 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatio-temporal detachment of homogeneous electron transfer in controlling selectivity in mediated organic electrosynthesis†
Jack W.
Hodgson
 ,
Ana A.
Folgueiras-Amador
,
Derek
Pletcher
,
David C.
Harrowven
,
Guy
Denuault
* and
Richard C. D.
Brown
*
,
Ana A.
Folgueiras-Amador
,
Derek
Pletcher
,
David C.
Harrowven
,
Guy
Denuault
* and
Richard C. D.
Brown
*
School of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: rcb1@soton.ac.uk
First published on 6th June 2023
Abstract
In electrosynthesis, electron transfer (ET) mediators are normally chosen such that they are more easily reduced (or oxidised) than the substrate for cathodic (or anodic) processes; setting the electrode potential to the mediator therefore ensures selective heterogeneous ET with the mediator at the electrode, rather than the substrate. The current work investigates the opposite, and counter intuitive, situation for a successful mediated electroreductive process where the mediator (phenanthrene) has a reduction potential that is negative to that of the substrate, and the cathode potential is set negative to both (Eele < EM < Es). Simulations reveal a complex interplay between mass transport, the relative concentrations of the mediator and substrate as well as the heterogeneous and homogeneous rate constants for multiple steps, which under suitable conditions, leads to separation of the homogeneous chemistry in a reaction layer detached from the electrode. Reaction layer detachment is a spatio-temporal effect arising due to opposing fluxes of the mediator radical anion M˙− and the substrate 1, which ultimately prevents 1 from reaching the electrode, thereby affording a different reaction pathway. Simulations representative of unstirred batch (1D) and flow (2D) electrolysis are presented, which qualitatively reproduce the experimental selectivity outcomes for mediated and unmediated electroreductive cyclisation of aryl iodide 1. The potential to use highly reducing homogeneous ET agents, possessing reduction potentials beyond those of the substrates, offers exciting opportunities in mediated electrosynthesis.
Introduction
Aryl radical cyclisation is an established approach for the synthesis of carbocyclic and heterocyclic compounds, applied in research laboratories around the world towards a plethora of interesting and useful target molecules. A classical method involves heating an aryl bromide or iodide, such as 1, in the presence of Bu3SnH and a suitable initiator (e.g. AIBN) to give the cyclised product 2 and the corresponding tributyltin halide (Scheme 1). Although radical cyclisation using tin hydrides provides a reliable synthesis with broad scope, its appeal from environmental and safety perspectives is diminished somewhat due to the toxicity of organotin compounds, hazards associated with initiators, and the high reaction temperatures often required. Significant effort has been devoted to replacing Bu3SnH with other hydrides, developing conditions that are substoichiometric in organotin, and efficient methods for removing organotin impurities, but in spite of these developments the use of Bu3SnH in radical cyclisation often remains the method of choice for laboratory applications. Recent years have seen vigorous interest in alternative methods to access aryl radicals from a variety of precursors, particularly, using photochemical activation.1–3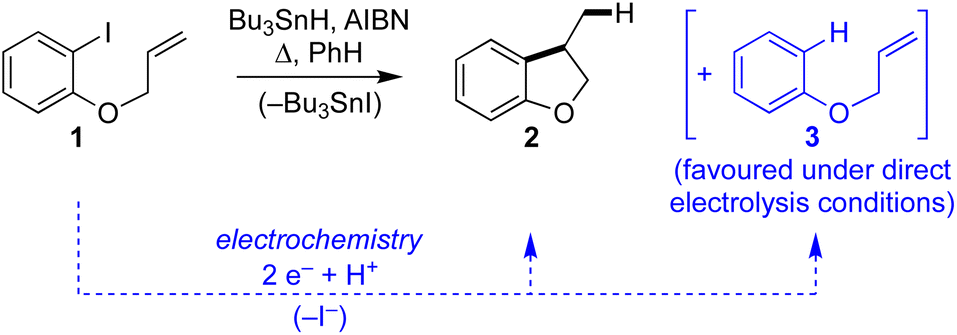 | ||
| Scheme 1 Classical Bu3SnH mediated radical cyclisation of aryl iodide 1 and electroreductive approach. | ||
Electrochemistry offers a more environmentally acceptable method to produce aryl radicals by reduction of aryl halides.4 However, a limitation is that direct cathodic electrolysis of aryl halides favours hydrogenolysis products (e.g.3) over radical cyclisation (e.g.2) for all but a limited sub-set of substrates (Scheme 1).5,6 The electrochemical mechanism involves electron transfer (ET) to aryl iodide 1, giving rise to a frangible radical anion 1˙− which very rapidly loses iodide ion to afford an aryl radical 4 (Scheme 2).7 The relative rates of the onward reactions of 4 affect the selectivity for either formal hydrogenolysis product 3 or cyclised product 2, and as will be discussed below, a crucial factor is the spatial proximity of the aryl radical to the cathode due to the highly favourable ET to 4 giving the aryl anion 5.8
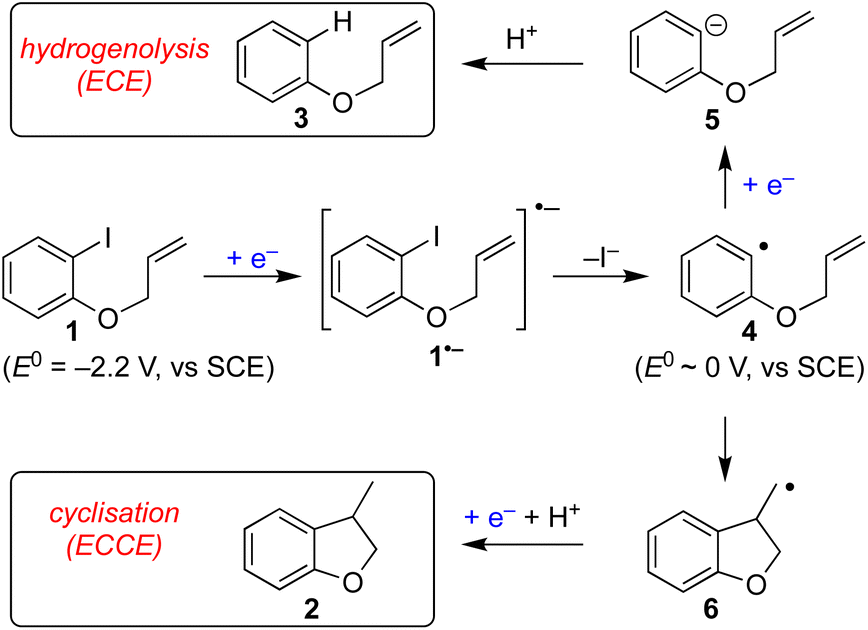 | ||
| Scheme 2 Competing hydrogenolysis and cyclisation pathways observed for direct electroreduction of aryl iodide 1. In the pathway acronyms (ECE or ECCE), E stands for a heterogeneous ET at the electrode while C stands for a homogeneous process in solution. | ||
The use of outer sphere electron transfer mediators in electrosynthesis is attracting increasing interest from organic chemists.9 In this context, the mediator (M) behaves as an electron shuttle between electrode surface and substrate such that ET to/from the substrate occurs as a homogeneous process. It is important to recognise that heterogeneous ET – focussing here on reduction of a mediator to its radical anion – occurs within molecular distances from the electrode, whereas homogeneous ET between mediator radical ions and substrate occurs anywhere both species meet, with the proviso that the reduction potential of M is sufficiently negative.9 The ability to move ET away from the electrode provides an opportunity for altered and useful selectivity, for example, in situations where ET is followed by a very fast chemical reaction leading to an intermediate that can either undergo a second ET (ECE pathway) or a chemical reaction (ECCE pathway). An illustrative case is the cathodic reduction of aryl halides highlighted below, where direct electrolysis favours hydrogenolysis (ECE), whereas, in the presence of suitable mediators the radical intermediate can undergo selective transformations such as cyclisation onto pendant unsaturation (ECC).7
Grimshaw et al. reported that direct electrolysis of aryl chloride 7 at a Hg cathode in a divided cell gives the hydrogenolysis product 9, whereas electrolysis of the same substrate in the presence of stilbene (10) affords the cyclised indolene 8 in good yield (Scheme 3a).5d In another study, Mitsudo et al. demonstrated that very high selectivity for the cyclised dihydrobenzofuran 12 is achieved from electrolysis of aryl bromide 11 conducted in the presence of 1 equivalent of diethylfluorene 14 in an undivided cell equipped with a sacrificial electrode.10 In the absence of a mediator, the selectivity is reversed yielding mainly hydrogenolysis product 13 (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 ∼ 1:1.7). At this juncture, it is worth highlighting that in the former case the mediator 10 has a reduction potential that is positive to the respective substrate, whereas the reverse is true for the reaction mediated by diethylfluorene (i.e. diethylfluorene is actually harder to reduce at the cathode than the aryl bromide substrate 11). Other examples of mediated reductive electrocyclization of aryl halides via the intermediacy of aryl radicals have been reported.11,12 The studies described thus far highlight the interplay of the two different reaction manifolds shown in Scheme 2, which are dependent on a number of factors, including homogeneous and heterogeneous ET rate constants and mass transport.13 The preferential formation of hydrogenolysis products in direct electrochemical reduction of aryl halides is well established (Scheme 2), arising because loss of halide ion from frangible radical anions such as 1˙− is so rapid that the aryl radical 4 is formed close to the cathode.7 Aryl radicals exhibit significantly more positive reduction potentials
:13 ∼ 1:1.7). At this juncture, it is worth highlighting that in the former case the mediator 10 has a reduction potential that is positive to the respective substrate, whereas the reverse is true for the reaction mediated by diethylfluorene (i.e. diethylfluorene is actually harder to reduce at the cathode than the aryl bromide substrate 11). Other examples of mediated reductive electrocyclization of aryl halides via the intermediacy of aryl radicals have been reported.11,12 The studies described thus far highlight the interplay of the two different reaction manifolds shown in Scheme 2, which are dependent on a number of factors, including homogeneous and heterogeneous ET rate constants and mass transport.13 The preferential formation of hydrogenolysis products in direct electrochemical reduction of aryl halides is well established (Scheme 2), arising because loss of halide ion from frangible radical anions such as 1˙− is so rapid that the aryl radical 4 is formed close to the cathode.7 Aryl radicals exhibit significantly more positive reduction potentials 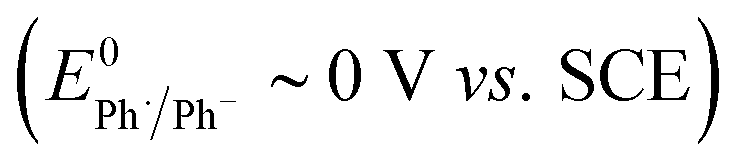 compared to aryl halides; they are thus readily reduced at the potential required to form the parent radical anion, and therefore the ECE pathway prevails. On the other hand, when a suitable mediator is present, ET from M˙− to the aryl halide 1 can occur wherever both radical anion and substrate are found in the electrolyte solution. When ET takes place away from the electrode, the ensuing rapid ejection of halide ensures that the aryl radical 4 is also formed away from the electrode.
compared to aryl halides; they are thus readily reduced at the potential required to form the parent radical anion, and therefore the ECE pathway prevails. On the other hand, when a suitable mediator is present, ET from M˙− to the aryl halide 1 can occur wherever both radical anion and substrate are found in the electrolyte solution. When ET takes place away from the electrode, the ensuing rapid ejection of halide ensures that the aryl radical 4 is also formed away from the electrode.
