
Artificial photosynthesis using metal/nonmetal-nitride semiconductors: current status, prospects, and challenges
M. G.
Kibria
and
Z.
Mi
*
Department of Electrical and Computer Engineering, McGill University, 3480 University Street, Montreal, Québec H3A 0E9, Canada. E-mail: zetian.mi@mcgill.ca; Tel: +1 514 398 7114
First published on 9th November 2015
Abstract
Artificial photosynthesis, i.e. the chemical transformation of sunlight, water and carbon dioxide into high-energy-rich fuels is one of the key sustainable energy technologies to enable a carbon-free, storable and renewable source of energy. Although significant progress has been made over the last four decades, the development of efficient, long-term stable, scalable, and cost-competitive photocatalysts has remained one of the key challenges for the large-scale practical application of this frontier technology. Over the last decade metal/nonmetal-nitrides have emerged as a new generation of photocatalyst materials for artificial photosynthesis owing to their distinct optoelectronic and catalytic properties. This article provides an overview of the state-of-the-art research activities on the development of metal/nonmetal-nitride semiconductor based photocatalysts and photoelectrodes for solar-fuel conversion.
 M. G. Kibria | Md Golam Kibria received B.Sc. in electrical and electronic engineering from Khulna University of Engineering and Technology, Bangladesh in 2006; MASc and Ph.D. in electrical and computer engineering from McMaster University and McGill University, Canada in 2010 and 2015, respectively. He received Green Talents Award, FRQNT Post-Doctoral Fellowship, Academic Gold Medal, Tomlinson Doctoral Fellowship, SPIE Education Scholarship, and Best Student Presentation Award at 30th NAMBE conference. At present, he is a visiting scholar at McGill University. He is interested in the broad area of artificial photosynthesis, i.e. solar water splitting and CO2 reduction for sustainable energy and green environment. |
 Z. Mi | Zetian Mi is an Associate Professor in the Department of Electrical and Computer Engineering at McGill University. He received the Ph.D. degree in Applied Physics from the University of Michigan, Ann Arbor in 2006. Prof. Mi’s teaching and research interests are in the areas of III-nitride semiconductors, low dimensional nanostructures, nanophotonics, and solar fuels. He has received many awards, including the Hydro-Québec Nano-Engineering Scholar Award in 2009, the William Dawson Scholar Award in 2011, the Christophe Pierre Award for Research Excellence in 2012, and the Engineering Innovation Award in 2015 at McGill University. He has also received the Young Scientist Award from the International Symposium on Compound Semiconductors in 2015 and the Young Investigator Award from the 27th North American Molecular Beam Epitaxy Conference in 2010. Prof. Mi served as the Associate Editor of IEEE J. Lightwave Technol. as well as the chair and organizer of many international conferences regularly. |
Introduction
Amongst various sustainable energy options, solar energy may be the ultimate solution to mitigate the current and future global energy demand.1 The currently available photovoltaic cells can convert sunlight to electricity with an efficiency up to 46%.2 However, because of the intermittency and variable atmospheric conditions, the major challenge remains in storing solar energy for short and long-term applications.3 At present, there are four forms of technologies available to store energy, including (1) potential energy in the form of pumped-hydroelectric, compressed air, and electric charge in super/ultracapacitors, (2) kinetic energy mainly in the form of flywheels, (3) thermal energy in the form of concentrated solar thermal and geothermal, and (4) chemical energy in the form of batteries or fuels.3 Unfortunately, all of the present energy storage technologies experience one or more of the key obstacles, including high cost, short time storage, and low energy density to be implemented for sustainable and large-scale applications.3 Alternatively, a potentially viable technology for solar energy storage is artificial photosynthesis, which refers to any scheme that mimics natural photosynthesis by which green plants convert sunlight, carbon dioxide (CO2) and water into carbohydrates to capture and store solar energy in the chemical bonds of a fuel (solar-fuel).4–9 There are commonly two schemes of artificial photosynthesis, including sunlight driven water splitting into hydrogen (H2) fuel and photoinduced CO2 reduction into various hydrocarbons.5,9 To date, research efforts on artificial photosynthesis are mostly directed towards sunlight-driven water splitting.10 Although H2 is an important fuel and chemical feedstock with the highest energy density by mass (∼140 MJ kg−1), it suffers from low volumetric energy densities. A practical alternative is hydrocarbon fuel with optimum volumetric energy density for better integration with existing energy infrastructure.3 Although there is an immense potential, major challenge remains in developing a cheap, efficient and stable artificial photosynthesis system that is capable of producing cost-competitive fuels (i.e., hydrogen, methanol, etc.) to replace fossil fuels.In an artificial photosynthesis system, the dye molecule or semiconductor photocatalyst captures solar energy and subsequently splits water into its constituents (i.e., H2 and O2) with a positive change in Gibbs free energy (i.e., uphill reaction):11
| H2O → ½O2 (g) + H2 (g), ΔG = +237.178 kJ mol−1 | (1) |
The hydrogen produced from this reaction can be the central energy carrier in a Hydrogen Economy.12,13 Alternatively, this renewable hydrogen could be used to reduce anthropogenic CO2 for the exothermic formation of useful energy-rich hydrocarbons i.e., methane, formic acid, methanol, etc. These renewable value-added hydrocarbons can replace conventional fuels used for transportation or can be used as the basic synthetic components for hundreds of chemicals.9,14–16
Although the concept of artificial photosynthesis was envisioned and proposed in 1874 by Verne (available at http://www.literature-web.net/verne/mysteriousisland, 1874), and in 1912 by Ciamician,17 respectively, the experimental demonstration was not reported until late 60's. In 1968 Boddy reported light-driven oxygen evolution at an n-type rutile (TiO2) electrode.18 Subsequently, in 1972 Fujishima and Honda applied this concept for water photoelectrolysis in a cell composed of an n-type rutile (TiO2) photoanode and a platinum cathode.19 On the other hand, the two earliest reports on light-driven CO2 reduction are Halmann's work on photoelectrochemical CO2 reduction in 1978,20 and Honda and co-workers' work on photocatalytic CO2 reduction in 1979.21 These seminal studies stimulated decades of international effort on the development of various efficient, stable and cost-effective photocatalysts for sunlight-driven water splitting and CO2 reduction.
Photocatalytic (also known as photochemical, Schottky-type, suspended photocatalyst or photoparticle system) and photoelectrochemical (photoelectrode system) are the two common schemes for solar-driven water splitting and CO2 reduction. In a photocatalytic water splitting approach, light absorption, charge carrier separation, and catalytic reactions proceed on an integrated photosystem, consisting of a host photocatalyst and one or more co-catalysts.22 The ideal limiting solar-to-hydrogen (STH) efficiency of such an integrated photosystem is 14.4% (single-bandgap photosystem), assuming a bandgap of 2.0 eV and energy loss of 0.8 eV per electron.23 A 10% STH efficiency in a photocatalytic system would provide H2 at a cost of $1.63 per kg, providing a cost-competitive alternative to gasoline.24 To date research has been largely focused on metal-oxide based photocatalysts owing to their photostability in aqueous solution.10,11,25 However, most of the metal-oxides suffer from inefficient light absorption due to their large bandgap (O2p orbital is located at ca. +3.0 eV or higher) and/or poor optoelectronic properties, i.e., their short electron–hole lifetimes and low mobility.26 Therefore, a number of band-engineering and nanostructuring strategies have been developed to overcome the limiting factors of metal-oxides, including metal/nonmetal ion doping (e.g., C4−, N3−, and S2−), solid solution (GaN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZnO, quantum efficiency = 5.9%), elemental substitution, sensitization, etc.27–31 However, owing to inefficient light absorption and limited carrier extraction, the achieved STH efficiency of such a band-engineered photocatalyst is far below the values of practical interest (∼10%). Although relatively high STH efficiencies (e.g., 5% from CoO,32 2% from CDots-C3N4,33 and 1.8% from p-GaN/p-InGaN34) have been reported by combinatorial approaches very recently, their long-term instability and high cost remain the key concerns for commercialization. Alternatively, a Z-scheme (also known as a tandem or two-step photoexcitation system) photosystem has been developed to utilize a wide variety of small bandgap materials.35–37 In this system, two or more photoabsorbers are connected via a redox shuttle. While the ideal limiting STH efficiency of such a Z-scheme is relatively high (24.4% for bandgaps of 2.25 and 1.77 eV),23 the achieved STH efficiency to date is extremely low (∼0.1%) due to greater system complexity.38
:ZnO, quantum efficiency = 5.9%), elemental substitution, sensitization, etc.27–31 However, owing to inefficient light absorption and limited carrier extraction, the achieved STH efficiency of such a band-engineered photocatalyst is far below the values of practical interest (∼10%). Although relatively high STH efficiencies (e.g., 5% from CoO,32 2% from CDots-C3N4,33 and 1.8% from p-GaN/p-InGaN34) have been reported by combinatorial approaches very recently, their long-term instability and high cost remain the key concerns for commercialization. Alternatively, a Z-scheme (also known as a tandem or two-step photoexcitation system) photosystem has been developed to utilize a wide variety of small bandgap materials.35–37 In this system, two or more photoabsorbers are connected via a redox shuttle. While the ideal limiting STH efficiency of such a Z-scheme is relatively high (24.4% for bandgaps of 2.25 and 1.77 eV),23 the achieved STH efficiency to date is extremely low (∼0.1%) due to greater system complexity.38
In the scheme of photoelectrochemical (PEC) water splitting, the oxidation and reduction reactions proceed on two different electrodes that are connected via an external circuit.39 In this case, some external bias is usually required for efficient carrier separation and to overcome the resistance between the electrodes in the solution. As the light absorption, charge separation, and catalytic reactions do not proceed at close proximity, PEC water splitting is more complex and is nearly one order of magnitude more expensive than a photochemical system at equal efficiency.40 To date the reported STH efficiencies of metal-oxide and other semiconductor-based photoelectrodes are low due to inefficient light absorption, limited carrier separation, and insufficient redox potentials.31,41 To overcome the efficiency bottleneck and reduce the cost, photovoltaic (PV) integration with a PEC system (PV + PEC) or with electrocatalyts (PV + EL) has achieved impressive success, with STH efficiencies from 3 to 22%.42–52 While the achieved efficiency is over half of the theoretical efficiency limit of these devices (i.e., 24.4% for a tandem and 30% for multi-junction),23,53,54 the long-term instability of the photoabsorbers, and their limited scalability due to high cost remain some of the major concerns.55 Therefore, a number of stable passivation materials (e.g., TiO2, Ir/TiO2, Ni, SrTiO3, MnO, etc.) have been developed to enhance the stability of Si and III–V; however, with limited success.56–60
