
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and application of a 19F-labeled fluorescent nucleoside as a dual-mode probe for i-motif DNAs†
Wen Ann Weea,
Ji Hye Yuma,
Shingo Hirashimaa,
Hiroshi Sugiyama *ab and
Soyoung Park*a
*ab and
Soyoung Park*a
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-oiwakecho, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: sugiyama.hiroshi.3s@kyoto-u.ac.jp; park.soyoung.3z@kyoto-u.ac.jp
bInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Yoshida-ushinomiyacho, Sakyo-ku, Kyoto 606-8501, Japan
First published on 10th March 2021
Abstract
Because of their stable orientations and their minimal interference with native DNA interactions and folding, emissive isomorphic nucleoside analogues are versatile tools for the accurate analysis of DNA structural heterogeneity. Here, we report on a bifunctional trifluoromethylphenylpyrrolocytidine derivative (FPdC) that displays an unprecedented quantum yield and highly sensitive 19F NMR signal. This is the first report of a cytosine-based dual-purpose probe for both fluorescence and 19F NMR spectroscopic DNA analysis. FPdC and FPdC-containing DNA were synthesized and characterized; our robust dual probe was successfully used to investigate the noncanonical DNA structure, i-motifs, through changes in fluorescence intensity and 19F chemical shift in response to i-motif formation. The utility of FPdC was exemplified through reversible fluorescence switching of an FPdC-containing i-motif oligonucleotide in the presence of Ag(I) and cysteine.
Introduction
By visually perceiving an object, understanding of its nature may be obtained more directly. Fluorescence-based imaging has led to advances in our ability to explore within cells through the interrogation of numerous biomolecules, including DNA, RNA, and proteins.1,2 In particular, fluorescent probes that interfere minimally with the native conformation and properties of targets are valuable in the tracking of freely moving assemblages containing structural heterogeneities, as well as to study their intermolecular interactions.3–6 One suitable class of candidates that fulfills the above condition is fluorescent nucleosides. As an advantage over conventional fluorescent dyes, the utility of fluorescent nucleosides is further amplified when incorporated sequence-specifically into programmable DNA/RNA oligonucleotides.7–9Fluorescent nucleosides have been shown to be powerful tools in the investigation of both biological and inorganic systems. A series of fluorescent isomorphic nucleosides (nucleotides) that we had previously reported on have exhibited excellent biophysical and photophysical properties.10–12 Isomorphic emissive DNA analogues based on a thieno[3,4-d]-pyrimidine core, thdG, 2′-OMe-thG, and thdT, have been applied to investigate DNA structures and protein interactions. In addition, a Watson–Crick base-pairable FRET system was established using thdG and tC and was successfully applied to the investigation of nucleosomes.13 Very recently, we also reported on the synthesis of dioxT, a dioxoloquinazoline-containing tricyclic thymine analogue with excellent photophysical properties.14 The highly selective and sensitive binding between dioxT and metal ions allowed us to demonstrate its utility as a heavy metal sensor.15
In parallel, 19F-labeled nucleosides have been developed as versatile probes due to the practicality of 19F NMR spectroscopy in investigating the structures and interactions of biomolecules.16 Unique features, such as the intrinsic sensitivity of the 19F nucleus and its absence from general biomolecules, make 19F NMR probes highly attractive.17,1819F-labeled bases have been used in diverse applications to explore DNA/RNA dynamics and heterogeneity.19–21 Fujimoto and co-workers synthesized oligonucleotides containing trifluoromethylcytidine or trifluoromethylthymidine and demonstrated the detection of the B–Z transition using 19F NMR chemical shifts.22 Furthermore, the 19F NMR approach for the investigation of DNA/RNA G-quadruplex conformations and enzymatic digestions has been reported with bis(trifluoromethyl)phenyl group-modified oligonucleotides.23–25
In order to exploit the properties of both techniques, innovative dual probes that merge the advantages of fluorescence and 19F NMR spectroscopy into a single nucleoside have been reported recently. Hocek and colleagues synthesized dual fluorophores of nucleic acids by the introduction of a fluorine-containing biaryl group to 2′-deoxyuridine and 7-deazapurine.26 Additionally, Srivatsan's group reported 5-fluorobenzofuran-2′-deoxyuridine as a dual probe, which they used to study the structural diversity of G-quadruplex-forming sequences under biological conditions.27 Despite its dual functionality, the quantum yield of the probe could be improved. Nevertheless, such dual-purpose probes facilitate the investigation of the structures and interactions of nucleic acids with other biomolecules. However, to the best of our knowledge, a cytidine-based dual probe has yet to be reported.
Here, we report the synthesis, characterization, and application of the first cytidine-based dual probe, trifluoromethylphenylpyrrolocytidine derivative (FPdC). By attaching a 19F NMR-active trifluoromethyl group to a pyrrolo-dC core, a marked increase in brightness was observed. Using the unprecedented quantum yield and highly sensitive 19F NMR signal, we employed FPdC in i-motif DNA structures and demonstrated its unique photophysical property as a Ag(I)-binding fluorophore in this specific DNA conformation.