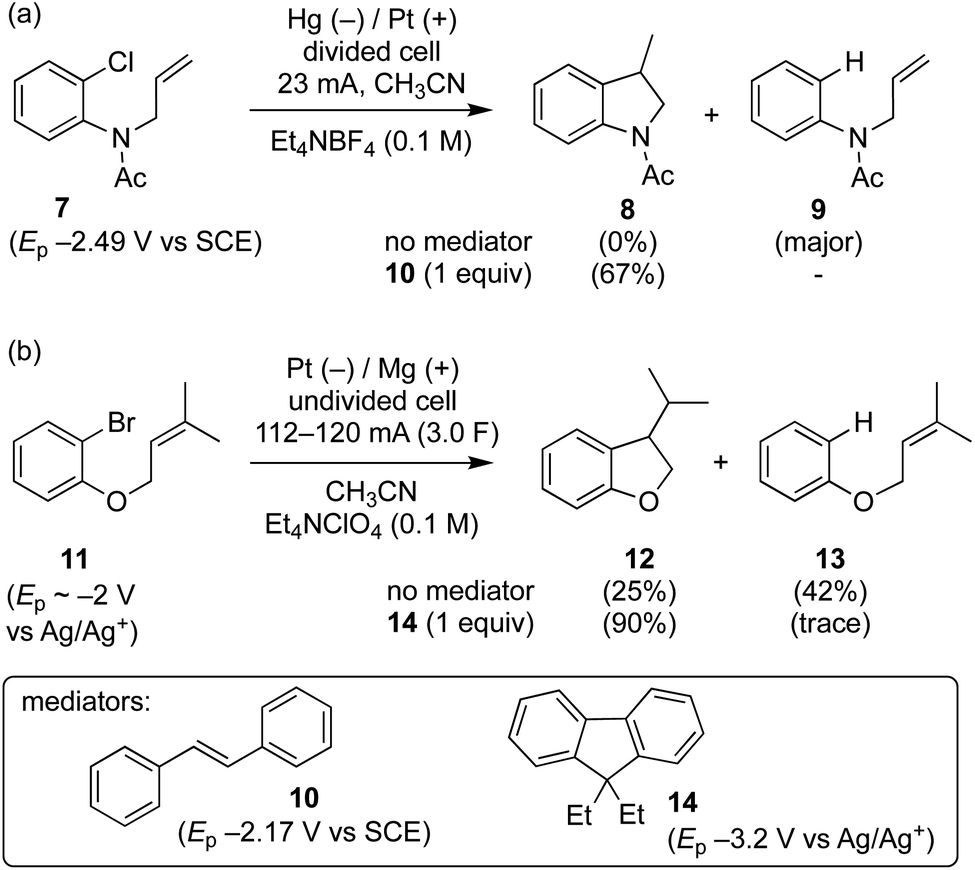 | ||
| Scheme 3 Direct and mediated electroreductions of aryl halides 7 and 11. | ||
A fast chemical reaction step (e.g. cyclisation to 6) outpaces reduction of the aryl radical, as the latter requires either mass transport to the cathode, or a homogeneous ET with M˙− (or ArI˙−, disproportionation). Therefore, in order for the radical cyclisation pathway (ECCE) to predominate, the aryl radical 4 should not be present close to the cathode where heterogeneous ET becomes the most favourable process.
In texts discussing mediated electrosynthesis it is stated that the mediator (M) must be more easily reduced or oxidised than the substrate in order to favour ET between the mediator and electrode, rather than the substrate and electrode, to allow continuous regeneration of the mediator in situ.9 In the context of mediated electroreduction, this follows a common observation that in situations where two or more electroactive species are present, that with the less negative reduction potential is reduced preferentially at the electrode, assuming sufficiently different reduction potentials. However, this neglects the important influence of mass transport, which limits the overall rate of electrochemical reactions involving very fast ET and chemical steps. Interestingly, several papers describe reductive cyclisation of aryl halides in the presence of mediators with reduction potentials that are negative to that of the substrate (i.e. the mediator is harder to reduce than the substrate). For example, phenanthrene (E0 = −2.5 V vs. SCE), and fluorene (Ep = −3.5 V vs. Ag/Ag+) are reported as mediators for cyclisations of aryl halides (E0 ∼ −2 V vs. SCE) (see Scheme 3b).10,11b Consequently, under the potential required to drive the electrochemical reduction of the mediator, direct cathodic reduction of the substrate is a highly favourable process. The authors did not propose a satisfactory explanation to account for the observed selectivity for cyclised products in the presence of mediators, and a rationale based only on reduction potentials is neither obvious nor sufficient.
Recently, we reported an electroreductive cyclisation of aryl halides, including 1, in the presence of substoichiometric amounts of a highly reducing mediator in an undivided flow cell (Scheme 4).14 Voltammetry clearly shows that the mediator, phenanthrene (M), has a reduction potential negative to that required to electrochemically reduce the substrate 1. When mediator is present, the major product is dihydrobenzofuran 2 (82%) with radical dimerisation product 15 (8%) and hydrogenolysis product 3 (2%) as minor by-products. In the absence of mediator, selectivity is reversed and 3 is the major product (48%, 2:3:15 ∼ 1:2:0). We proposed that, in the mediated process, homogeneous ET takes place in a reaction layer (or zone) that is detached from the cathode surface, and that the substrate 1 is prevented from reaching the electrode surface by the flux of highly reducing mediator radical anion M˙− diffusing outwards from the cathode and balancing the inward flux of aryl halide. In the work described here, we present simulations to illustrate and highlight the important role of mass transport in achieving selectivity in the mediated electroreduction of ArI 1 as a consequence of a time-dependent detachment of the homogeneous reaction layer, and discuss the factors influencing detachment in unstirred batch and laminar flow regimes. Significantly, we show that the reduction potential of the mediator does not need to be positive to that of the substrate to achieve a selective mediated electrosynthesis using catalytic mediator in situ. We believe that a better understanding and appreciation of the interplay between mass transport, electron-transfer and chemical steps will offer great opportunities in electrosynthesis, and will also account for other outcomes that cannot be rationalised by electrochemical mechanisms alone.
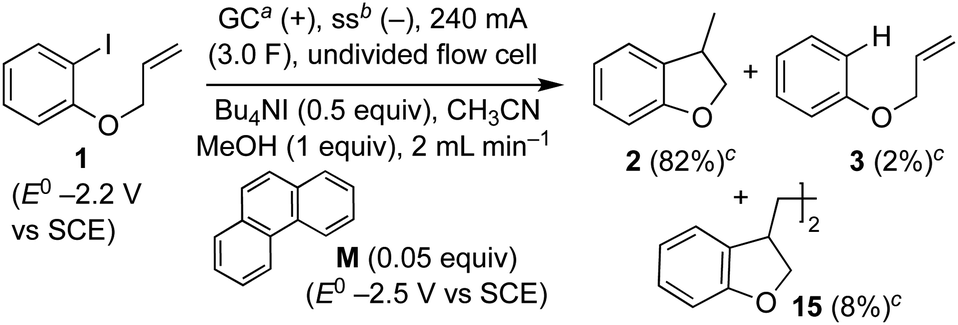 | ||
| Scheme 4 Selective electroreductive cyclisation of aryl iodide 1 in an undivided electrochemical flow cell, using a highly reducing catalytic mediator. a Glassy carbon anode. b Stainless steel cathode. c Yields are estimated using calibrated GC. | ||
Simulations
1D simulations were carried out using DigiElch v.8 and COMSOL Multiphysics v.6.0 whereas 2D simulations were performed with COMSOL Multiphysics v.6.0. The reaction steps (1)–(11) described in Schemes 5–7, were used in all cases, unless stated otherwise. In the 1D simulations, diffusion perpendicular to the electrode was the only form of mass transport considered. In contrast, 2D simulations involved convection (flow) and diffusion parallel to the electrode as well as diffusion normal to the electrode. For the 1D simulations, the cathode (1 cm2) was located 0.5 mm away from an inert wall.15 For the 2D simulations, the cathode (0.95 mm long, 2 mm deep) was located on the floor of a narrow channel flow cell (0.5 mm high, 1 mm long, 2 mm deep).15 For simplicity, solution velocities within the channel were computed assuming laminar flow. With DigiElch, adaptive meshing was used to allow the numerical procedure to track the rapidly changing concentration profiles. In COMSOL, an expanding grid with an adaptive mesh environment was used to confine a high density of mesh elements near the electrode surface where narrow distributions of species were expected. Without this, the numerical solver is not able to compute highly localised concentration gradients. However, the optimum mesh is difficult to predict as the reaction layer thickness and its distance away from the electrode depend on the rate constants of the homogeneous processes and on the concentrations and diffusion coefficients of the species involved.16 In COMSOL, the mesh was first optimised by comparing the results of the 1D simulations with those obtained with DigiElch. For the 2D simulations, the optimised 1D mesh was then used to constrain the size and growth of mesh elements along the normal to the electrode. The mesh was further refined near the upstream electrode edge where high current densities were expected. Along the channel, larger mesh elements were selected as concentrations were not expected to vary rapidly.Mechanism of direct and mediated electrochemical reduction of aryl iodide 1
The present work concerns simulations of direct and mediated electroreduction of aryl iodide 1, for which we have previously disclosed electroanalytical data and preparative results in batch and flow electrolysis reactors.14 A proposed mechanism for direct electrochemical reduction of 1 leading to hydrogenolysis product 3 incorporates reactions (1)–(3) (Scheme 5).7 Much effort has been devoted by others to the study of these mechanisms, including determination of kinetic and thermodynamic parameters for a variety of aryl halides that are not directly accessible by electrochemical techniques.7 Where necessary, the present work uses literature values for rate constants and reduction potentials from related compounds/intermediates where we were unable to determine them.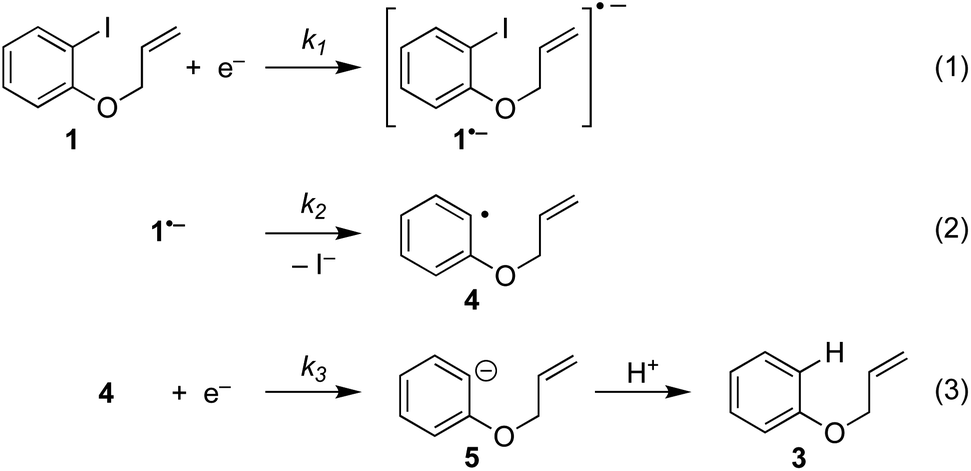 | ||
| Scheme 5 ECE(C) pathway: reaction steps involved in direct electrochemical reduction (hydrogenolysis) of ArI 1. | ||
The rate of ET of aryl iodide 1 at the electrode is primarily determined by the standard rate constant for ET, k1, which reflects the extent of reorganisation energy required to form the transition state, and by the electrode potential which sets the ET driving force. The latter is high because the electrode potential of −2.8 V vs. SCE, set in the simulation, is more negative than the E0 for aryl iodide 1; as a result, 1 is readily reduced at the electrode surface where its concentration is driven to zero. The standard potential for 1 (E0 = −2.2 V vs. SCE) and rate constant (k1 = 5 × 10−3 cm s−1) for heterogeneous electron transfer were obtained from simulated voltammograms fitted to the experimental voltammetry.14,17 The characteristic features of the voltammogram show a single irreversible wave due to a rate limiting electron transfer and very fast fragmentation of ArI˙− giving aryl radical 4 and I−, the former undergoing facile ET and protonation. A rate constant (k2) of 1010 s−1 or greater is expected for reaction (2) based upon published experimental and theoretical values for fragmentation of radical anions of aryl iodides and bromides.18 As discussed above, the heterogeneous ET in reaction step (3) is also assumed to be rapid as the electrode potential necessary for reduction of aryl halide 1 is well negative to that required for reduction of Ar˙ 4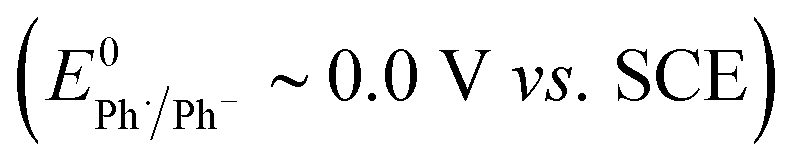 .8a An estimation of the heterogeneous electron transfer rate constant (k3 ∼ 0.03 cm s−1) for step (3) was taken from Andrieux et al.8a The final protonation of the aryl anion 5 by the solvent (CH3CN) is incorporated within step (3) as protonation of this highly basic species is under diffusion control, and in any case, anion 5 is not involved in any other reaction steps.