In the meantime, research on metal/nonmetal-nitride photocatalysts and photoelectrodes (e.g., GaN, InGaN, C3N4, T3N5, Ge3N4, W2N, InN, BCN, etc.) for water splitting has drawn considerable attention. As illustrated in Fig. 1, publications in this field have increased exponentially in the last decade. This rapid rise has been fuelled, to a certain extent, by the recent development of LED lighting technology, which has led to significantly improved material quality of metal-nitride semiconductors with dramatically reduced manufacturing cost.61Tables 1 and 2 summarize the major studies on metal/nonmetal-nitride based photocatalysts and photoelectrodes for solar water splitting, respectively. Metal/nonmetal-nitrides possess excellent catalytic, electrical and optical properties. For example, metal/nonmetal-nitride semiconductors often possess a narrow band gap due to the more negative potential of the N2p orbital compared to the O2p orbital in metal-oxides.62 As an example, the bandgap of metal-oxide Ta2O5 is 3.9 eV, whereas the bandgap of Ta3N5 is 2.1 eV.63 As illustrated in Fig. 2, the bandgap of most of the metal/nonmetal-nitrides straddle the redox potential of water, with sufficient kinetic overpotentials for water redox reactions and CO2 reduction to various hydrocarbons. In contrast, most of the metal-oxides do not possess a suitable conduction band edge required for water (or CO2) reduction to hydrogen (or hydrocarbons).10 One of the nitrides, GaN, possesses a direct energy bandgap that can be tuned from 3.4 to 0.65 eV across the ultraviolet, visible, and near-infrared spectrum by introducing indium (In),64,65 thereby offering the unique opportunity to harness nearly the entire solar spectrum.66 Additionally, nitrides possess a high absorption co-efficient and large charge carrier mobility, leading to excellent photon absorption and charge carrier extraction for efficient solar-fuel conversion.65,67,68 In contrast to traditional III–V compounds, wherein the chemical bonds are mostly covalent, the chemical bonds in III-nitrides are strongly ionic.69 Because of the strong ionicity of nitrides, the surface states are located mostly near the band edges, which prevent them from being non-radiative recombination centers. Consequently, the Fermi level is not pinned in the energy gap of III-nitrides, thereby suppressing the participation of these states in the self-oxidation process of the photoanode, resulting in photostability of the electrode.69 However, the presence of any surface defects, which often depends on the growth method, may lead to Fermi-level pinning in the bandgap and photodegrade the material. Indeed, recent studies have demonstrated excellent photostability of nearly defect-free metal-nitrides against photocorrosion in acidic and neutral pH electrolytes.70–72 Therefore, it is suggested that defect-free and high crystalline quality nitrides can function both as anodes and cathodes. However, in the presence of high density of defects, nitrides may be more suitable as photocathodes.73
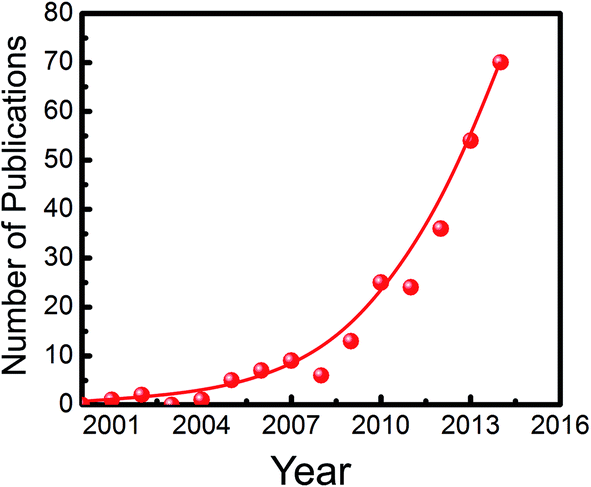 | ||
| Fig. 1 The rise of nitrides. Annual number of publications in Web of Science when a search for the topic “water splitting” and “nitride” was performed. | ||
| Photocatalyst | Co-catalyst | Light source | Reaction solution | Activity (μmol h−1 g−1) | Efficiency (%) | Ref. (year) | |
|---|---|---|---|---|---|---|---|
| H2 | O2 | ||||||
| BCN | Pt, RuO2, IrO2, Ni–Co LDH | 300 W Xe | Triethanolamine AgNO3 | 80 | 11 | AQE = 0.54 (405 nm) | 154 (2015) |
| CDots-C3N4 | 300 W Xe | Pure water | 575 | 287 | STH = 2% | 33 (2015) | |
| p-InGaN/p-GaN | Rh/Cr2O3 | 300 W Xe | Pure water | 3.46 mol h−1 g−1 | 1.69 mol h−1 g−1 | STH = 1.8% | 34 (2015) |
| InGaN/MC-540 | Rh | 300 W Xe | Acetonitrile and EDTA | 65 mmol h−1 g−1 | AQE = 0.3% (525–600 nm) | 119 (2015) | |
| GaN:Mg |
Rh/Cr2O3 | 300 W Xe | Pure water | 4 mol h−1 g−1 | 2 mol h−1 g−1 | IQE = 51% (200–365 nm) | 155 (2014) |
| g-C3N4 | 3 wt% Pt | ≥420 nm | Triethanolamine | 3327 | AQE = 26.5% (400 nm) | 150 (2014) | |
| Conjugated C3N4 | 3 wt% | ≥420 nm | 10 v% triethanolamine | 14800 |
AQE = 8.8% (420 nm) | 141 (2014) | |
| (Ga0.82Zn0.18)(N0.82O0.18) nanostructure | Rh2−xCrxO3 | >400 nm | H2SO4 (pH 4.5) | 1271 | 635.5 | AQE = 17.3% (400 nm) | 109 (2014) |
| mpg-C3N4 dye-sensitized C3N4 nanosheet | 1.25 wt% Pt | ≥420 nm | 5 v% triethanolamine | 6525 | AQE = 33.4% (460 nm) | 153 (2013) | |
| InGaN/GaN | Rh/Cr2O3 | 300 W Xe | Pure water | 92 mmol h−1 g−1 | 46 mmol h−1 g−1 | AQE = 1.86% (395–405 nm) | 118 (2013) |
| Ta3N5 | 2 wt% CoOx | ≥420 nm | 0.01 M AgNO3 | 4500 | AQE = 5.2% (500–600 nm) | 124 (2012) | |
| Hollow C3N4 nanospheres | 3 wt% Pt | >420 nm | 10 v% triethanolamine | 11200 |
AQE = 7.5% (420 nm) | 133 (2012) | |
| g-C3N4 | 1 wt% RGO, 1.5% Pt | >400 nm | 25% methanol | 451 | AQE = 2.6% | 151 (2011) | |
| GaN nanowire | Core/shell Rh/Cr2O3 | 300 W Xe | Pure water | 3.6 | 1.8 | AQE = 0.5% | 101 (2011) |
| g-C3N4 | 3 wt% Pt | >420 nm | 10 v% triethanolamine | ∼110 | AQE = 0.1% (420–460 nm) | 80 (2009) | |
| (Ga0.82Zn0.18)(N0.82O0.18) | Rh2−xCrxO3 | >400 nm | H2SO4 (pH 4.5) | 3090 | 1533 | AQE = 5.9 (420–440 nm) | 108 (2008) |
| GaN powder | Rh2−xCrxO3 | 450 W mercury | H2SO4 (pH 4.5) | 64 | 32 | AQE = 0.7% (300–340 nm) | 99 (2007) |
| GaN powder (Mg, Zn, Be doped) | RuO2 | 450 W mercury | Pure water | 750 | 375 | 98 (2007) | |
| β-Ge3N4 | RuO2 | 450 W mercury | 1 M H2SO4 | 3.6 mmol h−1 g−1 | 1.8 mmol h−1 g−1 | 94 (2007) | |
| β-Ge3N4 | RuO2 | 450 W mercury | H2SO4 (pH 0) | 1 mmol h−1 | 0.5 mmol h−1 | AQE = 9% (300 nm) | 93 (2005) |
| Photoelectrode | Co-catalyst | Light intensity (mW cm−2) | Electrolyte | Photocurrent (mA cm−2) | Efficiency | Ref. (year) |
|---|---|---|---|---|---|---|
| n+–p-Si/n-GaN/TJ/p-InGaN | Pt | 130, AM1.5G | 1 M HBr | −40.6 at 0.26 VNHE | ABPE = 8.7% at 0.33 VNHE | 197 (2015) |
| InN/InGaN QD | 100, xenon lamp | pH 3 H2SO4 and 0.5 M Na2SO4 | 12.7 at 0 VAg/AgCl | IPCE = 56% at 600 nm at 0 VAg/AgCl | 184 (2015) | |
| Coaxial InGaN/GaN MQW nanowire photoanode | 150, AM1.5G | 1 M HCl | 2.1 at 1 VCathode | ABPE = 0.3%, at 0.4 VCathode, IPCE = 15% at 350 nm at 1 VCathode | 181 (2015) | |
| GaN/AlN/n-GaN photocathode | 110, 220, 330 | 0.5 M H2SO4 | −2.0 (330 mW cm−2) at −0.5 VAg/AgCl/NaCl | 166 (2014) | ||
| InGaN/GaN MQW | 100 | 1 M HBr | 1.2 at VCE = 0 | STH = 1.5% at 0 VCE | 196 (2014) | |
| InGaN/GaN nanoporous | 100 | 1 M HCl | 0.4 at VCE = 1.0 | IPCE = 46% at 355 nm | 177 (2014) | |
| InGaN nanowall | 75 | 0.5 M HBr (pH 3) | 3.4 at 0 VAg/AgCl | IPCE = 16% at 350 nm | 176 (2014) | |
| n-InGaN planar photoanode | 2000 | 1 M HCl, 1 M NaCl | Max photo-conversion efficiency 0.23% | 195 (2013) | ||
| Ta3N5 nanorod | Co3O4/CO(OH)2 | 100, AM1.5G | 1 M NaOH | 3.64 at 1.23 VRHE | IPCE = 39.5% at 400 nm and 1.23 VRHE | 192 (2013) |
| InGaN planar photoanode | 500 W, Xe lamp | 1 M HBr, 0.5 M H2SO4, 1 M HCl | 2 at 1 VRHE | IPCE = 58% at 1.0 VRHE 400–430 nm (H2SO4) | 175 (2013) | |
| InGaN nanowire | Pt | 40 | 0.5 M H2SO4 | 5, −0.5 VNHE | IPCE = 40% at −0.45 VNHE 400–430 nm (H2SO4) | 182 (2013) |
| InGaN/GaN nanorod | NiO | 100, AM1.5 | 1 M NaOH | 0.3, 1 VCE | 183 (2013) | |
| InGaN/GaN core/shell nanowire | 300 W Xe, AM1.5G | 1 M HBr | 23 at 1.0 VAg/AgCl | IPCE = 27.6% at 350 nm and 1.0 VAg/AgCl | 70 (2013) | |
| GaN nanorod | 100 | 0.5 M HCl | 5.5 at VCE = 1.0 V | STH = 0.26% at 0.8 VCE | 167 (2013) | |
| GaN nanowire | 13.2 at 350 nm | 1 M HBr, 1 M KBr | 14 (HBr) at 0.0 VAg/AgCl | IPCE = 18% at 350 nm and 0.3 VAg/AgCl | 168 (2013) | |
| Ta3N5 films | Co(OH)x | 100, AM1.5G | 1 M NaOH | 5.5 at 1.23 VRHE | IPCE = 50% at 400–470 nm and 1.2 VRHE | 190 (2013) |
| Ta3N5 nanorod arrays | Co-Pi | 100, AM1.5G | 0.5 M Na2SO4 (pH 13) | 3.8 at 1.23 VRHE | IPCE = 41.3% at 440 nm | 193 (2013) |
| Ta3N5 films | Co3O4 | 100, AM1.5G | 1 M NaOH | 3.1 at 1.2 VRHE | IPCE = 36–40% at 400–500 nm and 1.2 VRHE | 194 (2012) |
| Si/InGaN core/shell nanowire | 350 without AM1.5 | pH 3 H2SO4 with 0.5 M of Na2SO4 | 62.6 μA cm−2 at VRHE = 1.23 | 178 (2012) | ||
| Ta3N5 nanotube | IrO2, Co3O4, Pt, Co-Pi | 110 | 0.1 M Na2SO4 | −1.2 (IrO2), −1.0 (Co3O4), −0.8 (Co-Pi), −0.25 (Pt), | IPCE = 10% 0.6 VAg/AgCl for Ta3N5/IrO2 | 188 (2012) |
| InGaN film | 500 W Xe lamp | 1 M HBr | 0.5 at 0.8 V vs. Ag/AgCl | IPCE = 42% at 400 nm 0.8 V vs. Ag/AgCl | 114 (2011) | |
| Ta3N5 porous | IrO2 | 0.1 M Na2SO4 (pH 6) | 3.8 at 1.15 VRHE | IPCE = 31% at 500 nm and 1.15 VRHE | 189 (2011) | |
| p-InGaN film | 132 | 1 M HBr | 1.2, at VCE = 1.2 V | 173 (2010) | ||
| Ta3N5 nanotube | 1 M KOH | IPCE 5.3% 0.5 VCE at 450 nm | 187 (2010) | |||
| W2N nanowire | 100, AM1.5 | 0.5 M, H2SO4 | 1.5 at VSCE = 1.2 | ABPE = 0.4% at 0.84 VSCE | 186 (2009) | |
| InGaN film | 500 W Xe lamp | 1 M HCl | 25 at VCE = 1.0 V | 172 (2008) | ||
| InGaN film | 500 W Xe lamp | 1 M HBr | 1.0 at VSCE = 0.8 | IPCE = 9% at 400–430 nm at VSCE = 0.8 | 174 (2008) | |
| GaN patterned | 4200 | 1 M NaOH | 17.34 at 0.50 VCE | ABPE = 0.3% at 0.50 VCE | 161 (2007) | |
| GaN film | 110 | 1 M HCl | 0.48 at 0 VCE | STH = 0.61% at 0.0 VCE | 159 (2007) |
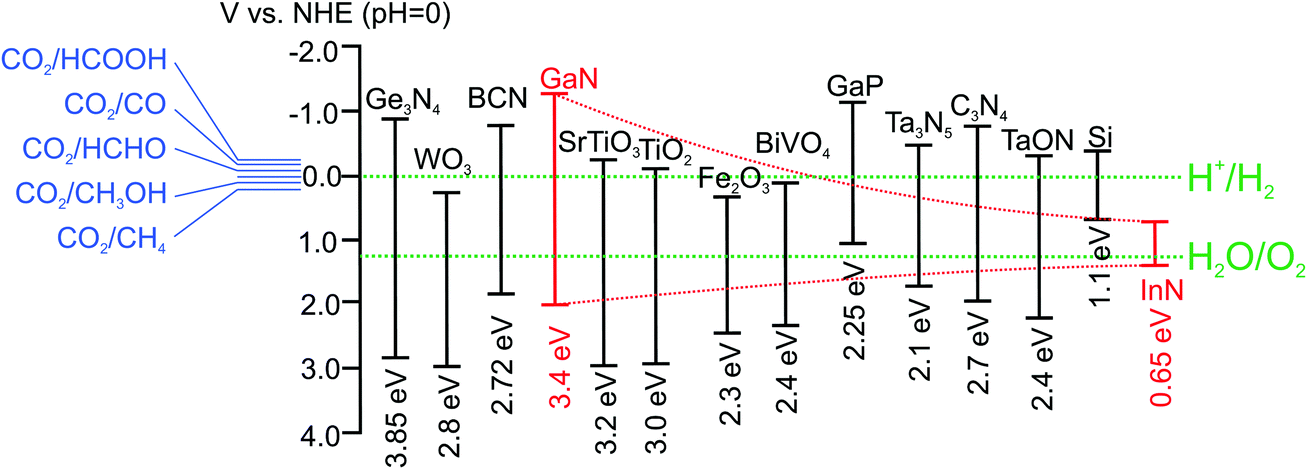 | ||
| Fig. 2 Band edge positions of commonly reported nitride photocatalysts. The oxidation and reduction potentials of water are also shown (green dotted lines). The red dotted line represents the band edge positions of InxGa1−xN with x increasing from left to right (0–1). The reduction potentials of CO2 to various value added products are also shown.10,11,25,63,85 | ||
In an effort to improve the performance of blue LEDs, the epitaxial growth technique of high crystalline quality III-nitrides has been substantially improved in the early 90's.61 Subsequently, in 1995, John Turner and co-workers demonstrated that high quality metal-nitride (n-GaN) functioned as a viable photoelectrode material for solar water splitting.74 Detailed photoelectrochemical characterization reveals that the bandgap of GaN straddles the redox potential of water with sufficient overpotentials, such that photolysis of water is possible on GaN without external bias.75–79 This work triggered the development of various nitride-based photocatalysts and photoelectrode materials (i.e., InGaN, T3N5, Ge3N4, and W2N) over the years. In 2009, a cheap, stable and earth abundant nonmetal nitride, i.e., polymeric carbon-nitride, has been developed by Antonietti's group, which can produce hydrogen from water under visible light irradiation.80 This seminal work has opened a new avenue for further research on carbon-nitrides to function as a viable catalyst for solar water splitting and CO2 reduction to energy rich hydrocarbons under visible light irradiation. In recent years, metal/nonmetal-nitrides in the form of nanostructures have been investigated by a number of research groups because of their excellent structural, optical and catalytic properties over their bulk counterparts.
Given the rapid development of nitride based photocatalysts and photoelectrodes for artificial photosynthesis, there is clearly an urgent need to provide a comprehensive overview of the recent progress and research activities in this area. Unlike other review articles,10,25,31,39,73,81–83 this article focuses only on metal/nonmetal-nitride based photocatalysts and photoelectrodes for solar powered artificial photosynthesis, including photocatalytic and photoelectrochemical water splitting and CO2 reduction. The rest of the article is organized as follows. First, the mechanism of solar water splitting is briefly discussed. Photocatalytic water splitting using UV and visible light sensitive metal/nonmetal-nitrides is then summarized. Subsequently, a discussion on efficiency enhancement in photocatalytic water splitting is provided. This is followed by a summary of photoelectrochemical water splitting using UV and visible light sensitive metal/nonmetal-nitrides. Photocatalytic and photoelectrochemical CO2 reduction using metal/nonmetal-nitrides are then discussed. Finally, the future prospects and challenges of nitrides for artificial photosynthesis are presented.
