
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplete deconstruction of SF6 by an aluminium(I) compound†
Daniel J.
Sheldon
 and
Mark R.
Crimmin
*
and
Mark R.
Crimmin
*
Molecular Sciences Research Hub, Department of Chemistry, Imperial College London, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 16th June 2021
Abstract
The room-temperature activation of SF6, a potent greenhouse gas, is reported using a monovalent aluminium(I) reagent to form well-defined aluminium(III) fluoride and aluminium(III) sulfide products. New reactions have been developed to utilise the aluminium(III) fluoride and aluminium(III) sulfide as a nucleophilic source of F− and S2− for a range of electrophiles. The overall reaction sequence results in the net transfer of fluorine or sulfur atoms from an environmentally detrimental gas to useful organic products.
Sulfur hexafluoride (SF6) is widely used as an electrical insulating gas in circuit breakers.1 SF6 possesses unique chemical and physical inertness and excellent thermal conductivity; properties that result from its high dielectric constant, high heat capacity and high density.1–3 However, SF6 is a potent greenhouse gas with a global warming potential (GWP100) 23
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 times greater than CO2 and a long atmospheric lifetime of 3200 years.4–7 As a result, its emission is restricted through the Kyoto protocol as one of the six most prominent greenhouse gases.6–8 Specific attention has been directed towards its control as in many cases there are no suitable alternatives or drop-in replacements for SF6.4,6,9 Typical methods for its destruction are energy intensive and often produce toxic and corrosive products.10–12 Efficient methods for recycling or destroying SF6 are therefore highly sought after.13
900 times greater than CO2 and a long atmospheric lifetime of 3200 years.4–7 As a result, its emission is restricted through the Kyoto protocol as one of the six most prominent greenhouse gases.6–8 Specific attention has been directed towards its control as in many cases there are no suitable alternatives or drop-in replacements for SF6.4,6,9 Typical methods for its destruction are energy intensive and often produce toxic and corrosive products.10–12 Efficient methods for recycling or destroying SF6 are therefore highly sought after.13
A challenge remains to transform SF6 into non-toxic, high-value compounds under mild reaction conditions.12 Not only does this offer an attractive method for its depletion, but it opens up the potential to use SF6 as a source of S and F atoms through the deconstruction of this molecule to its elemental components. In particular, there has been recent interest in using SF6 as a fluorinating agent in organic synthesis. Fluorinated building blocks are increasingly crucial in the pharmaceutical, agrochemical and materials industries, where fluorine substitution is used to improve the quality and efficiency of new products.14–16
The activation and chemical deconstruction of SF6 has been achieved with strong reducing agents or low-valent transition metal complexes.9,17–26 The latter approach results in the formation of transition metal fluorides and sulfides. Metal-free approaches have also been reported in which strong nucleophiles directly attack SF6.27 In recent years, these synthetic approaches have been developed further and reactions that allow the onwards use of the fluorine content of SF6 in organic synthesis have been targeted. Particular attention has been given to the use of SF6 in the deoxyfluorination of alcohols.18,28–34 In one example, Braun and co-workers developed a photochemical protocol in which SF6 is reduced by an NHC to form a difluoroimidazolidine, which was then successfully applied in the deoxyfluorination of a range of alcohols.30
For some time we have been interested in using main group nucleophiles to activate the C–F bonds in environmentally persistent fluorocarbons.35–42 Herein we report the extension of this methodology to the rapid, room temperature activation of SF6 by a monovalent aluminium(I) species. This reaction results in the complete deconstruction of SF6 to its reduced elemental components, forming well-defined aluminium(III) fluoride and sulfide products. The fluoride species can be used as a nucleophile in onward synthesis, while the sulfide species is shown to act as a sulfide source in the formation of a heterocycle, thus allowing the elemental fluorine and sulfur content of SF6 to be re-used.
An excess of sulfur hexafluoride (1 bar) was added to a C6D6 solution of [{(ArNCMe)2CH}Al] (1, Ar = 2,6-di-isopropylphenyl). The red solution rapidly turned pale yellow. Monitoring the reaction by 1H and 19F NMR spectroscopy revealed the complete consumption of 1 and the formation of [{(ArNCMe)2CH}AlF2] (2) within 15 min at 22 °C (Scheme 1).
