A highly selective G-quadruplex-based luminescent switch-on probe for the detection of nanomolar strontium(II) ions in sea water†
Ka-Ho
Leung‡
a,
Victor Pui-Yan
Ma‡
a,
Hong-Zhang
He
a,
Daniel Shiu-Hin
Chan
a,
Hui
Yang
a,
Chung-Hang
Leung
bc and
Dik-Lung
Ma
*a
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China. E-mail: edmondma@hkbu.edu.hk
bState Key Laboratory of Quality Research in Chinese Medicine, University of Macau, Macao SAR, China
cInstitute of Chinese Medical Sciences, University of Macau, Macao SAR, China
First published on 26th July 2012
Abstract
A G-quadruplex-selective luminescent iridium(III) switch-on probe has been developed for the selective detection of Sr2+ ions in buffered solutions and sea water. The method achieves high sensitivity towards Sr2+ ions with a detection limit in the nanomolar range and high selectivity for Sr2+ ions over other metal ions.
Alkaline earth metal ions play essential roles in the regulation of various biological processes. For example, Ca2+ ions are involved with neurotransmitter release, the contraction of cardiac muscle, the development of skeletal muscle and as cofactors of enzymes regulating the blood clotting cascade.1 The Sr2+ ion, as a congener of Ca2+, also plays an important role in bone growth and the prevention of bone fractures.2 However, evidence has suggested that excess strontium beyond a threshold concentration could lead to cardiac and neurological disorders,3 as cells uptake both Ca2+ and Sr2+ due to their similar properties. Therefore, a selective and reliable detection method for Sr2+ ions is of significant interest to the scientific community.
A number of techniques have been developed for the accurate determination of Sr2+ ions, including atomic absorption/emission spectroscopy (AAS/AES),4 inductively-coupled plasma mass spectrometry (ICP-MS/ICP-AES)5 and X-ray fluorescence spectroscopy (XRF).6 However, these techniques require time-consuming and extensive sample pre-treatment procedures and involve the use of sophisticated instrumentation. In recent years, small molecule fluorescent probes7 and nanoparticle-based colorimetric8 and spectroscopic9 methods have been reported for the sensitive detection of Sr2+ ions. Alternative methods using ion-selective electrodes for the selective detection of Sr2+ have also been proposed.10 However, the fluorescent organic probes and colorimetric methods reported thus far have displayed relatively poor selectivity for Sr2+ over other alkali earth metal ions, while ion-selective electrodes tend to be costly to manufacture and modify for small-scale applications.
There have been substantial reports of luminescent DNA probes for the detection of analytes such as DNA11 and metal ions.12 In particular, the G-quadruplex is a non-canonical DNA structure formed from guanine-rich sequences that is comprised of planar stacks of four guanine bases stabilized by Hoogsteen hydrogen bonding and monovalent cations.13 Interestingly, certain divalent ions have also been reported to induce or stabilize the G-quadruplex motif structure.14 Hardin et al. and Chen have demonstrated that certain telomeric DNA sequences can be induced to form G-quadruplex structures in the presence of Sr2+ ions.15 Recently, Qu et al.16 have utilized human telomeric G-quadruplex DNA (5′-AGGGTTAGGGTTAGGGTTAGGG-3′), single-wall carbon nanotubes (SWNTs) and thiazole orange (TO) to devise a platform for the detection of Sr2+ in a buffered system.
