
Introduction of a cyano group at the 2-position of an (R,S)-3-hydroxy-2-(phosphonomethoxy)propyl (HPMP) derivative of thymine elicits selective anti-HBV activity†
Shuai
Tan
a,
Elisabetta
Groaz
 ab,
Mark N.
Prichard‡
c,
Raj
Kalkeri
d,
Roger
Ptak
d and
Piet
Herdewijn
*a
ab,
Mark N.
Prichard‡
c,
Raj
Kalkeri
d,
Roger
Ptak
d and
Piet
Herdewijn
*a
aKU Leuven, Rega Institute for Medical Research, Medicinal Chemistry, Herestraat 49-Box 1041, 3000 Leuven, Belgium. E-mail: Piet.Herdewijn@kuleuven.be
bDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Via Marzolo 5, 35131 Padova, Italy
cDepartment of Paediatrics, University of Alabama at Birmingham, Birmingham, AL 35294-3412, USA
dDepartment of Infectious Disease Research, Drug Development, Southern Research Institute, 431 Aviation Way, Frederick, Maryland 21701, USA
First published on 29th April 2021
Abstract
The substantial impact of acyclic nucleoside phosphonates (ANPs) on human medicine encourages the synthesis of new ANP analogues with a potentially differentiated antiviral spectrum. Herein, we demonstrate the functionalization of the 2-position of the (R,S)-3-hydroxy-2-(phosphonomethoxy)propyl side-chain of an inactive ANP with a polar cyano group to generate a thymine analogue with selective inhibition of hepatitis B virus (HBV) replication (SI > 302; EC50 = 0.33 μM), without significant antiretroviral activity. These findings suggest new strategies to synthesize unique ANPs with a targeted antiviral profile.
Introduction
It is more than three decades since Holy and De Clercq discovered the broad-spectrum antiviral activity of acyclic nucleoside phosphonates (ANPs), unlocking a new era of antiviral drug development.1,2 Two notable features are common to all the members of this class of nucleoside derivatives: (1) the conformational flexibility of the acyclic pseudosugar chain that makes them more amenable to interact with their target enzymes; (2) the enzymatic and chemical stability of the phosphonate moiety combined with its disposition to undergo a step economic metabolic activation, especially allowing it to by-pass the first rate-limiting phosphorylation reaction in the nucleoside enzymatic cascade towards the formation of pharmacologically active species.3Among the earliest examples of ANPs to be synthesized are (S)-3-hydroxy-2-(phosphonomethoxy)propyl (HPMP) nucleosides, which were described to effectively inhibit the replication of a variety of DNA viruses including herpes-, pox-, polyoma-, adeno-, and papillomaviruses.1,2 In particular, the cytosine containing congener [1, (S)-HPMPC or cidofovir, Fig. 1] was the first ANP to receive FDA approval owing to its ability to suppress the progression of cytomegalovirus (HCMV) retinitis in immunocompromised patients.4 It is worth mentioning that only moderate activities against hepatitis B virus (HBV) were associated with (S)-HPMPs.5–7 However, it was subsequently demonstrated that the activity spectrum of ANPs could be significantly altered by simple structural simplifications at the side chain. The impact of this approach on drug discovery is highlighted by the widespread clinical use of orally available prodrugs including tenofovir disoproxil,8,9 tenofovir alafenamide,10 and adefovir dipivoxil11 (2–4, Fig. 1) to treat human immunodeficiency virus infection/acquired immunodeficiency syndrome (HIV/AIDS) and chronic hepatitis B. While compounds 1 and 2–3 feature a hydroxymethyl and methyl group at the 2-position, respectively, compound 4 lacks a 2-substituent.