Solar water splitting: mechanism
The fundamental processes involved in the solar water splitting reaction are shown in Fig. 3. The initial step involves the absorption of incident photons and the generation of electron–hole pairs. Depending on the carrier lifetime, diffusion length, crystalline quality and photocatalyst dimension, photogenerated carriers can either recombine radiatively/nonradiatively or they can diffuse toward the semiconductor–liquid interface to drive the redox reaction. Any defects can act as trapping and recombination centers between photogenerated electrons and holes, resulting in a decrease in the photocatalytic activity. The physical size of the photocatalyst also determines the activity of the photocatalyst. If the size is small, the photogenerated carriers will travel less distance to reach the surface and hence there will be less probability of carrier recombination.73 The use of nanostructures can thus significantly improve the performance of photocatalysts having short carrier lifetime and low mobility.73,84,85 Furthermore, the near-surface band structure also plays a critical role in charge carrier extraction, as discussed in the later part of this article. The final step involves the reduction and oxidation (redox) of water on the photocatalyst surface via the photogenerated electrons and holes, respectively. The overall water splitting (i.e., simultaneous oxidation and reduction of water) consists of two half-reactions, i.e., the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER).39 The OER is essentially the first step in the water splitting reaction. It oxidizes water to form O2, as described by eqn (2) below.| 2H2O ↔ O2 + 4e− + 4H+, Eanodic = 1.23 V − 0.059 (pH) V (NHE) | (2) |
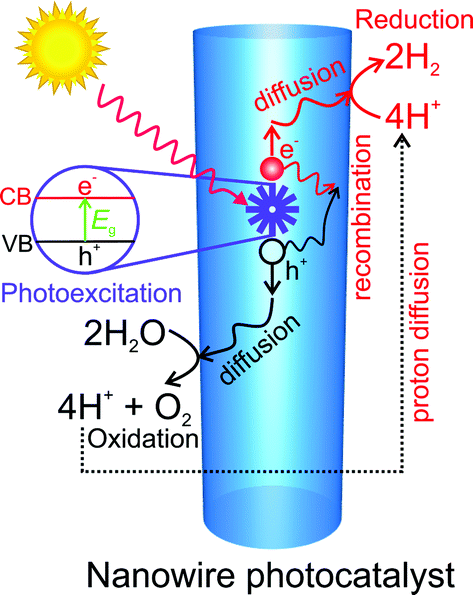 | ||
| Fig. 3 Schematic illustration of the main process steps in water splitting, including photoexcitation, carrier generation, diffusion, recombination, water oxidation, proton diffusion, and reduction reaction on the surface of nanowire photocatalysts. | ||
Since this reaction requires a high oxidizing potential, +1.23 V vs. NHE (pH = 0), the valence band maximum (VBM) has to be positioned at more positive potential than +1.23 vs. NHE (pH = 0). This reaction releases four protons (H+), which are reduced by the photogenerated electrons in the HER (eqn (3)).39
| 4H+ + 4e− ↔ 2H2, Ecathodic = 0 V − 0.059 (pH) V (NHE) | (3) |
Therefore, the conduction band minimum (CBM) of the semiconductor has to be positioned at more negative potential than 0 V vs. NHE at pH = 0. The theoretical minimum band gap for water splitting is 1.23 eV, which corresponds to a light wavelength of ∼1000 nm. In practice, however, the overall bandgap requirement rises to 1.5–2.5 eV to provide sufficient kinetic overpotentials to overcome entropic losses, OER and HER overpotentials, and other parasitic losses.39 Kinetically and energetically the OER is much more complex and slower than the HER as it requires multiple intermediate steps involving four photons.86,87 As a result, water oxidation, i.e., the primary reaction required for H2 production, is often the bottleneck that presents a significant difficulty in the development of an efficient catalyst. Kinetically, the water oxidation process competes with fast e–h bulk recombination, fast e–h surface recombination, surface O2 adsorption, and self-oxidation of the photocatalyst.86
In addition to the bandgap and band edge requirements, long-term stability of the photocatalyst in aqueous solution (in the dark and under illumination) as well as the cost and material availability are the key requirements. Furthermore, HER and OER co-catalysts often need to be incorporated on the photocatalyst surface to reduce the overpotentials required for enhanced photocatalytic activity.88 In this regard, a number of nitride based stable and efficient co-catalysts or electrocatalysts have been developed very recently, such as NiMoNx,89 Ni3N,90 Co0.6Mo1.4N2,91 and W2N.92
Photocatalytic water splitting using metal/nonmetal-nitrides
A number of UV and visible light responsive metal/nonmetal-nitrides have been developed in the last decade for photocatalytic water splitting, including UV light sensitive germanium nitride (β-Ge3N4) and GaN, and visible light sensitive InGaN, carbon nitride (g-C3N4) and Ta3N5. The following sections provide an overview of the recent progress in UV and visible light sensitive metal/nonmetal-nitride photocatalysts for photocatalytic water splitting.UV-light responsive metal/nonmetal-nitride photocatalysts
Sato et al. reported the first example of a nitride photocatalyst i.e., β-Ge3N4 for overall water splitting in 2005.93 It was demonstrated that the RuO2 nanoparticle dispersed β-Ge3N4 photocatalyst successfully decomposed water into H2 and O2 under UV light. Density functional theory (DFT) calculations suggested that the valence band of β-Ge3N4 consisted of N2p orbitals, and the photoexcited holes in such orbitals were able to oxidize water to form O2 without requiring an OER co-catalyst. The RuO2 nanoparticles served as an active HER co-catalyst to accelerate the overall water splitting. The stable photocatalytic activity of β-Ge3N4 in acidic solution (1 M H2SO4) was further confirmed by Domen's group.94 Subsequently, by reducing the density of defects via high-pressure ammonia treatment, a 4-fold enhancement in the photocatalytic activity of β-Ge3N4 was observed.95 The observation of overall water splitting using a nitride based β-Ge3N4 photocatalyst triggered further investigations on other nitride photocatalysts. Between 2005 and 2007, a number of reports demonstrated the thermodynamic and kinetic potentials of GaN for overall water splitting,76–79,96–99 which supports John Turner's observation74 in 1995. Stable and stoichiometric decomposition of H2O into H2 and O2 was demonstrated on a GaN particulate sample decorated with either RuO2 or Rh2−xCrxO3 HER co-catalysts.96,99 Interestingly, no OER co-catalyst was required for stoichiometric decomposition of water, suggesting that the surface of nitrides is inherently an active catalyst for the OER reaction. Further studies on GaN showed that divalent metal ion (Mg2+, Zn2+, and Be2+) doping significantly improved the stability and activity of GaN for overall water splitting.98On the other hand, the development of heterogeneous nanostructured photocatalysts has attracted tremendous attention in the last two decades owing to their immense potential for solar-fuel production, such as efficient light absorption, large reaction surface area, efficient charge carrier extraction, higher solubility, reduced recombination, and tunable electronic band structure.73,84,85,100 Because of these unique opportunities, a number of metal-nitride nanostructured photocatalysts have been developed recently. Plasma-assisted molecular beam epitaxially (MBE) grown GaN nanowires on a Si substrate have been utilized for photocatalytic water splitting, for the first time, by the authors' group.101 The Rh/Cr2O3 core/shell nanoparticle decorated GaN nanowires successfully dissociated neutral pH water into H2 and O2 in stoichiometric ratio under full arc illumination. Compared to GaN particulate and thin film samples, significantly enhanced photocatalytic activity of GaN nanowire was observed. This enhanced activity was attributed to the large surface-to-volume ratio of one-dimensional nanowires and significantly reduced defect densities. Additionally, it was revealed that the well-defined nonpolar (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surfaces of GaN nanowires are catalytically stable and active compared to its polar counterpart.102 Muckerman's group studied the nonpolar (100) surface of GaN using ab initio molecular dynamics simulation (AIMD), and found that the nonpolar (100) GaN surfaces were very reactive for spontaneous dissociation (H2O → H+ + OH−) of the majority (∼83%) of the water molecules.102,103 In contrast, many experimental and theoretical studies suggest that water molecules do not dissociate on the photoactive TiO2 anatase (101) and rutile (110) surfaces.104–106 Moreover, the AIMD study revealed that the photogenerated holes on nonpolar (100) GaN surfaces had sufficient standard free-energy to drive the four-step water oxidation reaction.102 In addition, the low effective free-energy barrier for proton diffusion on the GaN (100) surface facilitates enhanced migration of protons from the O2 evolution reaction sites to H2 evolution sites; thus improving the efficiency.107
0) surfaces of GaN nanowires are catalytically stable and active compared to its polar counterpart.102 Muckerman's group studied the nonpolar (100) surface of GaN using ab initio molecular dynamics simulation (AIMD), and found that the nonpolar (100) GaN surfaces were very reactive for spontaneous dissociation (H2O → H+ + OH−) of the majority (∼83%) of the water molecules.102,103 In contrast, many experimental and theoretical studies suggest that water molecules do not dissociate on the photoactive TiO2 anatase (101) and rutile (110) surfaces.104–106 Moreover, the AIMD study revealed that the photogenerated holes on nonpolar (100) GaN surfaces had sufficient standard free-energy to drive the four-step water oxidation reaction.102 In addition, the low effective free-energy barrier for proton diffusion on the GaN (100) surface facilitates enhanced migration of protons from the O2 evolution reaction sites to H2 evolution sites; thus improving the efficiency.107
The photocatalytic activity of GaN nanowires grown by Ni catalyst-assisted metal–organic chemical vapor deposition (MOCVD) has also been reported.71 Such GaN nanowires showed better activity in photodegrading dye solution compared to GaN submicron dots or thin films owing to the larger surface area and better crystallinity of the nanowires. Stable and enhanced photocatalytic activity of GaN nanowires was observed at acidic pH, with much better performance than TiO2 and ZnO nanowires. Because of the large bandgap of β-Ge3N4 (3.8 eV) and GaN (3.4 eV), only UV light can be harnessed, which consists of ∼4% of the solar spectrum. As a result, significant research efforts have been devoted to developing visible light sensitive and efficient metal/nonmetal-nitride photocatalysts.
Visible-light responsive metal/nonmetal-nitride photocatalysts
In an effort to extend the light absorption of UV light sensitive wurtzite GaN and ZnO, Domen's group developed a solid solution of GaN and ZnO (Ga1−xZnx)(N1−xOx) with an absorption edge at ∼510 nm.29 The Rh2−yCryO3 HER co-catalyst decorated solid solution (Ga1−xZnx)(N1−xOx) demonstrated successful dissociation of water under visible light (up to ∼510 nm) with an apparent quantum efficiency (AQE) of 2.5% at 420–440 nm. In a follow-up study, an AQE of 5.9% at 420–440 nm was demonstrated by post-calcination treatment of the as-synthesized (Ga1−xZnx)(N1−xOx) solid solution.108 Recently, by forming nanostructures of (Ga1−xZnx)(N1−xOx), an AQE of 17.3% at 400 nm was demonstrated by Li et al.109 The reduced bandgap of the solid solution is attributed to the p–d repulsion (i.e., N2p–Zn3d), which provides the top of the valence band formed by N2p atomic orbitals with higher potential energy.Kibria et al. demonstrated a viable defect-engineering approach to extend the absorption edge of GaN nanowires up to 450 nm using Mg doping.110 In order to reduce the bandgap, nitrogen vacancy related donor states and Mg impurity related acceptor states were simultaneously introduced into the bandgap of GaN nanowires during the epitaxial growth process, as illustrated in Fig. 4. Using such Mg-doped GaN nanowires, successful overall neural water splitting was demonstrated under violet light with intra-gap excitation up to 450 nm. An energy conversion efficiency of ∼1.34% was demonstrated under violet light (375–450 nm).
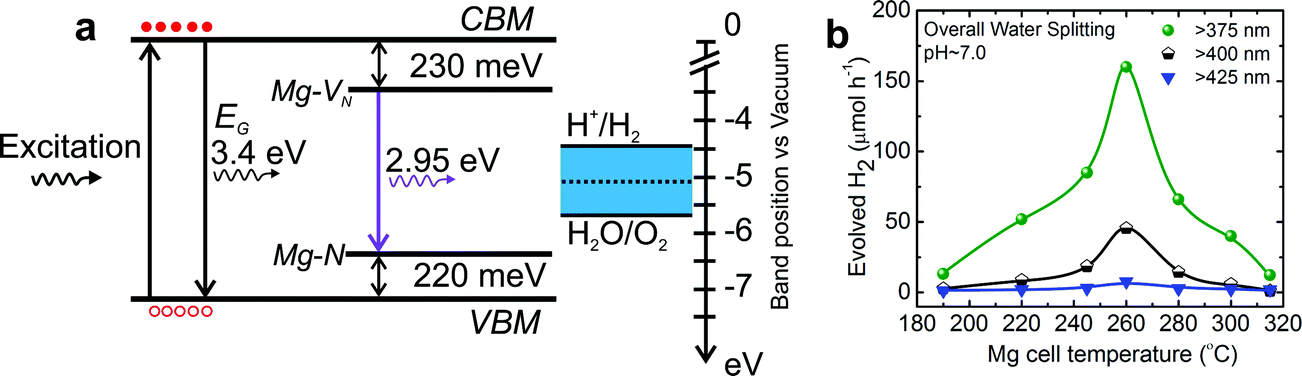 | ||
| Fig. 4 (a) Schematic energy band diagram of GaN:Mg nanowires illustrating the formation of nitrogen vacancy (VN) and Mg acceptor related intra-gap states, along with the redox potential of water (vs. vacuum level). (b) H2 evolution rate from overall neutral water splitting from different Mg doped GaN (GaN:Mg) samples with different intra-gap excitations using a 300 W xenon lamp and long-pass optical filters. Reprinted with permission from ref. 110. Copyright 2015, AIP Publishing LLC. | ||
As illustrated in Fig. 2, the bandgap of GaN can be tuned from 3.4 to 0.65 eV by introducing In, providing a viable approach to capture the visible and near-infrared solar spectrum. Recent DFT studies have shown that the conduction and valence band edge of InGaN can straddle the water redox potentials for In compositions up to ∼50%, which suggests that photocatalytic overall water splitting can be possibly realized under red and even near-infrared light irradiation.66 However, the growth of high crystalline quality InGaN with high In content has been extremely challenging for a number of reasons.111,112 For instance, the large lattice mismatch (11%) between InN and GaN results in a solid phase miscibility gap,113 and the high vapor pressure of In over Ga leads to low In incorporation in InGaN.112 Additionally, the difference in formation enthalpies between InN and GaN causes strong In surface segregation, which creates In rich clusters.114 These factors lead to a large number of non-radiative recombination centers and strong carrier localization, which limit the photocatalytic performance of InGaN. Furthermore, TEM studies on InGaN/GaN quantum wells reveal the presence of misfit dislocations in InGaN, when InGaN is grown beyond a critical thickness.115 This critical thickness decreases drastically with increasing In content. Therefore, the realization of high crystalline quality and high In content InGaN with sufficient thickness for efficient light absorption is quite challenging.116 For these reasons, there have been very few studies on the photocatalytic activities of InGaN. Among different growth techniques, MBE promises to grow In-rich InGaN with superior crystalline quality.112 Recently, the author's group has achieved nearly defect-free metal-nitride nanowires by PAMBE to function as a visible light active photocatalyst.117 By performing OER and HER half reactions in the presence of respective sacrificial reagents, Kibria et al. demonstrated the thermodynamic and kinetic potentials of InGaN nanowires for overall water splitting with In compositions up to 32%.118 The bandgap tunability of metal-nitrides from 6.2 eV (AlN) to 0.65 eV (InN) combined with the epitaxial growth of nearly defect-free nanowire structures allows for developing monolithically integrated multi-band nanowire photocatalysts to minimize the thermalization loss of energetic electrons.23 None of the previously reported photocatalysts can function as a single material platform to harness effectively the solar spectrum using a multi-band approach. In this context, Kibria et al. developed triple-band InGaN/GaN nanowires with bandgaps of 3.4, 2.96, and 2.22 eV, which led to overall neutral pH water splitting under UV, blue, and green light irradiation (up to 560 nm), as illustrated in Fig. 5.118 A maximum AQE of ∼1.86% was demonstrated for overall neutral water splitting at 400 nm. Further extension of the absorption edge to deep-visible and near-infrared requires the growth of high (>40%) In content InGaN. As an alternative approach, Kibria et al. developed dye-sensitized InGaN nanowires to extend the solar absorption in the deep-visible spectrum.119 It was demonstrated that Merocyanine-540 dye-sensitized and Rh nanoparticle incorporation on In0.25Ga0.75N nanowire arrays (absorption edge ∼500 nm) can produce hydrogen from ethylenediaminetetraacetic acid (EDTA) and acetonitrile mixture solution under green, yellow and orange light irradiation (up to 610 nm). An AQE of 0.3% was demonstrated in the wavelength range of 525–600 nm, providing a viable approach to harness deep-visible and near-infrared solar energy for efficient and stable water splitting.
