
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal-based catalysts for the electrochemical CO2 reduction: from atoms and molecules to nanostructured materials
Federico
Franco
,
Clara
Rettenmaier
,
Hyo Sang
Jeon
 and
Beatriz
Roldan Cuenya
*
and
Beatriz
Roldan Cuenya
*
Department of Interface Science, Fritz-Haber Institute of the Max Planck Society, 14195 Berlin, Germany. E-mail: roldan@fhi-berlin.mpg.de
First published on 25th August 2020
Abstract
The electrochemical reduction of carbon dioxide (CO2) powered by renewable energy is an attractive sustainable approach to mitigate CO2 emissions and to produce fuels or value-added chemicals. In order to tackle the challenges related to selectivity, activity, overpotential and durability, transition metal-based catalysts have been widely investigated in the last decades. In an effort to bridge the gap between the fields of homogeneous and heterogeneous catalysis, this review aims to survey the main strategies explored for the rational design of a wide variety of different metal catalysts, ranging from molecular systems to single-atom and nanostructured catalysts. Transition metal complexes containing heme and non-heme ligands have been selected to discuss the recent advances in the understanding of the structure–function relationship in molecular homogeneous catalysis as well as to summarize the main approaches proposed for the heterogenization or confinement of molecular catalysts on conductive surfaces. The main strategies to minimize catalyst cost are also presented, leading to atomically dispersed molecular-like M–Nx moieties embedded on 2D conducting materials. The superior performances of single-atom catalysts (SACs) and the structural similarity with their molecular analogs, suggest that transition metal catalysts containing well-defined sites may be intrinsically more active and selective towards CO2 conversion than the bulk heterogeneous materials. Finally, design approaches for metal nanoparticles (NPs) based on size, shape, and support tuning are summarized and compared to novel strategies based on the interaction with surface-bonded organic molecules. The studies herein presented show that the basic principles in molecular catalysis and organometallic chemistry can be effectively used to design new efficient and selective heterogeneous catalysts for CO2 reduction.
 Federico Franco | Federico Franco earned a PhD in Chemistry and Material Sciences at the University of Turin in 2016 under the supervision of Prof. Roberto Gobetto. Until 2019, he worked as a postdoctoral researcher at the Institute of Chemical Research of Catalonia (ICIQ) in the group of Prof. Julio Lloret-Fillol, where he focused on the mechanistic understanding of molecular catalysts for the electro- and photochemical activation of small molecules. He is currently a postdoctoral researcher in the Department of Interface Science at the Fritz-Haber Institute of the Max Planck Society, where he is studying heterogeneous electrocatalysts for energy conversion processes. |
 Clara Rettenmaier | Clara Rettenmaier received her master's degree in physical chemistry from the Technical University of Munich (Germany) in 2018. She is now a PhD candidate in the Department of Interface Science at the Fritz-Haber Institute of the Max Planck Society, Germany. Her current research focuses on in situ and operando electrocatalytic studies of carbon dioxide reduction. |
 Hyo Sang Jeon | Hyo Sang Jeon received his PhD in Clean Energy and Chemical Engineering from the Korea University of Science and Technology (UST-KIST). He is now a postdoctoral researcher in the Department of Interface Science of the Fritz-Haber Institute of the Max Planck Society, Germany. His research mainly focuses on the understanding of structure-chemical state and reactivity correlations during the electrochemical reduction of carbon dioxide. |
 Beatriz Roldan Cuenya | Beatriz Roldan Cuenya is a Professor and Director of the Interface Science Department of the Fritz-Haber Institute of the Max-Planck Society in Berlin Germany since 2017. She received her PhD in solid state physics from the University of Duisburg-Essen (Germany) in 2001. She was a postdoctoral scholar at the Chemical Engineering Department of the University of California Santa Barbara (USA) from 2001–2003. She joined the University of Central Florida (USA) as an Assistant Professor of Physics in 2004, and became a full professor in 2012. From 2013–2017 she worked as Professor of Physics at the Ruhr University Bochum (Germany). |
1. Introduction
The dependence of anthropogenic activities on fossil fuels has contributed to a dramatic rise in the atmospheric CO2 levels leading to severe environmental issues in our modern society, including global warming and serious pollution problems.1 In this scenario, the implementation of technologies based on CO2 utilization to produce carbonaceous fuels and commodity chemicals is highly desirable to drive the transition towards a new green economy.2–4 Among the proposed approaches, the electrochemical conversion of CO2 powered by renewable sources is an attractive sustainable alternative to the massive utilization of fossil resources.5–8 It can occur at ambient conditions and the efficiency of the process can be potentially controlled by the applied bias. Furthermore, this strategy is compatible with an on-site production of a wide variety of organic C1- and C2-building blocks from a non-toxic, abundant and inexpensive greenhouse gas, mitigating the limitations related to their storage and distribution (especially for toxic gaseous products, like CO).9The electrocatalytic CO2 reduction reaction (CO2RR) may undergo several multi-electron multi-proton pathways, leading to a wide range of carbon-based products.10 Selectivity is therefore a major challenge in CO2RR, since competitive undesired CO2RR mechanisms and/or the hydrogen evolution reaction (HER) may interfere with the formation of a specific product. Due to the extreme inertness and stability of the CO2 molecule, both thermodynamic and kinetic barriers hinder an efficient electrochemical CO2 activation.11 In the absence of a catalyst, the direct outer-sphere single-electron reduction of CO2 to form the CO2˙− radical anion is energetically demanding (E0 = −1.90 V vs. NHE), mainly due to the high reorganizational energy required to bend the linear CO2 molecule.12 Proton-assisted multi-electron steps generally lead to thermodynamically more favorable pathways.13 A transition metal-based catalyst is often required to lower the activation barrier of the reaction and to drive the process at acceptable rates at specific potentials. The specific interaction of the catalytic metal center with H+/CO2 or CO2RR intermediates is crucial to determine the selectivity of the overall process.14 In addition, a proper kinetic control of the whole catalytic system is essential to achieve the desired selectivity, and factors such as the applied potential, the proton concentration in the reaction medium, and the solubility of CO2 (e.g. about 0.03 M in common aqueous electrolytes, 0.31 M in CH3CN and 0.19 M in DMF15) must be also carefully assessed. In the last decades, a plethora of transition metal-based catalysts have been reported for efficient CO2RR, ranging from molecular catalysts16–22 to metal nanoparticles23,24 and bulk heterogeneous materials.25–27 In parallel, the increasing interest for in situ and operando microscopic and spectroelectrochemical (SEC) techniques applied to both, the homogeneous and heterogeneous systems has led to a strong improvement in the fundamental understanding of the factors governing the CO2RR process.28–32 In spite of this, further improvement of the catalyst performance in terms of activity, selectivity and stability is highly desired to match the requirements for practical applications.
In this perspective, we aim to provide an overview of the major advances and trends in the broad field of the electrochemical CO2RR, covering the different explored areas from molecular catalysis to nanostructured materials. In addition to the conventional distinction between molecular and heterogeneous catalysts, here we will feature some innovative promising approaches to design novel hybrid catalysts for CO2RR with intermediate properties between molecules and bulk materials (Fig. 1).33–38
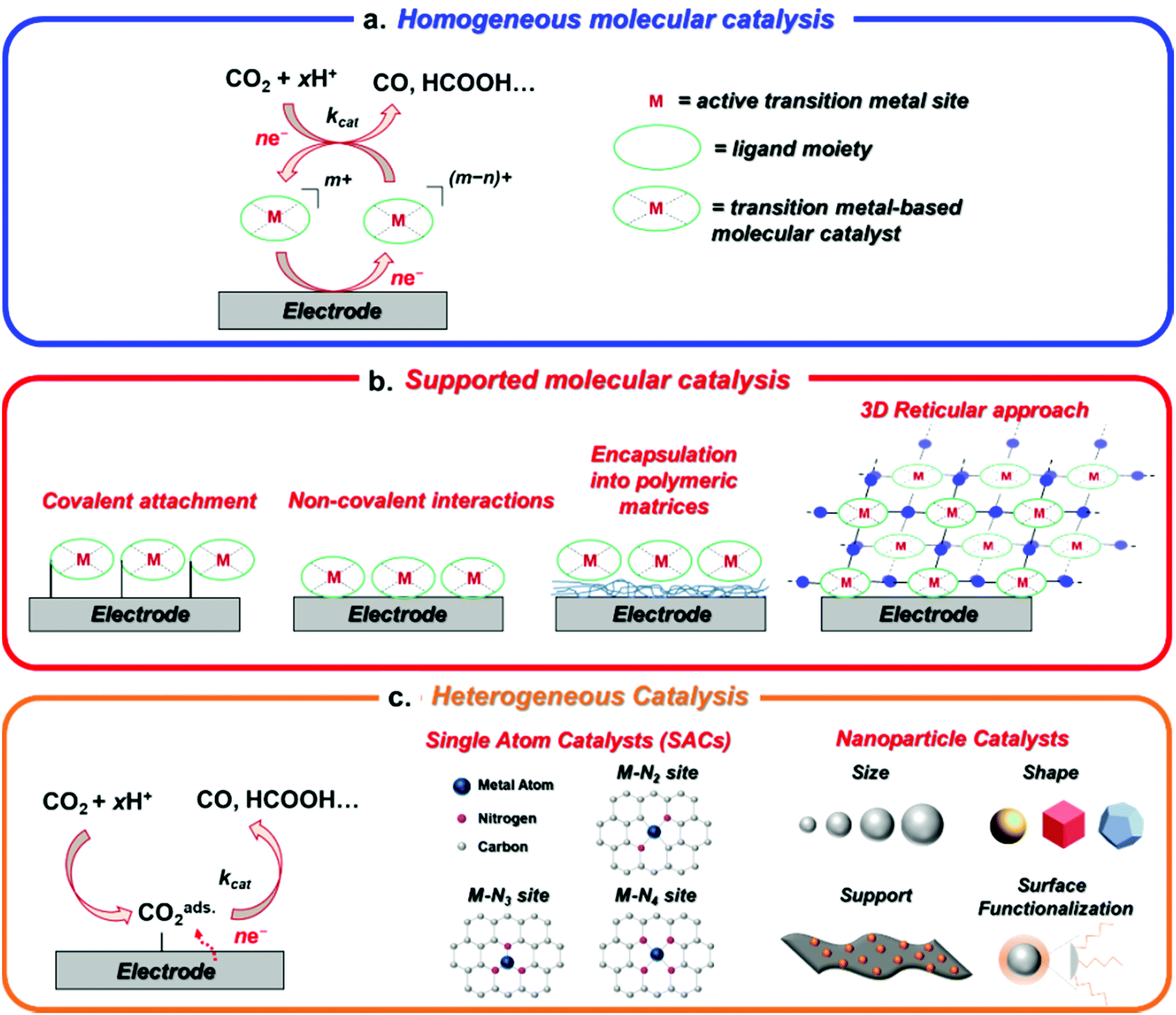 | ||
| Fig. 1 Different types of transition metal catalysts for CO2RR: (a) homogeneous molecular catalysis, (b) surface-anchored molecular catalysis by using several immobilization strategies and (c) heterogeneous catalysis. | ||
A rational catalyst design primarily requires the elucidation of the structure–activity relationship. The present work will focus on the main reported strategies to control the structure, morphology and chemical composition at the atomic, molecular, supramolecular and nano levels, highlighting how these aspects affect the reactivity towards CO2RR. Some aspects related to the modeling of the electrical interface in a broader sense, such as the effect of the local pH, electrolyte or ionic strength will be also briefly discussed.39–41 In the attempt to link molecular and heterogeneous systems, a recurrent topic will be the contribution from the fundamental principles of molecular catalysis and coordination chemistry to the design of new efficient materials with tunable activity and selectivity. To emphasize this concept, the CO2RR catalytic properties of molecular systems and materials containing active metal sites with structurally similar well-defined coordinative environments will be correlated to each other. Given the huge number of metal complexes reported over the last decades as efficient CO2RR catalysts, the discussion about molecular catalysis will be herein mainly limited to metal complexes containing heme (including metalloporphyrins and phthalocyanines) and non-heme (cyclam, cyclam-like and others) macrocyclic ligands as representative examples. There are several reasons supporting this choice. Firstly, they represent one of the most widely studied and relevant families of molecular catalysts reported up to date, being able to promote a highly efficient and selective CO2 reduction to CO under electrochemical conditions. They also showed an extreme versatility and long-term durability as both homogeneous and supported catalysts, which make them very appealing systems for scale-up applications. From a fundamental perspective, the mechanistic understanding of the catalytic CO2RR for these systems possesses a solid experimental and theoretical basis, providing a broad overview for the critical assessment of structural effects in molecular catalysis, as well as of the strategies for heterogenization of molecular catalysts on solid electrodes. Furthermore, the active M–N4 moiety of metallo-porphyrinoid derivatives or non-heme macrocyclic complexes represents an ideal model to describe the active sites of several reported single-atom electrocatalysts for CO2RR. In this regard, we have also included a brief discussion about some representative examples of Ni, Fe and Co molecular catalysts containing non-macrocyclic tetradentate nitrogen ligands, which help us to establish a more intimate connection between the molecular and heterogeneous systems. It is worth reminding that several remarkable classes of molecular catalysts based on 1st/2nd/3rd row transition metals and non-heme ligands (polypyridyl, aminopyridyl, organometallic ligands, etc.) have been widely studied for efficient CO2RR to HCOOH or CO and comprehensively reviewed in recent works.16–18,20,42–46 Nevertheless, this work aims to highlight the main concepts and findings from molecular catalysis that can be useful for the design of new efficient catalysts, rather than providing an exhaustive overview of all the molecular families reported in the literature for CO2RR.
After briefly defining the general structural features of the materials discussed in the review (Section 2), the main examples of molecular catalysts containing heme and non-heme macrocyclic ligands for electrocatalytic CO2 conversion (in homogeneous and surface-immobilized forms, reticular materials and supramolecular assemblies) will be presented in Sections 3 and 4, respectively. The latter address a critical discussion on the different approaches for the heterogenization of molecular catalysts to shed light on the similarities and differences with the homogeneous counterparts. The state-of-the-art of the emerging fields of single-atom catalysts for CO2RR will be examined in Section 5, whereas Section 6 will be focused on the recent developments regarding the use of nanostructured catalysts.
2. CO2RR catalysts based on transition metals
Following a traditional scheme, the transition metal-based catalysts for CO2RR are roughly divided into two main categories, corresponding to molecular and heterogeneous catalysts. In molecular catalysis, a well-defined active site is generally proposed, whereby a metal center is surrounded by a specific organic ligand framework (Fig. 1a and b). The coordination environment (first and second coordination sphere) of the catalytic site determines its electronic and steric properties, providing a specific catalyst–substrate interaction under given external conditions (solvent, pH, electrolyte). Thus, the control over the intrinsic activity and selectivity for CO2RR is achieved through ligand design by using conventional synthetic methods of organic/organometallic chemistry.47–50 An accurate investigation of the mechanistic details of the catalytic reaction is generally obtained by using electrochemical and spectroscopic tools, leading to a deep understanding of CO2RR mechanistic details. Most of the reported molecular catalysts for CO2RR are capable to promote the two-electron (2e) CO2 conversion to carbon monoxide (CO) or formic acid (HCOOH) with high efficiency and selectivity. On the other hand, clear examples of molecular systems able to produce more than 2e CO2RR reduction products are still rare.51–53 Furthermore, molecular systems generally suffer from low current densities, which limit their implementation in real devices for scale-up applications.In sharp contrast, bulk heterogeneous catalysts usually contain a large number of poorly characterized active surface states, without a full structural control at the molecular or atomic levels (Fig. 1c). These aspects typically result in considerably higher current densities in comparison with the molecular systems, at the expense of a lower selectivity and a more difficult rationalization of the process. Nevertheless, although the HER is generally favored over CO2RR on several metal electrodes (e.g. Ni, Fe, Ti, Pt) in aqueous media, a number of transition metals have been shown to produce CO (e.g. Au, Ag, Zn)54 or HCOOH (e.g. In, Sn, Cd) with high faradaic yields.55 The product distribution is generally determined by the relative binding energies of adsorbed *CO, *COOH and *H intermediates (* indicates the adsorption site on the metal surface), which are strongly dependent on the metal's electronic structure (i.e. density of states).10,56–58 Weak *CO binding on the metal surface facilitates CO desorption and evolution (Au), whereas too large *CO adsorption energies are detrimental for CO2RR, leading to catalyst poisoning and a preference for HER (Pt). For intermediate *CO binding energies (e.g. Cu), *CO is not easily desorbed and undergoes further reductive and/or alternative chemical steps (protonation, coupling reactions) enabling the formation of the target >2e CO2RR products, such as hydrocarbons or alcohols.14,59–65
In the last years, the rigid dualism between molecules and heterogeneous catalysts has been gradually overcome through the investigation of novel families of materials with specific molecularly or atomically defined properties. As such, emerging classes of hybrid heterogeneous catalysts showed superior CO2RR performances than the molecular counterparts albeit preserving the integrity of the molecular catalytic site (Fig. 1). The main families of transition metal-based catalysts investigated by the current research in the CO2RR field can be summarized as follows:
• Homogeneous molecular catalysts, which are typically organometallic complexes able to promote electrochemical CO2RR when dissolved in common organic electrolytes (CH3CN, THF, DMF, NMP). In this case, the catalyst is a molecule freely-diffusing in solution with a well-defined standard potential and acts as a redox mediator between the electrode and the substrate (Fig. 1a). In most instances, the real active species is electrogenerated in situ: when the applied bias matches the redox potential of the catalyst, the catalyst precursor gets reduced at the electrode surface forming the catalytic species which, in turn, reacts with the substrate with kinetics (kcat) depending on its intrinsic activity and the external conditions (e.g. applied potential, medium). In homogeneous electrocatalysis, only a small fraction (confined in the diffusion layer) of the total amount of catalyst present in solution (≈mM concentration) is directly involved in the catalytic process. The catalytic behavior can be accurately studied by electrochemical (cyclic voltammetry) and complementary in situ spectro-electrochemical methods (UV-Vis, FTIR, EPR, Raman) to identify the main intermediate species involved in the process. In some cases, the reaction intermediates can be even chemically synthesized and isolated. Homogeneous catalysts are ideal candidates to investigate the catalytic CO2RR mechanism at a fundamental level, but deactivation pathways occurring in solution or on the electrode surface affect their long-term stability.
• Heterogenized molecular catalysts, whereby the catalytic species no longer diffuses in solution but is immobilized on the electrode, thus improving the electrical contact between the electrode and the catalyst at the interface (Fig. 1b).45,66,67 Although a number of water-soluble molecular catalysts for CO2RR have been reported so far,68–71 the electrode functionalization approach allows to overcome the solubility limitations in aqueous electrolytes often reported for homogeneous catalysts (most of them are soluble in organic solvents or organic–water mixtures), and helps to facilitate the catalyst recycling and the separation of liquid CO2RR products. Moreover, the physicochemical interactions at the catalyst–electrode interface may induce drastic reactivity changes, including altering the conventional reaction pathways occurring in the homogeneous phase. In this perspective, the fundamental understanding of these factors would represent a powerful tool to tune the reactivity, activity and selectivity of molecular catalysts. The heterogenized electrocatalysts consist of thin films containing a number of active sites (10−7–10−12 mol cm−2) which is highly dependent on the catalyst loading and the deposition method. The strategies used for molecular catalyst immobilization can be divided into three main classes: (1) electropolymerization, which leads to the formation of electroactive polymer films with high surface densities and porosity.72 Depending on the number of voltammetric cycles, a precise control over the thickness and surface coverage may be obtained; (2) covalent attachment to the surface through the presence of different functional groups (amino, diazonium, thiol, carboxylic acids, etc.), which typically entails the formation of robust films with low surface coverage (≈10−10–10−12 mol cm−2 for a monolayer);73,74 (3) immobilization via non-covalent (π–π stacking) interactions, which generally provides higher catalyst densities (≈10−8 mol cm−2) but also a less controlled surface functionalization.75 In analogy with the homogeneous case, surface spectroscopic techniques are used to gain mechanistic insight into the catalytic process. Low catalyst loading, partial catalyst detachment during operation, poor electrochemical contact and the need for elaborated synthetic strategies are the main limitations of classical functionalization methods.
• Reticular 3D materials, in which the active molecular unit is encapsulated into porous organic or metalorganic ordered architectures with tunable structure and properties. Among them, covalent organic frameworks (COFs), metal organic frameworks (MOFs) or porous supramolecular assemblies are versatile platforms used to significantly improve the robustness of a catalyst under electrochemical conditions. In principle, the combination of the characteristic physical properties of MOF and COF materials (e.g. high surface area, adjustable pore size) with suitable integrated molecular building blocks enables to fine-tune their activity and selectivity for CO2RR.33 However, an accurate control over the thickness of the layers grown on the electrode is essential to optimize the electron and mass transport properties of the material and, therefore, to maximize the efficiency of the process.76 Furthermore, the catalytic performance of these materials is typically limited by an inefficient charge carrier mobility, which leads to slow electron kinetics. In this regard, imine-based COFs have shown enhanced electronic properties, improving the electrical connection between the active sites of the layered material.77–79 Moreover, in comparison with conventional heterogenization methods, the imine (or amine) groups contained in the microporous matrix, together with its specific porous morphology favor the capture of CO2, leading to an increase in the local CO2 concentration at the catalytic sites (especially in aqueous media).80–82 Homogeneous, heterogenized and reticular systems can be defined as molecular materials, since the metalorganic active site preserves its fundamental structural and electronic properties at a molecular level even after the heterogenization process.83
• Single-atom catalysts (SACs), a rapidly emerging unique class of materials bridging the molecular and heterogeneous catalysis fields. In SACs, transition metal atoms are atomically dispersed on a 2D support and stabilized through the coordination to heteroatom (usually N, C, P or S) dopants. Indeed, the active sites are homogeneously distributed on a conductive surface, mimicking the coordination environment of molecular complexes. This feature enables an accurate structural characterization of the catalyst at an atomic scale and, at the same time, maximizes the number of free coordination sites per metal atom. Unlike the molecular materials, the SACs are generally obtained by thermal treatment of generic metal precursors (inorganic salts) and heteroatom-rich organic molecules which, depending on the synthetic conditions, give rise to isolated molecular-like surface units. A challenge from this approach is however to demonstrate that in fact only SAC motifs are available on the as prepared samples (as opposed to a mixture of single atoms and small clusters or nanoparticles) as well as the difficulty of keeping such SAC motifs stable under reaction conditions. Doped carbon-based materials are generally used as suitable supports for SACs, due to their high specific surface area and tunable surface properties.
• Nanostructured materials, which comprise metal clusters (MCs) and nanoparticles (NPs), differing from each other by the number of atoms. These materials are extremely attractive for CO2RR, due to their high surface-to-volume-ratios and the presence of several low-coordinated reactive surface sites. Their properties are strongly dependent on their size (from approximately 1–5 to hundreds of nanometers), aggregation level, shape and composition (mono- or multimetallic). The nanostructured materials represent the last frontier between the molecular materials and the extended (mainly metallic) surfaces, which include single-crystal and polycrystalline materials.
It is worth mentioning that the classification reported above is meant to exemplify the rationalization of the enormous number of CO2RR catalysts reported in the literature, in an effort to give a general perspective of the multi-faceted CO2RR field. Some interesting limiting cases that lay out of this classification, including the in situ decomposition of homogeneous catalysts under electrochemical conditions to form active nanostructured deposits on the electrode, are beyond the scope of the present review.84 Nonetheless, the case of Ni-cyclam catalyst will be discussed as an example of in situ heterogenization of molecular systems, highlighting the critical role that the specific interactions between the adsorbed molecular reduced species and the electrode surface may play in catalysis. Furthermore, a discussion on CO2RR promoted by organic or metal-free catalysts will not be included in this review.
3. Molecular catalysts for CO2RR containing heme ligands
As anticipated above, here we discuss the main examples of molecular materials for CO2RR reported in the literature based on metal complexes with heme ligands, including porphyrin and phthalocyanine derivatives. The first paragraphs describe the most relevant systems based on Fe and Co, highlighting the main strategies for catalyst optimization in both homogeneous and heterogenized approaches. The last paragraph will instead summarize the main findings reported for analogous systems containing other transition metals.3.1 Iron
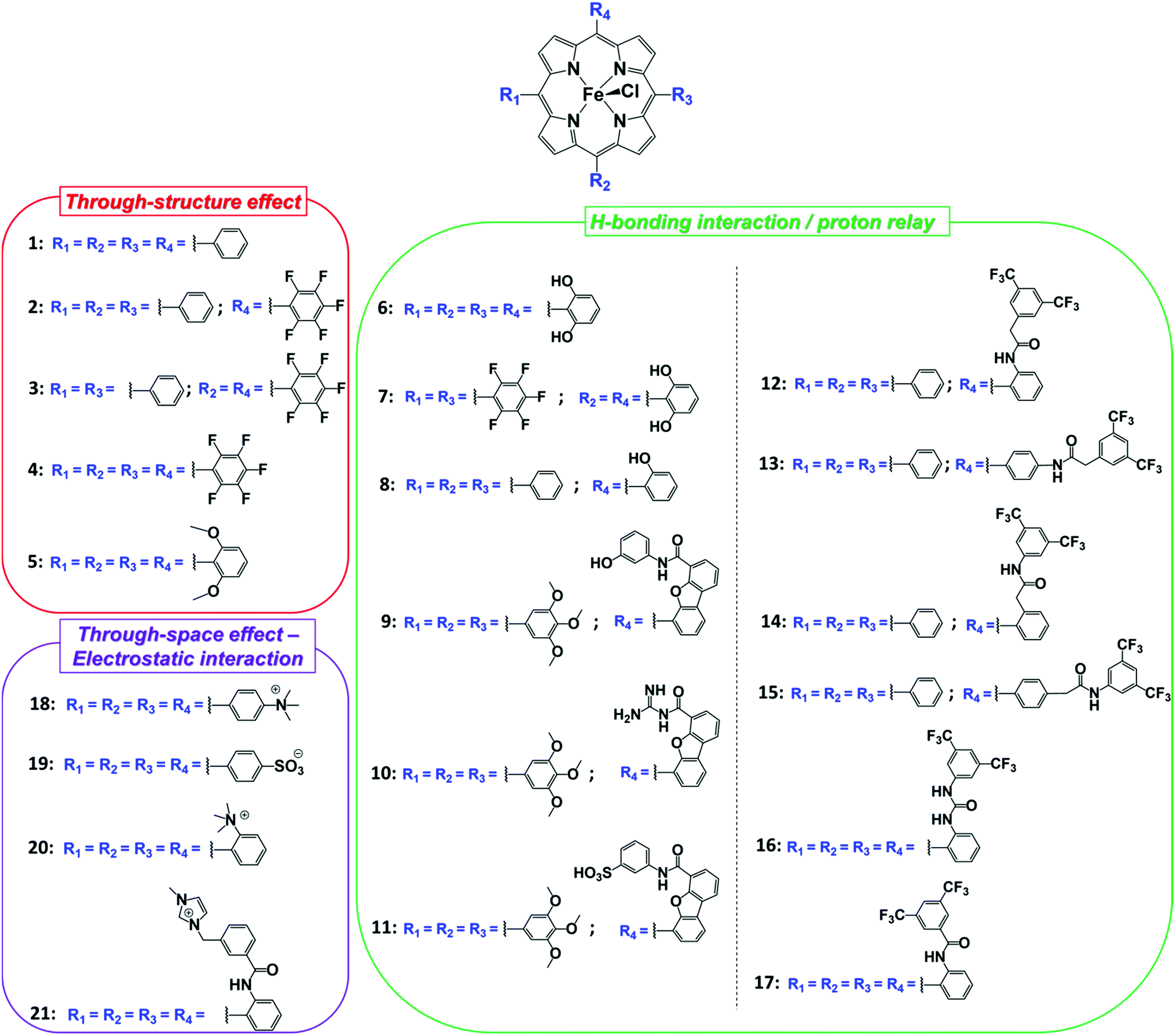 | ||
| Chart 1 Synthetic molecular Fe porphyrin systems reported as homogeneous catalysts for CO2RR. | ||
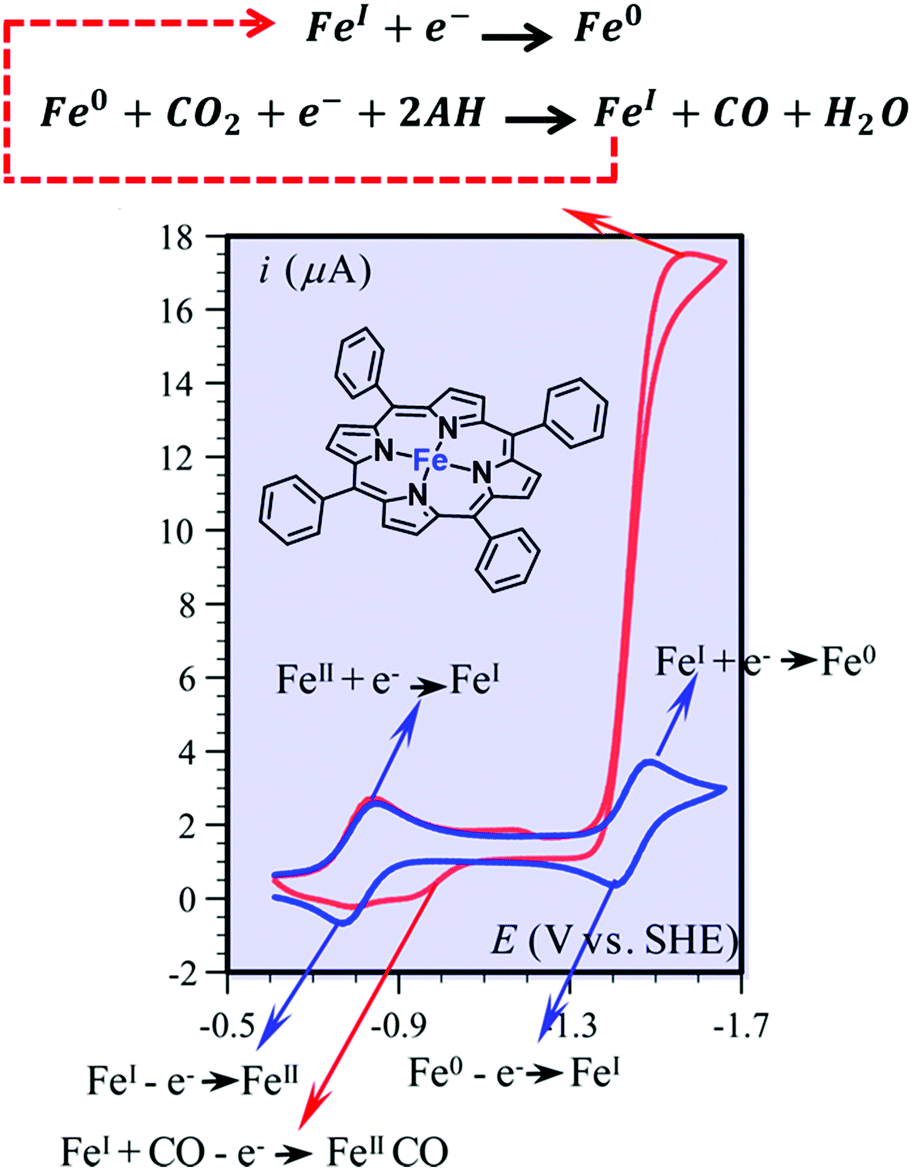 | ||
| Fig. 2 Cyclic voltammetry of FeTPP (1) (1 mM) in DMF + 0.1 M n-Bu4NPF6, in the absence (blue) and presence of 0.23 M CO2 and of 10 mM PhOH (red). Sketch of the electrochemical reactions. Adapted with permission from ref. 86. Copyright 2013 American Chemical Society. | ||
The electrochemical catalytic CO2-to-CO conversion mediated by FeTPP in the presence of Lewis or Brønsted acids has been proposed to occur via a two-electron “push–pull mechanism”.94,96 In the first step, the electrogenerated active [Fe(TPP)]2− species interacts with CO2 to form a key [Fe(CO2)(TPP)]2− adduct, in which the electron density is “pushed” from the nucleophilic metal center to the electrophilic CO2 molecule. The asymmetric [FeI(CO2˙−)]2− resonance form has been proposed to be the predominant structure, in agreement with DFT calculations (Scheme 1).86,87,98 Through an ion-pair (Lewis) or H-bonding (Brønsted) formation, the acid stabilizes the intermediate and helps to “pull” the electron density out of the substrate, thereby facilitating the C–O cleavage step. Notably, an additional stabilization of the key intermediates due to an intramolecular H-bonding allowed the experimental detection of a [FeIICO2]2− adduct and its protonated [FeIICO2H]− derivative at cryogenic temperatures using vibrational spectroscopy.99 This scheme is also consistent with the reported catalytic behavior of a dimeric Fe porphyrin system, whereby the catalytic response of the catalytic Fe site is enhanced by the intramolecular assistance of the second Fe porphyrin unit.100
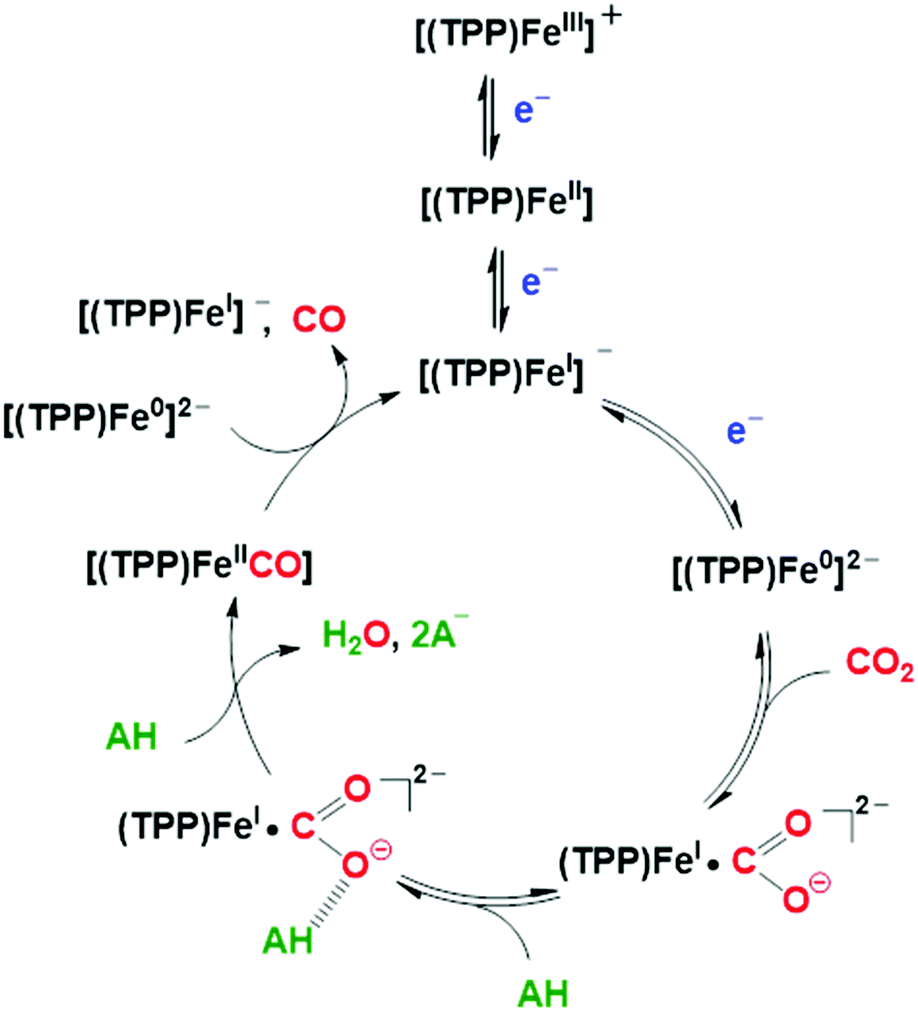 | ||
| Scheme 1 Proposed catalytic mechanism for the electrochemical CO2-to-CO mediated by [Fe(TPP)]Cl (1) in organic media in the presence of Brønsted acids.86,102 | ||
The role of added Brønsted acids in the proton-assisted mechanism of FeTPP was systematically investigated in detailed kinetic studies, based on the “foot-of-the-wave” analysis (FOWA) of the voltammetric profiles.85,86,101 In the initial stage of the process, an acid molecule (AH) undergoes an H-bonding stabilization of the [Fe(CO2)(TPP)]2− primary intermediate ([FeI(CO2˙−)]2− predominant resonance form, vide supra), followed by a successive dehydration step involving another AH molecule which leads to the cleavage of the C–O bond (Scheme 1).86,102 At moderate acid concentrations, a proton-coupled intramolecular electron transfer from the iron center to CO2, concerted with the breaking of the C–O bond, was unambiguously determined to be the rate-determining step of the catalytic process.86,98,102,103 The subsequent [FeII(CO)(TPP)] intermediate undergoes fast CO release, closing the catalytic cycle upon homogeneous single electron transfer by another formal Fe0 species (Scheme 1). The extreme robustness, efficiency and selectivity of 1 towards CO formation for a wide range of concentrations of various mild proton donors is appealing for several applications. Recently, Skrydstrup and collaborators have developed a low-cost and scalable setup to use the CO generated by the FeTPP-mediated CO2 electroreduction in homogeneous phase as reactant in Pd-catalyzed carbonylation reactions for the synthesis of pharmaceutically relevant molecules.104,105
The acidity of the reaction medium primarily affects the overpotential and selectivity of FeTPP (1) (Chart 1). When strong acids are employed (e.g. Et4NH+, CH3COOH), metal protonation occurs and the competitive HER becomes the main pathway. On the other hand, weak proton donors, like 1-propanol (PrOH), generally induce sluggish kinetics for CO2RR with non-selective formation of CO (FE ≈ 60%) and formate (FE ≈ 35%) mixtures.96 A recent report suggests that the addition of tertiary amines is an effective strategy to drive the selectivity of 1 towards HCOOH formation in the presence of weak acids.106 The strong trans-coordination of tertiary amines to the Fe center increases the basicity of the central C-atom of the substrate in the [Fe(CO2)(TPP)]2− adduct, thereby facilitating its protonation with the subsequent release of formate. Following this approach, FEs as high as 68% or 72% for HCOOH were obtained by adding 40 mM of quinuclidine or trimethylamine to a 40 mM PrOH solution of 1, respectively.106 In contrast with the conventional pathway for HCOOH formation based on net CO2 insertion into a reactive M–H bond (a common intermediate to HER),107,108 this alternative strategy uses weak acids to produce HCOOH, circumventing the formation of undesired hydride species.
On the basis of the accurate elucidation of the CO2RR catalytic mechanism of the parent system and the extreme versatility of the porphyrin moiety, in the last decade numerous efforts have been directed to the rational design of novel iron porphyrin-based catalysts. In particular, a number of studies have focused on the correlation between the electronic and steric effects of different substituents on the porphyrin ring and the CO2RR activity of the catalyst (Chart 1). The substituent effects can be divided into two categories, defined as through-structure and through-space effects, respectively (Fig. 3 and 4).47,109 The former term mainly refers to the use of mesomeric or inductive effects promoted by electron-withdrawing or electron-donating substituents to modify the electronic structure of the catalyst.110,111 To investigate the influence of the through-bond inductive effect on CO2RR, the electrochemical behavior of the unsubstituted FeTPP compound (1) was systematically compared with a series of Fe tetraphenylporphyrins, containing different degrees of substitution with perfluoro (2–4) and o,o′-methoxy meso aryl groups (5) (see Chart 1 and Fig. 3).110,112 The correlation between the catalytic rate (defined as maximum turnover frequency or TOFmax) and the standard redox potential of a catalyst (E0cat) enables a rational evaluation of the electronic substitution effect on the catalytic performance within the same family of molecular catalysts.113–117 The analysis of the inductive effects in the series 1–4 gives rise to a linear scaling relationship, which highlights two opposite trends (Fig. 3a, right): for electron-withdrawing groups, the positive impact on the E0cat is partially balanced by lower catalytic rates, as a consequence of a diminished nucleophilicity of the Fe0 center. In turn, electron-donating substituents tend to enhance the reactivity of the catalytic center at the expense of a less favorable catalytic potential. In other words, considerations solely focused on electronic effects (i.e. the so-called “redox innocence” and “non-innocence” of the ligand frameworks89) leave little room for the optimization of the catalyst.
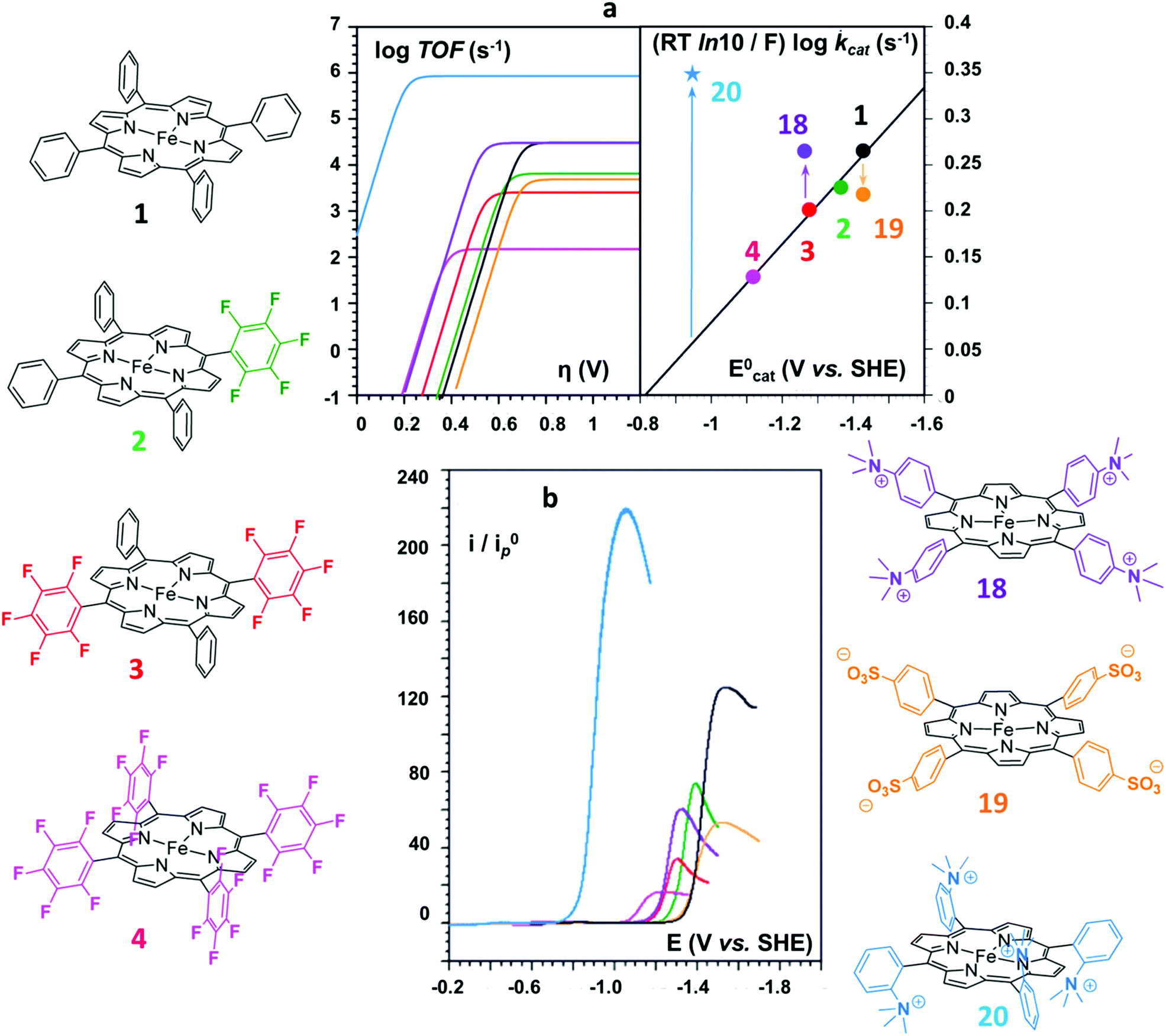 | ||
| Fig. 3 Structure substituent effects and Coulombic interaction effects of positively and negatively charged substituents on electrocatalytic CO2 conversion to CO by the series of homogeneous Fe porphyrin catalysts 1–4 and 18–20: (a) catalytic Tafel plots (left) and correlation between TOFmax = kcat and E0cat (right); (b) cyclic voltammetry of 20 (light blue), 4 (magenta), 3 (red), 18 (purple), 2 (green), 1 (black), and 19 (orange) in the potential domain of the catalytic CO2 reduction wave in DMF + 0.1 M n-Bu4NPF6 + 0.1 M H2O + 3 M PhOH, at 0.1 V s−1 under 1 atm CO2 (catalyst conc.: 1 mM). The current, i, is normalized against the peak current of the one-electron FeII/FeI reversible wave, i0p, obtained at the same scan rate (0.1 V s−1). Adapted with permission from ref. 112. Copyright 2016 American Chemical Society. | ||
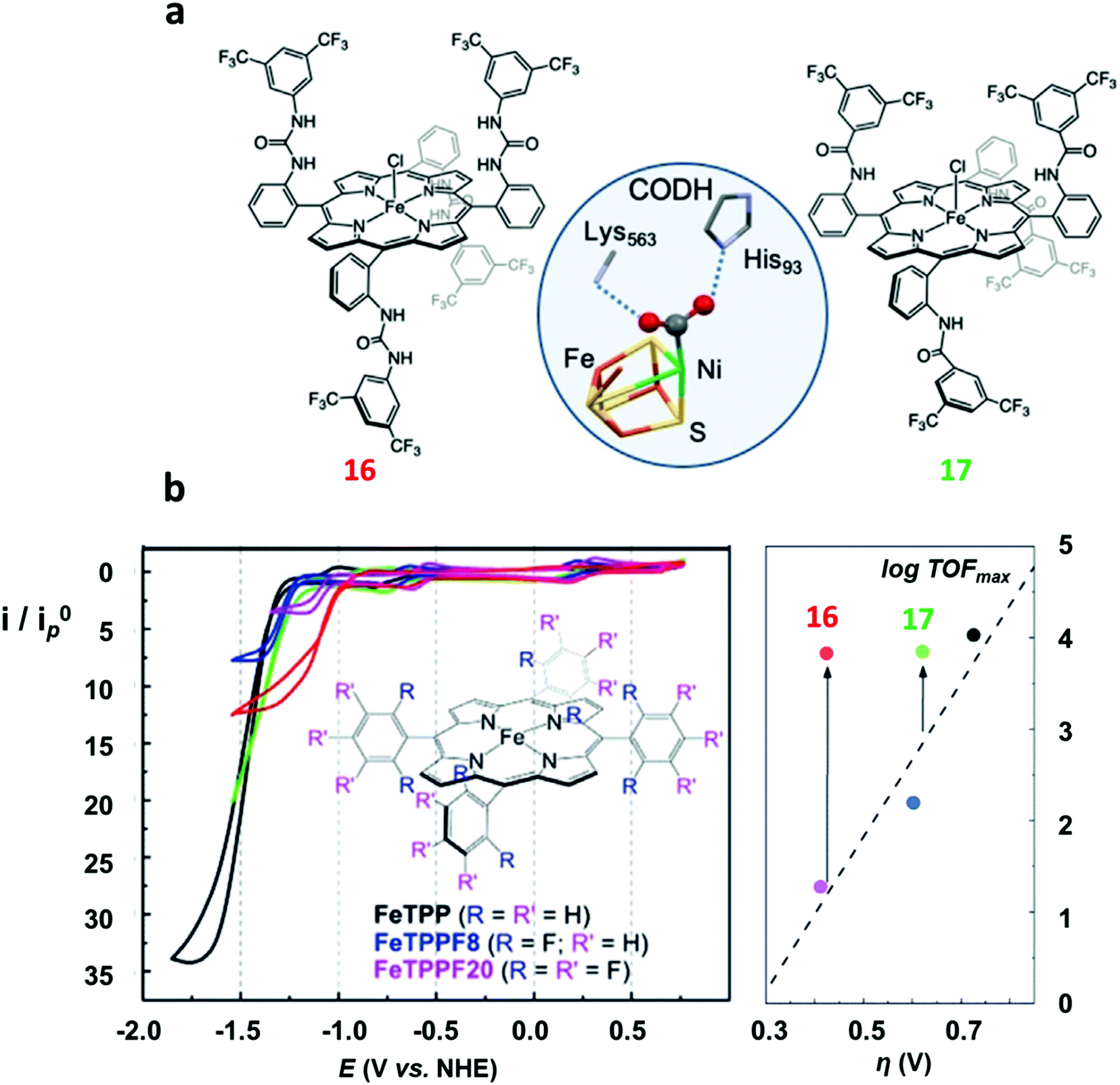 | ||
| Fig. 4 (a) Molecular drawing of 16 (left) and 17 (right). The X-ray structure of the FeNi active site (C-cluster) of CO dehydrogenase (CODH)134 (center) is shown for comparison. (b) Cyclic voltammograms of 1 mM FeTPP (1, black) and its modified analogues (blue = FeTPPF8, pink = FeTPPF20 (4), green = 17, red = 16) in DMF containing 0.1 M n-Bu4NPF6 at 25 °C under argon (top left) and under CO2 with 5.5 M water as proton source (bottom left). Plot of calculated TOFmax (from FOW analysis) as a function of the catalytic overpotential (right). Adapted with permission from ref. 126, Copyright 2019, Wiley-VCH. | ||
The modeling of through-space interactions between specific residues in the second or outer coordination sphere and the metal center may activate alternative mechanistic pathways for a catalytic reaction, giving rise to new scaling relations. This strategy represents a powerful tool for catalyst optimization and has been effectively used in molecular catalysis to circumvent the tradeoff between rate and overpotential forced by the scaling relationships observed with substituents displaying bare inductive effects.112,118,119 A first common approach consists in the use of local proton sources or H-bond donors on the porphyrin moiety. Depending on the specific spatial orientation of the pendant groups, they may induce a strong boosting effect on the CO2 reduction catalysis, due to a stabilization of the Fe–CO2 adduct by intramolecular H-bonding and/or an increase of the local concentration of proton donors. For instance, the introduction of eight phenolic functionalities in ortho, ortho′ positions of the porphyrin phenyls (6) led to a dramatic enhancement of the catalytic properties of 1, resulting in a durable and selective CO production (FECO = 94%, FEH2 = 6%) at −1.16 V vs. NHE (η = 0.466 V) in DMF/0.1 M NBu4PF6 + 2 M H2O.120 It clearly outperformed the corresponding methoxy derivative (5), thus unambiguously confirming the crucial role played by the pre-positioned OH groups in 6 on CO2RR. The latter act both as intramolecular proton relays, facilitating the successive protonation and C–O bond cleavage steps, and as H-bonding stabilizers of the primary Fe–CO2 intermediate formed between the electrogenerated formal Fe0 and CO2. As a consequence of the additional H-bonding stabilization induced by the pendant hydroxyl groups, catalysis requires a second electron uptake after the Fe–CO2 adduct formation, which is more difficult than the first one, unlike for the unsubstituted FeTPP catalyst (1, Scheme 1).87,98,102 In the case of the catalyst 6, the second electron transfer, required to close the catalytic cycle, was suggested to be concerted with the breaking of one of the two C–O bonds of CO2 and with proton transfer.87 A combination of the inductive and spatial effects may lead to catalysts with superior activity. As an example, the perfluorination of two opposite phenyl rings of 6 gave rise to the catalyst 7 with improved CO2RR performances.121 The choice for a suitable reaction medium is another crucial aspect to consider for catalyst optimization.122,123 An asymmetric tetraphenylporphyrin iron catalyst 8 containing only a single proton relay revealed to be a poor CO2RR catalyst in DMF, whereas a strong enhancement in the catalytic response was observed in CH3CN.122
In an attempt to mimic the key stabilizing role of amino acid residues by H-bonding interaction in enzymatic systems, a number of varying H-bond donor groups have been incorporated in the structure of iron porphyrin complexes.99,124–127 The effect of different hanging proton donors (phenol (9), guanidine (10) and sulfonate (11) groups) at a dibenzofuran scaffold has been explored in a series of iron hangman porphyrins.124 Although an excellent selectivity to CO was obtained in all three cases (FEs > 93%), the CO2RR catalytic rate was found to follow the 9 > 10 > 11 order, highlighting the detrimental effect of the deprotonated pendant sulfonate group on catalysis due to unfavorable electrostatic interactions. While the hanging guanidine group in 10 could potentially mimic the role played by arginine in several CO2 binding proteins,128 it was found to interact via H-bonding more favorably with the porphyrin platform than with CO2, resulting in a minor activity of 10 in comparison with 9. Recently, Chang and co-workers have systematically investigated a series of four positional isomers (12–15) with amide functionalities in the ortho- or para-positions of the meso phenyl ring, exploring the effect of both proximal and distal N–H configurations with respect to the porphyrin platform.125 CO2 reduction analysis revealed that the presence of the amide group in the ortho position is required to observe a significant increase in CO2RR activity, with the distal isomer (14) being the most active across the series. This resulted from a more favorable spatial location of the amide group which leads to an enhanced H-bonding stabilization of the Fe–CO2 intermediate. The presence of local urea functional groups, able to establish multipoint H-bonding with the metal-bound CO2 molecule, was reported to have an even stronger effect on catalysis in comparison with the amide groups.126 In particular, due to the cooperative effect of four urea groups, the biomimetic complex 16 exhibited a considerably higher intrinsic CO2RR activity than the amide-containing derivative 17, resulting in a comparable TOF value as to 1 at ca. 300 mV more positive overpotential (Fig. 4).