Experimental
Preparation of compound 7
The detailed synthetic route and characterization data for compounds 2–6 were reported by Hudson and co-workers.28 Compound 7 was synthesized from intermediate 6 respectively through the same general procedure. CuI (0.3 equiv.) followed by Et3N (10 equiv.) were added to a THF solution of compound 6 before introduction of 4-ethynyl trifluorotoluene (2.3 equiv.) then Pd(PPh3)Cl2 (0.1 equiv.). The resultant reaction mixture was stirred in the dark at 50–55 °C for 18 h and the solvent was removed. After filtering the mixture through Celite and washing with ethyl acetate, the filtrate was collected and the solvent evaporated. The crude intermediate was dissolved in EtOH and Et3N (10 equiv.) was added to the solution. The resultant reaction mixture was stirred at 55 °C for 18 h. Purification of the resultant reaction mixure by column chromatography afforded compound 7 (72%).Preparation of compound 10
Compound 10 was prepared by dropwise addition of DIPEA at 0 °C to a solution of compound 7 in CH2Cl2 followed by stirring at 0 °C for 10 min. Phosphoramidochloridous acid was subsequently added and the mixture was stirred for a further 10 min at 0 °C before warming to room temperature and stirring for another 1 h. After removal of the solvent, the residue was dissolved in MeCN and used immediately without purification for solid-phase DNA synthesis.Oligonucleotide synthesis
Solid-phase oligonucleotide synthesis was performed on an ABI DNA synthesizer (Applied Biosystem, Foster City, CA) or M-2-MX DNA/RNA synthesizer (Nihon Techno Service Co., Ltd, Tsukuba, Japan). 1C and IM-CT were synthesized on solid support using FPdC phosphoramidite (compound 10) and commercially available O5′-dimethoxytrityl-2′-deoxyribonucleoside O3′-phosphoramidites. Cleavage from the solid support and deprotection, followed by purification with Glen-PakTM cartridges yielded the desired oligonucleotides. After HPLC purification (COSMOSIL 5C18 AR-II, Nacalai Tesque, Inc., Kyoto, 150 × 10 mm id), the identity of the products were confirmed by MALDI-TOF MS using a Bruker microflex-KSII (Bruker Corporation, Billerica, MA) (Table S2, ESI†). DNA concentrations were determined by using NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE).Preparation of cell lysate
HeLa cells at 90% confluence were fixed with 270 μL of 37% formaldehyde and 500 μL of 2.5 M glycine. After collection of the cells and centrifugation to obtain the pelleted nuclei, the cell pellet was resuspended in nuclear lysis buffer on ice. Ultra-sonication and centrifugation yielded the nuclear lysate. For acidification of the nuclear lysate (pH 7.8 to pH 5.5), 10 μL of 6 M HCl was added to 300 μL of nuclear lysis buffer.UV-visible absorbance spectroscopy and UV-melting assays
UV-vis absorbance spectra were measured at 20 °C on a JASCO V-750 spectrophotometer equipped with a JASCO PAC-743R thermocontrolled cell changer and a JASCO CTU-100 thermocirculator. Melting temperatures were determined by measuring changes in absorbance at 260 nm as a function of temperature. Absorbance was recorded from 15 to 90 °C at a rate of 1 °C min−1. All samples were prepared in a total volume of 120 μL containing 2.5 μM oligonucleotide and 10 mM Na cacodylate buffer (various pH).CD spectroscopy
CD spectra of oligonucleotide solutions collected in 1 nm steps from 360 nm or 320 nm to 220 nm were measured using JASCO J-805LST Spectrometer in a 1 cm quartz cuvette. Each spectrum shown is the average of two individual scans. All samples were prepared in a total volume of 110 μL containing 2.5 μM oligonucleotide and 10 mM Na cacodylate buffer (various pH).Fluorescence, quantum yield and lifetime measurements
Fluorescence measurements were obtained using 3 mm path length JASCO FMM-100 quartz microcells on a JASCO FP-6300 Spectrofluorometer equipped with a JASCO EHC-573 temperature controller. The emission spectra were recorded from 220 nm to 600 nm at a scan rate of 100 nm min−1 with an excitation wavelength of 374 nm for FPdC and 1C, and 365 nm for IM-CT. Quantum yields were measured with a Hamamatsu Photonics Quantaurus-QY C11347-11 Absolute PL Quantum Yield Spectrometer and calculated with the integrated measurement software. Fluorescence lifetime measurements were obtained using TCSPC (time-correlated single photon counting) on a Horiba Scientific Fluorohub equipped with a Horiba Scientific FluoroCube 3000U-SHK and a 375 nm Horiba Scientific NanoLED pulsed diode light source. Emission wavelength was set at the emission maximums found through fluorescence spectroscopy of each ODN. TAC range was 200 ns and Repetition rate was 1 MHz. The data was fitted to exponential functions using DAS6 (HORIBA).19F NMR of oligonucleotides