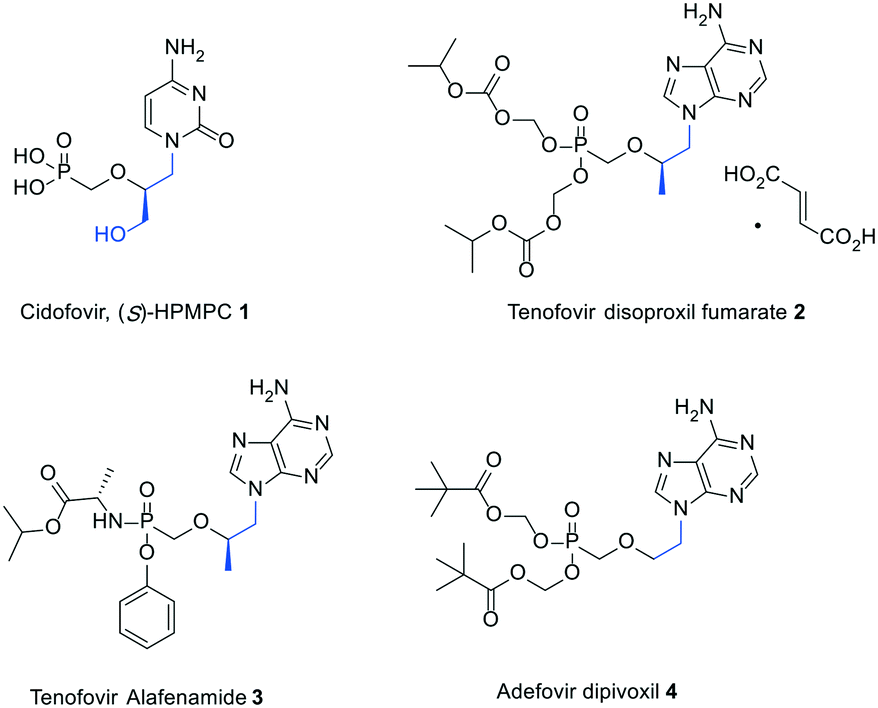 | ||
| Fig. 1 Clinically approved ANPs for the treatment of CMV, HBV, and HIV infections. | ||
By closely inspecting historic structure–activity relationship (SAR) of ANPs, it becomes apparent that potent antiretroviral nucleosides often exert concomitant anti-HBV activity and vice versa.12,13 Such antiviral effect may be partly due to the structural similarities shared by the binding sites of HIV reverse transcriptase (RT) and HBV DNA polymerase, which are the molecular targets of ANPs. Up to now, however, there is a little understanding of the different structural requirements for ANPs to act as selective inhibitors of either HIV or HBV. Discernment of the factors at the origin of the potential binding to a single enzymatic target would thus provide additional insights to design novel specifically targeted antiviral ANP molecules.
Here, we modified the HPMP side chain by introducing a cyano group at the 2-position (Fig. 2) in order to determine the influence of this additional polar group on the SAR of ANPs through the biological evaluation of a newly synthesized series of racemic 2,2-disubstituted analogues (2-cyano-3-hydroxy-2-(phosphonomethoxy)propyl or CHPMP). The inspiration for the present study was the observation that the previous introduction of a nitrile substituent of the 4′-C-position of furanose nucleosides resulted in enhanced anti-HBV activity.14 Based on the structural analogy depicted in Fig. 2, it was envisaged that HPMPs could provide an attractive platform for endorsing such approach and potentially giving rise to a different antiviral effect.
 | ||
| Fig. 2 Comparison of the structure of 4′-C-cyano-nucleosides with HPMP and CHPMP (this study) derivatives. | ||
Results and discussion
Preliminary efforts were directed towards the preparation of the key racemic CHPMP side chain synthon 11 for further nucleobase insertion. As shown in Scheme 1, the synthesis started with 1,3-dihydroxyacetone 5, whose primary hydroxy functionalities were successively protected with a tert-butyldiphenylsilyl (TBDPS) and methoxymethyl (MOM) group to furnish compounds 6 and 7, respectively, according to a literature procedure.15 Ketone 7 was then converted to racemic tertiary alcohol 8 upon treatment with NaCN in a NH4Cl solution. The subsequent