.8a An estimation of the heterogeneous electron transfer rate constant (k3 ∼ 0.03 cm s−1) for step (3) was taken from Andrieux et al.8a The final protonation of the aryl anion 5 by the solvent (CH3CN) is incorporated within step (3) as protonation of this highly basic species is under diffusion control, and in any case, anion 5 is not involved in any other reaction steps.
The electrochemical and chemical steps (4)–(8) are involved in the mediated reductive cyclisation of aryl iodide 1 giving rise to the experimentally observed cyclisation and radical dimerisation products 2 and 15, respectively (Scheme 6). Reactions (5) and (7) involve homogeneous ET, and their rate constants (k5 = 4.0 × 105 M−1 s−1, and k7 = 1 × 109 M−1 s−1) are approximated from literature values of related processes.19,20 Step (4) is a heterogeneous ET to the mediator and values for the ET rate constant (k4 = 3 × 10−2 cm s−1), and 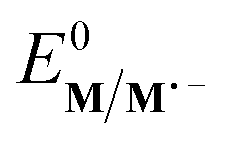 (−2.5 V vs. SCE) were obtained from the simulated voltammetry fitted to our experimental data.14,17 The rate constant (k6 = 8 × 109 s−1) for cyclisation step (6) is taken from experimental values estimated for the same reaction.21 As protonation of highly basic alkyl anionic species by a component of the electrolysis medium is rapid, and has no bearing on onwards reactions, it is incorporated in step (7).22 Previous studies in deuterated solvents provide strong evidence that dihydrobenzofuran is formed principally by step (7) rather than through H-atom abstraction. Therefore, abstractions of a hydrogen atom from solvent by aryl and alkyl radicals have not been included in the present simulations. The rate constant (k8 = 109 M−1 s−1) for dimerisation of alkyl radical 6 (step (8)) is approximated to that reported for the 2-phenylethyl radical.23
(−2.5 V vs. SCE) were obtained from the simulated voltammetry fitted to our experimental data.14,17 The rate constant (k6 = 8 × 109 s−1) for cyclisation step (6) is taken from experimental values estimated for the same reaction.21 As protonation of highly basic alkyl anionic species by a component of the electrolysis medium is rapid, and has no bearing on onwards reactions, it is incorporated in step (7).22 Previous studies in deuterated solvents provide strong evidence that dihydrobenzofuran is formed principally by step (7) rather than through H-atom abstraction. Therefore, abstractions of a hydrogen atom from solvent by aryl and alkyl radicals have not been included in the present simulations. The rate constant (k8 = 109 M−1 s−1) for dimerisation of alkyl radical 6 (step (8)) is approximated to that reported for the 2-phenylethyl radical.23
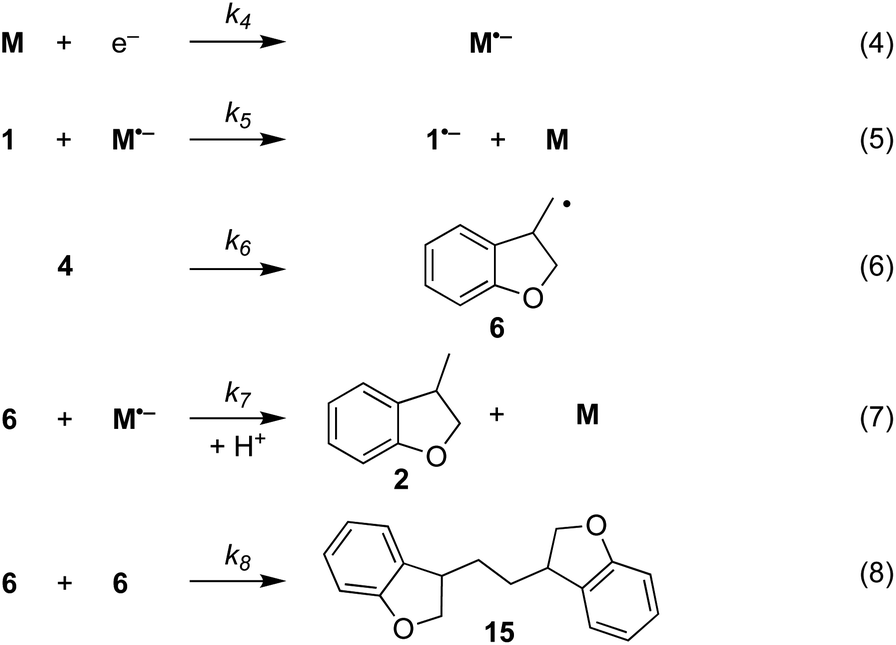 | ||
| Scheme 6 Reaction steps in the mediated electroreductive cyclisation of aryl iodide 1, including dimerisation of alkyl radical 6. | ||
Additional reactions included in simulations are heterogeneous reduction of the cyclised radical 6 (step (9)), homogeneous reduction of aryl radical 4 by M˙− (step (10)) or through disproportionation step (11) with 1˙− (Scheme 7). While all the products of these reactions are observed experimentally, heterogeneous reduction of the cyclised radical is only expected to be important in the direct mechanism. Bimolecular reactions (10) and (11) are unlikely to contribute significantly toward the formation of 3 as both radical anion 1˙− and aryl radical 4 are consumed in very fast unimolecular reaction steps (2) and (6).
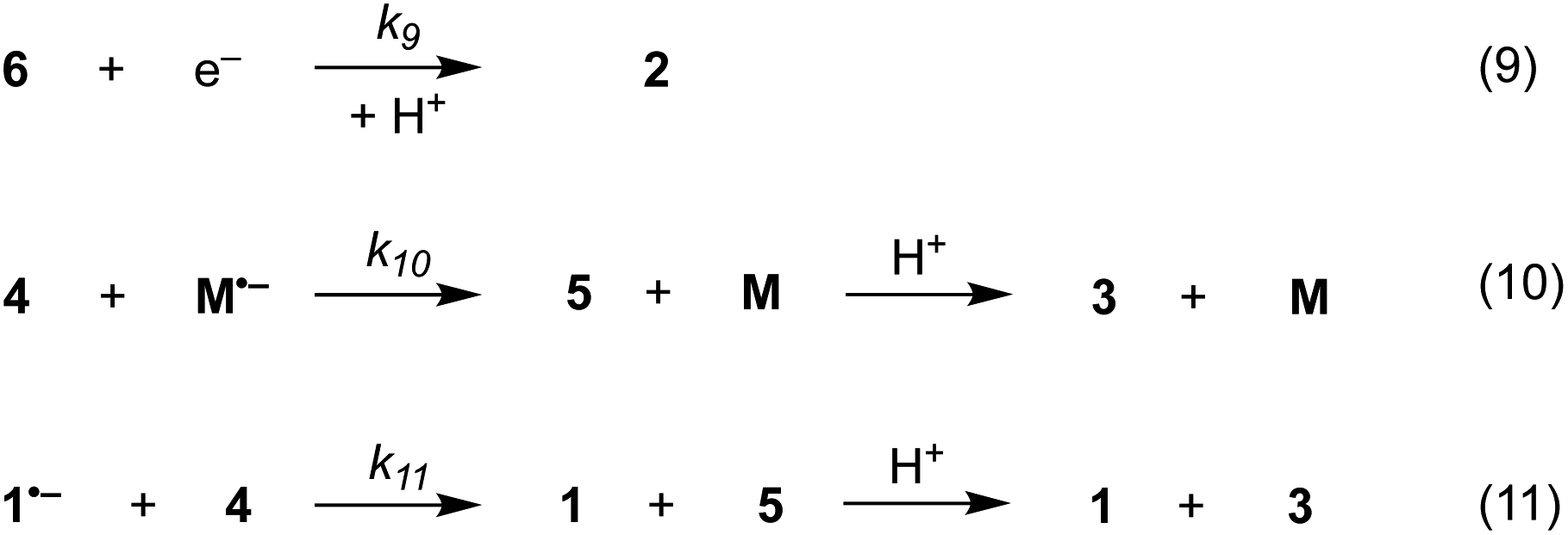 | ||
| Scheme 7 Additional reaction steps (9)–(10) included in simulations. | ||
Diffusion coefficients for the substrate (1, 3.3 × 10−5 cm2 s−1) and phenanthrene (M, 2.0 × 10−5 cm2 s−1) were determined using voltammetry.14 The diffusion coefficients for intermediates and mediator radical anions are approximated to those of their respective parent compounds.
Results
Reaction product profiles
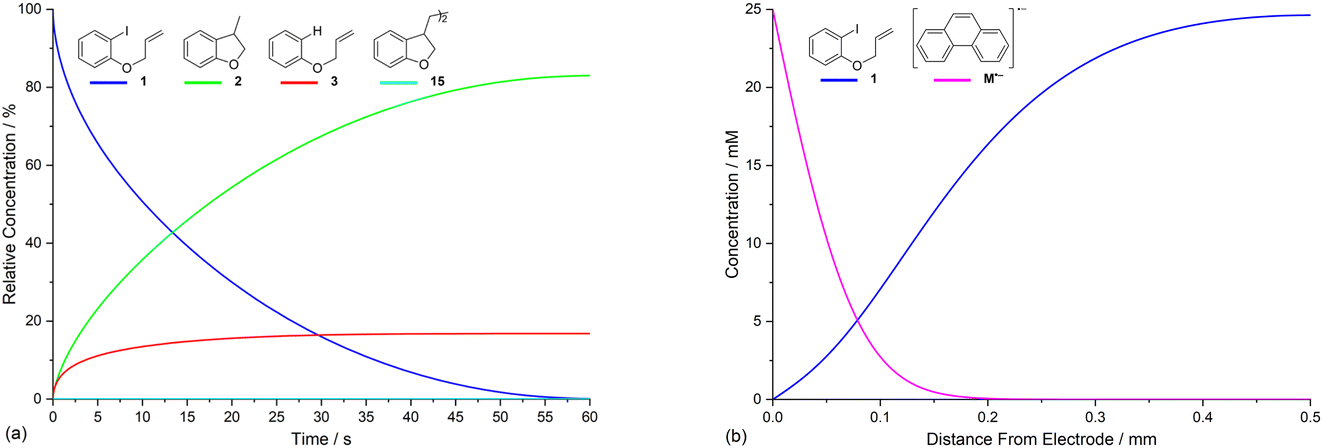
Electrolysis of ArI 1 was first simulated in one dimension (1D) combining the electrochemical steps (1)–(11) using COMSOL Multiphysics. The results were validated with simulations performed in DigiElch for identical conditions. This allowed optimisation of the COMSOL mesh elements along the electrode normal. Subsequently, the same mechanism was simulated in COMSOL in the presence of fluid flow (see below). The 1D simulations, where mass transport is only through diffusion perpendicular to the electrode, are representative of unstirred batch electrolysis conditions, whereas the 2D flow simulations are representative of channel flow cells as found in the Ammonite 8 electrochemical reactor,24 where mass transport involves convection and diffusion parallel to the electrode surface as well as diffusion perpendicular to the electrode. The fluid dynamics models do not include migration of charged species under the influence of electric field, which will be a topic of further work. Fig. 1a shows percentage conversions for electrolysis of ArI (t = 0 s, [1] = 0.025 M) from the 1D simulation over 60 s in the absence of the mediator ([M] = 0 M) with the electrode potential set to −2.8 V vs. SCE, a value that is negative to both substrate and phenanthrene. As conversion of ArI proceeds, cyclisation and hydrogenolysis products (2 and 3, respectively) are both formed with a modest selectivity for 3 (2:3 ∼ 1:1.2), which qualitatively is comparable with the experimental results for direct electrolysis in a stirred batch cell (2:3 ∼ 1:2).14 In the unmediated reaction, the balance between hydrogenolysis and cyclisation pathways is influenced by the relative rates of steps (3) and (6). Cyclisation of aryl radical 4 is a 5-exo-trig process with a high rate constant (k6 = 8 × 109 s−1),21 whereas the rate of heterogeneous ET step (2) is determined by both the standard rate constant, k2, and the electrode potential. At conditions of high cathode overpotential, heterogeneous ET to Ar˙ (E0 ∼ 0 V vs. SCE) is a very fast process, giving rise to the observed selectivity.