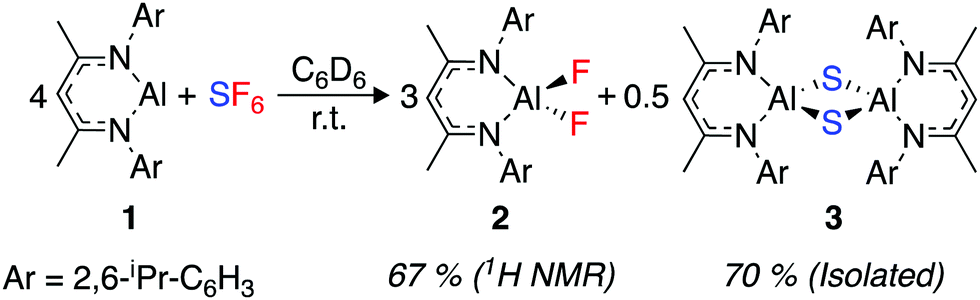 | ||
| Scheme 1 Reaction scheme for SF6 activation by 1. 1H NMR yields are reported against a ferrocene internal standard. | ||
2 is a known compound and the data match that reported in the literature.37 Although no further products were detected by NMR spectroscopy, the reaction was accompanied by the production of a colourless precipitate, suggesting the formation of an insoluble by-product. Repeating the reaction with slow diffusion of the SF6 into a C6D6 solution of 1 led to the formation of single crystals of the insoluble product suitable for X-ray diffraction. The side-product was determined as [{(ArNCMe)2CH}Al(μ-S)]2 (3) (Scheme 1).433 is also a known compound, and crystallised as a polymorph (monoclinic, C2/c) of a previously reported structure (monoclinic, C2/m). Crystalline samples of 3 were found to be insoluble in common laboratory solvents.
The mechanism for SF6 activation was investigated by DFT calculations (Fig. 1). The reaction sequence is likely initiated by nucleophilic attack of 1 at a fluorine atom of SF6, proceeding viaTS-1 (ΔG1‡ = 10 kcal mol−1), to give 2 and SF4 (Int-1). A further equivalent of 1 then reacts with SF4 in a similar nucleophilic manner viaTS-2 (ΔG2‡ = 11 kcal mol−1) to form SF2 and 2 (Int-2). SF4 possess a see-saw structure where the axial and equatorial fluorine atoms are inequivalent. The calculations suggest that the most favourable pathway involves attack of 1 at the equatorial fluorine of SF4 as this is the most electrophilic (least electronegative) site. Another equivalent of 1 then reacts in a similar fashion with SF2viaTS-3 (ΔG3‡ = 11 kcal mol−1) to form Int-3. Int-3 is subsequently attacked by a final equivalent of 1, leading to Int-4viaTS-4 (ΔG4‡ = 3 kcal mol−1). A rearrangement to form the experimentally observed