As an alternative strategy to a directional intramolecular H-bonding interaction, the stabilization of negatively charged iron–CO2 bound intermediates may also be obtained via through-space Coulombic interaction with positively charged pendant groups spatially oriented in the proximity of the active site.47 This effect was first demonstrated by the enhanced CO2RR activity of complex 18, in which the presence of four trimethylanilinium groups in a para position of the TPP phenyl rings resulted in a marked upper left deviation from the linear through-structure effect relationship between catalytic rate and catalyst redox potential (Fig. 3a, right).112 A comparable effect but opposite in sign was observed by replacing the trimethylanilinium substituents with negatively charged sulfonate groups (19), owing to unfavorable electrostatic interactions. Furthermore, the derivative bearing the quaternary ammonium groups in ortho positions (compound 20) exhibited a significant decrease of the catalytic overpotential while dramatically increasing the TOF (Fig. 3). This effect also leads to a parallel improvement of the molecular catalytic Tafel plot, which correlates the catalyst efficiency (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) TOF) and the overpotential (defined as the difference between the applied potential and the standard potential of the reaction to be catalyzed, in general A → B, η = E0AB − E) (Fig. 3a, left). Despite the fact that there is some debate on the value of the standard potential for the CO2/CO couple in organic solvents,49,129,130 complex 20 represents the most efficient homogeneous molecular catalyst for selective CO2-to-CO electroreduction reported to date, displaying an unprecedented maximum TOF value of ≈106 s−1 at a 220 mV overpotential. Remarkably, the selectivity to CO is nearly quantitative even in the presence of high concentration of phenol (3 M). Moreover, 20 showed an excellent durability, without any selectivity loss after 84 h electrolysis under CO2 on different working electrodes.112 Taking advantage of the ionic character of the porphyrin, the trimethylammonium derivative 18 was also employed to drive homogeneous CO2 conversion to CO in an aqueous electrolyte.68,98 At a close to neutral pH (6.7), CO was electrocatalytically produced with a FE of 90%, with only a minor amount of H2 (7%) at −0.97 V vs. NHE.68 Analogously to trimethylammonium cations, pendant methylimidazolium moieties in the complex 21 displayed a positive effect on CO2RR catalysis in a DMF/H2O mixture due to the electrostatic stabilization of the reaction intermediate.131 In a homogeneous 0.1 M KCl aqueous solution, 21 showed an excellent selectivity to CO (91%) at low overpotential (−0.948 V vs. NHE, η = 418 mV).
TOF) and the overpotential (defined as the difference between the applied potential and the standard potential of the reaction to be catalyzed, in general A → B, η = E0AB − E) (Fig. 3a, left). Despite the fact that there is some debate on the value of the standard potential for the CO2/CO couple in organic solvents,49,129,130 complex 20 represents the most efficient homogeneous molecular catalyst for selective CO2-to-CO electroreduction reported to date, displaying an unprecedented maximum TOF value of ≈106 s−1 at a 220 mV overpotential. Remarkably, the selectivity to CO is nearly quantitative even in the presence of high concentration of phenol (3 M). Moreover, 20 showed an excellent durability, without any selectivity loss after 84 h electrolysis under CO2 on different working electrodes.112 Taking advantage of the ionic character of the porphyrin, the trimethylammonium derivative 18 was also employed to drive homogeneous CO2 conversion to CO in an aqueous electrolyte.68,98 At a close to neutral pH (6.7), CO was electrocatalytically produced with a FE of 90%, with only a minor amount of H2 (7%) at −0.97 V vs. NHE.68 Analogously to trimethylammonium cations, pendant methylimidazolium moieties in the complex 21 displayed a positive effect on CO2RR catalysis in a DMF/H2O mixture due to the electrostatic stabilization of the reaction intermediate.131 In a homogeneous 0.1 M KCl aqueous solution, 21 showed an excellent selectivity to CO (91%) at low overpotential (−0.948 V vs. NHE, η = 418 mV).
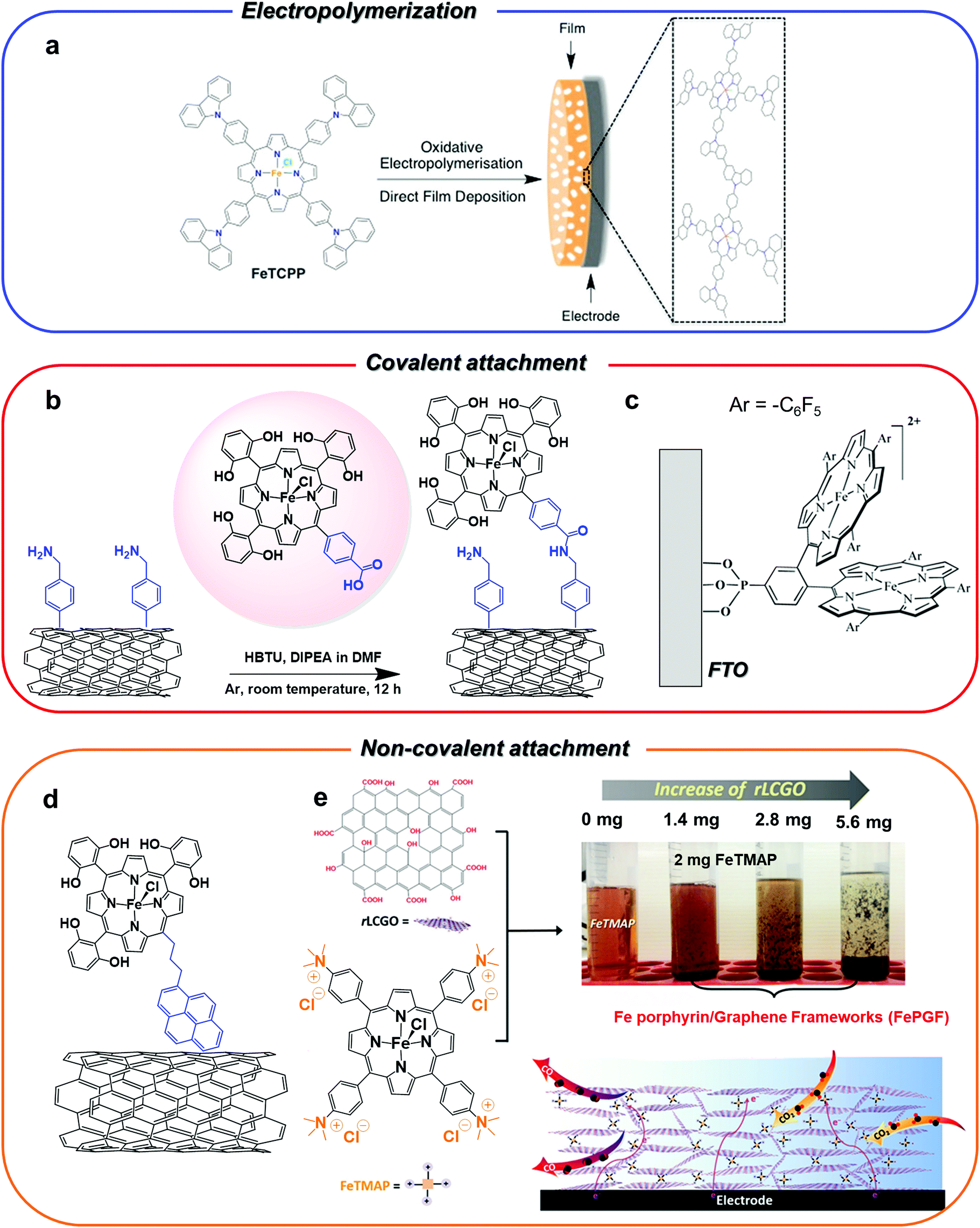 | ||
| Fig. 5 Different strategies for heterogenization of Fe porphyrin CO2RR catalysts on carbon-based electrodes. The panels show pictorial representations of the catalysts reported in the ref. 72 (a), 132 (b), 136 (c), 133 (d) and 138 (e), respectively. Figure a is adapted with permission from ref. 72. Copyright 2016, Royal Society of Chemistry. Figure b is adapted with permission from ref. 132. Copyright 2016, Royal Society of Chemistry. Figure c is adapted with permission from ref. 136. Copyright 2017 American Chemical Society. The figure e is adapted with permission from ref. 138. Copyright 2018, Wiley-VCH. | ||
Analogous results were obtained for a similar system, whereby the pyrene-terminal linker on the porphyrin moiety was replaced by a phenyl ring with a carboxylic acid at the para position (Fig. 5b). The coupling between the –COOH group of the catalyst and the surface –NH2 groups of the MWCNT electrode led to a covalent attachment of the catalyst through the formation of an amide linkage.132 At a slightly more negative applied potential (−1.06 V vs. SHE, η = 510 mV), the functionalized electrode showed an excellent selectivity for CO production (80–90%) over 3 hours electrolysis. Remarkably, a drastic drop of the activity and selectivity to CO (FE going from 77% to 51% over 3 h) was observed for the immobilized unsubstituted iron porphyrin derivative without pendant OH groups.132 A strong chemical binding of molecular catalysts to metal oxide surfaces may be also obtained.135 For instance, a co-facial Fe porphyrin dimer was successfully immobilized as a monolayer (≈10−12 mol cm−2) on a fluorine-doped tin oxide (FTO) surface via a phosphonic acid anchoring group (Fig. 5c).136 The FTO/Fe porphyrin assembly exhibited good electrocatalytic activity and stability for CO2-to-CO conversion in both non-aqueous and aqueous (pH 7.0) solutions. The immobilization of the catalyst on a thin layer of SnO2 or TiO2 NPs on FTO increased the catalyst loading (≈10−9–10−10 mol cm−2), whereas the co-modification of the catalyst-free FTO surface with hydrophobic n-butyl phosphonic acid groups allowed to suppress the non-innocent behavior of the bare FTO electrode.
In some cases, the interaction of the porphyrin unit itself with the electrode surface was found to be strong enough to guarantee a stable functionalization without the need for a chemical modification of the ligand structure. For instance, the positively charged trimethylammonium molecular derivative 18, was effectively anchored to a carbon support by simply drop-casting a suspension of the catalyst with the Nafion binder and carbon powder.137 The modified electrode was integrated into a home-made electrolyzer for CO2/H2O splitting into CO/O2 and was employed as a cathode for selective CO production (FE 90%) at neutral pH. The device showed good performances, providing an overall 50% energy efficiency and current densities of about 1 mA cm−2 over 30 h electrolysis at a 2.5 V cell voltage.137 In a recent report, a highly porous 3D hierarchical composite (FePGF) was fabricated by mixing complex 18 with reduced liquid crystalline graphene oxide (rLCGO), as a result of the agglomeration due to the π–π stacking and electrostatic interactions between the positive charges on 18 and the negative charges of rLCGO (Fig. 5e).138 The FePGF electrode (obtained by drop-casting a suspension of FePGF on a carbon support) showed an enhanced CO production and stability in comparison with the homogeneous derivative 18. Moreover, the catalyst–graphene interaction contributed to improve the electron delocalization and facilitate the electron transfer, inducing a ca. 100 mV positive shift in the catalytic onset potential. At neutral pH and 430 mV overpotential (−0.54 V vs. RHE), the FePGF electrode sustained a highly selective production of CO (FE 99%, TOF = 2.9 s−1, TON = 104400) over 10 h electrolysis, with negligible formation of H2.138 By a slight modification of this system, a simple and facile self-assembly hydrothermal method was developed to prepare a 18–graphene hydrogel (FePGH) deposited on a reticulated vitreous carbon (RVC) support.139 The FePGH/RVC electrode displayed similar performances as the FePGF/CFP system in terms of selectivity and long-term stability, but at a lower overpotential (−0.39 V vs. RHE, η = 280 mV).
In the attempt to increase the amount of catalyst incorporated in the thin film, modified Fe porphyrin complexes were used as structural and functional building blocks of porous hybrid architectures. Some Fe porphyrin-based MOF materials containing Zr6 clusters as nodes were reported to be able to mediate CO2RR to CO in both organic and aqueous electrolytes. More specifically, the Fe-MOF-525 system produced a mixture of CO (FE 54%) and H2 (FE 45%) in CH3CN solution,140 whereas the PCN-222(Fe) MOF catalyst mixed with carbon black afforded a selective conversion of CO2 to CO (FECO 91%) in aqueous solution at −0.60 V vs. RHE (η = 494 mV).141 Notably, the amount of electroactive Fe-TPP units deposited on the electrode surface in the Fe-MOF-525 catalyst is considerably higher than the conventional heterogenization methods.140 A novel Fe porphyrin-based COF material, prepared by a straightforward solvent-free method, was also recently reported as a fairly stable catalyst for CO2RR, producing CO in good yields in organic media. Nevertheless, catalytic tests in aqueous conditions resulted in major H2 production.142 In addition to MOFs and COFs, supramolecular assemblies accommodating iron porphyrin moieties served as competent catalysts for efficient CO production. In comparison with direct non-covalent functionalization of molecular FeTPP, its encapsulation into a rhombicuboctahedral porous organic cage (POC) substantially increased the electroactive surface area, leading to superior catalytic rates and durability.143 In neutral water, the supramolecular catalyst was able to produce 55250 turnovers of CO with FEs close to 100% at −0.63 V vs. RHE (η = 510 mV) over 24 hours. Remarkably, a kinetic analysis suggested that the porous POC framework enhances the catalytic response by facilitating CO2 diffusion and increasing the local concentration of CO2, albeit not altering the catalytic mechanism of FeTPP at a molecular level.143
3.2 Cobalt
In contrast with the Fe analogues, the Co complexes bearing porphyrin-like ligands have received considerably less attention as homogeneous CO2RR catalysts in both organic and aqueous electrolytes.144–146 Instead, they were found to be very efficient catalysts once immobilized on the surface of conductive carbon-based electrodes using a wide variety of functionalization strategies. Here we aim to focus on the molecular Co catalysts containing the most popular porphyrin and phthalocyanine moieties (see Chart 2). A comparative study between an immobilized Co phthalocyanine and other porphyrin-like Co–N4 complexes highlighted that the former possesses an ideal platform for catalytic CO production, favoring the rapid formation of the key intermediate *COOH and the desorption of CO.147 These findings suggest that not only the metal coordinative environment, but also the bulk structure of the ligand plays an important role in catalysis. A variety of other Co porphyrinoids have been proposed for CO2RR so far, including chlorins,148 corroles149,150 and corrins.151 Among them, it is worth mentioning a CoII chlorin complex (22) adsorbed on MWCNTs, which was shown to be able to catalyse the CO2-to-CO electroreduction with high FE (89%) at low pH values.148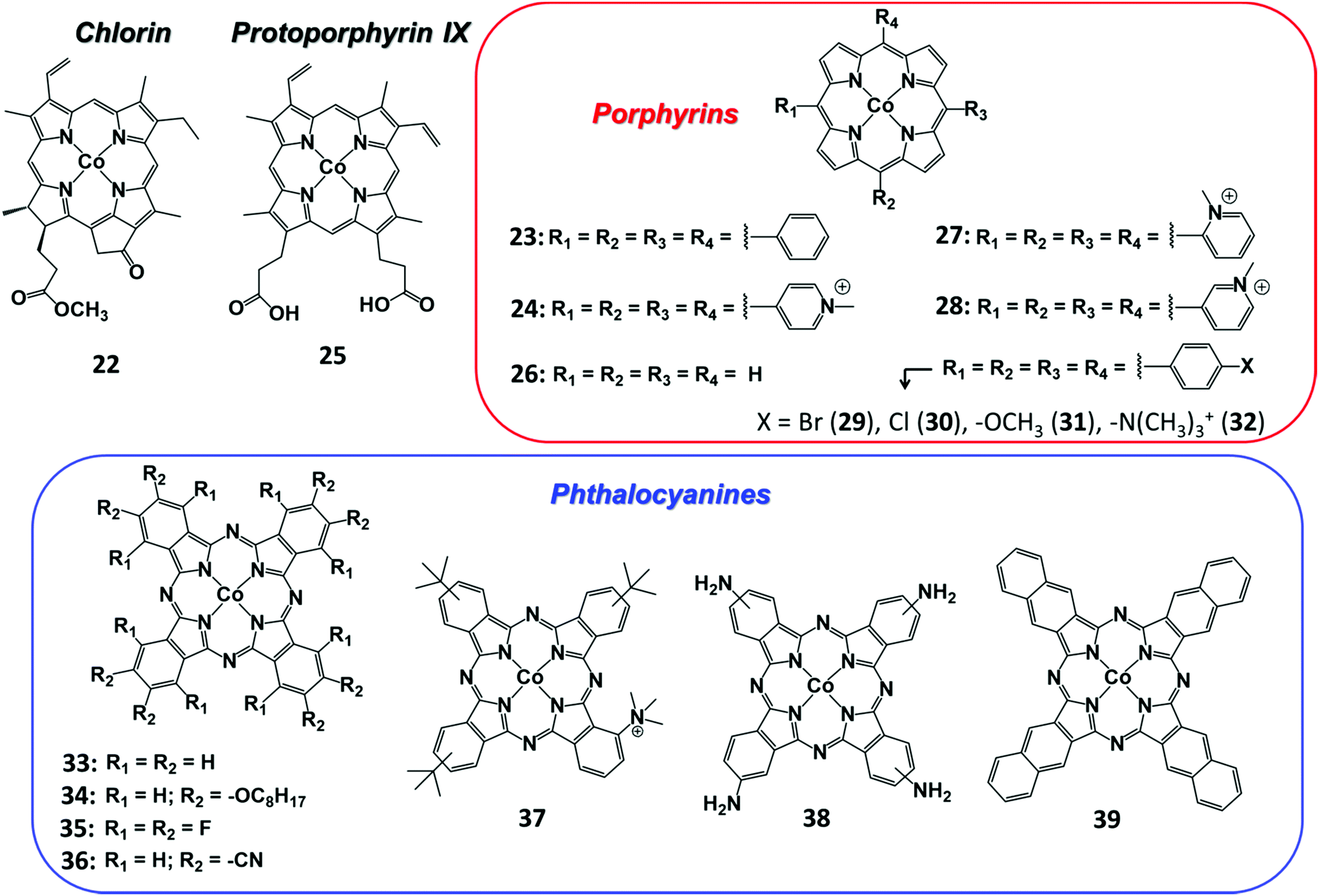 | ||
| Chart 2 Selected molecular Co systems containing porphyrinoid structures reported for CO2RR. | ||
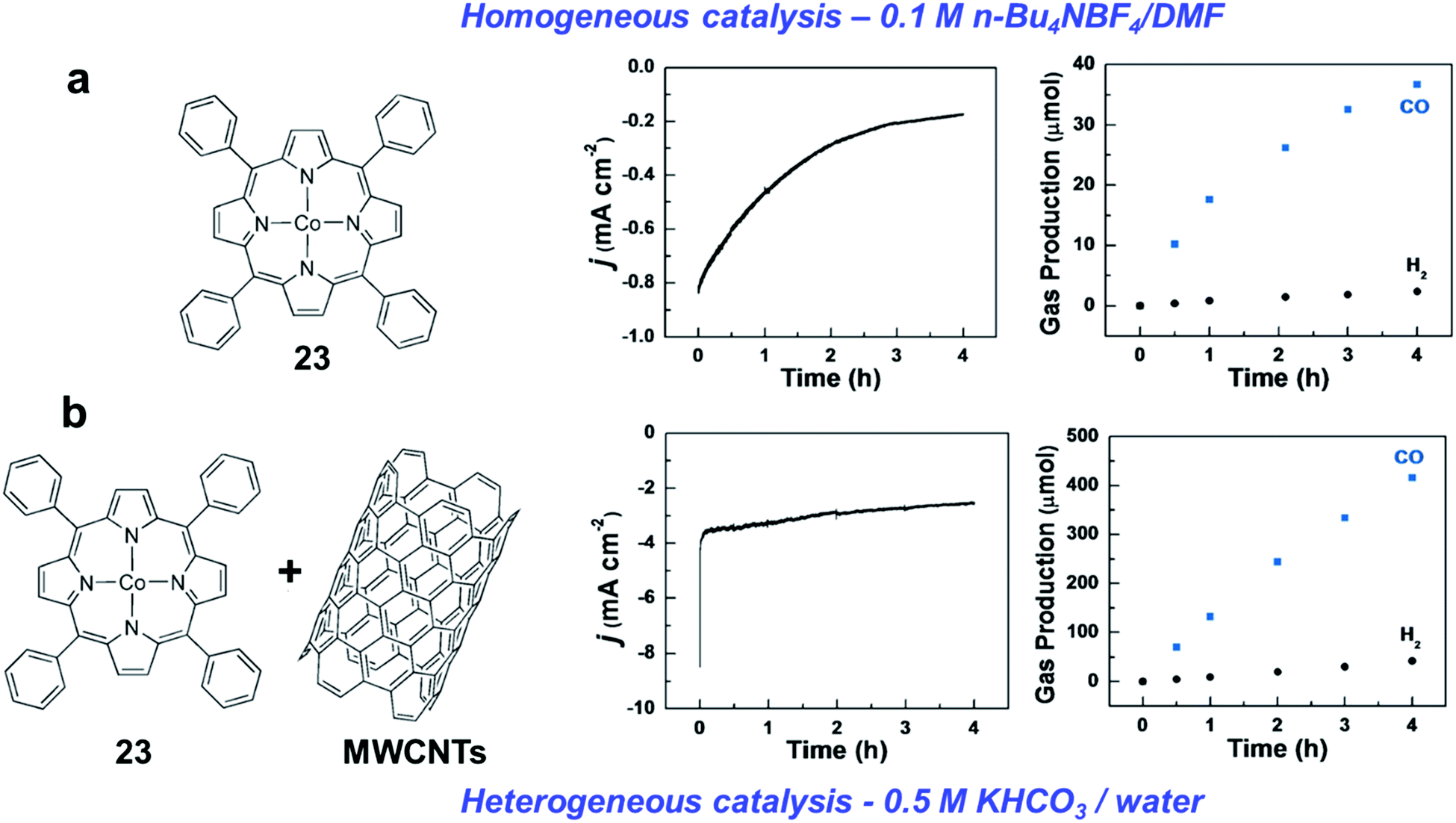 | ||
| Fig. 6 Comparison of the CO2RR catalytic behavior of CoTPP (23) as homogeneous catalyst in (a) organic medium (DMF), and (b) heterogenized catalyst supported on MWCNTs in aqueous electrolyte, respectively. (a) Current density recorded for an electrolysis at −2.05 V vs. SCE at a GC plate on 1 mM CoTPP (23) in the presence of CO2 (left) and the ensuing production of CO and H2 (right). The electrolyte solution is 0.1 M n-Bu4NBF4/DMF. (b) Current density for a 4 h electrolysis at −1.35 V vs. SCE with CoTPP–CNT on a GC plate (Γ = 1.7 × 10−7 mol cm−2) in the presence of CO2, and the ensuing production of CO and H2. The electrolyte is 0.5 M KHCO3. Adapted with permission from ref. 152. Copyright 2017, Wiley-VCH. | ||
In addition to the interaction between the catalyst and the support, the pH is a crucial factor to control selectivity. A recent study of a Co protoporphyrin (25) immobilized on PG (CoPP/PG) revealed that small amounts of CH3OH (6e) and CH4 (8e) can be formed in addition to CO in water at moderate overpotential (ca. 500 mV), being the product distribution highly dependent on pH.51 In addition to gas-chromatographic measurements during electrolysis experiments, online electrochemical mass spectrometry (OLEMS) was used to detect the gaseous products during slow voltammetric scans (1 mV s−1). At pH 3, production of CO (major, FE ≈ 40%) and CH4 (FE < 0.5%) was observed at less negative potentials than HER, whereas H2 is the dominant product at pH 1, with FEs below 1% for both CO and CH4 (traces of HCOOH and CH3OH were also detected). In spite of the very low yields, the FE for CH4 was found to be slightly higher at pH 1, due to a fast CO reduction to CH4 occurring in these conditions simultaneous to HER. The pH-dependent CO2RR behaviour of CoPP/PG is consistent with the DFT mechanism proposed for the simple Co porphine complex (CoP, 26):154 the initial [Co(P)(CO2)]− formation, due to the CO2 binding by the active [CoI(P)]− is followed by an intramolecular electron transfer which leads to a catalyst-bound CO2˙− radical anion. This adduct acts as a strong Brønsted base abstracting a proton by a water molecule to give the neutral [Co(P)(COOH)]0 intermediate. Then, the next neutral [Co(P)(CO)]0 carbonyl species can undergo either CO release or further reduction to CH4 through a series of concerted PCET steps.154
A fine-tuning of the electronic properties of the ligand scaffold is another key aspect to consider for the rational design of an optimal heterogeneous molecular catalyst for CO2RR. Recently, a rigorous study on a series of immobilized Co porphyrin catalysts containing varying peripheral aryl substituents (24, 27–32, Chart 2) showed that both inductive and electrostatic substituent effects impact the catalytic CO2 electroreduction to CO in neutral aqueous media.158 In order to minimize the aggregation effects, the catalytic properties were probed at low catalyst loadings using TOFCO as a descriptor of the CO2RR activity. As a major finding, the logTOFCO was found to linearly increase with the electron-donating character of the substituent (lower Hammett parameter, σ) across the series, in agreement with a rate-determining step involving an electron transfer from the Co center to CO2. Furthermore, the immobilized Co complexes bearing cationic functionalities displayed an enhanced CO2RR catalytic response, likely due to an additional electrostatic stabilization of the key intermediate (analogously to homogeneous Fe porphyrin catalysts in non-aqueous electrolyte112).
Several strategies for covalent attachment of Co porphyrins to carbon-based electrodes have been developed in alternative to the non-covalent approach (Fig. 7). In most cases, the molecular catalyst undergoes a chemical reaction or coordinates to specific functional groups or organic molecules introduced on the electrode surface. For instance, the functionalization of GC electrodes with 4-aminopyridine was obtained by direct anodic oxidation of the amine group (Fig. 7a)159 or through the formation of an amide linkage (Fig. 7b).160 The axial coordination of the pendant pyridine group to the unsubstituted CoTPP complex ensured a stable immobilization of the molecular catalyst on the surface. The so-formed catalytic films produced CO with moderate FEs (>50%) at −1.2 V vs. SCE in phosphate buffer, corresponding to ca. 105 turnovers.160 More recently, a robust covalent linkage of an alkyne-functionalized Co porphyrin to a boron-doped diamond electrode was achieved via a CuI-catalyzed “click” reaction with superficial azide-terminal groups (Fig. 7c).73 The CO2RR process in CH3CN was monitored by FTIR, providing evidence of a reductive disproportionation of CO2 into CO and CO32−, even though the evolved products were not quantified. In another report, an unmodified protoporphyrin IX (25) was covalently grafted to the O-atoms of hydroxyl-functionalized CNTs by reflux in the presence of a tertiary amine (Fig. 7d).161 In comparison with conventional physical methods, this procedure increased the catalyst loading, maintaining a high level of dispersion. In terms of CO2RR performance, the modified electrode exhibited an excellent selectivity to CO (FE 98%) and durability (TONCO = 6 × 104) at a 490 mV overpotential, with a stable current density of ca. 25 mA cm−2 over 12 hours. Recently, a direct electroreductive covalent grafting of a diazonium-modified CoTPP to carbon cloth also revealed to be an effective way to obtain high catalytic CO turnovers (3.9 × 105) and good selectivity (FECO 81%) (Fig. 7e).162 In this case, the π-conjugated phenylene linker contributes to enhance the electron transfer from the electrode to the attached molecular catalyst. Lastly, the electropolymerization of substituted Co porphyrins containing aminophenyl163 or vinyl164 groups also resulted in the formation of catalytic films for CO2RR.
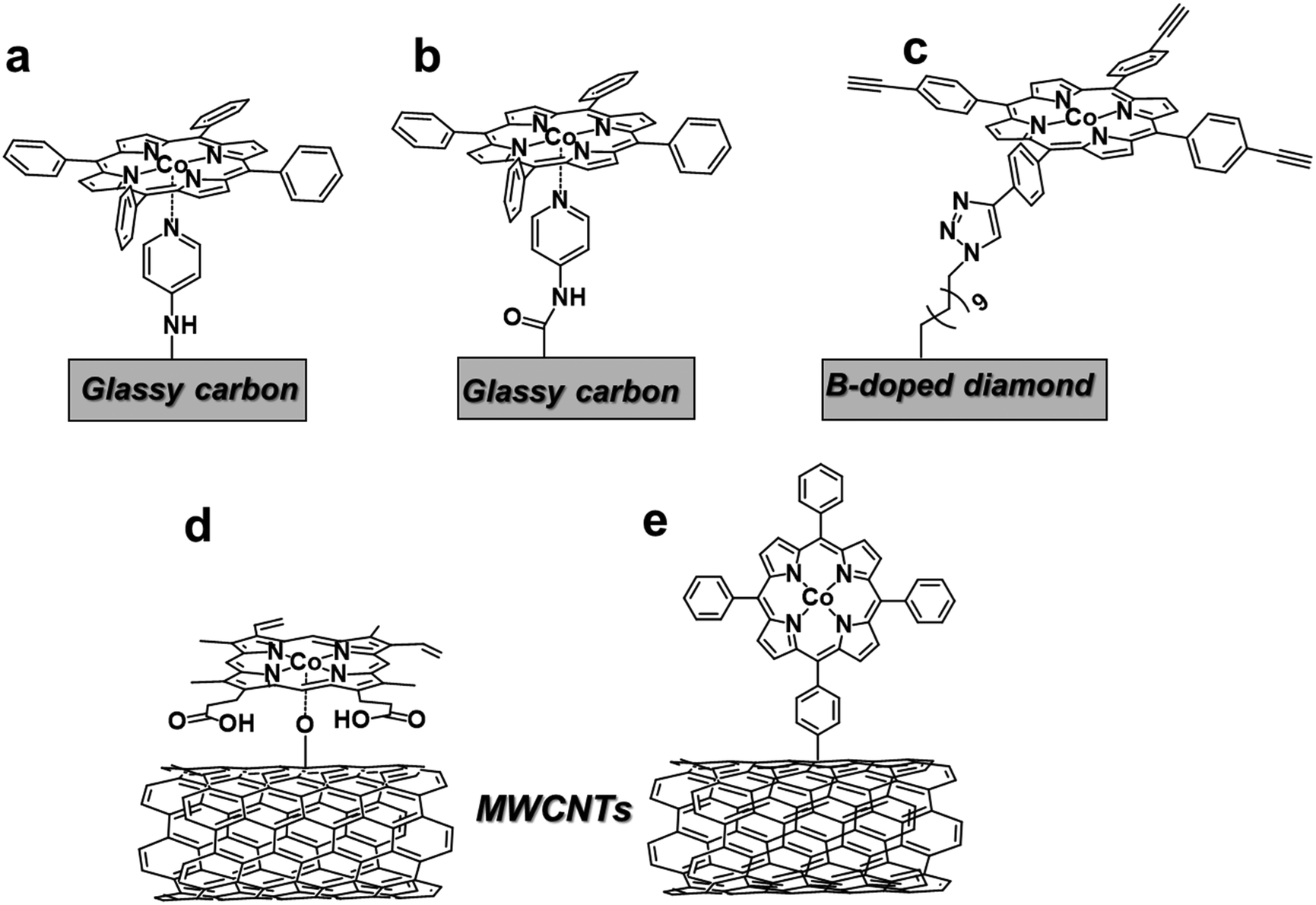 | ||
| Fig. 7 Different proposed strategies for covalent attachment of Co porphyrin catalysts for CO2RR. Pictorial representation of catalysts reported in ref. 73 and 159–162. | ||
The reticulation of Co porphyrin-like molecular catalysts into porous networks has represented a significant step forward towards the design of new electrocatalytic materials for CO2RR.76,78,165 In a first example, a modified CoTPP was used as the catalytic linker unit assembled into a porous MOF structure, namely Al2(OH)2TCPP-Co (TCPP-H2 = 4,4′,4′′,4′′′-(porphyrin-5,10,15,20-tetrayl)tetrabenzoate).76 In an attempt to maximize the amount of Co centres electrically connected to the electrode, thin films of MOF were grown directly on the surface, resulting in a sustained electrocatalytic conversion of aqueous CO2 to CO with current selectivity up to 76% at −0.7 V vs. RHE. By directly growing the MOF on a transparent conductive fluorine-doped tin oxide (FTO) plate, an estimation of the formal redox potential (E1/2) for the CoII/I transition was made by means of in situ UV-Vis spectroelectrochemistry.76 Recently, the structure of 2D MOF nanosheets [TCPP(Co)/Zr-BTB] was reported to facilitate the exposure of the Co porphyrin active sites to CO2, showing comparable results as to Al2(OH)2TCPP-Co.166 The post-modification of the unsaturated coordination sites of Zr6 clusters with different organic molecules allowed to tune the micro-environment around TCPP(Co), favouring CO2RR over HER. Following another strategy, the combination of a Co porphyrin linker with a reductive polyoxometalate (POM) unit gave rise to a MOF electrocatalyst for CO2RR to CO with improved electrical properties, resulting in excellent FECO (99%) and durability (>36 h) at −0.8 V vs. RHE.167
A remarkable improvement in the catalytic performance was obtained by the incorporation of Co porphyrin building blocks into COF structures, whereby a dialdehyde organic linker is connected to the catalytic unit (5,10,15,20-tetrakis[(4-aminophenyl)porphinato]-cobalt, CoTAP) by imine condensation (Fig. 8).78 Unlike MOFs, the presence of organic linkers offers the opportunity to easily tune the pore size and, in turn, the CO2 adsorption properties of the material. In order to study this effect on CO2RR, 1,4-benzenedicarboxaldehyde (BDA) and biphenyl-4,4′-dicarboxaldehyde (BPDA) were employed as struts for the synthesis of two different COF materials, namely COF-366-Co and COF-367-Co, respectively. Albeit an excellent selectivity to CO (FE ca. 90%) was obtained for both catalysts in neutral aqueous conditions at −0.67 V vs. RHE (η = 550 mV), the latter exhibited an increased TONCO in comparison with COF-366-Co over a 24 h electrolysis period. The catalytic turnover of COF-367-Co was further improved by adopting a multivariate approach (>2.9 × 105 CO turnovers for COF-367-Co(1%)), consisting in diluting the electroactive Co porphyrin active sites in the lattice with isostructural metalloporphyrins that are catalytically inactive for CO2RR (e.g. Cu). It was also found that growing thin films of COFs on highly oriented pyrolytic graphite led to a 9-fold improvement in the catalytic activity of COF-366-Co over the microcrystalline COF powder deposited on a carbon fabric support, due to an improved electrical contact between the catalytic centers and the electrode.165
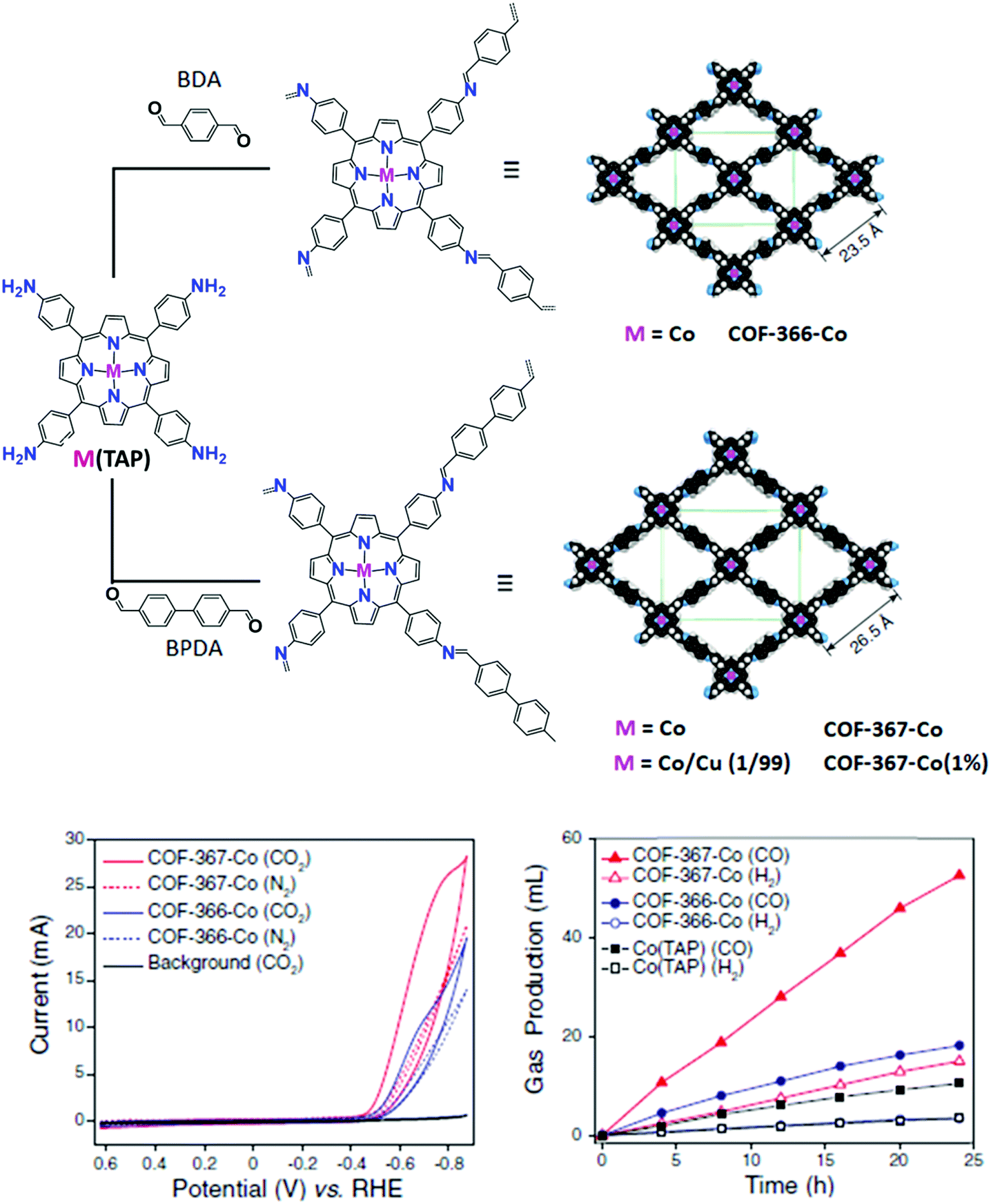 | ||
| Fig. 8 Design and synthesis of metalloporphyrin-derived 2D covalent organic frameworks reported in ref. 78. The space-filling structural models of COF-366-M and COF-367-M were obtained using Materials Studio 7.0 and refined with experimental PXRD data. Bottom left: Cyclic voltammograms of COF-366-Co and COF-367-Co in a CO2-saturated medium (blue and red solid lines, respectively) or N2-saturated medium (blue and red dotted lines, respectively). The black solid line shows background (bare carbon electrode) CV responses in the CO2-saturated medium. The medium was pH 7.2 aqueous potassium phosphate buffer (0.2 M) with additives: 0.5 M KHCO3 under CO2 atmosphere to maintain a neutral pH, or 0.5 M NaClO4 under N2 atmosphere to match the ionic strength. Bottom right: Long-term bulk electrolyses at −0.67 V (vs. RHE), showing the volume of CO produced by COF-367-Co (red solid triangles), COF-366-Co (blue solid circles), or Co(TAP) (black solid squares) and the volume of H2 produced by COF-367-Co (red open triangles),COF-366-Co (blue open circles), or Co(TAP) (black open squares). Adapted with permission from ref. 78. Copyright 2015, American Association for the Advancement of Science. | ||
In addition to the physical properties of the COF material, the electronic effect exerted by the framework on the metal center was also investigated. Interestingly, the observation of an additional pre-edge feature in the X-ray absorption spectra (XAS) of the COF catalysts absent in the molecular CoTAP counterpart suggested that an electronic communication takes place between the lattice and the metal center.78 To investigate the inductive effect of the framework in more detail, a series of COFs were prepared using struts with different electron-withdrawing or electron-donating groups. Even though the experimental trend deviates from the expected order based on basic inductive effect considerations, XAS and CV measurements showed clear differences across the series, providing a direct observation of the effect of the framework functionalization on the metal center.165
Besides the electronic effect related to the framework, reticular materials are ideal systems to investigate the role of secondary interactions on catalysis, offering the opportunity to tune the local microenvironment of molecularly defined catalytic sites. The boosting effect of such interactions on CO2RR electroreduction in aqueous media was recently demonstrated for Co protoporphyrin (CoPP) molecular units immobilized on 2D metal–organic layer (MOL) scaffolds (Fig. 9).168 Following a post-synthetic approach, CoPP moieties were incorporated into two different MOL backbone derivatives, built from benzenetribenzoate (BTB) and 4′-(4-benzoate)-(2,2′,2′′-terpyridine)-5,5′′-dicarboxylate (TPY) linkers, respectively. In the TPY–MOL–CoPP material, the pyridine/pyridinium pendant groups adjacent to the CoPP active sites were found to engage a cooperative synergistic boosting effect on catalysis (Fig. 9a), resembling the second or outer coordination sphere effects above discussed for the homogeneous heme catalysts (see Section 3.1.1). In particular, the presence of pendant pyridine residues resulted in a significant improvement of the CO2RR selectivity over HER for TPY–MOL–CoPP (FECO > 90%, jCO/jH2 = 11.8) in comparison with the BTB–MOL–CoPP derivative containing only phenyl rings (jCO/jH2 = 2.7), Fig. 9b. The crucial role of the preassembled pyridine/pyridinium groups was demonstrated by the FECO decay observed upon addition of divalent cations (Ca2+ or Zn2+) to the solution, due to the blockage of the terpyridyl units. Moreover, a possible electronic effect of axial coordination of the pendant pyridine rings was ruled out by the low faradaic yields obtained upon addition of exogenous pyridine. Notably, during reduction of TPY–MOL–CoPP under CO2 (pH 6.8), in situ electrochemical diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) provided evidence of pyridine protonation to form pyridinium units (increasing band at 1438 cm−1), while these spectral changes were not observed under N2 neither with BTB–MOL–CoPP under CO2 (Fig. 9c).168 DFT calculations suggested that the pyridinium moieties of the framework exert a cooperative boosting effect by lowering the energy barrier for both, CO2 adsorption and C–O bond cleavage, the latter being the rate-determining step of the process. The proposed mechanism is analogous to the one above mentioned for the Co porphine system,154 whereby the one-electron reduction of CoPP to [CoPP]− is followed by CO2 binding and protonation steps, leading to the formation of the key [pyH–O2C–Co(PP)]0 adduct, stabilized by a pre-positioned pyridine moiety to favor CO2RR over HER. A second electron uptake is then followed by the C–O bond cleavage to form the [Co(PP)(CO)]0 species which undergoes CO release.168
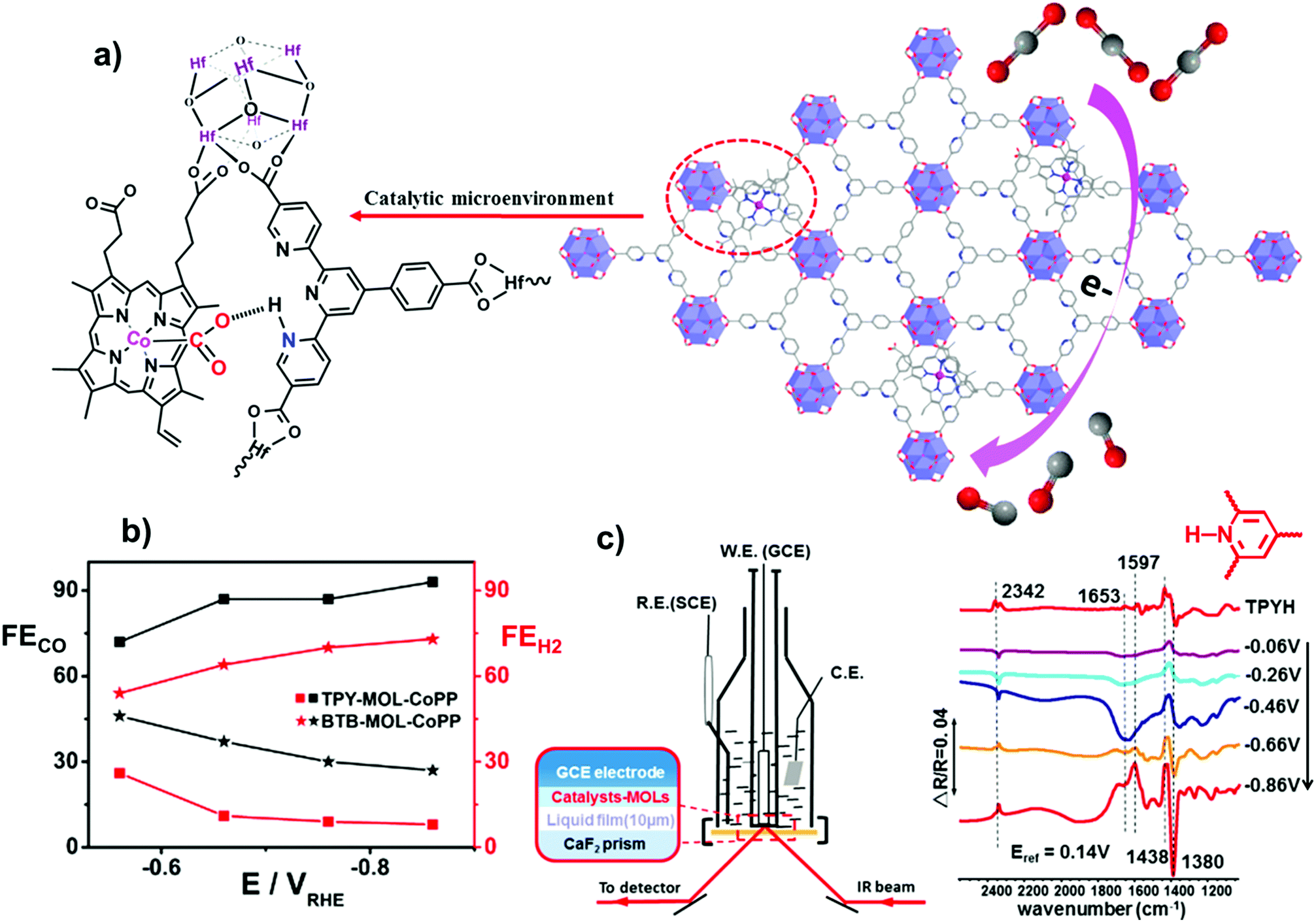 | ||
| Fig. 9 (a) Schematic showing the structure of the TPY–MOL–CoPP and the cooperative activation of CO2 by CoPP and pyH+. (b) FE for CO and H2 at varying electrolysis potentials for different catalysts in CO2-saturated 0.1 M NaHCO3. (c) On the left it is shown a schematic of the thin-layer IR cell for the in situ DRIFTS measurements. On the right, the IR spectrum of TPYH+–MOL (pH 4.0 with HClO4, red line), and DRIFTS of TPYMOL–CoPP in the potential scan range of −0.06 to −0.86 V vs. RHE in a CO2-saturated aqueous solution of 0.1 M NaHCO3. The reference spectrum was taken at 0.14 V vs. RHE. Adapted with permission from ref. 168. Copyright 2019 American Chemical Society. | ||
Finally, the efficiency of electron transport to the catalytic site is a crucial limiting factor to the usage of reticular systems for efficient CO2RR.33 In this regard, the incorporation of electron carriers as building blocks of the framework has revealed to be an effective strategy to improve electron migration in COFs. For instance, crystalline COFs obtained through the assembly of Co porphyrin catalytic units and tetrathiafulvalene struts, serving as an electron donors, resulted in enhanced durability, activity and selectivity to CO.79,169 Moreover, the exfoliation of bulk COFs into 2D ultrathin nanosheets (ca. 5 nm thickness) led to further improved CO2RR performances compared to the unexfoliated COF material, resulting in excellent CO selectivity in a wide potential range.79
The functionalization of the CoPc structure with electron-withdrawing substituents displayed beneficial effects on catalysis. For example, a perfluorinated CoPc complex (35) adsorbed on carbon cloth served as a robust catalyst for simultaneous CO2/CO conversion and H2O/O2 splitting.183 At the cathode, 35 was able to produce CO with high selectivity (FE 93%) at −0.8 V vs. RHE at neutral pH. It has been hypothesized that the fluorine substituents not only induce a positive shift of the CoII/I redox potential, but also facilitate the CO release step, thus accelerating the product removal and catalytic turnover. An analogous enhancing catalytic effect was observed by introducing –CN groups to the CoPc molecule (Fig. 10). The CN-functionalized CoPc complex (36) supported on MWCNTs exhibited higher CO selectivity at a lower overpotential than the parent CoPc catalyst, producing CO in high yields (FE 98%) and current densities (≈15 mA cm−2) at −0.63 V vs. RHE (η = 520 mV) at near-neutral pH.184 The preparation method and the use of MWCNTs had a positive impact on the CO2RR activity, ensuring uniform and robust catalyst distribution on the electrode. Notably, a 36/CNT cathode was successfully implemented in a microflow cell with a CoOx/CNT anode, sustaining a selective CO production (FE 94% with jCO = 31 mA cm−2) at a cell voltage of 1.9 V in 1 M KOH aqueous electrolyte.185 A stable CO evolution (FE ≈ 90%) was observed for 10 h at a constant cell voltage of 2.0 V.