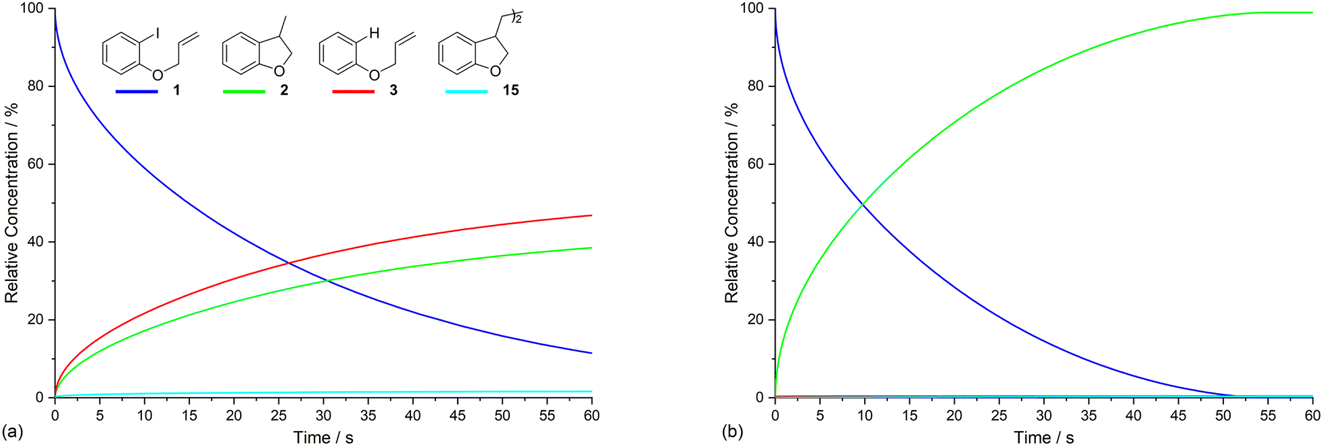 | ||
| Fig. 1 1D simulation (representative of unstirred batch conditions) showing relative concentrations of ArI 1, and products 2, 3 and 15, respectively, as a function of time for: (a) direct electrolysis of ArI 1; (b) electrolysis of 1 in the presence of phenanthrene (1 equiv.). Starting concentration of ArI 1 is 0.025 M. Electrolysis time = 0 to 60 s under constant potential of −2.8 V vs. SCE. | ||
The product 15 from the dimerisation of alkyl radical 4 (step (8)) is seen in small amount (2%) under the simulation conditions where the cathode potential is set to −2.8 V vs. SCE. The influence of cathode potential on product selectivity is discussed below. Repeating the simulation in the presence of 1 equivalent of phenanthrene ([M] = 0.025 M) shows a dramatic switch in the selectivity (Fig. 1b), now strongly in favour of cyclised product 2, with minor amounts of hydrogenolysis and dimerisation products. After 60 s, the simulated selectivity profile (2:3:15 ∼ 99:0.5:0.5) again displays a similar trend to the experimental outcome from our batch electrolysis (2 (74%):3 (3%):15 (1%) ∼ 95:4:1). Another interesting feature of the simulation in the presence of phenanthrene is that very little ArI remains after 50 s, compared to ∼10% at 60 s when the mediator is absent. The various factors influencing the selectivity outcome of the electrolysis are discussed in the ensuing sections.
Time dependence of concentration profiles of the reaction components
Defining the spatial distribution of reactive species is central to this work and is presented here using concentration profiles with respect to distance from the cathode for the 1D simulations. Consumption of species at the cathode gives rise to concentration gradients between the electrode surface and the inert wall, and these are seen to evolve in a time dependent fashion. In the absence of phenanthrene, concentration profiles for ArI 1 show its concentration to be highest near the inert wall and dropping to zero at the cathode surface where it is consumed by heterogeneous ET step (1) to give Ar˙− (Fig. 2a). Concentration profiles for 1 flatten from t = 1–60 s as its total concentration decreases by its progressive conversion to products 2 and 3. Simulated profiles for 3 and 2 show concentrations that are highest at the cathode, where they are formed by the direct mechanism steps (1)–(3), or (1), (2), (6) and (9), respectively (Fig. 2b, only the profile for 3 is shown as the one for 2 behaves similarly). The steepest concentration profile gradients are at short reaction times (1 s) and close to the electrode where conversion is driven by fast heterogeneous ET and chemical reactions, and relatively little diffusion has occurred. Profiles for the reactive intermediates 1˙− and 4 are not shown as these species do not accumulate, or escape the proximity of the cathode in the direct process, due to their consumption in very fast onward reaction steps (2) and (3)/(6), respectively.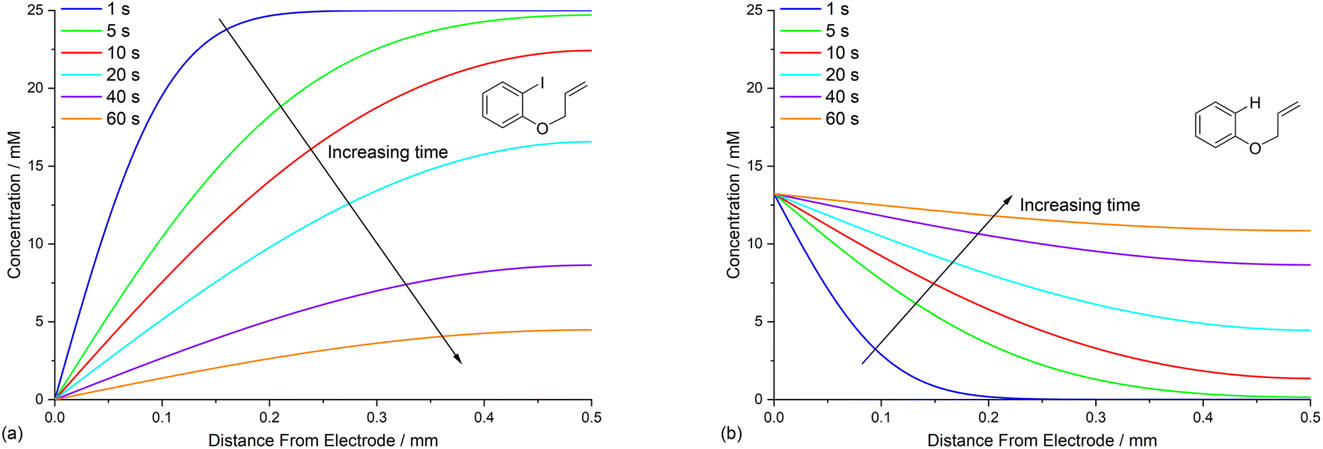 | ||
| Fig. 2 Time dependent profiles for direct electrolysis of ArI 1 showing: (a) ArI 1; (b) hydrogenolysis product 3. Starting concentration of 1 is 0.025 M. Electrolysis time = 0 to 60 s under constant potential of −2.8 V vs. SCE. | ||
The simulated profiles for electrolysis of 1 in the presence of phenanthrene (1 equiv.) at a cathode potential of −2.8 V vs. SCE reveal a more interesting time dependent behaviour, particularly in the early stages of the process (Fig. 3). Under the conditions Eede < E0M < E01, both 1 and M are electrochemically reduced (steps (1) and (4)) at the onset of electrolysis, with their concentrations tending to zero at the cathode (Fig. 3a and b). However, as M˙− diffuses away from the electrode it reacts homogeneously with 1 thereby accelerating the depletion of 1 near the electrode. Even after a short time (t < 1 s), 1 is sufficiently depleted in the region close to the cathode for M to act as the charge shuttle and the mechanism for reduction of 1 switches from heterogeneous (direct) step (1) to homogeneous (mediated) step (5). As time progresses, the region depleted in 1 grows from the cathode as 1 is consumed by the strongly reducing mediator radical anion M˙− as it diffuses outwards. The concentration profiles for M˙− show that its flux from the cathode surface overcomes the inwards flux of ArI 1 (Fig. 3c). Considering hydrogenolysis product 3, its profiles at the beginning of electrolysis show concentration highest close to the cathode (Fig. 3d), which arises from the initial direct (ECE) process. As electrolysis time progresses, the concentration of 3 at the cathode decreases, tending towards a constant concentration across the cell showing that no further 3 is being produced and the direct mechanism is no longer contributing significantly. Thus, the hydrogenolysis product 3 that is present in the reaction solution is only produced during the very early stages of the process, highlighting the switch from direct to mediated mechanism. Formation of 3 by homogeneous reactions (10) and (11) does not appear to contribute significantly as already discussed above. Direct visualisation of the detached reaction layer is challenging as intermediates 1˙− and Ar˙ 4 do not accumulate due to very rapid onward reactions. However, the concentration profile for alkyl radical 6 – formed from 1˙− by fragmentation and cyclisation steps (5) and (6) – provides insight into the zone where 1˙− is produced (and consumed). In Fig. 4 the maximum, albeit low, concentration of 4 can be seen to move progressively outwards from the cathode with time, which is a manifestation of homogeneous ET step (5) occurring away from the cathode, i.e. reaction layer detachment.