products 2 and 3 is calculated to be thermodynamically feasible. When following the reaction by NMR spectroscopy, no reaction intermediates could be detected and the reaction proceeds to completion within 15 minutes at room temperature. These observations are consistent with the small activation barriers calculated for these elementary steps.
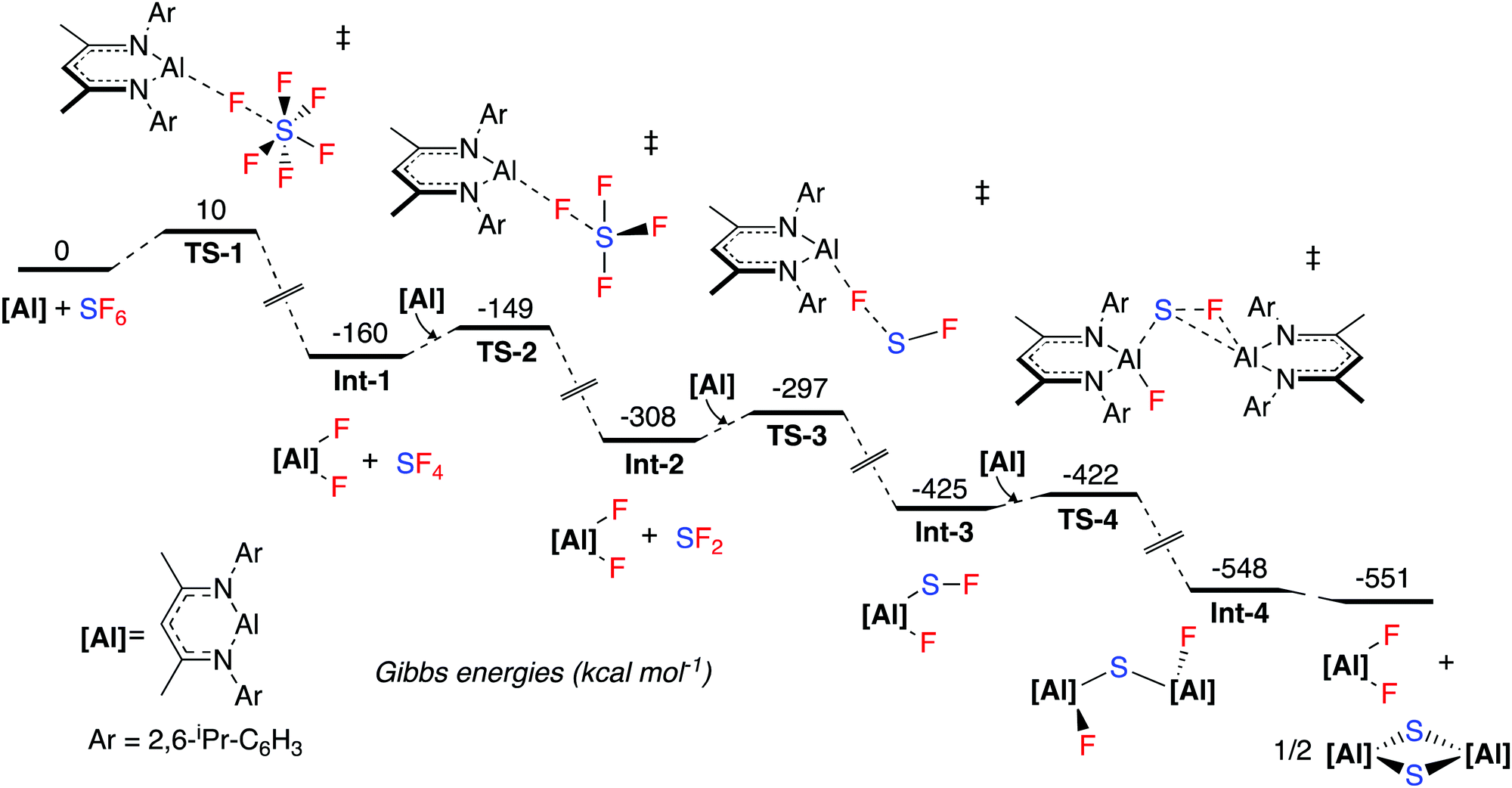 | ||
| Fig. 1 Calculated potential energy surface for SF6 activation. The M06-2X functional was used with a hybrid basis set, 6-31g**(C,H)/6-311+g*(N,F,S). The SDDAll pseudopotential was used for Al. Dispersion and solvent effects were included via single-point corrections, using Grimme's D3 correction for dispersion and the PCM (solvent = benzene) model for solvent. | ||
Numerous mechanistic analyses of SF6 activation propose a first step involving single electron transfer to SF6 from a transition metal, alkali metal or photocatalyst.9,19,23,25,28–30 Dielmann and co-workers have proposed an alternative mechanism involving nucleophilic attack at the fluorine atom of SF6 by a strongly nucleophilic phosphine, in a pathway similar to the one calculated here.27
NBO analysis of the transition states was carried out. TS-1, TS-2 and TS-3 are calculated to involve the nucleophilic attack of 1 at a fluorine atom of SFx (x = 6, 4, 2). The NPA charges show a trend of increasing negative charge at the sulfur atom as the maxima associated with the transition state is traversed, and conversely an accumulation of positive charge at aluminium. This implies electron density is transferred from aluminium to sulfur, consistent with nucleophilic attack, rather than a fluoride abstraction mechanism (see ESI† for NBO data). Wiberg Bond Indices are consistent with a decrease in the S–F bond order in TS-1 relative to SF6 itself (ESI† Table S3).