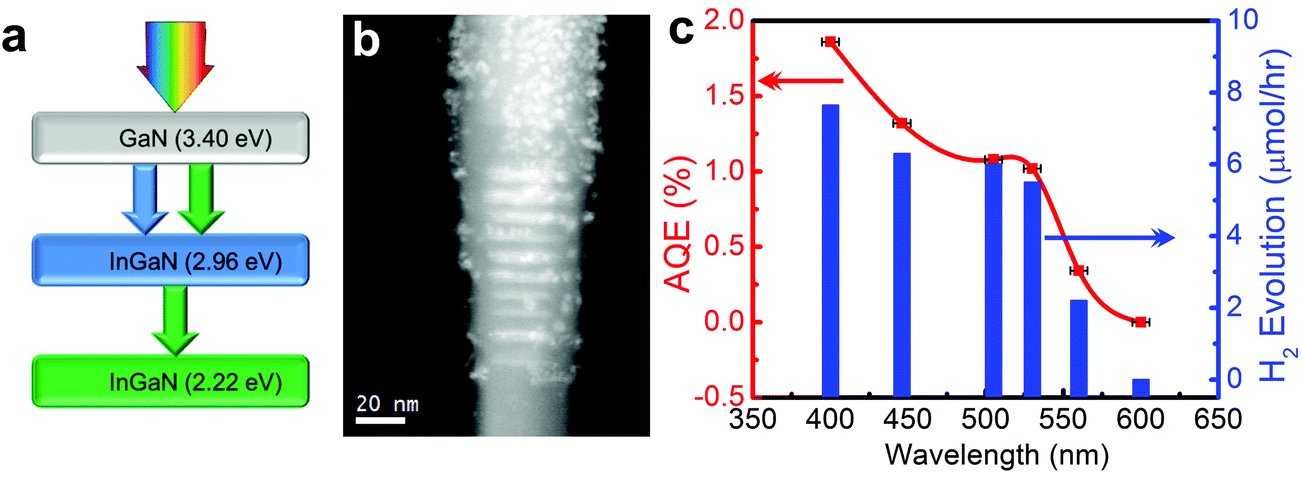 | ||
| Fig. 5 (a) Schematic of a triple-band InGaN/GaN nanowire heterostructure, illustrating the light absorption process. (b) Electron energy loss spectroscopy (EELS) spectrum image of the Rh/Cr2O3 core/shell nanoparticle decorated triple-band InGaN/GaN nanowire heterostructure. (c) Apparent quantum efficiency (AQE) and rate of H2 evolution vs. excitation wavelength. The experiment was performed in neutral pH water under 300 W xenon lamp irradiation with different band-pass filters without any other energy input. The horizontal error bars represent the full-width-half-maximum of the bandpass filters. The red solid line is a guide to the eye. Reprinted with permission from ref. 118. Copyright 2013, American Chemical Society. | ||
In the year of 2002, Domen's group reported a promising visible light (<590 nm) active transition metal-nitride semiconductor i.e., Ta3N5; the bandgap (2.1 eV) of which is well positioned to straddle the redox potential of water.120,121 Hydrogen or oxygen generation under visible light in the presence of a respective sacrificial reagent confirmed the thermodynamic and kinetic potentials of Ta3N5 for the HER and OER. While Ta3N5 has been utilized as an O2 evolution photocatalyst in a two-step Z-scheme photosystem,122 overall water splitting has not been reported in a Ta3N5 based single-step photosystem to the best of our knowledge. To date the reported quantum efficiency of the Ta3N5 photocatalyst is still very low, despite its near-perfect band edge position and visible light absorption.123 This is attributed to the fact that the commonly used thermal nitridation process of the oxide precursor (Ta2O5) leads to insufficient crystallization with the presence of extensive charge recombination centers, thereby limiting the quantum efficiency. In order to improve the performance of Ta3N5, Domen's group modified the surface of the starting precursor (Ta2O5) with a small amount of alkali metal salt.124 Compared to conventional nitridation derived Ta3N5, Ta3N5 nitrided from alkali metal salt (Na2CO3) modified Ta3N5 exhibited higher crystallinity and smaller particles; demonstrating a 6-fold improvement in the photocatalytic activity for O2 evolution under visible light. By incorporating CoOx OER co-catalysts, an AQE of 5.2% at 500–600 nm was reported for the O2 half reaction. Very recently, Chen et al. demonstrated that a MgO nanolayer (2–5 nm) surface coating not only improves the interfacial contact between the hydrophilic CoOx co-catalyst and hydrophobic Ta3N5, but also decreases the defect density of Ta3N5 through a passivation effect.125 This interface engineering significantly improves interfacial charge transfer, leading to a relatively high AQE of 11.3% at 500–600 nm for the O2 half reaction.
More recently, the use of nonmetal-nitrides for solar-fuel generation has also been studied. Wang et al. demonstrated the first example of a visible light sensitive metal-free nitride as a new material platform to enable earth-abundant photocatalysts for water splitting.80 Graphitic carbon nitride (g-C3N4) was synthesized by thermal polycondensation of common organic monomers, as illustrated in Fig. 6. g-C3N4 is a soft polymeric nitride with a conjugation structure and possesses very high thermal and chemical stability, and excellent optoelectronic properties, including a direct energy bandgap of 2.7 eV, which straddles the redox potential of water, as shown in Fig. 2.126,127 The g-C3N4 photocatalyst showed stable photocatalytic activity to generate H2 from water under visible light (up to 540 nm) in the presence of electron donors without using any noble metal co-catalyst. This study has triggered intensive research efforts on g-C3N4 for solar-fuel conversion.126,128 Nevertheless, the photocatalytic activity of as-synthesized g-C3N4 is substantially low, which is attributed to insufficient sunlight absorption, inefficient carrier separation, and fast recombination of charge carriers.126,129 Therefore, a number of strategies have been developed, including nanostructure design in the form of porous structures,130,131 nanospheres,132,133 helical nanostructures,134 1D nanostructures (nanorods, nanowires, nanobelts, and nanotubes),135 bandgap engineering through structural-distortion,136 non-metal doping (i.e. S, F, B, P),137,138 metal-doping (i.e. Pt, Pd, Fe, Zn, Cu),139,140 molecular doping/copolymerization (dicyandiamide-barbituric acid, dicyandiamide-2-aminobenzonitrile, dicyandiamide-diaminomaleonitrile, dicyandiamide-3-aminothiophene-2-carbonitrile, and urea-phenylurea),141,142 dye-sensitization,143 and construction of various semiconductor–semiconductor heterojunctions.126,129,144etc. In order to promote charge carrier separation and migration for enhanced photocatalytic activity, g-C3N4 has been hybridized with various nanocarbon composites (i.e. highly conductive graphene, multi-walled carbon nanotubes, or reduced graphene oxide),129,145,146 and with polymers to form all-polymeric nanocomposites. Additionally, incorporation of noble metal co-catalysts (i.e. Pt, Au, Ag) on the g-C3N4 photocatalyst is found to accelerate charge separation and reduce the overpotential required for the water redox reaction.126 As noble metals are rare and expensive, a number of non-noble-metal co-catalysts have also been developed, such as Ni(OH)2, NiS, NiS2, MoS2, CoSe2, etc.144,147–149 It should be noted that depending on the precursor used for the synthesis of g-C3N4, the photocatalytic performance is found to be different due to the structural variations of the as-synthesized material.126 Martin et al. reported a highly efficient g-C3N4 photocatalyst synthesized from a low cost precursor, urea, which exhibited an excellent hydrogen evolution rate of nearly 20 mmol h−1 g−1 in the hydrogen half reaction under full arc irradiation with a QE of 26.5% at 400 nm.150 The reported QE was claimed to be one order of magnitude higher than any existing g-C3N4 photocatalysts.141,151–153 The excellent activity of urea-derived g-C3N4 was attributed to the more negative conduction band edge position, and improved exciton distribution over its structure.
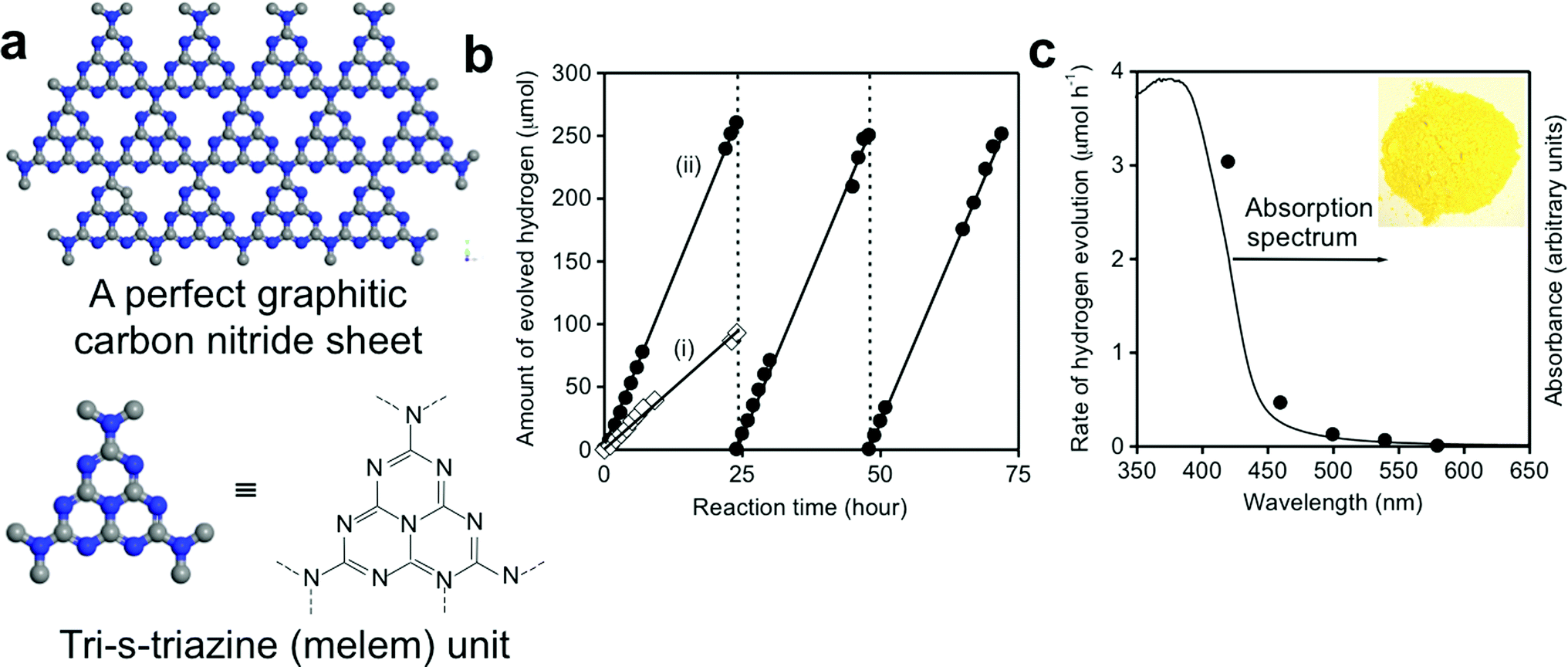 | ||
| Fig. 6 (a) Schematic illustration of a perfect g-C3N4 sheet constructed from melem units. (b) H2 production from water in the presence of a sacrificial electron donor (10 vol% triethanolamine) under visible light (>420 nm) by (i) unmodified g-C3N4 and (ii) 3.0 wt% Pt-deposited g-C3N4 photocatalyst. (c) H2 production rate from water in the presence of a sacrificial electron donor (10 vol% methanol) by a 0.5 wt% Pt-deposited g-C3N4 photocatalyst vs. wavelength of the incident light. UV-visible absorption spectrum of the g-C3N4 catalyst is also shown for comparison. Reprinted with permission from Macmillan Publishers Ltd: Nature Materials (ref. 80), copyright 2008. | ||
Very recently, Wang and co-workers reported another metal-free and visible light sensitive two-dimensional (2D) nitride photocatalyst. By carbon doping into hexagonal boron nitride (h-BN), ternary alloy boron carbon nitrides (BCN) with band gaps of 2.08, 2.56, and 2.72 eV were synthesized using the pyrolysis method.154 The band edge positions of BCN alloys were found to straddle the redox potential of water, and the bandgap can be tuned by controlling the amount of carbon doping. The as-synthesized BCN alloy was shown to drive the HER without any noble co-catalysts. However, Ni–Co layered double hydroxides (Ni–Co LDHs) were used as co-catalysts to promote O2 evolution. The demonstration of visible light sensitive 2D metal-free nitrides i.e., C3N4 and BCN will stimulate further research on other earth abundant and stable 2D material families for photocatalysis.
Enhanced efficiency by engineering the near-surface band structure
The near-surface band structure plays a critical role in enhancing the efficiency and stability of photocatalysts. In the case of nanostructured photocatalysts, the charge carrier extraction at the semiconductor–liquid interface is no longer diffusion limited, as the diffusion lengths of the photoexcited carriers are often larger than the physical dimension of the photocatalysts.73 It has been reported by Kibria et al. that one of the major obstacles for achieving high efficiency and stable overall water splitting over the emerging nanostructured photocatalysts is directly related to the near-surface band bending.155 The presence of upward (or downward) band bending is commonly measured for n (or p-type) semiconductors.156 Such near-surface band bending is required for efficient charge carrier separation in a PEC system, wherein oxidation and reduction reactions occur on different electrodes. However, in the case of photocatalytic water splitting, the presence of any surface band bending suppresses either electron (in the case of n-type semiconductors) or hole (in the case of p-type semiconductors) diffusion towards the semiconductor–liquid interface. Therefore, the overall water splitting reaction is hampered. In an effort to enhance the efficiency and stability of metal-nitride nanowire photocatalysts, Kibria et al. demonstrated that with a controlled amount of Mg dopant incorporation during the epitaxial growth process, the near-surface band bending can be precisely tuned.155Fig. 7a illustrates the estimated EF − EV from X-ray photoelectron spectroscopy valence band spectrum (shown in the inset). It is seen that, with increasing Mg dopant incorporation, EF − EV, i.e., the near-surface band bending approximately, can be tuned from 2.6 eV to 0.5 eV, and the near-surface region can be transformed from n-type to weakly p-type; providing a viable approach to control the charge properties and charge carrier transfer at the semiconductor–liquid interface. By tuning the band bending on the nonpolar (100) surfaces of GaN nanowires using p-type Mg doping, an internal quantum efficiency (IQE) or absorbed photon conversion efficiency (APCE) of ∼51% was achieved under UV light, which was nearly two orders of magnitude higher than undoped GaN, as shown in Fig. 7b.155 Stable and stoichiometric dissociation of neutral pH water was further demonstrated on p-type GaN nanowire arrays. In order to enhance the efficiency under visible light, p-type Mg doping was further optimized in InGaN nanowires, as shown in Fig. 8.34,157 Subsequently, a double-band p-GaN/p-InGaN nanowire heterostructure was developed, wherein the near-surface band bending was optimized for GaN and InGaN using p-type Mg doping. The APCE can reach ∼69% under visible light (up to 475 nm), which is the highest value ever reported under visible light. Using a double-band p-GaN/p-In0.2Ga0.8N nanowire heterostructure and Rh/Cr2O3 core/shell HER co-catalyst loading, an STH efficiency of ∼1.8% was demonstrated under concentrated sunlight (26 suns).34 Ultrafast exciton and charge-carrier dynamics studies on such p-GaN/p-In0.2Ga0.8N nanowires further revealed that the Rh/Cr2O3 core/shell HER co-catalysts significantly accelerated the carrier extraction at the nanowire–co-catalyst interface.158
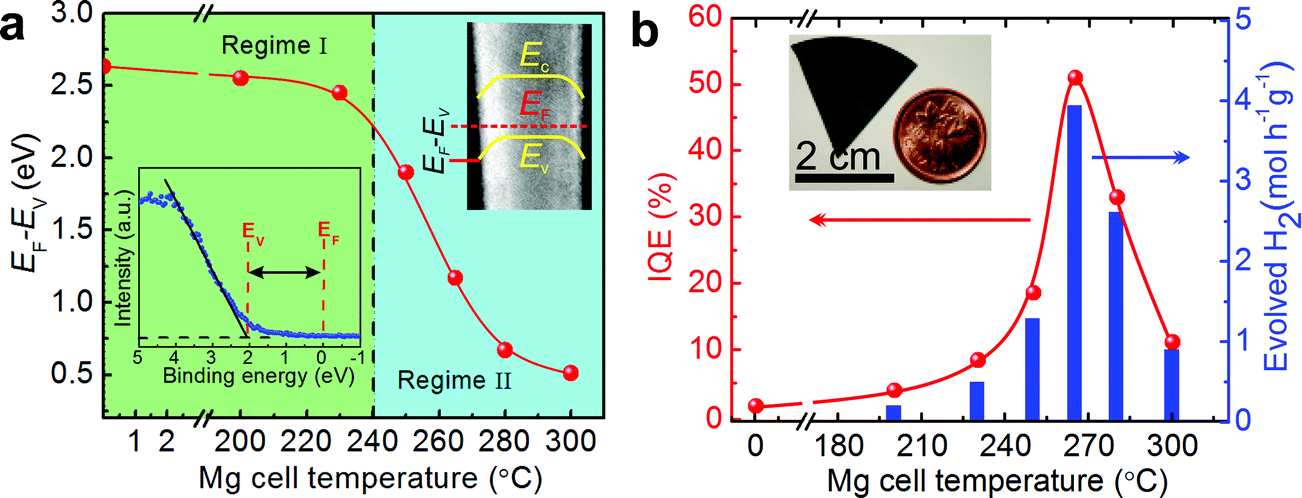 | ||
| Fig. 7 (a) Estimated EF − EV from angle resolved X-ray photoelectron spectroscopy valence spectrum (lower left inset) for different Mg doped GaN nanowire samples. The upper right inset shows the downward band bending and EF − EV on the TEM image of a single GaN nanowire. The dotted vertical line separates regime I (n-type surface) from regime II (p-type surface). (b) Internal quantum efficiency (IQE) and rate of H2 evolution from overall neutral water splitting by an ∼0.387 mg GaN:Mg nanowire catalyst with different Mg doping concentrations under 300 W xenon lamp irradiation. The Mg incorporation in GaN nanowires is directly proportional to the Mg cell temperature. Reproduced from ref. 155 with permission from Macmillan Publishers Ltd: Nature Communications, copyright 2014. | ||
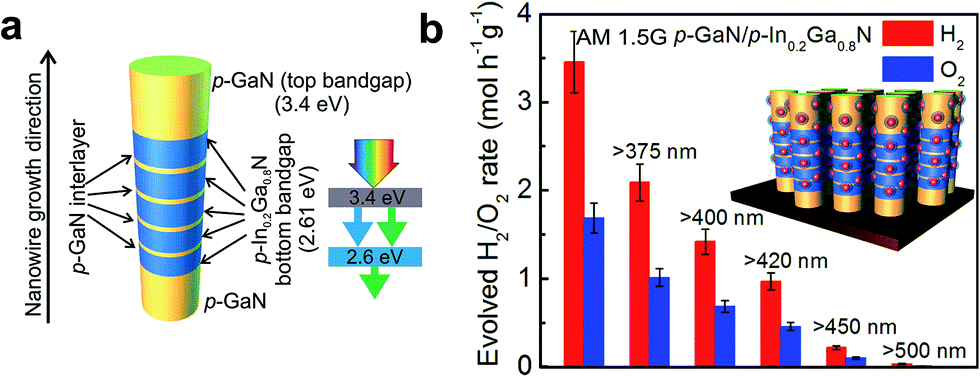 | ||
| Fig. 8 (a) Schematic of a double-band GaN/In0.2Ga0.8N nanowire photocatalyst. Five p-InGaN nanowire segments are incorporated along the growth axis of the p-GaN nanowire for visible light absorption. The inset shows the light absorption by the double-band structure. (b) Rate of H2 and O2 evolution from the Rh/Cr2O3 core/shell nanoparticle decorated double band GaN/InGaN nanowires. The reaction was performed using an ∼0.48 mg catalyst in neutral pH water under 300 W xenon lamp irradiation with an AM1.5 filter and various long-pass filters without any other energy input. The inset shows core/shell Rh/Cr2O3 nanoparticle decorated GaN/InGaN nanowires. Reproduced from ref. 34 with permission from Macmillan Publishers Ltd: Nature Communications, copyright 2015. | ||
Photoelectrochemical water splitting using metal/nonmetal-nitride photoelectrodes