alkylation of 8 with diisopropylphosphonomethyl tosylate [(iPrO)2POCH2OTs] under various conditions afforded only unreacted starting material. However, the replacement of (iPrO)2POCH2OTs with diisopropylphosphonomethyl triflate [(iPrO)2POCH2OTf], which was freshly prepared following a reported method, yielded the desired diisopropyl ester phosphonate 9 in 72% yield.16 TBAF mediated removal of the silyl protecting group of intermediate 9 proceeded smoothly to afford compound 10 in 64% yield. Treatment of sterically encumbered primary alcohol 10 with triflyl chloride (TfCl) successfully generated the unstable aliphatic chain precursor 11, which was directly used in the next step without purification.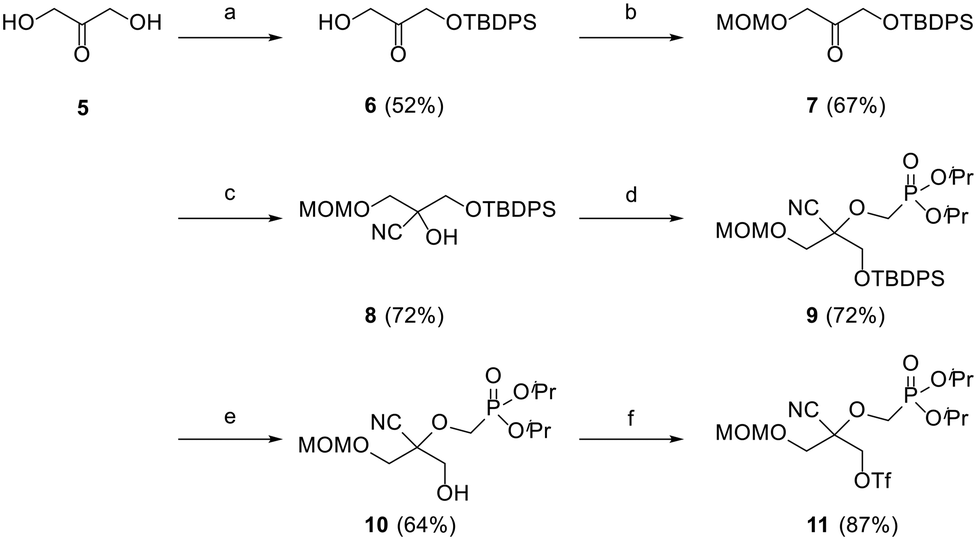 | ||
| Scheme 1 Reagents and conditions: (a) TBDPSCl, imidazole, DMF, rt, 18 h; (b) MOMCl, DIPEA, DCM, rt, 18 h; (c) NaCN, NH4Cl, H2O, Et2O, rt, 18 h; (d) n-BuLi, (iPrO)2POCH2OTf, −78 °C to rt, 4 h; (e) TBAF, THF, 0 °C to rt, 1 h; (f) n-BuLi, TfCl, Et2O, −78 °C, 2 h. | ||
The assembly of 2,2-disubstituted CHPMP nucleoside analogues 14a–c involved an initial coupling reaction between fragment 11 and suitably protected nucleobases, i.e., N3-benzoylthymine (TBz), N3-benzoyluracil (UBz), and 6-chloropurine, under basic conditions to afford intermediates 12a–c (Scheme 2). Next, removal of the benzoyl and MOM groups of 12a–b was accomplished by successive treatment with 7 N methanolic ammonia and 2 N HCl to afford alcohols 13a and 13b in 85 and 77% yield, respectively.
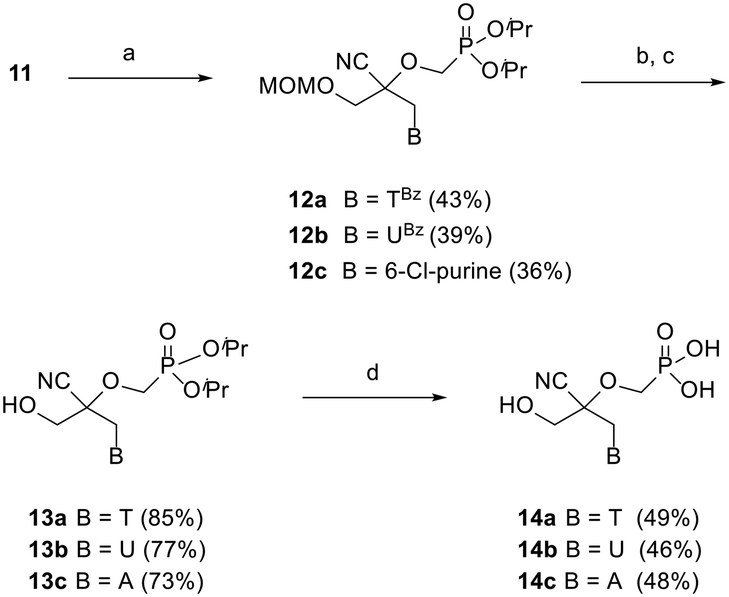 | ||
| Scheme 2 Reagents and conditions: (a) N3-benzoylthymine, Cs2CO3, DMF, rt, 20 h, for 12a; N3-benzoyluracil, Cs2CO3, DMF, rt, 20 h, for 12b; 6-Cl-purine, Cs2CO3, DMF, rt, 20 h, for 12c; (b) 7 N NH3 in MeOH, rt, 2 h for 13a/b; sat. NH3 in EtOH, 70 °C, 18 h for 13c; (c) 2 N HCl, THF, 50 °C, 1 h; (d) TMSBr, 2,6-lutidine, MeCN, 0 °C to rt, 18 h. | ||
The chlorine atom of 12c was converted to an amino group by refluxing in a saturated solution of ammonia in ethanol, followed by acid hydrolysis of the MOM group to provide alcohol 13c. The last deprotection step was performed by exposure of 13a–c to trimethylsilyl bromide (TMSBr), which proceeded smoothly to furnish thymine, uracil, and adenine containing phosphonate acids 14a–c in 46–49% yield.