Luminescent metal complexes have received considerable attention in photochemistry,17 organic optoelectronics18 and luminescent sensing.19 In the context of luminescent sensing, transition metal complexes offer several distinctive advantages which render them suitable candidates as selective probes for the G-quadruplex. Firstly, most transition metal complexes exhibit emission in the visible region with a long phosphorescence lifetime, allowing them to be readily distinguished from background fluorescence arising from endogenous fluorophores in the sample matrix. Secondly, the precise and versatile arrangement of co-ligands on the metal centre allows metal complexes to interact selectively with biomolecules. Thirdly, these metal complexes often possess interesting photophysical properties, which are strongly affected by subtle changes of their local environment and facilitate the luminescent visualization of G-quadruplex DNA. Lastly, the photophysical properties of metal complexes can be fine-tuned without the need for lengthy synthetic protocols due to the modular nature of inorganic complex synthesis. In recent years, square-planar platinum(II) complexes,19b,20 zinc porphyrins21 and ruthenium(II) polypyridyl complexes22 have been previously reported as metal-based luminescent probes of G-quadruplex DNA. However, the application of Group 9 octahedral complexes such as iridium(III) for G-quadruplex detection has been comparatively less explored.23
Our group has recently reported the discovery and application of complex 1 as a luminescent “switch-on” probe for the detection of gene deletion using a bimolecular “split” G-quadruplex-based system (Fig. 1).23 Thus, we were interested to explore whether the G-quadruplex-selective luminescent response of 1 could be applied to detect other types of relevant G-quadruplexes for potential sensing applications. We report herein the application of the iridium(III) complex 1 as a luminescent G-quadruplex-selective probe for the oligonucleotide-based sensing of Sr2+ ions in aqueous buffer and sea water samples.
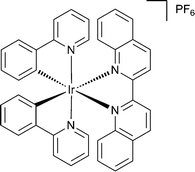 | ||
| Fig. 1 Chemical structure of cyclometallated iridium(III) complex bearing the 2,2′-biquinoline ligand (1). | ||
The mechanism of the G-quadruplex-based method for Sr2+ determination using Ir(III) complex 1 as a luminescent probe is depicted in Scheme 1. In the absence of Sr2+, the Tetrahymena telomeric sequence13 T2 (5′-G4T2G4T2G4T2G4-3′) adopts a random coil structure. Complex 1 interacts weakly with single-stranded DNA and the resulting emission signal is low, presumably due to non-radiative decay of the excited state by complex-solvent interactions. The addition of Sr2+ induces the formation of a tetramolecular G-quadruplex structure, which is strongly bound by complex 1. The binding of the iridium(III) complex 1 to the G-quadruplex terminus partially shields the metal complex from the bulk solvent environment, which suppresses non-radiative decay of the excited state emission thus resulting in a “switch-on” luminescence response to Sr2+ ions. For maximum efficiency, the DNA was annealed after the addition of Sr2+ ions to ensure that the folding response of the oligonucleotides to the analyte would not be limited by kinetic barriers to interconversion.
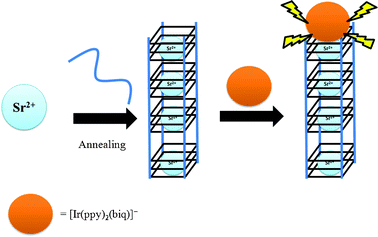 | ||
| Scheme 1 Schematic representation of the G-quadruplex-selective probe for the detection of Sr2+ ions. The Sr2+ ion induces a conformational change of the oligonucleotide T2 from a random coil structure to an intermolecular G-quadruplex,15b allowing the interaction of complex 1 with the G-quadruplex and resulting in increased emission intensity at λ = 638 nm. | ||
The luminescence intensity of 1 with T2 increased with the Sr2+ concentration, and reached saturation at 10 μM of Sr2+ ions in buffered solution. A 7.2-fold increase in luminescence intensity was observed with T2 at saturating concentrations of Sr2+ ions (Fig. 2A). The luminescence response of the system against Sr2+ ions was linear up to 1 μM (R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 = 0.97) (Fig. 2B), and the detection limit of this assay for Sr2+ ions at a signal-to-noise ratio of 3 was estimated to be 13 nM (3σ). In addition, the luminescence enhancement was readily observable by the naked eye under UV-irradiation (Fig. S1, ESI†). These results suggest that the Sr2+-induced formation of the T2 G-quadruplex structure could be transduced into measurable luminescent output by iridium(III) complex 1.