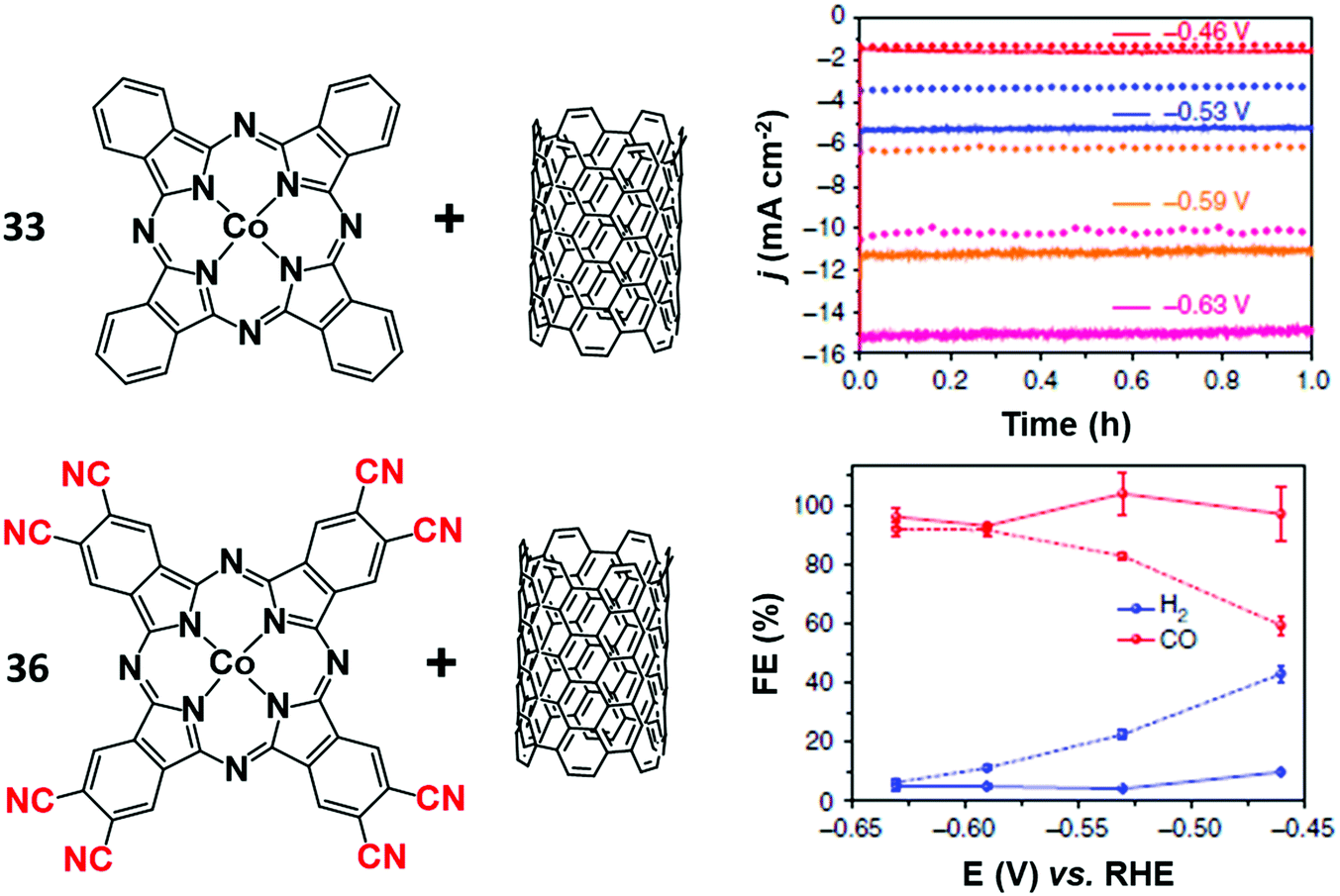 | ||
| Fig. 10 Comparison of the CO2RR performances of CoPc (33) and the CN-functionalized derivative 36 anchored to a MWCNT electrode in aqueous electrolyte (0.1 M KHCO3). Chronoamperograms (top right) and FEs (bottom right) of CO2RR products at different potentials for CoPc-CN/CNT (solid line) in comparison with CoPc/CNT (dotted line). Adapted with permission from ref. 184. Copyright 2017, Nature Publishing Group. | ||
Another remarkable example of high-performance heterogeneous catalyst for CO2-to-CO conversion is represented by a novel Co complex bearing a trimethylammonium group and three tert-butyl substituents installed on the phthalocyanine moiety (37) (Chart 2).186 The structure of the latter is reminiscent of the highly active Fe porphyrin systems containing positively charged pendant groups.112,137–139 Remarkably, porous films of 37 with carbon black or MWCNTs on carbon paper showed an excellent stability and nearly quantitative selectivity to CO in a wide range of pH (4–14). At neutral pH, 37 was found to outperform CoPc (33), resulting in an average current density of ca. 18 mA cm−2 for CO production (93% selectivity) at −0.676 V vs. RHE (η = 539 mV). Even more importantly, 37 provided excellent results once supported on a gas-diffusion cathode and used in a flow cell setup under alkaline conditions (1 M KOH): at a very low overpotential (−0.3 V vs. RHE, η = 200 mV) a jCO = 22.2 mA cm−2 was obtained, while reaching an impressive maximum partial current density of 165 mA cm−2 at −0.92 V vs. RHE (η = 810 mV) (Fig. 11).186 In a related work, the readily available, low-cost parent CoPc complex (33) was able to sustain a selective CO2-to-CO conversion at 50 mA cm−2 for >100 hours in a flow reactor.187 The implementation of 33 in a tandem flow cell with a Ni foam OER catalyst led to an excellent selectivity for CO production at commercially relevant current densities (≥150 mA cm−2). Furthermore, the FECO drop observed upon increasing the current density from 150 to 200 mA cm−2 was due to a depletion in proton concentration, rather than to a real degradation of the molecular catalyst. Indeed, a FECO ≈ 88% was maintained at 200 mA cm−2 upon the addition of PhOH during the preparation of the catalyst ink.187 The latter may also act as a local pH buffer, alleviating the issues related to precipitation of insoluble KHCO3 crystals at the cathode. This improvement led to an overall cell voltage of ca. 2.5 V, outperforming an Ag solid-state CO2RR catalyst at a comparable CO partial current density.188 These results demonstrate that earth-abundant metal-based molecular catalysts can be efficiently implemented in real devices for selective CO2 conversion to CO.
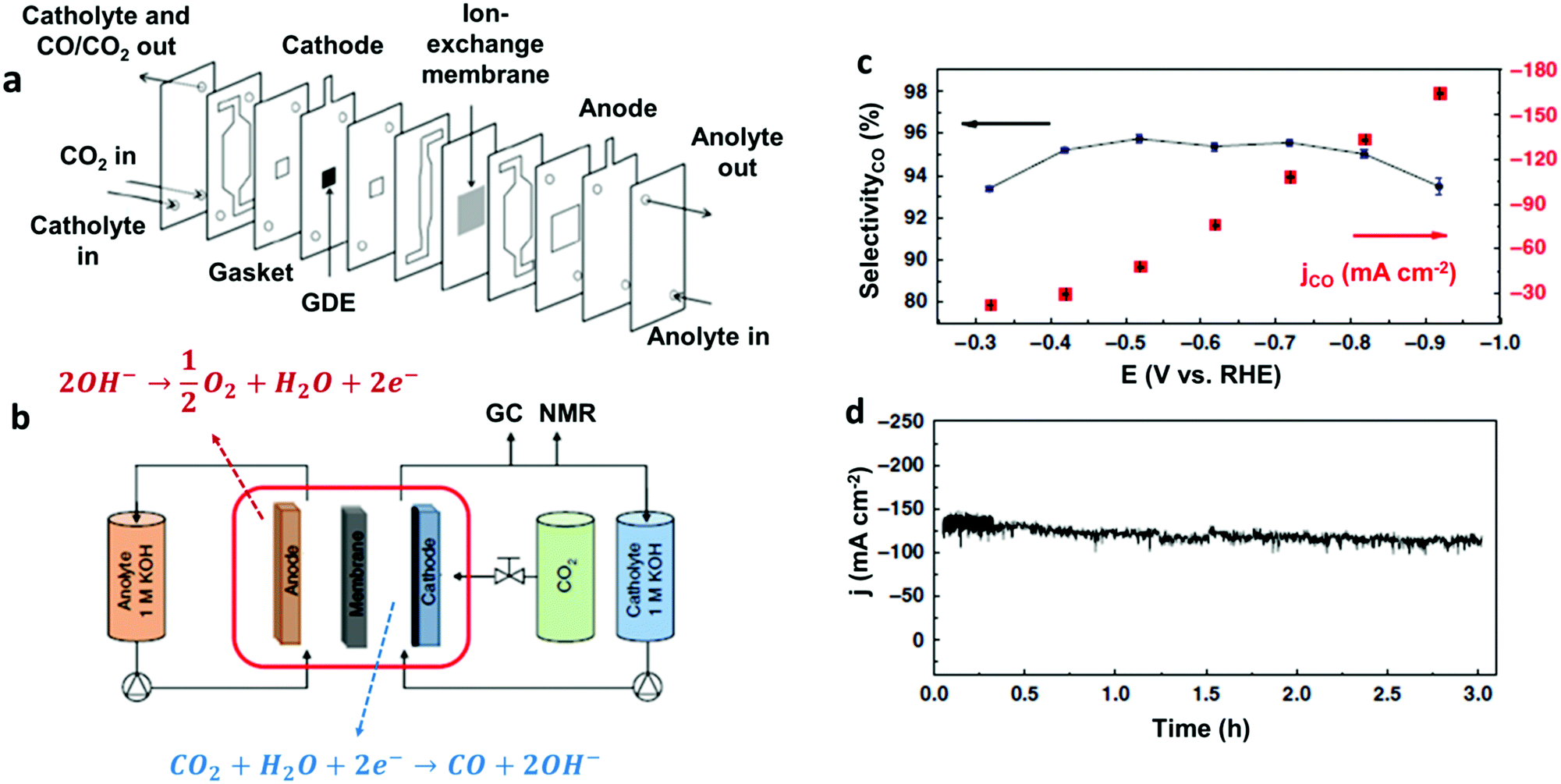 | ||
| Fig. 11 (a) Cross-sectional view of the CO2 electrolyzer flow cell and (b) general scheme of the entire experimental set-up used in ref. 186. (c) Current density (right) and selectivity for CO production (left) as a function of the potential, and (d) bulk electrolysis at a fixed potential (E = −0.72 V vs. RHE) for 37@carbon black deposited onto a carbon paper as cathodic material, in 1 M KOH. Adapted with permission from ref. 186. Copyright 2019, Nature Publishing Group. | ||
Besides the outstanding stability and efficiency for CO production, their extreme versatility is another attractive feature of Co phthalocyanines. Very recently, it has been demonstrated that CoPc/MWCNT electrodes are able to catalyse the electrochemical CO2 reduction to CH3OH in aqueous media via a CO intermediate.52 Electrolyses performed under CO-saturated and highly basic (pH 13) conditions reached a FE ≈ 14% for CH3OH production at −0.64 V vs. RHE (η = 740 mV). In addition to CH3OH, a small amount (ca. 3%) of formaldehyde, HCHO, was also detected in solution after an electrolysis under CO at −0.54 V vs. RHE. In strongly alkaline conditions, a non-faradaic disproportionation of HCHO to a mixture of CH3OH and HCOO− (Cannizzaro reaction) may occur in solution and it should be considered.189 In order to demonstrate that the Cannizzaro reaction accounts only for a small part of the detected CH3OH after electrolysis under CO, the ratios CH3OH/HCOO− were calculated to be 16 and 27 at pH 13 and 12, respectively. Furthermore, an electrolysis experiment under Ar in the presence of HCHO led to a considerable amount of CH3OH (FE > 18%) at −0.54 V vs. RHE, confirming that HCHO is an intermediate for CH3OH formation. These findings open the door to a new sequential two-step CO2RR strategy for fuels production using the same CoPc molecular catalyst: at neutral pH, CoPc can efficiently and selectively convert CO2 into CO, which can be further reduced to CH3OH using the same catalyst under basic conditions (pH 12–13). The capability of the immobilized molecular CoPc system to catalyse multi-electron CO2RR has been further confirmed by a recent study, whereby a CoPc/CNT composite was found to convert CO2 to CH3OH with FE > 40% at −0.94 V vs. RHE in a near-neutral electrolyte.53 The major drawback relies in the low durability of the system, likely due to a partial hydrogenation of the Pc ligand, causing a dramatic drop of the CH3OH production after a few hours of electrolysis. However, the introduction of four amino groups onto the Pc moiety resulted in a substantial improvement of the catalyst (compound 38, Chart 2) robustness, leading to a sustained production of CH3OH with average FE ca. 28% for 12 h of electrolysis.
In alternative to non-covalent functionalization methods, CoPc can be effectively anchored to a carbon-based electrode surface via the incorporation within a deposited layer of a coordination polymer such as poly-4-vinylpyridine (P4VP) (Fig. 12).190,191 The dangling-free pyridyl groups act as the anchoring points for CoPc through an axial coordination to the Co center (Fig. 12a). In acidic conditions, the CoPc-P4VP electrode is a competent catalyst for the selective CO production (FE ≈ 90%) at −0.73 V vs. RHE (η = 610 mV), outperforming the parent CoPc system.192 The catalytic boosting effect of pyridine coordination on CoPc has been also reported elsewhere.193 Two major parameters were found to affect the catalytic response: (i) the axial coordination of pyridine (primary coordinative environment), which increases the nucleophilicity of the CoI center facilitating the CO2 binding step; (ii) the interaction with partially protonated peripheral pyridyl residues of the polymeric film that may undergo secondary coordination sphere effects (e.g. stabilizing H-bonding interactions, proton relays), as previously seen for a Co porphyrin catalyst incorporated into a MOF structure (see Section 3.2.1).192 In order to evaluate these factors independently from each other, the electrocatalytic CO2RR behaviour of CoPc and CoPc-P4VP was systematically compared with the following three systems (Fig. 12b): the five-coordinate CoPc(py), in the absence of the P4VP membrane; CoPc encapsulated into a non-coordinating poly-2-vinylpyridine layer (CoPc-P2VP), whereby the axial coordination of the pyridine is prevented by steric hindrance; the five-coordinate CoPc(py)-P2VP, derived by embedding CoPc(py) within the P2VP polymer.192 A detailed kinetic analysis highlighted a change in the rate determining step across the series, corresponding to the CO2 binding step for the four coordinate derivatives (CoPc, CoPc-P2VP) and to a subsequent protonation of the coordinated CO2 intermediate for the five-coordinated systems (CoPc(py), CoPc-P4VP, CoPc(py)-P2VP), confirming the pivotal role of the secondary proton relays in controlling the proton delivery to the CoPc active sites.194 Other surface functionalities than the pyridyl groups may also contribute to enhance the catalytic properties of CoPc-type species. For example, the preferential immobilization of planar Co 2,3-naphthalocyanine (39) on doped graphene via axial Co–O coordination to the terminal sulfoxide groups rather than to the carboxyl ones, improved the electronic communication between the catalyst and the conductive surface, leading to a 3-fold increase of TOF for CO production and a FE up to 97%.195
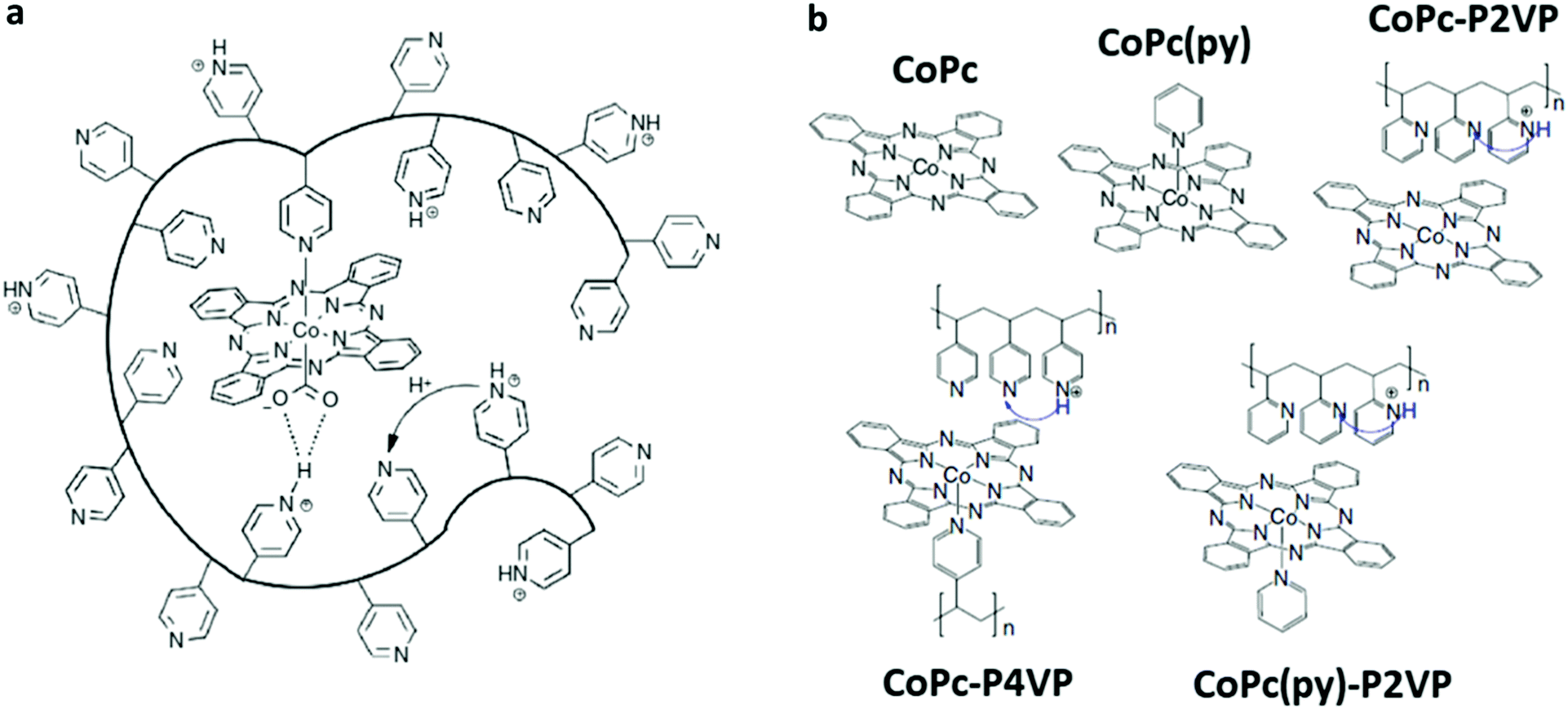 | ||
| Fig. 12 (a) An illustration of a cobalt phthalocyanine (CoPc) encapsulated within a hydrophobic poly-4-vinylpyridine (P4VP) membrane highlighting the postulated primary-, secondary-, and outer-coordination sphere effects.192,194 (b) Selected catalyst and polymer–catalyst composite systems investigated in ref. 192 and 194 along with their postulated coordination environment and proton relays. Adapted with permission from ref. 194. Copyright 2019, Nature Publishing Group. | ||
Finally, Co phthalocyanine derivatives have been employed to design new hierarchical 3D materials for efficient CO2RR. In a recent report, Co phthalocyanine catechol building blocks have been implemented into a novel metal–catecholate framework, namely MOF-1992.196 The system displayed an original topology with more accessible CoPc sites, as well as improved charge transfer properties, leading to significantly higher electroactive surface area than the previously reported reticular Co/Fe catalysts.76,78,140,141,165 At neutral pH, MOF-1992 was able to mediate a selective conversion of CO2 to CO (FE 80%) at −0.63 V vs. RHE (η = 520 mV) with relevant current densities (>16 mA cm−2). Among the other proposed reticular or supramolecular approaches, it is worth to mention a hybrid approach, based on the decoration of the external surface of the zeolite ZIF-90 with active Co tetraminonaphthalocyanine units for selective CO2-to-CO conversion.80 In this case, the electronic structure of the Co center was not altered by the zeolite framework.
3.3 Other transition metals
4. Molecular catalysts for CO2RR containing non-heme macrocyclic and polydentate nitrogen ligands
In this section, the most relevant molecular systems based on non-heme macrocyclic and polydentate nitrogen ligands will be reviewed. We will focus on Fe, Co and Ni systems, which cover the majority of the molecular catalysts with this type of ligands reported so far for CO2 electroreduction. Section 4.1 is devoted to the discussion of the widely studied NiII and CoII tetraazamacrocycle complexes. The main molecular catalysts with alternative nitrogen-containing macrocyclic frameworks will be summarized in Section 4.2. Finally, Section 4.3 briefly summarizes some relevant recent studies carried out on molecular systems based on macrocyclic-like nitrogen ligands, comprising aminopyridyl, polypyridyl and tripodal N4 systems.4.1 Tetraazacyclam macrocycles
:H2O 4:1 mixture.213
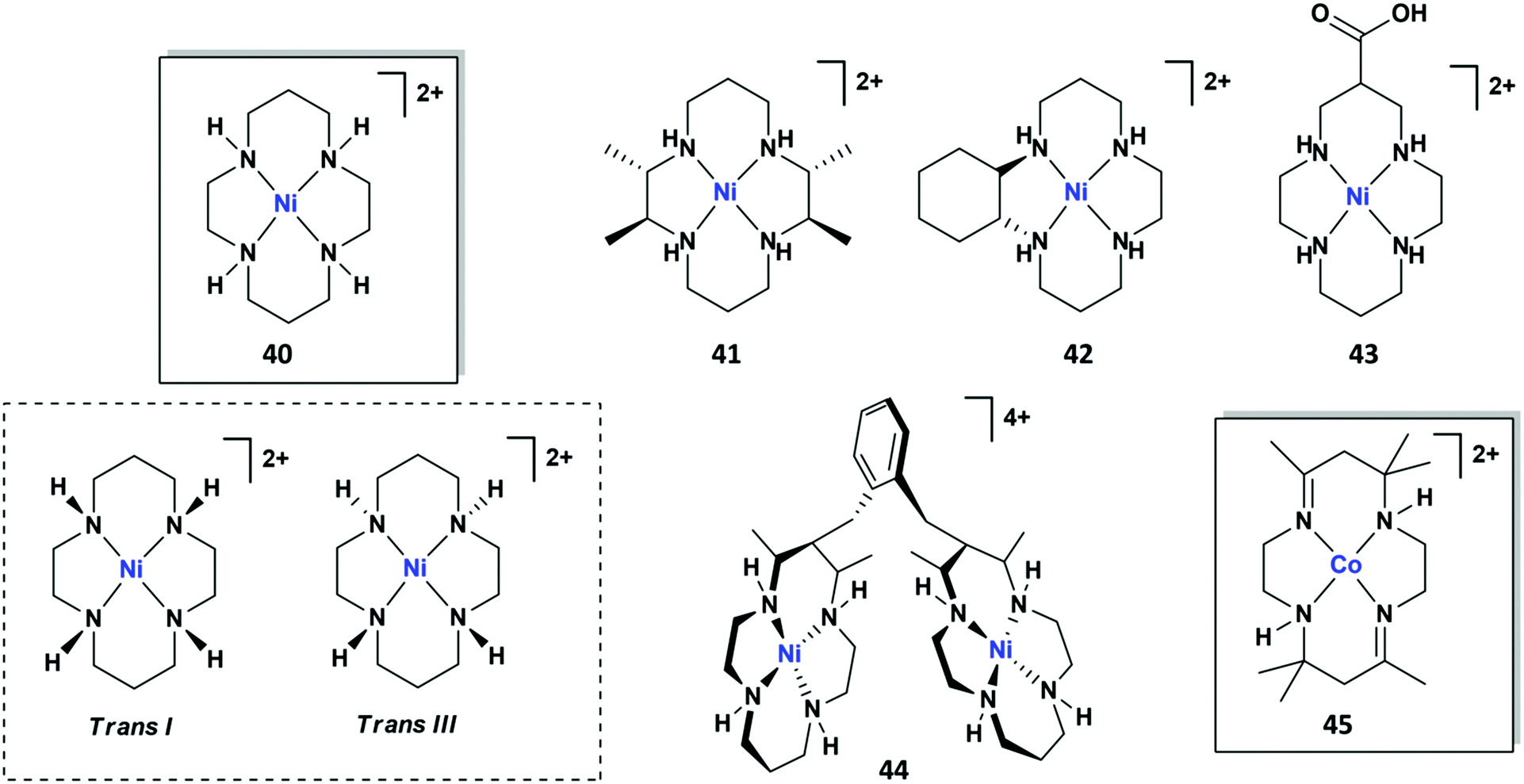 | ||
| Chart 3 Structures of selected molecular Ni and Co tetrazamacrocycles reported for CO2RR. | ||
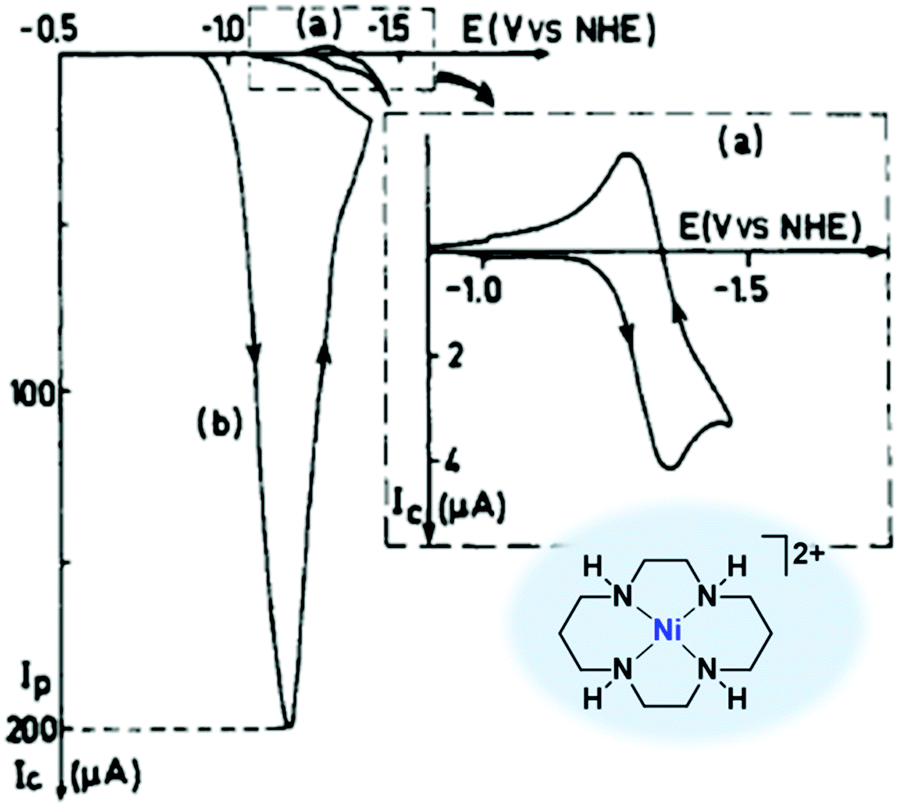 | ||
| Fig. 13 Cyclic voltammetry of [Ni(cyclam)]2+ (40, 1 mM) in 0.1 M KClO4 (pH 4.5) under N2 (curve a) or CO2 (curve b) on a hanging Hg electrode. Scan rate: 0.1 V s−1. Reprinted with permission from ref. 210. Copyright 1986 American Chemical Society. | ||
The exclusive selectivity of 40 toward CO2RR over H2 evolution in water has been attributed to the unfavorable formation of the [Ni(H)(cyclam)]2+ hydride intermediate (pKa ∼ 1.8) in the operating pH conditions (pH ∼ 4).214 In the proposed mechanism for CO2 conversion to CO mediated by 40 (Scheme 2), the electrogenerated [Ni(cyclam)]+ species readily adsorbs on the electrode surface, undergoing CO2 binding to form a [Ni(CO2)(cyclam)]+ adduct in a η1-CO2 binding mode.215 Analysis of the molecular orbitals suggests that the electronic structure of this intermediate can be described by the NiI–CO02 ↔ NiII–CO2˙− resonance forms, featuring only moderate metal-to-ligand charge transfer from the nucleophilic Ni center to the bound CO2. The moderate affinity of the [Ni(cyclam)]+ species for CO2 binding is consistent with the relatively low values of the CO2 binding constant (KCO2 ∼ 100–101 M−1) experimentally obtained in various solvents.214,216,217 In the next step, in the presence of H3O+ or H2CO3 as proton donor, the Ni–CO2 adduct converts into the [Ni(CO)(cyclam)]2+ species via a proton-coupled electron transfer concerted to C–O bond cleavage, similar to the mechanism proposed for the Fe porphyrins (Scheme 1). The final exergonic CO release step from [Ni(CO)(cyclam)]2+ completes the catalytic cycle.215 However, under catalytic conditions the competitive one-electron reduction of [Ni(CO)(cyclam)]2+ may occur, leading to the formation of the [Ni(CO)(cyclam)]+ complex. The endergonic character of the CO dissociation step from this species, due to an increased π-backdonation from the more nucleophilic metal center to the bound CO, suggests that [Ni(CO)(cyclam)]+ may be accumulated during catalysis.215 Despite the low CO solubility in aqueous and organic solvents, the gaseous CO evolved during catalysis may also recombine with [Ni(cyclam)]+, depleting the catalyst at the interface, owing to the high values experimentally obtained for the CO binding constant (KCO ∼ 105 M−1).218,219 At more negative applied potentials, [Ni(CO)(cyclam)]+ can be further reduced to produce the unstable neutral [Ni(CO)(cyclam)]0 species, which undergoes rapid ligand loss, degrading to the Ni(CO)4 deactivation product. The formation of both, the [Ni(CO)(cyclam)]+ and Ni(CO)4 species was experimentally detected by in situ FTIR-spectro-electrochemistry (SEC) on a GC electrode under CO2 in CH3CN, confirming the product inhibition of the catalyst.217 In order to contrast catalyst poisoning by CO coordination, Kubiak and coworkers reported an alternative strategy based on the addition of a sacrificial carbonylation substrate to a solution of 40.217 The [Ni(TMC)]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetra-decane) complex was chosen for this purpose, due to its higher affinity for CO binding and lower CO2RR ability on GC electrodes as compared to 40.213 The presence of an excess amount of [Ni(TMC)]2+ effectively led to an observable current increase under CO2. The role of [Ni(TMC)]+ as a CO scavenger was further confirmed by FTIR-SEC, demonstrating the formation of [Ni(CO)(TMC)]+ during CO2RR catalyzed by 40. These results indicate the CO dissociation from the [Ni(CO)(cyclam)]+ species to be the rate-limiting step of CO2RR on GC electrodes.
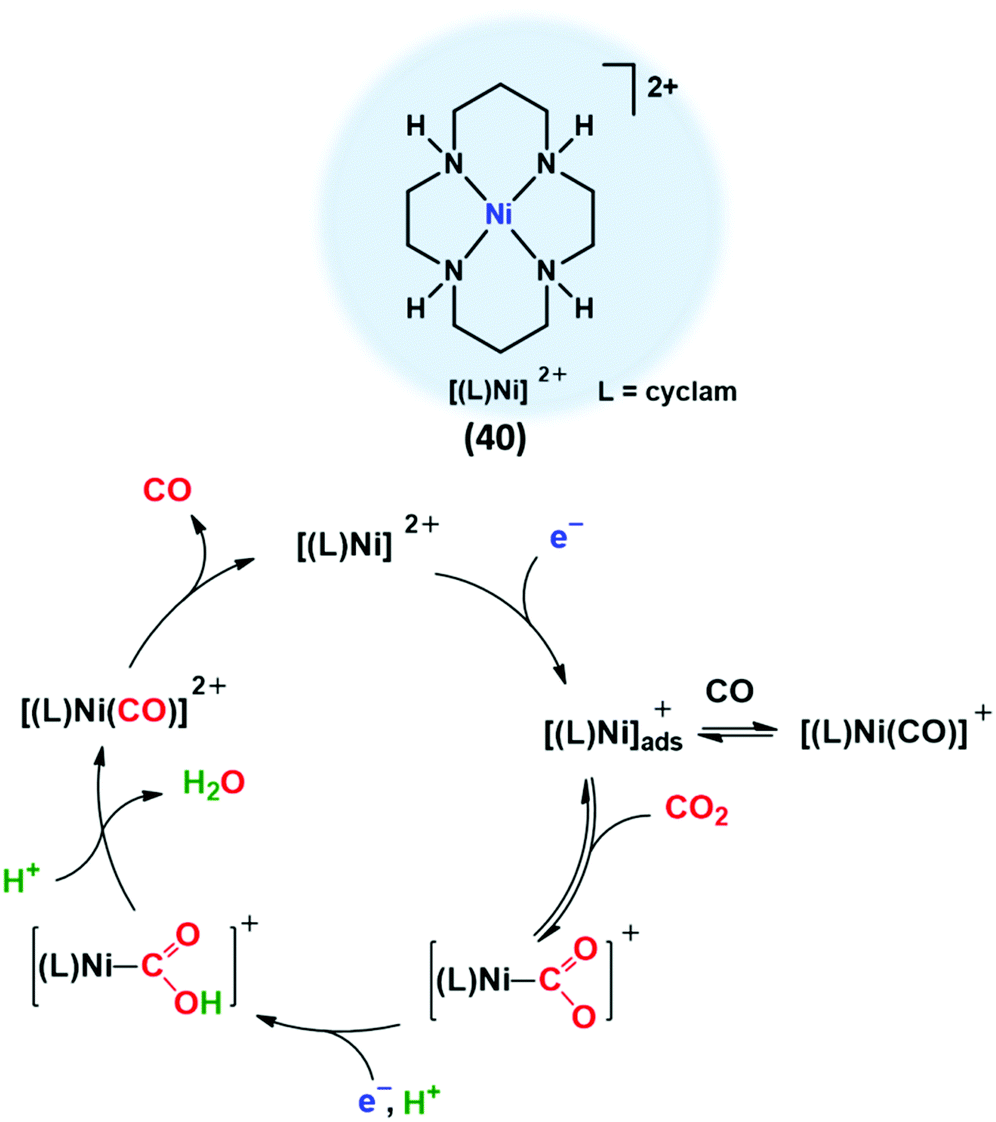 | ||
| Scheme 2 Proposed mechanism for the electrochemical CO2-to-CO catalyzed by [Ni(cyclam)]2+ (40).210,215 | ||
Despite the fact that significant synthetic efforts have been made over the past 30 years, most of the structural modifications to the cyclam macrocycle did not result in any improvement of the CO2RR catalytic performance of the parent NiII complex 40. A plethora of related NiII complexes, comprising tetraaza open-chain ligands, alternative macrocyclic structures or substituted cyclam rings showed poorer CO2RR activity, selectivity or durability under electrochemical conditions.208,210 The origin of the unique electrocatalytic behavior of [Ni(cyclam)]2+ under CO2 has been proposed to rely on a combination of structural and experimental factors, such as electrode material and electrolyte. The kinetic inertness of the macrocyclic [Ni(cyclam)]2+ even in acidic media may explain the unique durability in comparison with other acyclic or cyclic analogous complexes.210 Moreover, the redox-innocent character of the cyclam backbone increases the nucleophilicity of the NiI center and, thus, its reactivity toward CO2. Lower affinity for CO2 binding was observed for some NiII derivatives bearing unsaturated 14-membered tetraazamacrocycles (“cyclam-like”).11,220 The N-alkylation of the amine group of the cyclam ligand also led to a significant drop in the catalytic activity. This trend has been explained by a progressively diminished reducing power of the electrogenerated NiI species by increasing the N-alkylation substitution level, consistent with the positive shift experimentally observed for the E0(NiII/I) redox potential.213,215,221 Steric hindrance of the bulky alkyl groups may also hamper the CO2 binding, as well as influence the adsorption of the catalyst on the electrode surface.222 Furthermore, the pendant amine functionalities installed on the cyclam moiety have been proposed to play a crucial role in the H-bonding stabilization of the η1-CO2 adduct through an outer coordination sphere effect.11,213 Notably, the attachment of an additional pendant NH3+ group to the cyclam structure provided an enhanced catalytic production of CO for the corresponding NiII complex.223
The specific geometrical conformation of the cyclam ring was found to be critical for the efficiency of the catalyst, strongly affecting the stability of key molecular intermediates adsorbed on the electrode surface. In aqueous solution, [Ni(cyclam)]2+ (40) exists as a mixture of two dominant different conformational isomers, namely Trans-I and Trans-III, respectively in a 15% and 85% equilibrium (see Chart 3).224 Although the reductive NiII/I process has been proposed to be accompanied by a rapid conformational change of the [Ni(cyclam)]+ complex adsorbed on the Hg electrode,212,222,225 the identity of the surface-bound conformer that plays the active role in catalysis is still under debate. Some experimental and computational studies suggested the Trans-I isomer of [Ni(cyclam)]+ to be the preferred geometry for CO2 binding compared to the Trans-III derivative, presumably due to a more favorable spatial orientation of the pendant N–H groups which would contribute to stabilize the Ni–CO2 adduct by H-bonding interaction.11,213,215,222 However, DFT optimized geometries of [Ni(CO)(cyclam)]+ indicate that, in the homogeneous case, the Trans-I [Ni(cyclam)]+ isomer binds CO much more strongly that the Trans-III conformer.217 These findings suggest that a square-planar geometry is desirable to facilitate CO detachment from the carbonyl species, whereas out-of-plane distortions of the [Ni(CO)(cyclam)]+ structure would contribute to further stabilize the metal–CO bond. In a recent integrated experimental–computational study, Kubiak and coworkers demonstrated that the Trans-III [Ni(cyclam)]+ conformer is preferentially adsorbed on a Hg surface through dispersive interactions, thus representing the active species involved in CO2RR.226 Importantly, the Hg surface was found to facilitate the CO desorption kinetics of the adsorbed Trans-III [Ni(CO)(cyclam)]+ complex, by weakening the Ni–CO σ interactions. The origin of this effect was ascribed to the flattened geometry of the adsorbed Ni macrocycle, which reduces the CO binding affinity. These findings shed light on the decisive role played by the catalyst–electrode interactions to explain the catalytic efficiency of [Ni(cyclam)]2+ (40) on Hg under CO2, suggesting that the stability of the adsorbed [Ni(CO)(cyclam)]+ intermediate influences the efficiency of the overall process.
Only a few molecular Ni derivatives containing tetraazamacrocyclic ligands have been reported to outperform the parent complex 40. Among them, the RRSS-[Ni(HTIM)]2+ (HTIM = 2,3,9,10-tetramethyl-1,4,8,11-tetraazacyclotetradecane) (41) and [Ni(MTC)]2+ (MTC = 2,3-trans-cyclohexano-1,4,8,11-tetraazacyclotetradecane) (42) complexes described by Fujita et al. revealed to be excellent electrocatalysts for selective CO production on a Hg pool electrode at pH 5 and −0.96 V vs. NHE (Chart 3).216,225 At very low pH values (pH < 2), CO/H2 mixtures were produced depending on the applied potential. Analogously to the parent [Ni(cyclam)]2+ complex, the remarkable activity of these systems was ascribed to their favorable geometry and stereochemistry. For instance, the RRSS isomer of the [Ni(HTIM)]2+ complex (41) features a flat geometry analogous to the Trans-III conformer of [Ni(cyclam)]2+, suggesting an optimal ligand structure for adsorption on Hg. Moreover, the RRSS isomer was considerably more active than the RSSR isomer, since an unfavorable orientation of the bulky methyl groups in the latter hinders CO2 binding at the Ni center. More recently, the functionalization of the reference [Ni(cyclam)]2+ catalyst with a carboxylic acid led to a more efficient electrocatalyst, 43, for selective CO2-to-CO conversion in aqueous media over a wide pH range.70 The most remarkable aspect of this system consists in its significant selectivity toward CO production (FECO = 66%; FEH2 = 15%) at −0.99 V vs. NHE in very acidic aqueous conditions (pH < 2), while the parent 40 catalyst predominantly produced H2 (FECO = 13%; FEH2 = 73%). Finally, it is worth mentioning that a number of di-211,227 or trinuclear228 Ni macrocyclic systems have been reported for CO2RR so far, showing comparable or inferior catalytic properties than 40. However, a remarkable activity was recently shown for a dinuclear NII macrocycle complex (44, Chart 3),229 displaying a synergistic cooperative effect between the two Ni centers analogous to a previously mentioned iron porphyrin system.100,136 The catalyst exhibited an almost exclusive selectivity toward CO production on a GC electrode in a CH3CN:H2O (4:1) mixture as well as in pure water, outperforming both, a mononuclear NII derivative and a synthetic dinuclear system featuring a longer spatial distance between the Ni centers.