Next, the cytosine CHPMP derivative (CHPMPC) 18 was obtained from protected uracil intermediate 12b, as illustrated in Scheme 3. The benzoyl group of 12b was removed by ammonolysis, followed by a two-step conversion of the uracil nucleobase to the corresponding cytosine. Standard protecting group manipulations further led to the desired phosphonic acid 18 upon hydrolysis of the MOM and diester groups of 16.
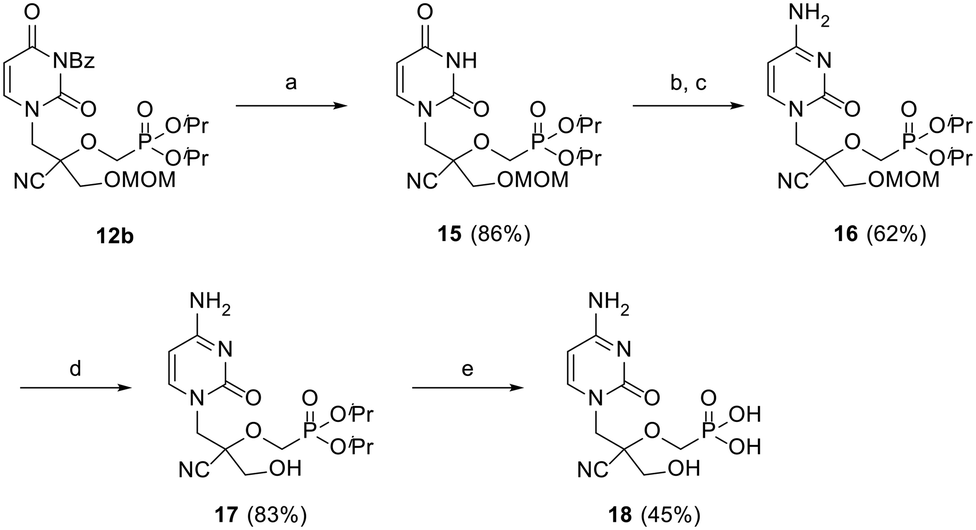 | ||
| Scheme 3 Reagents and conditions: (a) 7 N NH3 in MeOH, rt, 6 h; (b) TPSCl, DIPEA, MeCN, rt, 6 h; (c) 25% NH3 solution, rt, 12 h; (d) 2 N HCl, THF, rt, 4 h; (e) TMSBr, 2,6-lutidine, MeCN, 0 °C to rt, 18 h. | ||
As shown in Scheme 4, triflate synthon 11 was reacted with 6-chloro-2-(di-Boc-amino)-9H-purine in the presence of Cs2CO3 to afford protected intermediate 19 in 41% yield. Cleavage of the diester protecting groups was then achieved by treatment with TMSBr, followed by nucleobase hydrolysis upon treatment with 2 N HCl at 80 °C to furnish guanine derivative (CHPMPG) 20. All final CHPMPs were purified by reversed-phase HPLC (RP-HPLC) and isolated as their corresponding triethylammonium salts.
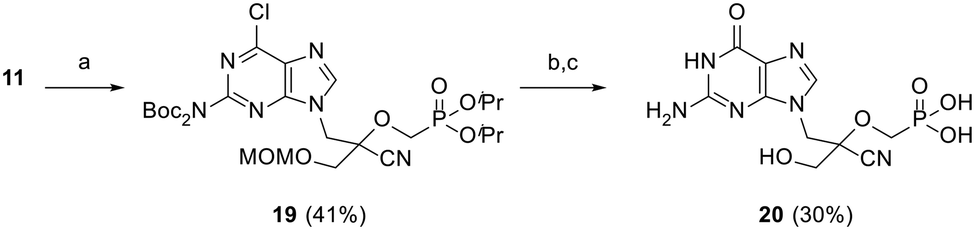 | ||
| Scheme 4 Reagents and conditions: (a) 6-chloro-2-(di-Boc-amino)-9H-purine, Cs2CO3, DMF, rt, 20 h; (b) TMSBr, MeCN, 0 °C to rt, 18 h; (c) 2 N HCl, 80 °C, 6 h. | ||
As mentioned above, the prime pathogens targeted by ANPs are DNA viruses and retroviruses. Therefore, all synthesized CHPMP nucleoside analogues were subjected to antiviral profiling by performing a series of standard cell based antiviral assays against a select set of herpesviruses, such as HCMV and varicella zoster virus (VZV), hepadnaviruses (e.g., HBV), as well as retroviruses (e.g., HIV). At first, we evaluated compounds 14a–c, 18, and 20 for antiviral activity in human foreskin fibroblasts (HFFs) infected with the HCMV AD-169 strain, along with the determination of their cytotoxic effects (CC50).17,18 Ganciclovir was included in the experiment as reference nucleoside analogue, and the corresponding results are shown in Table 1. As it can be observed, among the CHPMP series only purine containing analogues demonstrated micromolar inhibition of HCMV. In particular, adenine derivative 14c (CHPMPA) exhibited an EC50 of 1.1 μM. This antiviral response is comparable to that originally described for parent (S)-HPMPA in human embryonic lung cells infected with the same HCMV strain (EC50 = 1.0 μM),1 and similar to ganciclovir. Guanine congener 20 (CHPMPG) was also endowed with antiviral activity against HCMV (EC50 = 2.0 μM), albeit slightly lower compared to that of both (S)-HPMPG (EC50 = 0.66 μM) and (R)-HPMPG (EC50 = 0.56 μM).19 Overall, although the introduction of a 2-cyano group did not significantly affect the anti-HCMV activity of HPMPA and HPMPG, it drastically reduced activity in comparison to FDA-approved (S)-HPMPC. Surprisingly, the 2-cyano derivative 18 (CHPMPC) did not demonstrate antiviral activity in the HCMV assay. Furthermore, thymine and uracil congeners 14a (CHPMPT) and 14b (CHPMPU) were also inactive against HCMV, concurring with the previous report demonstrating the inactivity of parent HPMPT and HPMPU against HCMV.2
| Compound | HCMV | VZV | ||||||
|---|---|---|---|---|---|---|---|---|
| EC50a (μM) | EC90b (μM) | CC50c (μM) | SI50d | EC50a (μM) | EC90b (μM) | CC50c (μM) | SI50d | |
| a Effective concentration required to reduce viral replication by 50%. b Effective concentration required to reduce viral replication by 90%. c Cytotoxic concentration required to reduce cell viability by 50%. d SI50: CC50/EC50. e Not determined. | ||||||||
| 14a | >150.0 | >150.0 | >150.0 | 1 | >150.00 | >150.00 | >150.00 | 1 |
| 14b | >150.0 | >150.0 | >150.0 | 1 | >150.00 | >150.00 | >150.00 | 1 |
| 14c | 1.1 | >150.0 | >150.0 | >137 | 106.44 | >150.00 | >150.00 | 1 |
| 18 | >150.0 | >150.0 | >150.0 | 1 | >150.00 | >150.00 | >150.00 | 1 |
| 20 | 2.0 | >30.0 | 101.8 | 50 | 2.08 | >30.00 | 97.56 | 47 |
| Ganciclovir | 0.9 | 74.2 | >150.0 | >175 | NDe | NDe | NDe | NDe |
| Acyclovir | NDe | NDe | NDe | NDe | 4.17 | 30.23 | >150.00 | >36 |
In a subsequent assay, HFF cells were infected with the Ellen strain of VZV and used to evaluate the antiviral effect of the synthesized CHPMP derivatives, with acyclovir as positive control (Table 1).17,18 While the parent compounds HPMPA, HPMPG, and HPMPC are highly potent anti-VZV agents,2 the corresponding CHPMPs did not retain good potency against VZV with the exception of guanine congener 20 which exhibited an EC50 value of 2.08 μM, exhibiting higher potency than acyclovir.
Next, the anti-HBV activity of CHPMPs was evaluated in a human hepatoblastoma cell line (HepG2 2.2.15), which contains two copies of the HBV wild-type (strain ayw) genome and constitutively produces high levels of HBV.20–22 Real-time qPCR (TaqMan) was used to measure the extracellular HBV copy number associated with virions released from the cells. The cell viability and CC50 values were measured by using a tetrazolium dye uptake assay. The results are summarized in Table 2; Lamivudine [(−)-L-2′,3′-dideoxy-3′-thiacytidine, 3TC], which acts as a dual inhibitor of HIV and HBV, was included as a positive control.
| Compound | EC50a (μM) | EC90b (μM) | CC50c (μM) | SI50d |
|---|---|---|---|---|
| a Effective concentration required to reduce viral replication by 50%. b Effective concentration required to reduce viral replication by 90%. c Cytotoxic concentration required to reduce cell viability by 50%. d SI50: CC50/EC50. | ||||
| 14a | 0.33 | 0.79 | >100 | >302 |
| 14b | >100 | >100 | >100 | 1 |
| 14c | 3.6 | 9.9 | >100 | >28 |
| 18 | >100 | >100 | >100 | 1 |
| 20 | >100 | >100 | >100 | 1 |
| 3TC | 0.04 | 0.13 | 2124 | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 571 571 |
Among CHPMPs, adenine containing congener 14c exhibited moderate activity (EC50 = 3.6 μM) against HBV, in agreement with previously reported EC50 values of around 1.0 μM for (S)-HPMPA in different cell lines.5–7 Early studies demonstrated that (S)-HPMPT was largely inactive against HBV (EC50 > 400 μM in HB611 cells).6 Moreover, the replacement of the hydroxy group of (S)-HPMPT with a fluorine atom led to 3-fluoro-2-(phosphonomethoxy)propyl thymine analogue (S)-FPMPT that also showed no anti-HBV activity when tested up to 100 μM.23 In striking contrast, not only did thymine analogue 14a (CHPMPT) display potent activity (EC50 = 0.33 μM) but also its selectivity index (SI50) exceeded 302, suggesting 14a to be the most promising ANP candidate among the tested compounds. On the other hand, the weak to moderate anti-HBV activities of (S)-HPMPC (cidofovir) and HPMP-guanine (HPMPG) were totally lost owing to the cyano modification.
The synthesized CHPMPs were further evaluated for antiretroviral activity in an MTT assay using MT-4 cells infected with wild type strains IIIB (HIV-1 strain) and ROD (HIV-2 strain).24,25 In parallel, their cytotoxicity was assessed in the MT-4 cell line. The results are summarized in Table S1† relative to lamivudine (3TC) as reference compound. Overall, the synthesized CHPMP derivatives were either inactive or very weakly active (e.g., CHPMPG showed EC50 values of 51.04 and 33.69 μM against HIV-1 and HIV-2, respectively) against HIV, similar to their parent HPMP derivatives.26
Remarkably, compound 14a is active against HBV, while exerting no activity against HIV-1 or HIV-2. This HBV specific antiviral activity can be considered as a crucial attribute not shared by other ANPs, thus providing the opportunity for the development of selective anti-HBV ANPs. As compound 14a is a mixture of two enantiomers, additional work is further being conducted to obtain enantiomerically pure compounds and their prodrugs.
Conclusions
We synthesized a novel series of racemic 2,2-disubstituted acyclic nucleoside phosphonates by introducing a cyano group at the 2-position of the HPMP side-chain, which exhibited a significantly altered antiviral activity spectrum relative to their parent ANPs. While the presence of a 2-cyano functionality did not profoundly influence the anti-HCMV inhibitory effect of purine containing derivatives, a marked loss of activity was observed for the cytosine derivative (CHPMPC) compared to cidofovir. On the other hand, CHPMPs proved to be largely inactive against VZV except for the guanine containing congener (CHPMPG) that showed equipotent activity as compared to reference anti-herpes compound acyclovir.Surprisingly, the thymine containing analogue (CHPMPT) exhibited low micromolar antiviral activity as well as a favorable selectivity index (SI > 302) against hepatitis B virus (HBV), without affecting HIV replication. This is the first report of ANP with potent HBV specific antiviral activity, but without activity against HIV, supporting the view that minor structural changes can differentially impact interaction with either HIV or HBV polymerases. As a further note, our work demonstrates how distinctive antiviral profiles can arise from the combination of specific pseudosugar scaffolds with nucleobases that generally account for poor or no antiviral activity among ANPs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. T. wishes to acknowledge the China Scholarship Council (CSC) for funding (grant number 201606220077). The authors wish to thank Prof. Christophe Pannecouque (KU Leuven) for providing the anti-HIV data.Notes and references
- E. Declercq, A. Holy, I. Rosenberg, T. Sakuma, J. Balzarini and P. C. Maudgal, Nature, 1986, 323, 464–467 CrossRef CAS PubMed.
- E. Declercq, T. Sakuma, M. Baba, R. Pauwels, J. Balzarini, I. Rosenberg and A. Holy, Antiviral Res., 1987, 8, 261–272 CrossRef CAS.
- E. Groaz and S. De Jonghe, Front. Chem., 2021, 8, 616863 CrossRef PubMed.
- G. L. Plosker and S. Noble, Drugs, 1999, 58, 325–345 CrossRef CAS PubMed.
- T. Yokota, S. Mochizuki, K. Konno, S. Mori, S. Shigeta and E. Declercq, Antimicrob. Agents Chemother., 1991, 35, 394–397 CrossRef CAS PubMed.
- T. Yokota, K. Konno, S. Shigeta, A. Holy, J. Balzarini and E. Declercq, Antiviral Chem. Chemother., 1994, 5, 57–63 CrossRef CAS.
- R. A. Heijtink, J. Kruining, G. A. Dewilde, J. Balzarini, E. Declercq and S. W. Schalm, Antimicrob. Agents Chemother., 1994, 38, 2180–2182 CrossRef CAS PubMed.
- H. B. Fung, E. A. Stone and F. J. Piacenti, Clin. Ther., 2002, 24, 1515–1548 CrossRef CAS PubMed.
- A. Kuo, J. L. Dienstag and R. T. Chung, Clin. Gastroenterol. Hepatol., 2004, 2, 266–272 CrossRef CAS PubMed.
- A. S. Ray, M. W. Fordyce and M. J. M. Hitchcock, Antiviral Res., 2016, 125, 63–70 CrossRef CAS PubMed.
- P. Marcellin, T. Chang, S. G. Lim, M. J. Tong, W. Sievert, M. L. Shiffman, L. Jeffers, Z. Goodman, M. S. Wulfsohn, S. Xiong, J. Fry, C. L. Brosgart, N. Afdhal, C. O'Conner, P. Andreone, C. Cursaro, P. Angus, R. Vaughan, V. Bain, K. Gutfreund, K. Barange, M. Duffant, E. Barnes, M. Bennett, J. Pressman, D. Bernstein, F. Bonino, B. Coco, M. Borum, S. Schuck, M. Bourliere, S. Benali, N. Boyer, C. Castelnau, R. Brown, S. Scales, P. Buggisch, J. Peterson, G. Cooksley, G. MacDonald, P. Couzigou, D. Foucner, D. Crawford, A. Der, P. Desmond, A. Boussioutas, A. DiBisceglie, B. Bacon, D. Dieterich, D. Goldman, G. Dusheiko, J. Enriquez, A. Gallego, S. Esposito, J. Lemieszewski, R. Esteban, M. Buti, T. Faust, K. Wherity, A. Francavilla, F. Malcangi, M. Fried, C. Nakayama, R. Gilson, M. Lascar, R. Gish, H. Trinh, S. Gordon, S. Colar, M. Gregor, S. Kaiser, J. Heathcote, D. Imagawa, I. Jacobson, J. Rooney, C. James, R. Fallis, A. Jain, S. Chen, J. Ma, A. Hsing, S. Nonaka-Wong, M. Kraus, C. M. Jen, K. Kaita, G. Koval, H. Parrish, K. Kowdley, I. Kronborg, A. Nicoll, P. Kullavanijaya, J. Amonrattanakosol, J. Lao-Tan, L. Garcia, Y. F. Liaw, R. N. Chien, A. Lok, P. Richtmyer, P. Luengrojanakul, T. Tanwandee, M. Manns, A. Schueler, P. Martin, V. Peacock, G. McCaughan, S. Strasser, J. McHutchison, P. Pockros, I. Merican, S. Lachmanan, R. Mohamed, R. Naccarato, S. Fagiuoli, M. Nelson, C. Higgs, G. Pastore, R. Perrillo, C. Denham, S. Pol, H. Fontaine, C. Riely, D. Litley, M. Rizzetto, M. Lagget, M. Rodriguez, M. Espiga, V. Rustgi, P. Lee, S. Sacks, J. Farley, D. Samuel, C. Feray, J. Sasadeusz, M. Gioupouki, D. Shaw, M. Le Mire, D. Shelton, M. Sherman, A. Bartolucci, E. Schiff, A. Siebert, J. Sollano, F. Dy, P. Thuluvath, L. Tong, C. Trepo, M. Maynard, J. C. Trinchet, N. Carrie, D. Vetter, S. Metzger, J. Vierling, J. Clarke-Platt, E. Wakil, N. Bzowej, T. Warnes, T. Wright, A. Kwong, Y. Y. Young, J. P. Zarski, V. Leroy, G. Adefovir Dipivoxil 437 Study and G. Royal Free Viral Hepatitis, N. Engl. J. Med., 2003, 348, 808–816 CrossRef CAS PubMed.
- L. G. Gurtler, Intervirology, 2014, 57, 212–217 CrossRef PubMed.
- Y. Yasutake, S. Hattori, N. Tamura, K. Matsuda, S. Kohgo, K. Maeda and H. Mitsuya, Sci. Rep., 2020, 10, 3021 CrossRef CAS PubMed.
- S. Hayashi, N. Higashi-Kuwata, D. Das, K. Tomaya, K. Yamada, S. Murakami, D. J. Venzon, S. Hattori, M. Isogawa, S. G. Sarafianos, H. Mitsuya and Y. Tanaka, Antiviral Res., 2020, 176, 104744 CrossRef CAS PubMed.
- B. M. Loertscher, P. R. Young, P. R. Evans and S. L. Castle, Org. Lett., 2013, 15, 1930–1933 CrossRef CAS PubMed.
- S. G. Dumbre, M. Y. Jang and P. Herdewijn, J. Org. Chem., 2013, 78, 7137–7144 CrossRef CAS PubMed.
- M. N. Prichard, K. A. Keith, D. C. Quenelle and E. R. Kern, Antimicrob. Agents Chemother., 2006, 50, 1336–1341 CrossRef CAS PubMed.
- M. N. Prichard, D. C. Quenelle, C. B. Hartline, E. A. Harden, G. Jefferson, S. L. Frederick, S. L. Daily, R. J. Whitley, K. N. Tiwari, J. A. Maddry, J. A. Secrist and E. R. Kern, Antimicrob. Agents Chemother., 2009, 53, 5251–5258 CrossRef CAS PubMed.
- J. Balzarini, A. Holy, J. Jindrich, L. Naesens, R. Snoeck, D. Schols and E. Declercq, Antimicrob. Agents Chemother., 1993, 37, 332–338 CrossRef CAS PubMed.
- M. A. Sells, M. L. Chen and G. Acs, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 1005–1009 CrossRef CAS PubMed.
- B. E. Korba and G. Milman, Antiviral Res., 1991, 15, 217–228 CrossRef CAS PubMed.
- R. Kalkeri, J. Z. Peng, C. S. Huang, Z. H. Cai, R. G. Ptak and M. J. Suto, Adv. Virol., 2020, 2020, 8844061 Search PubMed.
- M. Luo, E. Groaz, G. Andrei, R. Snoeck, R. Kalkeri, R. G. Ptak, T. Hartman, R. W. Buckheit, D. Schols, S. De Jonghe and P. Herdewijn, J. Med. Chem., 2017, 60, 6220–6238 CrossRef CAS PubMed.
- R. Pauwels, J. Balzarini, M. Baba, R. Snoeck, D. Schols, P. Herdewijn, J. Desmyter and E. Declercq, J. Virol. Methods, 1988, 20, 309–321 CrossRef CAS PubMed.
- C. Pannecouque, D. Daelemans and E. De Clercq, Nat. Protoc., 2008, 3, 427–434 CrossRef CAS PubMed.
- R. Pauwels, J. Balzarini, D. Schols, M. Baba, J. Desmyter, I. Rosenberg, A. Holy and E. Declercq, Antimicrob. Agents Chemother., 1988, 32, 1025–1030 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00086a |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2021 |