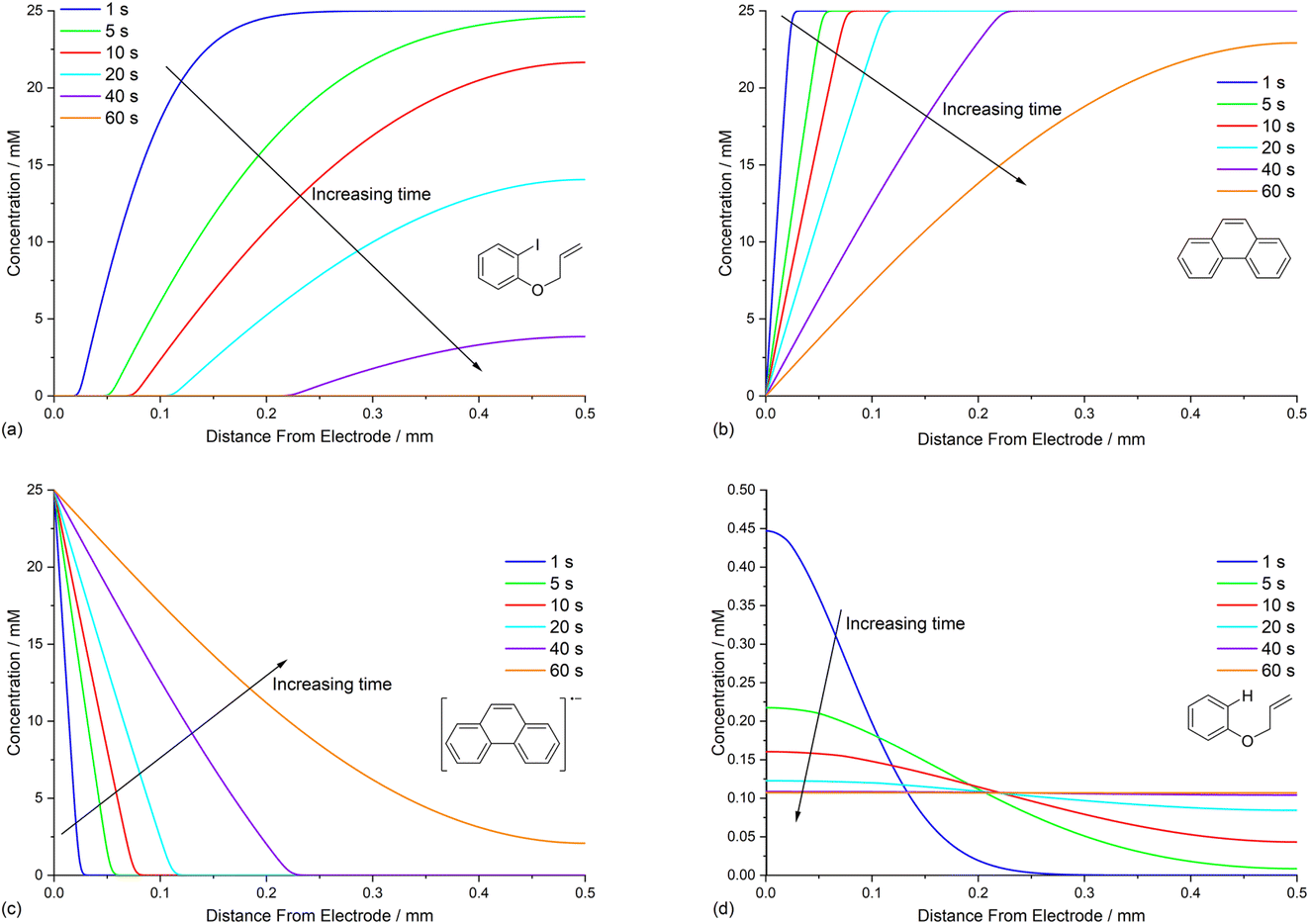 | ||
| Fig. 3 Time dependent concentration profiles for electrolysis of ArI 1 in the presence of phenanthrene (1 equiv.) showing: (a) ArI 1. (b) Phenanthrene (M). (c) Phenanthrene radical anion (M˙−). (d) Hydrogenolysis product 3. Starting concentration of ArI 1 is 0.025 M. Electrolysis time = 0 to 60 s under constant potential of −2.8 V vs. SCE. | ||
 | ||
| Fig. 4 Illustrative visualisation of reaction layer detachment showing simulated concentration profiles for: (a) the cyclised radical 4 between 0.1 s and 25 s of electrolysis, moving progressively away from the cathode. (b) 1 and M˙− after 5 s of electrolysis intersecting where their fluxes are matched. | ||
It is important to emphasise that under the simulated conditions in the presence of phenanthrene, the process starts as direct plus mediated before becoming fully mediated. The time taken to establish the mediated pathway depends on several parameters, including, heterogeneous and homogeneous rate constants, diffusion coefficients, and bulk concentrations. The influence of these, and other variables, will be explored through simulations presented and discussed in the ensuing sections.
Diffusion coefficient of mediator
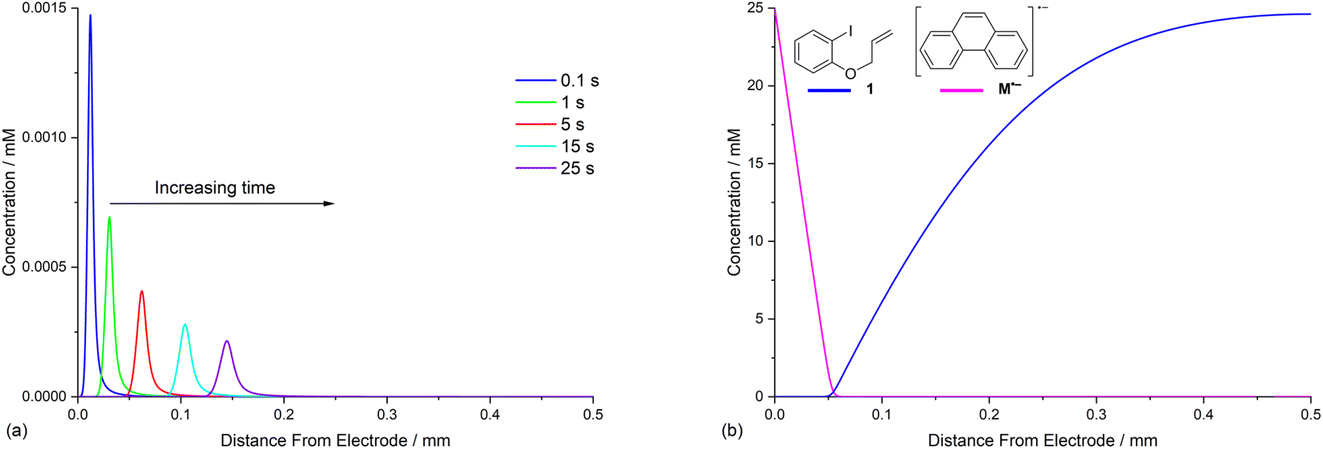
The diffusion coefficients for phenanthrene (2.0 × 10−5 cm2 s−1) and aryl iodide 1 (3.3 × 10−5 cm2 s−1) were established experimentally from voltammetry, and values for reaction intermediates are assumed to be the same as their parent molecules.14,17 Simulations of concentration profiles allowed us to visualise the influence of varying diffusivity of mediator M using three different values (1, 2, and 4 × 10−5 cm2 s−1), and in all cases time dependent reaction layer detachment was observed (Fig. 5). Detachment progresses more quickly for a mediator with a higher diffusion coefficient, and at t = 5 s the maximum concentration of cyclised intermediate 6 occurs further from the cathode (Fig. 5a). Greater diffusivity of M/M˙− supports an increased flux of reducing species perpendicular from the cathode, pushing homogeneous ET outwards into solution, which is also indicated by the depletion of 1 at an increased distance from the electrode (Fig. 5b). These results are consistent with previous work using microelectrodes.16 It is also apparent that the larger diffusion coefficient also increases the rate of conversion, indicated from the higher concentration of cyclised intermediate (integral of peak area from Fig. 5a).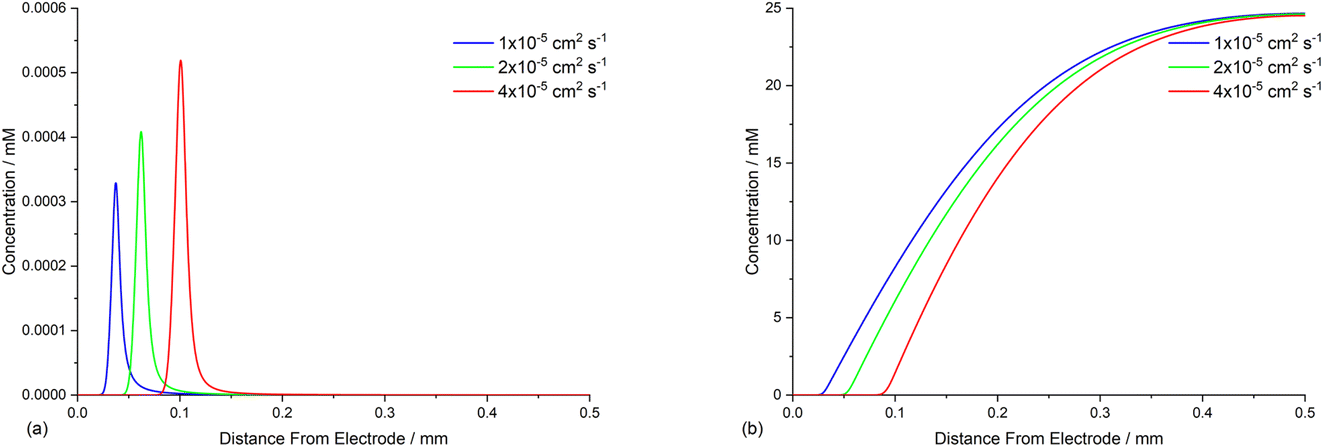 | ||
| Fig. 5 Influence of mediator (E0 = −2.5 V vs. SCE) with different diffusion coefficients (1 × 10−5 cm2 s−1, 2 × 10−5 cm2 s−1, and 4 × 10−5 cm2 s−1) on reaction layer detachment. Concentration profiles of: (a) cyclised radical 6 at t = 5 s; (b) ArI 1 at t = 5 s. | ||
Mediator stoichiometry
Opposing fluxes of mediator radical anion and ArI 1 play a key role in determining the region where homogeneous ET reaction (5) takes place, and a sufficient flux of M˙− is required for detachment of the reaction layer. As the flux is the product of the diffusion coefficient and concentration gradient, the stoichiometry of mediator is an important parameter. Fig. 6a–c show the effect of varying amounts of mediator (phenanthrene, 1.0, 0.5, and 0.05 equiv.) relative to the substrate 1 (0.025 M). Under the conditions of the simulation, detachment of the reaction layer is observed even with catalytic electron transfer mediator (0.05 equiv.) at short time (1 s of simulation). Detachment is realised more quickly for higher mediator loadings (Fig. 6a and b), which is evident from the smaller amount of hydrogenolysis product (area under the red line). It should be stressed that no further hydrogenolysis product is being produced under any of the mediator loadings at 1 s, and all simulations show that the mediated mechanism has taken over at this time. The simulated product distribution after electrolysis for 60 s shows high selectivity for the cyclised product 2 at all mediator loadings investigated (Fig. 6d), which is in good qualitative agreement with experimental results using catalytic (0.05 equiv.) and stoichiometric (1 equiv.) mediator.14 As the simulated conditions only include mass transport by diffusion, they cannot be considered truly representative of the experimental conditions in batch (stirred) or flow. None the less, the similar trends in the experimental and simulated results are noteworthy. Simulations of mediated electrolysis under flow conditions will be discussed in later parts of this paper.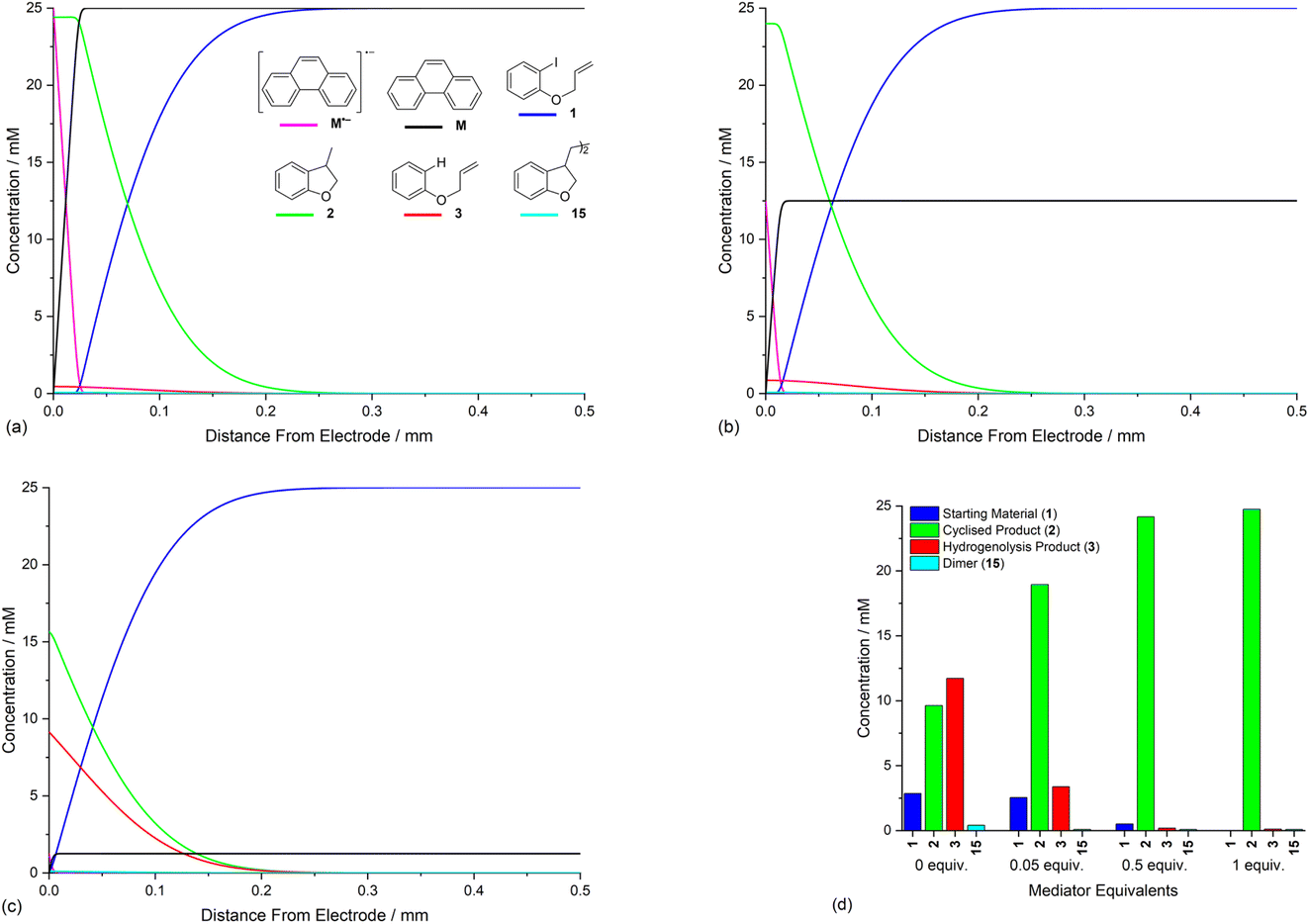 | ||
| Fig. 6 Influence of mediator (phenanthrene, M) stoichiometry on reaction layer detachment. Concentration profiles for species 1, 2, 3, 15, M and M˙− for simulated electrolysis under constant potential of −2.8 V vs. SCE at t = 1 s. (a) 1 equiv. of M. (b) 0.5 equiv. of M. (c) 0.05 equiv. of M. (d) Product selectivity profile from simulations with different mediator loadings after electrolysis for 60 s. | ||
Influence of mediator