In addition to the electrode material, the catalytic CO2RR performances of NiII tetrazamacrocyclic complexes were found to be extremely sensitive to the reaction medium. For [Ni(cyclam)]2+, a proper choice of solvent and electrolyte is vital to achieve an efficient and selective process, with water being a particularly suitable solvent. In an early report, the stability of the [Ni(CO)(cyclam)]+ complex was found to be significantly lower in aqueous media compared to DMF, suggesting that water may play an active role in promoting a fast decomposition of the deactivation carbonyl species.210 Furthermore, the solvation properties of water molecules are crucial to stabilize the Ni–CO2 adduct by H-bonding interactions. Recently, an electron-deficient organo-urea additive was reported to act as a multipoint H-bond donor, boosting the electrocatalytic CO2RR of [Ni(cyclam)]2+ in wet CH3CN (up to 1 M H2O), without altering the exclusive selectivity of the catalyst to CO over H2 (Fig. 14).230 In this system, the added urea acts as a co-catalyst rather than a stoichiometric additive, contributing to stabilize the key intermediates involved in the CO2RR process. The superior promotional catalytic effect observed for the Schreiner's urea additive (two-point H-bond donor) compared to other single-point H-bond donors or acids, indicates that the effect is not due to acidity alone.
 | ||
| Fig. 14 Cyclic voltammograms showing [Ni(cyclam)]2+ (40, 1 mM) under Ar (black dashed line) and under CO2 (colored lines) with 0–5 equivalents of bis(3,5-trifluoromethyl)-phenylurea (Schreiner's urea, see the inset) as an additive. Reprinted with permission from ref. 230. Copyright 2019 American Chemical Society. | ||
The use of different electrolytes or buffer systems in aqueous media also led to drastic changes in activity and selectivity of [Ni(cyclam)]2+.210 The influence of the buffer identity on the catalytic CO2RR activity of [Ni(cyclam)]2+ was recently studied, revealing the buffer charge to be the main factor affecting the activity and selectivity of the reaction.231 In particular, small-sized non-coordinating cationic buffers were found to be beneficial for CO2RR over HER, suggesting the involvement of pseudo outer coordination sphere effects (electrostatic, protonation, H-bonding interactions, etc.) on the key reaction intermediates. Albeit more disordered than the local effect previously discussed for molecular catalysts, the role of the electrolyte or buffer should be carefully taken into account for the optimization of a catalytic system, offering the opportunity to design local microenvironments for efficient CO2RR. The catalytic behavior of [Ni(cyclam)]2+ was also explored in ionic liquids, showing better activity and selectivity for CO production by using the hydrophilic 1-butyl-3-methylimidazolium tetrafluoroborate (BMImBF4) solvent/electrolyte system.232 On the other hand, an unexpected change of selectivity was observed by using DMF with low water content as reaction medium, resulting in the production of CO/HCOO− mixtures depending on the applied potential (FEHCOO− up to 75%).211 This apparently counterintuitive behavior was ascribed to the formation of an alternative Ni–η1-OCO adduct, which however was predicted to be energetically less favored than the η1-CO2 one.215
Although [Ni(cyclam)]2+ is a very efficient and low-cost electrocatalyst for CO production operating in aqueous media, the need for hazardous mercury pool-based electrodes strongly limits its usage for practical applications. As above mentioned, owing to the key role played by the electrode material in the catalytic reaction, the replacement of Hg with more environmentally friendly solid electrodes while maintaining a comparable catalytic performance is a challenge. In this perspective, some attempts have been described in order to covalently attach Ni tetrazamacrocycle molecular systems to carbon-based electrodes233 or metal oxide-based photoelectrodes.234 For example, some [Ni(alkynyl-cyclam)]2+ catalysts anodically electrografted to the surface of a GC electrode, displayed a predominant H2 evolution activity (FEH2 = 89%; FECO = 7%) in a CO2 saturated CH3CN/water solution, attributing the low performance to a possible steric hindrance of the catalyst conformation detrimental for CO2RR.233 A more efficient system was obtained by incorporating the [Ni(cyclam)]2+ complex into a poly-(allylamine) (PALA) matrix, through Schiff's base condensation via axial coordination of 4-pyridinecarboxaldehyde.235 Both, the axial pyridine coordination and the encapsulation into the polymer backbone contributed to lower the overpotential, enabling a durable CO production with high faradaic yields (79–92%) over 24 h at −0.78 V vs. Ag/AgCl (pH 8, 50 mM Tris buffer). As previously discussed, analogous beneficial effects of pyridine coordination and/or polymer encapsulation on CO2RR have been observed for several cobalt phthalocyanine or porphyrin systems (see Section 3). This approach is particularly attractive to incorporate molecular catalysts into biological scaffolds for the synthesis of bioinspired systems that can be used for catalytic purposes. In particular, proteins are robust platforms featuring a well-defined hosting environment which offers the opportunity to modulate the activity/selectivity of molecular catalysts through secondary coordination sphere interactions. As an example, the axial coordination of the Ni-cyclam catalyst at a pendant histidine residue of azurin led to an artificial metalloenzyme active for CO2RR, displaying a positive shift in the onset catalytic potential under CO2 compared to free [Ni(cyclam)]2+.236 Although bulk electrolysis data were not reported, photocatalytic quantitative data showed an increased selectivity to CO for the azurin-[Ni(cyclam)]2+ scaffold in comparison with free [Ni(cyclam)]2+, suggesting a critical role played by the protein environment. Moreover, the presence of the redox-active Cu center also had a remarkable impact on improving the selectivity towards CO2RR, mimicking the role of iron–sulfur clusters in CODH. Owing to the extremely conformation-sensitive CO2RR process mediated by [Ni(cyclam)]2+, it was also speculated that the protein environment may induce constraints to geometrical distortions of the cyclam ligand, favoring CO2RR over HER.236
Finally, Machan and co-workers demonstrated the possible implementation of homogeneous systems for CO2RR using a flow-cell technology.237 More specifically, [Ni(cyclam)]2+ was employed as a benchmark homogeneous electrocatalyst in a non-aqueous electrolyzer for CO2RR to CO based on a continuous flow-cell configuration (Fig. 15). In this setup, ferrocene served as a sacrificial electron donor, whereas NH4PF6 was used as both exogenous proton donor and supporting electrolyte. The flow-cell system afforded CO production with FE > 80% and current densities up to 50 mA cm−2 using a graphite felt cathode in CH3CN in the presence of 0.5 M NH4PF6. Compared to the behavior in a conventional H-type cell setup, the [Ni(cyclam)]2+ homogeneous catalyst showed extended durability and better selectivity in the flow-cell configuration. Moreover, it was found that the solvent plays a key role in the efficiency of the process, with CH3CN providing the best results owing to its high CO2 solubility and favorable H-bonding ability (compared to DMF, as aforementioned for Fe porphyrins122). As expected, the system was limited by the formation of inactive [Ni(CO)(cyclam)]+ and Ni(CO)4 species, which were detected by UV-Vis and FTIR spectroscopy.237 Albeit achieving lower current densities compared to the flow-cell setups developed for immobilized molecular catalysts in aqueous media, this approach holds promise and, together with further engineering optimization, can be extended to other types of molecular catalysts, or used to pair CO2RR with oxidative processes using organic substrates.
 | ||
| Fig. 15 Non-aqueous flow cell electrolyzer with [Ni(cyclam)]2+ (40) as homogeneous electrocatalyst for CO2 electroreduction to CO. (a) Schematic plot of the recirculating flow setup. (b) Picture of the flow electrolyzer during operation. (c) FE with various catalyst concentrations at 1.6 V cell potential; (d) current density profiles at various cell potentials; (e) current density contribution measured by gaseous products at various cell potentials. Conditions: 5 cm2 of graphite felt as electrode, 10 mM [Ni(cyclam)]2+ (40) (or varied as noted in (d) and (e)), 0.5 M NH4PF6 as supporting electrolyte in CH3CN, 0.1 M Fc as sacrificial electron donor, flow rate 8.0 mL min−1, three layers of Celgard film. All experiments in a two-electrode configuration with cathode and anode electrodes made of graphite felt (SIGRACELL, GFD4.6 EA, 4.6 mm thickness, 5 cm2 area). Current density is calculated as the average of the first hour of electrolysis, and the measured faradaic efficiency corresponds to the product analysis after 1 h of electrolysis. Adapted with permission from ref. 237. Copyright 2020 American Chemical Society. | ||
:1 mixture for 45 at −1.6 V vs. SCE in pure water or a H2O/CH3CN mixed solvent.208 The lower selectivity for CO2RR shown by 45 as compared to 40 is related to the different reactivity of the [Co(HMD)]+ and [Ni(cyclam)]+ active species towards H+. As derived by a comparison of the CO2/H+ binding constants for the two complexes, the protonation of the [Co(HMD)]+ complex is favored over CO2 binding at considerably higher pH values than [Ni(cyclam)]+, resulting in the ability for the latter to selectively catalyze CO2RR in a wider pH range.214,241
Due to the flexibility of the macrocyclic ligand, the [Co(HMD)]2+ complex exists in the N-rac and N-meso isomeric forms. At room temperature, the isomerization reaction was found to occur slowly for the CoII complexes but much faster for the singly reduced CoI species, rapidly converting the N-meso into the N-rac isomer.11 The proposed mechanism for CO2RR is analogous to the one previously discussed for the [Ni(cyclam)]2+ catalyst, involving the CO2 binding at the electrogenerated CoI species. However, the [Co(HMD)]+ complex exhibited a much higher affinity for CO2 in DMSO as compared to the Ni analogue.220 In contrast to the latter, the reversible CO2 binding to [Co(HMD)]+ resulted in an observable positive shift of the voltammetric CoII/I wave, which allowed the estimation of a KCO2 as high as 7 × 104 M−1. Owing to the large value of the CO2 binding constant, Fujita and co-workers afforded to isolate the [Co(CO2)(HMD)]+ adduct and thoroughly investigated its electronic structure in CH3CN by using a number of spectroscopic techniques, including UV-Vis, FTIR, 1H-NMR, XANES, laser-flash photolysis and pulse radiolysis.11,241–246 Unlike the case of the similar [Ni(CO2)(cyclam)]+ adduct, spectroscopic evidences of a strong charge transfer from the CoI center to the bound CO2 were found for [Co(CO2)(HMD)]+, suggesting a [CoII(CO2˙−)(HMD)]+ structure.11,246 At low temperature, as an effect of trans axial coordination of a solvent CH3CN molecule to form the six-coordinate [Co(CH3CN)(CO2)(HMD)]+ adduct, an even stronger metal-to-ligand charge transfer was observed, suggesting the formation of the [CoIII(CH3CN)(CO22−)(HMD)]+ complex.11,246 The stereochemical features of the Co–CO2 adduct were found to be critical for the CO2 binding step, including the conformational geometry of the macrocyclic ligand and the steric hindrance provoked by the methyl groups.242 Furthermore, an additional stabilization due to intramolecular H-bonding interaction with pendant NH groups was proposed to have a remarkable impact on the CO2 binding properties of the CoI species.245 The pivotal role of H-bonding stabilization of the Co–CO2 intermediate is also consistent with the experimental observation that polar solvents, and in particular water, favorably impact the value of the CO2 binding constant.241,247
As aforementioned for the Ni derivatives, CoI tetraazamacrocycles also generally show higher affinity for CO than for CO2, owing to a strong π-backdonation from the metal to coordinating CO. The [Co(CO)(HMD)]+ complex was isolated and characterized, featuring a five-coordinated square-pyramidal geometry with a significant out-of-plane distortion of the Co center.248 As previously discussed for [Ni(CO)(cyclam)]+, the flexibility of the HMD ligand contributes to further stabilize the Co–CO bond. It is worth mentioning that diluted CH3CN solutions of freshly prepared [Co(CO2)(HMD)]+ partially decomposed after several days, resulting in the formation of traces of CO and H2 (derived from adventitious water) in the vessel headspace.243 A binuclear Co species containing a Co–COOH–Co motif was isolated from the solution and characterized by X-ray crystallography, thus suggesting the possibility that the cooperative effect of a second Co center may be involved in the CO2 binding step.249
4.2 Pyridyl-based macrocyclic ligands
This section focuses on the main reported molecular catalysts for CO2RR based on non-cyclam macrocyclic ligands, highlighting some fundamental electronic and structural aspects related to the role of the ligand framework in the catalytic CO2RR process. A first representative example is given by the Co systems with the N4H ligand (N4H = 2,12-dimethyl-3,7,11,17-tetraazabicyclo-[11.3.1]-heptadeca-1(7),2,11,13,15-pentaene), which contains the potentially redox-active pyridyldiimine platform (see Chart 4). In some early studies, Co and Ni complexes of the N4H ligands were found to be relatively poor catalysts for CO2RR, suffering from low efficiencies toward CO production and/or a severe HER competition.208,240,250 Several years later, Peters and co-workers investigated the electrocatalytic behavior of the [CoIII(N4H)(Br)2]+ complex (46, see Chart 4), with voltammetric data showing a catalytic current for CO2 reduction upon the formal CoI/0 wave.251 Bulk electrolysis under CO2 in aqueous CH3CN (10 M H2O) resulted in moderate faradaic yields for CO production (FECO = 45%) together with H2 evolution (FEH2 = 30%). Albeit non selective for CO2RR to CO, this result is remarkable since the same complex was well-known to efficiently catalyze HER in the absence of CO2.252,253 The five-coordinated [Co(N4H)(CH3CN)]+ complex, containing a formal CoI state was chemically generated and found to be a pre-catalyst for CO2RR. Owing to the redox non-innocent character of the N4H moiety,254 DFT calculations suggested its electronic structure to be best described as a low-spin CoII antiferromagnetically coupled to a ligand radical anion, which was hypothesized to contribute in steering the selectivity towards CO2RR over HER.251 In a later study, in situ FTIR spectroscopy was used to detect the [CoI(N4H)(CO2)]+ species, selectively generated upon visible-light irradiation of the starting [CoII(N4H)(CH3CN)]2+ complex in the presence of a sacrificial electron donor and a [Ru(bpy)3]2+ photosensitizer.255 Intramolecular H-bonding interaction between the N–H of the macrocycle ligand and the oxygen atom of bound CO2 was proposed to strongly contribute to the stabilization of this adduct, whose formation was found to be critical for the selectivity of the overall CO2RR process. This shed light on the crucial role played by the metal–ligand cooperation for CO2 activation. Moreover, a photochemical treatment of the initial [CoII(N4H)(CH3CN)]2+ complex with [Ir(ppy)3] sensitizer, which has stronger reducing power than [Ru(bpy)3]2+, led to the formation of the transient two-electron-reduced [CoI(N4H)(CO2−)] intermediate, detected by using rapid-scan FTIR spectroscopy.256 This adduct underwent spontaneous CO dissociation and catalyst regeneration on the timescale of seconds at room temperature, allowing to monitor the bond breaking step leading to CO evolution.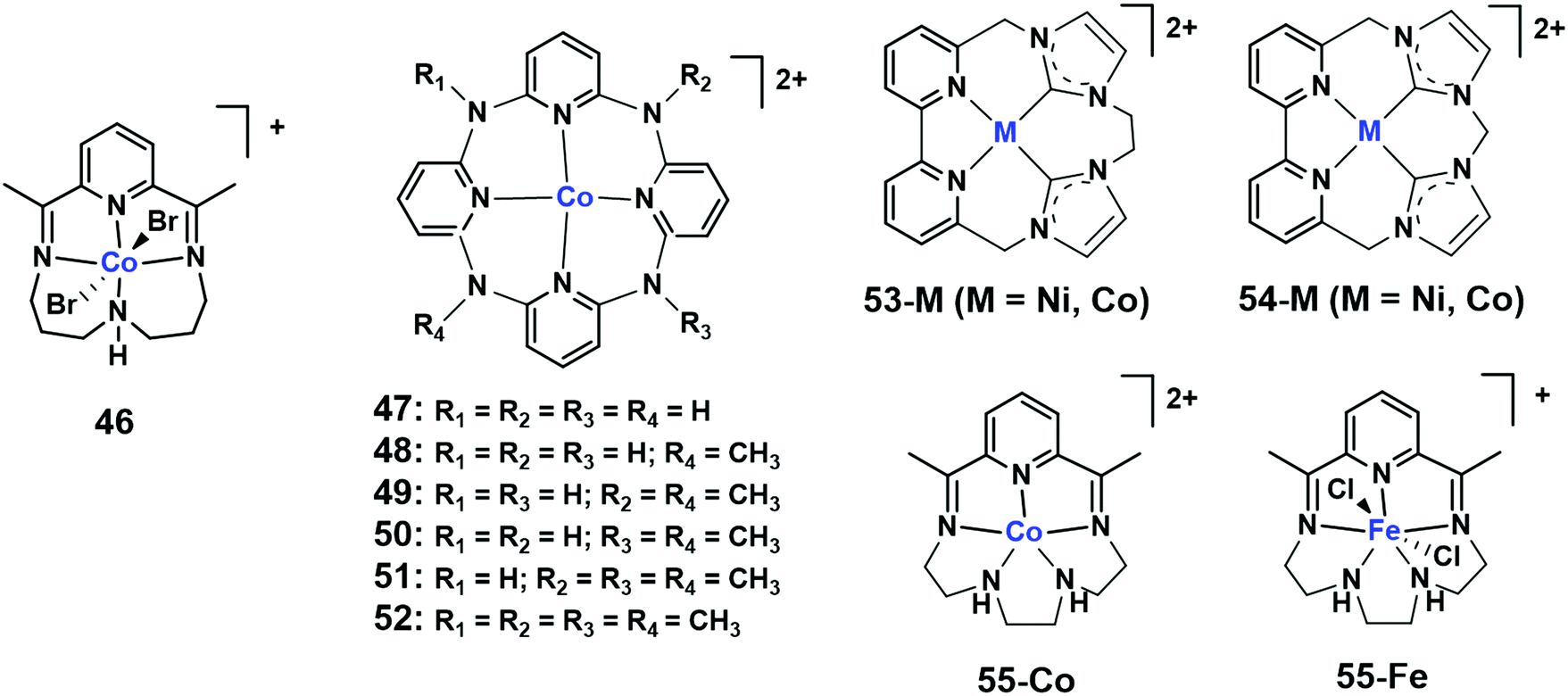 | ||
| Chart 4 Structures of selected molecular Ni, Co and Fe catalysts for CO2RR supported by pyridyl-based macrocyclic ligands. | ||
In an effort to rationalize the second coordination sphere effect of pendant amines on the CO2RR capability of Co macrocycles, Marinescu and co-workers recently investigated a series of Co complexes based on the macrocyclic azacalix[4](2,6)pyridine framework with different alkyl substituents on the four pendant N–H groups (Chart 4).119,257 In DMF solution, the complex 47, which contains four secondary amines, showed a catalytic current increase under CO2 atmosphere upon the CoI/0 wave, resulting in an excellent faradaic yield for CO production (98%) at −2.8 V vs. Fc+/0 in the presence of 1.2 M TFE as exogenous proton source.119 In sharp contrast, the N-alkylated derivatives (methyl and allyl groups, respectively) containing only tertiary amines displayed negligible catalytic current for CO2 reduction, despite the fact that the presence of the electron-donating alkyl groups causes a negative shift of the CoI/0 redox potential as compared to 47. Bulk electrolysis data confirmed the latter to be poor CO2RR electrocatalysts with low CO faradaic efficiencies, probing the crucial effect of the secondary N–H macrocycle groups on both, the catalytic overpotential and efficiency. An additional experimental–theoretical kinetic analysis on a series of Co complexes featuring varying secondary and tertiary (N-methyl substituents) macrocyclic amines (47–52, see Chart 4), was carried out to further investigate the origin of such boosting effect (Fig. 16a).257 Unlike 52, the complexes 47–51 showed high CO current densities and FEs (≥90%), resulting in a linear correlation between the catalytic rate constant and the number of secondary amines across the series (Fig. 16b). An analogous rate measured for isomers 49 and 50 indicates that the spatial orientation of the pendant N–H groups has only a minimal effect on the rate, suggesting a non-cooperative effect of the pendant amines. The first step of the proposed catalytic mechanism consists in the two-electron reduction of the initial CoII state to form the formal Co0 species responsible for CO2 binding (Fig. 16c). This aspect, which was confirmed by both experimental and theoretical data, is in contrast with the behavior shown by the cyclam-like Co systems, whereby the nucleophile CoI species is able to bind to the substrate.11 Moreover, owing to the conformational strain of the azacalix[4](2,6)pyridine macrocycle, the pendant N–H groups were predicted to exert an unfavorable intramolecular H-bonding to the bound CO2 molecule. Instead, the steric repulsion of the N-methyl groups was found to have a major effect on CO2 binding. In analogy with the tetrazamacrocycle Co systems,246 DFT calculations suggest that an intramolecular charge transfer from the Co center to CO2 stabilizes the Co–CO2 adduct. The CO2 binding is followed by two sequential protonation steps (EECC mechanism), with the latter being the rate-limiting step, forming H2O and a CoII–CO intermediate, which undergoes a facile release of CO. Theory confirmed the involvement of the secondary pendant amines in the second rate-limiting protonation step, assisting the intermolecular H+ transfer from the acid to the bound COOH, rather than via an intramolecular H+ transfer from the N–H group to COOH.257
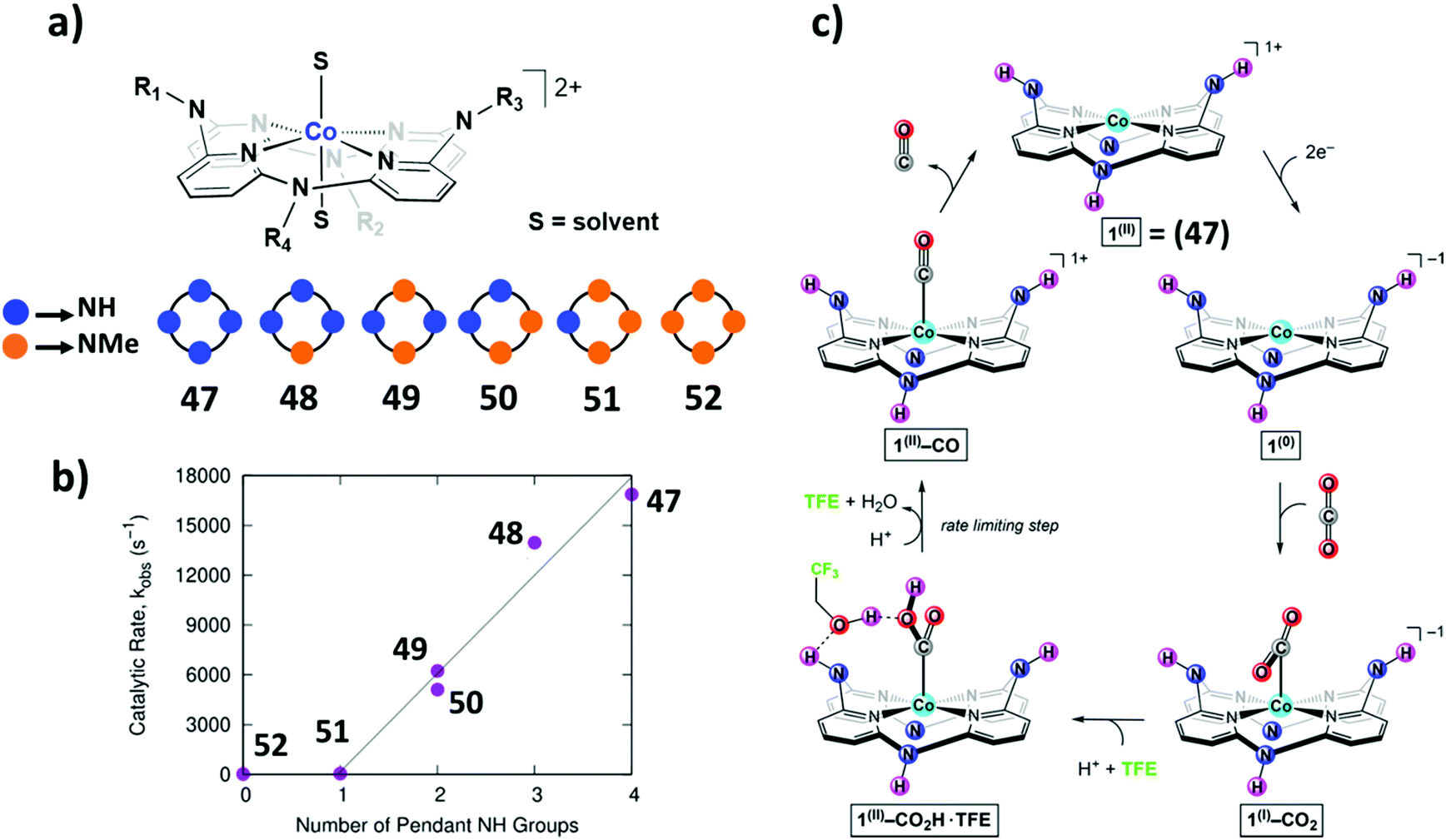 | ||
| Fig. 16 (a) Sketch of the Co complexes 47–52. (b) Experimental catalytic rate constants, kobs (s−1), as a function of the number of pendant secondary amines for complexes 47–52 measured in the presence of 1.5 M TFE and under CO2 saturation at a scan rate of 0.1 V s−1. Rates are obtained from the plateau current. A linear fit (R2 = 0.97) is shown in gray for complexes 47–52. (c) Proposed EECC catalytic cycle illustrated with complex 1(II) = 47 where E = electrochemical, and C = chemical step. Adapted with permission from ref. 257. Copyright 2018 American Chemical Society. | ||
As previously discussed for heme and tetrazamamacrocyclic complexes, a planar and rigid geometry of the ligand framework is particularly suitable for a molecular catalyst to promote an efficient CO2RR. In particular, the beneficial role of macrocyclic platforms, also known as “macrocyclic effect”, generally results in an improved catalyst durability and overall efficiency of the process. Jurss and co-workers have recently investigated the effect of structural rigidity on the electronic and catalytic properties of the metal center in a series of Co and Ni complexes bearing redox-active ligands based on bipyridyl-N-heterocyclic carbene (NHC) moieties (Chart 4 and Fig. 17).258,259 In these studies, Ni and Co complexes based on 16- and 15-membered macrocyclic ligands (53-X and 54-X with X = Ni, Co, respectively, see Chart 4) displaying different rigidity were explored for electrocatalytic CO2RR and compared with the corresponding non-macrocyclic analogues (indicated as bpy-NHC-X, X = Ni, Co, see Fig. 17). In the Ni series, the bpy-NHC-Ni complex exhibited the most flexible geometry, whereas distorted square-planar configurations were observed for 53-Ni and 54-Ni, with the smaller 54-Ni macrocycle being the most planar.258 A marked difference was shown when comparing the long-term electrocatalytic CO2RR-to-CO performances in wet CH3CN across the series, following the order bpy-NHC-Ni < 53-Ni < 54-Ni: the most rigid 15-membered 54-Ni derivative exhibited high faradaic yields for CO production (87%) while the bpy-NHC-Ni system predominantly produced H2 (Fig. 17b). The different catalytic behavior was rationalized in terms of the nature of the 1e− reduced species, which was calculated to be metal-centered for the flexible bpy-NHC-Ni system and ligand-based for the more constrained 54-Ni complex. In other words, the flexible open platform leads to a more nucleophilic Ni center upon the first electron uptake, favoring the formation of a Ni-hydride intermediate responsible for the observed selectivity loss.258
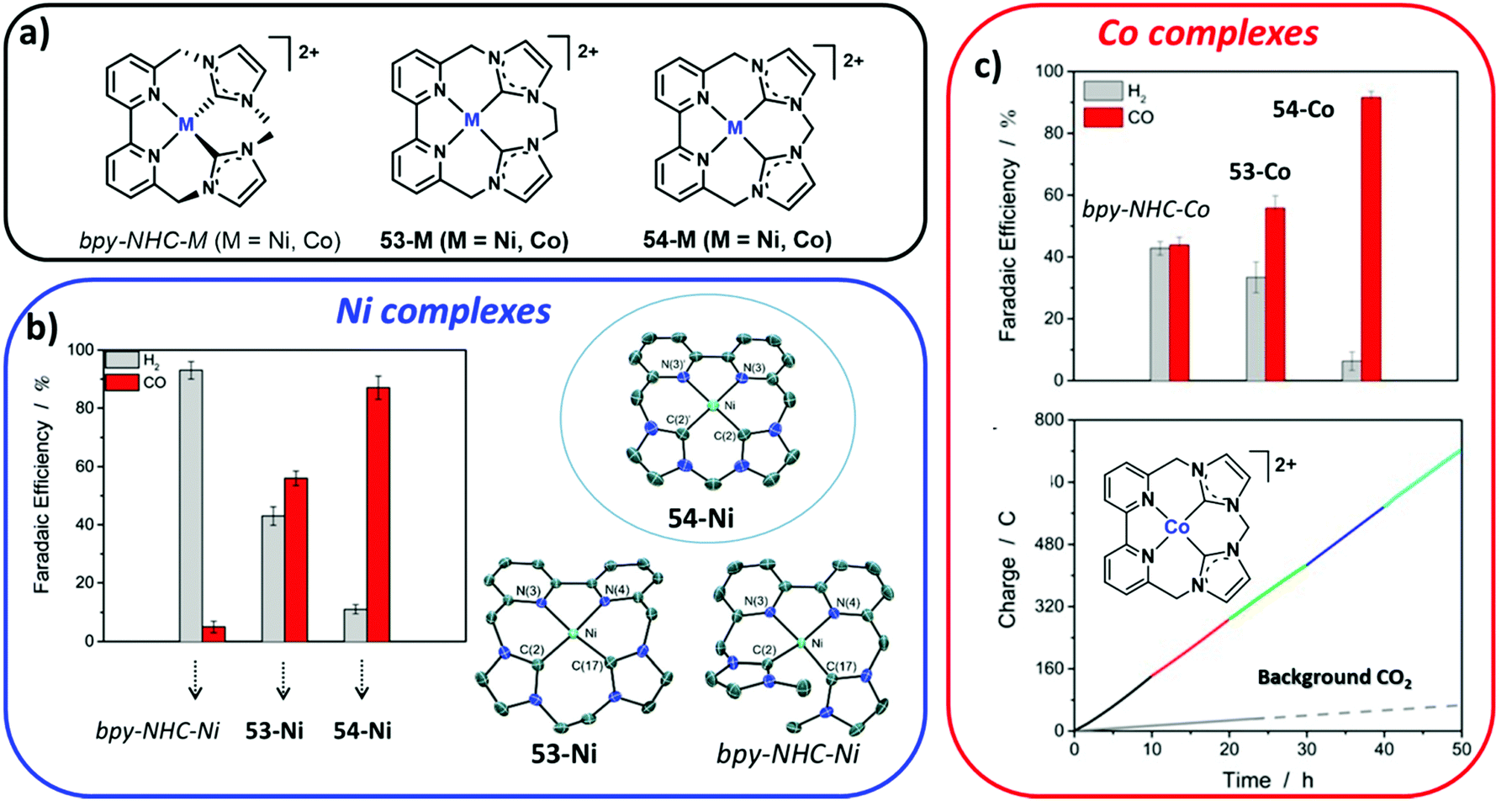 | ||
| Fig. 17 (a) Sketch of the families of NiII and CoII catalysts supported by non-macrocyclic and macrocyclic bipyridyl-NHC ligands. (b) FEs for H2 (grey) and CO (red) obtained during controlled-potential electrolysis with 0.2 mM of 53-Ni, 54-Ni, bpy-NHC-Ni, respectively. Electrolyte: CO2-saturated CH3CN/0.1 M n-Bu4NPF6 solutions containing 2% H2O at a GC rod. The inset shows the ORTEP diagrams of cations in 53-Ni, 54-Ni and bpy-NHC-Ni. (c) (top) FEs of H2 (grey) and CO (red) from electrolysis experiments with 10 μM Co catalyst in CO2-saturated H2O/0.1 M NaClO4 at a Hg pool (4 cm dia.) working electrode. On the bottom, a charge vs. time plot of consecutive CPEs is reported with 10 μM 54-Co (see inset) in CO2-saturated aqueous 0.1 M NaClO4. Conditions: Eappl = −1.04 V vs. NHE, Hg pool. CO2 was re-saturated every 10 h. Adapted with permission from ref. 258 and 259. Copyright 2018–2019 Royal Society of Chemistry and American Chemical Society, respectively. | ||
The same reactivity trend was also observed for the Co series, with enhanced FE, TOF and activity for CO production found for more rigid macrocyclic platforms.259 In particular, a stark difference was observed by comparing the bulk electrolysis data obtained in aqueous media (pH 4.2) on a Hg pool electrode (Fig. 17c). The more planar 54-Co system exhibited excellent CO selectivity over HER, in contrast to the open-chained bpy-NHC-Co catalyst which resulted in the production of an approximately 1:1 CO:H2 gas mixture. In a similar fashion as [Ni(cyclam)]2+, a reductive adsorption of the molecular catalysts over the Hg electrode surface was found to occur. As for the Ni cogeners, the structural features of the ligand scaffolds directly affected the electronic structure and reactivity of the reduced Co complexes, allowing to build-up a structure–reactivity relationship. However, unlike the Ni counterparts, the first electron uptake was found to be ligand-based for all of the Co complexes. On the contrary, the nature of the second reduction markedly differs across the series, being ligand-based for bpy-NHC-Co and metal-centered for 54-Co. Thus, for the Co series, more rigid ligand frameworks contribute to increase the nucleophilicity of the Co center in the 2e− reduced species, thus favoring the CO2 binding step and increasing the selectivity towards CO2RR. In contrast with the trend observed for the Ni and Co tetraazamacrocycles, the Co derivatives with bipyridyl-NHC ligands revealed to be better catalysts for CO2RR to CO compared to their Ni analogues in terms of overpotential, activity and selectivity in the presence of H2O. From one side, these examples highlight the crucial role of the redox non-innocent character and geometry of the ligand in catalysis. On the other hand, the differences in the electronic structure observed for Ni and Co macrocycles bearing the same ligand platform suggests that the nature of the metal center also has a strong influence on the reactivity towards CO2RR. In some cases, a simple change of the metal center may lead to a drastic change in the product distribution for CO2RR. For example, the Co complex bearing a pentadentate N5 pyridine-diimine macrocycle (55-Co) was reported as a selective catalyst for CO2 electroreduction to CO in DMF, whereas the Fe counterpart (55-Fe) produced HCOO− in high faradaic yields.260 The origin of such a drastic change of selectivity was ascribed to the poor π-donating properties of the active Fe site, which stabilizes a η1-OCOH adduct.
4.3 Non-macrocyclic tetradentate nitrogen ligands
In addition to the heme and non-heme macrocyclic systems, some representative classes of Fe, Co and Ni organometallic complexes based on non-macrocyclic N4 ligands have been recently reported to be competent catalysts for CO2RR.Lu and co-workers recently described a non-cyclam NiII tripodal homogeneous electrocatalyst (56, see Chart 5) exhibiting a remarkable selectivity toward CO production in DMF/H2O mixtures even in the presence of relevant amounts of added water.261 The origin of the high intrinsic affinity for CO2 over H+ and robustness of 56 compared to other analogous molecular complexes, was ascribed to the strong basicity of the ligand, containing the methylated benzimidazole donors. The redox-innocent character of the ligand strongly enhances the nucleophilicity of the NiI center which readily binds CO2, as suggested by an observable positive shift of the voltammetric NiII/I wave under CO2-saturated conditions. The product distribution could be tuned in a controlled manner by modulating the applied potential, affording syngas mixtures with a CO:H2 ratio of 1:1 or 1:2 at more negative potentials, likely due to the partial contribution of the less CO2RR selective NiI/0 wave.261
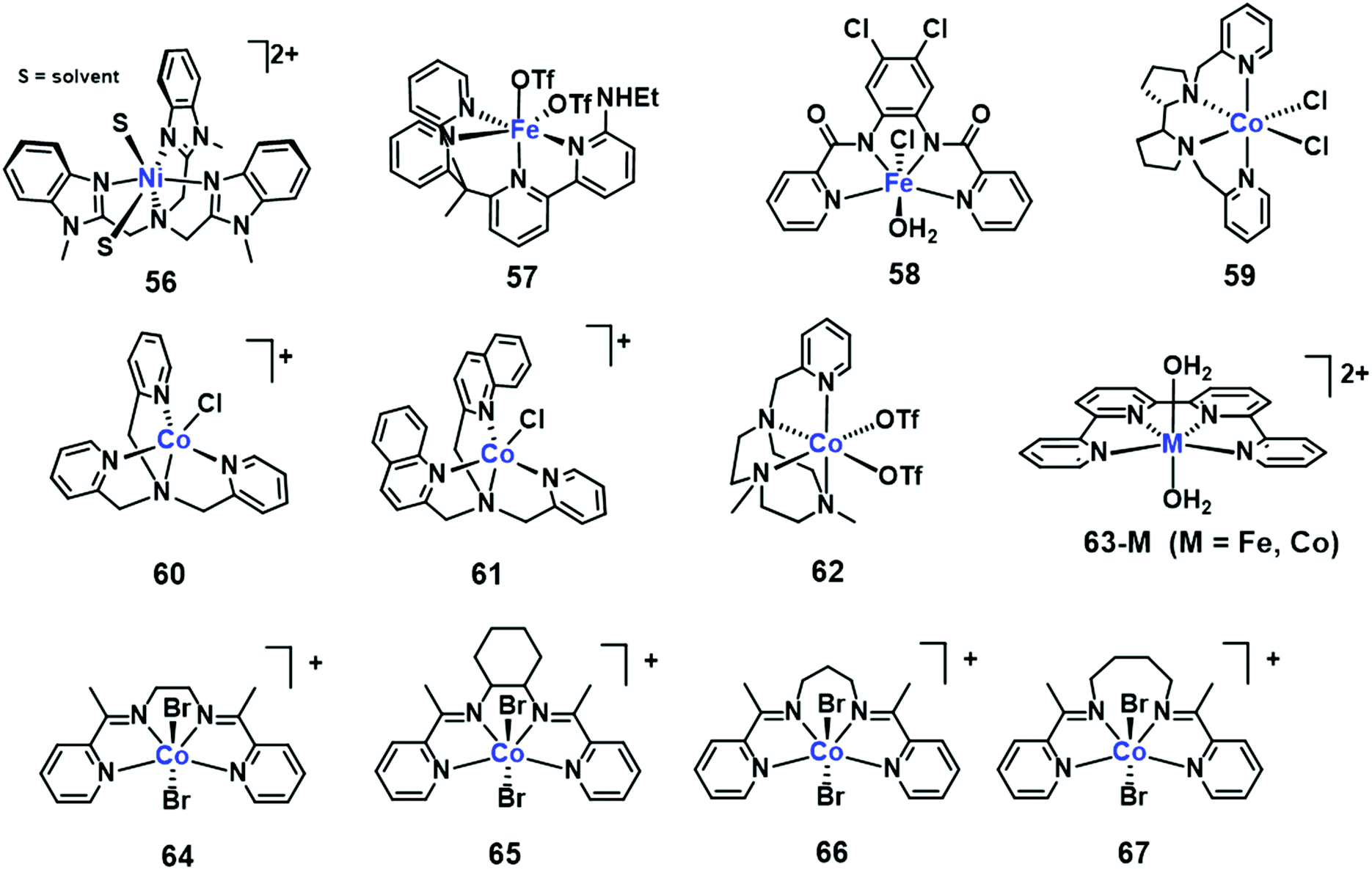 | ||
| Chart 5 Structures of selected molecular Ni, Co and Fe catalysts for CO2RR supported by non-macrocyclic tetradentate nitrogen ligands. | ||
The majority of the non-heme Fe electrocatalysts studied for CO2RR displayed high or moderate faradaic efficiencies for formate production, including a highly selective iron carbonyl cluster catalyst69,262,263 and Fe complexes with bipyridine-containing Schiff base ligands,264,265 phenanthroline derivatives266 or macrocyclic nitrogen platforms.260 Recently, a series of FeII complexes supported by polydentate bipyridyl-based platforms bearing different functional groups in the second coordination sphere, were explored for CO2RR.267 Among them, the only system showing a high selectivity for CO production in aqueous CH3CN (FECO = 81%; FEH2 = 11%) was a derivative featuring a pendant secondary amine group (57). An increased acidity of the intramolecular proton relay (phenolic functionality) was found to dramatically enhance the H2 evolution process. It is noteworthy that changes in the CO2RR selectivity induced by appended groups in the second coordination sphere have been recently reported also for molecular Mn catalysts.268–271 Moreover, a non-heme Fe complex with a tetradentate dicarboxamide N4 in-plane ligand (58), recently showed a high selectivity for CO2 electroreduction to CO as a homogeneous electrocatalyst in DMF/2% H2O solvent mixture.272 In addition, the complex was immobilized on N-doped graphene (N-G) by refluxing a DMF solution of 58 in the presence of suspended N-G platelets. It was suggested that this procedure induced immobilization of 58 on N-G via axial coordination by N dopants of the graphene support to the Fe centers, promoted by the π–π stacking interaction between the planar ligand framework of the complex and the graphene layer. The heterogeneous catalyst supported on a GC electrode resulted in an efficient and selective CO2 conversion to CO (FECO = 90%) in aqueous media (pH = 7.3) at −0.58 V vs. RHE.272
Among the earth-abundant transition metal molecular catalysts containing alternative polydentate platforms, the polypyridyl, aminopyridyl or tripodal cobalt complexes are perhaps the most widely studied systems. In general, the reported activity and selectivity of these catalysts strongly varies depending on the ligand framework and the specific operating reaction conditions. For instance, Che and co-workers recently reported a series of Co complexes with tetradentate N donor ligands for the electro- and photocatalytic CO2RR.273 Among those systems, only the cis-[Co(PDP)Cl2] complex (59, PDP = 1,1′-bis(2-pyridinylmethyl)-2,2′-bipyrrolidine) displayed a selective electrocatalytic CO production in CH3CN, albeit the FECO was found to progressively decrease over time during electrolysis. Contrariwise, the other complexes of the series supported by similar ligands displayed very poor electrocatalytic properties for CO2RR, resulting in the formation of only traces of CO.273 In another comparative study, a family of tripodal CoII complexes containing pyridyl or less basic quinolyl groups displayed variable ability for a selective CO2-to-CO conversion under electrochemical conditions, with the complexes 60 and 61 showing the best performances (FECO = 58–72% and 84%, respectively).274 Furthermore, a number of polypyridyl Co complexes were reported to suffer from very low faradaic yields for CO production (≤30%),275,276 which are not related to competitive HER or to alternative CO2RR pathways. In addition, moderate durability in long-term electrolysis is generally reported even for some CO-selective Co systems, suggesting a possible CO deactivation pathway.119
In an effort to rationalize the electrocatalytic CO2RR behaviour of aminopyridyl Co catalysts, Lloret-Fillol and co-workers recently reported a compelling experimental–computational mechanistic study on the model [Co(LN4)](OTf)2 complex (62, LN4 = 1-[2-pyridylmethyl]-4,7-dimethyl-1,4,7-triazacyclononane), elucidating its reactivity towards CO2 at different redox potentials.277 Owing to the highly basic character of the LN4 ligand, both experimental and computational data indicate that the nucleophilic CoI species is able to undergo CO2 binding and C–O bond cleavage steps at the CoII/I redox potential in anhydrous CH3CN, leading to the formation of the key [Co(CO)(LN4)]+ complex. This pivotal intermediate, analogously to the deactivating carbonyl species observed in the case of [Ni(cyclam)]2+ (vide supra), was in situ detected under electrochemical conditions and thoroughly characterized by a combination of spectroscopic and SEC techniques (UV-Vis, FTIR). However, the endergonicity of the CO release step from [Co(CO)(LN4)]+ prevented the observation of an electrocatalytic CO2RR behavior at low overpotentials. Under pure electrochemical conditions, a catalytic CO production can be achieved only at more negative potentials (close to the formal CoI/0 couple), required for further reduction of [Co(CO)(LN4)]+, giving rise to a proposed alternative pathway based on the binding of another CO2 molecule at the Co center (Fig. 18, blue). In contrast, an efficient catalysis was observed at the CoII/I redox potential under photochemical conditions, in which the more favorable CO release from the [Co(CO)(LN4)]2+ intermediate was proposed to be kinetically favored over the competitive diffusion-controlled electron transfer to form [Co(CO)(LN4)]+ (Fig. 18, green).277 The beneficial effect of visible-light irradiation on catalysis was further demonstrated by a substantial improvement of the observed FE for CO production during bulk electrolysis upon irradiation, suggesting that light-assisted electrocatalysis is an effective strategy to alleviate CO inhibition limitation. It is also worth noting that several reported Co aminopyridyl catalysts showed optimal performances under photochemical273,274,278,279 or photoelectrochemical280 conditions compared to pure electrochemical conditions.
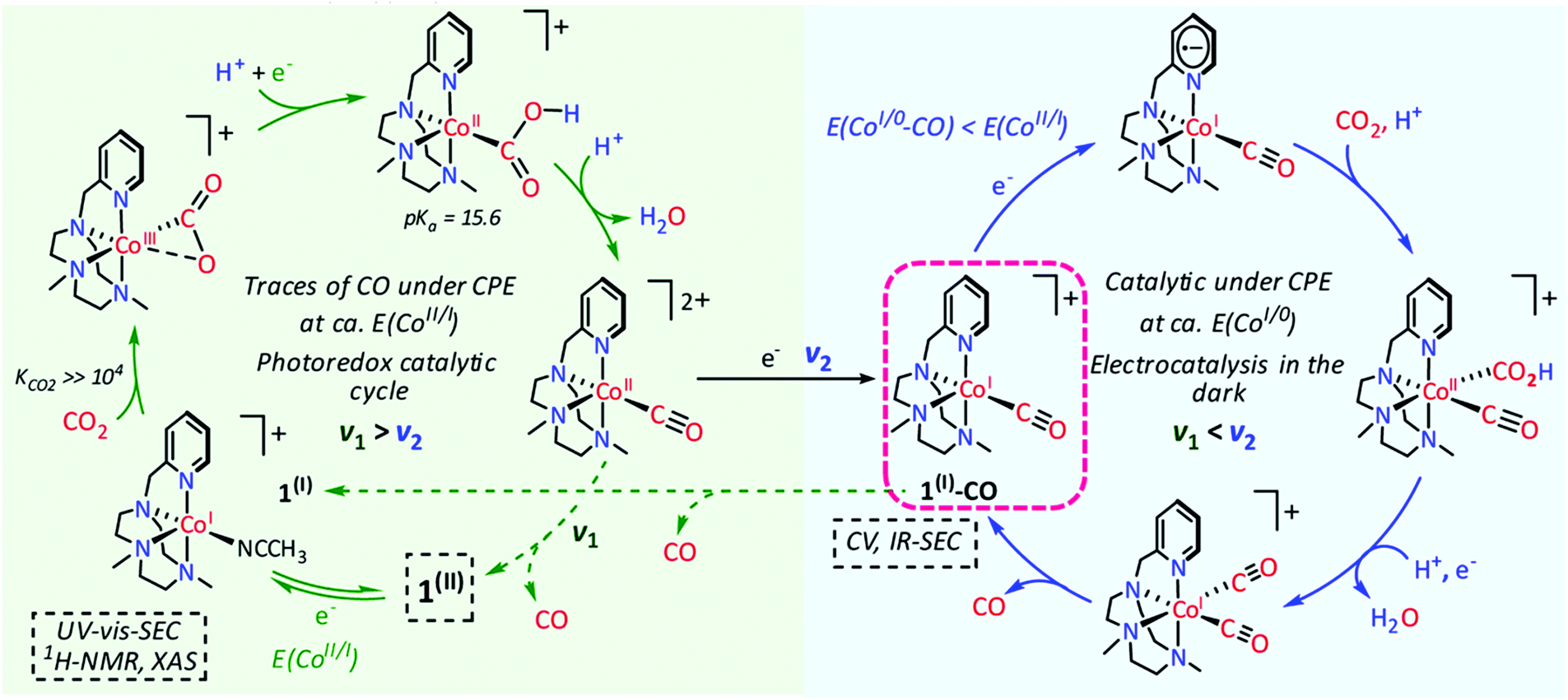 | ||
| Fig. 18 Proposed unified mechanism for photo- and electrochemical CO2 reduction to CO catalyzed by the molecular 1(II) = [Co(LN4)(CH3CN)2]2+ complex. The labile triflate ligands of compound 62 shown in Chart 5 are rapidly exchanged with coordinating solvent molecules in CH3CN, forming the doubly charged [Co(LN4)(CH3CN)2]2+ complex. The picture shows the main proposed catalytic intermediates based on experimental evidence (dotted boxes) and DFT calculations. CPE: Controlled-potential electrolysis. Adapted with permission from ref. 277. Copyright 2020 American Chemical Society. | ||
A strikingly similar CO inhibition process has been reported to affect also the electrocatalytic performances of the non-heme [Fe(qpy)]2+ complex (63-Fe, qpy = 2,2′:6′,2′′:6′′,2′′′-quaterpyridine).281 In CH3CN, the electrochemically generated FeI species engages a fast and irreversible CO2 binding and, after protonation and reductive C–O bond cleavage, leads to the formation of a [Fe(CO)(qpy)]+ adduct. Analogously to 62, the CO release from [Fe(CO)(qpy)]+ was found to compete with its further one-electron reduction to form the inactive neutral [Fe(CO)(qpy)]0 species, detected by UV-Vis and FTIR-SEC. Such a deactivation process led to a low FECO (48%) obtained during controlled-potential electrolysis at −1.2 V vs. SCE in the presence of 1 M PhOH, even though the system displayed an excellent CO selectivity over HER. A substantial improvement of the faradaic yield (70%) was gathered by using the light-assisted strategy.281 Moreover, 63-Fe revealed to be a very efficient and selective catalyst for visible light-driven CO2 conversion to CO in pure homogeneous photochemical conditions282 as well as in a hybrid system mixed with mesoporous graphitic carbon nitride.283
Notably, the Co quaterpyridine derivative, [Co(qpy)]2+ (63-Co), displayed excellent electrocatalytic performances without any evidence of inhibition by CO poisoning.281 In the presence of a large excess of added exogenous acid (3 M PhOH), the one-electron reduced CoI species reversibly binds two molecules of PhOH in axial position, forming the [CoI(qpy)(PhOH)2]+ complex. Further reduction of the latter leads to the doubly-reduced active [CoI(qpy˙−)(PhOH)2] species (the second electron is likely to be delocalized on the qpy moiety), responsible for a selective CO production (FECO = 94%) at a very low overpotential (−1.1 V vs. SCE, η = 140 mV). Owing to the high observed catalytic rates and low overpotential, 63-Co favorably compares with the most active reported molecular catalysts for CO2RR to CO and closely matches the performance of the highly efficient Fe0 porphyrins (see Fig. 19).281 In a related study, the use of high scan-rate cyclic voltammetry (up to 500 V s−1) shed light on the presence of an alternative reaction pathway dominant at low PhOH concentrations (<1.5 M).284 This mechanism, occurring at higher overpotential (η = 600 mV), is based on the formation of the active three-electron reduced [Co(qpy)]− species, which may undergo hydride formation with subsequent selectivity loss. Furthermore, the precursor [CoI(qpy˙−)] species readily adsorbed on the electrode surface, as already reported in earlier studies.285 Overall, high concentrations of added PhOH were found to enhance CO selectivity over HER, decrease the overpotential and prevent intermediate adsorption on the electrode surface. An analogous beneficial effect of the added proton source on the catalyst stability was also observed for other classes of molecular Co catalysts.286 Owing to the flat, conjugated geometry of the qpy ligand, [Co(qpy)]2+ was successfully immobilized on MWCNTs, leading to one of the most efficient heterogenized molecular catalysts reported so far for CO2RR to CO in aqueous media.287 At close to neutral pH (7.3), the [Co(qpy)]2+/MWCNTs electrode was able to quantitatively produce CO from CO2 with excellent durability and at a very low applied overpotential (−0.35 V vs. RHE, η = 240 mV).