An IRC calculation connects TS-1 directly to 2 and SF4 (Int-1), where a second fluorine transfer has also occurred. This suggests that the second fluorine transfer step is a barrierless process somewhere on the pathway between TS-1 and Int-1. A very similar process is found for TS-2. Second-order perturbation analysis of TS-1 reveals donation of electron density from the aluminium lone pair into σ*(S–F) (17 kcal mol−1), with simultaneous donation of electron density from the same fluorine atom into the empty p-orbital of the aluminium atom (14 kcal mol−1). Similar donor–acceptor interactions, albeit of slightly different magnitudes are found for TS-2, TS-3 and TS-4.
ETS-NOCV calculations were performed to further probe the postulated nucleophilic attack mechanism.44 The largest contributor (Δρ1) to the orbital interaction (ΔEorb) for TS-1, TS-2 and TS-3 involves donation from the aluminium lone pair to σ* (S–F) (Fig. 2).
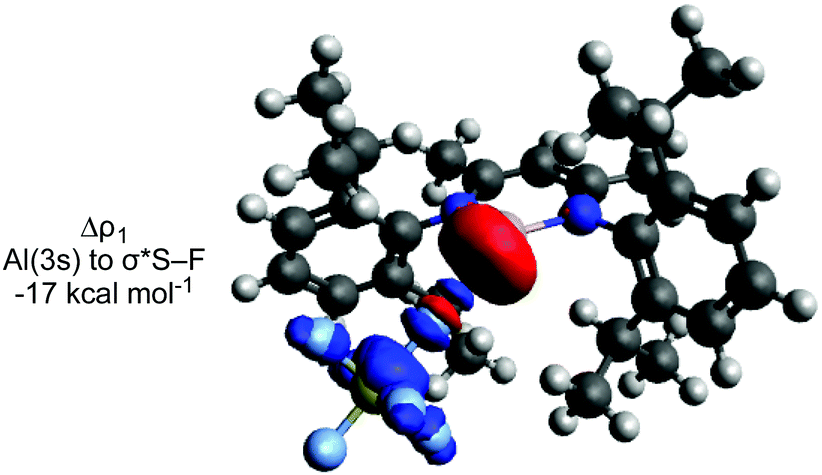 | ||
| Fig. 2 ETS-NOCV deformation density plot for TS-1. Charge flow is from red to blue. | ||
It is evident that attack of the aluminium occurs at the fluorine atom of the S–F bond. Along with the orbital interactions discussed, this is likely also due to the electrostatic interaction between Al and F (see ESI† for NPA charges), and the fluorophilic nature of aluminium. There is a contrast here to halocarbon reactivity where ‘frontside’ SN2X attack at the halogen atom is rare, although has been proposed in some recent examples with other fluorophilic nucleophiles.38,45,46
We were interested in the utility of the fluorinated aluminium species 2 as a nucleophile for onward synthesis. Organoaluminium fluorides have been the subject of previous reviews.47,48 The use of these compounds as a nucleophilic source of fluorine is very rare owing to the thermodynamic stability of the Al–F bond.49,50 We report here a fluoride metathesis reaction of 2 with various electrophiles (Fig. 3).
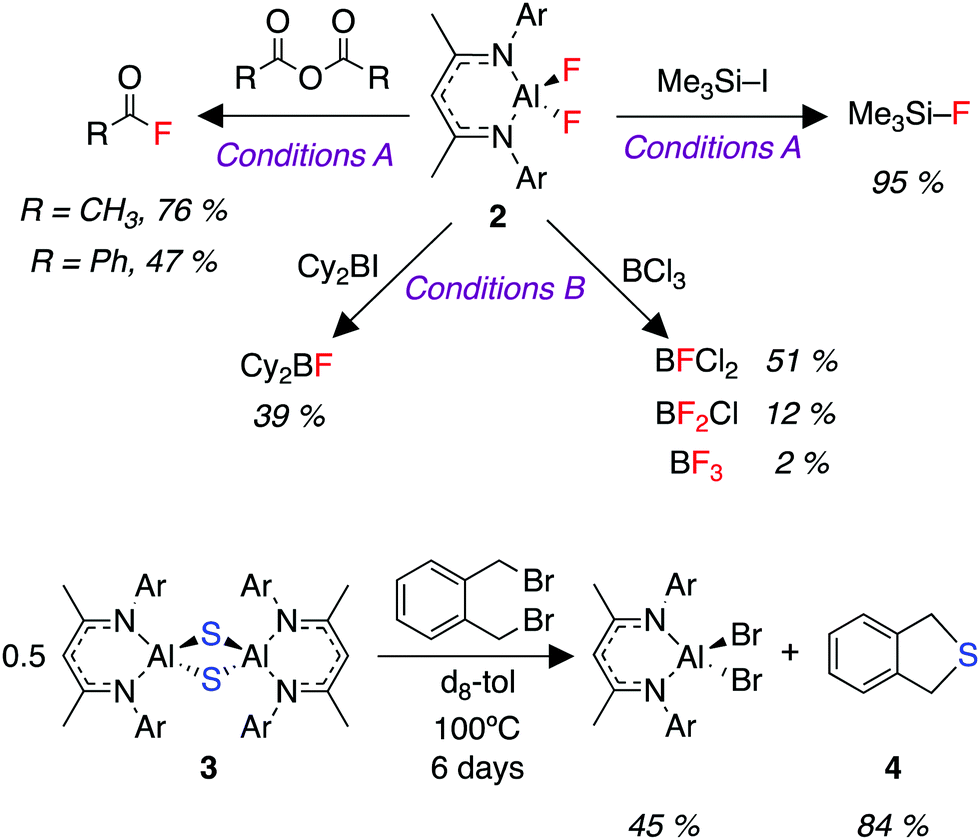 | ||
| Fig. 3 Fluoride and sulfide transfer reactions. Conditions (A): heat at 150 °C for 16 hours in m-xylene solvent. Conditions (B): room temperature for 10 minutes in m-xylene solvent. 10 equiv. of electrophile used for all the above reactions. Yields are determined by quantitative 1H or 19F NMR spectroscopy against a ferrocene, 1,3,5-trimethoxybenzene or 1,2-difluorobenzene internal standard. | ||
Reaction of 2 with organic anhydrides resulted in the formation of acyl fluorides. Acyl fluorides are becoming increasingly important and valuable fluorinating agents due to their unique balance of stability and reactivity.51–54 Furthermore, the reaction of 2 with trimethysilyl iodide produces trimethylsilyl fluoride, a silylating agent for ketones, alcohols, terminal alkynes and various lithiated precursors.55–592 also reacted with BCl3 to produce a series of commercially relevant Lewis acids.60,61 Finally, despite its lack of solubility, we were able to demonstrate the transfer of sulfide (S2−) from 3 to α,α′-dibromo-o-xylene to form the sulfur heterocycle 4 (Fig. 3).62 These fluoride (F−) and sulfide (S2−) transfer reactions represent a formal re-use of the atoms derived from SF6, and thus the overall reaction sequence describes the transfer of fluorine and sulfur from a potent greenhouse gas to highly useful organic products.
In conclusion, we have developed a transition metal free process to deconstruct the potent greenhouse gas SF6 to its elemental components (F− and S2−) using a monovalent aluminium(I) compound under ambient conditions. The aluminium(III) fluoride and sulfide products of the reaction are well-defined and easy to separate by virtue of their differing solubilities. We have undertaken DFT calculations to propose a viable pathway for SF6 activation through nucleophilic attack by the Al(I) fragment at the σ*(S–F) orbital of an S–F bond. We have demonstrated the utility of the aluminium difluoride product (2) as a nucleophilic source of fluorine for organic substrates, and we have shown the ability of 3 to transfer it's sulfide content. Overall, the complete activation of SF6 to its elemental components has been developed in a system where the fluorine and sulfur content can be re-used in the synthesis of valuable compounds.
We are grateful to ERC for generous funding (Fluorofix:677367), to the EPSRC and Imperial College London for DTP studentship funding (Daniel Sheldon), and Richard Kong is thanked for help with crystallography.
Conflicts of interest
There are no conflicts to declare.References
- L. G. Christophorou, J. K. Olthoff and R. J. Van Brunt, IEEE Electr. Insul. Mag., 1997, 13, 20–24 Search PubMed.
- J. R. Case and F. Nyman, Nature, 1962, 193, 473 CrossRef CAS.
- K. Seppelt, Chem. Rev., 2015, 115, 1296–1306 CrossRef CAS PubMed.
- M. Rigby, J. Mühle, B. R. Miller, R. G. Prinn, P. B. Krummel, L. P. Steele, P. J. Fraser, P. K. Salameh, C. M. Harth, R. F. Weiss, B. R. Greally, S. O’Doherty, P. G. Simmonds, M. K. Vollmer, S. Reimann, J. Kim, K.-R. Kim, H. J. Wang, J. G. J. Olivier, E. J. Dlugokencky, G. S. Dutton, B. D. Hall and J. W. Elkins, Atmos. Chem. Phys., 2010, 10, 10305–10320 CrossRef CAS.
- M. Maiss and C. A. M. Brenninkmeijer, Environ. Sci. Technol., 1998, 32, 3077–3086 CrossRef CAS.
- K. S. Betts, Environ. Sci. Technol., 1998, 32, 487A–488A CrossRef CAS PubMed.
- G. P. Stiller, T. von Clarmann, M. Höpfner, N. Glatthor, U. Grabowski, S. Kellmann, A. Kleinert, A. Linden, M. Milz, T. Reddmann, T. Steck, H. Fischer, B. Funke, M. López-Puertas and A. Engel, Atmos. Chem. Phys., 2008, 8, 677–695 CrossRef CAS.
- A. A. Lindley and A. McCulloch, J. Fluorine Chem., 2005, 126, 1457–1462 CrossRef CAS.
- L. Zámostná and T. Braun, Angew. Chem., Int. Ed., 2015, 54, 10652–10656 CrossRef PubMed.
- R. Kurte, H. M. Heise and D. Klockow, J. Mol. Struct., 1999, 480–481, 211–217 CrossRef CAS.
- C.-H. Tsai and J.-M. Shao, J. Hazard. Mater., 2008, 157, 201–206 CrossRef CAS PubMed.
- W.-T. Tsai, J. Fluorine Chem., 2007, 128, 1345–1352 CrossRef CAS.
- S. Bouvet, B. Pégot, S. Sengmany, E. Le Gall, E. Léonel, A.-M. Goncalves and E. Magnier, Beilstein J. Org. Chem., 2020, 16, 2948–2953 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed.
- H. C. Cowen, F. Riding, E. Warhurst, A. D. Buckingham, R. J. W. Le Fèvre, G. D. Meakins, R. N. Haszeldine, J. Jander, G. R. Clemo, B. W. Fox, R. Raper, J. F. W. McOmie, I. M. White, O. L. Brady, P. E. Halstead, A. G. Sharpe, G. W. Gray, B. Jones, J. Cast, T. S. Stevens, F. Bell, J. W. Cook, L. Hunter, R. M. Barrer, N. Mackenzie, D. MacLeod, C. C. Barker, F. D. Casson, G. F. Laws, W. Carruthers, E. S. Lane and C. Williams, J. Chem. Soc., 1953, 4168–4188 RSC.
- C. Berg, T. Braun, M. Ahrens, P. Wittwer and R. Herrmann, Angew. Chem., Int. Ed., 2017, 56, 4300–4304 CrossRef CAS PubMed.
- G. C. Demitras and A. G. MacDiarmid, Inorg. Chem., 1964, 3, 1198–1199 CrossRef CAS.
- H. Deubner and F. Kraus, Inorganics, 2017, 5, 68 CrossRef.
- P. Holze, B. Horn, C. Limberg, C. Matlachowski and S. Mebs, Angew. Chem., Int. Ed., 2014, 53, 2750–2753 CrossRef CAS PubMed.
- B. G. Harvey, A. M. Arif, A. Glöckner and R. D. Ernst, Organometallics, 2007, 26, 2872–2879 CrossRef CAS.
- R. Basta, B. G. Harvey, A. M. Arif and R. D. Ernst, J. Am. Chem. Soc., 2005, 127, 11924–11925 CrossRef CAS PubMed.
- L. Zámostná, T. Braun and B. Braun, Angew. Chem., Int. Ed., 2014, 53, 2745–2749 CrossRef PubMed.
- M. Wozniak, T. Braun, M. Ahrens, B. Braun-Cula, P. Wittwer, R. Herrmann and R. Laubenstein, Organometallics, 2018, 37, 821–828 CrossRef CAS.
- D. Dirican, N. Pfister, M. Wozniak and T. Braun, Chem. – Eur. J., 2020, 26, 6945–6963 CrossRef CAS PubMed.
- F. Buß, C. Mück-Lichtenfeld, P. Mehlmann and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 4951–4955 CrossRef PubMed.
- M. Rueping, P. Nikolaienko, Y. Lebedev and A. Adams, Green Chem., 2017, 19, 2571–2575 RSC.
- T. A. McTeague and T. F. Jamison, Angew. Chem., Int. Ed., 2016, 55, 15072–15075 CrossRef CAS PubMed.
- P. Tomar, T. Braun and E. Kemnitz, Chem. Commun., 2018, 54, 9753–9756 RSC.
- P. Tomar, T. Braun and E. Kemnitz, Eur. J. Inorg. Chem., 2019, 4735–4739 CrossRef CAS.
- D. Rombach and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2020, 59, 300–303 CrossRef CAS PubMed.
- D. Rombach and H.-A. Wagenknecht, ChemCatChem, 2018, 10, 2955–2961 CrossRef CAS.
- S. Kim, Y. Khomutnyk, A. Bannykh and P. Nagorny, Org. Lett., 2021, 23, 190–194 CrossRef CAS PubMed.
- M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994–15996 RSC.
- C. Bakewell, A. J. P. White and M. R. Crimmin, J. Am. Chem. Soc., 2016, 138, 12763–12766 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2018, 57, 6638–6642 CrossRef CAS PubMed.
- G. Coates, B. J. Ward, C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. – Eur. J., 2018, 24, 16282–16286 CrossRef CAS PubMed.
- G. Coates, H. Y. Tan, C. Kalff, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2019, 58, 12514–12518 CrossRef CAS PubMed.
- G. Coates, F. Rekhroukh and M. R. Crimmin, Synlett, 2019, 2233–2246 CAS.
- N. A. Phillips, G. J. Coates, A. J. P. White and M. R. Crimmin, Chem. – Eur. J., 2020, 26, 5365–5368 CrossRef CAS PubMed.
- D. J. Sheldon, G. Coates and M. R. Crimmin, Chem. Commun., 2020, 56, 12929–12932 RSC.
- V. Jancik, M. M. Moya Cabrera, H. W. Roesky, R. Herbst-Irmer, D. Neculai, A. M. Neculai, M. Noltemeyer and H.-G. Schmidt, Eur. J. Inorg. Chem., 2004, 3508–3512 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- C. Strohmann, M. Bindl, V. C. Fraaß and J. Hörnig, Angew. Chem., Int. Ed., 2004, 43, 1011–1014 CrossRef CAS PubMed.
- L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279–292 CrossRef CAS PubMed.
- J. Pinkas and H. W. Roesky, J. Fluorine Chem., 2003, 122, 125–150 CrossRef CAS.
- S. Singh and H. W. Roesky, J. Fluorine Chem., 2007, 128, 369–377 CrossRef CAS.
- O. Mó, M. Yáñez, M. Eckert-Maksić, Z. B. Maksić, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2005, 109, 4359–4365 CrossRef PubMed.
- K. K. K. Goh, A. Sinha, C. Fraser and R. D. Young, RSC Adv., 2016, 6, 42708–42712 RSC.
- Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS PubMed.
- J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 3268–3269 CrossRef CAS PubMed.
- Y. Ogiwara, S. Hosaka and N. Sakai, Organometallics, 2020, 39, 856–861 CrossRef CAS.
- Y. Liang, Z. Zhao, A. Taya and N. Shibata, Org. Lett., 2021, 23, 847–852 CrossRef CAS PubMed.
- N. J. Sisti, Encyclopedia of Reagents for Organic Synthesis, 2001 Search PubMed.
- B. Fredelake, C. Ebker, U. Klingebiel, M. Noltemeyer and S. Schmatz, J. Fluorine Chem., 2004, 125, 1007–1017 CrossRef CAS.
- G. J. Sherborne, A. G. Gevondian, I. Funes-Ardoiz, A. Dahiya, C. Fricke and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 15543–15548 CrossRef CAS PubMed.
- J. H. Song, T. Nabeya, Y. Adachi, Y. Ooyama and J. Ohshita, J. Organomet. Chem., 2019, 883, 47–51 CrossRef CAS.
- W. B. Farnham, US5268511(A), 1993.
- R. J. Brotherton, C. J. Weber, C. R. Guibert and J. L. Little, Boron Compounds, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, pp. 237–255 Search PubMed.
- H. Heaney, Boron Trifluoride, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001 Search PubMed.
- J. A. Gladysz, V. K. Wong and B. S. Jick, Tetrahedron, 1979, 35, 2329–2335 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, computational and crystallographic details. CCDC 2084888. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc02838c |
| This journal is © The Royal Society of Chemistry 2021 |