Recently, photoelectrochemical water splitting using metal/nonmetal-nitrides has also gained considerable attention. Planer and nanostructured GaN has been employed as UV light active photoelectrodes. For visible light activity, InGaN and Ta3N5 have been commonly studied.UV light responsive metal/nonmetal-nitride photoelectrodes
Fujii et al. observed H2 gas generation using an n-type GaN photoelectrode at 1.0 VCE in 2005.76 While n-GaN was found to be stable in HCl, photocorrosion was observed in KOH electrolyte.78 Fujii et al. also reported that the band-edge potentials of p-GaN were identical to that of n-GaN.77 The incorporation of a suitable dopant into the GaN photoelectrode is one of the strategies to enhance the device efficiency. M. Ono et al. reported the carrier concentration dependent PEC properties of the n-GaN photoanode.159 The maximum photocurrent was measured with a carrier concentration of 1.7 × 1017 cm−3. Fujii et al. reported that the presence of alcohol in the NaOH electrolyte increased the H2 evolution rate nearly twice compared to that without alcohol.160 The higher H2 production activity in the presence of alcohol was attributed to easier oxidation of alcohol compared to that of water. The existence of alcohol further suppressed the photocorrosion of GaN caused by self-oxidation. By patterning a planar n-type GaN surface with metal stripes and n-GaN ridges, Waki et al. achieved higher photocurrent density in 1 M NaOH.161 This higher photocurrent was attributed to the eradication of the current crowding effect and the enhancement in effective surface area for photoelectrolysis. The current crowding effect can also be minimized by placing immersed finger-type indium tin oxide (IF-ITO) ohmic contacts on n- and p-GaN photoelectrodes to achieve an enhanced photocurrent and H2 generation rate.162,163 Kikawa et al. measured the flat-band potentials of Ga-face and N-face n-GaN, and found that the conduction band edge of the N-face was ∼0.3 eV more negative than that of Ga-face GaN.164 As a consequence, more hydrogen bubbles and cathodic current were observed on the N-face than that of Ga-face GaN. In contrast, from Mott–Schottky analysis, Lin et al. concluded that the conduction band edge of Ga-polar GaN was ∼0.4 eV more negative than that of N-polar GaN.165 While the Ga-polar surface was found to have higher photoconversion efficiency at negative bias (vs. Pt counter electrode), the N-polar surface exhibited higher photoconversion efficiency at positive bias (vs. Pt counter electrode). By utilizing the spontaneous and piezoelectric polarization in wurtzite III-nitrides, Nakamura et al. demonstrated a polarization-engineered GaN/AlN/GaN photocathode without p-type doping.166 This novel photocathode showed enhanced cathodic photocurrent compared to p-GaN in H2SO4 electrolyte.Nanostructuring the photoelectrode can lead to enhanced light absorption, suppressed carrier recombination, and efficient carrier extraction. Up to 6 times enhancement in photocurrent density was demonstrated with GaN nanorod arrays compared to the GaN planar photoelectrode.167 The H2 evolution rate increased from 0.1 to 0.73 ml h−1 cm−2 and STH conversion efficiency increased from 0.04% to 0.26% using the nanorod arrays. The PEC properties of PAMBE grown undoped and Si-doped GaN nanowire arrays were studied in HBr and KBr electrolyte by AlOtaibi et al.168 Maximum IPCE values of ∼15% and ∼18% were measured for undoped and Si-doped GaN nanowires at −0.1 and 0.3 V vs. Ag/AgCl under 350 nm excitation, respectively. It was further demonstrated that the electrochemical properties of GaN nanowires can be tuned with controlled doping and external bias via the electrolyte.169
Visible light responsive metal/nonmetal-nitride photoelectrodes
A number of strategies have been developed to enhance the light absorption of nitrides in the visible spectral range. Liu et al. demonstrated the visible light (400–600 nm) activity of the Mn-doped GaN photoelectrode with an IQE of 61% at 450 nm.170 This visible light response was attributed to the Mn-related intermediate band formed in the bandgap of GaN. However, photocorrosion of the electrode was inevitable owing to the presence of Mn-related structural defects in GaN.Fujii et al. studied the PEC properties of InGaN for H2 generation in 2005.171 At 1 VCE, the In0.02Ga0.98N photoelectrode showed higher photoactivities compared to GaN photoelectrodes in 1 M HCl. Later on, Li et al. studied the PEC properties of a 200 nm thick n-InGaN epilayer grown by MOCVD.172 A drastic enhancement in photocurrent density and hydrogen evolution rate was demonstrated by increasing the In content from 20 to 40%. Aryal et al. reported the excellent stability of p-InxGa1−xN (0 ≤ x ≤ 0.22) epilayers in aqueous HBr solution.173 In another study, Luo et al. demonstrated the good photostability and visible light response of a MOCVD grown 60 nm thick In0.2Ga0.8N electrode in aqueous HBr solution.174 The turnover number reached 847 after 4000 s irradiation, and the incident photon conversion efficiency (IPCE) was nearly 9% under 400–430 nm at 0.8 V vs. SCE. In a subsequent report, in order to enhance the IPCE, the authors grew 250 nm thick In0.2Ga0.8N. However, the In-rich InGaN phases caused by In segregation on the surface reduced the photocurrent owing to the presence of surface recombination centers.114 By removing these In-rich InGaN phases using 1 M HCl aqueous solution, the IPEC was found to increase from 15% to 42% at 400 nm at 0.8 V vs. Ag/AgCl with enhanced stability of the photocurrent. The authors further demonstrated an IPCE of 53% and 58% under 400–430 nm at 1 V vs. RHE after surface treatment of In0.3Ga0.7N in HBr and H2SO4 aqueous solution, respectively.175
A number of metal-nitride nanostructured photoelectrodes have been developed for efficient water splitting. Nearly 57% enhancement in H2 evolution rate was revealed in the case of PAMBE grown In rich (40–50%) InGaN nanowall structures compared to that of planar InGaN.176 Benton et al. fabricated nanoporous structures of GaN and InGaN/GaN by photoelectrochemical etching in KOH solution.177 An IPCE of 32% and 46% at 355 nm was demonstrated for GaN and InGaN/GaN nanoporous structures, respectively, which were nearly 4-fold higher than as-grown planer devices. In order to enhance the effective surface area, Si/InGaN core/shell hierarchical nanowires were synthesized using photolithography and CVD to function as a photoanode for water splitting.178 Nearly 5 times enhancement in photocurrent density was demonstrated for hierarchical Si/InxGa1−xN (x = 0.08–0.1) nanowires compared to that of InGaN nanowires on a planar Si substrate reported by the same group. However, the photocurrent recorded from Si/InxGa1−xN (x = 0.08–0.1) nanowire electrodes was very small (∼10 μA cm−2 at 1 VRHE under 1 sun), which was attributed to fast carrier recombination and inefficient charge transfer at the semiconductor/electrolyte interface. On the other hand, AlOtaibi et al. reported an n-type In0.3Ga0.7N/GaN core/shell double-band nanowire photoanode grown by PAMBE on a Si (111) substrate, as illustrated in Fig. 9.70 The core/shell double-band nanostructures provided efficient light absorption and a stable photoelectrochemical reaction in HBr electrolyte. Stable PEC water splitting and H2 generation under UV and visible light (up to 600 nm) were demonstrated with an IPCE of 27.6% under 350 nm excitation at 1 V vs. Ag/AgCl. Caccamo et al. compared the photocurrent density of single crystalline n-type GaN/In0.3Ga0.7N (core/shell) nanorods with that of GaN nanorods synthesized by selective area growth metal organic vapour phase epitaxy (MOVPE).179 While the photocurrent was the same for both electrodes for applied potentials up to 1 V vs. RHE, nearly 10-fold higher photocurrent density was observed in the case of GaN/In0.3Ga0.7N (core/shell) nanorods at 1.35 V vs. RHE. Ebaid et al. recently synthesized coaxial InGaN/GaN multiple quantum well (MQW) nanowire heterostructure photoanodes using MOCVD.180 With careful optimization of the In content and number of QWs, such a MQW nanowire heterostructure enabled stable water splitting in 1 M HCl with a maximum IPCE of 8.6% at 350 nm at 1 Vcathode and an applied bias photon-to-current efficiency (ABPE) of 0.21% at 0.4 Vcathode. The same group further studied the carrier dynamics on InGaN/GaN MQW coaxial nanowires to improve the efficiency.181 Defect-induced recombination and strong localization of excitons were revealed in samples with thin QWs (up to 3 nm). In contrast, strong band-to-band transitions and negligible localization were observed in samples with thick QWs (∼6 nm). By carefully engineering the InGaN QW thickness to reduce the carrier localization and defect density in coaxial nanowires, an IPCE of 15% at 350 nm at 1 Vcathode was achieved.
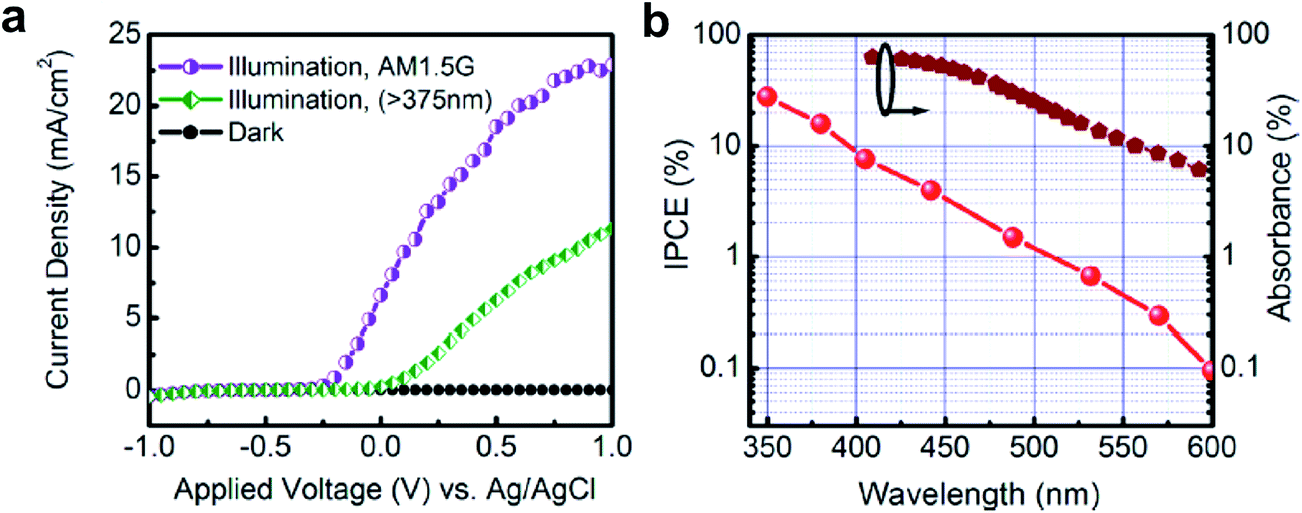 | ||
| Fig. 9 (a) Current density vs. applied voltage (vs. Ag/AgCl) in 1 M HBr under 300 W xenon lamp irradiation. The visible light response from InGaN nanowires is confirmed by adopting a long-pass (>375 nm) filter. The measured current density is ∼23 mA cm−2 using a 300 W Xe lamp with AM1.5G filter at a bias of 1 V (vs. Ag/AgCl). (b) Incident-photon-to-current conversion efficiency (IPCE) of InGaN/GaN nanowire photoelectrodes measured at 1 V (vs. Ag/AgCl) in 1 M HBr in a semi log scale. The calculated spectral absorbance by the InGaN segments is also shown for comparison. Reprinted with permission from ref. 70. Copyright 2013, American Chemical Society. | ||
The PEC properties of n- and p-type InGaN nanowires grown by PAMBE for water splitting were studied by in situ electrochemical mass spectroscopy (EMS) in 0.5 M H2SO4.182 An IPCE of 40% at a potential of −0.5 V vs. NHE was measured in the visible spectrum. Stable photocurrent and H2 evolution were observed for 60 min. The PEC properties of the InGaN/GaN nanorod LED structure grown by MOCVD was studied by Benton et al.183 The photochemical etching of the nanowires owing to self-oxidation by the photogenerated holes in aqueous NaOH solution was significantly suppressed by incorporating NiO nanoparticles onto the nanowires. The NiO nanoparticles help to suppress carrier recombination and promote the oxidation reaction on its surfaces rather than on the nanowire surface. Rodriguez et al. recently demonstrated that PAMBE grown InN quantum dot decoration doubled the PEC efficiency of In0.54Ga0.46N photoelectrodes, with a maximum IPCE of up to 56% at 600 nm at 0 V vs. Ag/AgCl and stable photocurrent for over 10 h.184 Rajaambal et al. reported InGaN QDs on ZnO for efficient visible light absorption with high photostability for solar light harvesting.185
Apart from group-III metal-nitrides, a few other metal-nitride photoelectrode materials have also been reported. Chakrapani et al. studied the PEC properties of tungsten nitride (W2N) nanowire arrays.186 While W2N showed n-type behavior with good photoactivity at moderate bias, prolonged photolysis resulted in photocorrosion of the nanowires due to the presence of defects. However, mixed phase W2N–WO3 showed improved photo-stability. Feng et al. synthesized highly oriented Ta3N5 nanotube arrays for visible light responsive photoelectrolysis.187 In a two-electrode arrangement, an IPCE of 5.3% was achieved at 450 nm with 0.5 VCE bias in KOH solution. Cong et al. studied the PEC water oxidation properties of Ta3N5 nanotubes decorated with IrO2, Co3O4, Co-Pi, and Pt nanoparticles under visible light.188 The Ta3N5 nanotube showed three times higher photocurrent than the regular Ta3N5 film. The PEC water oxidation on Ta3N5 nanotubes was improved by IrO2, Co3O4, and Co-Pi nanoparticles. A maximum IPCE of ∼10% at 400 nm was demonstrated for IrO2 decorated Ta3N5 nanotube arrays at 0.6 VAg/AgCl. Higashi et al. demonstrated an efficient Ta3N5 photoanode for overall water splitting into H2 and O2 under visible light. By performing a necking treatment (TaCl5 treatment + NH3 treatment) and loading IrO2·nH2O OER co-catalyst nanoparticles on a Ta3N5 photoanode, an IPCE of 31% at 500 nm with 1.15 V vs. RHE in aqueous Na2SO4 solution was demonstrated.189 Li et al. reported that by thermal or mechanical exfoliation of surface recombination centers and loading Co(OH)x OER co-catalysts, the IPCE of Ta3N5 photoanodes can be significantly improved.190 A highest photocurrent (among all currently available Ta3N5 photoanodes to our knowledge) of 5.5 mA cm−2, which corresponds to an IPCE of 50% under 400–470 nm at 1.23 V vs. RHE in aqueous NaOH solution, was demonstrated.189–194 However, the photocurrent was reduced to 55% in 2 h of illumination, which is attributed to oxidation of Ta3N5 by the photogenerated holes. To address the poor photostability of Ta3N5 for water oxidation, Liu et al. reported a ferrihydrite (Fh) passivation layer that allows stable water oxidation for over 6 h with a benchmark photocurrent over 5.2 mA cm−2 at 1.23 V vs. RHE.191 The remarkably enhanced photostability of Fh/Ta3N5 is attributed to the hole storage capability of the Fh layer.