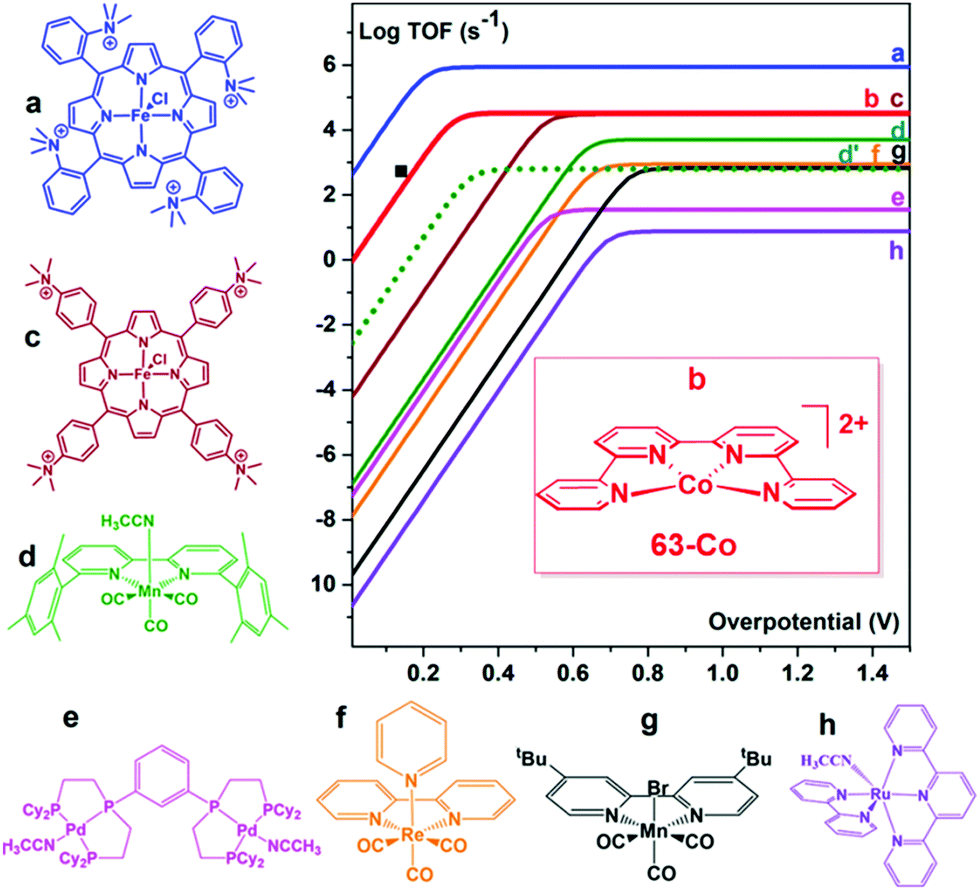 | ||
| Fig. 19 Catalytic Tafel plots of selected heme and non-heme molecular catalysts reported in the literature for the CO2-to-CO electrochemical conversion in DMF or CH3CN (a,112 c,112 d/d′,289 e,290 f,291 g,292 h293). The catalysts a and c correspond to the catalysts 20 and 18 shown in Chart 1, respectively. The data for catalyst b (63-Co) refer to ref. 281 (■: TOF value for 63-Co obtained from electrolysis data). Adapted with permission from ref. 281. Copyright 2018 American Chemical Society. | ||
The examples of macrocyclic and non-macrocyclic molecular systems discussed in this section suggest that both, electronic and structural factors should be taken into consideration to avoid CO inhibition, since flexible ligand geometries and electron-rich metal centers both contribute to stabilize CO adducts. In a recent study, McCrory and co-workers explored the catalytic behavior of a series of Co complexes with bis-(pyridylmonoimine)-based ligands of varying degrees of flexibility, suggesting a direct correlation between structural flexibility and catalytic CO2RR activity.288 In particular, a structurally rigid and quasi-planar catalyst (complex 64 in Chart 5) was shown to feature a more negative onset potential for the catalytic wave but also the highest efficiency toward CO production in the presence of high amounts of an added proton source. On the other hand, derivatives with structurally more flexible architectures (65–67) resulted in a more positive onset potentials for catalysis, but at the expense of considerably lower FECO values. As indicated by the higher CO binding equilibrium constants, lower CO dissociation constants and decreased activity under CO2/CO gas mixtures, the flexible Co systems were found to be more prone to CO poisoning during catalysis through the formation of stable CoI–CO carbonyl species, which was ascribed as the origin of the diminished overall activity.288
5. Single-atom catalysts
Single-atom catalysts (SACs) typically define a class of materials based on highly dispersed monometallic active sites embedded on a 2D conductive surface. The active site of SACs comprises not only the isolated single transition metal atom but also its three- or four-fold anchoring sites (typically C or N atoms), which mimic the role of the first coordination shell in molecular catalysis.294 So far, the overwhelming majority of the SACs reported for CO2RR are based on M–N–C materials, whereby atomically dispersed 1st row transition metals are incorporated onto N-doped carbon supports.295 The nature of the metal center and the structural features of the coordinative environment were found to strongly affect the CO2RR performance in terms of activity, selectivity and overpotential. The compact M–N3 or M–N4 configurations confined on a single carbon plane are the most commonly proposed moieties for the catalytic site of SACs, being reminiscent of the structures of molecular complexes with macrocyclic ligands employed as efficient catalysts for CO2 conversion to CO (Chart 6). However, although the effect of the surrounding carbon matrix has received considerably less attention, some groups have hypothesized a non-innocent role of the carbon atoms spatially close to the active site in the catalytic process.296,297 | ||
| Chart 6 Schematics of the main M–Nx–Cx coordinative configurations explored for single-site catalysts for CO2RR to CO atomically dispersed catalysts on a 2D carbon support. | ||
Hence, the SAC approach represents a promising strategy to tailor the electronic structure of the active site as means of controlling the reactivity toward CO2RR. In comparison with the corresponding metal-based bulk materials or NPs,14,298–300 SAC systems generally possess unique catalytic properties, selectively promoting CO2RR over the competing HER in aqueous media. Recently, single-metal porphyrin-like sites were predicted to hinder the HER by forcing an on-top *H adsorption as compared to the more energetically favorable hollow site binding of hydrogen on a bulk metal catalyst surface.301 Mixing bulk materials and SACs can even be exploited to controllably adjust the H2/CO ratio, which could be used as further feedstock for the Fischer–Tropsch process.
Despite the major benefits of SACs revealing high selectivity with maximal metal atom utilization, challenges related to the synthesis and structural characterization emerge. The availability, exclusivity and distribution of the SAC motifs on the prepared sample are critical for the development of SACs. Advanced characterization techniques like electron microscopy, X-ray absorption spectroscopy (XAS), Fourier-transform infrared (FTIR) spectroscopy or scanning tunneling microscopy (STM) can assist the design and the investigation of single-atom structures. In particular, aberration corrected TEM and high-angle annular dark-field STEM (HAADF-STEM) are common techniques used for the determination of the size and distribution of isolated metal atoms and to extract the local structural information.36 Moreover, synchrotron radiation techniques allow the investigation of the overall chemical environment of the single metal atoms. A further challenge is the possible non-uniform functionalization of the carbon structure, which leads to a variety of active sites and their irregular distribution on the support.302 In the case of M–N–C materials, a number of different nitrogen atom functionalities (e.g. pyrrolic, pyridinic, graphitic, etc.) are potential competitive coordination sites for the metal or may act themselves as metal-free catalytic sites for CO2RR, changing neutrality, altering charge and spin densities and promoting thermodynamic stability.296,303–305 Furthermore, aggregation phenomena should be considered in the investigation of SACs. Indeed, SACs tend to aggregate into clusters or NPs during the catalytic process or storage due to their thermodynamic instability, high surface free energies and low coordination. Therefore, their stability under reaction conditions should be demonstrated. Other factors related to the reaction conditions, such as pH and electrolyte, may also have a strong influence on their catalytic response.
In a recent study, a series of M–N–C model catalysts with different earth-abundant transition metals (M = Mn, Fe, Co, Ni, Cu) have been explored for CO2RR in aqueous media, in order to systematically rationalize the effect of the nature of the metal center on the catalytic activity and selectivity.306 All the materials featured atomically dispersed in-plane M–N4 moieties, with negligible structural and morphological differences across the series. This allowed a direct correlation between the CO2RR performances and the type of metal (alternative configurations with one or two adsorbed axial H2O molecules have also been considered, M–N4–H2O). The catalytic CO2RR tests at −0.5 V and −0.6 V vs. RHE (pH 6.7), resulted in a volcano-like diagram, with the Fe–N–C and Co–N–C catalysts being the most active (partial jCO), depending on the applied potential (Fig. 20a and b). However, the Fe-, Mn- and Ni–N–C catalysts revealed to be the most selective toward CO production across the series (FECO > 80%), whereas the high current densities for the Co–N–C counterpart predominantly accounted for H2 evolution (Fig. 20a). Operando XANES was also performed to monitor the changes in the oxidation state under catalytic conditions, suggesting that it remained unchanged during CO2RR for Co(2+) and Mn. It was partially reduced for Fe and Ni (from 3+/2+ to 2+ for Fe, and from 2+ to 1+ for Ni), while it was strongly reduced to Cu0 in the case of Cu.306 The experimental results were rationalized by using DFT calculations, which modeled the binding energies of the key intermediates involved in the CO2RR process, according to four elementary steps: (i) CO2 + e− → *CO2−; (ii) *CO2− + H+ → *COOH; (iii) *COOH + H+ + e− → *CO + H2O; (iv) *CO → CO. The rate-determining step (RDS) was found to be highly dependent on the nature of the metal center, being the *CO desorption step (step iv) for Mn–N4 and Fe–N4 moieties or the CO2 activation and first electron transfer for Ni–N4 and Cu–N4 (step i). In the series, the Co–N4 sites showed an optimum balance between the energy barriers for CO2 activation and CO desorption. In particular, the CO2RR activity across the series was rationalized in terms of the Gibbs free energy change at the rate-determining step (ERDS), resulting in a volcano trend analogous to the experimental one (Fig. 20b).306 According to this diagram, M2+–N4–H2O sites were proposed as the most active centers in Fe–N–C and Co–N–C, whereas the Ni1+–N4 configuration was predicted to be the most active in Ni–N–C, in analogy to the macrocyclic Ni molecular catalysts (see Section 4.1.1). Moreover, the difference between the binding energies for *CO2− and *H was proposed as a suitable descriptor to predict selectivity toward CO production, showing a direct correlation to the number of d-antibonding electrons, which is determined by the basic principles of coordination chemistry according to the crystal field splitting theory (Fig. 20c and d).306 Following this approach, the high selectivity experimentally observed for Fe and Ni catalysts could be reproduced, as well as the more favourable HER promoted by Co–N–C.306
 | ||
| Fig. 20 (a) FE at −0.6 V (up) and −0.5 V (bottom) vs. RHE obtained over the M–N–C catalysts (M = Mn, Fe, Co, Ni, and Cu) for 90 min electrolysis under CO2-saturated 0.1 M KHCO3 aqueous electrolyte (pH 6.7) at room temperature. (b) Comparison of experimental CO partial current density at −0.6 V vs. RHE for pyrolyzed M–N–C materials and DFT calculated trends (U = −0.6 V vs. RHE). ERDS is the calculated free reaction energy at the specific RDS of CO2RR to CO for each active site. (c) Octahedral, square pyramid and square planar symmetry (orange indicates the antibonding states). (d) The experimental selectivity at −0.6 V vs. RHE over Fe–N–C, Co–N–C, and Ni–N–C vs. the DFT-simulated E(*CO2−) − E(*H).306 Adapted with permission from ref. 306. Copyright 2019 American Chemical Society. | ||
This representative example shows the crucial role played by the metal center in determining the catalytic activity and selectivity of a series of single-site catalysts featuring similar coordinative environments. In the next paragraphs, we aim to explore the different types of metal coordination proposed for the main families of SACs (Chart 6).
5.1 Ni–N–C catalysts
An indicator of the growing interest in Ni–N–C catalysts is given by the increasing amount of studies published in the last years. Most of these reports mainly focus on the development of novel synthetic procedures to obtain efficient Ni–N–C catalysts, aiming to preserve coordinatively unsaturated Ni–N sites confined into N-doped 2D materials.307–309 The most common synthetic approach consists in a simple pyrolysis of a Ni salt with N-doped carbon materials obtained from N-rich organic molecules. For instance, the dispersion of Ni sites in a N-doped carbon matrix obtained from dicyandiamide and 2-methylimidazole led to a catalyst displaying maximum FECO 97% at −0.9 V vs. RHE.309 In another study, an increased content of pyrrolic-N centers in N-doped carbon hollow spheres was obtained by modulating the concentration of the melamine precursor, remarkably increasing the density of Ni sites and, in turn, the catalytic CO2RR activity.310 Metal–organic Ni complexes containing nitrogen ligands, such as [Zn(Ni)-bidppz] (bidppz = 11,11′-bis(dipyrido[3,2-a:2′,3′-c]phenazinyl))308 or Ni(NH3)6I2,311 were also employed as suitable precursors to achieve robust Ni–N–C SACs for selective CO2-to-CO reduction. Remarkably, Wu and coworkers recently reported a Ni SAC containing well-defined Ni–N4 sites synthesized through a topo-chemical transformation of a Ni-doped g-C3N4 sample with a carbon layer. A combination of XAS data and TEM/HAADF-STEM images proved the retention of the preformed Ni–N4 structures (Ni units coordinated to pyridinic-N) upon the thermal treatment and the lack of agglomeration of the Ni atoms to form particles.312 The Ni–N4 moieties, which are reminiscent of the structure of the Ni cyclam molecular catalyst, were supposed to be responsible for the excellent CO2RR activity, producing CO in high faradaic yields (99%) at −0.81 V vs. RHE.Further research provided a more in-depth structural characterization of the Ni–N4 active site and their role in the CO2RR, revealing several similarities with the molecular Ni-based N4 macrocyclic systems. Recently, a detailed XAS and XPS study allowed the elucidation of the structural features of the active moiety in a robust and highly CO-selective Ni SAC dispersed on an N-doped graphene matrix (A-Ni-NG, see Fig. 21).313 The isolated Ni–N4 moieties displayed a distorted square-planar geometry which deviates from the canonical D4h symmetry typical of the NiPc molecular derivative. Moreover, XAS and EPR measurements confirmed the presence of monovalent paramagnetic NiI centers with a 3d9, S = 1/2 electronic configuration, which possess a high intrinsic reactivity due to the partially filled 3dx2−y2 orbitals. In analogy with the molecular Ni cyclam-like derivatives, the initial NiI state is responsible for the CO2 binding step, as confirmed by operando XAS data, Fig. 21. Just by adding CO2 to the electrolyte solution at open-circuit voltage (OCV), the Ni K-edge was found to shift to higher energy, ca. 0.4 eV with respect to the spectrum recorded under Ar atmosphere, indicating an increase of the Ni oxidation state, Fig. 21a. These results are consistent with a charge transfer from the NiI center to the C 2p orbital of the bound CO2 molecule to form an adsorbed *CO2δ− species, similarly to what it was observed for Co cyclam-like complexes (Fig. 21c).246 Nonetheless, there was no clear evidence of charge transfer from the Ni center to CO2 in the case of the [Ni(CO2)(cyclam)]+ adduct (see above).11 When a bias potential (at −0.7 V vs. RHE) is applied under CO2, a shift of the Ni K-edge back to lower energies was observed, suggesting the regeneration of the starting NiI state after the completion of a single two-electron CO2-to-CO turnover, Fig. 21a. Interestingly, an observable shift of the main EXAFS peak to longer lengths during CO2RR (at −0.7 V vs. RHE) is indicative of an out-of-plane distortion from the planar Ni–N4 graphene configuration, probably due to a redistribution of the electron density in the 3d orbitals of Ni between the 4 Ni–N bonds and the Ni–C bond, Fig. 21b. As previously discussed, significant distortions in the macrocyclic geometry have been predicted to stabilize the axial Ni–CO bond and disfavor CO dissociation from the [Ni(cyclam)(CO)]+ intermediate, and are supposed to be at the origin of the CO inhibition phenomenon affecting the homogeneous Ni-cyclam system.217 The geometrical constraints forced by the graphene framework may prevent an excessive distortion of the square-planar cyclam-like Ni active site, ensuring a more efficient CO release step as compared to the Ni-cyclam homogeneous counterpart. As above mentioned, the electrode-flattened cyclam structure of the adsorbed [Ni(cyclam)(CO)]+ complex has been proposed to be responsible for the enhanced catalytic rates observed for [Ni(cyclam)]2+ on a Hg electrode in comparison with the homogeneous derivative.217,226 Analogous trends correlating the planarity of the ligand scaffold to a more efficient CO release step were also previously discussed for other families of molecular catalysts (see Section 4).288
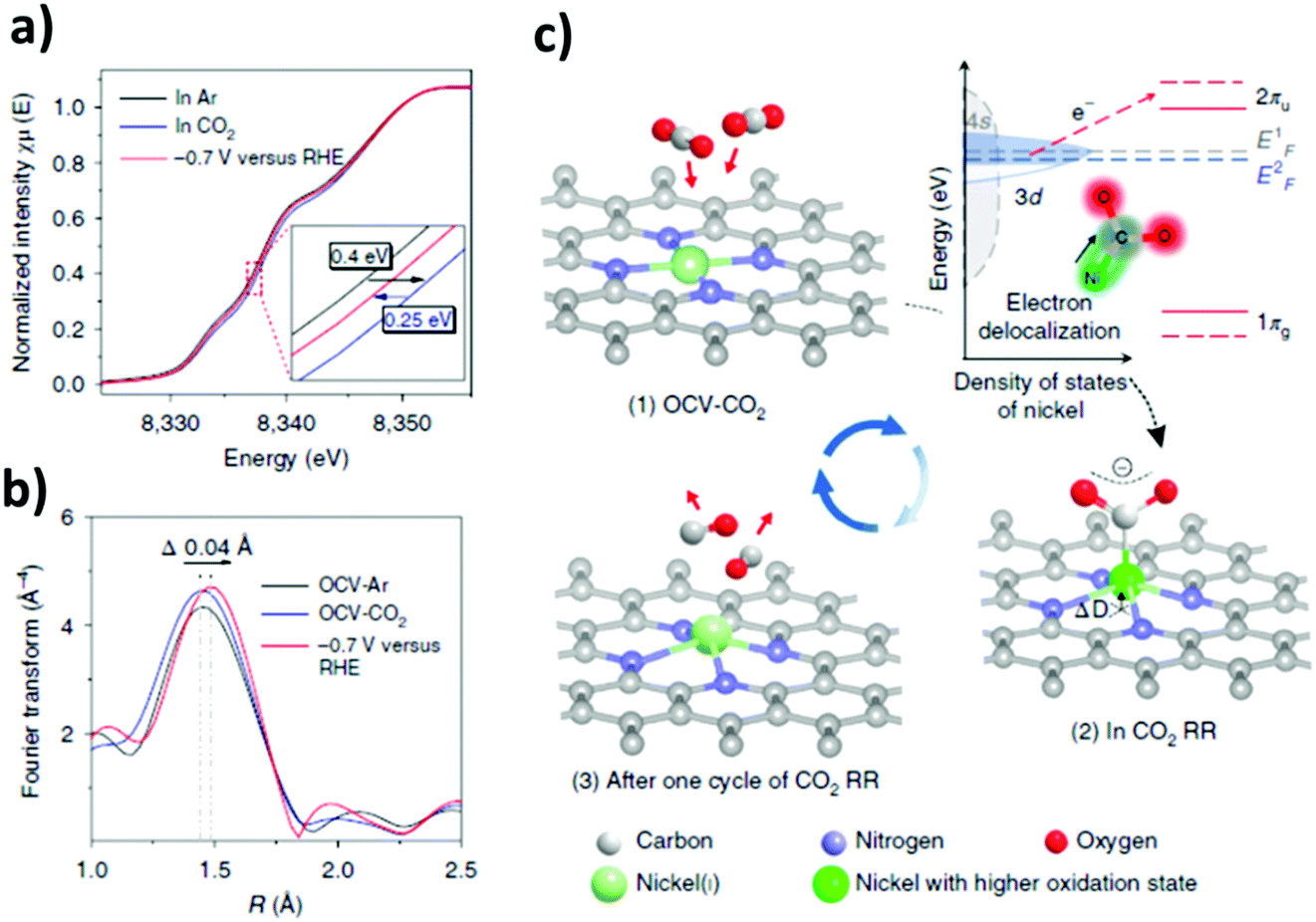 | ||
| Fig. 21 Operando XAS data from ref. 313. (a) Normalized Ni K-edge XANES spectra (enlarged in the inset) and (b) Fourier transform magnitudes of EXAFS spectra (without phase correction) of A-Ni-NG. (c) Dynamic structural changes of the active site during CO2 activation and electroreduction. ΔD: out-of-plane displacement of Ni atom resulting from charge transfer from the NiI atom to CO2. E1F and E2F: Fermi levels of A-Ni-NG before and after formation of Ni–CO2δ−, respectively. 1πg and 2πu: CO2 molecular orbitals.321 Adapted with permission from ref. 313. Copyright 2018 Nature Publishing Group. | ||
The unique properties of Ni–N4 motifs in promoting an efficient CO2RR have been demonstrated in several other recent reports. These materials are generally obtained by facile adsorption of Ni2+ ions on N-doped graphene314 or a porous carbon matrix.315 However, well-defined organometallic Ni complexes containing pre-formed Ni–N4 coordination units can be adopted as single-site precursors of SACs, being the source of both, metal and nitrogen at a time. Notably, the local coordination environment of the active site in the Ni SAC is highly dependent on the structure of the original complex precursor and the type of support material. For instance, the annealing of a Ni complex with a flexible tripodal nitrogen ligand led to a SAC containing distorted square-planar NiII–N4 species uniformly dispersed over GO sheets, which displayed superior CO2RR performance than the molecular derivative.316 The presence of GO in the synthetic process was found to play an essential role in preventing the formation of metallic bulk NPs which reduce the catalytic activity.
In another study, a Ni SAC synthesized by C–C coupling of a Ni phthalocyanine precursor (NiTAPc) with CNTs served as a model system to investigate the mechanism for electrochemical CO2RR.317 This approach resulted in the formation of uniformly distributed Ni–N4 moieties with a canonical D4h symmetry and NiII oxidation state, almost identical to the original sites of the NiTAPc precursor. A combination of operando XAS, operando Raman and NAP-XPS provided insights into the dynamic changes of the electronic state of Ni during CO2RR, indicating that the initial NiII species does not interact with CO2, yet the low-valent reduced NiI state is the species responsible for CO2 activation. These results are consistent with those previously discussed for another Ni SAC (vide supra) and establish a direct correlation with the behavior of molecular Ni cyclam-like systems whereby the NiI state is the catalytically active species.11 The proposed pathway for CO2RR to CO on the Ni SAC is analogous to the one proposed for molecular Ni catalysts with N4 macrocyclic ligands (see Scheme 2).215 This involves the initial reduction of NiII to NiI, which then undergoes an electrophilic addition of CO2 to form the adsorbed *CO2− intermediate, with electron delocalization from the Ni center to the bound CO2 molecule. As aforementioned, only moderate charge transfer from the NiI center to coordinated CO2 was observed for the molecular [Ni(CO2)(cyclam)]+ adduct.11 In agreement with the experimental Tafel slope and the reaction orders for H+/CO2, the next hydrogenation step, *CO2− + H+ → *COOH, was found to be rate-limiting, followed by the formation of the adsorbed *CO intermediate and the final release of gaseous CO from the surface.
The control over the coordination environment of the Ni–N–C material has been suggested to be critical to achieve an efficient CO2RR. Although “cyclam-like” macrocyclic Ni–N4 moieties are generally proposed as the active sites in several reported Ni–N–C SACs, alternative configurations have been recently predicted to display high intrinsic CO2RR activity. Moreover, the high annealing temperature usually required for the synthesis of Ni–N–C materials may generate defects in the carbon matrix, making the identification of the active site even more challenging. In particular, the role of C species in the coordination of the Ni atoms has been considered. Wang and co-workers explored the CO2RR performances of different Ni sites in graphene vacancies containing various Ni–C/Ni–N coordination environments.318 They observed that the incorporation of N-dopants induced remarkable changes in the morphology of the Ni sites but also greatly improved the selectivity towards CO production. More recently, a general host–guest cooperative protection strategy based on the introduction of polypyrrole into bimetallic MgNi-MOF-74, followed by a pyrolysis step, allowed to obtain Ni SACs with tunable N/C coordination by controlling the annealing temperature.319 The hybrid NiSA–N2–C material with the lowest N coordination number (2) showed the best performance across the series, affording a maximum FECO 98% at −0.8 V vs. RHE. By using theoretical and experimental tools, Yamauchi and co-workers systematically investigated a series of hybrid Ni–NxC4−x (x = 0–4) moieties containing different combinations of coordinating N or C atoms in an attempt to elucidate the role of C atoms on the structural and catalytic properties of Ni N–C SACs.320 For each configuration, the thermodynamic barrier associated to each of the three elementary steps of the CO2RR pathway were compared, i.e. the *COOH formation (* + CO2 + H+ + e− → *COOH), the *CO formation (*COOH + H+ + e− → *CO + H2O) and CO desorption (*CO → * + CO). Looking at the electronic structure, it was found that C-rich configurations tend to lead to an increase in the electron density over the Ni atom, favouring the CO2 activation step at the expense of a stronger interaction with adsorbed CO. On the other hand, an increase in the N coordination number facilitates the CO desorption step, yet limiting the nucleophilicity of the Ni center. Overall, the Ni–N4 sites are limited by the first electrochemical step, showing the highest barrier for *COOH formation, whereas CO desorption is energetically unfavorable for Ni–C4 moieties. Therefore, the Ni–N2C2 configuration was predicted to be the optimum structure for CO2RR, displaying the best compromise between these opposite tendencies. The experimental data corroborated the theoretical predictions, since Ni–N–C materials containing mixed N/C coordination synthesized at intermediate carbonization temperatures provided the best results in terms of activity and selectivity towards CO production.320 Interestingly, the coordinative motif of Ni–N2C2 moieties homogeneously dispersed on a 2D graphene layer shows structural analogies with an efficient homogeneous Ni catalyst bearing a macrocyclic ligand based on a redox-active 2,2′-bipyridyl core and electron-rich N-heterocyclic carbene (NHC) donors (54-Ni), previously discussed in the Section 4.2.258 In particular, the optimal 15-membered macrocyclic configuration forced the molecular Ni system 54-Ni to a distorted square-planar arrangement which is consistent with synchrothron-based experimental data on analogous Ni–N2C2 SACs.320 For the molecular Ni derivative 54-Ni, the contribution of the quasi-planar ligand environment to extra-electron delocalization revealed to be decisive to prevent the formation of a metal-hydride species.
The question on the optimal coordination structure in Ni–N–C SACs is however still under debate, due to the possible heterogeneities that can be found in the real materials, as well as the extreme difficulty to unambiguously characterize experimentally the local coordinative environment of a catalyst and to precisely control its structure/composition through synthetic parameters. Moreover, DFT calculations based on the so called Computational Hydrogen Electrode (CHE) model may also lead to an oversimplified description of the system.322 Recently, the kinetic barriers for the CO2RR pathway promoted by several hybrid Ni–N/C coordination sites were calculated by using ab initio molecular dynamics (AIMD), proposing the mixed Ni–N1C3 configuration to be the most active and selective site for CO2RR.322 Importantly, the study shed light on the site charge capacity and the H-bonding interactions as the crucial factors determining the activity and selectivity of a given SAC. The former describes the number of charges the site can carry and contributes to lower the barrier for the electrochemical steps, yet might be detrimental for an efficient CO desorption. Furthermore, the H-bonding interactions with water molecules of the medium are essential to assist the initial chemisorption of CO2 on the surface, stabilize the intermediates and facilitate the protonation steps. It is remarkable to note that the beneficial effect of cooperative H-bonding interactions on CO2RR was demonstrated for the [Ni(cyclam)]2+ molecular catalyst in water and in organic electrolytes (Section 4.1).230 In particular, the presence of an urea-based exogenous additive led to a significant enhancement of the catalytic response of [Ni(cyclam)]2+ due to its ability to form cooperative multipoint hydrogen-bond interactions (Fig. 14), without altering the selectivity of the process. Moreover, we have already discussed the beneficial role of water or polar solvents on CO2 binding, activation and conversion observed for Ni and Co tetraazamacrocyclic complexes (Section 4.1).11,210,243,247
In contrast with the predominant literature on Ni N–C SACs, some reports have suggested that coordinatively unsaturated Ni–N moieties are preferential catalytic sites for CO2 binding and conversion, decreasing the affinity for HER.323,324 For example, some Ni–N active sites confined on porous carbon with different coordination numbers were synthesized by pyrolysis of a Zn/Ni bimetallic zeolitic imidazolate framework-8 (ZIF-8), displaying a high CO selectivity (FEs 92–98%) over a broad potential window (−0.53 V to −1.03 V vs. RHE). In agreement with the low Ni coordination numbers experimentally observed by EXAFS analysis, computational data suggested that unsaturated Ni–N2V2 (V stands for coordination vacancy) possess a lower free energy for the formation of the *COOH intermediate as compared to the Ni–N4 sites.324 An impregnation-pyrolysis method using a highly-defective sponge-templated GO support was suggested to favor the formation of analogous coordinatively unsaturated Ni sites, due to the trapping of Ni atoms in the material defects.325 In another study, a pyrrolic Ni–N3 moiety was proposed as the catalytically relevant configuration for a highly active Ni SAC confined within a porous N-doped carbon sheath.326 Nakanishi and co-workers adopted a Covalent Triazine Framework (CTF), consisting of microporous conjugated polymers with 1,3,5-triazine linker units, as a suitable platform to favor the formation of coordinatively unsaturated metal SACs.327 Upon the impregnation of Ni2+ ions into the reticular structure, the resulting porous material (Ni-CTF) displayed a lower Ni–N coordination number (close to 3) than the Ni-TPP reference, with Ni sites analogous to terpyridyl or iminopyridyl molecular Ni units embedded into a polymeric matrix.328–330 As corroborated by DFT calculations, which suggest a more favorable free energy barrier for *COOH formation for Ni–N3 sites as compared to Ni–N4 moieties, Ni-CTF was found to outperform Ni-TPP for CO2RR, leading to a FECOca. 90% at −0.8 V vs. RHE.327
The influence of the carbon matrix should be also taken into account to rationalize the CO2RR performances of Ni–N–C materials. For instance, pendant residues located in the second or outer coordination sphere may actively participate in the catalytic CO2RR pathway of the SAC, as widely demonstrated for several enzymatic systems and artificial molecular electrocatalysts (see Sections 3 and 4).48 In this regard, Ni–N2+2–C8 structures located at the edge between two adjacent carbon sheets were predicted to promote an energetically more favorable pathway for CO2 reduction to CO, as compared to the isolated bulk-hosted Ni–N4–C10 sites confined within a single graphitic layer.296 This is due to the capability of uncoordinated carbon atoms with dangling bonds close to the catalytic site to assist the dissociation of the *COOH intermediate. Moreover, the optimization of the physicochemical properties of the support material revealed to be an effective approach to enhance the catalytic performance of Ni–N–C catalysts. More specifically, an increased porosity and a high density of defects on the carbon support have been shown to facilitate fast CO2 mass transport and enhance the incorporation of the single metal atoms, contributing to boost the catalytic activities of the Ni–N–C catalysts.323,331–333 Yang and co-workers explored a top-down strategy via thermal atomization to prepare N–C materials enriched with Ni single atoms, by trapping the latter on the surface defects of the N-doped carbon matrix. This approach was found to improve the atomic distribution of the active sites on the support surface, positively impacting the CO2RR activity, selectivity to CO (FECO > 90% between −0.6 V and −1.0 V vs. RHE) and durability of the catalyst.331 In another report, a highly porous and surface defect-rich microwave exfoliated graphene oxide was used as support to disperse N-coordinated Ni atoms.332 The abundant defects on the pore edges favored the anchoring of the Ni atoms, leading to high selectivity and mass activity.
By virtue of the promising catalytic properties of Ni SACs, some efforts have been recently done to implement them in real devices by using Gas Diffusion Electrodes (GDE) in order to achieve commercially relevant current densities. A reactive three-phase interface is prepared by feeding gaseous CO2 from the back of a GDE to optimize the accessibility of CO2 to the catalyst surface. This setup overcomes the limitation of the low CO2 solubility and mass transfer in aqueous electrolyte. A few recent reports afforded high current densities over 300 mA cm−2 with selective CO production,334–336 reaching a maximum of FECO 90% between 100 and 200 mA cm−2.334 Albeit exhibiting similar performances as the benchmark AgOx catalyst in an H-type cell (Fig. 22a), the Ni–N–C catalysts showed higher current densities and less dramatic FE decrease for CO production in the electrolyzer setup (Fig. 22b). Recently, commercially relevant current densities for CO production (308 mA cm−2) with excellent selectivity (FECO 88%) could be sustained for up to 120 hours by using a flexible and robust Ni SAC-decorated porous carbon membrane as GDE.315 The highly hydrophobic structure was found to favor CO2 adsorption and possess higher electrochemical active surface area as well as low electric resistance.
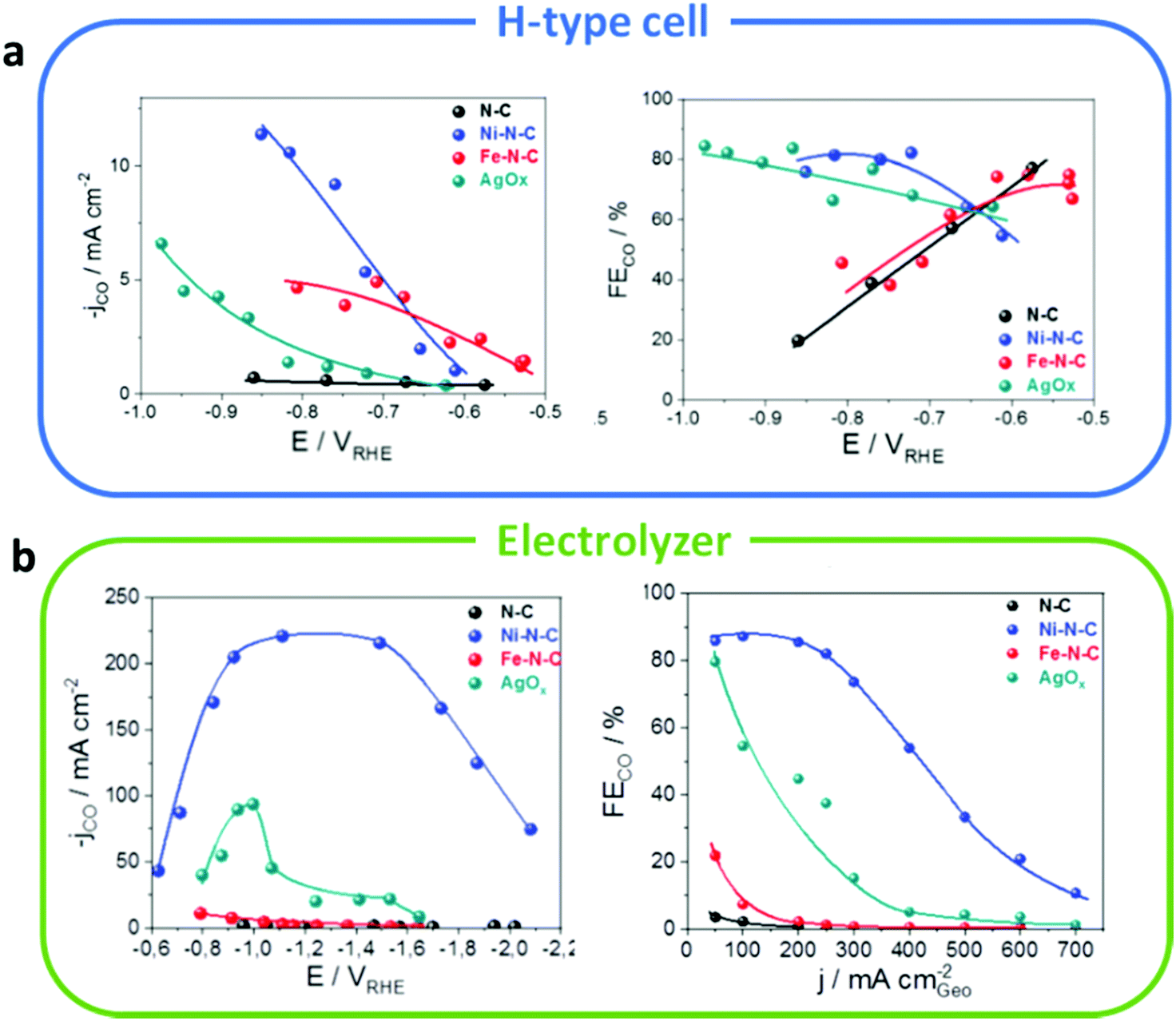 | ||
| Fig. 22 (a) Catalytic performance (geometric CO current density) and product analysis (FE) of N–C (black), Fe–N–C (red), Ni–N–C (blue) and AgOx (cyan) catalysts measured in a H-type cell displayed as a function of the applied electrode potential. (b) Electrolyzer test results under controlled current densities (galvanostatic operation). The CO partial current density as a function of the electrode potential and the faradaic CO efficiency as a function of the applied electrolyzer current density are shown. All tests were performed in a CO2-saturated 1 M KHCO3 electrolyte and 1 mg cm−2 catalyst loading. Adapted with permission from ref. 334. Copyright 2019, Royal Society of Chemistry. | ||
5.2 Fe–N–C catalysts
Due to its extreme earth-abundance, the utilization of catalysts based on Fe for efficient CO2 conversion to CO is highly desirable. Jaouen, Fontecave and co-workers studied the effect of different ratios of Fe-SACs and Fe-NPs on the catalytic performance in a series of Fe–N–C materials, establishing a correlation between the catalyst structure and CO2RR activity/selectivity.298 Depending on the synthesis, different amounts of atomically dispersed Fe atoms or particles were obtained in the N–C materials, which was found to strongly affect the CO2RR performance. In particular, isolated square-planar Fe–N4 moieties were found to be the active species for a selective CO production (FECO up to 80% at −0.5 V vs. RHE), whereas heterogeneous Fe-based species (e.g. NPs or carbides) mainly produced H2 under similar conditions. Interestingly, the higher the fraction of crystalline Fe, the higher the FE for H2. These results showed that the Fe speciation within the Fe–N–C material strongly affects the CO/H2 ratio, being attractive in the perspective of using non-expensive catalysts to produce CO/H2 mixtures useful for Fischer–Tropsch technologies. Although the crucial role of Fe on CO2RR in Fe–N–C catalysts has been verified by passivating the metal center with the coordinative SCN− ion leading to a decreased activity,337 the identification of the real active site responsible for the catalytic behavior of Fe–N–C systems is still under debate. For instance, some studies reported that a high CO selectivity can be even achieved by using metal-free N–C materials, albeit exhibiting considerably lower current densities than those obtained in the presence of Fe.338 Moreover, the mismatch experimentally observed between the CO2RR activity trends for a series of M–N–C (M = Fe, Co, Ni) and the corresponding molecular metalloporphyrins, suggests that ideal M–N4 active sites are only a rough approximation of the real catalytic units in M–N–C materials and that the interactions with the surrounding carbon matrix may play a relevant role in catalysis.338By using a combined experimental and theoretical approach, Pan et al. investigated the structural features of isolated Fe–N4 moieties on a carbon matrix synthesized by thermal activation of Fe-doped ZIF-8.297 Synchrotron-based XAS measurements confirmed the presence of discrete Fe–N4 sites containing Fe centers predominantly in 3+ oxidation state. However, Mössbauer spectroscopy suggested the co-existence of two different types of Fe–N4 structures within the Fe–N–C sample: planar bulk-hosted Fe–N4–C10 units fully embedded in a 2D graphitic layer, and out-of-plane distorted edge-hosted Fe–N2+2–C8 sites (see Chart 6). The latter bridge two armchair-like N-doped graphitic layers through the coordination between a Fe atom and 2 N heteroatoms exposed at the edges. According to computational DFT data, CO2RR is predicted to occur more favorably at the edge-hosted Fe–N2+2–C8 configuration, due to the beneficial role of the neighboring C atoms with dangling bonds to facilitate the key *COOH dissociation step.297 These data emphasize the importance of the specific interaction between the active site and the hosting environment in controlling the activity and selectivity of Fe–N–C SACs, suggesting analogous outer coordination sphere effects observed in homogeneous Fe systems with heme112,120,124–126,131 or non-heme ligands267 (see Sections 3 and 4).
An in situ infrared spectroscopy study in attenuated total reflection mode (ATR-IR) on a model Fe–N–C catalyst containing Fe–N4 sites provided useful insights into the nature of the catalytically relevant units in Fe–N–C materials and the main factors limiting their activity (Fig. 23a).339 When the applied potential matched the foot of the catalytic wave, an absorption band near 1900 cm−1 started to grow, indicating the formation of a *CO species adsorbed on the surface, as further confirmed by control experiments under CO atmosphere (Fig. 23b). In agreement with a *CO inhibition process, its intensity did not decrease significantly after purging the solution with Ar and applying more negative potentials, suggesting the formation of a spectator deactivation species rather than an on-cycle intermediate (Fig. 23c and d). In order to determine which sites could be affected by *CO poisoning, DFT calculations suggested that bulk-hosted Fe–N4 and edge-hosted Fe–N2+2 structures are catalytically inactive, since both of them displayed a prohibitively strong *CO adsorption. Contrariwise, Fe–N4 units within graphitic pores of a defect-rich Fe–N–C material are considered the active sites for CO2RR to CO, due to a local electronic effect of the pore environment which contributes to weaken the Fe–C bond.339 As previously discussed (Section 4), the formation of stable metal carbonyl deactivating species during CO2RR has been experimentally detected for several homogeneous Ni and Co systems based on non-heme N4 macrocyclic217 or aminopyridyl277,288 ligands. A molecular CO scavenger217 or light irradiation277 were successfully used as alternative strategies to alleviate the CO inhibition and favor catalyst regeneration in molecular systems (see Section 4). In a similar fashion, the formation of an inactive Fe carbonyl species (detected by in situ UV-Vis and FTIR SEC) was also found to limit the electrocatalytic CO2RR performance of the 63-Fe homogeneous catalyst in organic electrolytes at low overpotentials, leading to low faradaic efficiencies for CO production (Section 4.3). It is worth noting that this complex contains a well-defined Fe–N4 moiety, which is structurally similar to the sites accessible in the Fe–N–C SACs.281 Similarly to other molecular systems, visible-light irradiation significantly improved the catalytic performance.281–283
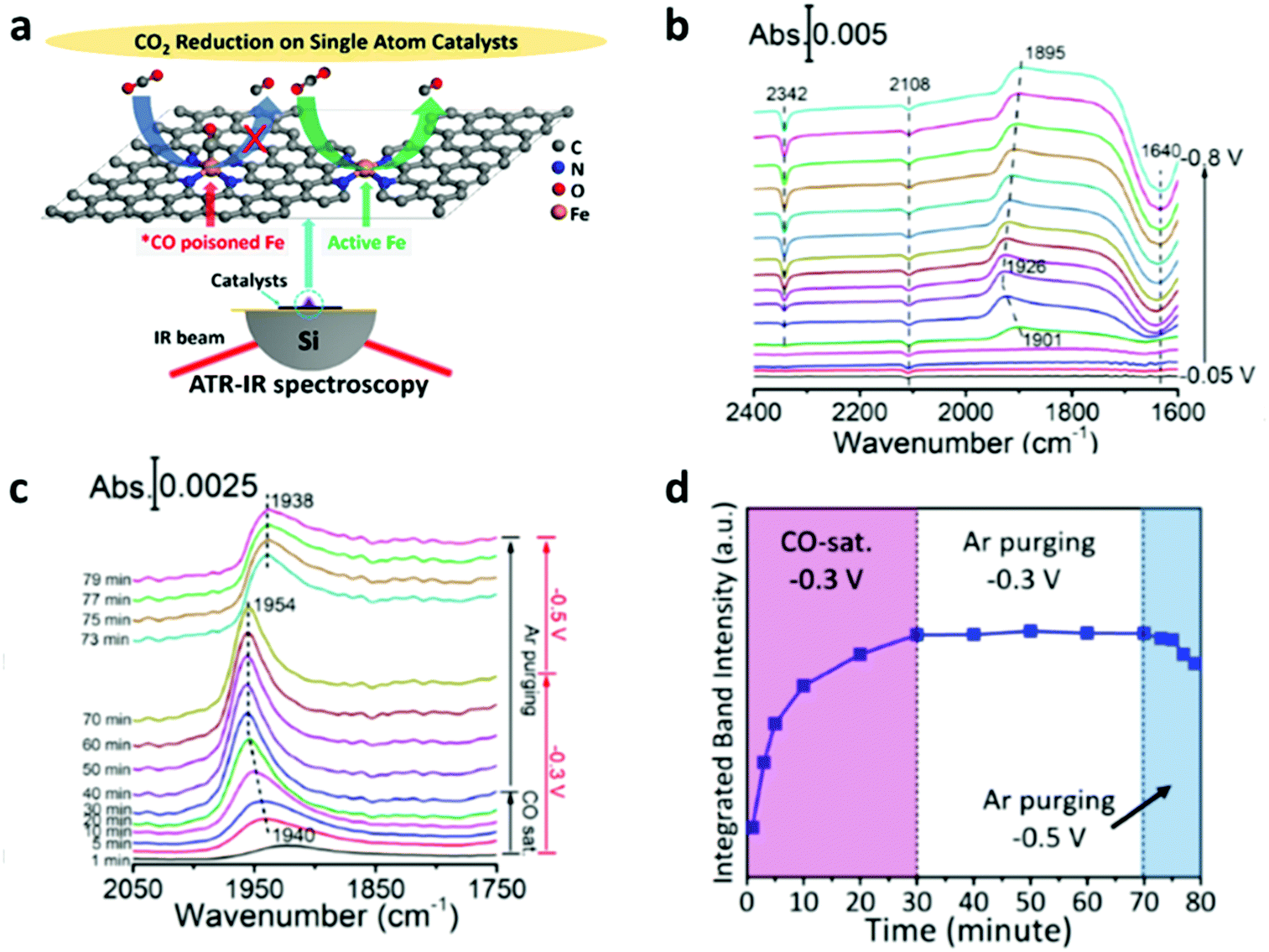 | ||
| Fig. 23 (a) Scheme of the setup. (b) Real-time ATR-IR spectra recorded while stepping the potential of the Fe–N–C-loaded Au/Si prism in the CO2-saturated 0.5 M KHCO3. (c) Real-time IR spectra and (d) the evolution of the integrated *CO band intensity when switching the CO/Ar atmosphere and potential. IR references were taken at 0 V for panel (b) and 0.1 V for panel and (c). Adapted with permission from ref. 339. Copyright 2019 American Chemical Society. | ||
A recent experimental study carried out by Chen, Hu and co-workers provided a significant improvement in the elucidation of the structure–activity relationship in Fe N–C SACs, establishing a direct correlation between coordinative environment, metal oxidation state and catalytic CO2RR activity.340 In this work, a Fe–N–C material featuring a porphyrin-like Fe3+–N4 pyrrolic coordination environment, displayed an outstanding activity for CO2RR to CO in a flow-cell setup, affording jCO = 94 mA cm−2 at −0.45 V vs. RHE with FECO > 90%. Operando XAS at −0.4 V vs. RHE revealed that the oxidation state of Fe remained unchanged during sustained CO2RR, likely due to a strong electronic coupling with the conductive support, as recently reported for some electrode-conjugated molecular sites.341,342 However, at more negative applied potentials (<−0.5 V vs. RHE) a decreased stability of the catalytic system was observed concomitantly with an apparent shift to lower energies of the Fe K-edge, indicating a Fe3+/2+ reduction. Moreover, the EXAFS spectra also revealed that the catalyst deactivation was accompanied by a drastic change in the local structure of the first coordination shell of the Fe atom, resulting in a decreased nitrogen coordination number (from 4 to 3) due to the loss of a pyrrolic N. It is worth noting that a partial hydrogenation or carboxylation of porphyrin rings under electrochemical conditions has been reported in several Fe molecular systems.72,93 The crucial role of the coordinative environment on the CO2RR performance was further demonstrated by the dramatic activity loss and faster deactivation observed for a reference Fe2+–N4 sample containing 4 pyridinic N atoms, synthesized from the [Fe(phen)3]2+ precursor. Importantly, unlike the case of the Fe3+–N4 catalyst, the CO2RR reaction at Fe2+–N4 sites was found to be rate-limited by CO desorption at high overpotentials. Taken together, these results show that the Fe3+–N4 (pyrrolic) configuration is able to promote an extremely durable and efficient CO2RR to CO: the pyrrolic N atoms stabilize the high Fe3+ oxidation state, which reduces the π-backdonation to adsorbed CO, resulting in a weaker CO binding. On the other hand, the Fe2+–N4 (pyridinic) sites catalyze CO2RR less efficiently due to stronger CO binding on Fe2+ center stabilized by pyridinic N.340 Notably, these findings match well with the different catalytic behavior commonly reported for homogeneous Fe catalysts bearing heme and non-heme ligands, suggesting an intimate correlation between some specific local metal-coordinative features in Fe–N–C SACs and the corresponding structurally similar Fe-based homogeneous counterparts. For instance, Fe porphyrin molecular systems, whose structure is reminiscent of the Fe3+–N4 (pyrrolic) sites, are known to be very efficient electrocatalysts for CO2RR to CO in organic electrolytes as well as in water, and are not generally affected by severe CO poisoning under CO2RR. On the other hand, in analogy with Fe2+–N4 (pyridinic) single sites, the Fe quaterpyridine homogeneous catalyst (63-Fe) suffers from CO deactivation under pure electrocatalytic conditions with the formation of stable Fe carbonyl species.281 Such a fascinating comparison between Fe SACs and molecular systems is a clear example that unveiling the relationships between structural features of a molecular ligand framework (e.g. rigidity vs. flexibility, macrocyclic vs. open structures) and the electronic properties of key intermediates involved in CO2RR may pave the way for a rational design of both, homogeneous and heterogeneous catalysts.