NMR spectra were obtained on a JEOL JNM ECA-600 spectrometer operating at 565 MHz for 19F NMR. Samples were prepared with 50 μM oligonucleotide and 20% D2O in 10 mM Na cacodylate buffer (pH as indicated) doped with trifluoroacetic acid as an internal standard for i-motif-forming sequences.Computational modelling
The geometry of modified nucleobases/nucleoside were optimized using DFT, and their optical spectra were calculated using DFT methods as implemented in the Gaussian 16W program package. All the calculations were done using B3LYP (Becke, three-parameter, Lee–Yang–Parr) hybrid exchange and correlation energy functional, default spin, with the 6-311G+(d,2p) basis set for all atoms. The DFT calculations were performed in the gas phase. After geometry optimization, frequency calculations were done to remove any vibrational unstable modes.Result and discussion
Photophysical properties of FPdC
Synthesis of the dual-probe cytosine analogue, FPdC, was conducted based on a previous report by Hudson and co-workers (Scheme 1).28 To incorporate the trifluoromethyl group into the molecule, ethynyl trifluorotoluene was introduced through a Sonogashira cross-coupling reaction. Subsequent cyclization provided the 5′-DMTr-protected nucleoside 7 as a precursor for solid-phase DNA synthesis. The highly emissive deoxyribonucleoside was also prepared and used to characterize the photophysical properties of FPdC (Scheme S1, ESI†).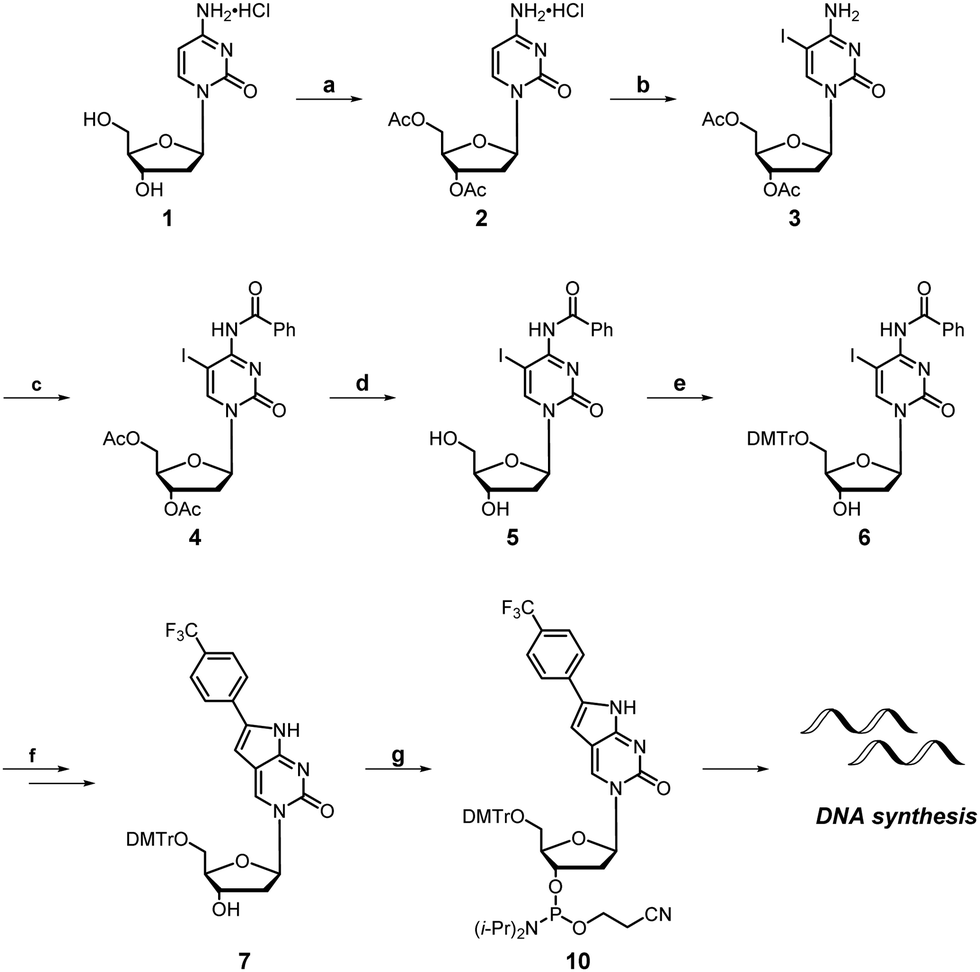 | ||
| Scheme 1 Synthesis of FPdC and FPdC-containing DNA. Reagents and conditions: (a) CH3COCl, AcOH, CHCl3, 50–60 °C, 3 days; (b) I2, HIO3, AcOH, 40 °C, 24 h; (c) Bz2O, pyridine, 85 °C, 3 h; (d) K2CO3, MeOH, r.t., 1 h; (e) DMTrCl, pyridine, r.t., 4 h; (f) 4-ethynyl trifluorotoluene, Pd(PPh3)Cl2, CuI, Et3N, THF, 50–55 °C, 18 h, then, Et3N, EtOH, 55 °C 18 h; (g) (iPr2N)2PO(CH2)2CN, DIPEA, CH2Cl2, 0 °C to r.t, 1.5 h. | ||
The photophysical properties of FPdC were examined under various solvent conditions (Fig. 1B). In water and dioxane, FPdC had an absorption band around 362 nm. In contrast, the absorption band was slightly red-shifted to 369 nm in methanol. FPdC was also found to exhibit the highest and lowest fluorescence intensity in methanol (λem = 450 nm) and dioxane (λem = 454 nm) respectively (Fig. 1B). To make sense of this disparity, the lifetime (τ), quantum yield (Φ), and molar absorptivity (ε) of FPdC in the three solvents were investigated (Fig. 1C and D). Calculation of the radiative (kr) and non-radiative (knr) relaxation rates from τ and Φ revealed that unsurprisingly, methanol had the highest kr/knr ratio of 1.2, compared to 1.0 in water and 0.06 in dioxane. Nevertheless, FPdC had an emission maximum at 454 nm with exceptional brightness (εΦ = 12048) in water, which represented a sixfold improvement compared to PhPyrdC (1894) and fourfold improvement compared to tC° (2700).29,30 Remarkably, the brightness exhibited by FPdC was also higher than that of the recently reported pA (10100) (Fig. 1A).31
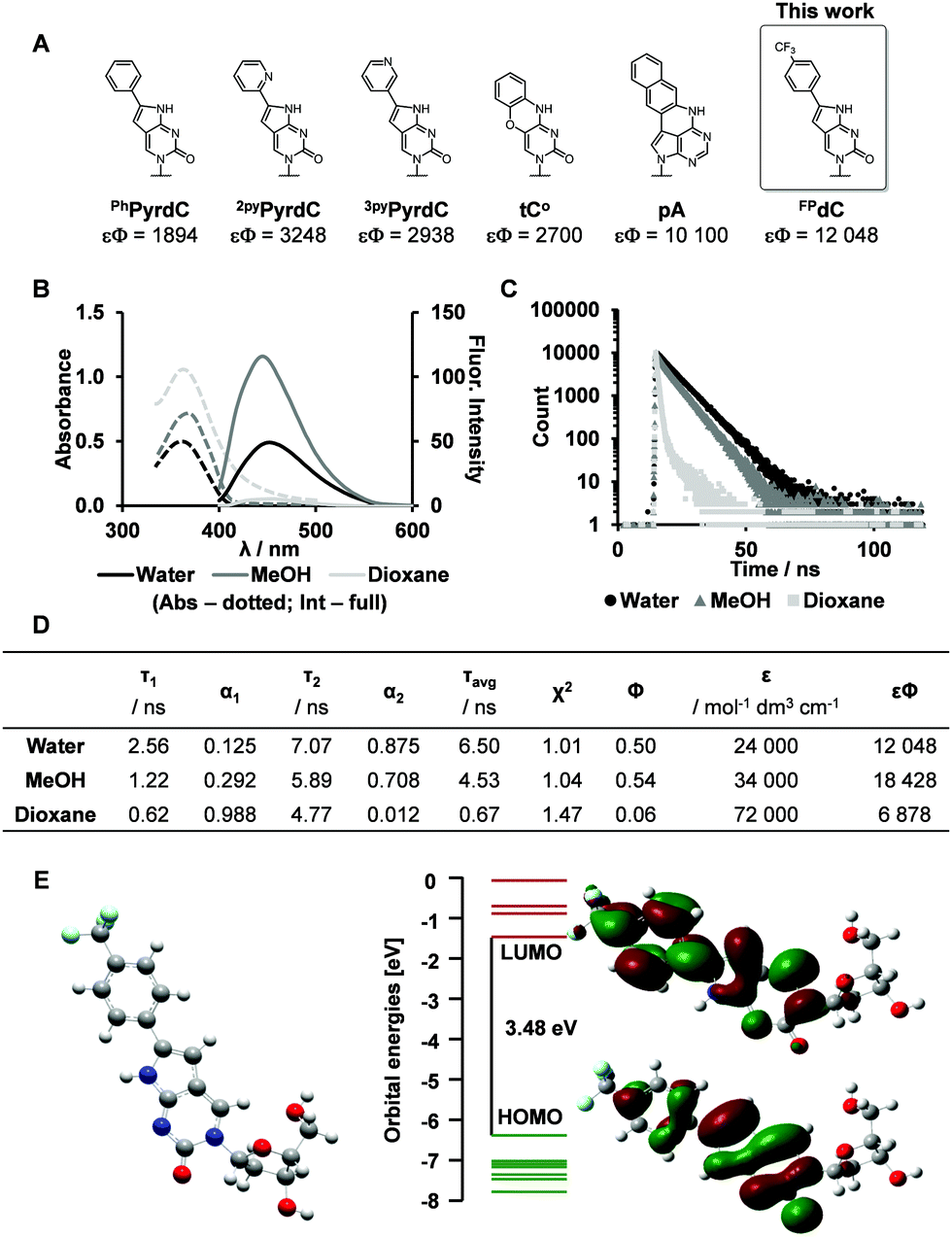 | ||
| Fig. 1 Synthesis and photophysical properties of FPdC. (A) Reported cytidine analogues and their brightness (εΦ) in buffer.29–31 (B and C) Absorbance and emission spectra, as well as fluorescence decay of FPdC (dotted lines: Absorbance, full lines: Fluorescence). (D) Quantified photophysical properties of FPdC. (E) DFT minimized geometry of FPdC and its HOMO and LUMO orbitals. | ||
To obtain deeper insight into its photophysical properties, gas phase DFT calculation of FPdC was performed. The HOMO and LUMO orbitals are depicted in Fig. 1E. The shapes of the frontier orbitals of FPdC were similar to those of PhPyrdC. However, compared with PhPyrdC, the optimized geometry of FPdC comprised a less distorted and more planar ring system (Fig. S24–S26, ESI†). This may account for the excellent photophysical properties of FPdC.