In an effort to reduce the required external bias for PEC water splitting, photovoltaic devices have been integrated with nitride photoelectrodes for efficient solar energy conversion. An n-InGaN working electrode has been developed which is biased by a GaAs solar cell.195 By optimizing the electrolyte and incident light intensity, and introducing immersed ITO ohmic contacts on the n-InGaN working electrode, the operating point of the device was tuned to match the maximum power point of the GaAs solar cell. A photoconversion efficiency of 0.18–0.23% was demonstrated under simulated sunlight with optimized conditions. In another study, Dahal et al. realized a monolithic solar-PEC device based on the InGaN/GaN MQW solar cell.196 An STH efficiency of 1.5% was reported at zero bias (VCE = 0 V). Excellent chemical stability was further demonstrated for a prolonged period of time (7 days) in aqueous HBr solution. Fan et al. recently synthesized a dual absorber photocathode, consisting of p-InGaN/tunnel junction/n-GaN nanowire arrays on a Si solar cell wafer using PAMBE, as illustrated in Fig. 10.197 Such monolithically integrated dual absorber nanowire photocathodes can operate efficiently without strict current matching between different absorbers, which is required for conventional planar tandem photoelectrodes. Unlike planar tandem photoelectrodes, wherein carrier extraction is possible only from the front surface, the one-dimensional nanowire architecture allows for lateral carrier extraction from different absorber layers for efficient redox reactions. Additionally, the insertion of the n++-GaN/InGaN/p++-GaN polarization-enhanced tunnel junction (shown in Fig. 10a) allows for efficient carrier transport along the axial direction of the nanowires. While the platinized n+–p Si solar cell wafer produced a photocurrent of −20 mA cm−2 at 0 VNHE, the platinized p-InGaN/tunnel junction/n-GaN nanowire on the n+–p Si solar cell wafer generated a photocurrent of −40 mA cm−2 at 0 VNHE. The Pt nanoparticle decorated monolithically integrated photocathode exhibited an ABPE of 8.7% at 0.33 VNHE and nearly unity faradic efficiency for H2 generation. The InGaN/GaN photocathode also exhibited stable photoactivity over 3 h. Very recently, AlOtaibi et al. demonstrated a III-nitride nanowire based dual-photoelectrode device to enhance the efficiency of conventional 2-photon tandem devices under parallel illumination by splitting the solar spectrum spatially and spectrally,198 as illustrated in Fig. 11. The dual-photoelectrode, consisting of a GaN nanowire photoanode and an InGaN nanowire photocathode, exhibited an open circuit potential of 1.3 V and nearly 20-fold enhancement in the power conversion efficiency under parallel illumination (400–600 nm), compared to that of individual photoelectrodes. Furthermore, a dual-photoelectrode, consisting of parallel-connected metal-nitride nanowire photoanodes and a monolithically integrated single-junction-Si/InGaN nanowire photocathode, exhibited an ABPE of 2% at ∼0.6 V vs. the photocathode, as illustrated in Fig. 11c.
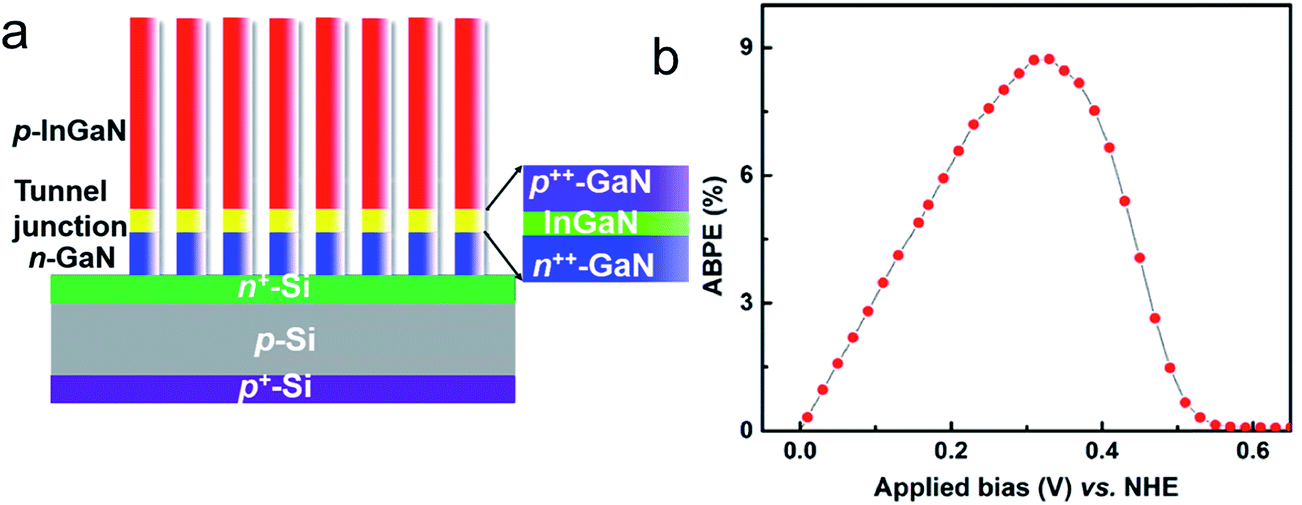 | ||
| Fig. 10 (a) Schematic of the photocathode grown on an n+–p Si solar cell substrate. The n-GaN and p-InGaN nanowire segments are connected by an n++-GaN/InGaN/p++-GaN polarization-enhanced tunnel junction, as shown in the inset. (b) Applied-bias-to-photon-conversion-efficiency (ABPE) vs. applied bias (vs. NHE) of the photocathode in 1 M HBr electrolyte under 1.3 sun illumination. Reprinted with permission from ref. 197. Copyright 2015, American Chemical Society. | ||
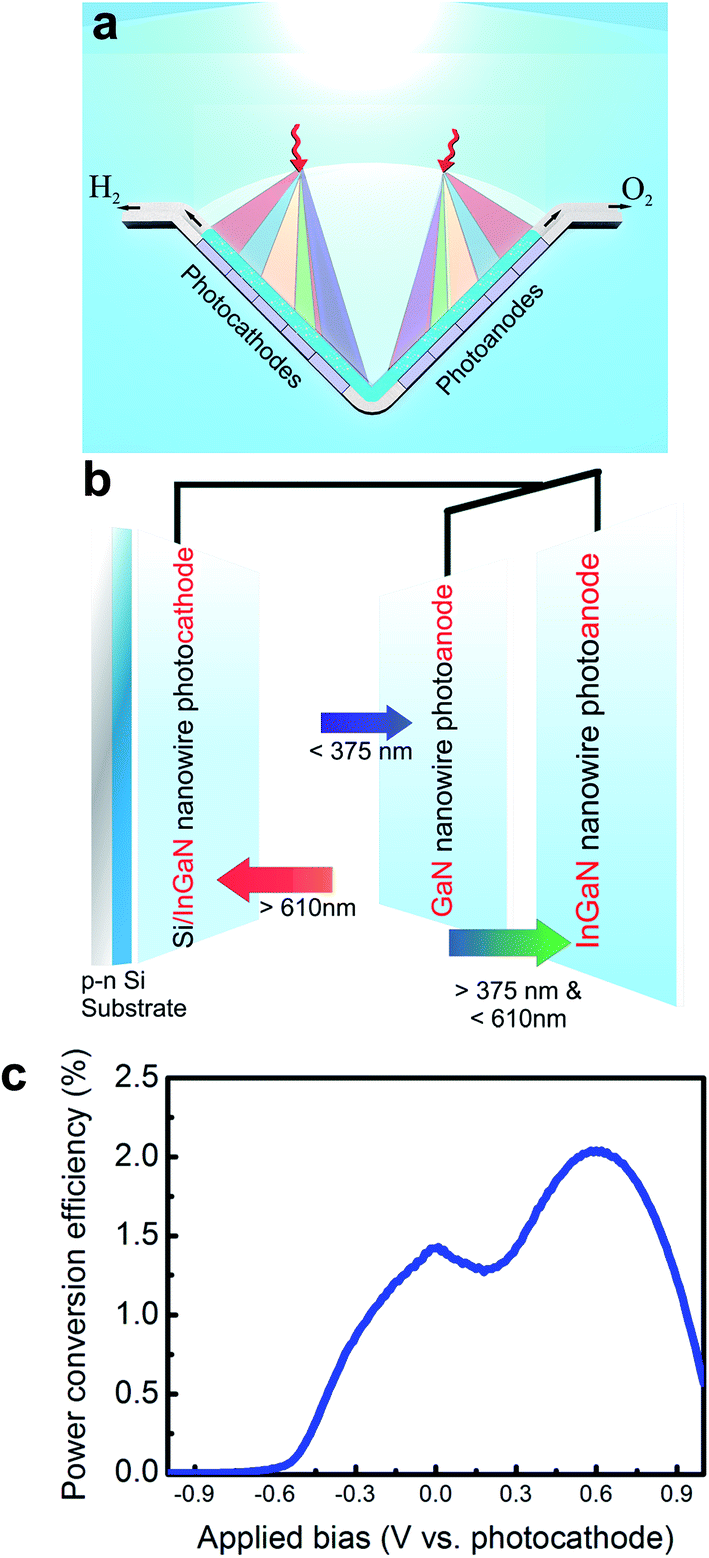 | ||
| Fig. 11 (a) Conceptual view of a dual-photoelectrode system under parallel illumination, in which the incident solar spectrum is spatially and spectrally split on the photoanode and photocathode. The photoanode (or photocathode) may be formed by parallel-connected anodes (or cathodes), each of which is illuminated with certain portion of the solar spectrum. (b) Schematic of a parallel-connected GaN and InGaN nanowire photoanode, and Si/InGaN photocathode. The incident sunlight is spectrally and spatially split among the photoelectrodes. (c) The power conversion efficiency of the dual-photoelectrode device (as shown in b) vs. applied bias under AM1.5G 1 sun illumination. The maximum power conversion efficiency is estimated to be ∼2% at ∼0.6 V vs. photocathode. Reprinted with permission from ref. 198. Copyright 2015, American Chemical Society. | ||
Photocatalytic and photoelectrochemical CO2 reduction using metal/nonmetal-nitrides
To date research efforts on artificial photosynthesis are mostly directed towards sunlight-driven water splitting to produce H2 fuel. Although H2 is an important fuel and chemical feedstock, it suffers from low volumetric energy densities.3 In contrast, hydrocarbon fuels with optimum volumetric energy density can be a practical alternative for better integration with existing energy infrastructure. Therefore, photocatalytic and photoelectrochemical CO2 reduction to high-energy-rich fuels is of tremendous interest. This process not only produces value-added fuels from abundant natural resources, but also provides an alternative approach to capture, sequestrate, and store anthropogenic CO2. For photocatalytic CO2 reduction, the bandgap of the semiconductor needs to straddle the oxidation potential of water and reduction potential of CO2 to various hydrocarbons. Upon bandgap irradiation, photoholes in the valence band of a semiconductor oxidize water to generate O2 and H+, and photogenerated electrons in the conduction band reduce CO2 by a sequence of reactions to produce CO, CH4 or CH3OH, etc.16Since the first demonstration of photoelectrochemical and photocatalytic CO2 reduction to hydrocarbons (HOOCH, CH2O, CH3OH, CH4) by Halmann20 in 1978 and Inoue21 in 1979, respectively, many groups have investigated the use of different semiconductors to achieve enhanced catalytic activities.16 Although a number of UV light sensitive photocatalysts (e.g., GaN, ZnS, SiC, SrTiO3, and TiO2) have been found that are catalytically active, the development of visible light sensitive photocatalysts is still very limited.15 As illustrated in Fig. 2, in contrast to most of the commonly used metal-oxides, the bandgaps of nitrides straddle the oxidation potential of water and reduction potential of CO2. Therefore, the development of various metal/nonmetal-nitride based photocatalysts for CO2 reduction has attracted significant attention recently.
AlOtaibi et al. have recently demonstrated photocatalytic CO2 reduction to CH4 and CO using GaN nanowire arrays with light being the only energy input.199 While the bare GaN nanowire was found to have higher photoactivity for CO production over CH4, the Rh/Cr2O3 core/shell nanoparticle decorated GaN nanowires provided higher activity and selectivity towards CH4 over CO production. Additionally, Pt nanoparticle decorated GaN nanowires showed an order of magnitude higher photoactivity for CH4 production than bare GaN nanowires, with over 24 h of stability. Yotsuhashi et al. demonstrated that the n-type Si doped GaN epilayer photoanode grown by MOCVD could produce HCOOH with 3% faradic efficiency with light being the only energy input.200 Later on, the faradic efficiency of the GaN epilayer was improved to 9% by enhancing the water oxidation of GaN with NiO co-catalysts.201 In a follow-up study, the efficiency of the photoanode was improved by using AlGaN/GaN heterostructures.202,203 In a subsequent study, a tandem photoanode of InGaN with two Si p–n junctions was developed for CO2 reduction to HCOOH with an energy conversion efficiency of 0.97%.204 While InGaN enhanced the light absorption of the photoanode, the embedded Si p–n junction raised the cathode potential for CO2 reduction and enhanced the reaction capability of the cathode.