The rate constants for homogeneous and heterogeneous ET steps (4), (5), (7) and (10) are related to the structure and reduction potential of the mediator. Furthermore, the rate of heterogeneous ET step (4) is affected by the potential of the cathode, and the diffusion coefficient of the mediator also directly influences concentration profiles as has been exemplified above. In view of these complex relationships, and challenges obtaining accurate rate constants for all ET steps, the present study takes, for the purpose of illustration, a single hypothetical mediator (M′) with a standard potential (−2.0 V vs. SCE) that is positive to that of the model substrate 1. Now ET from the mediator to 1 is thermodynamically unfavourable, however, the E0 for the mediator should remain sufficiently negative such that the rate of homogeneous ET step (5), when followed by a very rapid irreversible reaction (2), leads to a reasonable rate for aryl radical formation. In the simulation, a value of k5 = 100 M−1 s−1 is used for ET step (5), which is 250 times slower than when using phenanthrene.19 The ET rate constants for heterogeneous step (4) and homogeneous step (7) are also adjusted accordingly (k4 = 7 cm s−1 and k7 = 108 M−1 s−1), and all other conditions are unchanged from those used in Fig. 3.20,25Simulation of the product selectivity profile with M′ (E0 = −2.0 V vs. SCE) retains selectivity for the cyclised product 2, even under conditions where the electrode potential is negative to both mediator and substrate (Fig. 7a). However, an increased amount of hydrogenolysis product 3 is produced compared to the simulation with the more strongly reducing mediator, phenanthrene. Inspection of concentration profiles for the reducing species M′˙− and 1 show that some of the substrate now reaches the electrode surface due to the slower rate of homogeneous ET step (5), which effectively leads to a broadening of the homogeneous reaction zone such that it does not detach from the electrode completely under the simulated conditions. Although the simulation implies an advantage of using the more strongly reducing mediator phenanthrene, some caution should be exercised due to the uncertainty in the ET rate constants used. It should also be highlighted that, for a selective mediated synthesis using M′, the electrode potential would normally be set such that E01 < Eede < E0M′ to decrease the rate of the unmediated reaction.
 | ||
| Fig. 7 Influence of mediator M′ (E0 = −2.0 V vs. SCE) on product selectivity and reaction layer. (a) Relative concentrations of ArI 1 and 2, 3 and 15 as a function of time. (b) Concentration profiles for species 1 and M′˙− for simulated electrolysis under constant potential of −2.8 V vs. SCE at t = 5 s. Starting concentration of ArI 1 is 0.025 M. Electrolysis time = 0 to 60 s under constant potential of −2.8 V vs. SCE. | ||
Electrode potential
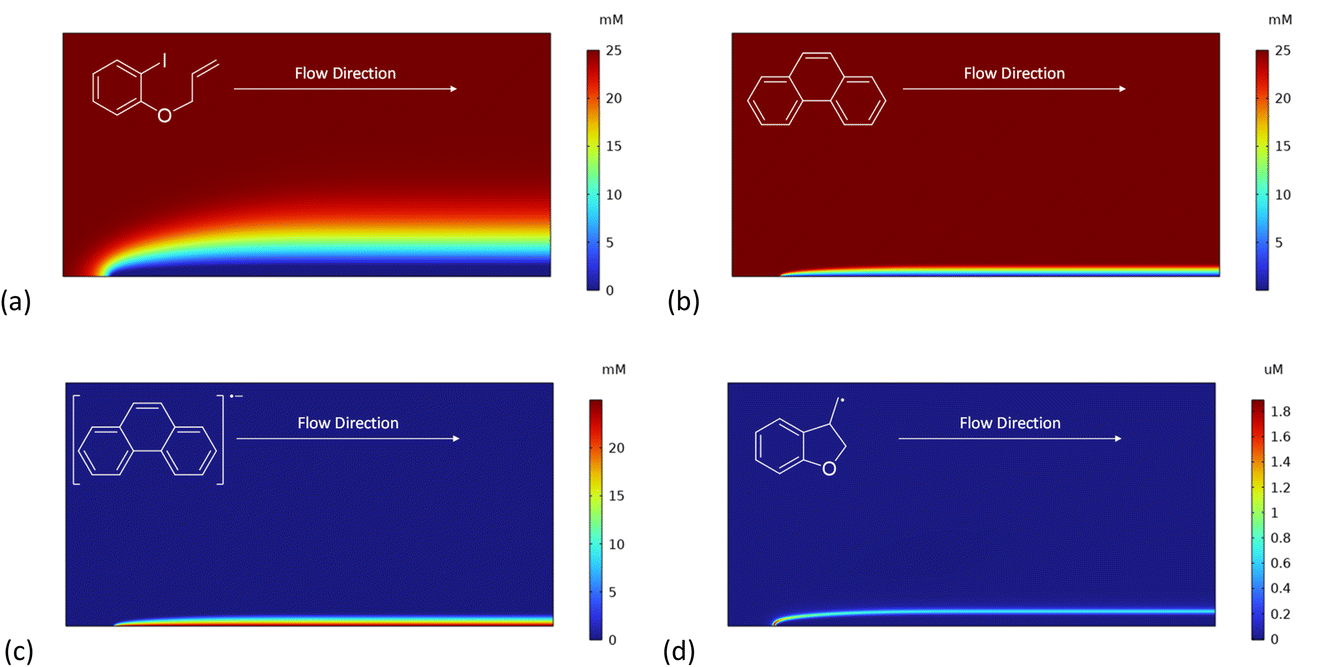
The effect of varying electrode potential on the electroreduction of aryl iodide 1 in the presence of phenanthrene (1 equiv.) as mediator is shown in Fig. 8. At cathode potentials that are negative or equal (−2.8 V and −2.5 V vs. SCE) to the standard potential for phenanthrene the mediated mechanism occurs already 1 s after electrolysis commences (Fig. 8a and b). The profiles at a cathode potential of −2.8 V are discussed above (see Fig. 3, 4 and 6a), and show reaction layer detachment. When the electrode potential is −2.5 V – close to E0 for phenanthrene – the mediated mechanism is again in operation, although the detached reaction layer takes longer to establish and is closer to the cathode at corresponding times (Fig. 8b). More hydrogenolysis product 3 is present, formed during a short period at the onset of electrolysis by the direct mechanism. The slower progress of detachment at −2.5 V can be explained by the lower heterogeneous rate constant for ET to the mediator, lowering the concentration gradient and flux of M˙− from the cathode.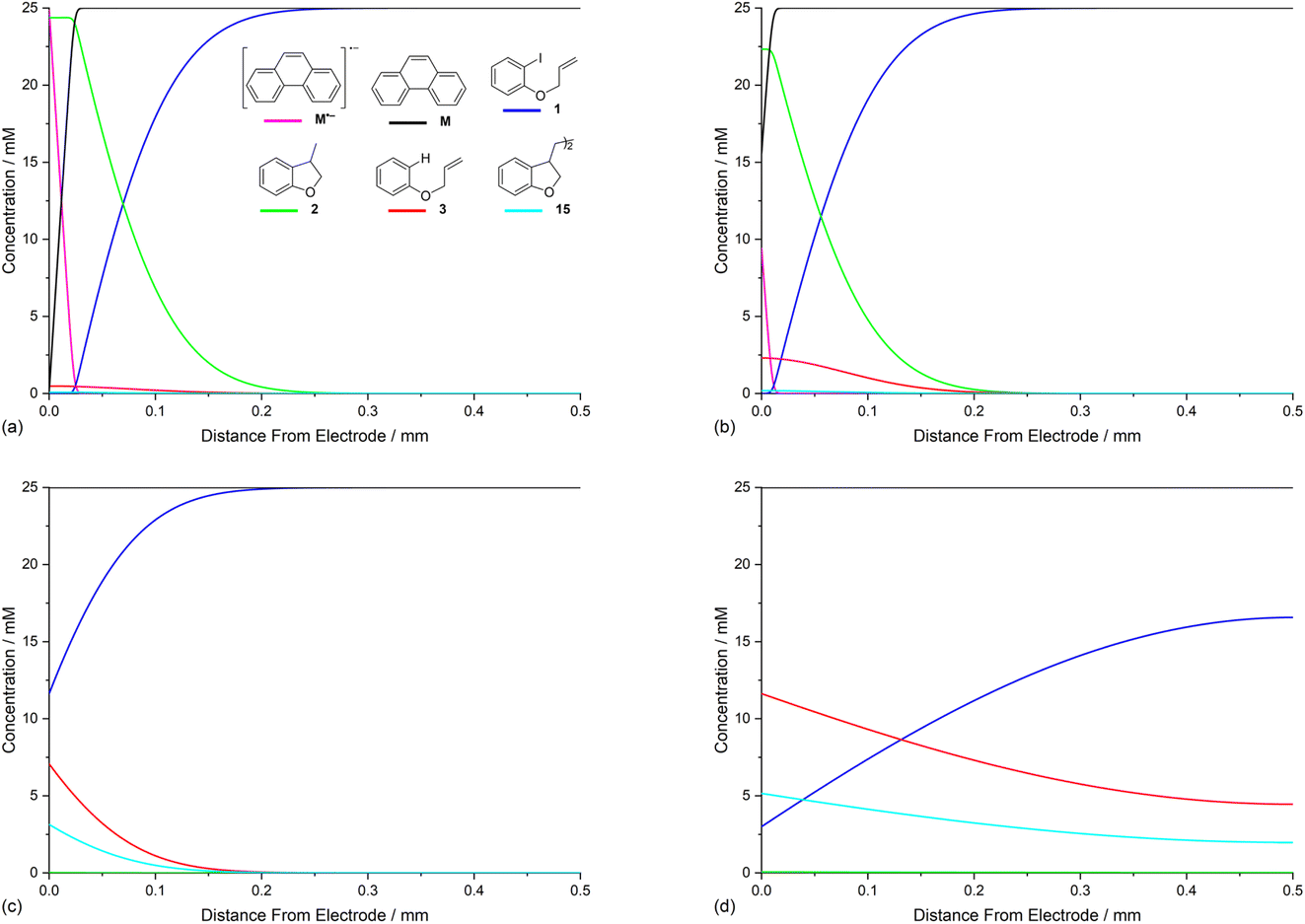 | ||
| Fig. 8 Influence of electrode potential on reaction mechanisms for electroreduction of 1 in the presence of phenanthrene (M, 1 equiv.). Concentration profiles for species 1, 2, 3, 15, M and M˙− at electrode potentials of: (a) −2.8 V vs. SCE at t = 1 s. (b) −2.5 V vs. SCE at t = 1 s. (c) −2.2 V vs. SCE at t = 1 s. (d) −2.2 V vs. SCE at t = 25 s. | ||
A further positive shift of the electrode potential to −2.2 V – now positive to phenanthrene and close to E0 for ArI – returns the mechanism to the direct one as very little M˙− is produced at the cathode (Fig. 8c). Even after an extended time (25 s) no detachment is seen at an electrode potential of −2.2 V, with the direct mechanism prevailing (Fig. 8d). An interesting feature of the simulated profiles at −2.2 V is the formation of dimer 15, rather than 2, which is explained by the relatively slow heterogeneous reduction of alkyl radical 6 at the more positive electrode potential.26 This should be investigated experimentally, as other reactions of cyclised alkyl radical 6 that are not included in the simulation may become significant at the more positive potential. Finally, and unsurprisingly, adjustment of the electrode potential to −2.0 V – positive to standard potentials for both ArI and phenanthrene – results in the rate of electrochemical reduction becoming very small indeed (not shown).