Observation of pH-Induced i-motif formation with FPdC
DNA is a dynamic biopolymer with a high order of structural heterogeneity.32–34 Apart from the canonical duplexes, DNA is known to form several noncanonical structures. For example, i-motifs are four-stranded structures formed from hemiprotonated cytosine pairs through intercalated hydrogen bonds.35 Furthermore, a recent study reported the direct visualization of i-motif structures in the nuclei of human cells.36 Consequently, the role of the i-motif as a regulator in the genome has received much attention in recent times. In this study, we synthesized sequences comprising four C-tracts separated by loop regions, 1C and IM-CT (Fig. 2A and Table S2, ESI†).37,38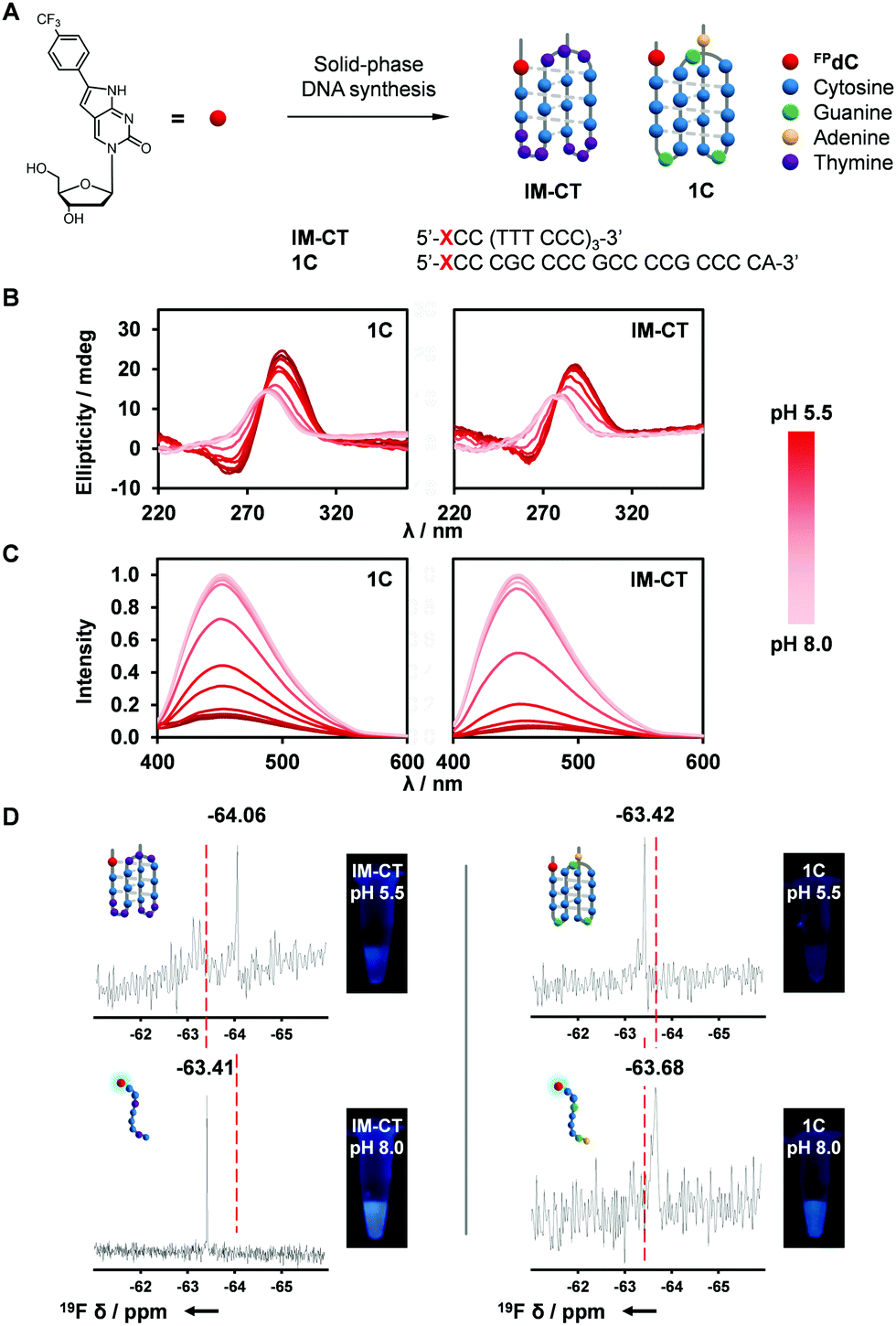 | ||
| Fig. 2 Characterization of FPdC-containing i-motif structures. (A) I-Motif-forming DNA sequences, 1C and IM-CT, containing FPdC. (B) CD and (C) normalized fluorescence spectra of 1C and IM-CT with pH. (D) 19F NMR spectra and images of 1C and IM-CT. | ||
Because i-motif structures exhibit a characteristic positive peak around 285 nm and a negative band around 260 nm in their CD spectra, CD spectroscopy was used to investigate the effect of FPdC incorporation on the i-motif-forming sequences.39,40 i-Motif formation was observed for both 1C and IM-CT sequences at pH 5.5. As the pH was increased to pH 8.0, the CD spectra reflected the unfolding of the i-motif to randomly coiled single-stranded DNA (Fig. 2B).41 Notably, the pKa between FPdC-containing and native sequences differed minimally (difference of −0.15 for IM-CT and −0.03 for 1C; Fig. S10, ESI†), suggesting that incorporation of FPdC did not majorly affect the pH-sensitivity of the i-motif sequences. In addition, UV-melting studies revealed that the presence of FPdC in the sequences did not destabilize i-motif formation (Fig. S5–S8, ESI†).