Since the first demonstration in 2009, polymeric C3N4 has been widely studied as a visible light sensitive catalyst for CO2 reduction.126,205–211 It has been demonstrated that the CO2 photoreduction activity and selectivity of the products is highly dependent on the structure of the g-C3N4 and the co-catalysts used. For example, melamine hydrochloride precursor derived g-C3N4 effectively photocatalyzes CO2 reduction into CO under visible light without any co-catalyst.212 In another study, urea derived g-C3N4 was found to be more effective than melamine-derived g-C3N4 due to the improved reaction surface area, and small crystal size.209 The incorporation of the Pt co-catalyst on g-C3N4 was reported to enhance photocatalytic activity and selectivity for CO2 reduction into CH3OH, CH4, HCHO, etc.213 Bai et al. reported that the Pd{111} facets of the Pd co-catalyst were more active than Pd{100} facets for CO2 photoreduction.214 Lin et al. demonstrated an inexpensive system consisting of the g-C3N4 photocatalyst, CoOx oxidative co-catalyst, and Co-bipyridine complex (Co(bpy)32+) electron mediator, that can reduce CO2 to CO under visible light.206 Heterojunctions of g-C3N4/In2O3,215 and g-C3N4/red-phosphor216 were also found to be effective for CO2 photoreduction to CH4 under visible light. Maeda and co-workers demonstrated that molecular ruthenium complex coupled polymeric C3N4, as shown in Fig. 12, can photocatalytically reduce CO2 to HCOOH under visible light with a high turnover number (200 for 20 h) and high selectivity (80%).208 A follow-up work on this hybrid photocatalyst revealed that the introduction of mesoporosity into C3N4 structures can increase the specific surface area and hence the activity.207 In a subsequent study, by carefully designing the catalytically active site and the reaction environment, a substantial improvement in the photocatalytic conversion of CO2 into formic acid was achieved, leading to a relatively high TON (>1000) and AQY of 5.7% at 400 nm.205 Very recently, 2D BCN has been developed, which can photocatalytically reduce CO2 to CO under visible light (>420 nm).154
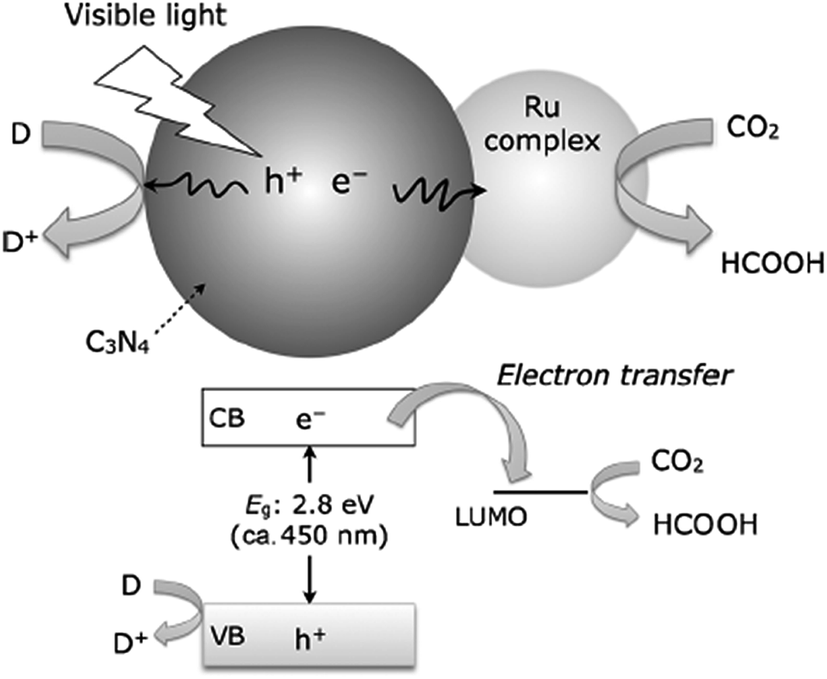 | ||
| Fig. 12 Schematic of photochemical CO2 reduction to HCOOH using a Ru complex/C3N4 hybrid photocatalyst. Reproduced from ref. 207 with permission from the Royal Society of Chemistry. | ||
Conclusions and perspectives
In summary, we have provided an overview of the current research status of metal/nonmetal-nitride based photocatalysts and photoelectrodes for artificial photosynthesis. Over the last decade, metal/nonmetal-nitrides have emerged as a new generation of photocatalysts owing to their unique optoelectronic and photocatalytic properties. A number of nitride based photocatalysts and photoelectrodes have been developed in recent years that can function effectively for solar water splitting and CO2 reduction under UV and visible light. Nearly 10 years of research efforts on nitrides have seen more success, to some extent, than what has been achieved from nearly 40 years of dedicated research on metal-oxide based materials for solar-fuel conversion.While STH efficiency of a few percentages and a few hours of stability have been achieved to date, it is far from the target STH efficiency of >10% with long-term stability (5000 h) to meet the DOE's target cost of $2–4 per kg H2 by 2018.217 Therefore, the synthesis of new materials and development of new technologies are needed. Although significant research efforts have been made to develop UV and visible light sensitive photocatalysts, little attention is paid to harness infrared light, which constitutes about 52% of the solar spectrum. Synthesis of defect-free In rich InGaN (In ∼ 50–100%) photocatalysts could harness a large part of the solar spectrum. For near-infrared (up to 2000 nm) light absorption, InN with a bandgap of 0.65 eV can be utilized. Since InN does not possess sufficient potential for the HER, it can be utilized in combination with another small bandgap semiconductor with sufficient HER potential in a Z-scheme to capture ∼80–90% of the solar spectrum. The tunable bandgap of InGaN enables the synthesis of a quadri-band photocatalyst, which can be one of the potential options for better utilization of the entire solar spectrum. Utilization of other potential technologies, such as multiple exciton generation,218 hot electron transfer, and up-conversion219 may help overcome the efficiency bottleneck as well. Moreover, because of their earth-abundance, excellent photovoltaic properties, strong photon absorption, and large carrier diffusion lengths, perovskite based PV (or their integration with Si based PV) may contribute to the long waited breakthrough in the solar hydrogen race.44,220–222 However, the stability of perovskite materials remains a major concern to achieve this goal. Apart from novel material development, the reaction kinetics, i.e., detailed thermodynamics and kinetics in interfacial carrier transfer in water splitting and CO2 reduction, need to be further understood to improve the efficiency and stability of the existing photocatalyst materials. An optimum photocatalyst should possess enhanced light absorption, rapid carrier collection/extraction, and wider solar spectrum absorption, lower cost and toxicity, and enhanced stability. Because of their tunable bandgaps, and unique optoelectronic and catalytic properties, it is expected that metal/nonmetal-nitrides will stimulate further research to overcome the efficiency and reliability bottleneck of metal-oxides, Si, and other commonly used photocatalysts for commercially viable artificial photosynthetic devices in the near future.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Climate Change and Emissions Management (CCEMC) Corporation.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2015, 23, 1–9 Search PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- M. Graetzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef CAS.
- R. L. House, N. Y. M. Iha, R. L. Coppo, L. Alibabaei, B. D. Sherman, P. Kang, M. K. Brennaman, P. G. Hoertz and T. J. Meyer, J. Photochem. Photobiol., C, 2015 DOI:10.1016/j.jphotochemrev.2015.08.002.
- S. Bensaid, G. Centi, E. Garrone, S. Perathoner and G. Saracco, ChemSusChem, 2012, 5, 500–521 CrossRef CAS PubMed.
- X. Li, J. G. Yu, J. X. Low, Y. P. Fang, J. Xiao and X. B. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J. P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- J. Mao, K. Li and T. Peng, Catal. Sci. Technol., 2013, 3, 2481–2498 CAS.
- L. Yuan and Y. J. Xu, Appl. Surf. Sci., 2015, 342, 154–167 CrossRef CAS.
- G. Ciamician, Science, 1912, 36, 385–394 CAS.
- P. Boddy, J. Electrochem. Soc., 1968, 115, 199–203 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495–500 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. B. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. L. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- L. Vayssieres, On Solar Hydrogen and Nanotechnology, John Wiley & Sons, 2010 Search PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- J. Q. Chen, D. H. Yang, D. Song, J. H. Jiang, A. B. Ma, M. Z. Hu and C. Y. Ni, J. Power Sources, 2015, 280, 649–666 CrossRef CAS.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- M. G. Kibria, F. A. Chowdhury, S. Zhao, B. AlOtaibi, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 6797 CrossRef CAS PubMed.
- A. Kudo, MRS Bull., 2011, 36, 32–38 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327–12333 CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- F. E. Osterloh, Top. Curr. Chem., Springer, 2015, pp. 1–38 Search PubMed.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- S. Licht, J. Phys. Chem. B, 2001, 105, 6281–6294 CrossRef CAS.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- J. S. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 Search PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- J. Brillet, J.-H. Yum, M. Cornuz, T. Hisatomi, R. Solarska, J. Augustynski, M. Graetzel and K. Sivula, Nat. Photonics, 2012, 6, 824–828 CrossRef CAS.
- S. A. Bonke, M. Wiechen, D. R. MacFarlane and L. Spiccia, Energy Environ. Sci., 2015, 8, 2791–2796 CAS.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057–14061 CrossRef CAS PubMed.
- K. Fujii, S. Nakamura, M. Sugiyama, K. Watanabe, B. Bagheri and Y. Nakano, Int. J. Hydrogen Energy, 2013, 38, 14424–14432 CrossRef CAS.
- G. Peharz, F. Dimroth and U. Wittstadt, Int. J. Hydrogen Energy, 2007, 32, 3248–3252 CrossRef CAS.
- O. Khaselev, A. Bansal and J. A. Turner, Int. J. Hydrogen Energy, 2001, 26, 127–132 CrossRef CAS.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- O. K. Varghese and C. A. Grimes, Sol. Energy Mater. Sol. Cells, 2008, 92, 374–384 CrossRef CAS.
- H. Gerischer, Faraday Discuss. Chem. Soc., 1980, 70, 137–151 RSC.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed.
- N. C. Strandwitz, D. J. Comstock, R. L. Grimm, A. C. Nichols-Nielander, J. Elam and N. S. Lewis, J. Phys. Chem. C, 2013, 117, 4931–4936 CAS.
- Y. W. Chen, J. D. Prange, S. Dühnen, Y. Park, M. Gunji, C. E. D. Chidsey and P. C. McIntyre, Nat. Mater., 2011, 10, 539–544 CrossRef CAS PubMed.
- L. Ji, M. D. McDaniel, S. Wang, A. B. Posadas, X. Li, H. Huang, J. C. Lee, A. A. Demkov, A. J. Bard, J. G. Ekerdt and E. T. Yu, Nat. Nanotechnol., 2015, 10, 84–90 CrossRef CAS PubMed.
- Y. Nanishi, Nat. Photonics, 2014, 8, 884–886 CrossRef CAS.
- K. Domen, Electrochem. Soc. Interface, 2013, 57–62 Search PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS.
- S. Arafin, X. H. Liu and Z. T. Mi, J. Nanophotonics, 2013, 7, 074599 CrossRef.
- J. Wu, J. Appl. Phys., 2009, 106, 011101 CrossRef.
- P. G. Moses and C. G. V. d. Walle, Appl. Phys. Lett., 2010, 96, 021908 CrossRef.
- A. G. Bhuiyan, A. Hashimoto and A. Yamamoto, J. Appl. Phys., 2003, 94, 2779–2808 CrossRef CAS.
- O. Ambacher, J. Phys. D: Appl. Phys., 1998, 31, 2653 CrossRef CAS.
- T. D. Moustakas, Phys. Status Solidi A, 2013, 210, 169–174 CrossRef CAS.
- B. AlOtaibi, H. P. T. Nguyen, S. Zhao, M. G. Kibria, S. Fan and Z. Mi, Nano Lett., 2013, 13, 4356–4361 CrossRef CAS PubMed.
- H. S. Jung, Y. J. Hong, Y. Li, J. Cho, Y.-J. Kim and G.-C. Yi, ACS Nano, 2008, 2, 637–642 CrossRef CAS PubMed.
- D. Zhuang and J. H. Edgar, Mater. Sci. Eng., R, 2005, 48, 1–46 CrossRef.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- S. S. Kocha, M. W. Peterson, D. J. Arent, J. M. Redwing, M. A. Tischler and J. A. Turner, J. Electrochem. Soc., 1995, 142, L238–L240 CrossRef CAS.
- J. Beach, R. Collins and J. Turner, J. Electrochem. Soc., 2003, 150, A899–A904 CrossRef CAS.
- K. Fujii, T. Karasawa and K. Ohkawa, Jpn. J. Appl. Phys., 2005, 44, L543 CrossRef CAS.
- K. Fujii and K. Ohkawa, Jpn. J. Appl. Phys., 2005, 44, L909 CrossRef CAS.
- K. Fujii and K. Ohkawa, Phys. Status Solidi C, 2006, 3, 2270–2273 CrossRef CAS.
- K. Fujii and K. Ohkawa, J. Electrochem. Soc., 2006, 153, A468–A471 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Q. Huang, Z. Ye and X. Xiao, J. Mater. Chem. A, 2015, 3, 15824–15837 CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. J. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- A. Kudo, Int. J. Hydrogen Energy, 2007, 32, 2673–2678 CrossRef CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- X. Chen, C. Li, M. Graetzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- L. Yang, H. Zhou, T. Fan and D. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 6810–6826 RSC.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- W.-F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- M. Shalom, D. Ressnig, X. Yang, G. Clavel, T. P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2015, 3, 8171–8177 CAS.
- B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186–19192 CrossRef CAS PubMed.
- H. X. Zhong, H. M. Zhang, Y. M. Liang, J. L. Zhang, M. R. Wang and X. L. Wang, J. Power Sources, 2007, 164, 572–577 CrossRef CAS.
- J. Sato, N. Saito, Y. Yamada, K. Maeda, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi, K. Domen and Y. Inoue, J. Am. Chem. Soc., 2005, 127, 4150–4151 CrossRef CAS PubMed.
- K. Maeda, N. Saito, D. Lu, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 4749–4755 CAS.
- Y. Lee, T. Watanabe, T. Takata, M. Hara, M. Yoshimura and K. Domen, J. Phys. Chem. B, 2006, 110, 17563–17569 CrossRef CAS PubMed.
- N. Arai, N. Saito, H. Nishiyama, Y. Inoue, K. Domen and K. Sato, Chem. Lett., 2006, 35, 796–797 CrossRef CAS.
- T. Kida, Y. Minami, G. Guan, M. Nagano, M. Akiyama and A. Yoshida, J. Mater. Sci., 2006, 41, 3527–3534 CrossRef CAS.
- N. Arai, N. Saito, H. Nishiyama, K. Domen, H. Kobayashi, K. Sato and Y. Inoue, Catal. Today, 2007, 129, 407–413 CrossRef CAS.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, Bull. Chem. Soc. Jpn., 2007, 80, 1004–1010 CrossRef CAS.
- A. Kubacka, M. Fernandez-Garcia and G. Colon, Chem. Rev., 2011, 112, 1555–1614 CrossRef PubMed.
- D. Wang, A. Pierre, M. G. Kibria, K. Cui, X. Han, K. H. Bevan, H. Guo, S. Paradis, A.-R. Hakima and Z. Mi, Nano Lett., 2011, 11, 2353–2357 CrossRef CAS PubMed.
- X. Shen, Y. A. Small, J. Wang, P. B. Allen, M. V. Fernandez-Serra, M. S. Hybertsen and J. T. Muckerman, J. Phys. Chem. C, 2010, 114, 13695–13704 CAS.
- X. Shen, P. B. Allen, M. S. Hybertsen and J. T. Muckerman, J. Phys. Chem. C, 2009, 113, 3365–3368 CAS.
- U. Aschauer, Y. He, H. Cheng, S.-C. Li, U. Diebold and A. Selloni, J. Phys. Chem. C, 2010, 114, 1278–1284 CAS.
- U. Diebold, N. Ruzycki, G. S. Herman and A. Selloni, Catal. Today, 2003, 85, 93–100 CrossRef CAS.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Gratzel, Phys. Rev. Lett., 1998, 81, 2954–2957 CrossRef CAS.
- J. Wang, L. S. Pedroza, A. Poissier and M. V. Fernández-Serra, J. Phys. Chem. C, 2012, 116, 14382–14389 CAS.
- K. Maeda, K. Teramura and K. Domen, J. Catal., 2008, 254, 198–204 CrossRef CAS.
- Y. Li, L. Zhu, Y. Yang, H. Song, Z. Lou, Y. Guo and Z. Ye, Small, 2015, 11, 871–876 CrossRef CAS PubMed.