2 = 0.97) (Fig. 2B), and the detection limit of this assay for Sr2+ ions at a signal-to-noise ratio of 3 was estimated to be 13 nM (3σ). In addition, the luminescence enhancement was readily observable by the naked eye under UV-irradiation (Fig. S1, ESI†). These results suggest that the Sr2+-induced formation of the T2 G-quadruplex structure could be transduced into measurable luminescent output by iridium(III) complex 1.
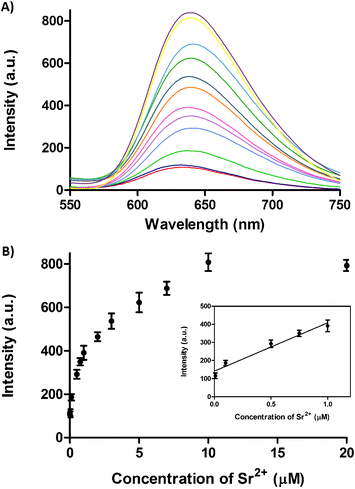 | ||
| Fig. 2 A) Emission spectra of the complex 1 (2 μM) and T2 (5 μM) in the presence of increasing concentrations of Sr2+ ions (0, 0.01, 0.1, 0.5, 0.75, 1, 2, 3, 5, 7, 10 and 20 μM). B) Luminescence response of complex 1 (2 μM) and T2 (5 μM) at λ = 638 nm versus Sr2+ ion concentration. Inset: Linear plot of the change in luminescence intensity at λ = 638 nm versus Sr2+ concentration. Error bars represent the standard deviations of the results from three independent experiments. | ||
The selectivity of this detection method for various concentrations of Sr2+ ions over twelve different metal ions (K+, Li+, Na+, Ba2+, Ni2+, Ca2+, Zn2+, Mg2+, La3+, Cr3+, Al3+ and Ti3+) was evaluated. At 0.1–10 μM of Sr2+ ions, the luminescence response of the system was significantly stronger than that for ten-fold excess concentrations of twelve other metal ions (Fig. 3). Furthermore, an excellent selectivity for Sr2+ was observed even at a 100-fold excess of other common ions naturally occurring in sea water, including K+, Na+, Ca2+ and Mg2+ ions (Fig. S2, ESI†). These results demonstrate the surprisingly high selectivity of our system towards Sr2+ ions over other metal ions, including its congeners Ca2+, Mg2+ and Ba2+. We propose that the selectivity of this system for Sr2+ ions arises from the size complementarity of Sr2+ ions within the central ionic channel of T2 G-quadruplex structure. For example, K+ and Na+ ions, with substantially smaller ionic radii compared Sr2+, have been demonstrated to be less effective at stabilizing the T2 G-quadruplex structure.15b
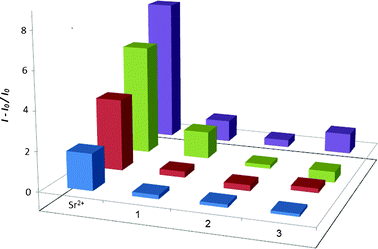 | ||
| Fig. 3 Relative luminescence intensity of complex 1 (2 μM) and T2 (5 μM) in the presence of Sr2+ (0.1, 1, 5, 10 μM) or 10-fold excess of groups 1–3 of metal ions (1, 10, 50, 100 μM; group 1: Ti3+, K+, Mg2+, Li+; group 2: Na+, Ba2+, Ni2+, La3+; group 3: Ca2+, Cr3+, Zn2+, Al3+) in Tris buffer (20 mM, pH 7.0). | ||
To verify that the luminescence response of complex 1 originated from the specific interaction with the Sr2+ ion-induced G-quadruplex, we examined the luminescent response of the complex to two DNA mutants designated T2m1 (5′-G![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif) GTTGGTTGGTTGG-3′) and T2m2 (5′-GGTTGGTTGGTTGG-3′) (base mutants underlined) that cannot form G-quadruplexes. No significant luminescence enhancement was observed at Sr2+ concentrations from 0.01 to 10 μM (Fig. S3, ESI†), suggesting the “switch-on” response of 1 is not due to the direct interaction of the complex with the Sr2+ ions.