Another recurrent topic in the available literature on Fe–N–C SACs is the beneficial effect of additional nitrogen doping on the CO2RR catalytic performances.305,343–345 An increase in the level of N doping was first predicted by first-principle calculations to facilitate the *COOH formation and *CO desorption steps in Fe–N4 SACs.346 In a similar fashion, the presence of additional N atoms near the Fe–N4 was calculated to exert a synergistic effect which contributes to lower the barrier for the protonation of the bound *CO2−, resembling the stabilizing effect of pendant amine,245,257,267 amide125 or urea126 groups in the second coordination sphere in molecular catalysis (see Sections 3 and 4).339 From an experimental perspective, Yang et al. studied the effect of the oxidation treatment and found improved CO2RR activity resulting from an increased amount of pyrrolic N.345 In another recent investigation, the annealing of hemin (Fe source) in the presence of an excess of melamine (N-rich additive) and defective graphene led to atomically dispersed Fe–N5 sites stabilized by pyrrolic N-doped graphene, acting as an additional axial ligand coordinated to Fe–N4 moieties (Fig. 24a).344 This unique configuration was found to outperform a Fe–N4 catalyst in terms of robustness and selectivity to CO (FE 97% at −0.46 V vs. RHE, Fig. 24b). Theory and the analysis of the local density of states (LDOS) suggested that the additional axial ligand contributes to reduce the electron density from the 3d orbitals of the metal center, alleviating the π-backdonation to coordinating CO and favoring the rapid CO release step. Following a similar concept, Yang and co-workers developed an alternative core–shell strategy to improve the *CO desorption step in Fe–N–C SACs, based on the encapsulation of Fe nitride nanoparticles (FexN) with graphene layers containing atomically dispersed Fe–N4–C units.347 Owing to the additional coordination to the N atom of the FexN core, the surface single-site assumed a Fe–N5–C configuration, displaying a much more efficient *CO desorption step as compared to the bare Fe–N4–C configuration. An analogous trans effect, based on the influence of a trans donor ligand on the CO2 activation bond, was recently observed for a homogeneous Ru catalyst bearing a 2,2′:6′,2′′-terpyridine and an asymmetric pyridyl-N-heterocyclic carbene (NHC) ligands.348 A stereochemical control was found to be essential to efficiently catalyze CO2RR to CO, since the presence of the strong σ-donating NHC ligand in trans to the site for CO2 bonding significantly boosted the kinetics of the CO dissociation step. Owing to the extreme versatility of this effect, trans coordination of a tertiary amine led to a change in the selectivity of the Fe porphyrins towards formate production, contributing to facilitate the protonation of reduced CO2 as well as the formate release step (vide supra).106 It is worth noting that, as discussed above, a beneficial effect of a pyridine or oxygen donor axial coordination was experimentally observed for electrode-functionalized molecular Co complexes with macrocyclic ligands.161,192–195 In a Fe–N–C single-site catalyst, the presence of a H2O molecule axially adsorbed on Fe–N4 moieties was found to lower the reaction barrier for the first electron transfer to form *CO2−.306 Moreover, in a tetraazamacrocyclic Co molecular complex, the trans axial coordination of a solvent CH3CN molecule to a five-coordinated Co–CO2 adduct led to an increase of the observed metal-to-ligand charge transfer (Section 4.1.2).11,246
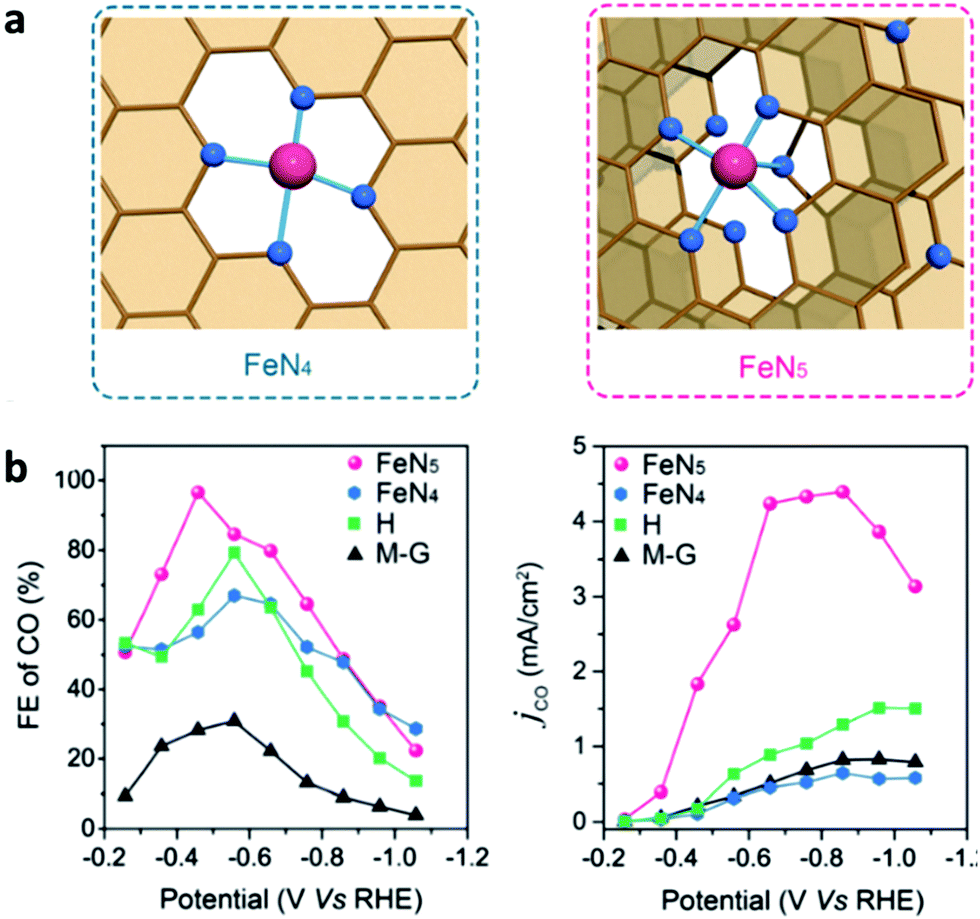 | ||
| Fig. 24 (a) Scheme of the FeN4 and FeN5 catalysts. CO2RR catalytic performance of the as-synthesized catalysts. (b) Faradaic efficiency and partial current density for FeN5, FeN4, and the precursors hemin (H) and melamine with graphene (M–G). Adapted with permission from ref. 344. Copyright 2019, Wiley-VCH. | ||
In spite of the numerous studies focusing on the effect of the coordinative environment of the Fe center on the CO2RR activity of Fe–N–C catalysts, the number of systematic reports concerning the influence of the reaction conditions (other than the applied potential) on catalysis is relatively scarce. In this regard, Strasser and co-workers investigated the role of the pH and electrolyte on CO2RR activity and selectivity of Fe–N–C catalysts, containing Fe–Nx moieties.349 Unlike H2 evolution, which was strongly favored at acidic pH values, the catalytic CO production was found to be independent of the pH on the normal hydrogen electrode (NHE) potential scale. This implies that the CO/H2 ratio may be fine-tuned by controlling the electrolyte pH, achieving high CO selectivity at high pH values. Moreover, the CO formation rate as a function of the pH on the RHE scale suggested a decoupled electron–proton transfer (DEPT) mechanism for CO production (Fig. 25a), whereby the rate-determining step is the formation of the key (Fe–N–C)–CO2− intermediate, followed by an irreversible protonation step (Fig. 25b). Notably, an analogous mechanism has been described for an immobilized Co protoporphyrin catalyst by Koper and co-workers (see Section 3.2.1), in which the formation of the key [Co(P)(CO2)]− adduct occurs after the initial one-electron reduction to form the active [CoI(P)]− species.51,154 Analogous reaction pathways have also been proposed for homogeneous macrocyclic catalysts,215 strongly suggesting that the molecularly defined solid-state Fe–Nx motifs are the primary active sites in Fe–N–C. These moieties may contribute to stabilize the (Fe–N–C)–CO2− intermediate giving rise to a solid-state heterogeneous charge-transfer reaction mechanism strikingly similar to that occurring in macrocyclic metal–Nx molecular catalysts complexes.349 The K+ cations were also proposed to provide an additional stabilization of this intermediate,349,350 exerting a beneficial effect on catalysis which resembles the synergistic boosting effect observed upon the addition of Lewis acids in several heme93,94 (see Section 3.1.1) and non-heme289,351,352 molecular catalysts. However, it is worth noting that the mechanism can be affected by the pH.353 These results are extremely important since they contribute to unify some fundamental concepts of the fields of heterogeneous and homogeneous catalysis.
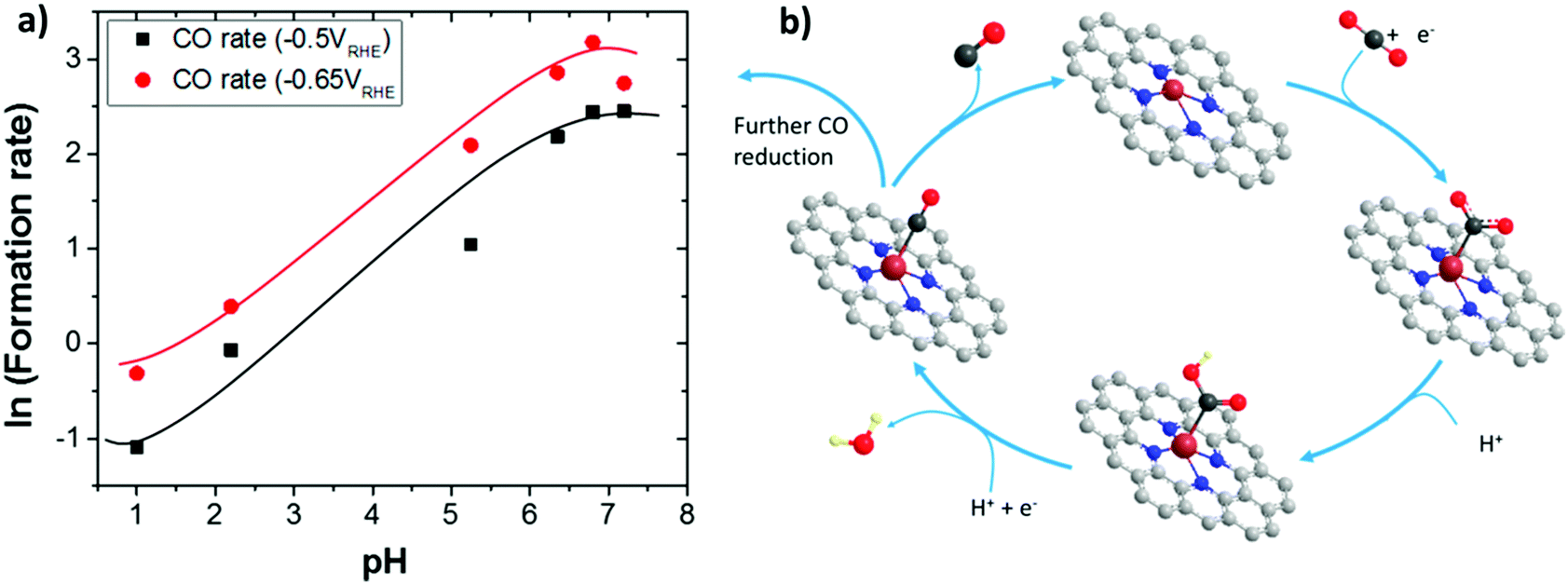 | ||
| Fig. 25 (a) pH dependence of the CO formation rate at a constant potential on the RHE scale (black, −0.5 VRHE; red, −0.65 VRHE); (b) scheme of the proposed catalytic cycle of the CO2RR on Fe–N–C catalysts. Adapted with permission from ref. 349. Copyright 2018 American Chemical Society. | ||
An interesting aspect of Fe–Nx moieties in Fe–N–C SACs relies in their ability to promote the reduction of CO2 to beyond 2e products, representing one of the few reported examples of solid-state non-Cu-based electrocatalysts able to produce hydrocarbons. In this perspective, it is supposed that the strong CO binding occurring at the Fe sites plays an important role, and that moderate *H adsorption may favor further protonation of the adsorbed *CO to generate >2e products. The rate for CH4 production was found to increase at low pH, suggesting that CO protonation is the rate-limiting step. Nevertheless, the concomitant enhancement of HER resulted in low FEs for CH4 production (<1%).349 In agreement with theoretical predictions,57,301 Fe–Nx sites of Fe–N–C materials were found to produce small amounts of CH4 in addition to the predominant CO. Moreover, operando EXAFS data revealed that an unusual change in the Fe oxidation state from Fe2+ to Fe1+ is likely involved in the CH4 production process.354 Interestingly, a molecular iron porphyrin system (complex 18, see Chart 1) was recently shown to catalytically generate CH4 from CO2 or CO under homogeneous photochemical conditions in the presence of a photosensitizer and a sacrificial electron donor.355 In this report, CO is a key intermediate in the CO2-to-CH4 process and the FeII/I process is pivotal, via the formation of a Fe-formyl intermediate. It is worth noting also that the Fe quaterpyridine complex 63-Fe was used as a molecular precursor for the electrochemical conversion of CO2 to CH4 (FE ca. 2%) in CH3CN in the presence of TEOA as a proton source.356 In this case, in situ formed Fe particles derived from the electrochemical decomposition of the starting molecular complex are likely the responsible species for CH4 production. In a recent integrated experimental–computational study, Ju et al. also elucidated the possible pathways for CO2RR to CH4 at the Fe–Nx sites as well as the role of the possible intermediate products formed during the CO2-to-CH4 cascades.357 As a result, CO was found to be the key intermediate toward CH4, CH3OH and CH2O production, yet CH2O (but not CH3OH) could be further reduced to CH4. Moreover, the isolated nature of atomically dispersed Fe–Nx units was identified as the main origin for low hydrocarbon selectivity, suggesting “dual-site” or “hybrid-tandem” catalysts, featuring cooperative sites for *CO and *H adsorption, as viable promising strategies.
5.3 Co–N–C catalysts
In contrast to the superior CO2RR performances displayed by the heterogenized molecular Co phthalocyanine complexes in comparison with derivatives containing other metals,204 Co–N–C SACs (especially Co–N4 sites) are typically intrinsically less selective for CO2RR than their Ni and Fe analogs, resulting in lower faradaic yields for CO production.297,337 This behavior is generally due to the predominant competitive HER process on Co–N–C catalysts,316,358 related to unfavorable *H adsorption energies and higher energetic barriers for *CO desorption as compared to the Fe and Ni analogues.306,314,318 Notably, the lower selectivity displayed by atomically dispersed Co–N4 sites versus the Ni counterparts reflects the different reactivity of molecular Ni and Co systems based on tetraazamacrocyclic ligands. As previously discussed in Section 4.1, the molecular [Ni(cyclam)]2+ catalyst is able to promote a selective CO2-to-CO conversion in aqueous media over a wide pH range, due to the unfavorable protonation of the active NiI species even at very acidic pH under CO2-saturated conditions.214 On the contrary, macrocyclic CoI species are generally protonated much easier, forming Co hydride intermediates which favor the competitive HER process over CO2RR (see Section 4.1).11 Furthermore, albeit CO inhibition has been experimentally observed also for the [Ni(cyclam)]2+ homogeneous system,217 CoI non-heme macrocycles are known to possess a great affinity for CO binding.11 The in situ formation of stable Co carbonyl species, hindering an efficient CO2RR, has been reported also for homogeneous Co systems bearing tetradentate aminopyridyl nitrogen ligands (see Section 4.3).277Nevertheless, the catalytic properties of Co–N–C materials towards CO2RR have been reported to be extremely dependent on the local coordination environment of the Co centers, with promising results obtained for alternative configurations respect to the conventional Co–N4 sites. A common strategy to control the nitrogen coordination number of single Co atoms consists in the variation of the pyrolysis temperature, since increasing annealing temperatures tend to favor a progressive disruption of the Co–N bonds. Following this approach, the CO2 electroreduction behavior of a series of Co–N–C SACs containing Co–N2, Co–N3 and Co–N4 moieties, respectively, was systematically investigated (Fig. 26a–c).359 As a general trend, the selectivity towards CO production was found to increase upon progressively decreasing the Co–N coordination number from 4 to 2, with Co–N2 showing the highest value for maximum FECO (95% at −0.68 V vs. RHE, Fig. 26d and e). In agreement with these results, the Co–N2 configuration showed the lowest charge-transfer resistance from the catalyst to the CO2 molecule, thereby favoring the activation of the latter to form CO2˙−. Interestingly, first-principles calculations suggest that a strong *CO binding occurs at the Co–N2 moieties. It was proposed that the coordination of a second CO molecule on the unsaturated Co site would help to promote the *CO desorption step.359
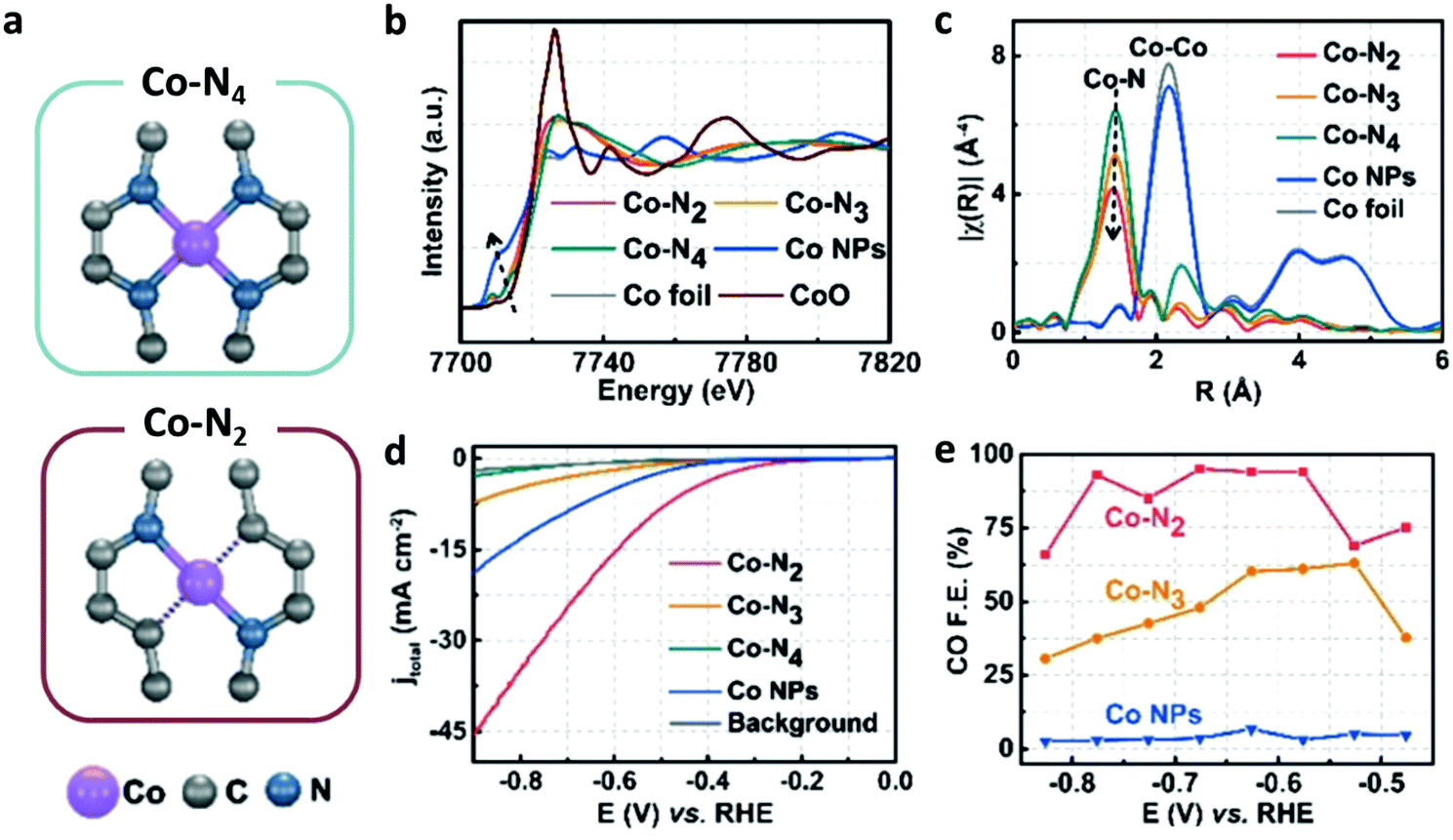 | ||
| Fig. 26 (a) Schemes of Co–N4 and Co–N2. (b) XANES (c) EXAFS spectra suggesting the atomic dispersion of Co atoms in Co–N2, Co–N3, and Co–N4, and the lowest N coordination number in Co–N2. (d) LSV of Co–N2, Co–N3, Co–N4, and Co NPs and pure carbon paper as background. (e) CO faradaic efficiencies at different applied potentials. Adapted with permission from ref. 359. Copyright 2018, Wiley-VCH. | ||
Nonetheless, an opposite trend was reported for some Co–N–C catalysts, suggesting that an increase in the N coordination number is beneficial for CO2RR. For example, atomically dispersed Co–N5 sites embedded into polymer-derived hollow N-doped porous carbon spheres exhibited remarkable CO2RR activity and stability, reaching a FECO 99% at −0.79 V vs. RHE.360In situ XAS measurements further confirmed the catalytic relevance of the Co–N5 units during CO2 electroreduction. Under similar conditions, the molecular CoPc metal precursor, containing well-defined planar Co–N4 moieties, resulted in lower efficiencies and current densities for CO production. In an attempt to decrease the Co–N coordination number in the original Co–N5 sample, the latter was pyrolyzed at increasingly higher temperatures, leading to a progressive drop in the observed FECO, partly due to formation of inactive Co NPs. The unique Co–N5 configuration was predicted to favor the *COOH formation and the *CO desorption steps.361 As mentioned above, analogous positive effects on catalysis due to the presence of axial nitrogen donors were found for Co molecular systems with heme macrocyclic ligands192,194 and in Fe–N–C SACs.344,347 In another study, a Co–N–C SAC containing isolated Co–N4 moieties provided a considerably improved selectivity for CO2-to-CO conversion in comparison with a Co–N4−x−Cx derivative accommodating Co sites with lower Co–N coordination number (FEs 82% vs. 47% at −0.8 V vs. RHE for Co–N4 and Co–N4−x–Cx, respectively).362
5.4 Other transition metals (Zn, Cu, Mn, Sn, Bi)
Albeit less studied than the Ni, Fe and Co analogs, a few single-atom M–N–C materials containing transition metals such as Zn, Cu, Mn, Sn or Bi, have been employed as efficient catalysts for CO2RR. Among them, the Mn–N–C systems have attracted growing interest due to the earth-abundant character of Mn, but also to the promising results shown by molecular Mn complexes bearing polypyridyl ligands.42,363–365 Mn–N–C catalysts were first reported in comparative studies on a series of M–Nx–C catalysts containing different transition metals, showing limited catalytic performances for the Mn–N–C materials as compared with the Ni and Fe counterparts.366,367 However, high-performance for CO2RR was subsequently reported for atomically dispersed Mn sites on an N-doped carbon matrix with a halogen and nitrogen dual-coordination, namely (X, N)–Mn/G (X = Cl, Br, I).368 More specifically, the (Cl, N)–Mn/G catalysts exhibited an excellent selectivity to CO with a maximum FE of 97% at −0.6 V vs. RHE, representing one of the best-performing heterogeneous catalysts reported so far for CO2RR. X-ray absorption spectroscopy analysis of (Cl, N)–Mn/G revealed a unique out-of-plane distorted Mn–N4Cl configuration for the active site, in which the low-valent Mn center (<2+) is coordinated to four N atoms and to one axial Cl atom. Under CO2-saturated atmosphere, in situ XAS spectra showed a higher energy shift for the Mn K-edge of (Cl, N)–Mn/G, consistent with an increased oxidation state of the Mn centers due to charge transfer from the low-valent Mn to the 2p orbitals of CO2 to generate adsorbed *CO2δ−. As mentioned above for the operando XAS data on a Ni–N–C catalyst,313 during CO2RR at −0.6 V vs. RHE, the Mn K-edge shifted back to lower energy. Notably, only little changes were observed for the Cl-free N–Mn/G sample, suggesting that the halogen atom plays a crucial role in the CO2 activation process. The computational data shed light on the crucial role played by the axial Cl coordination in the catalytic reaction. First, it was predicted to promote a distortion of the Mn site structure in agreement with the experimental results, facilitating the adsorption of CO2 and *COOH formation. Moreover, it displayed a stabilizing electronic effect on the *COOH intermediate increasing the electron density at the Mn–C bond, as well as facilitating the *CO desorption step. As previously discussed, analogous effects have been observed for several molecular systems192,194 and atomically dispersed M–Nx active sites344,347,361 coordinated to axial nitrogen donors.Copper is another interesting metal for heterogeneous CO2RR catalysis, widely studied for its unique ability to produce C2/C3 chemicals.39,369,370 However, in contrast with bulk heterogeneous Cu-based materials, Cu–N–C single-atom catalysts have been predicted to be quite unstable under electrochemical conditions. In particular, DFT calculations have shown low thermodynamic stability of the Cu–Nx sites under strongly reducing conditions (<−0.7 V vs. RHE) resulting in spontaneous decomposition of the N-coordinated Cu ions into metallic Cu NPs.367 Furthermore, the d-orbital of the Cu atom is filled with 9 (one singly occupied orbital) or 10 (fully occupied) electrons, indicating that the covalent bonds between copper and nitrogen are quite unlikely.57,302 In spite of these considerations, unsaturated Cu–N2 coordinated moieties anchored into a graphene matrix (Cu–N2/GN) have been recently reported to efficiently convert CO2 to CO with FECO 81% at −0.5 V vs. RHE.371 The high selectivity was attributed to the unsaturated environment of copper, as suggested by a relatively low calculated potential barrier between CO2 and the Cu–N2 moieties. However, the structural changes of the active sites occurring during the catalytic reaction remain unclear.
Zn has been studied in bulk materials for CO production but with limited performance and unclear underlying reaction mechanism.372 In contrast, some Zn–N–C materials prepared by different synthetic methods have been reported as highly selective catalysts towards CO production.373,374 From XPS and EXAFS analysis, the active site was suggested to involve Zn–Nx moieties, with pyridinic N atoms as possible Zn-coordination sites.374 The presence of Zn–N4 moieties as active sites was further corroborated by experimental and DFT results, showing a lower free energy barrier for the rate-limiting formation of *COOH than the comparing Zn–C4 and N4/C structures.373 Lastly, a few examples of SACs based on Sn and Bi have been reported. In particular, atomically dispersed Sn in N-doped catalysts were found to switch their selectivity at low overpotentials from HCOO− to CO with a FE of 91% at −0.6 V vs. RHE.375 Another interesting study revealed an excellent CO selectivity for Bi–N4 sites on carbon networks, reaching a FECO of 97% at −0.5 V vs. RHE.376
6. Metal nanoparticles for CO2RR
In order to develop heterogeneous metal catalysts for highly efficient and selective CO2RR, a rational design of the catalytic surface is required to control the stability of surface-bound intermediate species. In this scenario, by controlling the morphological, electronic, and surface chemical properties of metal nanoparticles (NPs) (or nanoclusters (NCs)), it is possible to understand the key factors influencing the performance of nanostructured CO2RR catalysts.369,377–383 For example, the size and shape of NPs can be used to control the number of low-coordinated sites and the ratio between the different crystal facets on the surface, which can affect the binding strength of the reaction intermediates (Section 6.1). In addition to structural effects related to the metal NP itself, the interaction between the NP surface and either the support material or small surface-bonded molecular stabilizers may play a role in determining the adsorption properties and electron transfer kinetics of the catalysts, thus influencing the stability of the intermediates involved in the catalytic reaction. These effects will be discussed in the Sections 6.2 and 6.3, respectively.6.1 Size and shape effects
A systematic investigation of the metal NP size-dependence of CO2RR was carried out for Au NPs. Sun et al.384 explored the electrocatalytic CO2RR behavior of monodispersed Au NPs with various sizes (4, 6, 8 and 10 nm, respectively), showing that the selectivity of CO2RR to CO tends to decrease with decreasing NP size. To account for this effect, DFT was used to calculate the free energies of reaction steps for CO and H2 production on certain crystal sites (i.e. Au(111), Au(211) and a 13-atom Au cluster) present on the NPs surface (Fig. 27a). The results revealed that catalytic CO evolution preferentially occurs at the Au(211) sites (denoted as “edge sites”), while Au(111) and 13-atom Au clusters (denoted as “corner sites”) are more active sites for H2 production. Hence, the increase in the H2 selectivity observed with decreasing NP size was ascribed to an increased content of corner sites on the NP surface. Analogous results were also obtained for micellar Au NPs within the size range of 1–8 nm.385 Specifically, small Au NPs (1.1 nm in size) displayed an activity that is more than two orders of magnitude higher than larger Au NPs (7.7 nm in size) (Fig. 27b). However, the higher activity was mainly associated to HER rather than CO production (Fig. 27c), consistent with the weaker binding of the COOH* intermediates and the presence of large H* coverages on the small Au NPs.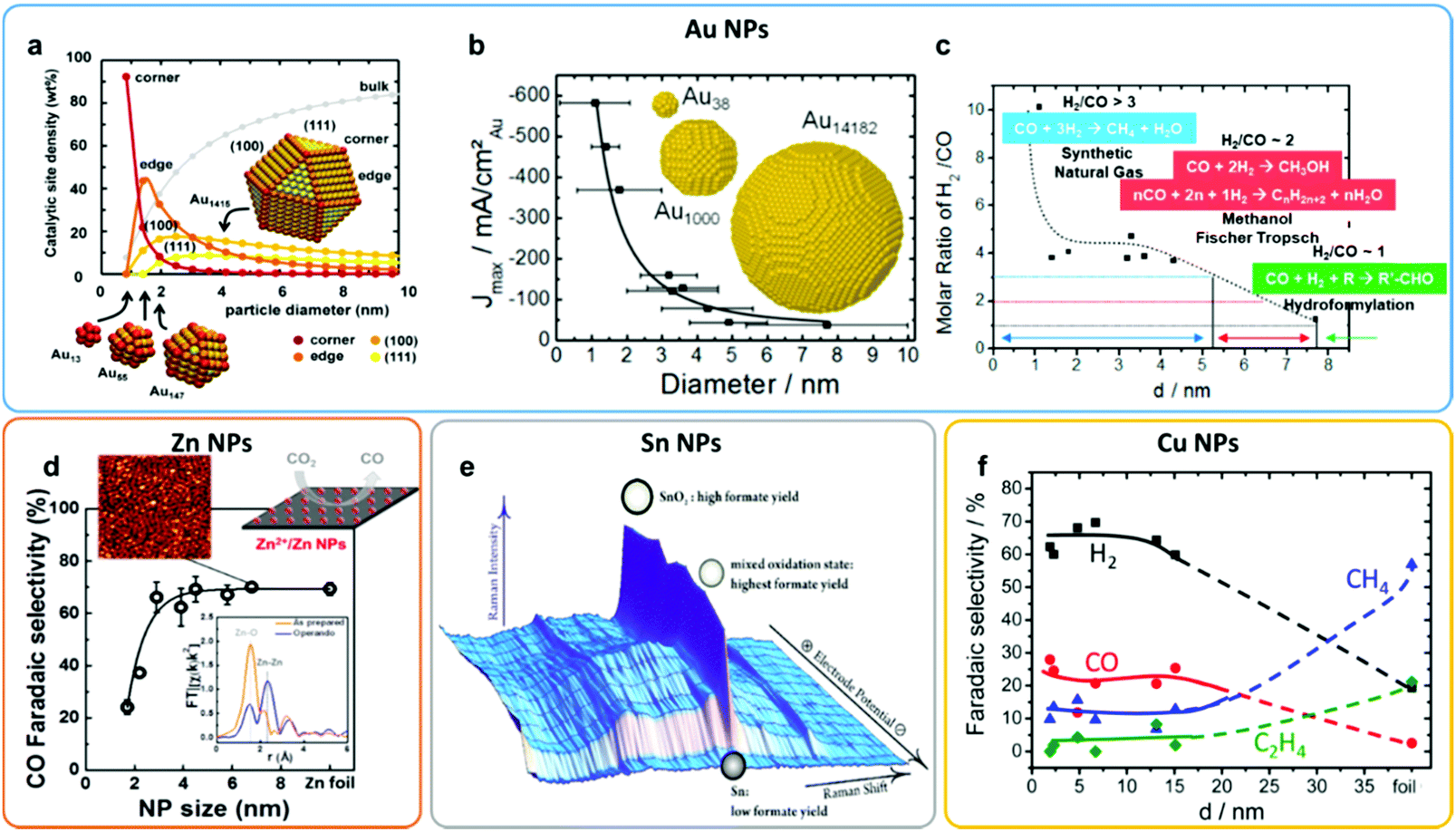 | ||
| Fig. 27 Size-dependent effects in CO2RR for (a–c) Au, (d) Zn, (e) Sn, and (f) Cu NPs. (a) Catalytic site density on closed-shell cuboctahedra Au clusters as a function of the particle diameter. Reproduced with permission from ref. 384. Copyright 2013 American Chemical Society. (b) CO2RR current densities at −1.2 V vs. RHE as a function of the Au NP size. (c) Molar ratio of H2 and CO produced at −1.2 V vs. RHE as a function of the Au NP size. Panels b and c reproduced with permission from ref. 385. Copyright 2014 American Chemical Society. (d) Faradaic selectivity toward CO measured as a function of the Zn NP size at −1.1 V vs. RHE. The inset displays an AFM image and operando EXAFS data of 6.8 nm Zn NPs. Reproduced with permission from ref. 389. Copyright 2018 American Chemical Society. (e) Operando Raman spectra tracking the surface oxidation state of Sn NPs as a function of the potential. Reproduced with permission from ref. 390. Copyright 2015 American Chemical Society. (f) Faradaic selectivity toward H2, CO, CH4 and C2H4 measured at −1.1 V vs. RHE as a function of Cu NP size. Reproduced with permission from ref. 391. Copyright 2014 American Chemical Society. | ||
In addition to Au NPs, various other metal NPs such as Bi,386 Sn,387 Ag,388 and Zn389 have shown a similar structural effect on CO2RR, displaying a significant drop of CO2RR efficiency below a certain NP size. However, depending on the specific metal NPs, a variety of different sizes showed the optimal catalytic performances for CO2 reduction to CO (or formate for Sn NPs). For instance, size-controlled Zn NPs ranging from 3 to 5 nm exhibited high CO2RR activity and selectivity to CO (FE ∼ 70%), whereas a drastic increase of H2 evolution was observed for NPs smaller than 3 nm, presumably due to a larger content of low-coordinated sites.389 In a similar fashion, the maximum FE for CO2 conversion to formate on Sn NPs was achieved for ∼5 nm NPs, representing a balance between surface stabilization of the CO2˙− intermediate and its activation via further protonation/reduction steps.387 As indicated by operando spectroscopic studies (XAS, Raman), oxidized/hydroxylated species were found to be stable during CO2RR for pre-oxidized Zn and Sn NPs. These cationic species are likely involved in the catalytic reaction and may lead to differences in the adsorption strength of the reaction intermediates (Fig. 27d and e).389,390 Moreover, it is worth noting that other factors related to the preparation method of the NPs (presence of surface-anchored organic agents, surface oxidation degree, defects, etc.) or the reaction conditions were found to have a strong influence of the CO2RR activity, contributing to alter the electronic structure and the physicochemical properties of the metal NPs (see next paragraph). A representative example is given by monodispersed Bi NPs with average sizes of 36 nm and 7 nm, whose catalytic properties for CO2RR to CO were explored in non-aqueous 1-butyl-3-methylimidazolium trifluoromethane-sulfonate ([bmim][OTf])/CH3CN electrolyte.386 After a hydrazine-based surface activation treatment, the Bi NPs showed an excellent catalytic activity, reaching a FECO ∼ 96% for the 36 nm NPs. Furthermore, a negligible size or morphology effect was observed on CO2RR efficiency in these conditions, likely due to a change of the CO2RR mechanism in aprotic media, which occurs via a direct H+ transfer between the adsorbed CO2 molecule and the electrolyte rather than via pre-adsorbed protons on the catalyst surface.386 On the other hand, the Bi NPs displayed a size-dependent behaviour after air exposure, resulting in a decreased efficiency for CO production with smaller NPs. These results suggest that the reduction of surface metal oxides to the metallic state is dependent on the size and morphology of the particle, being much faster for larger NPs.
In the case of Cu NPs,391 an increased population of low-coordinated sites also results in a preferred H2 production. However, for large Cu NPs and due to the unique multi-electron CO2RR properties of Cu, the product distribution is mainly based on the formation of hydrocarbons rather than CO (or formate) (Fig. 27f).391 While the adsorbed *CO and *COOH on the NP surface are considered as the key intermediates to form CO (or formate), Cu NPs possess the ability to further protonate *CO to form *HCO or *COH species, due to the fact that *CO is not easily desorbed from the Cu NPs.392–394 Thus, the studies on Cu catalysts have been focused on producing higher yields of hydrocarbons by exploring the way to efficiently stabilize *HCO or *COH intermediates. Indeed, the inherent selectivity of Cu has been considered as one of the main research topics of CO2RR, and many reviews and research articles are available.40,380,395–401 Further investigation of nanostructured Cu catalysts for CO2RR is outside of the scope of the present work.
In contrast with the reports mentioned above, an opposite trend for size-dependent CO2RR has been observed for a series of Pd NPs402 ranging from ∼2 to 10 nm. The efficiency and selectivity towards CO production was found to increase upon decreasing the NP size, leading to faradaic efficiencies between 5.8% (10.3 nm NPs) and 91.2% (3.7 nm NPs) at −0.89 V vs. RHE. In order to explain the experimental results, computational data highlighted that CO2 reduction is considerably favored at the corner and edge sites in comparison with the terrace sites, whereas the H2 evolution process is similar over all the three types of sites.
In addition to the size-dependence, shape modifications of NPs can also influence their CO2RR catalytic performance by controlling the preferential exposure of specific surface sites. In an attempt to design a catalyst with more exposed edges on the surface (Fig. 28a),403 Au nanowires (2 nm wide and 500 nm in length) were synthesized and found to display very low onset potentials (−0.2 V vs. RHE) and high FE for CO production (94%) (Fig. 28b). DFT calculations suggested that the high density of edge sites in the Au nanowires stabilizes the *COOH intermediate and facilitates the *CO release step. The catalytic performance for CO2RR has also been shown to be strongly dependent on the crystal facet located on the NP surface. For instance, triangular Ag nanoplates showed selective CO formation with a FE of 96.8% at −0.86 V vs. RHE,404 outperforming the spherical NPs with similar size as well as the bulk metallic Ag surface. These results were interpreted with the aid of theoretical data in terms of an optimum edge-to-corner ratio and predominant Ag(100) facets in the nanoplates (Fig. 28c). Shape engineering was also successfully adopted by Chen and co-workers405 to increase the catalytic CO2RR-to-CO activity and stability of Pd NPs. In this study, Pd octahedra dominated by (111) facets showed higher CO selectivity (FE up to 95%) and improved activity compared to Pd cubes with (100) facets (Fig. 28d). Interestingly, in situ XAS revealed that regardless of the original facet, a Pd hydride species (PdH) is formed under reaction conditions. Based on DFT calculations, the PdH(111) species was proposed as the active site, being able to promote CO desorption.
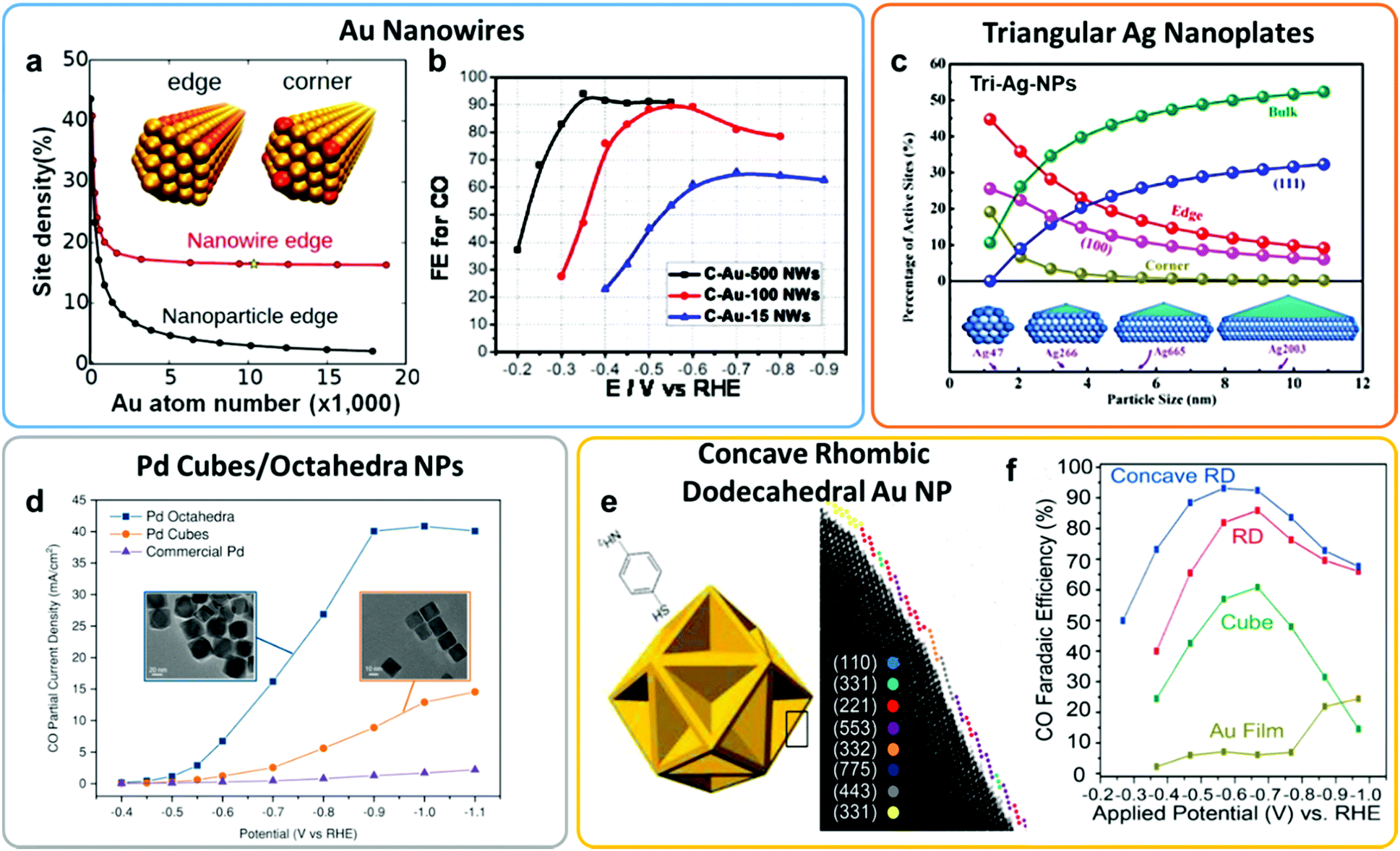 | ||
| Fig. 28 Shape-effect on CO2RR: (a and b) Au nanowires, (c) triangular Ag nanoplates, (d) Pd cubes and octahedra NPs, and (e and f) concave rhombic dodecahedral Au NP. (a) Edge site weight percentage for a 2 nm wide Au NW and an Au NP as a function of the number of Au atoms. (b) CO FE of 500, 100, and 15 nm-long Au nanowires. Panels a and b reproduced with permission from ref. 403. Copyright 2014 American Chemical Society. (c) Active adsorption site density on triangular Ag nanoplates as a function of the NP size. Reproduced with permission from ref. 404. Copyright 2017 American Chemical Society. (d) CO partial current density of Pd cubes and octahedra NPs and their TEM images. Reproduced with permission from ref. 405. Copyright 2019 Wiley-VCH. (e) Schematic illustration and atomic structures of a concave rhombic dodecahedron Au NP. (f) CO FE of Au film, Au cube, Au RD, and Au concave RD as a function of applied potential. Panels e and f reproduced with permission from ref. 406. Copyright 2015 American Chemical Society. | ||
Lastly, nanoparticles enclosed by high-index facets have been experimentally reported to further inhibit H2 production during CO2RR. As an example, concave rhombic dodecahedron (RD) Au NPs with various high-index surfaces406 including (331), (221) and (553) were found to exhibit an onset potential for CO2RR to CO at −0.23 V vs. RHE, 200 mV positively shifted relative to that of an Au film. A FECO ∼ 70% was observed at −0.37 V vs. RHE, which is approximately three times higher than that of Au NPs exposing low-index facets (Au nanocubes) (Fig. 28e and f).
6.2 Support effect
Since the electrochemical CO2 reduction process over heterogeneous catalysts takes place at a solid/liquid interface, a control over the charge transfer between the catalyst and the surface intermediate species is critical to enhance the efficiency of the process. In this sense, the selection of a suitable support material is essential to ensure fast CO2RR reaction kinetics and to adjust the binding strength of intermediates species. For example, the CO production over Ag NPs deposited on carbon black can be doubled by using a TiO2 support.407 Based on the analysis of the CV curves, the TiO2 support was found to stabilize the reaction intermediates through the involvement of TiIV/III redox couples, also acting as a redox carrier to improve the CO2 reduction kinetics (Fig. 29a). Recently, Au NPs on a C3N4 support were also reported to exhibit a better performance than carbon-supported Au NPs.408 The strong interaction between the Au NPs and the C3N4 support was predicted to induce the formation of a negatively charged Au surface, which could stabilize the key *COOH intermediate. Along this line, the effect of the C3N4 support can be further improved by incorporating carbon quantum dots (CDots), as demonstrated by the enhanced performances shown by the ternary Au NPs–CDots–C3N4 configuration409 (Fig. 29b). In the latter case, DFT calculations revealed that the combination of CDots with C3N4 results in an enhanced conductivity and CO2/H2 adsorption, facilitating the CO2 reduction to CO. A number of other carbon- or metal oxide-based materials displayed beneficial effect on CO2RR when used as supports in combination with Au NPs. Among them, graphene nanoribbons (GNR) decorated with Au NPs improved the performances of Au NPs supported on carbon black (Fig. 29c),410 likely due to the strong d–π interaction that can serve to modulate the electronic structure of the Au NP, leading to the acceleration of the charge transfer from the CNR to the Au NPs. Interestingly, it is worth noting that similar results were observed for the non-covalent functionalization of molecular catalytic systems based on π–π stacking and electrostatic interactions.138 In fact, the strategy of adjusting the electronic structure of the active catalysts to improve the CO2RR performance can be widely applied from molecular to nanostructured catalysts. Furthermore, an Au NPs/CeOx assembly led to a substantial improvement in the efficiency towards CO production in comparison with the single bare components411 (Fig. 29d). As suggested by in situ scanning tunneling microscopy and photoemission spectroscopy, the enhanced reactivity towards CO2RR stems from the stabilizing interaction of hydroxyl groups on ceria terraces with key intermediates involved in the catalytic reaction.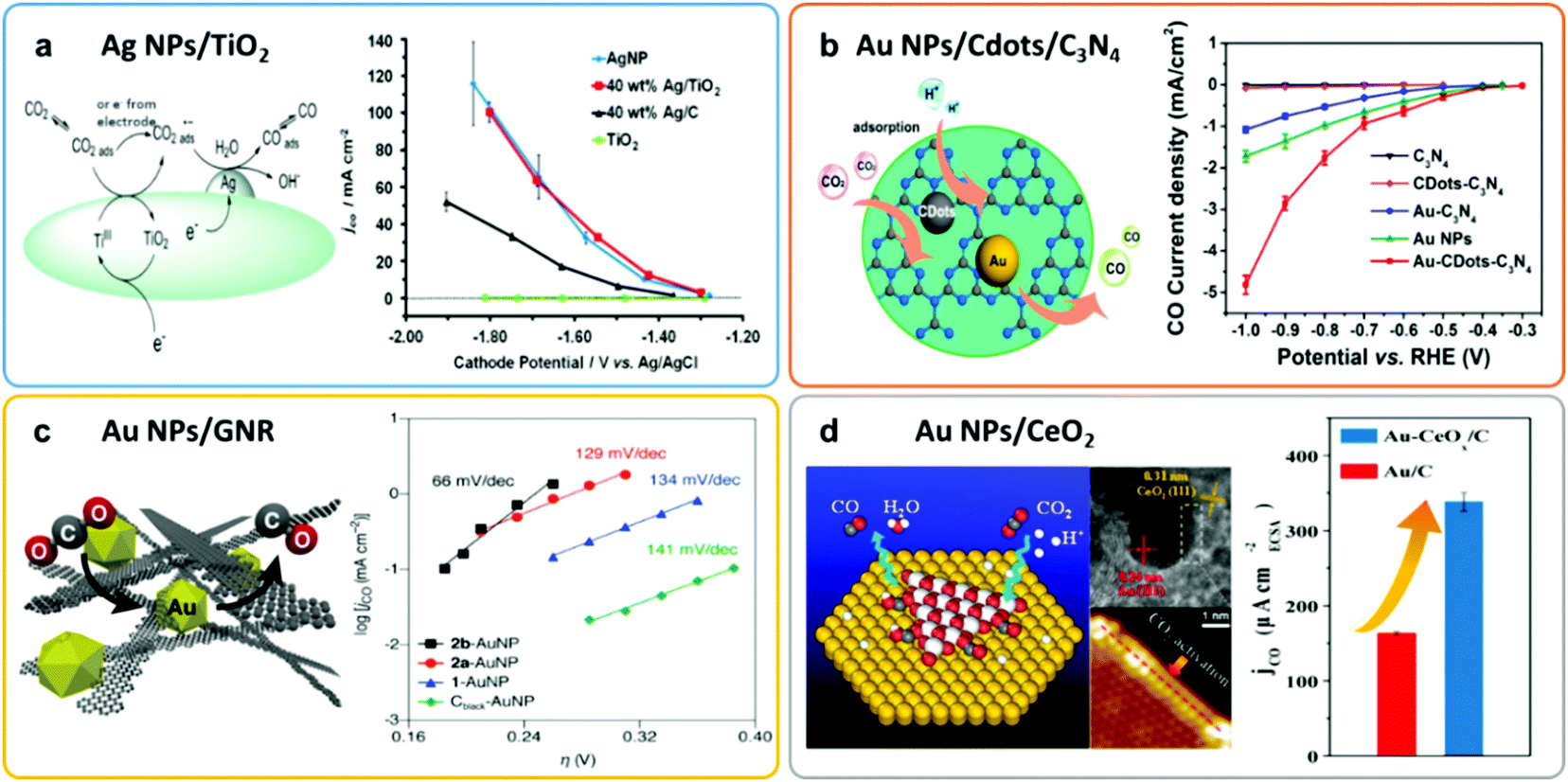 | ||
| Fig. 29 Studies on the support effect on (a) Ag NPs/TiO2, (b) Au NPs/Cdots/C3N4, (c) Au NPs/CeO2, and (d) Au NPs/GNR. (a) Schematic illustration of the CO2RR reaction mechanism to CO on Ag/TiO2. Partial current density for CO production as a function of the cathode potential with Ag/TiO2, Ag/C, Ag NP, and TiO2. Reproduced with permission from ref. 407. Copyright 2014 Wiley-VCH. (b) Schematic illustration of carbon dots (CDots) and Au NPs on C3N4 (Au–CDots–C3N4) for CO2RR. Partial current density for CO production as a function of the applied potential, catalyzed by C3N4 CDots–C3N4, Au–C3N4, Au NPs, and Au–CDots–C3N4. Reproduced with permission from ref. 409. Copyright 2018 American Chemical Society. (c) Schematic illustration of Au NPs embedded in GNR. Tafel slopes for CO activity by GNR– and carbon black–AuNP composite materials. Reproduced with permission from ref. 410. Copyright 2017 American Chemical Society. (d) Schematic illustration of Au NPs supported on CeOx for CO2RR. Partial current density for CO with Au–CeOx/C and Au/C catalysts. Reproduced with permission from ref. 411. Copyright 2017 American Chemical Society. | ||
The encapsulation of Ag NCs into a reticular MOF structure also revealed to be an effective strategy to enhance the stability and selectivity towards CO2RR.412 This was assigned to the interaction of the catalyst with the porous MOF which favors the electron and mass transfer during CO2RR over HER.412 In particular, the intimate interfacial contact between the Ag NCs and the MOF, brought about by the removal of the native ligands before the encapsulation, was rationalized to be crucial for the improved selectivity toward CO2RR.
6.3 Surface functionalization with organic molecules
A promising complementary strategy to particle size, shape, composition and support design consists in capping metal NPs with chelating organic molecules.413–415 This alternative approach applies the concepts and tools of conventional coordination chemistry to design novel metal nanocatalysts with improved catalytic properties. In comparison with the traditional strategies to control the structure of metallic NPs, this approach offers a number of additional advantages: (i) the organic capping agents usually act as NPs stabilizers, preventing or minimizing aggregation phenomena and NP oxidation upon air exposure; (ii) the catalytic properties of metal NPs can be tailored by modifying the electronic and structural features of the organic anchoring agent; (iii) tuning of the local environment around the nanocatalyst surface (e.g. increased hydrophobicity, activation of second coordination shell interactions, etc.) in order to alter the product distribution of CO2RR. A variety of synthetic methods and organic molecules (surfactants, organic ligands based on C, N, P, S or O atoms) have been adopted to functionalize metal NPs.416 Thiols, amines and NHC ligands represent the main families of organic ligands used to stabilize monometallic NPs for CO2RR, and the main advances for each class are discussed in the next paragraphs. NPs or NCs based on late transition metals, such as Au, Ag and Pd, cover the overwhelming majority of the reported organic-stabilized nanocatalysts for CO2RR, generally producing CO as the major product. A few specific studies on functionalized flat bulk metallic catalysts will be also discussed in the text, since they provide complementary information for the understanding of the role of the organic ligand on the CO2RR activity and selectivity of metallic heterogeneous catalysts. In general, nanomaterials showed enhanced activity compared to polycrystalline surfaces due to their improved reactivity, conductivity and stability under electrochemical conditions.414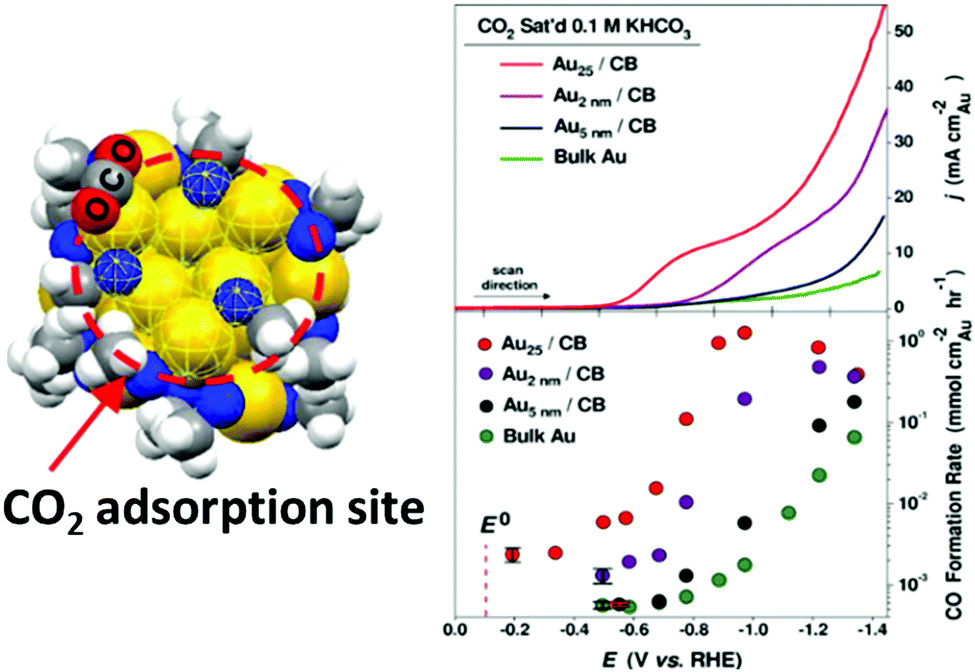 | ||
| Fig. 30 Schematic illustration of ligand protected Au25 nanoclusters. LSV and CO formation rate for the various Au catalysts. Reproduced with permission from ref. 417. Copyright 2012 American Chemical Society. | ||
The use of cysteamine as anchoring agent has revealed to be beneficial for the CO2RR performances of Au NPs and Ag NPs.388,419,420 For an optimal particle size of 5 nm, Ag NPs on carbon decorated with cysteamine molecules showed a considerably lower overpotential and a 4-fold enhanced CO faradaic efficiency at −0.75 V vs. RHE compared to a polycrystalline Ag foil.388 While Tafel slope analysis suggested the formation of the *CO2− adduct to be rate-limiting, DFT calculations highlighted an electronic effect on the Ag surface induced by the Ag–S interaction, which is reminiscent of the through-structure substituent effects observed in molecular catalysts (see Section 3). More specifically, the presence of cysteamine capping agents was predicted to increase the spatial localization of the unpaired electron at the Ag surface, contributing to stabilize the key *COOH intermediate. Interestingly, the cysteamine molecules had only a small impact on the CO binding energy to the surface, resulting in an overall enhanced catalytic activity.388
A follow-up experimental–computational study extended the mechanistic understanding of the cysteamine-Ag NPs system at a molecular level, proposing that the enhanced activity and selectivity may arise from a synergistic contribution of the electronic effect promoted by the Ag–S interaction and the role of the pendant NH2 group of anchoring cysteamine molecules, which would assist the CO2 chemisorption process on the Ag surface.419 In particular, ab initio molecular dynamics (AIMD) simulations suggested that the terminal amine group of the surface ligand may contribute to stabilize the chemisorbed CO2 molecule by H-bonding interaction (in cooperation with a water molecule, Fig. 31a), exerting an outer coordination sphere effect analogous to that shown by macrocyclic N4 molecular catalysts with pendant amine groups (see Sections 4.1–4.2). In agreement with this interpretation, the length of the alkyl chain of the anchoring ligand was found to be critical for the catalytic performances: Ag NPs functionalized with the 11-amino-1-undecanethiol (C11-Ag) did not display an analogous enhancement effect shown by the cysteamine-capped Ag NPs (C2-Ag) (Fig. 31b).419 In this case, the excessive length of the alkyl chain led to an unfavorable geometry which was not suitable to stabilize the surface-bound CO2 intermediate, being the terminal NH2 group too far away from the Ag surface. Moreover, unlike for C11-Ag, a shift of the N–H band to lower energy was observed by in situ ATR-IR spectroscopy for C2-Ag, suggesting the involvement of the NH2 group in the CO2 chemisorption event.419
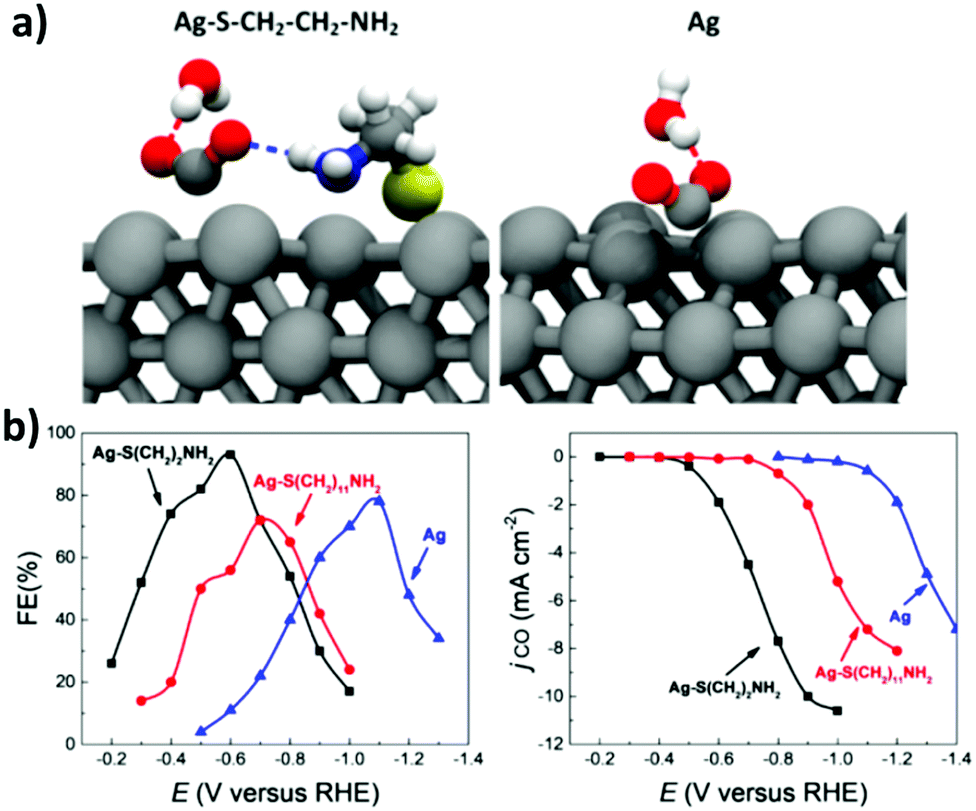 | ||
| Fig. 31 (a) Chemisorbed CO2 on cysteamine-functionalized (left) and on cysteamine-free (right) Ag(111) surface. Only the water molecules directly forming hydrogen bond (slashed line) are shown. The colors are: Ag in silver, C in gray, O in red, N in blue, S in yellow, and H in white. (b) Influence of the alkyl chain length on CO2RR in terms of FECO (left) or jCO (right) vs. potential. Reproduced with permission from ref. 419. Copyright 2018 American Chemical Society. | ||
An analogous boosting effect due to cysteamine capping agents was also recently reported for Au NPs, showing an exclusive CO selectivity at low overpotential and a 110-fold enhanced mass activity in comparison with the naked Au NPs.420 XAS spectroscopy confirmed the effect of the Au–S interactions on altering the electronic structure of the gold surface and EXAFS analysis confirmed a lower Au–Au coordination number for cysteamine-Au NPs in comparison with the ligand-free Au NPs. Furthermore, the replacement of cysteamine with 1-propanethiol as a capping agent (terminal CH3 group) resulted in a significant drop of the CO2RR catalytic activity, thereby probing the critical role played by the NH2 group of the former.420 Notably, outer coordination sphere effect on CO2RR activity and selectivity has been also observed on polycrystalline Au foil functionalized with thio-tethered ligands.421 Depending on the pKa of the terminal group of the thiolate ligand, the activity and faradaic efficiency toward CO, H2 and HCOO− could be tuned. In particular, cysteamine-modified Au electrodes exhibited a 2-fold increase in CO and H2 production respect to the bare Au foil, whereas the functionalization with 2-mercaptopropionic acid led to exclusive H2 evolution. Conversely, Au electrodes modified with 4-pyridylethylmercaptan (pKa = 5.2) showed a significantly higher production and FE for HCOO− as compared to the naked Au foil, owing to the role of the terminal pyridine/pyridinium groups. The latter were proposed to be involved in a proton-induced desorption mechanism leading to HCOO− formation.421 It is interesting to note that in Section 3 we have discussed several examples from molecular catalysis in which pendant pyridine/pyridinium groups in the second coordination sphere exerted a boosting effect for the catalytic CO2RR to CO.168,192,194 Moreover, a selectivity change induced by different groups in the second coordination sphere has been also observed in a number of homogeneous molecular catalysts.267–271
Surfactant molecules containing highly hydrophobic long alkyl chains are commonly used as stabilizing agents during metal NPs synthesis. In some cases however, they may partially block the surface active sites limiting the catalyst performance386 or be easily detached from the surface during prolonged electrolysis. In order to overcome this limitation, the surfactant molecules bound to Au NPs were replaced by chelating porphyrin-like tetradentate ligands with S-terminal anchoring groups.422 The latter act as hollow scaffolds on Au NPs, interacting with the metal catalyst but without hindering the accessibility of the catalytic sites. This structural modification led to a 110-fold current enhancement in comparison with the parent oleylamine-coated Au NPs, efficiently producing CO (FE up to 93%) at an overpotential of 340 mV (Fig. 32). Importantly, the chelation effect resulted in a remarkable stability, leading to only minor deactivation after 72 h electrolysis. Unlike the classical surfactant-based strategy, this alternative approach ensures a higher number of exposed sites, substantially increasing the current densities. In addition, the tetra-functionalized Au NPs were predicted to be intrinsically more active towards CO2RR with respect to both, bare Au(111) and oleylamine-coated Au NPs, showing an energetically more favorable *COOH formation. The electronic effect due to Au–S interactions was estimated to be negligible.422 In an earlier study, the same authors used tetrapodal S-terminal porphyrin platforms to functionalize polycrystalline Cu electrodes for electrochemical CO reduction (CORR).423 Based on DFT calculations, the different selectivity toward oxygenates production experimentally observed for various cage sizes was tentatively ascribed to a different stabilization of a key ketene intermediate via H-bonding interactions with the porphyrin cap.423 As discussed in Section 3.1.1, stabilizing the H-bonding interaction promoted by pendant amide groups displayed beneficial effects in CO2RR catalysed by homogeneous Fe porphyrins. Finally, we emphasize that this approach is particularly interesting for the design of bimetallic heterogeneous catalysts by metalation of the porphyrin moiety, merging concepts from molecular catalysis, supramolecular chemistry and surface science.
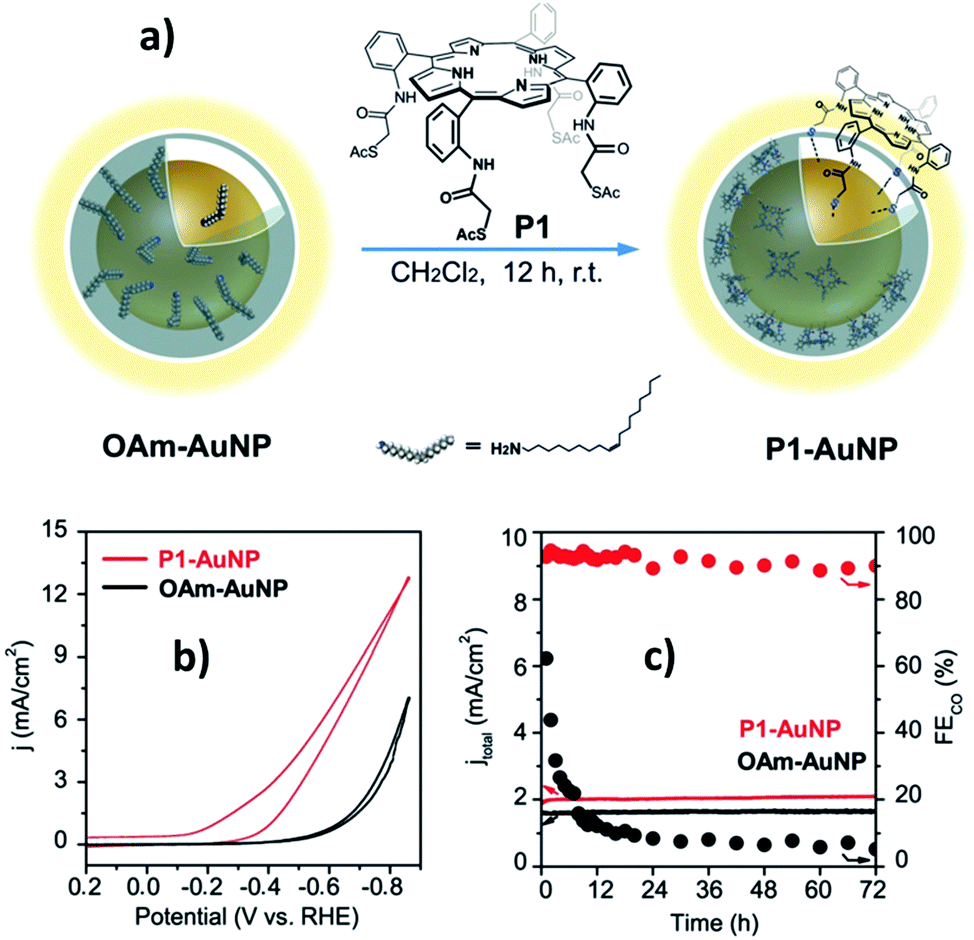 | ||
| Fig. 32 (a) Synthesis of P1-Au NPs. (b) CV scans of OAm-Au NP and P1-Au NP electrodes under CO2-saturated 0.5 M KHCO3 at pH 7.3. (c) Controlled-potential electrolysis of OAm-Au NP and P1-Au NP electrodes at −0.45 V vs. RHE over a 72 h time course. Reproduced with permission from ref. 422. Copyright 2018 Wiley-VCH. | ||
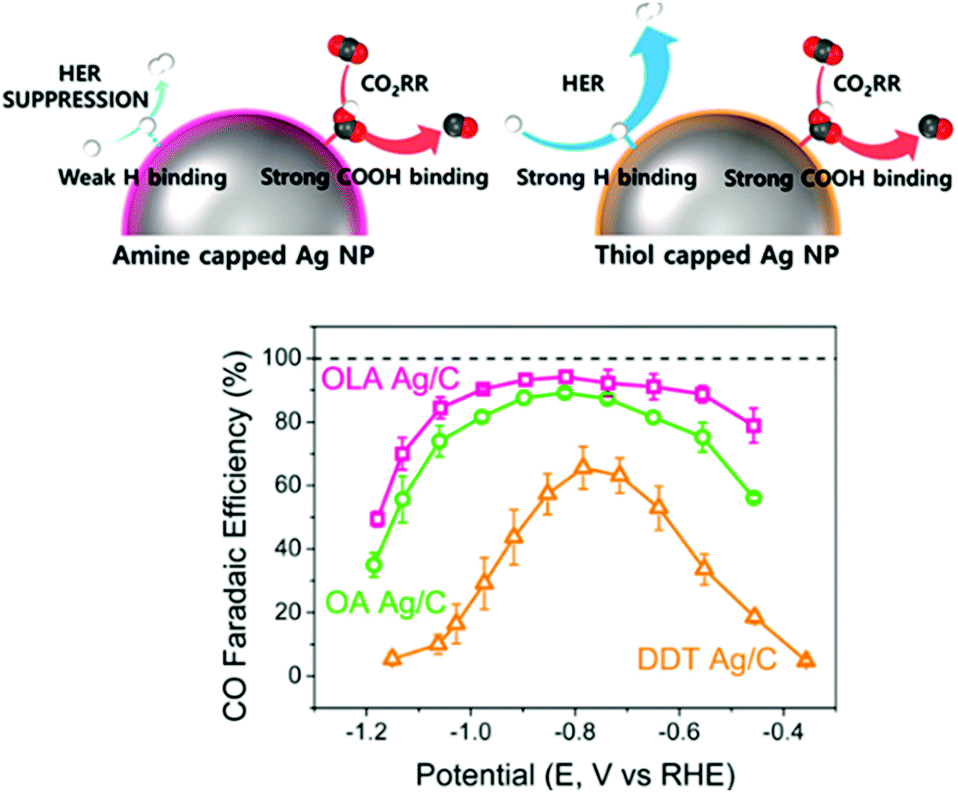 | ||
| Fig. 33 Schematic illustration of amine- and thiol-capped Ag NPs for CO2RR. CO faradaic efficiency as a function of the applied potential for oleylamine (OLA)-capped Ag/C, oleic acid (OA)-capped Ag/C, and dodecanethiol (DDT)-capped Ag/C. Reproduced with permission from ref. 424. Copyright 2017 American Chemical Society. | ||
Functionalization with oleylamine, a surfactant commonly used in the wet-chemistry synthesis of metal NPs, displayed an analogous boosting effect on CO2RR for Au NPs. In this regard, oleylamine-capping was recently reported to drastically enhance the selectivity of small Au NPs toward CO production over HER.425 The origin of the HER inhibition was ascribed to the preferential binding of oleylamine to the low-coordinated corner sites of the Au NPs, which are the ones favoring *H binding, thus leaving the edge sites free to engage *COOH formation and conversion to CO.425 The molecular structure of the capping agent was also found to play a role on the catalytic efficiency, resulting in a structure-dependent modification effect: linear amines revealed to be more suitable to promote CO2RR to CO as compared to the branched amines, with the CO selectivity increasing with the length of the alkyl chain.426 Across the series, oleylamine provided the best results in terms of either activity and selectivity, suggesting a correlation between the amine structure and the surface coverage. However, the stability of the capping agent under electrochemical conditions should be carefully assessed in order to reliably establish structure–function relationships. In the case of oleylamine-capped Au NPs, some reports indicate that catalyst degradation occurs at negative potentials, likely due to partial ligand detachment,426 as observed in some molecular systems (see Section 4.1.1).217 The applied bias-driven loss of surfactant molecules was shown to cause the aggregation of ultrasmall Au NPs, leading to bigger nanostructures that increase the selectivity toward CO production.427 Contrariwise, dendrimer-encapsulated Au NPs were found to be very stable under electrochemical conditions leading to predominant H2 evolution.
In addition to a direct functionalization of metal NPs with organic ligands, the outer coordinative environment of the metal NPs may cooperate with the latter to boost catalysis. For example, the adsorption of polyvinyl alcohol (PVA) on Au NPs led to a superior selectivity toward CO production compared to the naked Au NPs.428 It was proposed that the H-bond network at the metal–polymer interface may contribute to stabilize the key *COOH intermediate, through outer coordination sphere effects analogous to those displayed in homogeneous porphyrin catalysts bearing local phenolic groups (see Section 3.1.1).120 Moreover, the co-presence of organic molecules on the surface may contribute to the catalytic reaction. Some Au NPs supported on CNTs recently displayed a significantly improved catalytic CO2RR performances in the presence of axial pyridine groups covalently grafted on the CNT surface via the diazo-reaction.429 The hybrid catalysts showed excellent faradaic yield for CO production in a wide potential range at low overpotential, which could not be explained by the pyridine alone.430 Instead, it was proposed that the pyridine group might participate in the rate-limiting *COOH formation step in a similar manner as previously observed for several molecular systems (Section 3)168,192,194 and polycrystalline catalysts (see Section 6.3.1).421
In an effort to systematically investigate the effect of tailoring the organic component in a hybrid organic/inorganic interface on catalysis, Buonsanti and coworkers recently explored a series of colloidal Ag NCs functionalized with di-substituted imidazolium ligands with varying tail and anchoring groups.431 This interesting study focused on the key design guidelines to tune the organic component for NP functionalization, dissecting the contribution of each of the three fundamental elements (Fig. 34). The latter are: (i) the anchoring group to the Ag NC, which may affect the electronic structure of the metal surface by direct coordination, (ii) the imidazolium group, and (iii) the tail group. The probed integrity of the Ag NCs-coordinative organic platform under electrochemical conditions is another remarkable aspect of this study. In these systems, the presence of pendant imidazolium motifs was found to be crucial to achieve high selectivity for CO2 conversion to CO over HER. This is consistent with the role of co-catalysts in CO2RR reported for ionic liquids on metal electrodes432–435 and in homogeneous molecular catalysis.97,436 Moreover, in agreement with previous reports,433 the cationic character of the imidazolium moiety may play an important role in CO2RR. We note also that it may help to stabilize negatively charged surface intermediates formed during CO2RR via Coulombic through-space interactions, in analogy with the reported examples of homogeneous Fe porphyrin catalysts containing pendant imidazolium units131 or tetramethylammonio groups112 (Section 3.1.1). Unlike the imidazolium group, the chemical nature of the functional group anchoring on the Ag NC surface showed a less pronounced effect on selectivity.431 However, XPS analysis provided experimental evidence of an electronic effect of the substituents on the Ag surface, establishing a linear correlation between the specific activity for CO2RR to CO with the observed shifts in the Ag 3d5/2 peaks. As a general trend, electron-withdrawing substituents were found to be beneficial for CO2RR, with the NO2 group providing the best results. Although a reverse effect is generally reported for ligand-stabilized metal NPs (vide infra), a possible contribution of the imidazolium moiety may also play a role. Furthermore, the positive impact of electron-poor substituents on CO2RR has been reported for several heterogenized heme molecular catalysts (see Section 3.2.2).183,184 Finally, the tuning of the alkyl tail groups also displayed a strong effect on selectivity, with an intermediate tail length effectively suppressing HER due to a suitable interplay between an increased local hydrophobicity on the catalytic surface and without an excessive steric bulkiness.431 The steric hindrance of a long tail group reduces the accessibility of the electrolyte and CO2 to the metal surface, analogously as previously discussed for tetraazamacrocyclic Ni catalysts with N-alkyl substituents (see Section 4.1.1).
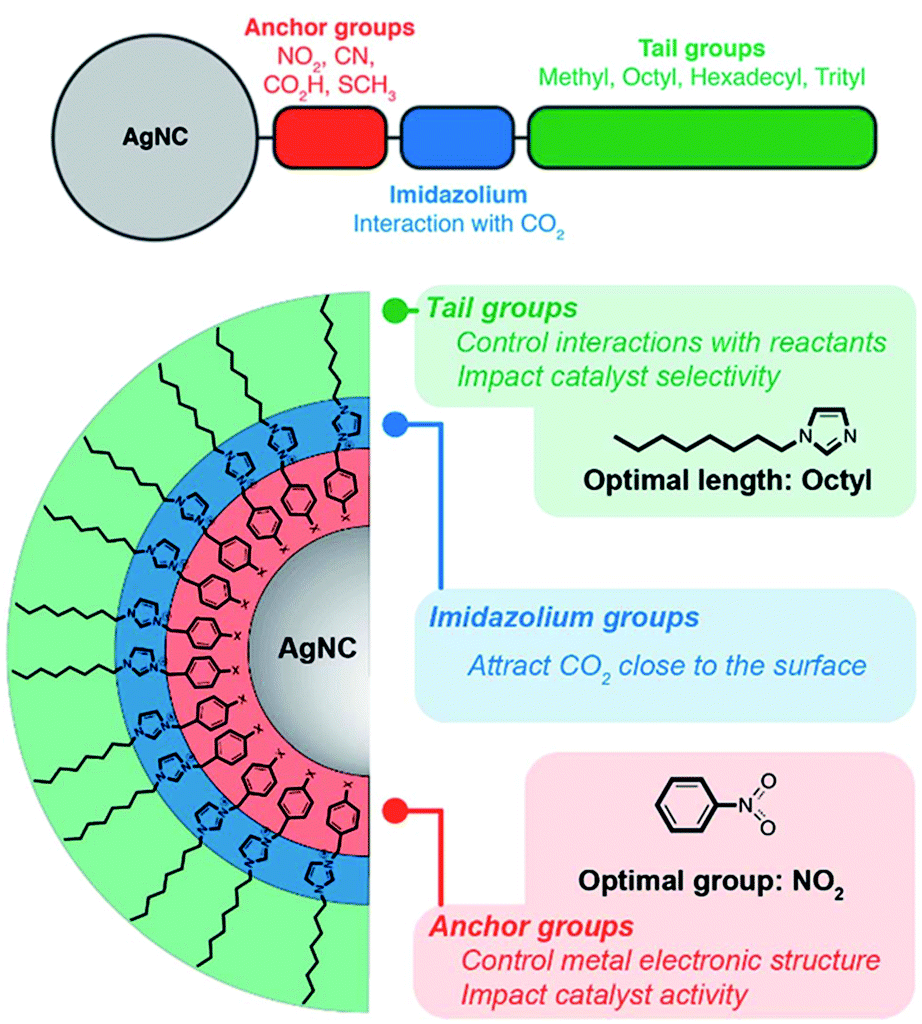 | ||
| Fig. 34 Overview of the effect of imidazolium ligands on an AgNC surface during the CO2RR. Reproduced with permission from ref. 431. Copyright 2019 Royal Society of Chemistry. | ||
These findings highlight how traditional concepts of organometallic chemistry (e.g., steric and electronic effects, second coordination sphere interactions, hydrophobic/hydrophilic interaction, etc.) can be effectively used to improve the understanding of the organic/inorganic interface at a molecular level and to tailor heterogeneous catalysts. Several representative examples of functional organometallic surfaces, based on bulk metal electrodes chemically interacting with nitrogen-based organic additives (e.g. ionic liquids,432–435 aminoacids,437N-arylpyridinium438–440 or N,N′-phenanthrolinium,441 benzimidazole,442 polyamines,443 polyaniline,444 triazole,445etc.) or supramolecular organic assemblies,446 have been also recently reported for CO2RR.447
Due to these favorable properties, Au NPs functionalized with monodentate NHC molecules exhibited a substantially improved CO2RR behavior as compared to the parent Au NPs in terms of FE (83% vs. 53%) and current densities (7.6-fold increase) for CO production at an overpotential of 460 mV, Fig. 35.454 The NHC ligation was found to alter the electronic structure of the Au surface, affecting the reaction pathway. For the bare Au NPs, kinetics analysis based on Tafel slope was consistent with a rate-determining step based on a single electron transfer to the adsorbed CO2 to form *CO2˙−. Contrariwise, the lower slope obtained for NHC-stabilized Au NPs suggested that it may undergo a pre-equilibrium one-electron transfer followed by a rate-limiting chemical step. Such a change of the mechanistic pathway was ascribed to the strong σ-donating electronic effect induced by the molecular ligand, which promotes a fast electron transfer prior to the rate-determining step.454 Notably, it was also proposed that the strong Au–C bond may destabilize the neighboring Au–Au surface bonds, causing a restructuring of the Au surface which resulted in improved CO2RR kinetics.
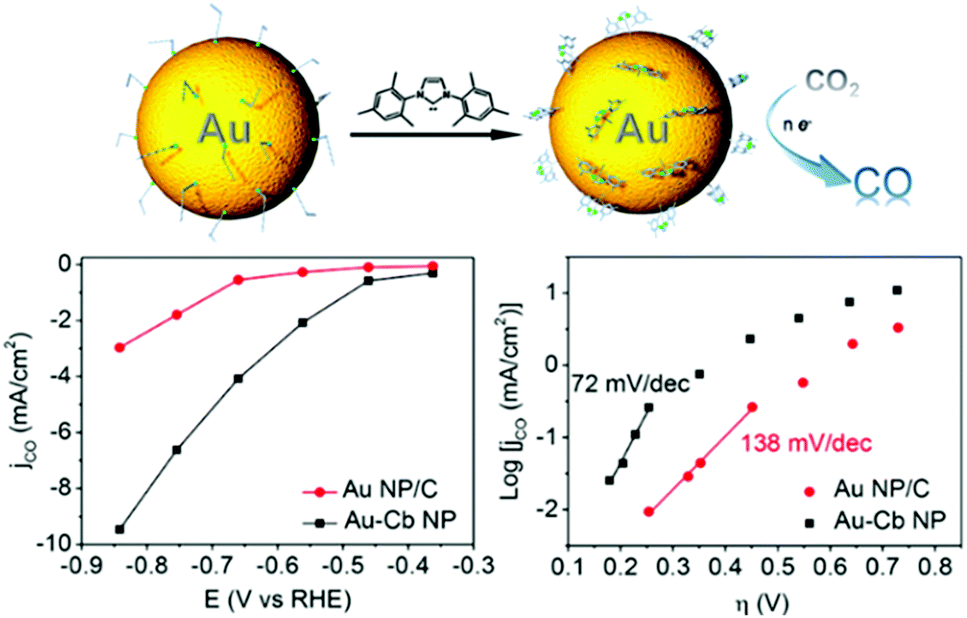 | ||
| Fig. 35 Schematic illustration of NHC-carbene functionalized Au NPs for CO2RR to CO. Partial current density and Tafel plot for CO production for Au–Cb NP and Au NP/C. Reproduced with permission from ref. 454. Copyright 2016 American Chemical Society. | ||
As mentioned above, the reported NHC-functionalized Au NPs454 or nanoclusters455 displayed good CO2RR selectivity and stability under electrochemical conditions. Recently, the encapsulation of metal NPs into monodentate or polydentate polymeric NHC matrices was explored as an effective strategy to further improve at the same time catalyst selectivity and long-term durability, preventing nanoclustering.456 The embedded Au NPs exhibited excellent FECO with a ca. 86% activity retention over 11 h electrolysis under CO2 atmosphere at −0.9 V vs. RHE, clearly outperforming the bare Au NPs which lost approximately 90% of their initial activity. The improved performance was ascribed to the electronic effect exerted by NHC ligands and the hydrophobicity of the polymer matrix, the latter helping to suppress HER and control the surface accessibility as observed for polymer-encapsulated molecular catalysts (Section 3). To further confirm the beneficial role played by the NHC polymer on catalysis, an analogous effect was also observed for Pd NPs, which showed an increase in the CO selectivity and stability as compared to commercial Pd/C.456
In addition to metal NPs, the functionalization of NHCs was recently reported to affect the catalytic properties of polycrystalline metal surfaces. In particular, Pd electrodes modified with tridentate NHC ligands displayed a 32-fold activity enhancement for CO2 conversion to C1 products as compared to an unmodified Pd foil, producing HCOO− in high faradaic yields.457 The electron-rich Pd surface induced by tris–NHC ligation led to a more favorable pathway for HCOO− production. While subtle differences were observed in catalysis upon tuning the alkyl N-substituents of the ligand, the NHC chelation showed a remarkable effect on the catalyst stability, with Tris–NHC ligands providing considerably higher durability than the bare Pd foil and the monodentate counterpart.457 Further investigation is needed to fully rationalize the influence of NHC ligands on the catalytic CO2RR as a function of the denticity and the type of substituents. Moreover, a spectroscopic investigation of the spatial orientation and coordination mode of NHC moieties on different metallic surfaces will provide useful insights to modulate the reactivity of the organic/inorganic interface towards CO2RR.458–462
7. Conclusions
In this work we have summarized the main advances achieved in the rational design of transition metal-based catalysts for the electrochemical conversion of CO2 into higher-energy carbon products. With a special focus on structural effects, we have provided a critical comparison of the main design principles directing the development of different types of catalysts, ranging from molecular systems to single-atom and nanostructured catalysts. For molecular systems based on heme and non-heme macrocyclic ligands, we presented detailed studies investigating the effect of the first and second coordination sphere on the catalytic activity, selectivity and overpotential of both, homogeneous and heterogenized catalysts. These studies revealed that molecular systems can be suitable catalysts for scaling-up a selective CO2RR under appropriate conditions. In addition to the conventional physical and chemical functionalization methods, alternative approaches for molecular catalyst confinement were explored, including the encapsulation into polymeric matrices, porous organic or metalorganic 3D frameworks and supramolecular assemblies. The main strategies adopted for the design of single-atom and nanostructured catalysts were also discussed. As compared to the corresponding bulk metallic surfaces, molecular-like catalysts show an intrinsically higher selectivity towards CO2 due to the specificity of their active site. For molecular and single-atom catalysts, earth-abundant 1st row transition metals (e.g. Mn, Fe, Ni, Co) are typically used for efficient and selective CO2RR in both, organic and aqueous media.We have reviewed the electrocatalytic CO2RR properties of the main families of molecular, single-atom and nanostructured catalysts on the basis of their specific structure–function relationships as well as in relation with their dependence on a number of experimental factors, including the electrode/support material, pH, buffer, electrolyte or the applied potential. In homogeneous systems, structural effects related to the metalorganic active moiety are predominant, with the electrode surface working as an electron collector and the molecular catalyst being usually dissolved in organic media. Contrariwise, for heterogeneous catalysts, the nature of the support material as well as the support–catalyst interaction strongly affect the catalytic properties.
Taken together, the studies reported herein suggest that a close connection between homogeneous and heterogeneous CO2RR electrocatalysis would be beneficial for both fields. From one side, the lessons of traditional coordination chemistry and the concepts developed in molecular catalysis may be extremely helpful to overcome the limits of the current heterogeneous catalysts, especially in terms of selectivity. On the other hand, the application of methods and tools used in materials science and heterogeneous catalysis to molecular systems would contribute to extend their applicability. We are confident that the principles for rational catalyst design summarized above will contribute to the further development of new efficient transition metal-based catalysts for efficient and selective electrochemical reduction of CO2.
8. Outlook
The studies presented herein serve to shed light on a variety of strategies adopted in the past to improve the efficiency of transition metal-based CO2RR catalysts. At different levels of complexity, the CO2RR activity and selectivity are extremely sensitive to the catalyst structure. In this regard, further development of electrochemical and in situ/operando spectro-electrochemical techniques is needed to improve the mechanistic understanding of the CO2RR process and to elucidate structure–function relationships as powerful tools for catalyst optimization. Moreover, new advances in the spectroscopic and microscopic techniques are required to achieve atomic or molecular structural details for the characterization of catalytic materials. In particular, single-atom catalysts show unique physico-chemical properties and an improvement in their structural characterization would allow to more accurately correlate their activity with specific structural features of the active site. This will be a major challenge in the field of energy conversion in the coming decades. Furthermore, an increase of the atomic aggregation level usually leads to a more favorable HER (especially in aqueous electrolyte) or to a catalytic inactivity, as indicated by the deactivation of molecular-like catalysts due to particle sintering. In this respect, monitoring the dynamic changes of the catalyst structure under operating conditions would also allow to elucidate the deactivation pathway, which is a crucial aspect for catalyst optimization in both homogeneous and heterogeneous catalysis.Another promising perspective for future research is a more in-depth investigation of the organic/inorganic interface of hybrid materials, by combining the basic principles and concepts of conventional coordination chemistry with the methods traditionally employed in surface science. Several examples reported in this work showed that the electronic properties of metal catalysts can be effectively tuned by modulating the stereo-electronic features of the surrounding organic or heteroatom-rich framework (e.g. reticular effect in molecular catalysts encapsulated into porous 3D frameworks or organic molecules anchored to metal NPs or polycrystalline metallic surfaces). However, systematic studies on the nature of these interactions as well as their effect on catalysis are rare, leaving much room for future investigation. The extreme versatility of this approach also offers the opportunity to explore several different strategies for catalyst optimization, based on chemical (coordination) or physical (electrostatic, hydrophobic) interactions at the metal–organic interface. In this scenario, the outer coordination sphere effects, reminiscent of the spatial interactions occurring in the active site of enzymes, deserve to be studied in more detail in both homogeneous and heterogeneous catalysts. For homogeneous molecular systems, they were rationalized to be an effective way to break scaling relationships, leading to an outstanding improvement of the catalytic rates and selectivity. In heterogeneous catalysis, these interactions have been less explored and can be described in terms of matrix effects, as recently reported for molecular catalysts embedded into conductive surfaces.194 The development of novel model systems and in-depth experimental kinetic analysis would be extremely important to further rationalize and dissect these effects.
In parallel with more detailed mechanistic studies and the investigation of new strategies for catalyst optimization based on structural effects, much effort should be directed to tackle the main technological and physical challenges related to the implementation of the heterogeneous and molecular catalysts into real devices. In particular, the use of standard electrolyzers or flow cell setups for testing the long-term performance of transition metal catalysts is highly encouraged to explore their practical applicability. This would also improve catalyst benchmarking, facilitating a systematic comparison of the different catalysts reported in the literature. For molecular homogeneous catalysts, the development of novel heterogenization strategies needs to be accompanied by their implementation in real devices, as suggested by some promising preliminary studies.187 At the same time, novel strategies based on the combination of CO2RR with other oxidative electrosynthetic processes (e.g. organic substrates) should be explored, as well as alternative approaches to utilize the CO2RR products (e.g. carbon monoxide) as precursors or building blocks for the synthesis of commodity chemicals.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work was funded by the European Research Council under grant ERC-OPERANDOCAT (ERC-725915) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project no. 406944504 – SPP 2080 and SFB 1316, subproject B1, as well as Germany's Excellence Strategy – EXC 2008/1 (UniSysCat) – 390540038. Open Access funding provided by the Max Planck Society.Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- A. Majumdar and J. Deutch, Joule, 2018, 2, 805–809 CrossRef.
- J. C. Abanades, E. S. Rubin, M. Mazzotti and H. J. Herzog, Energy Environ. Sci., 2017, 10, 2491–2499 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- R. Krause, D. Reinisch, C. Reller, H. Eckert, D. Hartmann, D. Taroata, K. Wiesner-Fleischer, A. Bulan, A. Lueken and G. Schmid, Chem. Ing. Tech., 2020, 92, 53–61 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2111–2112 CrossRef PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- M. König, J. Vaes, E. Klemm and D. Pant, iScience, 2019, 19, 135–160 CrossRef PubMed.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee, H. S. Ahn and W. Nam, Chem. Sci., 2018, 9, 6017–6034 RSC.
- K. Elouarzaki, V. Kannan, V. Jose, H. S. Sabharwal and J.-M. Lee, Adv. Energy Mater., 2019, 9, 1900090 CrossRef.
- F. Franco, S. Fernández and J. Lloret-Fillol, Curr. Opin. Electrochem., 2019, 15, 109–117 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- C. Jiang, A. W. Nichols and C. W. Machan, Dalton Trans., 2019, 48, 9454–9468 RSC.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- Z. Yin, G. T. R. Palmore and S. Sun, Trends Chem., 2019, 1, 739–750 CrossRef.
- G. Zhao, X. Huang, X. Wang and X. Wang, J. Mater. Chem. A, 2017, 5, 21625–21649 RSC.
- J.-H. Zhou and Y.-W. Zhang, React. Chem. Eng., 2018, 3, 591–625 RSC.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef CAS.
- T. E. Rosser and E. Reisner, ACS Catal., 2017, 7, 3131–3141 CrossRef CAS.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1, 0039 CrossRef CAS.
- A. Ge, B. Rudshteyn, P. E. Videla, C. J. Miller, C. P. Kubiak, V. S. Batista and T. Lian, Acc. Chem. Res., 2019, 52, 1289–1300 CAS.
- C. W. Machan, Curr. Opin. Electrochem., 2019, 15, 42–49 CrossRef CAS.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- P. Shao, L. Yi, S. Chen, T. Zhou and J. Zhang, J. Energy Chem., 2020, 40, 156–170 CrossRef.
- G. Yilmaz, S. B. Peh, D. Zhao and G. W. Ho, Adv. Sci., 2019, 6, 1901129 CrossRef CAS PubMed.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- Y. Zhu, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, Small Methods, 2019, 3, 1800438 CrossRef.
- J. A. Trindell, Z. Duan, G. Henkelman and R. M. Crooks, Chem. Rev., 2020, 120(2), 814–850 CrossRef CAS PubMed.
- R. M. Arán-Ais, D. Gao and B. Roldan Cuenya, Acc. Chem. Res., 2018, 51, 2906–2917 CrossRef PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- P. Sebastián-Pascual, S. Mezzavilla, I. E. L. Stephens and M. Escudero-Escribano, ChemCatChem, 2019, 11, 3626–3645 CrossRef.
- D. C. Grills, M. Z. Ertem, M. McKinnon, K. T. Ngo and J. Rochford, Coord. Chem. Rev., 2018, 374, 173–217 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- X.-M. Hu, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2019, 15, 148–154 CrossRef CAS.
- C. Costentin and J.-M. Savéant, Nat. Rev. Chem., 2017, 1, 0087 CrossRef CAS.
- A. W. Nichols and C. W. Machan, Front. Chem., 2019, 7, 397 CrossRef CAS PubMed.
- Y. Matsubara, ACS Energy Lett., 2019, 4, 1999–2004 CrossRef CAS.
- B. Das, A. Thapper, S. Ott and S. B. Colbran, Sustainable Energy Fuels, 2019, 3, 2159–2175 RSC.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- S. Mezzavilla, S. Horch, I. E. L. Stephens, B. Seger and I. Chorkendorff, Angew. Chem., 2019, 131, 3814–3818 CrossRef.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. Vayenas, R. White and M. Gamboa-Aldeco, Springer New York, 2008, ch. 3, vol. 42, pp. 89–189 Search PubMed.
- Y.-J. Zhang, V. Sethuraman, R. Michalsky and A. A. Peterson, ACS Catal., 2014, 4, 3742–3748 CrossRef CAS.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1988, 17–19, 10.1039/C39880000017.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS PubMed.
- Q. H. Low, N. W. X. Loo, F. Calle-Vallejo and B. S. Yeo, Angew. Chem., Int. Ed., 2019, 58, 2256–2260 CrossRef CAS PubMed.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- C. Reller, R. Krause, E. Volkova, B. Schmid, S. Neubauer, A. Rucki, M. Schuster and G. Schmid, Adv. Energy Mater., 2017, 7, 1602114 CrossRef.
- C. S. Le Duff, M. J. Lawrence and P. Rodriguez, Angew. Chem., Int. Ed., 2017, 56, 12919–12924 CrossRef CAS PubMed.
- R. M. Bullock, A. K. Das and A. M. Appel, Chem. – Eur. J., 2017, 23, 7626–7641 CrossRef CAS PubMed.
- M. Das Bairagya, R. J. Bujol and N. Elgrishi, Chem. – Eur. J., 2020, 26, 1–11 CrossRef.
- C. Costentin, M. Robert, J.-M. Savéant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882 CrossRef CAS PubMed.
- A. Taheri, E. J. Thompson, J. C. Fettinger and L. A. Berben, ACS Catal., 2015, 5, 7140–7151 CrossRef CAS.
- G. Neri, I. M. Aldous, J. J. Walsh, L. J. Hardwick and A. J. Cowan, Chem. Sci., 2016, 7, 1521–1526 RSC.
- J. J. Walsh, G. Neri, C. L. Smith and A. J. Cowan, Organometallics, 2019, 38, 1224–1229 CrossRef CAS.
- X.-M. Hu, Z. Salmi, M. Lillethorup, E. B. Pedersen, M. Robert, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Chem. Commun., 2016, 52, 5864–5867 RSC.
- S. A. Yao, R. E. Ruther, L. Zhang, R. A. Franking, R. J. Hamers and J. F. Berry, J. Am. Chem. Soc., 2012, 134, 15632–15635 CrossRef CAS PubMed.
- N. Elgrishi, S. Griveau, M. B. Chambers, F. Bedioui and M. Fontecave, Chem. Commun., 2015, 51, 2995–2998 RSC.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- Z. Yang, X. Zhang, C. Long, S. Yan, Y. Shi, J. Han, J. Zhang, P. An, L. Chang and Z. Tang, CrystEngComm, 2020, 22, 1619–1624 RSC.
- V. S. P. K. Neti, X. Wu, S. Deng and L. Echegoyen, Polym. Chem., 2013, 4, 4566–4569 RSC.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS.
- C. Costentin and J.-M. Savéant, Curr. Opin. Electrochem., 2019, 15, 58–65 CrossRef CAS.
- K. J. Lee, B. D. McCarthy and J. L. Dempsey, Chem. Soc. Rev., 2019, 48, 2927–2945 RSC.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2013, 135, 9023–9031 CrossRef CAS PubMed.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS PubMed.
- M. Hammouche, D. Lexa, J. M. Savéant and M. Momenteau, J. Electroanal. Chem. Interfacial Electrochem., 1988, 249, 347–351 CrossRef CAS.
- C. Costentin, J.-M. Savéant and C. Tard, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9104–9109 CrossRef CAS PubMed.
- C. Römelt, J. Song, M. Tarrago, J. A. Rees, M. van Gastel, T. Weyhermüller, S. DeBeer, E. Bill, F. Neese and S. Ye, Inorg. Chem., 2017, 56, 4745–4750 CrossRef PubMed.
- C. Römelt, S. Ye, E. Bill, T. Weyhermüller, M. van Gastel and F. Neese, Inorg. Chem., 2018, 57, 2141–2148 CrossRef PubMed.
- P. A. Davethu and S. P. de Visser, J. Phys. Chem. A, 2019, 123, 6527–6535 CrossRef CAS PubMed.
- M. Hammouche, D. Lexa, M. Momenteau and J. M. Saveant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Phys. Chem., 1996, 100, 19981–19985 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Saveant, J. Am. Chem. Soc., 1994, 116, 5015–5016 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS.
- J. Choi, T. M. Benedetti, R. Jalili, A. Walker, G. G. Wallace and D. L. Officer, Chem. – Eur. J., 2016, 22, 14158–14161 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2015, 48, 2996–3006 CrossRef CAS PubMed.
- B. Mondal, A. Rana, P. Sen and A. Dey, J. Am. Chem. Soc., 2015, 137, 11214–11217 CrossRef CAS PubMed.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- C. Costentin, M. Robert, J.-M. Savéant and C. Tard, Acc. Chem. Res., 2014, 47, 271–280 CrossRef CAS PubMed.
- M. T. Jensen, M. H. Rønne, A. K. Ravn, R. W. Juhl, D. U. Nielsen, X.-M. Hu, S. U. Pedersen, K. Daasbjerg and T. Skrydstrup, Nat. Commun., 2017, 8, 489 CrossRef PubMed.
- D. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- C. G. Margarit, N. G. Asimow, C. Costentin and D. G. Nocera, ACS Energy Lett., 2019, 72–78, DOI:10.1021/acsenergylett.9b02093.
- J. M. Barlow and J. Y. Yang, ACS Cent. Sci., 2019, 5, 580–588 CrossRef CAS PubMed.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- C. Costentin and J.-M. Savéant, J. Am. Chem. Soc., 2017, 139, 8245–8250 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- J. D. B. Koenig, J. Willkomm, R. Roesler, W. E. Piers and G. C. Welch, ACS Appl. Energy Mater., 2019, 2, 4022–4026 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- Y.-H. Wang, M. L. Pegis, J. M. Mayer and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 16458–16461 CrossRef CAS PubMed.
- C. M. Klug, A. J. P. Cardenas, R. M. Bullock, M. O’Hagan and E. S. Wiedner, ACS Catal., 2018, 8, 3286–3296 CrossRef CAS.
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, J. Am. Chem. Soc., 2018, 140, 16669–16675 CrossRef CAS PubMed.
- W. J. Shaw, M. L. Helm and D. L. DuBois, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1123–1139 CrossRef CAS PubMed.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS PubMed.
- S. Sinha and J. J. Warren, Inorg. Chem., 2018, 57, 12650–12656 CrossRef CAS PubMed.
- B. Zhao, H. Lei, N. Wang, G. Xu, W. Zhang and R. Cao, Chem. – Eur. J., 2020, 26, 4007–4012 CAS.
- C. G. Margarit, C. Schnedermann, N. G. Asimow and D. G. Nocera, Organometallics, 2019, 38, 1219–1223 CrossRef CAS.
- Eva M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Chem. Sci., 2018, 9, 2952–2960 RSC.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504–4509 CrossRef CAS PubMed.
- P. Sen, B. Mondal, D. Saha, A. Rana and A. Dey, Dalton Trans., 2019, 48, 5965–5977 RSC.
- T. R. Cundari, A. K. Wilson, M. L. Drummond, H. E. Gonzalez, K. R. Jorgensen, S. Payne, J. Braunfeld, M. De Jesus and V. M. Johnson, J. Chem. Inf. Model., 2009, 49, 2111–2115 CrossRef CAS PubMed.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS PubMed.
- Y. Matsubara, D. C. Grills and Y. Kuwahara, ACS Catal., 2015, 5, 6440–6452 CrossRef CAS.
- A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Chem. Commun., 2018, 54, 11630–11633 RSC.
- A. Maurin and M. Robert, Chem. Commun., 2016, 52, 12084–12087 RSC.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- F. Liu, T. Cardolaccia, B. J. Hornstein, J. R. Schoonover and T. J. Meyer, J. Am. Chem. Soc., 2007, 129, 2446–2447 CrossRef CAS PubMed.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Mater., 2017, 29, 7140–7150 CrossRef CAS.
- A. Tatin, C. Comminges, B. Kokoh, C. Costentin, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5526–5529 CrossRef CAS PubMed.
- J. Choi, P. Wagner, R. Jalili, J. Kim, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Adv. Energy Mater., 2018, 8, 1801280 CrossRef.
- J. Choi, J. Kim, P. Wagner, S. Gambhir, R. Jalili, S. Byun, S. Sayyar, Y. M. Lee, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Energy Environ. Sci., 2019, 12, 747–755 RSC.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- B.-X. Dong, S.-L. Qian, F.-Y. Bu, Y.-C. Wu, L.-G. Feng, Y.-L. Teng, W.-L. Liu and Z.-W. Li, ACS Appl. Energy Mater., 2018, 1, 4662–4669 CrossRef CAS.
- P. L. Cheung, S. K. Lee and C. P. Kubiak, Chem. Mater., 2019, 31, 1908–1919 CrossRef CAS.
- P. T. Smith, B. P. Benke, Z. Cao, Y. Kim, E. M. Nichols, K. Kim and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 9684–9688 CrossRef CAS PubMed.
- H. Kazuya, T. Katsuhiro, S. Hideo and T. Shinobu, Chem. Lett., 1977, 1137–1140 Search PubMed.
- T. Katsuhiro, H. Kazuya, S. Hideo and T. Shinobu, Chem. Lett., 1979, 305–308 Search PubMed.
- D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, J. Phys. Chem. A, 1998, 102, 2870–2877 CrossRef CAS.
- B. Hu, W. Xie, R. Li, Z. Pan, S. Song and Y. Wang, Electrochim. Acta, 2019, 135283, DOI:10.1016/j.electacta.2019.135283.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Chem. Commun., 2015, 51, 10226–10228 RSC.
- J. Grodkowski, P. Neta, E. Fujita, A. Mahammed, L. Simkhovich and Z. Gross, J. Phys. Chem. A, 2002, 106, 4772–4778 CrossRef CAS.
- A. Ogawa, K. Oohora, W. Gu and T. Hayashi, Chem. Commun., 2019, 55, 493–496 RSC.
- A. Ogawa, K. Oohora and T. Hayashi, Inorg. Chem., 2018, 57, 14644–14652 CrossRef CAS PubMed.
- X.-M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- K. Leung, I. M. B. Nielsen, N. Sai, C. Medforth and J. A. Shelnutt, J. Phys. Chem. A, 2010, 114, 10174–10184 CrossRef CAS PubMed.
- J. Shen, M. J. Kolb, A. J. Göttle and M. T. M. Koper, J. Phys. Chem. C, 2016, 120, 15714–15721 CrossRef CAS.
- G. Zhu, Y. Li, H. Zhu, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2016, 6, 6294–6301 CrossRef CAS.
- N. Sonoyama, M. Kirii and T. Sakata, Electrochem. Commun., 1999, 1, 213–216 CrossRef CAS.
- Y. Bochlin, E. Korin and A. Bettelheim, ACS Appl. Energy Mater., 2019, 2(12), 8434–8440 CrossRef CAS.
- M. Zhu, D.-T. Yang, R. Ye, J. Zeng, N. Corbin and K. Manthiram, Catal. Sci. Technol., 2019, 9, 974–980 RSC.
- H. Tanaka and A. Aramata, J. Electroanal. Chem., 1997, 437, 29–35 CrossRef CAS.
- T. Atoguchi, A. Aramata, A. Kazusaka and M. Enyo, J. Chem. Soc., Chem. Commun., 1991, 156–157, 10.1039/C39910000156.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- A. N. Marianov and Y. Jiang, Appl. Catal., B, 2019, 244, 881–888 CrossRef CAS.
- D. Quezada, J. Honores, M. García, F. Armijo and M. Isaacs, New J. Chem., 2014, 38, 3606–3612 RSC.
- J. E. Pander III, A. Fogg and A. B. Bocarsly, ChemCatChem, 2016, 8, 3536–3545 CrossRef.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- X.-D. Zhang, S.-Z. Hou, J.-X. Wu and Z.-Y. Gu, Chem. – Eur. J., 2020, 26, 1604–1611 CrossRef CAS PubMed.
- Y.-R. Wang, Q. Huang, C.-T. He, Y. Chen, J. Liu, F.-C. Shen and Y.-Q. Lan, Nat. Commun., 2018, 9, 4466 CrossRef PubMed.
- Y. Guo, W. Shi, H. Yang, Q. He, Z. Zeng, J.-y. Ye, X. He, R. Huang, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 17875–17883 CrossRef CAS PubMed.
- Q. Wu, R.-K. Xie, M.-J. Mao, G.-L. Chai, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, ACS Energy Lett., 2020, 5, 1005–1012 CrossRef CAS.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159, 10.1039/C39740000158.
- C. M. Lieber and N. S. Lewis, J. Am. Chem. Soc., 1984, 106, 5033–5034 CrossRef CAS.
- J. Zhang, W. J. Pietro and A. B. P. Lever, J. Electroanal. Chem., 1996, 403, 93–100 CrossRef.
- T. Abe, F. Taguchi, T. Yoshida, S. Tokita, G. Schnurpfeil, D. Wöhrle and M. Kaneko, J. Mol. Catal. A: Chem., 1996, 112, 55–61 CrossRef CAS.
- M. N. Mahmood, D. Masheder and C. J. Harty, J. Appl. Electrochem., 1987, 17, 1223–1227 CrossRef CAS.
- N. Furuya and K. Matsui, J. Electroanal. Chem. Interfacial Electrochem., 1989, 271, 181–191 CrossRef CAS.
- M. Zhu, R. Ye, K. Jin, N. Lazouski and K. Manthiram, ACS Energy Lett., 2018, 3, 1381–1386 CrossRef CAS.
- Z. Jiang, Y. Wang, X. Zhang, H. Zheng, X. Wang and Y. Liang, Nano Res., 2019, 12, 2330–2334 CrossRef CAS.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
- J. Chen, J. Li, W. Liu, X. Ma, J. Xu, M. Zhu and Y.-F. Han, Green Chem., 2019, 21, 6056–6061 RSC.
- H. Wu, M. Zeng, X. Zhu, C. Tian, B. Mei, Y. Song, X.-L. Du, Z. Jiang, L. He, C. Xia and S. Dai, ChemElectroChem, 2018, 5, 2717–2721 CrossRef CAS.
- M. Isaacs, F. Armijo, G. Ramírez, E. Trollund, S. R. Biaggio, J. Costamagna and M. J. Aguirre, J. Mol. Catal. A: Chem., 2005, 229, 249–257 CrossRef CAS.
- N. Morlanés, K. Takanabe and V. Rodionov, ACS Catal., 2016, 6, 3092–3095 CrossRef.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- X. Lu, Y. Wu, X. Yuan, L. Huang, Z. Wu, J. Xuan, Y. Wang and H. Wang, ACS Energy Lett., 2018, 3, 2527–2532 CrossRef CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- Y. Y. Birdja and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 2030–2034 CrossRef CAS PubMed.
- T. Yoshida, K. Kamato, M. Tsukamoto, T. Iida, D. Schlettwein, D. Wöhrle and M. Kaneko, J. Electroanal. Chem., 1995, 385, 209–225 CrossRef.
- T. Abe, T. Yoshida, S. Tokita, F. Taguchi, H. Imaya and M. Kaneko, J. Electroanal. Chem., 1996, 412, 125–132 CrossRef.
- W. W. Kramer and C. C. L. McCrory, Chem. Sci., 2016, 7, 2506–2515 RSC.
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y.-F. Han, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS.
- Y. Liu and C. C. L. McCrory, Nat. Commun., 2019, 10, 1683 CrossRef PubMed.
- J. Wang, X. Huang, S. Xi, J.-M. Lee, C. Wang, Y. Du and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 13532–13539 CrossRef CAS PubMed.
- R. Matheu, E. Gutierrez-Puebla, M. Á. Monge, C. S. Diercks, J. Kang, M. S. Prévot, X. Pei, N. Hanikel, B. Zhang, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 17081–17085 CrossRef CAS PubMed.
- J. Y. Becker, B. Vainas, R. Eger and L. Kaufman, J. Chem. Soc., Chem. Commun., 1985, 1471–1472, 10.1039/C39850001471.
- Y. Y. Birdja, J. Shen and M. T. M. Koper, Catal. Today, 2017, 288, 37–47 CrossRef CAS.
- A. J. Göttle and M. T. M. Koper, J. Am. Chem. Soc., 2018, 140, 4826–4834 CrossRef PubMed.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- Y. Wu, J. Jiang, Z. Weng, M. Wang, D. L. J. Broere, Y. Zhong, G. W. Brudvig, Z. Feng and H. Wang, ACS Cent. Sci., 2017, 3, 847–852 CrossRef CAS PubMed.
- J. Jiang, A. J. Matula, J. R. Swierk, N. Romano, Y. Wu, V. S. Batista, R. H. Crabtree, J. S. Lindsey, H. Wang and G. W. Brudvig, ACS Catal., 2018, 8, 10131–10136 CrossRef CAS.
- Y. Y. Birdja, R. E. Vos, T. A. Wezendonk, L. Jiang, F. Kapteijn and M. T. M. Koper, ACS Catal., 2018, 8, 4420–4428 CrossRef CAS PubMed.
- Z. Zhang, J. Xiao, X.-J. Chen, S. Yu, L. Yu, R. Si, Y. Wang, S. Wang, X. Meng, Y. Wang, Z.-Q. Tian and D. Deng, Angew. Chem., Int. Ed., 2018, 57, 16339–16342 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- D. Karapinar, A. Zitolo, T. N. Huan, S. Zanna, D. Taverna, L. H. Galvão Tizei, D. Giaume, P. Marcus, V. Mougel and M. Fontecave, ChemSusChem, 2020, 13, 173–179 CrossRef CAS PubMed.
- D.-D. Ma, S.-G. Han, C. Cao, X. Li, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2020, 264, 118530 CrossRef.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316, 10.1039/C39840001315.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- J. P. Collin, A. Jouaiti and J. P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1992, 322, 325–345 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- C. A. Kelly, Q. G. Mulazzani, M. Venturi, E. L. Blinn and M. A. J. Rodgers, J. Am. Chem. Soc., 1995, 117, 4911–4919 CrossRef CAS.
- J. Song, E. L. Klein, F. Neese and S. Ye, Inorg. Chem., 2014, 53, 7500–7507 CrossRef CAS PubMed.
- E. Fujita, J. Haff, R. Sanzenbacher and H. Elias, Inorg. Chem., 1994, 33, 4627–4628 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565–3573 CrossRef CAS PubMed.
- M. Fujihira, Y. Hirata and K. Suga, J. Electroanal. Chem. Interfacial Electrochem., 1990, 292, 199–215 CrossRef CAS.
- L. R. Furenlid, M. W. Renner, D. J. Szalda and E. Fujita, J. Am. Chem. Soc., 1991, 113, 883–892 CrossRef CAS.
- D. A. Gangi and R. R. Durand, J. Chem. Soc., Chem. Commun., 1986, 697–699, 10.1039/C39860000697.
- I. Zilbermann, M. Winnik, D. Sagiv, A. Rotman, H. Cohen and D. Meyerstein, Inorg. Chim. Acta, 1995, 240, 503–514 CrossRef CAS.
- K. Bujno, R. Bilewicz, L. Siegfried and T. A. Kaden, J. Electroanal. Chem., 1998, 445, 47–53 CrossRef CAS.
- E. Kimura, M. Haruta, T. Koike, M. Shionoya, K. Takenouchi and Y. Iitaka, Inorg. Chem., 1993, 32, 2779–2784 CrossRef CAS.
- P. J. Connolly and E. J. Billo, Inorg. Chem., 1987, 26, 3224–3226 CrossRef CAS.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 RSC.
- Y. Wu, B. Rudshteyn, A. Zhanaidarova, J. D. Froehlich, W. Ding, C. P. Kubiak and V. S. Batista, ACS Catal., 2017, 7, 5282–5288 CrossRef CAS.
- C. de Alwis, J. A. Crayston, T. Cromie, T. Eisenblätter, R. W. Hay, Y. D. Lampeka and L. V. Tsymbal, Electrochim. Acta, 2000, 45, 2061–2074 CrossRef CAS.
- E. Y. Lee, D. Hong, H. W. Park and M. P. Suh, Eur. J. Inorg. Chem., 2003, 3242–3249 CrossRef CAS.
- L.-M. Cao, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Green Chem., 2018, 20, 798–803 RSC.
- E. M. Nichols and C. J. Chang, Organometallics, 2019, 38, 1213–1218 CrossRef CAS.
- C. R. Schneider, L. C. Lewis and H. S. Shafaat, Dalton Trans., 2019, 48, 15810–15821 RSC.
- J. Honores, D. Quezada, M. García, K. Calfumán, J. P. Muena, M. J. Aguirre, M. C. Arévalo and M. Isaacs, Green Chem., 2017, 19, 1155–1162 RSC.
- A. Zhanaidarova, C. E. Moore, M. Gembicky and C. P. Kubiak, Chem. Commun., 2018, 54, 4116–4119 RSC.
- G. Neri, J. J. Walsh, C. Wilson, A. Reynal, J. Y. C. Lim, X. Li, A. J. P. White, N. J. Long, J. R. Durrant and A. J. Cowan, Phys. Chem. Chem. Phys., 2015, 17, 1562–1566 RSC.
- D. Saravanakumar, J. Song, N. Jung, H. Jirimali and W. Shin, ChemSusChem, 2012, 5, 634–636 CrossRef CAS PubMed.
- C. R. Schneider and H. S. Shafaat, Chem. Commun., 2016, 52, 9889–9892 RSC.
- C. Jiang, A. W. Nichols, J. F. Walzer and C. W. Machan, Inorg. Chem., 2020, 59, 1883–1892 CrossRef CAS PubMed.
- N. Sutin, C. Creutz and E. Fujita, Comments Inorg. Chem., 1997, 19, 67–92 CrossRef CAS.
- T. Ogata, Y. Yamamoto, Y. Wada, K. Murakoshi, M. Kusaba, N. Nakashima, A. Ishida, S. Takamuku and S. Yanagida, J. Phys. Chem., 1995, 99, 11916–11922 CrossRef CAS.
- A. H. A. Tinnemans, T. P. M. Koster, D. H. M. W. Thewissen and A. Mackor, Recl. Trav. Chim. Pays-Bas, 1984, 103, 288–295 CrossRef CAS.
- C. Creutz, H. A. Schwarz, J. F. Wishart, E. Fujita and N. Sutin, J. Am. Chem. Soc., 1991, 113, 3361–3371 CrossRef CAS.
- E. Fujita, C. Creutz, N. Sutin and D. J. Szalda, J. Am. Chem. Soc., 1991, 113, 343–353 CrossRef CAS.
- E. Fujita, D. J. Szalda, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1988, 110, 4870–4871 CrossRef CAS.
- T. Ogata, S. Yanagida, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 1995, 117, 6708–6716 CrossRef CAS.
- E. Fujita, C. Creutz, N. Sutin and B. S. Brunschwig, Inorg. Chem., 1993, 32, 2657–2662 CrossRef CAS.
- E. Fujita, L. R. Furenlid and M. W. Renner, J. Am. Chem. Soc., 1997, 119, 4549–4550 CrossRef CAS.
- M. H. Schmidt, G. M. Miskelly and N. S. Lewis, J. Am. Chem. Soc., 1990, 112, 3420–3426 CrossRef CAS.
- D. J. Szalda, E. Fujita and C. Creutz, Inorg. Chem., 1989, 28, 1446–1450 CrossRef CAS.
- E. Fujita and D. J. Szalda, Inorg. Chim. Acta, 2000, 297, 139–144 CrossRef CAS.
- C.-M. Che, S.-T. Mak, W.-O. Lee, K.-W. Fung and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1988, 2153–2159, 10.1039/DT9880002153.
- D. C. Lacy, C. C. L. McCrory and J. C. Peters, Inorg. Chem., 2014, 53, 4980–4988 CrossRef CAS PubMed.
- C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS PubMed.
- S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M.-N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544–17552 RSC.
- M. Ghosh, T. Weyhermüller and K. Wieghardt, Dalton Trans., 2010, 39, 1996–2007 RSC.
- M. Zhang, M. El-Roz, H. Frei, J. L. Mendoza-Cortes, M. Head-Gordon, D. C. Lacy and J. C. Peters, J. Phys. Chem. C, 2015, 119, 4645–4654 CrossRef CAS.
- H. Sheng and H. Frei, J. Am. Chem. Soc., 2016, 138, 9959–9967 CrossRef CAS PubMed.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef CAS PubMed.
- X. Su, K. M. McCardle, J. A. Panetier and J. W. Jurss, Chem. Commun., 2018, 54, 3351–3354 RSC.
- X. Su, K. M. McCardle, L. Chen, J. A. Panetier and J. W. Jurss, ACS Catal., 2019, 9, 7398–7408 CrossRef CAS.
- L. Chen, Z. Guo, X.-G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K.-C. Lau, T.-C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed.
- J.-W. Wang, H.-H. Huang, J.-K. Sun, D.-C. Zhong and T.-B. Lu, ACS Catal., 2018, 8, 7612–7620 CrossRef CAS.
- A. Taheri, C. R. Carr and L. A. Berben, ACS Catal., 2018, 8, 5787–5793 CrossRef CAS.
- N. D. Loewen, E. J. Thompson, M. Kagan, C. L. Banales, T. W. Myers, J. C. Fettinger and L. A. Berben, Chem. Sci., 2016, 7, 2728–2735 RSC.
- A. W. Nichols, S. Chatterjee, M. Sabat and C. W. Machan, Inorg. Chem., 2018, 57, 2111–2121 CrossRef CAS PubMed.
- A. W. Nichols, S. L. Hooe, J. S. Kuehner, D. A. Dickie and C. W. Machan, Inorg. Chem., 2020, 59, 5854–5864 CrossRef CAS PubMed.
- S.-N. Pun, W.-H. Chung, K.-M. Lam, P. Guo, P.-H. Chan, K.-Y. Wong, C.-M. Che, T.-Y. Chen and S.-M. Peng, J. Chem. Soc., Dalton Trans., 2002, 575–583, 10.1039/B108472K.
- D. Z. Zee, M. Nippe, A. E. King, C. J. Chang and J. R. Long, Inorg. Chem., 2020, 59, 5206–5217 CrossRef CAS PubMed.
- F. Franco, C. Cometto, F. Ferrero Vallana, F. Sordello, E. Priola, C. Minero, C. Nervi and R. Gobetto, Chem. Commun., 2014, 50, 14670–14673 RSC.
- F. Franco, C. Cometto, L. Nencini, C. Barolo, F. Sordello, C. Minero, J. Fiedler, M. Robert, R. Gobetto and C. Nervi, Chem. – Eur. J., 2017, 23, 4782–4793 CrossRef CAS PubMed.
- I. Fokin, A. Denisiuk, C. Würtele and I. Siewert, Inorg. Chem., 2019, 58, 10444–10453 CrossRef CAS PubMed.
- M. H. Rønne, D. Cho, M. R. Madsen, J. B. Jakobsen, S. Eom, É. Escoudé, H. C. D. Hammershøj, D. U. Nielsen, S. U. Pedersen, M.-H. Baik, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2020, 142, 4265–4275 CrossRef PubMed.
- E. A. Mohamed, Z. N. Zahran, Y. Tsubonouchi, K. Saito, T. Yui and M. Yagi, ACS Appl. Energy Mater., 2020, 3, 4114–4120 CrossRef CAS.
- F. Wang, B. Cao, W.-P. To, C.-W. Tse, K. Li, X.-Y. Chang, C. Zang, S. L.-F. Chan and C.-M. Che, Catal. Sci. Technol., 2016, 6, 7408–7420 RSC.
- J.-W. Wang, H.-H. Huang, J.-K. Sun, T. Ouyang, D.-C. Zhong and T.-B. Lu, ChemSusChem, 2018, 11, 1025–1031 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
- T. Shimoda, T. Morishima, K. Kodama, T. Hirose, D. E. Polyansky, G. F. Manbeck, J. T. Muckerman and E. Fujita, Inorg. Chem., 2018, 57, 5486–5498 CrossRef CAS PubMed.
- S. Fernández, F. Franco, C. Casadevall, V. Martin-Diaconescu, J. M. Luis and J. Lloret-Fillol, J. Am. Chem. Soc., 2020, 142, 120–133 CrossRef PubMed.
- S. L.-F. Chan, T. L. Lam, C. Yang, S.-C. Yan and N. M. Cheng, Chem. Commun., 2015, 51, 7799–7801 RSC.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- D. He, T. Jin, W. Li, S. Pantovich, D. Wang and G. Li, Chem. – Eur. J., 2016, 22, 13064–13067 CrossRef CAS PubMed.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, ACS Catal., 2018, 8, 3411–3417 CrossRef CAS.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S.-M. Ng, C.-C. Ko, G. Liu, L. Chen, M. Robert and T.-C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed.
- C. Cometto, R. Kuriki, L. Chen, K. Maeda, T.-C. Lau, O. Ishitani and M. Robert, J. Am. Chem. Soc., 2018, 140, 7437–7440 CrossRef CAS PubMed.
- C. Cometto, L. Chen, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, Organometallics, 2019, 38, 1280–1285 CrossRef CAS.
- K.-M. Lam, K.-Y. Wong, S.-M. Yang and C.-M. Che, J. Chem. Soc., Dalton Trans., 1995, 1103–1107, 10.1039/DT9950001103.
- W. Nie and C. C. L. McCrory, Chem. Commun., 2018, 54, 1579–1582 RSC.
- M. Wang, L. Chen, T.-C. Lau and M. Robert, Angew. Chem., Int. Ed., 2018, 57, 7769–7773 CrossRef CAS PubMed.
- W. Nie, Y. Wang, T. Zheng, A. Ibrahim, Z. Xu and C. C. L. McCrory, ACS Catal., 2020, 10, 4942–4959 CrossRef CAS.
- M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2016, 138, 1386–1393 CrossRef CAS PubMed.
- J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. DuBois, Organometallics, 2006, 25, 3345–3351 CrossRef CAS.
- K.-Y. Wong, W.-H. Chung and C.-P. Lau, J. Electroanal. Chem., 1998, 453, 161–170 CrossRef CAS.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- A. S. Varela, W. Ju and P. Strasser, Adv. Energy Mater., 2018, 8, 1703614 CrossRef.
- F. Pan, H. Zhang, Z. Liu, D. Cullen, K. Liu, K. More, G. Wu, G. Wang and Y. Li, J. Mater. Chem. A, 2019, 7, 26231–26237 RSC.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS.
- Y. Hori and A. Murata, Electrochim. Acta, 1990, 35, 1777–1780 CrossRef CAS.
- O. Koga and Y. Hori, Electrochim. Acta, 1993, 38, 1391–1394 CrossRef CAS.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Catal. Today, 2017, 288, 74–78 CrossRef CAS.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- A. Li, S. A. Nicolae, M. Qiao, K. Preuss, P. A. Szilágyi, A. Moores and M. M. Titirici, ChemCatChem, 2019, 11, 3602–3625 CrossRef CAS.
- W. Yang, S. Xu, K. Ma, C. Wu, I. D. Gates, X. Ding, W. Meng and Z. Gao, Nano Mater. Sci., 2020, 2, 120–131 CrossRef.
- J. Tuo, Y. Zhu, L. Cheng, Y. Li, X. Yang, J. Shen and C. Li, ChemSusChem, 2019, 12, 2644–2650 CrossRef CAS.
- J. Li, P. Pršlja, T. Shinagawa, A. J. Martín Fernández, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. Zhou, R. García-Muelas, N. López, J. Pérez-Ramírez and F. Jaouen, ACS Catal., 2019, 9, 10426–10439 CrossRef CAS.
- S. Zhao, G. Chen, G. Zhou, L. C. Yin, J. P. Veder, B. Johannessen, M. Saunders, S. Z. Yang, R. De Marco, C. Liu and S. P. Jiang, Adv. Funct. Mater., 2020, 30, 1906157 CrossRef CAS.
- C.-Z. Yuan, K. Liang, X.-M. Xia, Z. K. Yang, Y.-F. Jiang, T. Zhao, C. Lin, T.-Y. Cheang, S.-L. Zhong and A.-W. Xu, Catal. Sci. Technol., 2019, 9, 3669–3674 RSC.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal., B, 2019, 241, 113–119 CrossRef CAS.
- S. Ma, P. Su, W. Huang, S. P. Jiang, S. Bai and J. Liu, ChemCatChem, 2019, 11, 6092–6098 CrossRef CAS.
- F. Li, S. Hong, T.-S. Wu, X. Li, J. Masa, Y.-L. Soo and Z. Sun, ACS Appl. Energy Mater., 2019, 2, 8836–8842 CrossRef CAS.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1706617 CrossRef.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS.
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, J. Jo, H. Baik, V. Subramanian, M. Kim, U. Sim and K. T. Nam, Chem. – Eur. J., 2018, 24, 18444–18454 CrossRef CAS.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS.
- Z.-L. Wang, J. Choi, M. Xu, X. Hao, H. Zhang, Z. Jiang, M. Zuo, J. Kim, W. Zhou, X. Meng, Q. Yu, Z. Sun, S. Wei, J. Ye, G. G. Wallace, D. L. Officer and Y. Yamauchi, ChemSusChem, 2020, 13, 929–937 CrossRef CAS.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15, e1903668 CrossRef.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15, 1903668 CrossRef CAS.
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS.
- P. Su, K. Iwase, T. Harada, K. Kamiya and S. Nakanishi, Chem. Sci., 2018, 9, 3941–3947 RSC.
- C. Arana, S. Yan, M. Keshavarz-K, K. T. Potts and H. D. Abruna, Inorg. Chem., 1992, 31, 3680–3682 CrossRef CAS.
- C. Arana, M. Keshavarz, K. T. Potts and H. D. Abruña, Inorg. Chim. Acta, 1994, 225, 285–295 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CrossRef CAS.
- Y. Cheng, S. Zhao, H. Li, S. He, J.-P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan, M. F. Chisholm, S.-Z. Yang, C. Liu, J. G. Chen and S. P. Jiang, Appl. Catal., B, 2019, 243, 294–303 CrossRef CAS.
- Y. Zheng, J. Han, L. Takele, F. Xie, Y. Zhang, J. Sun, B. Han, J. Chen, Y. Gao and Z. Tang, Inorg. Chem. Front., 2019, 6, 1729–1734 RSC.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E.-s. Jeong, V. Subramanian, U. Sim and K. T. Nam, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- F. Pan, W. Deng, C. Justiniano and Y. Li, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- X.-M. Hu, H. H. Hval, E. T. Bjerglund, K. J. Dalgaard, M. R. Madsen, M.-M. Pohl, E. Welter, P. Lamagni, K. B. Buhl, M. Bremholm, M. Beller, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2018, 8, 6255–6264 CrossRef CAS.
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091 CrossRef CAS.
- M. N. Jackson, C. J. Kaminsky, S. Oh, J. F. Melville and Y. Surendranath, J. Am. Chem. Soc., 2019, 141, 14160–14167 CrossRef CAS PubMed.
- M. N. Jackson and Y. Surendranath, Acc. Chem. Res., 2019, 52, 3432–3441 CrossRef CAS.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- H.-J. Yang, J. Dong, Y.-H. Hong, W.-F. Lin, Z.-Y. Zhou and S.-G. Sun, Electrochem. Commun., 2018, 97, 82–86 CrossRef CAS.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- Q. Cheng, K. Mao, L. Ma, L. Yang, L. Zou, Z. Zou, Z. Hu and H. Yang, ACS Energy Lett., 2018, 3, 1205–1211 CrossRef CAS.
- S. Gonell, M. D. Massey, I. P. Moseley, C. K. Schauer, J. T. Muckerman and A. J. M. Miller, J. Am. Chem. Soc., 2019, 141, 6658–6671 CrossRef CAS.
- A. S. Varela, M. Kroschel, N. D. Leonard, W. Ju, J. Steinberg, A. Bagger, J. Rossmeisl and P. Strasser, ACS Energy Lett., 2018, 3, 812–817 CrossRef CAS.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- A. Zhanaidarova, H. Steger, M. H. Reineke and C. P. Kubiak, Dalton Trans., 2017, 46, 12413–12416 RSC.
- S. Sato, K. Saita, K. Sekizawa, S. Maeda and T. Morikawa, ACS Catal., 2018, 8, 4452–4458 CrossRef CAS.
- A. J. Göttle and M. T. M. Koper, Chem. Sci., 2017, 8, 458–465 RSC.
- N. Leonard, W. Ju, I. Sinev, J. Steinberg, F. Luo, A. S. Varela, B. Roldan Cuenya and P. Strasser, Chem. Sci., 2018, 9, 5064–5073 RSC.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- C. Cometto, L. Chen, D. Mendoza, B. Lassalle-Kaiser, T.-C. Lau and M. Robert, ChemSusChem, 2019, 12, 4500–4505 CrossRef CAS PubMed.
- W. Ju, A. Bagger, X. Wang, Y. Tsai, F. Luo, T. Möller, H. Wang, J. Rossmeisl, A. S. Varela and P. Strasser, ACS Energy Lett., 2019, 4, 1663–1671 CrossRef CAS.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal., B, 2019, 240, 234–240 CrossRef CAS.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- M. Stanbury, J.-D. Compain and S. Chardon-Noblat, Coord. Chem. Rev., 2018, 361, 120–137 CrossRef CAS.
- A. Sinopoli, N. T. La Porte, J. F. Martinez, M. R. Wasielewski and M. Sohail, Coord. Chem. Rev., 2018, 365, 60–74 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan, X. Cheng, B. Han, L. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119(12), 7610–7672 CrossRef CAS.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed.
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Adv. Funct. Mater., 2020, 30, 1907658 CrossRef CAS.
- H. Won da, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Angew. Chem., Int. Ed., 2016, 55, 9297–9300 CrossRef PubMed.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS PubMed.
- Z. Chen, K. Mou, S. Yao and L. Liu, ChemSusChem, 2018, 11, 2944–2952 CrossRef CAS PubMed.
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS.
- T. Bligaard and J. K. Nørskov, Electrochim. Acta, 2007, 52, 5512–5516 CrossRef CAS.
- B. Roldan Cuenya and F. Behafarid, Surf. Sci. Rep., 2015, 70, 135–187 CrossRef CAS.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- K. D. Yang, C. W. Lee, J. H. Jang, T. R. Ha and K. T. Nam, Nanotechnology, 2017, 28, 352001 CrossRef.
- F. Li, D. R. MacFarlane and J. Zhang, Nanoscale, 2018, 10, 6235–6260 RSC.
- R. M. Arán-Ais, D. Gao and B. Roldan Cuenya, Acc. Chem. Res., 2018, 51(11), 2906–2917 CrossRef.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. Roldan Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS.
- Z. Zhang, M. Chi, G. M. Veith, P. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Zhu, ACS Catal., 2016, 6, 6255–6264 CrossRef CAS.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS.
- H. S. Jeon, I. Sinev, F. Scholten, N. J. Divins, I. Zegkinoglou, L. Pielsticker and B. Roldan Cuenya, J. Am. Chem. Soc., 2018, 140, 9383–9386 CrossRef CAS.
- A. Dutta, A. Kuzume, M. Rahaman, S. Vesztergom and P. Broekmann, ACS Catal., 2015, 5, 7498–7502 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2019, 2, 86–93 CrossRef CAS.
- D. Ren, J. Fong and B. S. Yeo, Nat. Commun., 2018, 9, 925 CrossRef.
- K. D. Yang, C. W. Lee, K. Jin, S. W. Im and K. T. Nam, J. Phys. Chem. Lett., 2017, 8, 538–545 CrossRef CAS PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS.
- W. Zhu, S. Kattel, F. Jiao and J. G. Chen, Adv. Energy Mater., 2019, 9, 1802840 CrossRef.
- H.-E. Lee, K. D. Yang, S. M. Yoon, H.-Y. Ahn, Y. Y. Lee, H. Chang, D. H. Jeong, Y.-S. Lee, M. Y. Kim and K. T. Nam, ACS Nano, 2015, 9, 8384–8393 CrossRef CAS.
- S. Ma, Y. Lan, G. M. J. Perez, S. Moniri and P. J. A. Kenis, ChemSusChem, 2014, 7, 866–874 CrossRef CAS.
- L. Zhang, F. Mao, L. R. Zheng, H. F. Wang, X. H. Yang and H. G. Yang, ACS Catal., 2018, 8, 11035–11041 CrossRef CAS.
- S. Zhao, Z. Tang, S. Guo, M. Han, C. Zhu, Y. Zhou, L. Bai, J. Gao, H. Huang, Y. Li, Y. Liu and Z. Kang, ACS Catal., 2018, 8, 188–197 CrossRef CAS.
- C. Rogers, W. S. Perkins, G. Veber, T. E. Williams, R. R. Cloke and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 4052–4061 CrossRef CAS PubMed.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS.
- Y. T. Guntern, J. R. Pankhurst, J. Vávra, M. Mensi, V. Mantella, P. Schouwink and R. Buonsanti, Angew. Chem., Int. Ed., 2019, 58, 12632–12639 CrossRef CAS.
- J. A. Trindell, Z. Duan, G. Henkelman and R. M. Crooks, Chem. Rev., 2020, 120, 814–850 CrossRef CAS PubMed.
- Q. Lenne, Y. R. Leroux and C. Lagrost, ChemElectroChem, 2020, 7, 2345–2363 CrossRef CAS.
- D. Ung, I. A. Murphy and B. M. Cossairt, Dalton Trans., 2020, 49, 4995–5005 RSC.
- M. A. Ortuño and N. López, Catal. Sci. Technol., 2019, 9, 5173–5185 RSC.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef CAS PubMed.
- N. Austin, S. Zhao, J. R. McKone, R. Jin and G. Mpourmpakis, Catal. Sci. Technol., 2018, 8, 3795–3805 RSC.
- Z. Wang, L. Wu, K. Sun, T. Chen, Z. Jiang, T. Cheng and W. A. Goddard, J. Phys. Chem. Lett., 2018, 9, 3057–3061 CrossRef CAS.
- Z. Wang, K. Sun, C. Liang, L. Wu, Z. Niu and J. Gao, ACS Appl. Energy Mater., 2019, 2, 192–195 CrossRef CAS.
- Y. Fang and J. C. Flake, J. Am. Chem. Soc., 2017, 139, 3399–3405 CrossRef CAS.
- Z. Cao, S. B. Zacate, X. Sun, J. Liu, E. M. Hale, W. P. Carson, S. B. Tyndall, J. Xu, X. Liu, X. Liu, C. Song, J.-h. Luo, M.-J. Cheng, X. Wen and W. Liu, Angew. Chem., Int. Ed., 2018, 130, 12857–12861 CrossRef.
- M. Gong, Z. Cao, W. Liu, E. M. Nichols, P. T. Smith, J. S. Derrick, Y.-S. Liu, J. Liu, X. Wen and C. J. Chang, ACS Cent. Sci., 2017, 3, 1032–1040 CrossRef CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- M. Gao, Y. Zhu, Y. Liu, K. Wu, H. Lu, S. Tang, C. Liu, H. Yue, B. Liang and J. Yan, Chem. Commun., 2020, 56, 7021–7024 RSC.
- Y. Zhao, C. Wang, Y. Liu, D. R. MacFarlane and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1801400 CrossRef.
- J. A. Trindell, J. Clausmeyer and R. M. Crooks, J. Am. Chem. Soc., 2017, 139, 16161–16167 CrossRef CAS.
- L. Ma, W. Hu, Q. Pan, L. Zou, Z. Zou, K. Wen and H. Yang, J. CO2 Util., 2019, 34, 108–114 CrossRef CAS.
- Z. Ma, C. Lian, D. Niu, L. Shi, S. Hu, X. Zhang and H. Liu, ChemSusChem, 2019, 12, 1724–1731 CrossRef CAS.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X.-D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS.
- J. R. Pankhurst, Y. T. Guntern, M. Mensi and R. Buonsanti, Chem. Sci., 2019, 10, 10356–10365 RSC.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- S.-F. Zhao, M. Horne, A. M. Bond and J. Zhang, J. Phys. Chem. C, 2016, 120, 23989–24001 CrossRef CAS.
- G. P. S. Lau, M. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, J. Am. Chem. Soc., 2016, 138, 7820–7823 CrossRef CAS PubMed.
- H.-K. Lim, Y. Kwon, H. S. Kim, J. Jeon, Y.-H. Kim, J.-A. Lim, B.-S. Kim, J. Choi and H. Kim, ACS Catal., 2018, 8, 2420–2427 CrossRef CAS.
- D. C. Grills, Y. Matsubara, Y. Kuwahara, S. R. Golisz, D. A. Kurtz and B. A. Mello, J. Phys. Chem. Lett., 2014, 5, 2033–2038 CrossRef CAS PubMed.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2019, 123, 24453–24460 CrossRef CAS.
- A. Thevenon, A. Rosas-Hernández, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 58, 16952–16958 CrossRef CAS PubMed.
- S. Zhong, X. Yang, Z. Cao, X. Dong, S. M. Kozlov, L. Falivene, J.-K. Huang, X. Zhou, M. N. Hedhili, Z. Lai, K.-W. Huang, Y. Han, L. Cavallo and L.-J. Li, Chem. Commun., 2018, 54, 11324–11327 RSC.
- Y. Qiu, H. Zhong, W. Xu, T. Zhang, X. Li and H. Zhang, J. Mater. Chem. A, 2019, 7, 5453–5462 RSC.
- X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103–4111 CrossRef CAS.
- K. G. Schmitt and A. A. Gewirth, J. Phys. Chem. C, 2014, 118, 17567–17576 CrossRef CAS.
- A. Wagner, K. H. Ly, N. Heidary, I. Szabó, T. Földes, K. I. Assaf, S. J. Barrow, K. Sokołowski, M. Al-Hada, N. Kornienko, M. F. Kuehnel, E. Rosta, I. Zebger, W. M. Nau, O. A. Scherman and E. Reisner, ACS Catal., 2020, 10, 751–761 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- F. Franco, M. F. Pinto, B. Royo and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2018, 57, 4603–4606 CrossRef CAS PubMed.
- T. H. T. Myren, A. M. Lilio, C. G. Huntzinger, J. W. Horstman, T. A. Stinson, T. B. Donadt, C. Moore, B. Lama, H. H. Funke and O. R. Luca, Organometallics, 2019, 38, 1248–1253 CrossRef CAS.
- T. H. T. Myren, A. Alherz, J. R. Thurston, T. A. Stinson, C. G. Huntzinger, C. B. Musgrave and O. R. Luca, ACS Catal., 2020, 10, 1961–1968 CrossRef CAS.
- J. A. Therrien and M. O. Wolf, Inorg. Chem., 2017, 56, 1161–1172 CrossRef CAS.
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., 2018, 130, 5075–5079 CrossRef.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- L. Zhang, Z. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., 2019, 131, 15981–15987 CrossRef.
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 4981–4985 CrossRef CAS PubMed.
- H. Lu, Z. Zhou, O. V. Prezhdo and R. L. Brutchey, J. Am. Chem. Soc., 2016, 138, 14844–14847 CrossRef CAS PubMed.
- M. J. Trujillo, S. L. Strausser, J. C. Becca, J. F. DeJesus, L. Jensen, D. M. Jenkins and J. P. Camden, J. Phys. Chem. Lett., 2018, 9, 6779–6785 CrossRef CAS PubMed.
- A. Bakker, A. Timmer, E. Kolodzeiski, M. Freitag, H. Y. Gao, H. Mönig, S. Amirjalayer, F. Glorius and H. Fuchs, J. Am. Chem. Soc., 2018, 140, 11889–11892 CrossRef CAS PubMed.
- S. Dery, S. Kim, G. Tomaschun, I. Berg, D. Feferman, A. Cossaro, A. Verdini, L. Floreano, T. Klüner, F. D. Toste and E. Gross, J. Phys. Chem. Lett., 2019, 10, 5099–5104 CrossRef CAS PubMed.
- G. Lovat, E. A. Doud, D. Lu, G. Kladnik, M. S. Inkpen, M. L. Steigerwald, D. Cvetko, M. S. Hybertsen, A. Morgante, X. Roy and L. Venkataraman, Chem. Sci., 2019, 10, 930–935 RSC.
| This journal is © The Royal Society of Chemistry 2020 |