Simulations of electroreduction in a laminar flow regime
We recently described electroreductive cyclisation of ArI 1 in the presence of phenanthrene (1 equiv.) in a flow reactor, the Ammonite 8,14 which possesses a long path length (100 cm) and small interelectrode gap (0.5 mm) (see Scheme 4).24,27 Although it is beyond the scope of the present work to develop a simulation for mediated electrolysis in the 100 cm long spiral channel of this reactor, preliminary 2D simulations of the process have been performed over a short channel (1 mm) with an interelectrode gap of 0.5 mm using COMSOL Multiphysics and the conditions described above, but with introduction of parabolic laminar flow of the electrolyte solution parallel to the cathode.In batch simulations (1D), the concentration profiles of the different species only require the dimension perpendicular to the electrode, whereas in the flow reactor the concentrations of the different reaction components also vary along the channel length and it is beneficial here to consider 2D “heat maps” of the concentrations. The heat map in Fig. 9d, showing the spatial distribution of the cyclised radical intermediate 6, is a surrogate for the zone where the radical anion 1˙− is formed by either heterogeneous ET step (1) or homogeneous ET step (5), as 1˙− is converted to 6 very rapidly.
 | ||
| Fig. 9 Flow simulation for electroreduction of 1 in the presence of phenanthrene (M, 1 equiv.). Concentration heat maps for: (a) ArI 1. (b) Phenanthrene (M). (c) Phenanthrene radical anion (M˙−). (d) Cyclised radical intermediate 6. Starting concentration of ArI 1 is 0.025 M. Electrolysis time = 0 to 2.4 s under constant potential of −2.8 V vs. SCE. | ||
Furthermore, the radical 6 does not accumulate as it is consumed in onwards bimolecular reactions (7) and (8). The concentration heatmap for 6 shows that it is produced close to the cathode at the start of the electrode segment in the flow channel, but that its formation moves outwards from the electrode as the electrolyte flows downstream. While in unstirred batch reactors the reaction pathway switches from direct to mediated as time progresses, the 2D simulations demonstrate that under flow, the change in pathway from direct to mediated occurs as the solution progresses downstream from the electrode edge, a complex function of time and distance.
Another important aspect is that the flow yields a boundary layer where mass transport evolves from convection far from the electrode to diffusion near the electrode. At higher flow rates (not shown), the boundary layer will be thin and confine the diffusion region closer to the electrode. This will enhance the flux of 1 towards the electrode and it will take longer (both in time and distance) for the reaction pathway to switch from direct to mediated. At low flow rates, the boundary layer will extend further in solution, the diffusion region will widen, the flux of 1 will decrease, and the switch from direct to mediated pathway will occur sooner. A more detailed analysis of effect of the flow rate on the mechanism is the subject of ongoing work.
Discussion
A common logic applied in reductive electrosynthesis using ET mediators is that the redox potential of the mediator should be positive to the potential of the substrate to promote selective electron transfer from the cathode to the redox catalyst.9 Thus, for selective mediated electroreduction, the conditions E0M > Eede > E0S would be expected, where ede is the cathode, M is the mediator, and S is the substrate. However, this is not the situation seen in the mediated electroreductive cyclisations in Schemes 3b and 4, where E0S > E0M, and direct reduction of the substrate is expected to be favoured at electrode potentials required to reduce the mediator. We have proposed that the observed selectivity results from spatio-temporal effects where homogeneous ET occurs in a reaction layer that becomes progressively detached from the electrode surface.14 Under the conditions of detachment, and despite facile reduction of the substrate at the cathode, the substrate is not directly reduced because it does not reach the cathode surface. In view of the complex interplay between mass transport, time, and the rates of multiple steps – including chemical, homogeneous, and heterogeneous ET processes – we have established simulations in order to illustrate the key requirements for reaction layer detachment.The 1D simulations for the phenanthrene-mediated reduction of aryl iodide 1 support the proposed time dependent detachment of the homogeneous electrochemistry from the cathode, and are qualitatively consistent with the experimentally observed selectivities. Initially both 1 and M are reduced at the cathode, and the direct and mediated mechanisms operate simultaneously. However, as M˙− diffuses away from the electrode it reacts homogeneously with 1 thereby accelerating the depletion of 1 near the electrode. After a period of time, substrate 1 no longer reaches the cathode and the process becomes fully mediated where M acts as the charge shuttle between the electrode and the detached homogeneous reaction layer, favouring cyclisation product 2. The time taken to establish the mediated pathway depends on several parameters, primarily, heterogeneous and homogeneous rate constants, diffusion coefficients, and bulk concentrations. It is perhaps convenient to think of the position of the homogeneous reaction layer being determined by opposing fluxes of M˙− and 1, which are the products of their concentration gradients (time dependent) and diffusion coefficients.28 The thickness of the homogeneous reaction layer is dependent on the homogeneous rate constants and diffusion coefficients in the 1D simulation. Slower rates of homogeneous processes lead to broadening of the homogeneous reaction layer arising from longer lifetimes of reactive intermediates.
The flux of mediator radical anion M˙− from the cathode is affected by mass transport, concentration of M, and the rate of heterogeneous ET step (4). Although the latter is limited by the electron transfer rate constant (k4), and transfer coefficient (α), a sluggish ET is overcome by applying sufficient cathode over-potential (e.g. −2.8 V vs. SCE in the simulation). As the overall rate, i.e. the electrode current, is affected by the rate of ET and the rate of mass transport, the simulations also help to explain how the mediated mechanism prevails under galvanostatic conditions. When the applied current exceeds the rate at which the substrate 1 is replenished at the electrode surface by diffusion, the electrode potential adjusts to a value sufficiently negative to drive the electrochemical reduction of the mediator. This process not only provides the additional rate required to match the applied current but also promotes the mediated reduction of the substrate. Thus, reaction layer detachment can also be achieved under constant current conditions when E01 > E0M, providing that the current density exceeds the limiting current density for the substrate.
Reaction layer detachment is also seen in 2D simulations where mass transport by convection is included to represent conditions found in laminar flow cells. In the 2D simulations, detachment is seen as a function of distance along the flow channel, and the mechanism of reduction switches from direct plus mediated at the start of the electrode, to purely mediated after a short distance downstream from the electrode edge. Again, the initial results showing detachment are consistent with preparative work showing selectivity for the mediated process leading to the cyclised product 2 (Scheme 4). It should be emphasised that the current density and potential are not constant along the length of the flow channel,29 and 2D simulations in a longer channel require considerably greater computational power. A more detailed investigation is therefore beyond the scope of the current work and is the subject of ongoing work.
Finally, it should be recognised that our experimental work was conducted under constant current conditions, and without a reference electrode present in the flow cell to monitor the electrode potentials. It is, however, entirely reasonable for the cathode potential to be negative of E0M, and indeed, the experimental observation of the mediated pathway combined with the results of the simulation provide indirect support for this. The voltage across the flow cell is typically in the region of 5 to 6 V, and the measured cathode potential for the mediated electrolysis of 1 in a batch cell was −3.5 V vs. SCE under steady state conditions. On this basis we are confident that the electrode potential in the batch and flow cells are such that the simulated conditions of Eede < E0M < E01 are in operation.
Conclusions
Simulations presented herein support the proposed role of reaction layer detachment in electroreductive cyclisation of aryl halides in the presence of strongly reducing ET mediators. It is shown that mediators that are harder to reduce than their substrates – E0M < E01 – can be employed, providing that the conditions of homogeneous reaction layer detachment are achieved. The resulting highly reducing mediator radical anions promote kinetically accelerated and thermodynamically favourable homogeneous ET with substrates and intermediates, as opposed to classical application of mediators relying on thermodynamically uphill ET followed by irreversible coupled reactions.Reaction layer detachment is an example of a spatio-temporal effect arising due to opposing fluxes of two species – here the mediator radical anion M˙− and the substrate 1 – which undergo very rapid homogeneous ET and onwards reactions. The detachment results from a complex interplay between the relative concentrations and diffusion coefficients of the mediator and substrate as well as the rate constant for the homogeneous process between them. No detachment is observed when the substrate is not sufficiently depleted near the electrode i.e., when the homogeneous reaction between substrate and mediator is slow or when the flux of the substrate is much larger than that of the mediator. When detachment occurs, the width and location of the reaction zone are directly determined by mass transport, concentrations of M and 1, and the heterogeneous and homogeneous rate constants.
Experimental and computational work is underway to further investigate and understand the complex interplay of mass transport, heterogeneous and homogeneous ET and coupled chemistry that may lead to new opportunities in electrosynthesis using powerful homogeneous ET mediators.
Author contributions
JWH performed experiments and simulations. AAF performed experiments. DP, DCH, GD and RCDB conceptualised the project. RCDB, DCH and GD supervised the project. RCDB wrote the manuscript with assistance and contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the EPSRC (Photo-Electro Programme Grant EP/P013341/1 and EP/K039466/1), and the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work.Notes and references
- For general reviews of organic radicals and their reactions, see: (a) W. P. Neumann, Synthesis, 1987, 665–683 CrossRef CAS; (b) C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237–1286 CrossRef CAS; (c) Radicals in Organic Synthesis, Vol. 1 and 2, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 2001 Search PubMed; (d) S. Z. Zard, Radical Reactions in Organic Synthesis, Oxford University Press, Oxford, 2003 Search PubMed; (e) J. Lalevée and J. P. Fouassier, in Encyclopedia of Radicals in Chemistry, Biology and Materials, Vol 2: Synthetic Strategies and Applications, ed. C. Chatgilialoglu and A. Studer, John Wiley and Sons, Weinheim, 2012 Search PubMed.
- For a perspective on radicals in synthesis, see: M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- For selected reviews covering aryl radical chemistry from a variety of precursors, see: (a) C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS; (b) G. Pratsch and H. R. Heinrich, Top. Curr. Chem., 2012, 320, 33–59 CrossRef CAS PubMed; (c) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed; (d) N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC; (e) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed; (f) J. H. Lan, R. X. Chen, F. F. Duo, M. H. Hu and X. Y. Lu, Molecules, 2022, 27, 5364 CrossRef CAS PubMed.
- Electroreductive cyclisation of aryldiazonium salts has also been described: F. LeStrat, J. A. Murphy and M. Hughes, Org. Lett., 2002, 4, 2735–2738 CrossRef CAS PubMed.
- For some examples where selective cyclisations of aryl radicals have been achieved by direct electrolysis, see: (a) R. Munusamy, K. Samban Dhathathreyan, K. Kuppusamy Balasubramanian and C. Sivaramakrishnan Venkatachalam, J. Chem. Soc., Perkin Trans. 2, 2001, 1154–1166 RSC; (b) J. Grimshaw, R. J. Haslett and J. Trocha-Grimshaw, J. Chem. Soc., Perkin Trans. 1, 1977, 2448–2455 RSC; (c) S. Donnelly, J. Grimshaw and J. Trocha-Grimshaw, J. Chem. Soc., Chem. Commun., 1994, 2171–2172 RSC; (d) M. Dias, M. Gibson, J. Grimshaw, I. Hill, J. Trocha-Grimshaw and O. Hammerich, Acta Chem. Scand., 1998, 52, 549–554 CrossRef CAS; (e) R. Gottlieb and J. L. Neumeyer, J. Am. Chem. Soc., 1976, 98, 7108–7109 CrossRef CAS PubMed.
- For examples of cathodic reductive dehalogenation of aryl halides, see: (a) C. P. Andrieux, J. Badoz-Lambling, C. Combellas, D. Lacombe, J. M. Savéant, A. Thiebault and D. Zann, J. Am. Chem. Soc., 1987, 109, 1518–1525 CrossRef CAS; (b) K. Mitsudo, T. Okada, S. Shimohara, H. Mandai and S. Suga, Electrochemistry, 2013, 81, 362–364 CrossRef CAS; (c) J. Ke, H. Wang, L. Zhou, C. Mou, J. Zhang, L. Pan and Y. R. Chi, Chem. – Eur. J., 2019, 25, 6911–6914 CrossRef CAS PubMed; (d) C. B. Liu, S. Y. Han, M. Y. Li, X. D. Chong and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18527–18531 CrossRef CAS PubMed; (e) L. Lu, H. Li, Y. Zheng, F. Bu and A. Lei, CCS Chem, 2020, 2, 2669–2675 Search PubMed; (f) A. A. Folgueiras-Amador, A. E. Teuten, D. Pletcher and R. C. D. Brown, React. Chem. Eng., 2020, 5, 712–718 RSC.
- The reductive electrochemistry of aryl halides has been reviewed, see: (a) J.-M. Savéant, in Adv. Phys. Org. Chem., ed. D. Bethel, Academic Press, New York, 1990, vol. 26, pp. 1–130 Search PubMed; (b) J. Grimshaw, in Electrochemical Reactions and Mechanisms in Organic Chemistry, Elsevier Science, 2000, pp. 89–157 Search PubMed; (c) Z. Chami, M. Gareil, J. Pinson, J.-M. Savéant and A. Thiebault, J. Org. Chem., 1991, 56, 586–595 CrossRef CAS.
- ET to aryl radicals
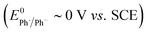 is a relatively facile process in comparison to the parent halides (E0 ∼ −2 V vs. SCE).
(a) C. P. Andrieux and J. Pinson, J. Am. Chem. Soc., 2003, 125, 14801–14806 CrossRef CAS PubMed;
(b) C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 126, 16051–16057 CrossRef CAS PubMed.
is a relatively facile process in comparison to the parent halides (E0 ∼ −2 V vs. SCE).
(a) C. P. Andrieux and J. Pinson, J. Am. Chem. Soc., 2003, 125, 14801–14806 CrossRef CAS PubMed;
(b) C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 126, 16051–16057 CrossRef CAS PubMed. - For selected reviews, see: (a) E. Steckhan, Angew. Chem., Int. Ed. Engl., 1986, 25, 683–701 CrossRef; (b) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (c) R. Francke, A. Prudlik and R. D. Little, in Science of Synthesis: Electrochemistry in Organic Synthesis, ed. L. Ackermann, Thieme: Stuttgart, 2021, vol. 1, pp. 293–324 Search PubMed; (d) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (e) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (f) W. Shao, B. Lu, J. Cao, J. Zhang, H. Cao, F. Zhang and C. Zhang, Chem. – Asian J., 2023, 18, e202201093 CrossRef CAS PubMed.
- K. Mitsudo, Y. Nakagawa, J. Mizukawa, H. Tanaka, R. Akaba, T. Okada and S. Suga, Electrochim. Acta, 2012, 82, 444–449 CrossRef CAS.
- For selected examples of electroreductive cyclisation using organic mediators, see ref. 5b and: (a) M. D. Koppang, G. A. Ross, N. F. Woolsey and D. E. Bartak, J. Am. Chem. Soc., 1986, 108, 1441–1447 CrossRef CAS; (b) N. Kurono, E. Honda, F. Komatsu, K. Orito and M. Tokuda, Tetrahedron, 2004, 60, 1791–1801 CrossRef CAS; (c) A. Katayama, H. Senboku and S. Hara, Tetrahedron, 2016, 72, 4626–4636 CrossRef CAS.
- For selected examples of Ni mediated electrochemical radical cyclisations of aryl halides, see: (a) S. Ozaki, H. Matsushita and H. Ohmori, J. Chem. Soc., Perkin Trans. 1, 1993, 2339–2344 RSC; (b) S. Olivero, J. C. Clinet and E. Duñach, Tetrahedron Lett., 1995, 36, 4429–4432 CrossRef CAS; (c) E. Duñach, M. J. Medeiros and S. Olivero, Electrochim. Acta, 2017, 242, 373–381 CrossRef; (d) C. Déjardin, A. Renou, J. Maddaluno and M. Durandetti, J. Org. Chem., 2021, 86, 8882–8890 CrossRef PubMed.
- C. Costentin and J. M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11147–11152 CrossRef CAS PubMed.
- A. A. Folgueiras-Amador, A. E. Teuten, M. Salam-Perez, J. E. Pearce, G. Denuault, D. Pletcher, P. J. Parsons, D. C. Harrowven and R. C. D. Brown, Angew. Chem., Int. Ed., 2022, 61, e202203694 CrossRef CAS PubMed.
- For further details of the simulations, see the ESI†.
- (a) G. Denuault, M. Fleischmann, D. Pletcher and O. R. Tutty, J. Electroanal. Chem., 1990, 280, 243–254 CrossRef; (b) O. R. Tutty and G. Denuault, IMA J. Appl. Math., 1994, 53, 95–109 CrossRef; (c) O. R. Tutty, J. Electroanal. Chem., 1994, 377, 39–51 CrossRef CAS.
- Experimental and simulated cyclic voltammetry data can be found in the supporting information for ref. 14.
- C. Amatore, M. A. Oturan, J. Pinson, J. M. Saveant and A. Thiebault, J. Am. Chem. Soc., 1985, 107, 3451–3459 CrossRef CAS.
- C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat and J. M. Saveant, J. Am. Chem. Soc., 1979, 101, 3431–3441 CrossRef CAS.
- D. Occhialini, J. S. Kristensen, K. Daasbjerg and H. Lund, Acta Chem. Scand., 1992, 46, 474–481 CrossRef CAS.
- (a) A. Annunziata, C. Galli, M. Marinelli and T. Pau, Eur. J. Org. Chem., 2001, 1323–1329 CrossRef CAS; (b) A. N. Abeywickrema and A. L. J. Beckwith, J. Chem. Soc., Chem. Commun., 1986, 464–465 RSC.
- Labelling studies provide strong evidence that dihydrobenzofuran is formed principally by step (7) rather than through H-atom abstraction. See ref. 14 and 10.
- P. Neta, J. Grodkowski and A. B. Ross, J. Phys. Chem. Ref. Data, 1996, 25, 709–1050 CrossRef CAS.
- R. A. Green, R. C. D. Brown, D. Pletcher and B. Harji, Electrochem. Commun., 2016, 73, 63–66 CrossRef CAS.
- A. Russell, K. Repka, T. Dibble, J. Ghoroghchian, J. J. Smith, M. Fleischmann, C. H. Pitt and S. Pons, Anal. Chem., 1986, 58, 2961–2964 CrossRef CAS.
- C. P. Andrieux, I. Gallardo and J. M. Saveant, J. Am. Chem. Soc., 1989, 111, 1620–1626 CrossRef CAS.
- The Ammonite 8 electrochemical reactor is available from Cambridge Reactor Design. https://www.cambridgereactordesign.com/ammonite/ammonite.html, accessed 30 Apr 2023.
- For examples of reaction layers, or zones in electrochemistry see ref. 13, 16 and: (a) M. E. G. Lyons, D. E. McCormack, O. Smyth and P. N. Bartlett, Faraday Discuss. Chem. Soc., 1989, 88, 139–149 RSC; (b) W. J. Albery and A. R. Hillman, J. Electroanal. Chem. Interfacial Electrochem., 1984, 170, 27–49 CrossRef CAS. For lead references to reaction-diffusion systems, see: (c) A. Comolli, A. De Wit and F. Brau, Phys. Rev. E, 2019, 100, 052213 CrossRef CAS PubMed; (d) A. Li, R. Chen, A. B. Farimani and Y. J. Zhang, Sci. Rep., 2020, 10, 3894 CrossRef CAS PubMed.
- R. A. Green, R. C. D. Brown and D. Pletcher, J. Flow Chem., 2015, 5, 31–36 CrossRef.
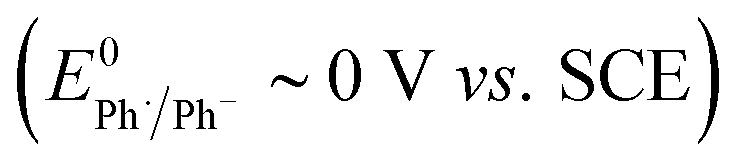
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fd00089c |
| This journal is © The Royal Society of Chemistry 2023 |