GTTGGTTGGTTGG-3′) and T2m2 (5′-GGTTGGTTGGTTGG-3′) (base mutants underlined) that cannot form G-quadruplexes. No significant luminescence enhancement was observed at Sr2+ concentrations from 0.01 to 10 μM (Fig. S3, ESI†), suggesting the “switch-on” response of 1 is not due to the direct interaction of the complex with the Sr2+ ions.
Finally, we examined the applicability of our method for testing real water samples collected from Lamma Island, Hong Kong. A syringe filter was used to remove any suspended particles or biological matter from the water sample. The filtered samples were spiked with known concentrations of Sr2+, and the emission spectra were recorded upon the addition of T2 and complex 1. The luminescence response of 1 in the real water sample was comparable with that in aqueous buffer (Fig. S4, ESI†). Additionally, the luminescence signal of 1 upon the addition of annealed T2 and Sr2+ attained steady-state within 5 min at room temperature (Fig. S5, ESI†). The results showed that our Sr2+ detection system can also be applied to a complex solution matrix such as a real water sample.
The detection limit of 13 nM for our present assay is comparable or better than most other recently reported analytical techniques for Sr2+ ion detection, with the exception of the ion-imprinted gold nanoparticles (NP) composites reported recently by Willner and co-workers for the detection of alkali earth metal ions.9 Their assay utilized an amplification mechanism involving coupling between the localized plasmon of the NPs and the surface plasmon wave to achieve femtomolar sensitivity for Sr2+ ions. However, our detection strategy has the advantages of simplicity and cost-effectiveness, and we have demonstrated the quantification of nanomolar Sr2+ ions in sea water without signal amplification or extensive prior preparation and derivatization of reagents. For ease of access, a comparison of the detection limit and linear range of detection for recently reported analytical techniques for Sr2+ ions have been summarized in Table S1, ESI.†
In summary, we have developed a label-free, G-quadruplex DNA-based switch-on detection methodology for Sr2+ ions in aqueous solution. This system is based on telomeric sequence T2 and iridium(III) complex 1, which exhibits a strong emission signal when bound to G-quadruplex DNA. The formation of G-quadruplex induced by Sr2+ ions is recognized by complex 1 with a switch-on luminescence response. The detection system is highly selective and is able to discriminate Sr2+ over other common metal ions, including its congeners Ca2+ and Ba2+. Our system is advantageous over other conventional methods due to its simple and low-cost nature and switch-on luminescence response.
Acknowledgements
This work is supported by Hong Kong Baptist University (FRG2/11-12/009), Environment and Conservation Fund (ECF Project 3/2010), Centre for Cancer and Inflammation Research, School of Chinese Medicine (CCIR-SCM, HKBU), the Research Fund for the Control of Infectious Diseases (RFCID/11101212) and the Research Grants Council (HKBU/201811), the University of Macau (Start-up Research Grant to C.-H. Leung), MYRG091(Y1-L2)-ICMS12-LCH and MYRG121(Y1-L2)-ICMS12-LCH).References
- (a) J. J. Anderson, Am. J. Clin. Nutr., 2000, 71, 1384–1386 CAS; (b) M. Lind and C. Bünger, Eur. Spine J., 2001, 10, S102–S109 CrossRef.
- P. J. Marie, P. Ammann, G. Boivin and C. Rey, Calcif. Tissue Int., 2001, 69, 121–129 CrossRef CAS.
- P. Patnaik, in Handbook of Inorganic Chemical Compounds; McGraw-Hill, 2003, pp. 77–78 Search PubMed.
