhorn, B. Weibert and N. Burzlaff, Eur. J. Inorg. Chem., 2003, 2003, 339 CrossRef; (f) A. Beck, B. Weibert and N. Burzlaff, Eur. J. Inorg. Chem., 2001, 2001, 521 CrossRef.
- (a) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717 CrossRef CAS PubMed; (b) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48 RSC; (c) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS.
- (a) A. Garcés, L. F. Sánchez-Barba, J. Fernández-Baeza, A. Otero, M. Honrado, A. Lara-Sánchez and A. M. Rodríguez, Inorg. Chem., 2013, 52, 12691 CrossRef PubMed; (b) M. Honrado, A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez, J. Martínez-Ferrer, S. Sobrino and A. M. Rodríguez, Organometallics, 2015, 34, 3196 CrossRef CAS.
- (a) C. Alonso-Moreno, A. Garcés, L. F. Sánchez-Barba, M. Fajardo, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez, A. Antiñolo, L. Broomfield, M. I. López-Solera and A. M. Rodríguez, Organometallics, 2008, 27, 1310 CrossRef CAS; (b) M. Honrado, A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2016, 35, 189 CrossRef CAS; (c) A. Garcés, L. F. Sánchez-Barba, J. Fernández-Baeza, A. Otero, I. Fernández, A. Lara-Sánchez and A. M. Rodríguez, Inorg. Chem., 2018, 57, 12132 CrossRef PubMed.
- (a) J. A. Castro-Osma, C. Alonso-Moreno, A. Lara-Sánchez, A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba and A. M. Rodríguez, Dalton Trans., 2015, 44, 12388 RSC; (b) J. A. Castro-Osma, C. Alonso-Moreno, I. Márquez-Segovia, A. Otero, A. Lara-Sánchez, J. Fernández-Baeza, A. M. Rodríguez, L. F. Sánchez-Barba and J. C. García-Martínez, Dalton Trans., 2013, 42, 9325 RSC.
- Z. Mou, B. Liu, M. Wang, H. Xie, P. Li, L. Li, S. Li and D. Cui, Chem. Commun., 2014, 50, 11411 RSC.
- (a) S. Harder, Alkaline-Earth Metal Compounds: Oddities and Applications, in Top. Organomet. Chem, Springer GmbH, 2013, vol. 45 CrossRef; (b) M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972 RSC.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168 CrossRef CAS PubMed.
- (a) M. G. Cushion, J. Meyer, A. Heath, A. D. Schwarz, I. Fernández, F. Breher and P. Mountford, Organometallics, 2010, 29, 1174 CrossRef CAS; (b) A. D. Schofield, M. L. Barros, M. G. Cushion, A. D. Schwarz and P. Mountford, Dalton Trans., 2009, 85 RSC; (c) H. R. Bigmore, J. Meyer, I. Krummenacher, H. Rüegger, E. Clot, P. Mountford and F. Breher, Chem. – Eur. J., 2008, 14, 5918 CrossRef CAS PubMed.
- R. G. Howe, C. S. Tredget, S. C. Lawrence, S. Subongkoj, A. R. Cowley and P. Mountford, Chem. Commun., 2006, 223 RSC.
- (a) A. R. Katritzky, A. E. Abdel-Rahman, D. E. Leahy and O. A. Schwarz, Tetrahedron, 1983, 39, 4133 CrossRef CAS; (b) E. Diez-Barra, A. de la Hoz, A. Sánchez-Migallón and J. Tejeda, J. Chem. Soc., Perkin Trans. 1, 1993, 1079 RSC; (c) A. Antiñolo, F. Carrillo-Hermosilla, E. Díez-Barra, J. Fernández-Baeza, M. Fernández-López, A. Lara-Sánchez, A. Moreno, A. Otero, A. Maria Rodríguez and J. Tejeda, J. Chem. Soc., Dalton Trans., 1998, 3737 RSC.
- (a) A. Otero, J. Fernández-Baeza, A. Antiñolo, F. Carrillo-Hermosilla, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, M. Fernández-López, A. M. Rodríguez and I. López-Solera, Inorg. Chem., 2002, 41, 5193 CrossRef CAS PubMed; (b) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, M. Sánchez-Molina, S. Franco, I. López-Solera and A. M. Rodríguez, Dalton Trans., 2006, 4359 RSC; (c) A. Otero, J. Fernández-Baeza, J. Tejeda, A. Lara-Sánchez, M. Sánchez-Molina, S. Franco, I. López-Solera, A. M. Rodríguez, L. F. Sánchez-Barba, S. Morante-Zarcero and A. Garcés, Inorg. Chem., 2009, 48, 5540 CrossRef CAS PubMed.
- (a) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, M. Sánchez-Molina, S. Franco, I. López-Solera and A. M. Rodríguez, Eur. J. Inorg. Chem., 2006, 2006, 707 CrossRef; (b) I. Bassanetti, M. Gennari, L. Marchiò, M. Terenghi and L. Elviri, Inorg. Chem., 2010, 49, 7007 CrossRef CAS PubMed.
- (a) A. Otero, J. Fernández-Baeza, J. Tejeda, A. Lara-Sánchez, S. Franco, J. Martínez-Ferrer, M. P. Carrión, I. López-Solera, A. M. Rodríguez and L. F. Sánchez-Barba, Inorg. Chem., 2011, 50, 1826 CrossRef CAS PubMed; (b) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. F. Sánchez-Barba, I. López-Solera and A. M. Rodríguez, Inorg. Chem., 2007, 46, 1760 CrossRef CAS PubMed.
- (a) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, A. M. Rodríguez and M. A. Maestro, J. Am. Chem. Soc., 2004, 126, 1330 CrossRef CAS PubMed; (b) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. F. Sánchez-Barba, M. Sánchez-Molina, A. M. Rodríguez, C. Bo and M. Urbano-Cuadrado, Organometallics, 2007, 26, 4310 CrossRef CAS.
- D. Adhikari, G. Zhao, F. Basuli, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2006, 45, 1604 CrossRef CAS PubMed.
- (a) L. F. Sánchez-Barba, A. Garcés, M. Fajardo, C. Alonso-Moreno, J. Fernández-Baeza, A. Otero, A. Antiñolo, J. Tejeda, A. Lara-Sánchez and M. I. López-Solera, Organometallics, 2007, 26, 6403 CrossRef; (b) L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, A. Otero, M. Honrado, A. Lara-Sánchez, A. M. Rodríguez and I. López-Solera, Eur. J. Inorg. Chem., 2014, 2014, 1922 CrossRef; (c) R. Han and G. Parkin, J. Am. Chem. Soc., 1992, 114, 748 CrossRef CAS.
- S. Trofimenko, J. C. Calabrese and J. S. Thompson, Inorg. Chem., 1987, 26, 1507 CrossRef CAS.
- R. Han and G. Parkin, Organometallics, 1991, 10, 1010 CrossRef CAS.
- P. J. Bailey, C. M. E. Dick, S. Fabre and S. Parsons, J. Chem. Soc., Dalton Trans., 2000, 1655 RSC.
- (a) R. Han, A. Looney and G. Parkin, J. Am. Chem. Soc., 1989, 111, 7276 CrossRef CAS; (b) J. L. Kisko, T. Fillebeen, T. Hascall and G. Parkin, J. Organomet. Chem., 2000, 596, 22 CrossRef CAS; (c) O. Michel, H. M. Dietrich, R. Litlabø, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Organometallics, 2012, 31, 3119 CrossRef CAS.
- (a) A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, J. Tejeda, M. Honrado, A. Garcés, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2012, 31, 4191 CrossRef CAS; (b) J. T. Hoffman and C. J. Carrano, Inorg. Chim. Acta, 2006, 359, 1248 CrossRef CAS; (c) B. S. Hammes, M. T. Kieber-Emmons, J. A. Letizia, Z. Shirin, C. J. Carrano, L. N. Zakharov and A. L. Rheingold, Inorg. Chim. Acta, 2003, 346, 227 CrossRef CAS; (d) B. S. Hammes and C. J. Carrano, Inorg. Chem., 1999, 38, 4593 CrossRef CAS PubMed.
- I. B. Gorrell, A. Looney and G. Parkin, J. Chem. Soc., Chem. Commun., 1990, 220 RSC.
- (a) Y. Huang, W. Wang, C.-C. Lin, M. P. Blake, L. Clark, A. D. Schwarz and P. Mountford, Dalton Trans., 2013, 42, 9313 RSC; (b) R. A. Collins, J. Unruangsri and P. Mountford, Dalton Trans., 2013, 42, 759 RSC; (c) L. Clark, G. B. Deacon, C. M. Forsyth, P. C. Junk, P. Mountford, J. P. Townley and J. Wang, Dalton Trans., 2013, 42, 9294 RSC; (d) M. G. Cushion and P. Mountford, Chem. Commun., 2011, 47, 2276 RSC; (e) M. P. Blake, A. D. Schwarz and P. Mountford, Organometallics, 2011, 30, 1202 CrossRef CAS; (f) A. D. Schwarz, K. R. Herbert, C. Paniagua and P. Mountford, Organometallics, 2010, 29, 4171 CrossRef CAS; (g) A. D. Schwarz, Z. Chu and P. Mountford, Organometallics, 2010, 29, 1246 CrossRef CAS; (h) H. E. Dyer, S. Huijser, N. Susperregui, F. Bonnet, A. D. Schwarz, R. Duchateau, L. Maron and P. Mountford, Organometallics, 2010, 29, 3602 CrossRef CAS; (i) L. Clark, G. B. Deacon, C. M. Forsyth, P. C. Junk, P. Mountford and J. P. Townley, Dalton Trans., 2010, 39, 6693 RSC; (j) L. Clark, M. G. Cushion, H. E. Dyer, A. D. Schwarz, R. Duchateau and P. Mountford, Chem. Commun., 2010, 46, 273 RSC; (k) A. D. Schwarz, A. L. Thompson and P. Mountford, Inorg. Chem., 2009, 48, 10442 CrossRef CAS PubMed; (l) H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32 RSC; (m) F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046 CrossRef CAS PubMed.
- H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286 CrossRef CAS.
- L. F. Sánchez-Barba, C. Alonso-Moreno, A. Garcés, M. Fajardo, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez, A. M. Rodríguez and I. López-Solera, Dalton Trans., 2009, 8054 RSC.
- A. Otero, J. Fernández-Baeza, A. Antiñolo, A. Lara-Sánchez, E. Martínez-Caballero, J. Tejeda, L. F. Sánchez-Barba, C. Alonso-Moreno and I. López-Solera, Organometallics, 2008, 27, 976 CrossRef CAS.
- (a) J. A. Castro-Osma, C. Alonso-Moreno, J. C. García-Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez and A. Otero, Macromolecules, 2013, 46, 6388 CrossRef CAS; (b) S. Milione, F. Grisi, R. Centore and A. Tuzi, Organometallics, 2005, 25, 266 CrossRef.
- (a) A. Rudin and H. L. W. Hoegy, J. Polym. Sci., Part A: Polym. Chem., 1972, 10, 217 CrossRef CAS; (b) I. Barakat, P. Dubois, R. Jérôme and P. Teyssié, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 505 CrossRef CAS; (c) J. R. Dorgan, J. Janzen, D. M. Knauss, S. B. Hait, B. R. Limoges and M. H. Hutchinson, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3100 CrossRef CAS.
- P. Dubois, N. Ropson, R. Jérôme and P. Teyssié, Macromolecules, 1996, 29, 1965 CrossRef CAS.
- W. M. Stevels, M. J. K. Ankoné, P. J. Dijkstra and J. Feijen, Macromolecules, 1996, 29, 8296 CrossRef CAS.
- C. M. Manna, H. Z. Kaplan, B. Li and J. A. Byers, Polyhedron, 2014, 84, 160 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863 CrossRef CAS.
- M. Honrado, A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2014, 33, 1859 CrossRef CAS.
- C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832 RSC.
- M. Westerhausen, Inorg. Chem., 1991, 30, 96 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Methods Enzymol, ed. C. W. Carter Jr., Academic Press, 1997, vol. 276, p. 307 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
Footnotes |
| † This paper is dedicated in admiration and friendship to Professor F. Geoff N. Cloke FRS. |
| ‡ Electronic supplementary information (ESI) available: Further experimental details, X-ray crystallographic details, CIF files and DFT calculation details. CCDC 1885899–1885910. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04985h |
| This journal is © The Royal Society of Chemistry 2019 |