Inspired by the favorable i-motif formation of FPdC-containing oligonucleotides and the remarkable photophysical properties of FPdC in water, we next used fluorescence and 19F NMR spectroscopy to explore FPdC as a dual-purpose probe for detecting i-motif structure formation. Fluorescence analysis of the oligonucleotides showed that FPdC exhibited a significant difference in fluorescence intensity between the folded i-motif structures and the single-stranded DNA (Fig. 2C). In contrast, fluorescence intensity of the FPdC mononucleoside was not affected by a change in pH, which suggests that the FPdC-containing oligonucleotides were able to act as probes for i-motif formation. Together with a change in emission, different chemical shifts corresponding to the folded and unfolded structures were also observed in their 19F NMR spectra (Fig. 2D). Notably, IM-CT and 1C showed a shielding and deshielding of 19F NMR signal respectively upon i-motif formation which may be attributed to the differences in electron density; FPdC in the IM-CT i-motif participates in hydrogen bonding while in 1C, FPdC is positioned outside the i-motif structure and hence the formation of the i-motif structure in 1C concomitantly prevents FPdC from interacting with other bases.38,41,42 The observed change in chemical shifts were comparable to those obtained by other 19F NMR nucleoside probes.43–45 In addition, the high resolution afforded by 19F NMR spectroscopy revealed the presence of minor structures formed by IM-CT, emphasizing the potential of FPdC as a 19F NMR-active dual fluorophore for nucleic acids.46
Both of the i-motif-forming DNA sequences, 1C and IM-CT, exhibited a change in fluorescence intensity after i-motif formation. However, IM-CT showed a more dramatic change with sevenfold higher emission compared to that of 1C at pH 8.0 (Fig. 3B). This finding was corroborated by the quantum yields and lifetimes measured for 1C and IM-CT (Fig. 3A). To investigate if the more constrained positioning of FPdC in i-motif structures could have contributed to their lower fluorescence intensities, increasing amounts of glycerol were added to methanol solutions of FPdC. However, FPdC exhibited a higher quantum yield with increasing viscosity (Fig. S22, ESI†). This indicated that conversely, the inhibition of large amplitude intramolecular motions could reduce non-radiative relaxation in FPdC.47 The effect of pH on the FPdC nucleoside was also examined to decouple the effect of pH on FPdC from the changes brought about by i-motif formation (Fig. S15, ESI†). As expected, and unlike 1C and IM-CT, no pH-dependent change in fluorescence was observed for the nucleoside. It should also be noted that although 1C and IM-CT both form i-motif structures, FPdC is expected to participate in base-pairing in the IM-CT structure but not in the 1C structure (Fig. 2A).38,41 Despite the lack of base-pairing in 1C, the structural changes that occurred during i-motif formation appeared to be sufficient for quenching to occur. In addition, the fluorescence changes associated with the different structures were consistent with the 19F NMR spectra.
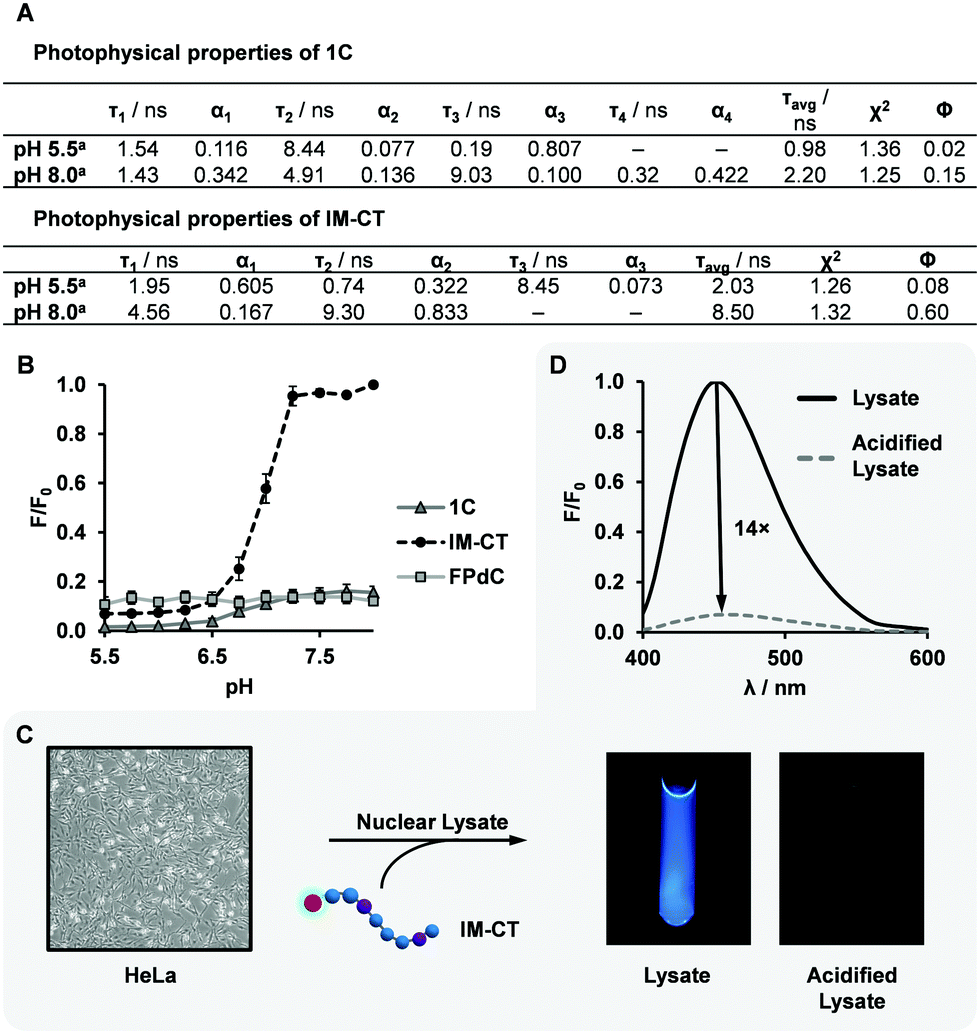 | ||
| Fig. 3 Photophysical properties of FPdC-containing i-motif sequences in buffer and in cell nuclear lysate. (A) Quantified photophysical properties of 1C and IM-CT. (B) Comparison of F/F0 values with pH, where F0 is the maximum intensity across all three species (n = 2, error bars indicate SE). (C and D) Fluorescence quenching of FPdC in HeLa cell nuclear lysate. | ||
Interestingly, oligonucleotide 1C had a quantum yield three times lower than the FPdC monomer at pH 8.0 (unfolded conformation), whereas the quantum yield of IM-CT (0.60) was comparable to that of the FPdC monomer (0.54). Accordingly, 1C and IM-CT had a kr/knr ratio of 0.2 and 1.5 respectively at pH 8.0. Unlike IM-CT, 1C contains guanine residues between tracts of cytosine residues. Guanine has been reported to quench fluorophores with high efficiency through an electron transfer process, and although FPdC was not be positioned adjacent to guanine in 1C, a recent study revealed that charge redistribution can occur in single-stranded DNA.48,49 The transfer of electrons from FPdC to guanine may thus account for the lower quantum yield observed in unfolded 1C. In addition, the lower quantum yield of FPdC exhibited in i-motif structures may be due to a higher conductivity and hence greater propensity for charge redistribution through the rigid structure, similar to the higher quenching observed in double-stranded DNA as compared to its single-stranded counterpart.48,50
It was also observed that unlike the well-established fluorescent nucleosides, 2AP and pA, which are quenched once incorporated into oligonucleotides, FPdC retains its high quantum yield in IM-CT.31,51 This observation demonstrates the versatility of FPdC as a highly sensitive molecular probe, and plans to characterize its properties and dependence on neighbouring bases are under way.52–55
To investigate the utility of IM-CT as a bifunctional probe in cellular conditions, we conducted fluorescence and 19F NMR measurements of IM-CT in HeLa cell nuclear lysate (Fig. 3C and D). When IM-CT was present in the buffered nuclear extract (pH 7.8), a high degree of fluorescence emission was observed. In addition, its 19F NMR shift of −63.42 ppm suggested that IM-CT was unfolded (Fig. S19, ESI†). Upon acidification of the sample with concentrated HCl, its fluorescence was visibly quenched, which suggested formation of the i-motif. This conclusion was corroborated by 19F NMR, which showed an upfield shift to –63.71 ppm. In addition, significant line broadening effects were not observed. Further studies are underway to evaluate the utility of FPdC as a dual probe in live cells.
Observation of Ag(I)-induced DNA folding with FPdC
The outstanding photophysical properties of FPdC led us to examine further applications. At neutral pH, it is known that the presence of Ag(I) can result in the formation of i-motif structures via the interaction between Ag(I) and cytosine.56 The Ag(I)-induced folded structures can be unfolded by chelation of the Ag(I) ions, and cysteine is often used for this purpose. In addition, pyrrolo-dC nucleotides were shown to possess Ag(I) capturing ability.29 Thus, we anticipated that FPdC-containing oligonucleotides could trace the reversible folding–unfolding process by Ag(I) and cysteine. With addition of 10 equivalents of Ag(I), the CD spectrum of IM-CT at pH 6.5 revealed a positive peak near 244 nm in addition to the typical i-motif band at 288 nm (Fig. 4A), suggesting the formation of a modified structure.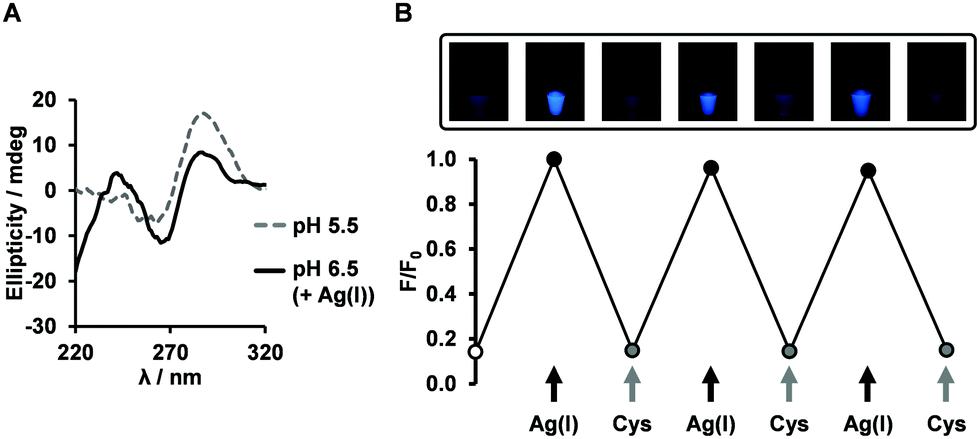 | ||
| Fig. 4 Application of FPdC as a fluorescent probe. (A) CD spectra of IM-CT at pH 6.5 upon the addition of Ag(I). (B) Fluorescence switching of IM-CT at pH 6.5 (n = 2, error bars indicate SE). | ||
We then proceeded to examine the fluorescence intensity change of IM-CT in the presence of Ag(I) or cysteine. We hypothesized that addition of Ag(I) will result in a decline of fluorescence through i-motif formation and subsequent addition of cysteine will recover the fluorescence. Surprisingly, the opposite tendency was observed when we conducted the experiments at pH 6.5. The addition of 10 equivalents of Ag(I) induced a fourfold increase in fluorescence emission and the addition of cysteine quenched the fluorescence. When the same experiment was performed on the FPdC nucleoside, no fluorescence switching was observed (Fig. S16, ESI†). Given that the CD spectra at both pH values mirrored the reversible changes in intensity and conformation, it is likely that structural changes were a major contributing factor to the fluorescence switching (Fig. 4B and Fig. S11, ESI†). To investigate if the structural change induced by Ag(I) played a role in the observed increase in fluorescence, the intensity of IM-CT was measured with increasing Ag(I) concentration. Indeed, fluorescence intensity followed a simple upward trend culminating in a plateau around nine equivalents of Ag(I) (Fig. S14, ESI†). It is thus highly likely that contrary to expectations, the difference in the Ag(I)-induced and pH-induced structures observed through CD spectroscopy was sufficient for the folding of the Ag(I)-induced structure to elicit an increase in emission instead.
To investigate the conformational changes of IM-CT in greater detail, 19F NMR spectroscopy was employed. Interestingly, 19F NMR suggested that the folded pH-induced i-motif structure (−64.04 ppm) and the unfolded single-stranded structure (−63.25 ppm) could coexist at pH 6.5. Upon addition of Ag(I), the peaks shifted downfield to −63.21 ppm, presumably, the Ag(I)-induced i-motif conformation. In line with the reversibility of the fluorescence and CD measurements, the initial state was restored upon removal of Ag(I) by cysteine chelation (Fig. S20, ESI†). Although further study is ongoing to investigate the Ag(I)-induced structure, this result demonstrates the potential of FPdC for use in bio-imaging and nanophotonic applications.
Conclusion
We have successfully synthesized a 19F-labeled fluorescent cytosine analogue (FPdC) and incorporated it into i-motif-forming DNA sequences. By introducing a trifluoromethyl moiety onto a previously characterized phenyl-pyrrolo-dC analogue, PhPyrdC, versatile 19F NMR functionality and a sixfold improvement in brightness were obtained. In addition, compared to the previously reported fluorescent tricyclic cytosine derivative tC°, FPdC provides improvements in brightness as a result of its high molar absorptivity (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mol−1 dm3 cm−1) and excellent quantum yield (0.50). When incorporated into oligonucleotides and upon formation of i-motif structures, the FPdC-containing oligonucleotides exhibited substantial changes in fluorescence intensity, as well as 19F NMR chemical shifts in buffer and in HeLa cell nuclear lysate. These changes in fluorescence intensity were shown to be highly reversible when the folding and unfolding of an i-motif-like structure was induced with Ag(I) ion and cysteine. Based on these results, FPdC shows promise as a Ag-binding fluorophore for specific DNA configuration. Given its high quantum yield and reversibility, this dual probe may also prove to be useful for a broad range of applications including spatial and temporal analysis of non-canonical DNA structures as well as DNA–protein interactions in cells. Further studies are underway to evaluate FPdC as a dual probe in live cells.
000 mol−1 dm3 cm−1) and excellent quantum yield (0.50). When incorporated into oligonucleotides and upon formation of i-motif structures, the FPdC-containing oligonucleotides exhibited substantial changes in fluorescence intensity, as well as 19F NMR chemical shifts in buffer and in HeLa cell nuclear lysate. These changes in fluorescence intensity were shown to be highly reversible when the folding and unfolding of an i-motif-like structure was induced with Ag(I) ion and cysteine. Based on these results, FPdC shows promise as a Ag-binding fluorophore for specific DNA configuration. Given its high quantum yield and reversibility, this dual probe may also prove to be useful for a broad range of applications including spatial and temporal analysis of non-canonical DNA structures as well as DNA–protein interactions in cells. Further studies are underway to evaluate FPdC as a dual probe in live cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We express sincere thanks for a Grant-in-Aid Priority Research (16H06356 for H. S) from Japan Society for the Promotion of Science (JSPS). We also thank KAKENHI program (Grant-in-Aid for scientific research C, 18K05315) for support to S. P. This work was also supported by AMED [JP20am0101101 (Platform Project for Supporting Drug Discovery and Life Science Research (BINDS) to H. S.)] and NIH awards R01CA236350 (to H. S.). We would like to thank Karin Nishimura (Graduate School of Engineering, Kyoto University) for technical assistance in obtaining the mass spectra of synthesized compounds.Notes and references
- J. W. Lichtman and J. A. Conchello, Nat. Methods, 2005, 2, 910–919 CrossRef CAS PubMed.
- T. Terai and T. Nagano, Curr. Opin. Chem. Biol., 2008, 12, 515–521 CrossRef CAS PubMed.
- W. K. M. Lai and B. F. Pugh, Nat. Rev. Mol. Cell Biol., 2017, 18, 548–562 CrossRef CAS PubMed.
- P. Kaur, M. J. Longley, H. Pan, W. Wang, P. Countryman, H. Wang and W. C. Copeland, J. Biol. Chem., 2020, 295, 5564–5576 CrossRef PubMed.
- B. Y. Michel, D. Dziuba, R. Benhida, A. P. Demchenko and A. Burger, Front. Chem., 2020, 8, 112 CrossRef CAS PubMed.
- J.-Y. Kim and H. S. Chung, Science, 2020, 368, 1253–1257 CrossRef CAS PubMed.
- M. Rist and J. Marino, Curr. Org. Chem., 2002, 6, 775–793 CrossRef CAS.
- L. M. Wilhelmsson, Q. Rev. Biophys., 2010, 43, 159–183 CrossRef CAS PubMed.
- A. A. Tanpure, M. G. Pawar and S. G. Srivatsan, Isr. J. Chem., 2013, 53, 366–378 CrossRef CAS.
- S. Park, H. Otomo, L. Zheng and H. Sugiyama, Chem. Commun., 2014, 50, 1573–1575 RSC.
- I. Okamura, S. Park, R. Hiraga, S. Yamamoto and H. Sugiyama, Chem. Lett., 2017, 46, 245–248 CrossRef CAS.
- J. H. Han, S. Yamamoto, S. Park and H. Sugiyama, Chem. – Eur. J., 2017, 23, 7607–7613 CrossRef CAS PubMed.
- J. H. Han, S. Park, F. Hashiya and H. Sugiyama, Chem. – Eur. J., 2018, 24, 17091–17095 CrossRef CAS PubMed.
- S. Hirashima, J. H. Han, H. Tsuno, Y. Tanigaki, S. Park and H. Sugiyama, Chem. – Eur. J., 2019, 25, 9913–9919 CrossRef CAS PubMed.
- J. H. Han, S. Hirashima, S. Park and H. Sugiyama, Chem. Commun., 2019, 55, 10245–10248 RSC.
- A. Olszewska, R. Pohl and M. Hocek, J. Org. Chem., 2017, 82, 11431–11439 CrossRef CAS PubMed.
- H. Chen, S. Viel, F. Ziarelli and L. Peng, Chem. Soc. Rev., 2013, 42, 7971–7982 RSC.
- S. L. Cobb and C. D. Murphy, J. Fluorine Chem., 2009, 130, 132–143 CrossRef CAS.
- L. Granqvist and P. Virta, J. Org. Chem., 2015, 80, 7961–7970 CrossRef CAS PubMed.
- A. Aro-Heinilä, T. Lönnberg and P. Virta, Bioconjugate Chem., 2019, 30, 2183–2190 CrossRef PubMed.
- T. Sakamoto, H. Hayakawa and K. Fujimoto, Chem. Lett., 2011, 40, 720–721 CrossRef CAS.
- S. Nakamura, H. Yang, C. Hirata, F. Kersaudy and K. Fujimoto, Org. Biomol. Chem., 2017, 15, 5109–5111 RSC.
- T. Ishizuka, A. Yamashita, Y. Asada and Y. Xu, ACS Omega, 2017, 2, 8843–8848 CrossRef CAS PubMed.
- H.-L. Bao, T. Ishizuka, T. Sakamoto, K. Fujimoto, T. Uechi, N. Kenmochi and Y. Xu, Nucleic Acids Res., 2017, 45, 5501–5511 CrossRef CAS PubMed.
- M. Chrominski, M. R. Baranowski, S. Chmielinski, J. Kowalska and J. Jemielity, J. Org. Chem., 2020, 85, 3440–3453 CrossRef CAS PubMed.
- J. Riedl, R. Pohl, L. Rulíšek and M. Hocek, J. Org. Chem., 2012, 77, 1026–1044 CrossRef CAS PubMed.
- S. Manna, D. Sarkar and S. G. Srivatsan, J. Am. Chem. Soc., 2018, 140, 12622–12633 CrossRef CAS PubMed.
- S. J. Cho, A. Ghorbani-Choghamarani, Y. Saito and R. H. E. Hudson, Curr. Protoc. Nucleic Acid Chem., 2019, e75 CrossRef PubMed.
- H. Yang, H. Mei and F. Seela, Chem. – Eur. J., 2015, 21, 10207–10219 CrossRef CAS PubMed.
- P. Sandin, K. Börjesson, H. Li, J. Mårtensson, T. Brown, L. M. Wilhelmsson and B. Albinsson, Nucleic Acids Res., 2007, 36, 157–167 CrossRef PubMed.
- M. Bood, A. F. Füchtbauer, M. S. Wranne, J. J. Ro, S. Sarangamath, A. H. El-Sagheer, D. L. M. Rupert, R. S. Fisher, S. W. Magennis, A. C. Jones, F. Höök, T. Brown, B. H. Kim, A. Dahlén, L. M. Wilhelmsson and M. Grøtli, Chem. Sci., 2018, 9, 3494–3502 RSC.
- G. Gupta, M. Bansal and V. Sasisekharan, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6486–6490 CrossRef CAS PubMed.
- R. D. Wells, D. A. Collier, J. C. Hanvey, M. Shimizu and F. Wohlrab, FASEB J., 1988, 2, 2939–2949 CrossRef CAS PubMed.
- J. Choi and T. Majima, Chem. Soc. Rev., 2011, 40, 5893–5909 RSC.
- K. Gehring, J. L. Leroy and M. Guéron, Nature, 1993, 363, 561–565 CrossRef CAS PubMed.
- M. Zeraati, D. B. Langley, P. Schofield, A. L. Moye, R. Rouet, W. E. Hughes, T. M. Bryan, M. E. Dinger and D. Christ, Nat. Chem., 2018, 10, 631–637 CrossRef CAS PubMed.
- K. Guo, A. Pourpak, K. Beetz-Rogers, V. Gokhale, D. Sun and L. H. Hurley, J. Am. Chem. Soc., 2007, 129, 10220–10228 CrossRef CAS PubMed.
- P. Školáková, D. Renčiuk, J. Palacký, D. Krafčík, Z. Dvořáková, I. Kejnovská, K. Bednářová and M. Vorlíčková, Nucleic Acids Res., 2019, 47, 2177–2189 CrossRef PubMed.
- G. Manzini, N. Yathindra and L. E. Xodo, Nucleic Acids Res., 1994, 22, 4634–4640 CrossRef CAS PubMed.
- J. Kypr, I. Kejnovská, D. Renciuk and M. Vorlícková, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- P. Bielecka and B. Juskowiak, Molecules, 2015, 20, 18511–18525 CrossRef CAS PubMed.
- C. Kasireddy, J. G. Bann and K. R. Mitchell-Koch, Phys. Chem. Chem. Phys., 2015, 17, 30606–30612 RSC.
- F. Seela and K. Xu, Org. Biomol. Chem., 2008, 6, 3552–3560 RSC.
- B. Puffer, C. Kreutz, U. Rieder, M. O. Ebert, R. Konrat and R. Micura, Nucleic Acids Res., 2009, 37, 7728–7740 CrossRef CAS PubMed.
- F. Sochor, R. Silvers, D. Müller, C. Richter, B. Fürtig and H. Schwalbe, J. Biomol. NMR, 2016, 64, 63–74 CrossRef CAS PubMed.
- M. A. S. Abdelhamid and Z. A. E. Waller, Front. Chem., 2020, 8, 40 CrossRef CAS PubMed.
- X. Liu, W. Chi, Q. Qiao, S. V. Kokate, E. P. Cabrera, Z. Xu, X. Liu and Y. T. Chang, ACS Sens., 2020, 5, 731–739 CrossRef CAS PubMed.
- H. Mao, G. Luo, Y. Zhan, J. Zhang, S. Yao and Y. Yu, Analyst, 2018, 143, 3292–3301 RSC.
- T. Latychevskaia, C. Escher, W. Andregg, M. Andregg and H. W. Fink, Sci. Rep., 2019, 9, 1–8 CAS.
- S. M. A. Daraghma, S. Talebi and V. Periasamy, Eur. Phys. J. E: Soft Matter Biol. Phys., 2020, 43, 1–9 CrossRef PubMed.
- C. T. Martin, A. Újvári and C. Liu, Methods Enzymol., 2003, 371, 13–33 CAS.
- J. M. Jean, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 37–41 CrossRef CAS.
- J. N. Wilson, Y. Cho, S. Tan, A. Cuppoletti and E. T. Kool, ChemBioChem, 2008, 9, 279–285 CrossRef CAS PubMed.
- C. P. Lawson, A. F. Füchtbauer, M. S. Wranne, T. Giraud, T. Floyd, B. Dumat, N. K. Andersen, A. H. El-Sagheer, T. Brown, H. Gradén, L. M. Wilhelmsson and M. Grøtli, Sci. Rep., 2018, 8, 13970 CrossRef PubMed.
- A. Dierckx, F.-A. Miannay, N. Ben Gaied, S. Preus, M. Björck, T. Brown and L. M. Wilhelmsson, Chem. – Eur. J., 2012, 18, 5987–5997 CrossRef CAS PubMed.
- H. A. Day, C. Huguin and Z. A. E. Waller, Chem. Commun., 2013, 49, 7696–7698 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00020a |
| This journal is © The Royal Society of Chemistry 2021 |