- (a) P. J. Slevin, E. Györy-Szebényi and G. Svehla, Talanta, 1972, 19, 307–315 CrossRef CAS; (b) Z. Arslan and J. F. Tyson, Talanta, 1999, 50, 929–937 CrossRef CAS.
- (a) M. Piette, B. Desmet and R. Dams, Sci. Total Environ., 1994, 141, 269–273 CrossRef CAS; (b) K. Usuda, K. Kono, S. Hayashi, T. Kawasaki, G. Mitsui, T. Shibutani, E. Dote, K. Adachi, M. Fujihara, Y. Shimbo, W. Sun, B. Lu and K. Nakasuji, Environ. Health Prev. Med., 2006, 11, 11–16 CrossRef CAS.
- (a) C. A. Lucchesi, Anal. Chem., 1957, 29, 370–373 CrossRef CAS; (b) A. Pejović-Milić, I. M. Stronach, J. Gyorffy, C. E. Webber and D. R. Chettle, Med. Phys., 2004, 31, 528–538 CrossRef.
- (a) H.-F. Ji, Y. Yang, X. Xu and G. Brown, Org. Biomol. Chem., 2006, 4, 770–772 RSC; (b) P. Kar, M. Suresh, D. Krishna Kumar, D. Amilan Jose, B. Ganguly and A. Das, Polyhedron, 2007, 26, 1317–1322 CrossRef CAS; (c) T. Banerjee, M. Suresh, H. N. Ghosh and A. Das, Eur. J. Inorg. Chem., 2011, 2011, 4680–4690 CrossRef CAS.
- J. Zhang, Y. Wang, X. Xu and X. Yang, Analyst, 2011, 136, 3865–3868 RSC.
- Y. Ben-Amram, R. Tel-Vered, M. Riskin, Z.-G. Wang and I. Willner, Chem. Sci., 2012, 3, 162–167 RSC.
- (a) A. Jain, V. Gupta and J. Raisoni, Sensors, 2004, 4, 115–124 CrossRef CAS; (b) M. A. Zanjanchi, M. Arvand, A. Islamnezhad and N. O. Mahmoodi, Talanta, 2007, 74, 125–131 CrossRef CAS; (c) H. A. Zamani, M. R. Ganjali, P. Norouzi and M. Adib, Mater. Sci. Eng., C, 2008, 28, 157–163 CrossRef CAS; (d) H. A. Zamani, M. Masrournia, H. Mohamadzadeh, M. R. Ganjali, M. Rahimizadeh and P. Ziaei, Mater. Sci. Eng., C, 2009, 29, 976–979 CrossRef CAS.
- (a) Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kotler and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430–7431 CrossRef CAS; (b) P. Conlon, C. J. Yang, Y. Wu, Y. Chen, K. Martinez, Y. Kim, N. Stevens, A. A. Marti, S. Jockusch, N. J. Turro and W. Tan, J. Am. Chem. Soc., 2007, 130, 336–342 CrossRef; (c) Y. Wu, C. J. Yang, L. L. Moroz and W. Tan, Anal. Chem., 2008, 80, 3025–3028 CrossRef CAS; (d) C. Wang, Z. Zhu, Y. Song, H. Lin, C. J. Yang and W. Tan, Chem. Commun., 2011, 47, 5708–5710 RSC; (e) J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS; (f) F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS.
- (a) D. S.-H. Chan, H.-M. Lee, C.-M. Che, C.-H. Leung and D.-L. Ma, Chem. Commun., 2009, 7479–7481 RSC; (b) B. Y.-W. Man, D. S.-H. Chan, H. Yang, S.-W. Ang, F. Yang, S.-C. Yan, C.-M. Ho, P. Wu, C.-M. Che, C.-H. Leung and D.-L. Ma, Chem. Commun., 2010, 46, 8534–8536 RSC; (c) D.-L. Ma, D. S.-H. Chan, B. Y.-W. Man and C.-H. Leung, Chem.–Asian J., 2011, 6, 986–1003 CrossRef CAS.
- (a) G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS; (b) K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef CAS.
- (a) B. I. Kankia and L. A. Marky, J. Am. Chem. Soc., 2001, 123, 10799–10804 CrossRef CAS; (b) D. Miyoshi, A. Nakao and N. Sugimoto, Nucleic Acids Res., 2003, 31, 1156–1163 CrossRef CAS; (c) I. M. Pedroso, L. F. Duarte, G. Yanez, A. M. Baker and T. M. Fletcher, Biochem. Biophys. Res. Commun., 2007, 358, 298–303 CrossRef CAS.
- (a) C. C. Hardin, E. Henderson, T. Watson and J. K. Prosser, Biochemistry, 1991, 30, 4460–4472 CrossRef CAS; (b) F. M. Chen, Biochemistry, 1992, 31, 3769–3776 CrossRef CAS.
- K. Qu, C. Zhao, J. Ren and X. Qu, Mol. BioSyst., 2012, 8, 779–782 RSC.
- (a) R. E. Holmlin, E. D. A. Stemp and J. K. Barton, J. Am. Chem. Soc., 1996, 118, 5236–5244 CrossRef CAS; (b) K. M.-C. Wong and V. W.-W. Yam, Coord. Chem. Rev., 2007, 251, 2477–2488 CrossRef CAS.
- (a) W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS; (b) S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923–925 CrossRef CAS.
- (a) T. J. Wadas, Q.-M. Wang, Y.-J. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef CAS; (b) D.-L. Ma, C.-M. Che and S.-C. Yan, J. Am. Chem. Soc., 2008, 131, 1835–1846 CrossRef; (c) M. H. Lim, H. Song, E. D. Olmon, E. E. Dervan and J. K. Barton, Inorg. Chem., 2009, 48, 5392–5397 CrossRef CAS; (d) D.-L. Ma, T. Xu, D. S.-H. Chan, B. Y.-W. Man, W.-F. Fong and C.-H. Leung, Nucleic Acids Res., 2011, 39, e67 CrossRef CAS; (e) D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012 DOI:10.1016/j.ccr.2012.07.005.
- (a) P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C.-M. Che, Chem.–Eur. J., 2009, 15, 13008–13021 CrossRef CAS; (b) P. Wang, C.-H. Leung, D.-L. Ma, S.-C. Yan and C.-M. Che, Chem.–Eur. J., 2010, 16, 6900–6911 CrossRef CAS.
- (a) J. Alzeer, B. R. Vummidi, P. J. C. Roth and N. W. Luedtke, Angew. Chem., Int. Ed., 2009, 48, 9362–9365 CrossRef CAS; (b) Z. Zhang, E. Sharon, R. Freeman, X. Liu and I. Willner, Anal. Chem., 2012, 84, 4789–4797 CrossRef CAS.
- (a) L. Xu, D. Zhang, J. Huang, M. Deng, M. Zhang and X. Zhou, Chem. Commun., 2010, 46, 743–745 RSC; (b) T. Wilson, M. P. Williamson and J. A. Thomas, Org. Biomol. Chem., 2010, 8, 2617–2621 RSC; (c) D. Sun, Y. Liu, D. Liu, R. Zhang, X. Yang and J. Liu, Chem.–Eur. J., 2012, 18, 4285–4295 CrossRef CAS; (d) D. Liu, Y. Liu, C. Wang, S. Shi, D. Sun, F. Gao, Q. Zhang and J. Liu, ChemPlusChem, 2012, 77, 551–562 CrossRef CAS; (e) M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- H.-Z. He, D. S.-H. Chan, C.-H. Leung and D.-L. Ma, Chem. Commun., 2012 10.1039/C2CC32253F.
Footnotes |
| † Electronic Supplementary Information (ESI) available: experimental details and supplementary spectral data. See DOI: 10.1039/c2ra21119j/ |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |