DOI:
10.1039/D4DT00514G
(Frontier)
Dalton Trans., 2024,
53, 7659-7668
The post-synthesis modification (PSM) of MOFs for catalysis
Received
22nd February 2024
, Accepted 5th April 2024
First published on 8th April 2024
Abstract
While there are myriad ways to construct metal–organic framework (MOF) based catalysts, the introduction of catalytic functionality via covalent post-synthesis functionalization (PSM) offers multiple advantages: (i) a wide range of different catalyst types are generated from a handful of well-known parent MOFs, (ii) MOF catalyst properties can be systematically tuned while changing few variables, and (iii) catalytically active functional groups that would otherwise interfere with MOF assembly can be introduced facilely. This last advantage is particularly crucial for our quest to generate MOF active sites that are decorated with multiple functional groups capable of cooperative activity, analogous to enzyme active sites.
Introduction
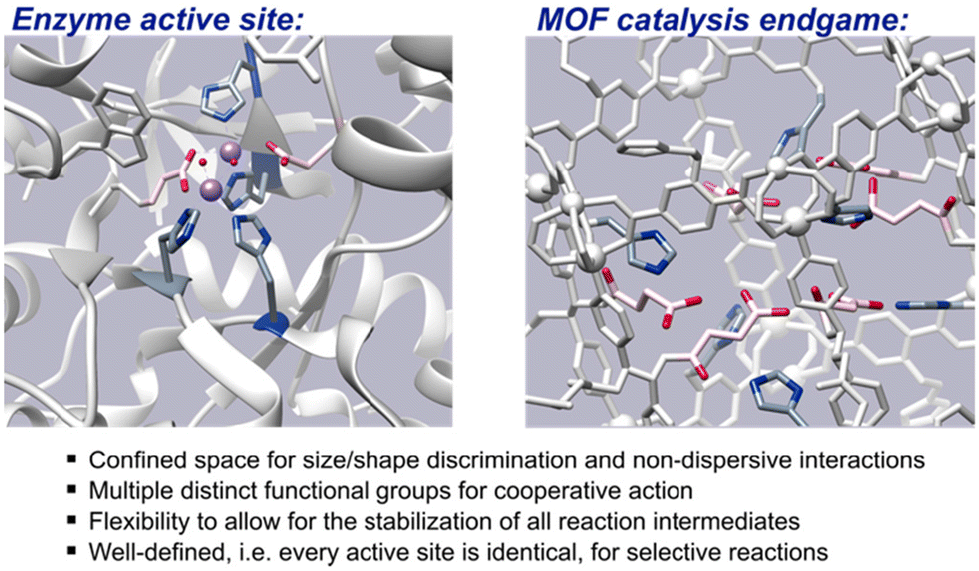
An oft-touted attribute of metal–organic framework (MOF) materials is their resemblance to enzyme active sites,1 largely because MOF-based catalysts transform reactants within the confines of their pores, much in the way that biochemical reactions are frequently restricted within enzyme cavities. However, the said confinement is often where the similarity between MOF catalysts and enzymes ends since, besides their constrained size and shape, enzyme active sites are also characterised by their decoration with multiple amino acid side chains. These multiple functional groups promote reactions depending on their chemical properties, e.g., acidity, basicity, hydrophobicity, nucleophilicity, flexibility etc. They not only define the confines of the active site, but also influence both the activity and selectivity of the enzymatic reaction by any combination of reacting covalently with substrates, stabilizing transition states, and perturbing, via non-covalent interactions, the physical properties of reacting species and/or other functional groups present in the cavity.
Thus, to more adequately mimic enzyme sites, thereby attaining more of their efficiency, the pores of MOF-based catalysts need to be decorated with multiple functional groups that can cooperatively orient and/or activate substrates and intermediates. Additional requirements for achieving more enzyme-like activity are that the cavities should be flexible and identically functionalised. Flexibility allows for dynamic binding, in which small adjustments of the cavity ensure favorable interactions for all species along the reaction pathway, while the uniformity of the functional groups present in each cavity is imperative for selective transformation. The difficulty in constructing such catalysts arises from the requirement that some of the functional groups have nucleophilic reactivity or are capable of supramolecular interactions such as hydrogen bonding and electrostatic interactions. The generation of MOFs in which such functionalities are present free and uncoordinated within the pore is challenging as these types of functional groups often coordinate to the metal building blocks, becoming a part of the framework vertices,2,3e.g. –OH in MOF-744 and –COOH in HKUST-1.5
Despite the difficulty of assembling MOFs bearing the desired functional groups as free substituents, there are several examples of generating such MOFs via traditional synthesis methods. We must note, however, that the functional groups in such frameworks are attached directly to the framework linkers, which are generally aromatic,2,6–9 rigid,10,11 or otherwise so sterically encumbered as to preclude their coordination to metal species during MOF assembly.12 This necessarily limits the ability of these functional groups to reorient themselves in the pores for dynamic binding. Unfortunately, the assembly of MOFs with linkers in which these ligating substituents are tethered to the linkers via aliphatic chains or other attachments that afford them conformational flexibility is more likely to yield structures in which those groups are coordinated to the metal vertices. Thus, the most reliable strategy for obtaining MOFs in which the desired functional groups are uncoordinated and available for synergistic catalysis is to introduce them into the pores after framework assembly.
Post-synthesis modification of MOFs
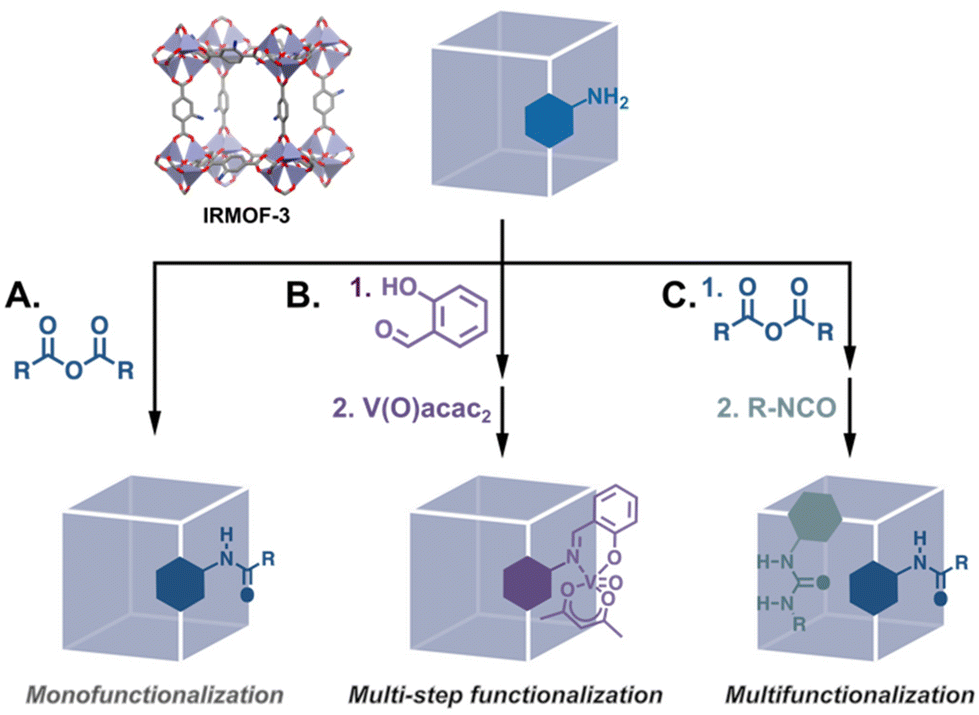
The post-synthesis modification (PSM) of MOFs spans a wide range of transformations, including the exchange of monotopic ligands at unsaturated metal clusters, transmetalation, linker exchange and insertion, etc.13 While multiple functionalities can be introduced using linker exchange/insertion and coordination at metal corners, in this article we focus on “traditional” PSM in which organic linkers are transformed via the breaking and/or forming of covalent bonds. This is because we are interested in catalytic MOFs that are well-defined and where there is a low risk of catalyst leaching under a variety of reaction conditions. In covalent PSM, a parent MOF with linkers bearing reactive substituents such as amines, hydroxyls, azides, terminal alkynes and alkenes, etc., is reacted with the corresponding substrate to yield a new MOF. Thus, a single parent MOF can be systematically modified to generate several mono- and multifunctionalised daughter MOFs using strategies such as single, tandem, and multi-step functionalization (Fig. 2). Perusal of the PSM literature reveals myriad examples of the covalent modification of MOFs to introduce functional groups that have supramolecular and/or nucleophilic capabilities that would otherwise interfere with MOF assembly. Of particular interest are reports in which the resulting daughter MOFs are applied to catalysed reactions.14
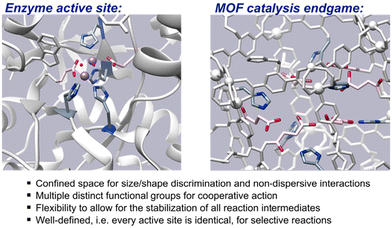 |
| | Fig. 1 Properties of enzyme active sites which multifunctional MOF-based catalysts aspire to. | |
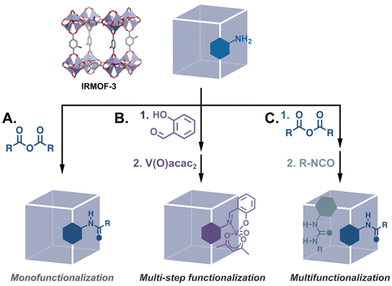 |
| | Fig. 2 PSM of a parent MOF can yield monofunctionalised daughter MOFs via (A) a single reaction15 and (B) a tandem process,16 or (C) multifunctionalised MOFs by reaction with multiple reactants.17 | |
The PSM of MOFs for catalysis
The first report of covalent PSM of a MOF was the alkylation of free pyridine groups in a homochiral MOF transesterification catalyst. It is worth mentioning that, while the parent framework demonstrated the first example of enantioselective catalysis by a MOF,6 the resulting methylpyridinum iodide MOF was itself not catalytically active. The next example of enantioselective catalysis was by a framework obtained by metalating a homochiral BINOL-based MOF. It should be noted that the construction of a large number of MOF catalysts involves this type of dative modification of frameworks assembled using “privileged” organic ligands, which highlights the importance of PSM in generating catalytic MOFs.18
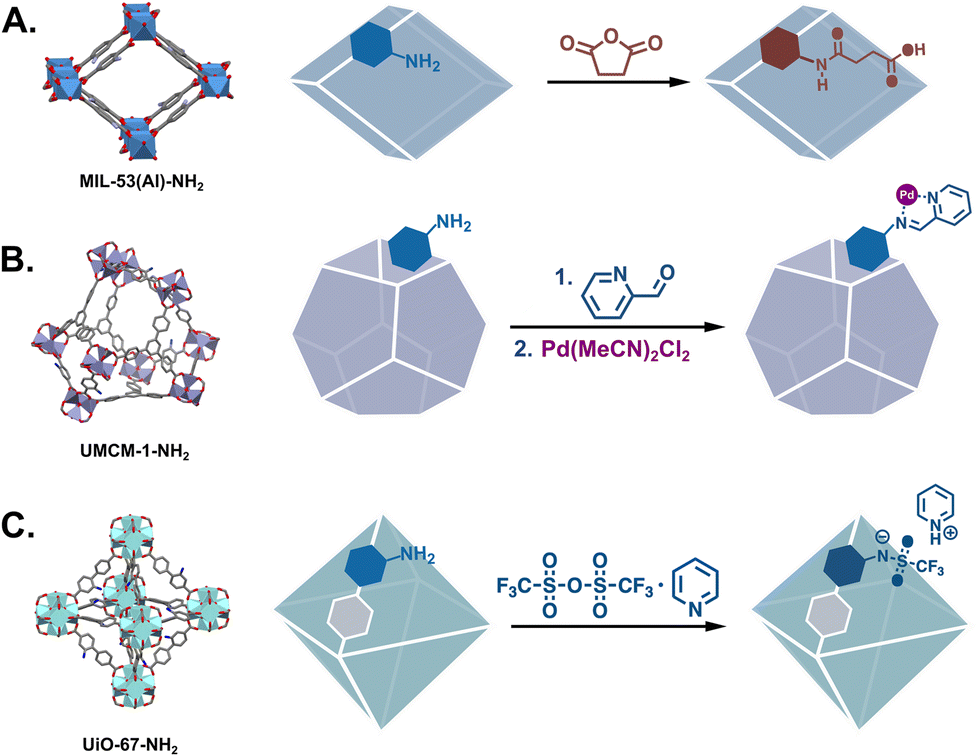
Though fewer, the examples of MOF catalysts produced via covalent modifications demonstrate the breadth of catalysts that can be produced from a single material. Just considering IRMOF-3, the archetypal modifiable MOF, we find that its –NH2 groups have been modified with numerous organic moieties that can be applied to an assortment of catalysed reactions. Many of these PSM-derived MOF catalysts have been metalated Schiff bases which are applied to a host of reactions (Fig. 2B).16,19 Eventually, catalysts were synthesised via the modification of other common –NH2 bearing MOFs such as UMCM-1-NH2 which has larger pores,20 and MIL-53(Al)-NH2 and UiO-66-NH2 which are chemically more stable.21,22 The framework structures of these MOFs and some representative PSM reactions are shown in Fig. 3.
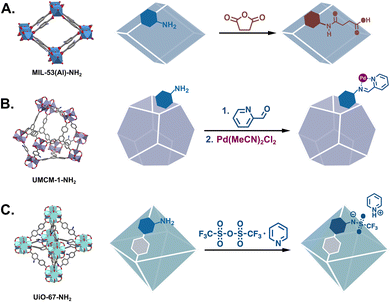 |
| | Fig. 3 Other common amine-tagged MOFs demonstrating PSM via nucleophilic substitution: (A) MIL-53(Al)-NH2,41 (B) UMCM-1-NH2,42 and (C) UiO-67-NH2.43 | |
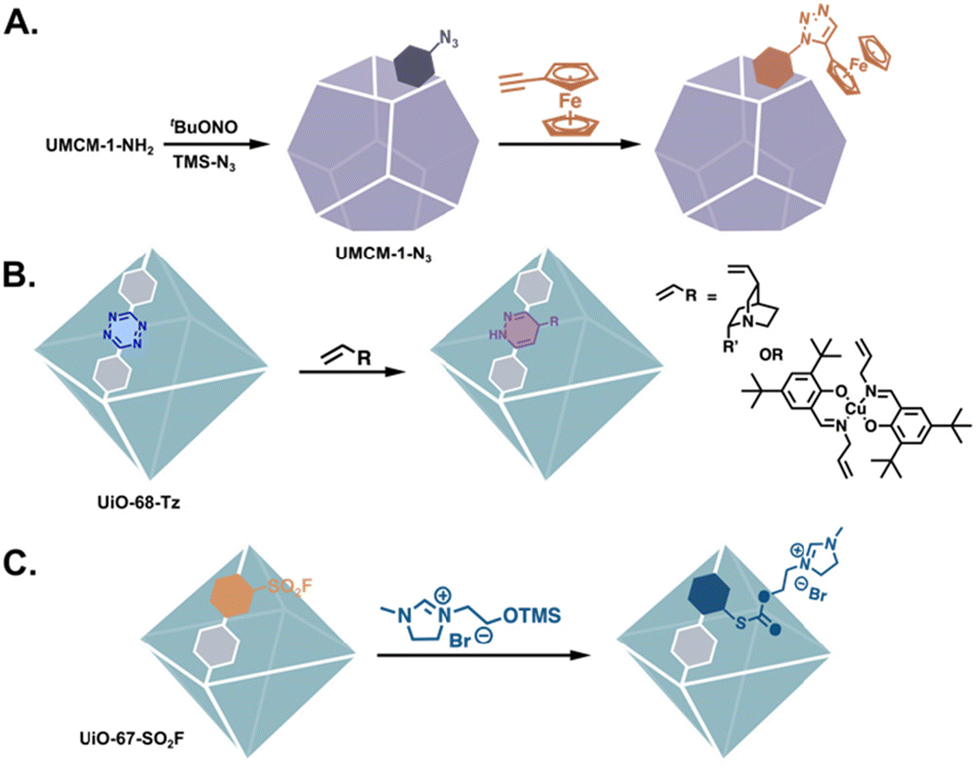
There are far fewer examples of MOF catalysts that have been generated via the PSM of non-amine reactive handles such as nitrogen heterocycles,6,23–26 aldehydes,27–29 acid anhydrides, etc.30 The advantage of non-amine reactive tags in the post-synthesis generation of catalysts is exemplified by “clickable” MOFs. Frameworks bearing either alkynes or azides can undergo the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC, Fig. 4A),31–37 a selective reaction occurring exclusively between azides and terminal alkynes regardless of other functional groups present. Other “click” reactions performed on MOFs include the tetrazine–alkene ligation and the sulfur(VI) fluoride exchange (SuFEx) reactions (Fig. 4B and C).38,39 These reactions allow for the introduction of functional groups that are more nucleophilic than –NH2,31,35,40 without requiring extra protection–deprotection steps, e.g. biologically relevant functionalities. Thus, the ability to functionalise MOFs with good nucleophiles is especially important when attempting to synthesise biomimetic MOF-based catalysts.
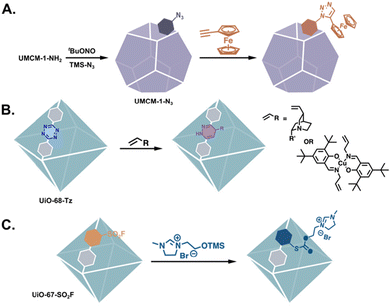 |
| | Fig. 4 MOFs tagged with non-amine functionalities for the introduction of complex functionality via (A) CuAAC,44 (B) tetrazine–alkene ligation,39 and (C) SuFEx “click” reactions.38 | |
Given the premise that PSM is an ideal way to generate enzyme-inspired environments in MOFs, the daughter MOFs of greatest interest are those that have been modified to bear functional groups that are reminiscent of amino acid side chains. Amino acid substituents possess any of several properties to tailor the chemical environments of enzyme cavities: acidity, basicity, nucleophilicity, hydrophobicity, etc. And, as the cavities are frequently decorated by several different side chains, many of these features are present simultaneously in enzyme active sites.42 Many enzymatic reactions depend on side chains with different properties acting synergistically to effect catalytic transformation (Fig. 1).45 For example, a common catalytic motif in enzymes is the catalytic triad consisting of an acidic, basic, and nucleophilic side chain.46 Promisingly, there are multiple examples of covalent MOF modification to introduce functionalities that have similar attributes.47
PSM to introduce nucleophilic catalysts
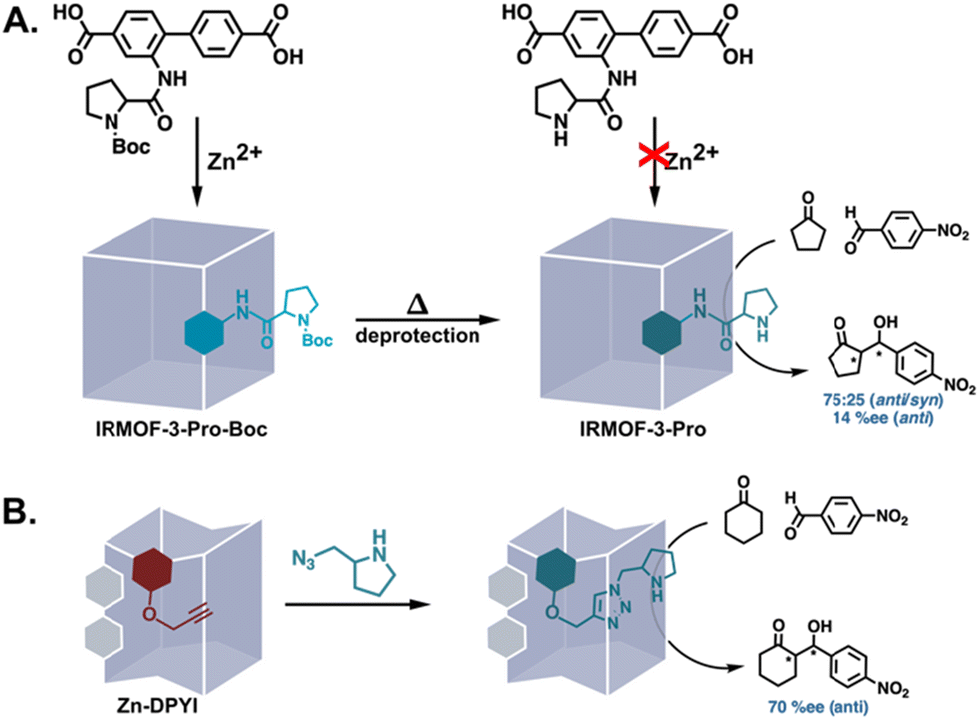
Nucleophilic side chains in enzymes partake in covalent catalysis via the formation of covalent bonds with substrates.48 While several amino acid residues can be nucleophilic, cysteine, serine, and threonine are the predominant nucleophiles.49 Though the amine of proline is not available for covalent catalysis as it forms the peptide backbone in enzymes, PSM has also been used to introduce analogues of this potent nucleophile into MOFs as an accessible catalyst. In most examples, the modification has involved the deprotection of MOFs assembled from linkers bearing protected prolines or pyrrolidines (Fig. 5A).50–56 However, the use of orthogonal reactions such as the CuAAC reaction has allowed the direct functionalization of alkyne and/or azide-tagged MOFs with unprotected proline analogues (Fig. 5B).31,35 Regardless of the PSM strategy employed for synthesizing MOFs decorated with proline analogues, these materials have been successfully applied to asymmetric aldol reactions, with some affording respectable enantioselectivities.31,53
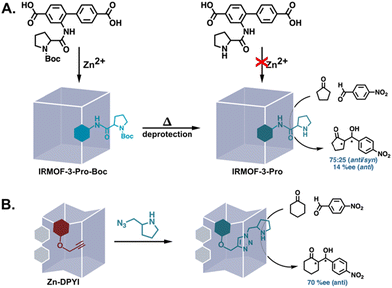 |
| | Fig. 5 Introduction of unprotected proline analogues into MOFs via PSM for the catalysis of aldol reactions. (A) The deprotection of a Boc-protected proline substituent.51 (B) CuAAC between an alkyne-tagged MOF and an azide-functionalised pyrrolidine.31 | |
PSM to introduce acid–base catalysts
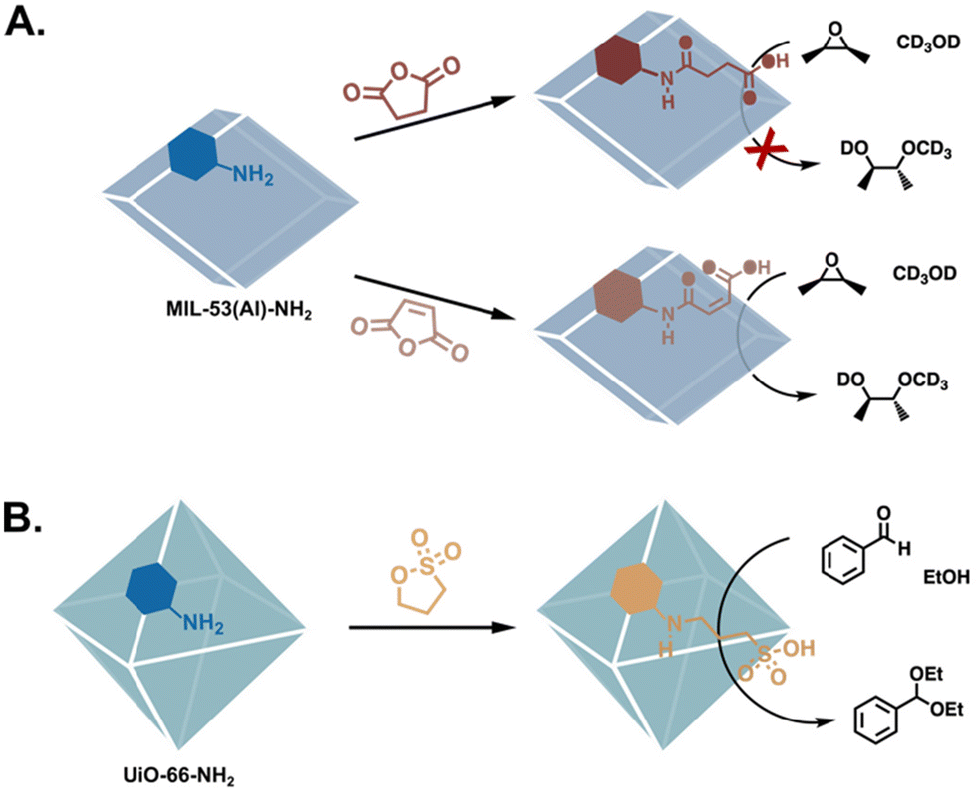
Another important mechanism in enzymatic reactions is acid–base catalysis, in which the reactions are promoted by the transfer of protons. MOF catalysts emulate these mechanisms by incorporating acidic functional groups (Fig. 6), basic substituents (Fig. 7), or hydrogen-bond donors (Fig. 8). Aspartic and glutamic acids primarily play an acidic role, though they can also be H-bond donors or acceptors, or nucleophiles in their deprotonated form. Aspartic and glutamic acid analogues have been grafted into MOF pores via the facile nucleophilic ring opening of cyclic anhydrides (Fig. 6A),17,57,58 as well as by the CuAAC reaction.59 In a demonstration of the efficiency of PSM, Garibay et al. synthesised several aliphatic carboxylic acid-bearing MOFs by reacting MIL-53(Al)-NH2 with different cyclic anhydrides. Via this facile modulation, they determined that the cis-maleic acid-based catalyst was the most effective in the methanolysis of several epoxides.41 Sulphonic acids have also been grafted onto MOFs post-synthetically,63 and their aptitude for catalysis has also been demonstrated in aldehyde acetalization, acid-catalysed epoxide ring-opening, and Morita–Baylis–Hillman reactions (Fig. 6B).60,64,65
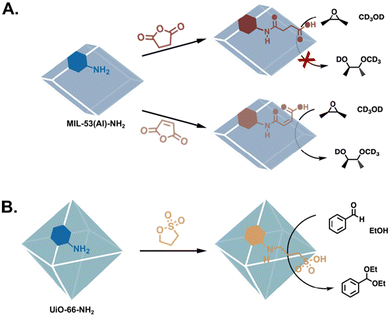 |
| | Fig. 6 PSM to functionalise MOFs with Brønsted acid catalysts. (A) Altering the stereoelectronic properties of the carboxylic acid substituents results in significantly different conversions in the methanolysis of epoxides.41 (B) A sulfonic acid-functionalised MOF is efficient in aldehyde acetalization.60 | |
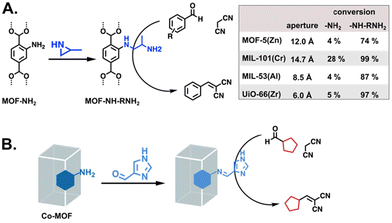 |
| | Fig. 7 Functionalization of MOFs with bases for the Knoevenagel reaction. (A) Ring-opening of aziridine by different –NH2-tagged MOFs to afford aliphatic primary amine MOF catalysts.61 (B) Imine condensation to introduce imidazole functionality.62 | |
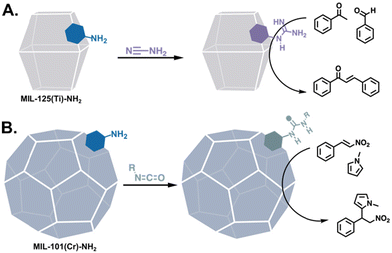 |
| | Fig. 8 PSM to afford MOFs with arginine-adjacent functionalities: (A) guanidine for the Claisen–Schmidt condensation reaction71 and (B) a series of ureas for the Friedel–Crafts alkylation reaction.72 | |
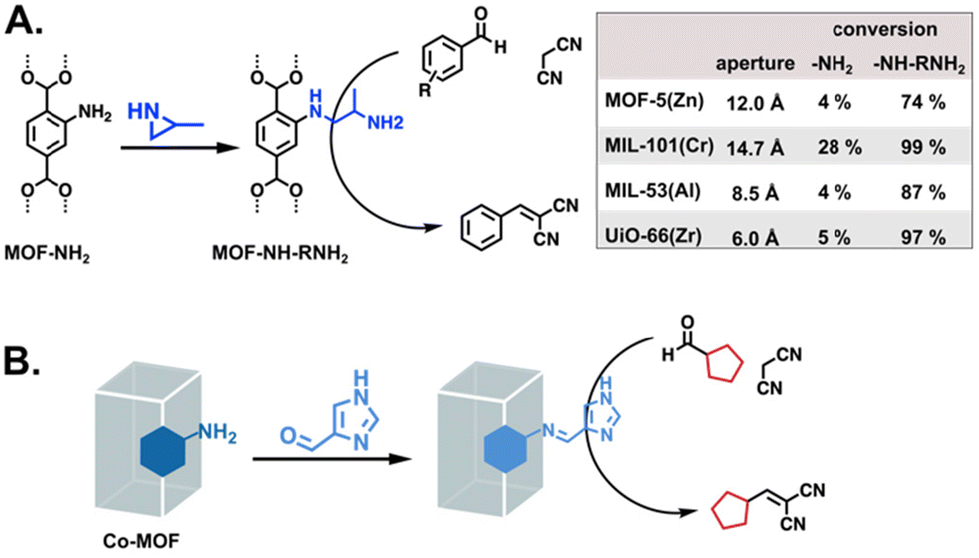
The primarily basic enzyme side chains belong to amino acids lysine, histidine, and arginine. Lysine-adjacent functionalities have been introduced as a variety of amines and have catalysed reactions such as transesterifications, Knoevenagel condensations, and Henry reactions.61,66,67 The report by Luan et al. highlighted the importance of the MOF scaffold, showing improved Knoevenagel reaction conversions with increasing MOF pore size (Fig. 7A). The analogue of histidine that is widely used in MOFs is imidazole, though it typically appears as part of the underlying framework rather than as a free substituent. Imidazole is an excellent coordinator of metals, with a large family of MOFs based on imidazolate SBUs,68 so, PSM is required to obtain free imidazoles.62,69,70 In the report by Liu et al., the imidazole functionalised MOF was successfully applied to the Knoevenagel reaction of furfural (Fig. 7B).62
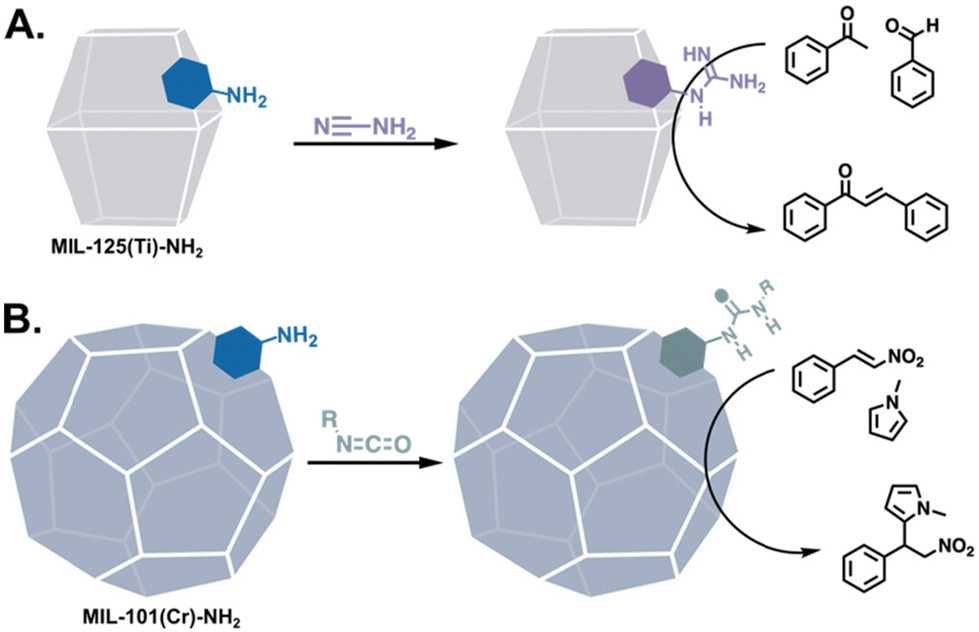
Arginine, the final primarily basic amino acid, bears a guanidine group that can be a base when neutral and a 2-point H-bond donor when protonated. Thus, MOFs have been modified with arginine-adjacent functionalities in the form of guanidine groups for the Claisen–Schmidt condensation (Fig. 8A)71 and CO2 fixation.73,74 Ureas and thioureas, the ubiquitous 2-point H-bonding organocatalysts, have also been generated in MOFs post-assembly75 and applied to Morita–Baylis–Hillman and Friedel–Crafts alkylation reactions (Fig. 8B).72,76
PSM for the hydrophobization of MOF catalysts
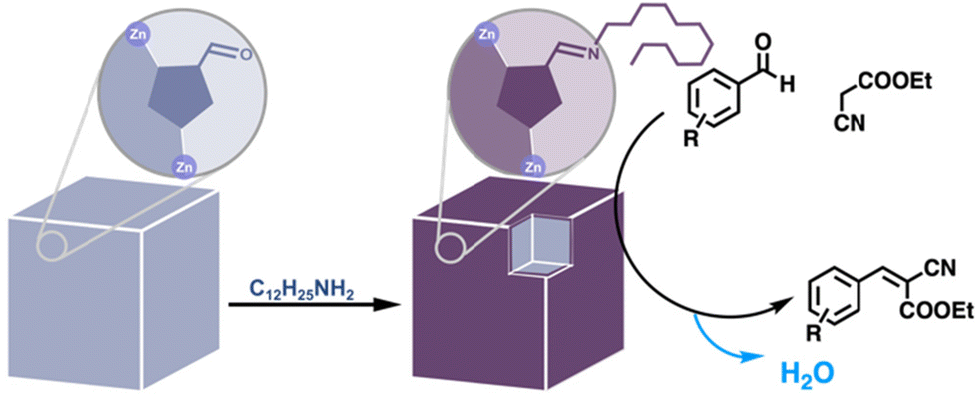
The final class of amino acids comprises those that are hydrophobic. The hydrophobicity of enzyme cavities is crucial for the binding of hydrophobic substrates in an aqueous environment,77,78 as well as for perturbing the pKas of residues involved in acid–base catalysis. A large percentage of MOF PSM reactions involve the introduction of hydrophobic, aliphatic and aromatic groups by a variety of reactions. In catalysis, hydrophobicity chiefly plays a stabilizing role, repelling water from moisture-sensitive frameworks.79,80 However, there are examples of hydrophobic groups accelerating condensation reactions by expelling water from MOF pores.81 For example, upon grafting dodecylamine onto an aldehyde-tagged framework, Canivet et al. observed a greater than ten-fold increase in the initial rate of a Knoevenagel condensation (Fig. 9).
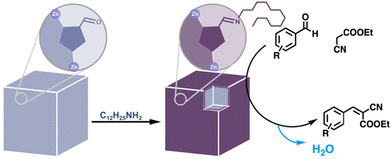 |
| | Fig. 9 Surface hydrophobization of a catalytic MOF to accelerate the Knoevenagel condensation reaction.81 | |
PSM of MOFs with amino acids
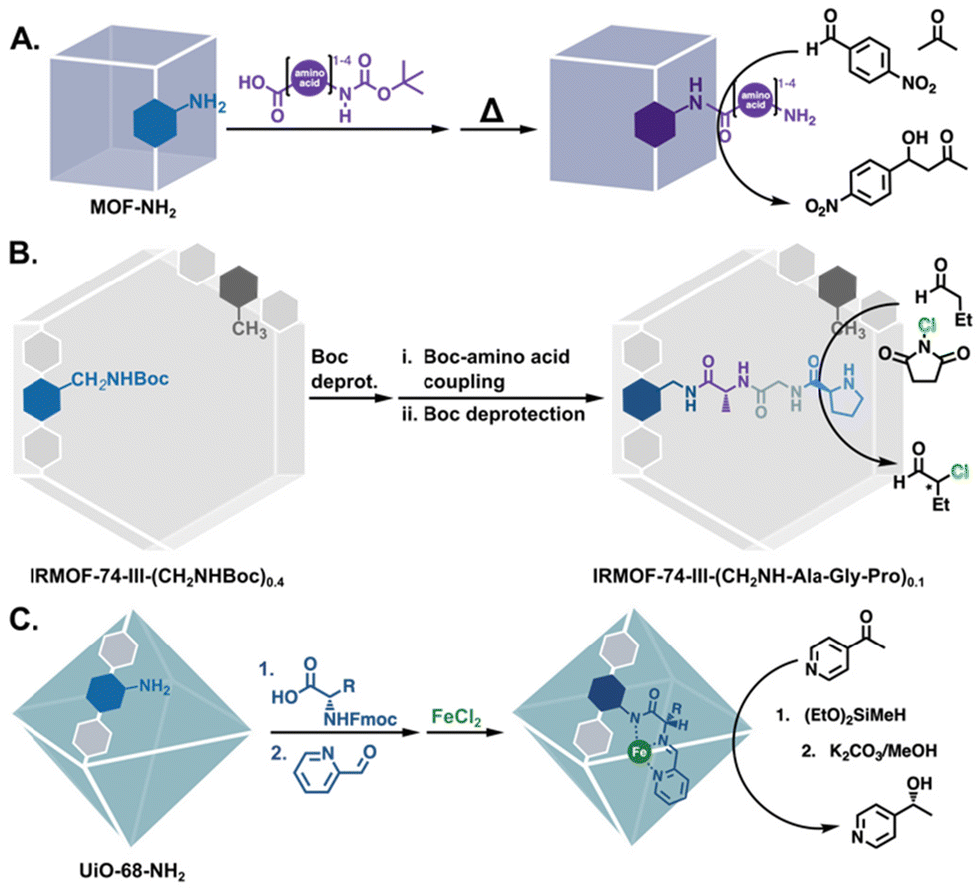
Aside from modifications with functional groups that resemble amino acids, MOFs have also been modified with actual amino acids for catalysis. In 2011, Farrusseng and coworkers reported the peptide coupling of amino acids to (In) MIL-68-NH2,50 and followed up with the grafting of oligopeptides (mono- to tetra-) onto –NH2 bearing MOFs (Fig. 10A).82 The proline-terminated mono- and dipeptide MOFs catalysed an asymmetric aldol reaction, giving 18% and 25% enantiomeric excess (ee), respectively. Using a different approach, Fracaroli et al. produced tripeptide-bearing MOF catalysts via a seven step sequence of peptide couplings and deprotection steps (Fig. 10B).83 The resulting MOF catalysts selectively cleaved a bond in an oligopeptide, while the solution phase tripeptide showed no such reactivity. Additionally, when functionalised with a proline-terminated tripeptide, the MOF catalyst achieved significantly higher ee than molecular proline (20% vs. 2%) in the α-chlorination of butyraldehyde. The authors postulated that the increased activity and selectivity were due to stereochemical constraints in the functionalised MOF pores.
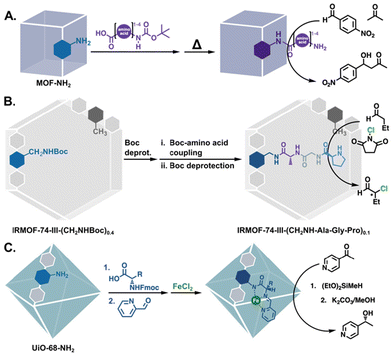 |
| | Fig. 10 PSM to functionalise MOFs with amino acids: (A) a series of mono-, di-, tri-, and tetrapeptides for the asymmetric aldol reaction.82 (B) Seven PSM steps to yield a tripeptide functionalised MOF for asymmetric α-chlorination.83 (C) Modified and metalated amino acids for the hydrofunctionalization of carbonyls.84 | |
More recently, Manna and co-workers also functionalised their MOFs with amino acids, but they went a few steps further by modifying the amino acids themselves to introduce additional functionality. Protected amino acids were coupled to UiO-68-NH2 and subsequently deprotected. The free amino acid was then condensed with 2-formylpyridine to form a bidentate pyridyl-imine moiety that was finally metalated with iron(II) (Fig. 10C).84 The resulting catalysts were active and selective in the hydrosilylation and hydroboration of carbonyls, with the valine-based catalyst, in particular, achieving excellent conversions and enantioselectivities (>95%) for most of the substrates.
Multifunctional MOF catalysts via PSM
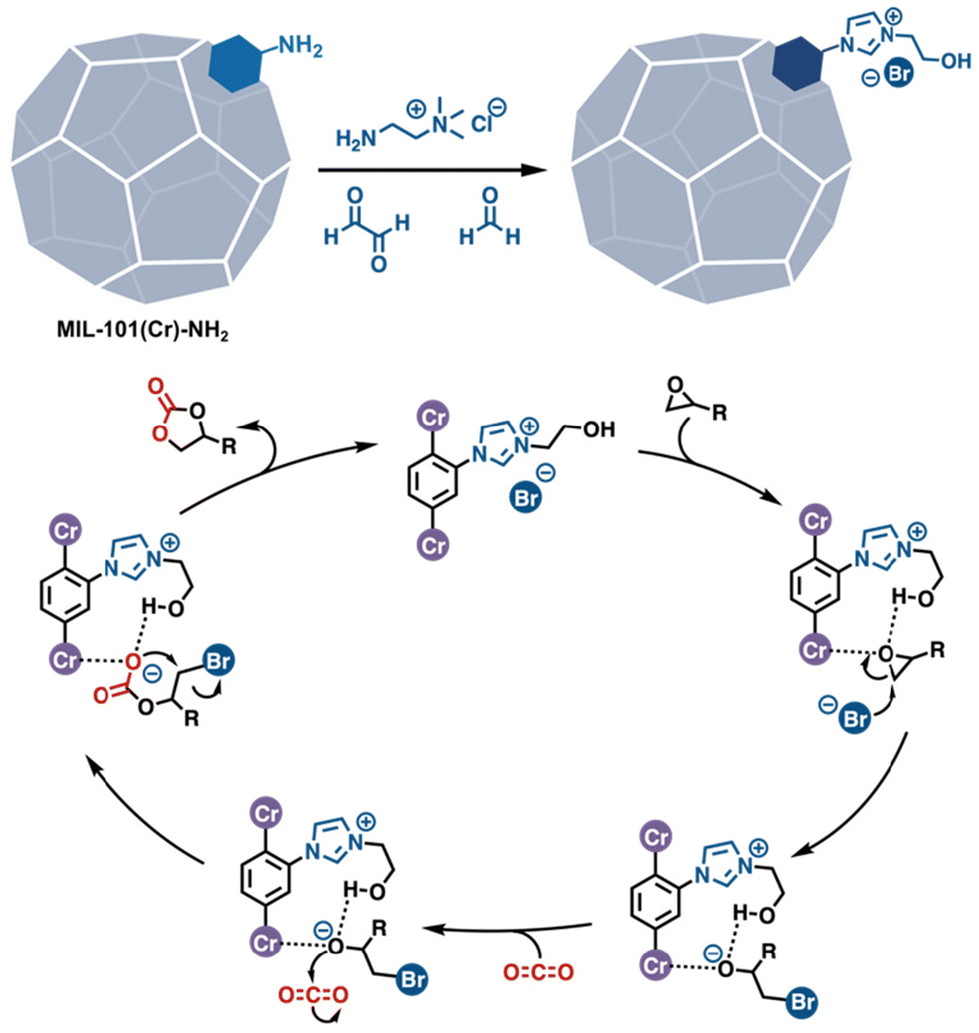
Given the extensive list of functional groups that emulate amino acid side chains that have already been grafted into MOFs, one would suppose that the introduction of multiple such groups into the confined spaces of MOF cavities would be a straightforward strategy for synthesizing enzyme inspired catalysts. Indeed, some of the previously mentioned PSM reports have resulted in, or involved, multifunctional catalysts. While most have been applied to tandem reactions in which each of the functional groups catalyses a different reaction,85,86 a few have demonstrated the promotion of reactions via the cooperative action of multiple groups. Such catalysis is most commonly observed in ionic MOFs where anionic counterions work synergistically with ammoniums,87 pyridiniums,23 phosphoniums,88 imidazoliums,89,90 triazoliums,36 guanadiniums,74etc., primarily for CO2 fixation (Fig. 11).
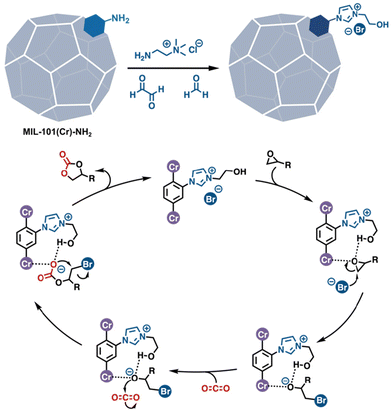 |
| | Fig. 11 A multifunctional MOF catalyst achieved by PSM. The Lewis acidic metal corner, the cationic imidazolium, and bromide work cooperatively for the fixation of CO2.90 | |
Other examples of multifunctional, PSM-derived MOF catalysts involve a catalytically active functionality together with one or more functionalities that tailor the pore environment for selectivity or further reaction acceleration. For example, via a 2-step diazotransfer/“click” reaction sequence, Savonnet et al. bifunctionalised an –NH2 bearing MOF with a basic trialkyl amine and a hydrophobic phenyl group (Fig. 12A).91 They found that, while the MOF solely functionalised with the trialkyl amine was active in the transesterification of ethyldecanoate with methanol, the hydrophobicised catalyst was significantly more active. The monofunctionalised (40% trialkyl amine) catalyst afforded 48% conversion after 20 h, while the bifunctionalised (30% trialkyl amine; 30% phenyl) catalyst had a conversion of 84%.
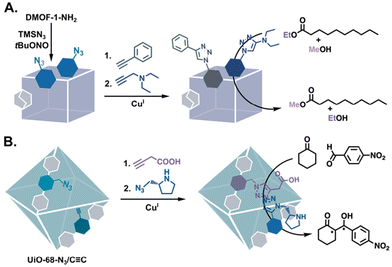 |
| | Fig. 12 Post-synthetic multifunctionalization of MOFs to generate bifunctional catalysts: (A) addition of an amine base and a hydrophobic substituent for transesterification91 and (B) addition of a proline covalent catalyst and an acid/base co-factor for the aldol reaction.35 | |
The benefits of multifunctional MOF catalysts in which the different groups are reminiscent of amino acid side chains are demonstrated in the PSM-derived, bifunctional MOFs reported by Zhang et al.35 The team synthesised a multivariate MIL-68 analogue in which the triphenyl linkers have azide or alkyne substituents (Fig. 12B). Following sequential CuAAC reactions to functionalise the MOF with both proline and carboxylic acid groups, the bifunctional MOF yielded 95% product in a proline-catalyzed aldol reaction. The bifunctional proline/carboxylic acid MOF produced four aldol products with a 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65 syn/anti ratio and 26% ee for the anti product. As evidence of the benefit of the secondary carboxylic functionality, when –COOH was replaced by –COOMe or –C
:65 syn/anti ratio and 26% ee for the anti product. As evidence of the benefit of the secondary carboxylic functionality, when –COOH was replaced by –COOMe or –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, yields of only 32% and 13%, respectively, were obtained.
CH, yields of only 32% and 13%, respectively, were obtained.
Outlook
While the last example demonstrated the benefits that can be obtained by having multiple distinct functional groups working cooperatively to turnover a reaction, there are drawbacks of using PSM to generate multifunctional MOFs for catalysis, namely, (i) the non-uniformity of the composition of the MOF pores and (ii) the reduction of pore size due to functionalization. At the first point, the blocking of the MOF pores by the additional functionality leads to reduced mass transport through the frameworks, resulting in lower apparent activity or no access to the interior active sites at all. Common strategies to circumvent such pore blockage include the use of mesoporous MOFs83 and macroporous–microporous hierarchical MOFs, and/or partial functionalization by, for example, synthesizing multivariate MOFs in which only a fraction of the linkers contain reactive groups (Fig. 10B).83,92
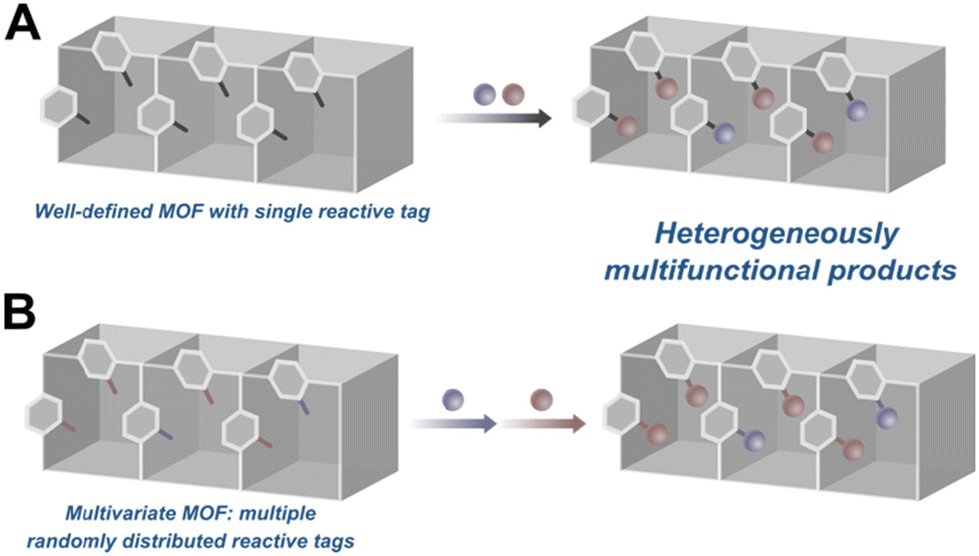
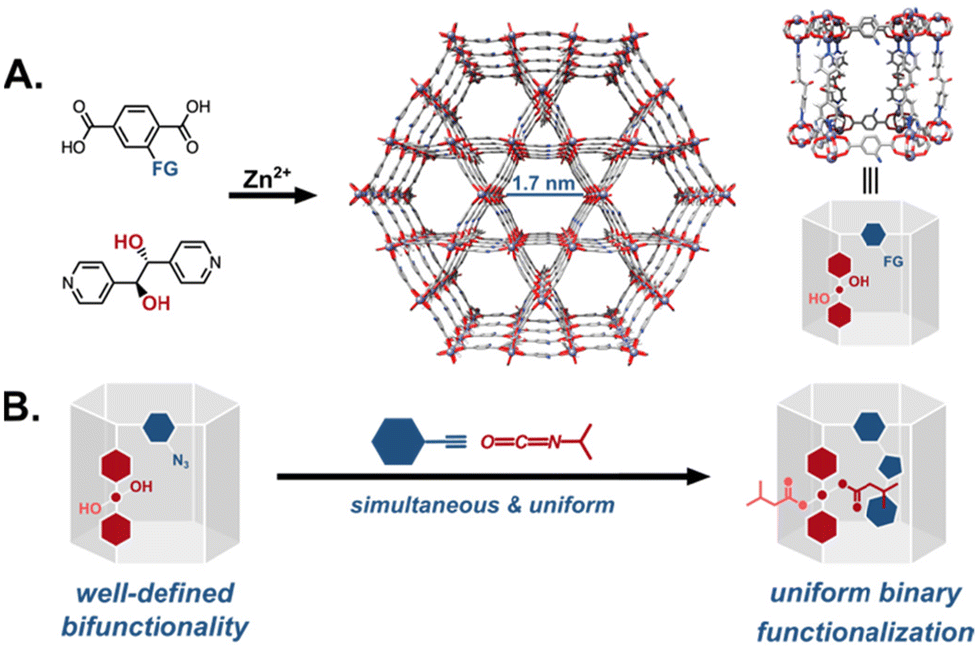
The previous example, however, leads to the second concern with multifunctionalized catalytic MOFs: non-uniformity due to the methods employed to introduce multiple functional groups into the active site. Two of the more prominent strategies are schematically represented in Fig. 13. In the first route, a MOF with a single reactive tag reacts with multiple reactants resulting in a random distribution of the moieties (Fig. 13A). Alternately, one can start with a MOF decorated with multiple reactive handles that can be independently functionalized with different reactants (Fig. 13B). In most examples of this strategy, however, the reactive tags in the parent MOF are also randomly distributed resulting in similarly multivariate daughter MOFs. This heterogeneity is difficult to characterize, requiring herculean efforts to map the distribution of the functional groups in the MOF. More importantly, in the context of catalysis, the heterogeneity of the MOF active sites likely leads to poor selectivities and sub-optimal activities. As a case in point, while the bifunctionalization of the proline MOF shown in Fig. 12B greatly improved the system's activity, the stereoselectivity was much lower compared to other proline MOFs performing the same reaction.31,53 We speculate that the construction of more homogeneously functionalised MOFs will deliver better selectivities for reactions that rely on the cooperative action of multiple functional groups. To this end, our group has spent the last few years developing strategies for the uniform covalent bifunctionalization of well-defined, mixed-linker MOFs with large pores and two disparate reactive functionalities (Fig. 14A).58,93,94 Taking advantage of the different reactivities of the tags, we have independently and quantitatively decorated MOFs with two different moieties, generating uniformly bifunctionalised MOF pores (Fig. 14B).
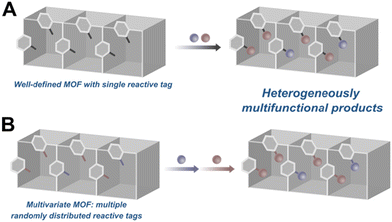 |
| | Fig. 13 Common strategies for the post-synthetic multifunctionalization of MOFs: (A) a well-defined MOF functionalized with two different moieties. (B) A multivariate MOF in which two different tags react independently with two different moieties. Both strategies result in multivariate MOFs. | |
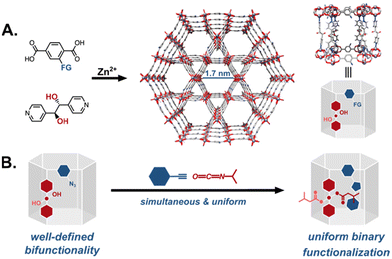 |
| | Fig. 14 Uniform post-synthetic multifunctionalization of MOFs: (A) well-defined, large-pore, pillared MOFs are constructed with two different linkers, each with independently reactive tags. For example, (B) a framework with orthogonally reactive tags yields a uniformly bifunctionalized MOF even in simultaneous reactions.93 | |
Conclusions
The myriad ways, discussed herein, of post-synthetically introducing catalytic functionality into MOFs (Table 1), coupled with the availability of strategies for uniform multifunctionalization, predict the construction of well-defined, confined, multifunctional MOF catalysts in the foreseeable future. Specifically, uniform MOF based catalysts in which the disparate functionalities are capable of cooperative action, thereby bringing us closer to our goal of synthesizing catalysts that possess the most salient features of enzymes.
Table 1 Summary of post-synthesis modification (PSM)-derived MOF catalysts
| Ref. |
Reactive tag |
MOF(s) |
PSM |
New functionality |
Catalysis |
|
16
|
–NH2 |
IRMOF-3 |
Imine condensation followed by metalation |
V(O) salicylidene |
Cyclohexene oxidation |
|
23
|
Pyridine |
UiO-66-Py |
N-Alkylation |
N-Methyl iodide and N-Methyl p-toluenesulfonate |
CO2 fixation with epoxide |
| UiO-67-Bpy |
|
24
|
Pyridine |
Pyridyl-MOF-1 |
N-Alkylation |
N-Methyl bromide |
CO2 fixation with epoxide |
|
25
|
Imidazolium |
Im-UiO-66 |
N-Alkylation |
N-Methyl iodide |
CO2 fixation with epoxide |
|
26
|
Pyridine |
1-Eu |
N-Alkylation |
N-Methyl halides |
CO2 fixation with epoxide |
|
27
|
–CHO |
UiO-67-CHO |
Imine condensation/reductive alkylation |
Alkyl amine |
Knoevenagel condensation |
|
28
|
–CHO |
ZIF-90 |
Imine condensation with aminopyridinium iodide |
Imino pyridinium iodide |
CO2 fixation with epoxide |
|
29
|
–CHO |
UiO-67-CHO |
Imine condensation followed by metalation |
Fe-metalated l-valinol |
Hydrofunctionalization |
| UiO-68-CHO |
|
30
|
–COOH/anhydride |
MIL-121 |
Decarboxylation/condensation followed by metalation |
Pt(NH3)4(OH)2 |
Oxygen reduction reaction (ORR) |
|
31
|
–CCH |
Zn-DPYI |
Cu-catalysed azide–alkyne cycloaddition (CuAAC) |
D or L pyrrolidine |
Asymmetric aldol reaction |
|
32
|
–N3 |
UiO-67-N3 |
CuAAC |
Alkyl amine |
Knoevenagel reaction |
|
33
|
–N3 |
MIL-101(Cr) |
CuAAC followed by metalation |
Terpyridyl(RuCl3) |
Alcohol oxidation |
|
35
|
–CCH/–N3 |
UiO-68-azide/alkyne |
CuAAC |
R-Pyrrolidine with carboxylic acid or methyl ester |
Aldol addition |
|
36
|
–N3 |
MIL-101-N3 |
CuAAC followed by N-alkylation |
3-Triazolium bromide |
CO2 fixation with epoxide |
|
37
|
–CCH |
UiO-66-alkyne |
Metalation |
Ni acetylide |
Knoevenagel condensation |
|
38
|
–SO2F |
UiO-67-SO2F |
Sulfur(VI) fluoride exchange (SuFEx) |
1H-Imidazolium bromide |
Benzoin condensation |
|
41
|
–NH2 |
MIL-53-(Al)-NH2 |
Nucleophilic acyl substitution |
Maleic acid |
Epoxide methanolysis |
|
60
|
–NH2 |
UiO-66-NH2 |
Propanesultone ring opening |
Sulfonic acid |
Benzaldehyde acetalization |
|
61
|
–NH2 |
UiO-66-NH2 |
Aziridine ring opening |
Alkyl amine |
Knoevenagel reaction |
| Cr-MIL-101-NH2 |
|
62
|
–NH2 |
Co-MOF |
Imine condensation |
Imidazole |
Knoevenagel reaction |
|
64
|
–NH2 |
UiO-66-NH2 |
Nucleophilic acyl substitution |
Sulfonic acid |
Acetalization and the Morita–Baylis–Hillman reaction |
|
65
|
–NH2 |
NH2-MIL-88-B (Fe) |
Propanesultone ring opening |
Sulfonic acid |
Epoxide ring-opening |
|
66
|
–NH2 |
MIL-53(Al)-NH2 |
Nucleophilic substitution |
Dimethyl amine |
Transesterification |
|
71
|
–NH2 |
NH2-MIL-125 |
Guanylation |
Guanidyl |
Claisen–Schmidt condensation |
|
72
|
–NH2 |
Cr-MIL-101-NH2 |
Nucleophilic addition to isocyanates |
Urea |
Friedel–Crafts alkylation |
|
76
|
–NH2 |
IRMOF-3 |
Nucleophilic addition to isocyanates |
Urea |
Morita–Baylis–Hillman reaction & acetalization |
|
81
|
–CHO |
SIM-1 |
Imine condensation |
Dodecylamine (exterior) |
Knoevenagel condensation |
|
82
|
–NH2 |
Al-MIL-101-NH2 |
Peptide coupling |
Mono-, di-, tri-, and tetrapeptides |
Asymmetric aldol reaction |
| In-MIL-68-NH2 |
| Zr-UiO-66-NH2 |
|
83
|
–CH2NHBoc |
MTV-IRMOF-74-III-(CH3)0.6(CH2NHBoc)0.4 |
Sequential peptide coupling |
Tripeptides |
Transesterification and α-chlorination |
|
84
|
–NH2 |
UiO-68-NH2 |
Peptide coupling followed by imine condensation and metalation |
Amino acid pyridylimine(Fe) |
Asymmetric hydrosilylation |
|
85
|
–NO2 and –SO3H |
MIL-101-NO2-SO3H |
NO2 reduction |
Amine and sulfonic acid |
Tandem deacetalization–nitroaldol reaction |
|
86
|
–NO2 |
MIL-101-NO2 |
NO2 reduction followed by partial propanesultone ring opening |
Amine and sulfonic acid |
Tandem deacetalization–Knoevenagel reaction |
|
87
|
–NH2 |
IRMOF-3 |
N-Alkylation of amine |
Methylammonium iodide |
CO2 fixation with epoxide |
|
88
|
–NH2 |
Cr-MIL-101-NH2 |
Nucleophilic substitution |
Triphenylalkylphosphonium bromide |
CO2 fixation with epoxide |
|
89
|
–Br |
MIL-101-Br |
Nucleophilic substitution by imidazole |
Imidazolium bromide |
CO2 fixation with epoxide |
|
90
|
–NH2 |
MIL-101-NH2 |
Debus–Radziszewski reaction |
Ethanol imidazolium |
CO2 fixation with epoxide |
|
91
|
–NH2 |
DMOF-NH2 |
Diazotransfer followed by CuAAC |
Alkyl amine and phenyl |
Transesterification |
Author contributions
T. G.: conceptualization (lead), writing – original draft (lead), and writing – review and editing (equal). P.M.: writing – review and editing (equal).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Science Foundation (Grant CHE-2240021) for providing partial support to both authors.
References
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed
 .
.
- B. N. Bhadra, I. Ahmed, H. J. Lee and S. H. Jhung, Coord. Chem. Rev., 2022, 450, 214237 CrossRef CAS .
- T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 6312–6313 CrossRef CAS PubMed .
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed .
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed .
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed .
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS PubMed .
- A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 2034–2036 CrossRef CAS PubMed .
- B. Rungtaweevoranit, C. S. Diercks, M. J. Kalmutzki and O. M. Yaghi, Faraday Discuss., 2017, 201, 9–45 RSC .
- K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866–3868 CrossRef CAS PubMed .
- M. S. Yazdanparast, V. W. Day and T. Gadzikwa, Molecules, 2020, 25, 697 CrossRef CAS PubMed .
- Y.-H. Kiang, G. B. Gardner, S. Lee, Z. Xu and E. B. Lobkovsky, J. Am. Chem. Soc., 1999, 121, 8204–8215 CrossRef CAS .
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS PubMed .
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC .
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368–12369 CrossRef CAS PubMed .
- M. J. Ingleson, J. P. Barrio, J.-B. Guilbaud, Y. Z. Khimyak and M. J. Rosseinsky, Chem. Commun., 2008, 2680–2682 RSC .
- S. J. Garibay, Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 7341–7349 CrossRef CAS PubMed .
- W. Gong, Z. Chen, J. Dong, Y. Liu and Y. Cui, Chem. Rev., 2022, 122, 9078–9144 CrossRef CAS PubMed .
- M. Kaur, S. Kumar, S. A. Younis, M. Yusuf, J. Lee, S. Weon, K.-H. Kim and A. K. Malik, Chem. Eng. J., 2021, 423, 130230 CrossRef CAS .
- Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296–306 CrossRef CAS PubMed .
- C. Volkringer and S. M. Cohen, Angew. Chem., 2010, 122, 4748–4752 CrossRef .
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC .
- H. Ji, K. Naveen, W. Lee, T. S. Kim, D. Kim and D.-H. Cho, ACS Appl. Mater. Interfaces, 2020, 12, 24868–24876 CrossRef CAS PubMed .
- J.-H. Xu, S.-F. Peng, Y.-K. Shi, S. Ding, G.-S. Yang, Y.-Q. Yang, Y.-H. Xu, C.-J. Jiang and Z.-M. Su, Dalton Trans., 2023, 52, 659–667 RSC .
- J. Liang, R.-P. Chen, X.-Y. Wang, T.-T. Liu, X.-S. Wang, Y.-B. Huang and R. Cao, Chem. Sci., 2017, 8, 1570–1575 RSC .
- W. Gao, C.-L. Wang, L. Chen, C.-Y. Zhu, P. Li, J.-Y. Li, J.-P. Liu and X.-M. Zhang, Appl. Organomet. Chem., 2022, 36, e6810 CrossRef CAS .
- F.-G. Xi, H. Liu, N.-N. Yang and E.-Q. Gao, Inorg. Chem., 2016, 55, 4701–4703 CrossRef CAS PubMed .
- J. Tharun, K.-M. Bhin, R. Roshan, D. W. Kim, A. C. Kathalikkattil, R. Babu, H. Y. Ahn, Y. S. Won and D.-W. Park, Green Chem., 2016, 18, 2479–2487 RSC .
- N. Antil, N. Akhtar, R. Newar, W. Begum, A. Kumar, M. Chauhan and K. Manna, ACS Catal., 2021, 11, 10450–10459 CrossRef CAS .
- S. Chen, Z. Song, J. Lyu, Y. Guo, B. E. G. Lucier, W. Luo, M. S. Workentin, X. Sun and Y. Huang, J. Am. Chem. Soc., 2020, 142, 4419–4428 CrossRef CAS PubMed .
- W. Zhu, C. He, P. Wu, X. Wu and C. Duan, Dalton Trans., 2012, 41, 3072–3077 RSC .
- X.-C. Yi, F.-G. Xi, Y. Qi and E.-Q. Gao, RSC Adv., 2014, 5, 893–900 RSC .
- S. Wu, L. Chen, B. Yin and Y. Li, Chem. Commun., 2015, 51, 9884–9887 RSC .
- B. Li, B. Gui, G. Hu, D. Yuan and C. Wang, Inorg. Chem., 2015, 54, 5139–5141 CrossRef CAS PubMed .
- Y. Zhang, B. Gui, R. Chen, G. Hu, Y. Meng, D. Yuan, M. Zeller and C. Wang, Inorg. Chem., 2018, 57, 2288–2295 CrossRef CAS PubMed .
- L.-J. Zhou, W. Sun, N.-N. Yang, P. Li, T. Gong, W.-J. Sun, Q. Sui and E.-Q. Gao, ChemSusChem, 2019, 12, 2202–2210 CrossRef CAS PubMed .
- H. Cheng, L. Ning, S. Liao, W. Li, S. Tang, J. Li, H. Chen, X. Liu and L. Shao, Appl. Catal., A, 2021, 623, 118216 CrossRef CAS .
- S. Park, H. Song, N. Ko, C. Kim, K. Kim and E. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 33785–33789 CrossRef CAS PubMed .
- M. Vinu, K. Sivasankar, S. Prabu, J.-L. Han, C.-H. Lin, C.-C. Yang and J. Demel, Eur. J. Inorg. Chem., 2020, 2020, 461–466 CrossRef CAS .
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354–14355 CrossRef CAS PubMed .
- S. J. Garibay, Z. Wang and S. M. Cohen, Inorg. Chem., 2010, 49, 8086–8091 CrossRef CAS PubMed .
- C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 9492–9493 CrossRef CAS PubMed .
- T. Kobayashi, K. Aoki and M. Sadakiyo, Inorg. Chem. Commun., 2021, 131, 108794 CrossRef CAS .
- G. Tuci, A. Rossin, X. Xu, M. Ranocchiari, J. A. van Bokhoven, L. Luconi, I. Manet, M. Melucci and G. Giambastiani, Chem. Mater., 2013, 25, 2297–2308 CrossRef CAS .
- D. Blow, Structure, 2000, 8, R77–R81 CrossRef CAS PubMed .
- G. Dodson and A. Wlodawer, Trends Biochem. Sci., 1998, 23, 347–352 CrossRef CAS PubMed .
- K.-Y. Wang, J. Zhang, Y.-C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R.-R. Liang, W. Shi and H.-C. Zhou, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed .
- L. J. Prins and P. Scrimin, Angew. Chem., Int. Ed., 2009, 48, 2288–2306 CrossRef CAS PubMed .
- R. Bischoff and H. Schlüter, J. Proteomics, 2012, 75, 2275–2296 CrossRef CAS PubMed .
- J. Canivet, S. Aguado, G. Bergeret and D. Farrusseng, Chem. Commun., 2011, 47, 11650–11652 RSC .
- D. J. Lun, G. I. N. Waterhouse and S. G. Telfer, J. Am. Chem. Soc., 2011, 133, 5806–5809 CrossRef CAS PubMed .
- C. Kutzscher, H. C. Hoffmann, S. Krause, U. Stoeck, I. Senkovska, E. Brunner and S. Kaskel, Inorg. Chem., 2015, 54, 1003–1009 CrossRef CAS PubMed .
- C. Kutzscher, G. Nickerl, I. Senkovska, V. Bon and S. Kaskel, Chem. Mater., 2016, 28, 2573–2580 CrossRef CAS .
- L. Liu, T.-Y. Zhou and S. G. Telfer, J. Am. Chem. Soc., 2017, 139, 13936–13943 CrossRef CAS PubMed .
- M. Sartor, T. Stein, F. Hoffmann and M. Fröba, Chem. Mater., 2016, 28, 519–528 CrossRef CAS .
- M. Zhou, E.-S. M. El-Sayed, Z. Ju, W. Wang and D. Yuan, Inorg. Chem. Front., 2020, 7, 1319–1333 RSC .
- T. Gadzikwa, O. K. Farha, K. L. Mulfort, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2009, 3720–3722 RSC .
- K. P. Samarakoon, C. S. Satterfield, M. C. McCoy, D. A. Pivaral-Urbina, T. Islamoglu, V. W. Day and T. Gadzikwa, Inorg. Chem., 2019, 58, 8906–8909 CrossRef CAS PubMed .
- H.-L. Jiang, D. Feng, T.-F. Liu, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 14690–14693 CrossRef CAS PubMed .
- Y. Luan, N. Zheng, Y. Qi, J. Yu and G. Wang, Eur. J. Inorg. Chem., 2014, 4268–4272 CrossRef CAS .
- Y. Luan, Y. Qi, H. Gao, R. S. Andriamitantsoa, N. Zheng and G. Wang, J. Mater. Chem. A, 2015, 3, 17320–17331 RSC .
- Z. Liu, L. Ning, K. Wang, L. Feng, W. Gu and X. Liu, J. Mol. Struct., 2020, 1221, 128744 CrossRef CAS .
- D. Britt, C. Lee, F. J. Uribe-Romo, H. Furukawa and O. M. Yaghi, Inorg. Chem., 2010, 49, 6387–6389 CrossRef CAS PubMed .
- Z. Miao, C. Qi, A. M. Wensley and Y. Luan, RSC Adv., 2016, 6, 67226–67231 RSC .
- M. Mohammadikish, Z. Valimohammadi and M. Masteri-Farahani, CrystEngComm, 2023, 25, 321–327 RSC .
- J. Chen, R. Liu, H. Gao, L. Chen and D. Ye, J. Mater. Chem. A, 2014, 2, 7205–7213 RSC .
- F.-G. Xi, W. Sun, Z. Dong, N.-N. Yang, T. Gong and E.-Q. Gao, Chem. Commun., 2020, 56, 13177–13180 RSC .
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed .
- H. A. Hamzah, W. J. Gee, P. R. Raithby, S. J. Teat, M. F. Mahon and A. D. Burrows, Chem. – Eur. J., 2018, 24, 11094–11102 CrossRef PubMed .
- D. P. Biswal, D. Singha, J. Panda and M. K. Rana, ChemPhysChem, 2023, 24, e202300311 CrossRef PubMed .
- S. Mukherjee, M. Singh, A. Ravani, A. Parekh, A. Shukla, S. Chaki, S. Neogi and M. K. Mishra, Microporous Mesoporous Mater., 2023, 359, 112636 CrossRef CAS .
- X.-W. Dong, T. Liu, Y.-Z. Hu, X.-Y. Liu and C.-M. Che, Chem. Commun., 2013, 49, 7681–7683 RSC .
- S. M. Masoom Nataj, S. Kaliaguine and F.-G. Fontaine, Catal. Today, 2023, 422, 114216 CrossRef .
- S. M. Masoom Nataj, S. Kaliaguine and F.-G. Fontaine, ChemCatChem, 2023, 15, e202300079 CrossRef CAS .
- A. Karmakar, S. Hazra and A. J. L. Pombeiro, Coord. Chem. Rev., 2022, 453, 214314 CrossRef CAS .
- Y. Luan, N. Zheng, Y. Qi, J. Tang and G. Wang, Catal. Sci. Technol., 2014, 4, 925–929 RSC .
- R. Breslow, J. Phys. Org. Chem., 2006, 19, 813–822 CrossRef CAS .
- L. Marchetti and M. Levine, ACS Catal., 2011, 1, 1090–1118 CrossRef CAS .
- L.-H. Xie, M.-M. Xu, X.-M. Liu, M.-J. Zhao and J.-R. Li, Adv. Sci., 2020, 7, 1901758 CrossRef CAS PubMed .
- L. Liu, Z.-P. Tao, H.-R. Chi, B. Wang, S.-M. Wang and Z.-B. Han, Dalton Trans., 2021, 50, 39–58 RSC .
- J. Canivet, S. Aguado, C. Daniel and D. Farrusseng, ChemCatChem, 2011, 3, 675–678 CrossRef CAS .
- J. Bonnefoy, A. Legrand, E. A. Quadrelli, J. Canivet and D. Farrusseng, J. Am. Chem. Soc., 2015, 137, 9409–9416 CrossRef CAS PubMed .
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352–8355 CrossRef CAS PubMed .
- R. Newar, N. Akhtar, N. Antil, A. Kumar, S. Shukla, W. Begum and K. Manna, Angew. Chem., Int. Ed., 2021, 60, 10964–10970 CrossRef CAS PubMed .
- Y.-R. Lee, Y.-M. Chung and W.-S. Ahn, RSC Adv., 2014, 4, 23064–23067 RSC .
- H. Liu, F.-G. Xi, W. Sun, N.-N. Yang and E.-Q. Gao, Inorg. Chem., 2016, 55, 5753–5755 CrossRef CAS PubMed .
- X. Zhou, Y. Zhang, X. Yang, L. Zhao and G. Wang, J. Mol. Catal. A: Chem., 2012, 361–362, 12–16 CrossRef CAS .
- W. Dai, P. Mao, Y. Liu, S. Zhang, B. Li, L. Yang, X. Luo and J. Zou, J. CO2 Util., 2020, 36, 295–305 CrossRef CAS .
- D. Ma, Y. Zhang, S. Jiao, J. Li, K. Liu and Z. Shi, Chem. Commun., 2019, 55, 14347–14350 RSC .
- S. Liu, M.-L. Gao, Y. Zhang, L. Liu and Z.-B. Han, Inorg. Chem., 2021, 60, 6152–6156 CrossRef CAS PubMed .
- M. Savonnet, A. Camarata, J. Canivet, D. Bazer-Bachi, N. Bats, V. Lecocq, C. Pinel and D. Farrusseng, Dalton Trans., 2012, 41, 3945–3948 RSC .
- L. Yan, T. Duan, T. Huang, B. Zhao and Y. Fan, Fuel, 2019, 245, 226–232 CrossRef CAS .
- K. P. Samarakoon, M. S. Yazdanparast, V. W. Day and T. Gadzikwa, Mol. Syst. Des. Eng., 2020, 5, 804–808 RSC .
- P. Matseketsa, D. Mafukidze, L. Pothupitiya, U. P. Otuonye, Y. Ç. Mutlu, B. B. Averkiev and T. Gadzikwa, Mol. Syst. Des. Eng. 10.1039/D3ME00185G .
|
| This journal is © The Royal Society of Chemistry 2024 |

 * and
Pricilla
Matseketsa
* and
Pricilla
Matseketsa