- M. G. Kibria, F. A. Chowdhury, S. Zhao, M. L. Trudeau, H. Guo and Z. Mi, Appl. Phys. Lett., 2015, 106, 113105 CrossRef.
- T. Stoica and R. Calarco, IEEE J. Sel. Top. Quantum Electron., 2011, 17, 859–868 CrossRef CAS.
- F. Yam and Z. Hassan, Superlattices Microstruct., 2008, 43, 1–23 CrossRef CAS.
- I. H. Ho and G. B. Stringfellow, Appl. Phys. Lett., 1996, 69, 2701–2703 CrossRef CAS.
- M. Li, W. Luo, B. Liu, X. Zhao, Z. Li, D. Chen, T. Yu, Z. Xie, R. Zhang and Z. Zou, Appl. Phys. Lett., 2011, 99, 112108 CrossRef.
- W. Lü, D. B. Li, C. R. Li, F. Shen and Z. Zhang, J. Appl. Phys., 2004, 95, 4362–4366 CrossRef.
- R. Singh, D. Doppalapudi, T. Moustakas and L. Romano, Appl. Phys. Lett., 1997, 70, 1089–1091 CrossRef CAS.
- Z. Mi and S. Zhao, Phys. Status Solidi B, 2015, 252, 1050–1062 CrossRef CAS.
- M. G. Kibria, H. P. T. Nguyen, K. Cui, S. Zhao, D. Liu, H. Guo, M. L. Trudeau, S. Paradis, A.-R. Hakima and Z. Mi, ACS Nano, 2013, 7, 7886–7893 CrossRef CAS PubMed.
- M. Kibria, F. Chowdhury, M. Trudeau, H. Guo and Z. Mi, Nanotechnology, 2015, 26, 285401 CrossRef CAS PubMed.
- G. Hitoki, A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, Chem. Lett., 2002, 736–737 CrossRef CAS.
- W.-J. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798–1803 CrossRef CAS.
- M. Tabata, K. Maeda, M. Higashi, D. Lu, T. Takata, R. Abe and K. Domen, Langmuir, 2010, 26, 9161–9165 CrossRef CAS PubMed.
- P. Zhang, J. Zhang and J. Gong, Chem. Soc. Rev., 2014, 43, 4395–4422 RSC.
- S. S. K. Ma, T. Hisatomi, K. Maeda, Y. Moriya and K. Domen, J. Am. Chem. Soc., 2012, 134, 19993–19996 CrossRef CAS PubMed.
- S. Chen, S. Shen, G. Liu, Y. Qi, F. Zhang and C. Li, Angew. Chem., Int. Ed., 2015, 54, 3047–3051 CrossRef CAS PubMed.
- S. W. Cao, J. X. Low, J. G. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS.
- Y. Cui, J. Huang, X. Fu and X. Wang, Catal. Sci. Technol., 2012, 2, 1396–1402 CAS.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 1139 CrossRef.
- Y. Zheng, L. Lin, X. Ye, F. Guo and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 11926–11930 CrossRef CAS PubMed.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef CAS PubMed.
- Z. Hong, B. Shen, Y. Chen, B. Lin and B. Gao, J. Mater. Chem. A, 2013, 1, 11754–11761 CAS.
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- Z. Ding, X. Chen, M. Antonietti and X. Wang, ChemSusChem, 2011, 4, 274–281 CAS.
- M. Zhang and X. Wang, Energy Environ. Sci., 2014, 7, 1902–1906 CAS.
- J. Zhang, M. Zhang, S. Lin, X. Fu and X. Wang, J. Catal., 2014, 310, 24–30 CrossRef CAS.
- K. Takanabe, K. Kamata, X. Wang, M. Antonietti, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 13020–13025 RSC.
- Y. Hou, A. B. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., Int. Ed., 2013, 52, 3621–3625 CrossRef CAS PubMed.
- A. Du, S. Sanvito, Z. Li, D. Wang, Y. Jiao, T. Liao, Q. Sun, Y. H. Ng, Z. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 134, 4393–4397 CrossRef CAS PubMed.
- L. Ge and C. Han, Appl. Catal., B, 2012, 117–118, 268–274 CrossRef CAS.
- J. Yu, S. Wang, B. Cheng, Z. Lin and F. Huang, Catal. Sci. Technol., 2013, 3, 1782–1789 CAS.
- L. Yin, Y.-P. Yuan, S.-W. Cao, Z. Zhang and C. Xue, RSC Adv., 2014, 4, 6127–6132 RSC.
- G. Zhang, S. Zang, Z.-A. Lan, C. Huang, G. Li and X. Wang, J. Mater. Chem. A, 2015, 3, 17946–17950 CAS.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 1139 CrossRef.
- Y. Wang, J. Hong, W. Zhang and R. Xu, Catal. Sci. Technol., 2013, 3, 1703–1711 CAS.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- M. G. Kibria, S. Zhao, F. A. Chowdhury, Q. Wang, H. P. T. Nguyen, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2014, 5, 3825 CAS.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- F. A. Chowdhury, Z. Mi, M. G. Kibria and M. L. Trudeau, APL Mater., 2015, 3, 104408 CrossRef.
- Y.-C. Pu, M. G. Kibria, Z. Mi and J. Z. Zhang, J. Phys. Chem. Lett., 2015, 6, 2649–2656 CrossRef CAS PubMed.
- M. Ono, K. Fujii, T. Ito, Y. Iwaki, A. Hirako, T. Yao and K. Ohkawa, J. Phys. Chem. Lett., 2007, 126, 054708 Search PubMed.
- K. Fujii, H. Nakayama, K. Sato, T. Kato, M.-W. Cho and T. Yao, Phys. Status Solidi C, 2008, 5, 2333–2335 CrossRef CAS.
- I. Waki, D. Cohen, R. Lal, U. Mishra, S. P. DenBaars and S. Nakamura, Appl. Phys. Lett., 2007, 91, 093519 CrossRef.
- S.-Y. Liu, Y.-C. Lin, J.-C. Ye, S. Tu, F. Huang, M. Lee, W. Lai and J. Sheu, Opt. Express, 2011, 19, A1196–A1201 CrossRef CAS PubMed.
- S.-Y. Liu, J. Sheu, M. Lee, Y.-C. Lin, S. Tu, F. Huang and W. Lai, Opt. Express, 2012, 20, A190–A196 CrossRef CAS PubMed.
- S. Kikawa, N. Kobayashi, J. Yamamoto, Y. Ban and K. Matsumoto, e-J. Surf. Sci. Nanotechnol., 2009, 7, 847–850 CrossRef CAS.
- Y.-G. Lin, Y.-K. Hsu, A. M. Basilio, Y.-T. Chen, K.-H. Chen and L.-C. Chen, Opt. Express, 2014, 22, A21–A27 CrossRef PubMed.
- A. Nakamura, K. Fujii, M. Sugiyama and Y. Nakano, Phys. Chem. Chem. Phys., 2014, 16, 15326–15330 RSC.
- J. Benton, J. Bai and T. Wang, Appl. Phys. Lett., 2013, 102, 173905 CrossRef.
- B. AlOtaibi, M. Harati, S. Fan, S. Zhao, H. Nguyen, M. Kibria and Z. Mi, Nanotechnology, 2013, 24, 175401 CrossRef CAS PubMed.
- J. Wallys, S. Hoffmann, F. Furtmayr, J. Teubert and M. Eickhoff, Nanotechnology, 2012, 23, 165701 CrossRef CAS PubMed.
- S.-Y. Liu, J. Sheu, Y.-C. Lin, S. Tu, F. Huang, M. Lee and W. Lai, Opt. Express, 2012, 20, A678–A683 CrossRef CAS PubMed.
- K. Fujii, K. Kusakabe and K. Ohkawa, Jpn. J. Appl. Phys., 2005, 44, 7433 CrossRef CAS.
- J. Li, J. Y. Lin and H. X. Jiang, Appl. Phys. Lett., 2008, 93, 162107 CrossRef.
- K. Aryal, B. N. Pantha, J. Li, J. Y. Lin and H. X. Jiang, Appl. Phys. Lett., 2010, 96, 052110 CrossRef.
- W. Luo, B. Liu, Z. Li, Z. Xie, D. Chen, Z. Zou and R. Zhang, Appl. Phys. Lett., 2008, 92, 262110 CrossRef.
- M. Li, W. Luo, Q. Liu, Z. Zhuang, Z. Li, B. Liu, D. Chen, R. Zhang, T. Yu and Z. Zou, J. Phys. D: Appl. Phys., 2013, 46, 345103 CrossRef.
- N. H. Alvi, P. E. D. Soto Rodriguez, P. Kumar, V. J. Gómez, P. Aseev, A. H. Alvi, M. A. Alvi, M. Willander and R. Nötzel, Appl. Phys. Lett., 2014, 104, 223104 CrossRef.
- J. Benton, J. Bai and T. Wang, Appl. Phys. Lett., 2014, 105, 223902 CrossRef.
- Y. J. Hwang, C. H. Wu, C. Hahn, H. E. Jeong and P. Yang, Nano Lett., 2012, 12, 1678–1682 CrossRef CAS PubMed.
- L. Caccamo, J. Hartmann, C. Fàbrega, S. Estradé, G. Lilienkamp, J. D. Prades, M. W. Hoffmann, J. Ledig, A. Wagner and X. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 2235–2240 CAS.
- M. Ebaid, J.-H. Kang, S.-H. Lim, J.-S. Ha, J. K. Lee, Y.-H. Cho and S.-W. Ryu, Nano Energy, 2015, 12, 215–223 CrossRef CAS.
- M. Ebaid, J.-H. Kang, S.-H. Lim, Y.-H. Cho and S.-W. Ryu, RSC Adv., 2015, 5, 23303–23310 RSC.
- J. Kamimura, P. Bogdanoff, J. Lähnemann, C. Hauswald, L. Geelhaar, S. Fiechter and H. Riechert, J. Am. Chem. Soc., 2013, 135, 10242–10245 CrossRef CAS PubMed.
- J. Benton, J. Bai and T. Wang, Appl. Phys. Lett., 2013, 103, 133904 CrossRef.
- P. E. D. S. Rodriguez, P. Aseev, V. J. Gómez, W. ul Hassan, M. Willander and R. Nötzel, Nano Energy, 2015, 13, 291–297 CrossRef.
- S. Rajaambal, M. Mapa and C. S. Gopinath, Dalton Trans., 2014, 43, 12546–12554 RSC.
- V. Chakrapani, J. Thangala and M. K. Sunkara, Int. J. Hydrogen Energy, 2009, 34, 9050–9059 CrossRef CAS.
- X. Feng, T. J. LaTempa, J. I. Basham, G. K. Mor, O. K. Varghese and C. A. Grimes, Nano Lett., 2010, 10, 948–952 CrossRef CAS PubMed.
- Y. Cong, H. S. Park, S. Wang, H. X. Dang, F.-R. F. Fan, C. B. Mullins and A. J. Bard, J. Phys. Chem., 2012, 116, 14541–14550 CrossRef CAS PubMed.
- M. Higashi, K. Domen and R. Abe, Energy Environ. Sci., 2011, 4, 4138–4147 CAS.
- M. Li, W. Luo, D. Cao, X. Zhao, Z. Li, T. Yu and Z. Zou, Angew. Chem., Int. Ed., 2013, 52, 11016–11020 CrossRef CAS PubMed.
- G. Liu, J. Shi, F. Zhang, Z. Chen, J. Han, C. Ding, S. Chen, Z. Wang, H. Han and C. Li, Angew. Chem., 2014, 126, 7295–7299 CrossRef PubMed.
- Y. Li, T. Takata, D. Cha, K. Takanabe, T. Minegishi, J. Kubota and K. Domen, Adv. Mater., 2013, 25, 125–131 CrossRef CAS PubMed.
- J. Hou, Z. Wang, C. Yang, H. Cheng, S. Jiao and H. Zhu, Energy Environ. Sci., 2013, 6, 3322–3330 CAS.
- M. Liao, J. Feng, W. Luo, Z. Wang, J. Zhang, Z. Li, T. Yu and Z. Zou, Adv. Funct. Mater., 2012, 22, 3066–3074 CrossRef CAS.
- S.-Y. Liu, J. Sheu, Y.-C. Lin, Y.-T. Chen, S. Tu, M. Lee and W. Lai, Opt. Express, 2013, 21, A991–A996 CrossRef PubMed.
- R. Dahal, B. N. Pantha, J. Li, J. Y. Lin and H. X. Jiang, Appl. Phys. Lett., 2014, 104, 143901 CrossRef.
- S. Fan, B. AlOtaibi, S. Y. Woo, Y. Wang, G. A. Botton and Z. Mi, Nano Lett., 2015, 15, 2721–2726 CrossRef CAS PubMed.
- B. AlOtaibi, S. Fan, S. Vanka, M. G. Kibria and Z. Mi, Nano Lett., 2015, 15, 6821–6828 CrossRef CAS PubMed.
- B. AlOtaibi, S. Fan, D. Wang, J. Ye and Z. Mi, ACS Catal., 2015, 5, 5342–5348 CrossRef CAS.
- S. Yotsuhashi, M. Deguchi, Y. Zenitani, R. Hinogami, H. Hashiba, Y. Yamada and K. Ohkawa, Appl. Phys. Express, 2011, 4, 117101 CrossRef.
- S. Yotsuhashi, M. Deguchi, Y. Zenitani, R. Hinogami, H. Hashiba, Y. Yamada and K. Ohkawa, Jpn. J. Appl. Phys., 2012, 51, 02BP07 CrossRef.
- S. Yotsuhashi, M. Deguchi, H. Hashiba, Y. Zenitani, R. Hinogami, Y. Yamada and K. Ohkawa, Appl. Phys. Lett., 2012, 100, 243904 CrossRef.
- S. Yotsuhashi, H. Hashiba, M. Deguchi, Y. Zenitani, R. Hinogami, Y. Yamada, M. Deura and K. Ohkawa, AIP Adv., 2012, 2, 042160 CrossRef.
- T. Sekimoto, S. Shinagawa, Y. Uetake, K. Noda, M. Deguchi, S. Yotsuhashi and K. Ohkawa, Appl. Phys. Lett., 2015, 106, 073902 CrossRef.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146–15151 CAS.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 CAS.
- T. Ohno, N. Murakami, T. Koyanagi and Y. Yang, J. CO2 Util., 2014, 6, 17–25 CrossRef CAS.
- J. Qin, S. Wang, H. Ren, Y. Hou and X. Wang, Appl. Catal., B, 2015, 179, 1–8 CrossRef CAS.
- G. Dong and L. Zhang, J. Mater. Chem., 2012, 22, 1160–1166 RSC.
- J. Yu, K. Wang, W. Xiao and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 11492–11501 RSC.
- S. Bai, X. Wang, C. Hu, M. Xie, J. Jiang and Y. Xiong, Chem. Commun., 2014, 50, 6094–6097 RSC.
- S.-W. Cao, X.-F. Liu, Y.-P. Yuan, Z.-Y. Zhang, Y.-S. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940–946 CrossRef CAS.
- Y.-P. Yuan, S.-W. Cao, Y.-S. Liao, L.-S. Yin and C. Xue, Appl. Catal., B, 2013, 140–141, 164–168 CrossRef CAS.
- DOE, Hydrogen Production Technical Team Roadmap, http://www1.eere.energy.gov/vehiclesandfuels/pdfs/program/hptt_roadmap_june2013.pdf.
- V. Sukhovatkin, S. Hinds, L. Brzozowski and E. H. Sargent, Science, 2009, 324, 1542–1544 CrossRef CAS PubMed.
- X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2013, 42, 173–201 RSC.
- W. Nie, H. Tsai, R. Asadpour, J.-C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak and M. A. Alam, Science, 2015, 347, 522–525 CrossRef CAS PubMed.
- T. Hamann, Science, 2014, 345, 1566–1567 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |