
Tuning molecular assembly to enhance azobenzene-based solar thermal fuel efficiency
Saugata
Sahu
 *a and
Santosh Kumar
Behera
*b
*a and
Santosh Kumar
Behera
*b
aResearch Institute for Electronic Science, Hokkaido University, Kita20, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0020, Japan. E-mail: Saugata255@gmail.com
bDepartment of Chemistry, Model Degree College, Nayagarh, Lathipada, 752079, Odisha, India. E-mail: santoshbeherau@gmail.com
First published on 17th January 2025
Abstract
Molecular solar thermal fuel (STF) systems harness solar energy from solar radiation and store it as chemical energy. The stored energy is released as heat in the presence of suitable stimuli. Recently, azobenzene and its several derivatives have largely been used to develop molecular solar thermal fuel systems. These molecules photoisomerize into a metastable state and store the solar energy. Various techniques are applied to tune the isomerization enthalpy, thermal back half-life and stability of the STF materials at the molecular level. In addition, the intermolecular assembly of the azo-molecules in an STF material plays an important role in altering the system's energy storage efficiency. A precise arrangement of photochromic compounds can be achieved by adjusting the chemical structures of the photoswitches, anchoring the photoswitches to a polymer/carbon-based material or attaching a phase-changing material to the photoswitches. These methodologies significantly alter the energy density and storage timing of the system. This review focuses on how suitable modulations of the molecular assembly nature of the photoswitches can be exploited to achieve highly efficient STF materials. Major factors, such as the structural design of the photochromes and different templating technologies, are addressed in detail. The proposed idea of tuning the molecular assembly in STF materials will provide rational guidance and facilitate the future development of efficient STF materials for large-scale applications in the field of renewable energy sources.
 Saugata Sahu | Saugata Sahu is an experimental photochemist specializing in the development of photoswitchable ligands and their applications in achieving spatiotemporal control of bio-functionality. He earned his BSc in Chemistry from the University of Calcutta in 2009, followed by an MSc in Chemistry from the Indian Institute of Technology (IIT) Guwahati in 2011. He completed his PhD in Organic Physical Chemistry at IIT Guwahati in 2016. Currently, he is a researcher in Professor Nobuyuki Tamaoki's group at Hokkaido University. His research focuses on leveraging molecular photoswitches for diverse applications, including controlling receptor–ligand interactions, host–guest chemistry, and molecular solar thermal fuels. |
 Santosh Kumar Behera | Santosh Kumar Behera earned his PhD in 2016, from the Department of Chemistry, Indian Institute of Technology (IIT) Guwahati, Assam, India. He was then a D. S. Kothari postdoctoral researcher at the Indian Institute of Science, Bangalore, India; a postdoctoral researcher at IMDEA Nanoscience, Madrid, Spain and a senior project associate at the CSIR-Institute of Minerals and Materials Technology, Bhubaneswar, India. He was a Marie Skłodowska-Curie Postdoctoral Fellow with Prof. Rubén D. Costa at the Chair of Biogenic Functional Materials, Technical University of Munich, Germany. He is currently an assistant professor in the Department of Chemistry, Govt. Model Degree College Nayagarh, Odisha, India. His research includes the design and development of luminescent small organic molecules and (nano)materials for optoelectronic and biological applications and the study of the dynamics of excited states through photophysical and optical spectroscopic studies. |
1. Introduction
Solar energy is considered one of the most sustainable renewable energy sources because of its huge and sustainable supply.1 The annual global energy consumption is equivalent to 0.05% of solar radiation reaching the earth's surface in a day.2 Molecular solar thermal fuel (STF) systems harness solar energy in a recyclable and cost-effective manner with zero greenhouse gas emission.3–6 These materials comprise a photoactive material that undergoes photoisomerization in the presence of solar radiation and stores solar energy as chemical energy within photoactive molecules. Normally, upon absorption of appropriate light irradiation, the photoactive material produces a high-energy metastable photoisomer. The stored energy in the metastable photoisomer can be accessed by triggering the back isomerization with suitable stimuli such as heat, light, or chemical catalysts. The advantages of the STF systems rely on simultaneous solar energy harvesting, storage, and stimuli-dependent energy-releasing efficiency. Although the first report on a STF material with the anthracene molecule was published by Luther and Weigert in the early 1900s,7 STF materials received significant attention in the late 2000s due to the fossil fuel crisis and alarming global warming.8,9 Since then, extensive research has been employed with various photoactive materials to enhance the efficiency of the STF materials.In principle, all photoactive molecules can function as solar energy storage materials in their metastable form (Fig. 1). However, the selection of photochromic materials is limited by considering solar radiation absorption efficiency, photoisomerization quantum yield, storage energy density, the stability of the metastable state and photodegradation behavior of the molecule. Stilbene and its derivatives were reported to absorb UV-visible light irradiation to generate a metastable Z-isomer.10,11 However, the storage energy density of stilbene-based materials is significantly low.10,11 Although various chemical modifications were attempted to increase the energy density of stilbene-based STF materials, a low to moderate energy density (5–105 kJ mol−1) could be achieved.10,11 This limits their potential application as energy storage materials. The dihydroazulene/vinylheptafulvene (DHA/VHF) couple, another type of photoactive material, was reported to exhibit low to moderate energy storage density (∼110–510 J g−1, theoretical value).12–14 DHA undergoes light-activated ring opening to yield a VHF photoisomer. Due to the significant separation between DHA and VHF absorption bands, the forward ring opening occurs almost quantitatively.15 The ring-closing reaction can be triggered thermally or by a chemical catalyst. The unidirectional photo-activation, high stability of the metastable state and good storage density make the DHA/VHF couple suitable for STF applications. Various molecular scaffolds of DHA/VHF were developed with chemical modification of the core photoswitching unit to enhance the energy storage performances of the photoactive materials. A few limitations of the DHA/VHF material are medium-dependent photoconversion, high photodegradation in polar medium, and unidirectional photoswitching.12,14,15 Norbornadiene/quadricyclane-based STF materials are some of the most promising STF materials because of their high energy storage density and long storage duration.16–18 The bicyclic hydrocarbon norbornadiene photo-isomerizes to a more strained polycyclic quadricyclane. Unsubstituted quadricyclane thermally back isomerizes to norbornadiene with a heat release of ∼96 kJ mol−1. A few major drawbacks of norbornadiene/quadricyclane-based STF materials are weak absorbance in the solar spectrum region (absorption λmax < 300 nm), low photoisomerization and photodegradation nature. Most recently, with a suitable donor–acceptor substitution at the sp2 carbon of norbornadiene, the absorption λmax could be increased to ∼360 nm with a high storage energy density (∼514 J g−1).5,16–21 Among all STF materials, azobenzene and its derivatives are the most studied systems and have attracted considerable attention due to their broad scope of chemical tunability to achieve desired physicochemical properties for suitable STF applications. Pristine azobenzene has an absorption maximum at ∼320 nm.22 The more stable E-isomer of azobenzene undergoes photoisomerization upon absorption of UV light (365 nm) to generate a metastable Z-isomer. The E-isomer is almost flat with no dipole moment. In contrast, the Z-isomer has a nonplanar staggered geometry with a dipole moment of 3.0 D.23 The molecular length contracts by ∼3.5 Å upon E-to-Z photoisomerization. The metastable state has a thermal back half-life of ∼1.4 days in benzene solution at 35 °C.24 The Z-to-E back isomerization can be accomplished by 400–450 nm light irradiation and heating, using chemical catalysts or electrochemically.22,25–28 The isomerization enthalpy of pristine azobenzene is 41.4 kJ mol−1.29 Notably, all of these photochemical parameters of azobenzene can be fine-tuned to achieve highly efficient STF materials. For example, the absorbance of azobenzene can be adjusted by suitable chemical modifications to achieve a better match with the solar spectrum.30–33 The isomerization enthalpy can be controlled by suitable ortho-substitution.34–36 The photoisomerization efficiency of azobenzene in a solid state can be controlled by adjusting the adjacent free volume surrounding the E-isomer.37,38 Furthermore, the ease of synthesis of various azobenzene derivatives, high photothermal stability, and the availability of multiple triggers for Z-to-E back isomerization make the azobenzene derivatives a clever choice for STF materials.39,40
 | ||
| Fig. 1 Molecular structures of different photoswitches and their isomers. | ||
Different design approaches are pursued to achieve ideal STF materials with (a) significant overlap of the absorption band with the solar spectrum, (b) quantitative photoisomerization and (c) long storage duration. A few recent review articles demonstrated how a suitable chemical modification on azobenzene or analogous substances could overcome those challenges.3,4,6,20,41 In the current scenario, primary research with STF materials focuses on achieving a high storage energy density, even more than the actual isomerization enthalpy. Molecular assembly of the azo-derivatives with suitable intermolecular interactions plays a pivotal role in achieving high energy density in solid-state STF materials (Scheme 1). Particularly, it relies on the enhanced intermolecular interaction between the E-isomers as compared to the Z-isomers in an ordered structure.42–44 A suitable assembly of the azo-derivatives strongly depends on the molecular structure of the STF material.43,45,46 The weak noncovalent interactions inside the assembled structure play a critical role in modulating photoisomerization efficiency and prolonging the storage duration of solid/condensed state STF materials. To this end, several structurally modified azo-derivatives have been reported to achieve highly efficient STF materials with different assembly natures. The present review mainly focuses on tuning molecular assembly through structural design and molecular templating to achieve highly efficient STF materials. Tuning of the molecular assembly towards highly efficient STF materials is discussed using photochemical phase changing materials, polymer-based materials, and carbon-based materials in the solid/condensed state.
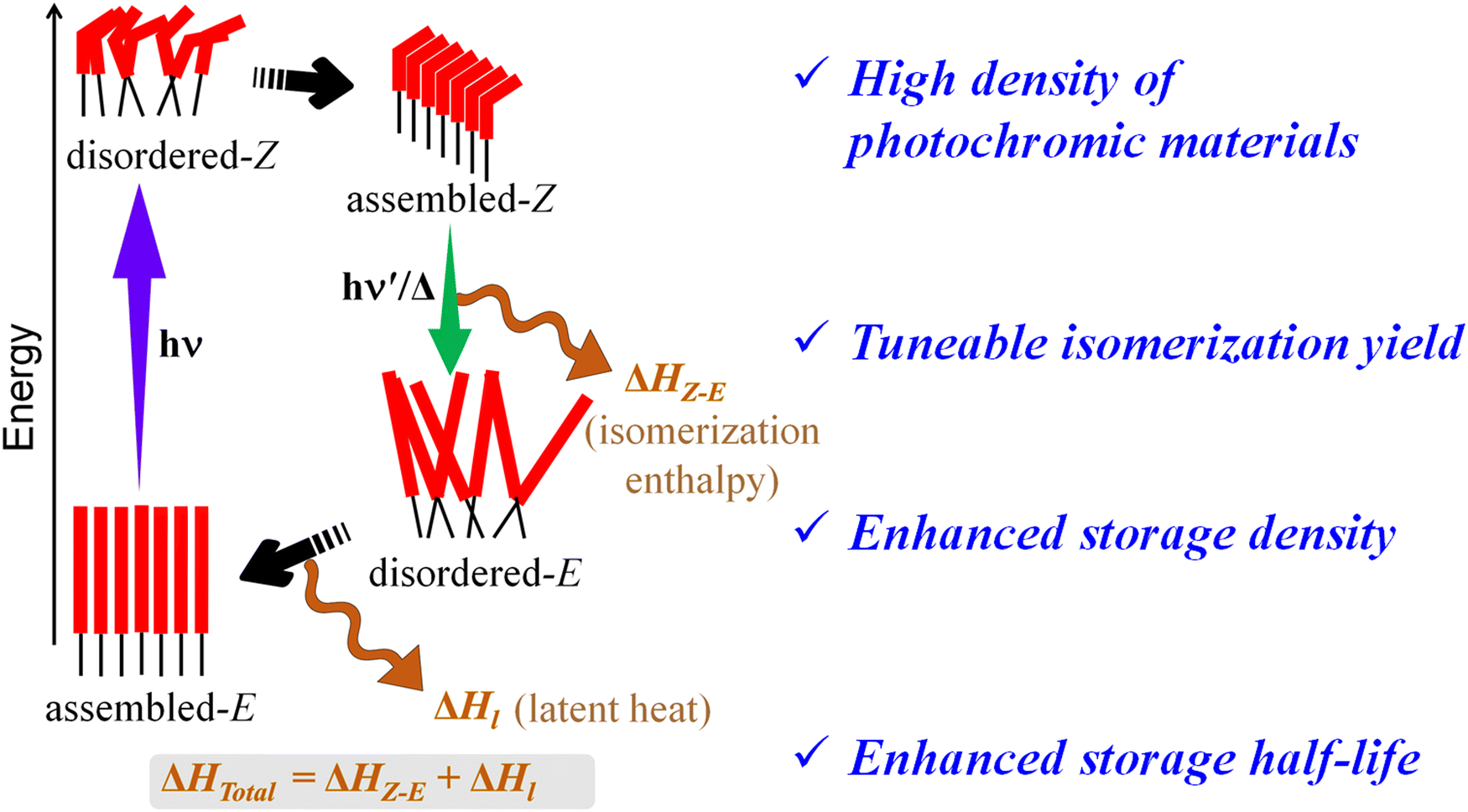 | ||
| Scheme 1 Assembled STF materials to enhance the fuel efficiency. | ||
2. Photochemical phase changing based STF materials
Very recently, photochemical phase-changing materials (PCM) have been developed as high-energy-density STF materials.44,45 These materials undergo a phase transition during photoisomerization.43 Their metastable Z-isomer possesses a lower liquid-to-crystalline phase-transition temperature (Tm) than the stable E-isomer. The Z-to-E photoisomerization not only releases the stored isomerization enthalpy (ΔHZ–E) but also the latent heat (ΔHl) due to the liquid-Z to crystalline-E phase transition. Thus, the photo-triggered release of the thermal energy, together with the isomerization enthalpy, significantly increases the energy density of the PCM-based STF materials. The main prerequisite for this preferential crystallization of one photoisomer over another lies in the significant structural difference between the two isomers. A general protocol for developing PCM materials is attaching a long alkyl chain with the photochromic moieties. The stable isomer with a planar aromatic structure favours the ordered crystalline state more than the metastable state with a nonplanar aromatic geometry.43,45 However, small alkyl chains attached to the azo-derivatives were also reported to obtain efficient PCM-STF materials.37,47,48 Herein, we have discussed a few critical structural designing strategies to obtain efficient PCM-STF materials.The molecular designing strategy to achieve photo liquefication of azobenzene derivatives was reported independently by various groups.49–53 Norikane et al. observed that the introduction of a methyl group at the 3-position of the 4,4′-bisalkoxy azo-derivative drastically altered the molecular packing behavior compared to the pristine azobenzene (Fig. 2).50 In contrast to 4,4′-alkoxy azo-derivatives (1 and 2), 3-methyl substituted derivatives (3–5) showed room temperature photo-liquefaction from a crystalline E-isomer to a liquid Z-isomer (Fig. 2(b) and (c)). The initial isomerization occurred only at the surface of the crystal structure. Further photoisomerization takes place at the crystal–liquid interface only if the melting temperature of the Z-isomer is lower than room temperature. In contrast, Z-isomers with higher melting temperatures did not melt at room temperature, and solid Z-isomers prevented further isomerization. The melting temperature (Tm) of the Z-isomer of compound 4 was −6 °C, whereas for the E-isomer Tm was 87 °C. In contrast, the Tm values of the Z- and E-isomers of compound 7 were 94 and 90 °C. Later, the scanning tunnelling microscopy study with the two-dimensional assembly revealed that different photo-liquefaction behaviors arise due to the existence of different conformers for the derivatives with the azobenzene core, 3-methyl azobenzene core and 3,3′-dimethyl azobenzene core.54 Although their work did not focus on the STF applications, their findings demonstrated an excellent strategy for developing room-temperature photo liquefiable azo-derivatives for STF applications.
 | ||
| Fig. 2 (a) Molecular structures of azo-derivatives 1–7. Photograph of the same (b) before and (c) after irradiation with 100 mW cm−2 365 nm light for 30 minutes. Reproduced from ref. 50 with permission from the American Chemical Society. | ||
The photo-liquefaction of the azobenzene derivative at room temperature for the STF application was pioneered by the Kimizuka group.55 They reported the cationic 4,4′-bisalkoxy azobenzene derivative possessing various alkyl tails (n), spacer methylene chains (m), and the oligo-(ethylene oxide) based ammonium group with counter ions (X−) (Fig. 3). The oligo-(ethylene oxide) based ammonium group was rationally introduced to lower the melting point of the ionic crystal. Compounds with long alkyl chains showed crystalline behaviour at room temperature compared to those with short alkyl chains. Also, by introducing the bis(trifluoromethylsulfonyl)amide (Tf2N−) anion, the intermolecular interaction was significantly lowered, resulting in an ionic liquid at room temperature. Upon UV irradiation, the crystalline E-isomer melted into a liquid phase Z-isomer within a few minutes, and the crystalline lamellar structure determined by X-ray diffraction and birefringence measurements using a polarising optical microscope disappeared (Fig. 3(b)). Visible light irradiation onto a liquid Z-isomer caused crystallization reversibly. A maximum energy density of 129 J g−1 was reported for 8(6,4)-Br.56 Later, Fuchter and co-workers demonstrated an efficient electrochemical Z-to-E isomerization with an azapyrazole derivative linked to a similar cationic ammonium group (Fig. 4).57 Both the E and Z isomers of the ionic molecule displayed a liquid phase at room temperature, probably due to the shorter alkyl chain length and the presence of the bis(trifluoromethylsulfonyl)amide (Tf2N−) counter ion in the azapyrazole derivative. The storage energy of the STF materials was limited to the Z-to-E isomerization enthalpy.
 | ||
| Fig. 3 (a) Molecular structures of cationic azobenzene derivatives with crystalline/liquid nature at room temperature. (b) X-ray diffraction pattern (left), picture of the sample (middle) and polarisation optical microscope image (right) before irradiation (top), after UV-illumination (middle) and after visible light irradiation (bottom) for 8(6,4)-Br. Reproduced from ref. 55 with permission from John Wiley and Sons. | ||
 | ||
| Fig. 4 Chemical structure and physical properties of azapyrazole-based ionic STF materials 11 and 12. Reproduced from ref. 57 with permission from the American Chemical Society. | ||
Simple azo-derivatives such as 4-methoxy azobenzene show significant differences in molecular packing in their E and Z-isomers.37 The melting points of the E and Z-isomers of 4-methoxy azobenzene are 53 and 25 °C, respectively. An STF material fabricated by thermal melting of 4-methoxy azobenzene on a cloth fabric showed UV-light mediated photo-liquefaction and photocharging to achieve a storage energy density of 201 J g−1 (Fig. 5).37 The STF material's energy density decreased at the high loading weight of the photochromic material. The loose dispersity of the azo-material in the fabric improved the energy density because the gap in the fabric template enabled more light exposure and provided more space for isomerization and phase transition. The thermal back half-life of the pristine azo-derivative and fabric STF material was 29 and 40 h, respectively. The intermolecular interaction between the fabric and the azo-material decreased the thermal back-isomerization rate and extended the storage half-life.
 | ||
| Fig. 5 Fabrication of a solid-state photo-liquefiable azobenzene derivative on a flexible fabric template. Reproduced from ref. 37 with permission from John Wiley and Sons. | ||
Asymmetric, alkoxy-substituted, phase-changing azobenzene derivatives were reported to demonstrate the importance of the molecular structure and the role of intermolecular interaction in the photoisomerization and crystallization processes.58 The intermolecular distance and van der Waals interaction were tuned with linear and branched alkoxy chains (Fig. 6). The π-bond stacking was regulated with an o-methyl substitution. Due to the good flexibility and low barrier of rotation in the linear alkoxy-chain compared to the branched alkoxy-chain, a regular crystalline arrangement was rapidly formed at higher crystallization temperature in E-14, facilitated by strong van der Waals interactions. The crystallization temperature was further reduced due to the perturbed π-stacking in E-16. On the other hand, Z-isomers exhibited a glass transition temperature (Tg) of −60.49 to −51.05 °C because of the non-planar geometry with very weak intermolecular interactions. The low Tg of Z-isomers made the STF materials suitable for low-temperature warming applications. Though the higher intermolecular interactions resulted in high enthalpy differences between the photoisomers, it lowered the E-to-Z photoisomerization yield. The storage lifetime improved for 16 because the methyl substitution stabilized Z-16 and increased the activation energy of the Z–E isomerization. The thermal back half-life of 14, 15 and 16 was 20.5, 15.3 and 30.6 h, respectively. A maximum energy density of 343 J g−1 was obtained with 14, which was utilized to raise the temperature by ∼6.3 °C in an annular device at −5 °C. It was also reported that the alkoxy chain length plays an important role in altering the melting and crystallization temperatures of the Z-rich state, energy density and heat release.59 A longer alkyl chain provides a greater entanglement and prevents structural transformations.
 | ||
| Fig. 6 Molecular structure of alkoxy-substituted azobenzene as the phase-changing STF material. Reproduced from ref. 58 with permission from Springer Nature. | ||
The roles of polarity and effective size of the functional groups in the phase behavior of E and Z isomers were investigated using various para-substituted azobenzene derivatives with different electron-donating and electron-accepting moieties (Fig. 7).47 Depending on the Hammett constant (σ) of the functional group, the phase transition temperature remarkably differs in the E and Z isomers. The difference in the E and Z-isomers’ melting point (ΔTm) linearly increased with σ (Fig. 6(b)). A more polar functional group on azobenzene substantially increased the melting point of the E-isomer and decreased the melting point of the corresponding Z-isomer. A lower crystallization enthalpy (ΔHc) of the E-isomer was observed for methoxy substitution (17) as compared to the chloro-substituted analogue (22) (Fig. 6(c)). Despite a smaller crystallization enthalpy (ΔHl), the poor π–π interactions of the aromatic core of the E-isomer in the p-methoxy (17) and p-ethoxy (18) derivatives yielded photo-liquefiable STF materials at room temperature (Fig. 6(d)).37 These compact STF materials with low molecular weight display a higher gravimetric energy density than the long-alkyl chain-bearing azobenzene-based STF materials.44 With simple p-ethoxy azobenzene (18) a maximum storage density of 349 J g−1 was obtained. A shorter thermal back half-life was observed for azo-derivatives with electron-donating and withdrawing groups. This finding reveals that a suitable substitution on compact azobenzene-based STF materials not only alters the Z-to-E isomerization enthalpy and thermal back isomerization lifetime but also modulates the ΔHl because of the difference in the molecular packing.
 | ||
| Fig. 7 (a) p-Substituted azobenzene derivatives (17–25) with different electron donating and accepting moieties and their corresponding Hammett constant (σ). (b) The difference between the E and Z melting points (ΔTm) with increasing σ. (c) Crystallization enthalpy (ΔHl) and isomerization energy (ΔHZ–E) for various p-substituted azobenzene derivatives. (d) Optical microscope image. The crystalline E-isomer of p-ethoxy azobenzene (18) shows room temperature UV-liquefaction and isomerization (left), whereas the crystalline E-isomer of p-chloroazobenzene (22) needs to melt with an external heat supply before UV mediated photo-charging (right). Reproduced from ref. 47 with permission from the Royal Society of Chemistry. | ||
ortho-Functionalized azobenzenes display E-to-Z switching with visible light irradiation and reverse photoisomerization with relatively shorter wavelength light irradiation.34,60,61ortho-Substituted azobenzenes with a tridecanoate appendant displayed photo-liquefaction and isomerization with filtered sunlight at room temperature (Fig. 8).61 The Z-isomer persisted in the liquid phase for a wide range of temperatures from −40 to 110 °C. In contrast, the melting and crystalline temperature of equivalent pristine Z-azobenzene was 60 and 10 °C, respectively.62 The melting and crystallization behavior of the E-isomer didn’t alter much compared to that of the pristine E-azobenzene. The steric hindrance imposed by the ortho-functionalization significantly hindered the molecular packing of the Z-isomer as compared to the E-isomer.61,63 A larger crystallization energy and isomerization enthalpy of the tetra-orthofluoro derivative (26) resulted in the highest energy-storage density, 150 J g−1, compared to those of the other derivatives. All the STF-materials (26–29) showed a very long (from days to years) storage half-life. The STF materials showed visible light-driven charging and discharging at room temperature (>0 °C) with a storage period of nearly two months. Later, visible-light responsive discotic nematic liquid crystals, derived from ortho-halo-substituted azobenzene and hexa(hexyloxy)triphenylene, were reported to function efficiently at below 0 °C.64 However, the ortho-substitution to achieve visible-light switching decreased isomerization enthalpy by 6–25 kJ mol−1 compared to the pristine azobenzene (41 kJ mol−1).29,34–36
 | ||
| Fig. 8 (a) Molecular structures of visible light-activated STF materials (26–29). (b) The melting temperature (Tm, °C) and crystallization temperature (Tc, °C) of the E and Z isomers of 26–29. Reproduced from ref. 61 with permission from the Royal Society of Chemistry. | ||
Different azo-derivatives with slow thermal back-isomerization characteristics are employed to develop PCM-STF materials with prolonged storage duration. Poulsen, Li and co-workers developed the pyrazolylazophenyl ether-based STF material that efficiently underwent a photochemical phase change from a crystalline E-isomer to a liquid Z-isomer at room temperature (Fig. 9).65 The half-life of the Z-isomer was 90 days. Simple alteration of the alkyl chain length attached to the phenyl ring of the pyrazolylazophenyl ethers provided a series of STF materials (30–50) with variable Tm (Fig. 9(b) and (c)). Upon discharging, a concurrent heat release via isomerization and crystallization was achieved at room temperature. The continuous crystallization of the E-isomer facilitated 100% Z-to-E photoisomerization. The pyrazolylazophenyl ether-based PCM-STF material provided an elongated heat storage time (∼90 days) with an energy density of 320–370 J g−1. These materials displayed a temperature increase >20 °C in a solar thermal battery and were applied for deicing at below 0 °C. Later, a similar PCM-STF material was developed with longer alkyl chains.66 Due to the high Tm of the Z-isomer, the charging process was performed at elevated temperatures (>Tm of Z). In addition, a high storage temperature was required to prevent the crystallization of the Z-isomer. These materials showed a similar storage energy density of ∼360 J g−1.
 | ||
| Fig. 9 (a) Molecular structures of pyrazolylazophenyl ether-based STF materials (30–50) that show a photochemical phase changing at room temperature. Liquid to crystal phase-transition temperature (Tm) with different alkyl chain lengths. Reproduced from ref. 65 with permission from the American Chemical Society. | ||
Phenylazopyrazole derivatives with a dodecanoate alkylester linkage at the pyrazole ring (Fig. 10) were reported as PCM-STF materials for heat release under extremely cold conditions (−30 °C).67 The alkyl ester group increased the crystallinity of the E-isomer while maintaining the liquid phase behaviour of the Z-isomer. The intermolecular π–π and hydrophobic interactions at the alkyl chain region in the E-isomer resulted in an effective liquid phase to crystalline phase transition at room temperature. In contrast, the Z-isomer preferably remained in the liquid phase due to the high polarity and bulky structure. For instance, Z-51, Z-53 and Z-54 remained in the liquid phase between −45 and 85 °C. Z-52 displayed a shorter temperature range −30 to 10 °C due to the cold crystallization at higher temperatures. A liquid film of Z-53 and Z-54 showed no crystallization at −20 °C even after two weeks. In contrast, a liquid film of Z-51 and Z-52 was stable for 24 and 7 h, respectively. Compound 52 showed a maximum storage density of 231 J g−1.56 Thus, the photoinduced phase-changing nature, heat storage duration and energy density of PCM-STF material can be fine-tuned with a rational molecular design for potential fuel and battery usage. Later, azobispyrazole derivatives with small terminal substituents were also reported as PCM-STF materials.48 Due to the unique crystal packing of the E-isomer, the liquefaction of the E-isomer upon UV light irradiation was achieved at room temperature, far below the melting temperature of the E-isomer. It was reported that the small terminal group instead of a long alkyl chain enhanced the UV light penetration over 1400 μm of thickness and facilitated the photo-liquefaction and solvation of the E isomer. The maximum storage energy density achieved with these azobispyrazole-based STF materials was 331 J g−1. The storage half-life of the material was 6–7 days.
 | ||
| Fig. 10 (a) Molecular structures of alkyl ester-functionalized arylazopyrazole-based STF materials (51–54) to achieve light-triggered heat release under extremely cold conditions (−30 °C). (b) Schematic illustration of azopyrazole derivatives that store latent heat in the stable liquid phase upon activation. (c) Thermal parameters of the corresponding E and Z isomers. Reproduced from ref. 67 with permission from the American Chemical Society. | ||
Another visible light-responsive azapyrazole-based STF material was reported to function below 0 °C.68 Usually, a lower melting temperature (Tm) of the Z-isomer than the working temperature is a prerequisite for the successful development of photo-liquefiable STF materials. To achieve visible-light photo-switchable PCM-STF materials rechargeable at below 0 °C, different 4-alkylthio phenylazapyrazole derivatives (55–74) were developed with various linear alkyl chains on the thioalkyl group with or without a terminal vinylic group (Fig. 11). The photoswitch exhibited a near-quantitative E-to-Z and Z-to-E photoisomerization with 400 and 532 nm irradiation wavelengths, respectively. Azo-compound 70 showed a Tm of the Z-isomer at −1 °C and the Tc (crystallization temperature) of the E-isomer at 34 °C. The X-ray crystal structure of 70 revealed an antiparallel packing and several weak alkyl–alkyl and alkyl–aryl interactions (Fig. 11(e)). The absence of strong intermolecular interactions in compound 70 accomplished the desired phase-changing behavior (Fig. 11(d)). The half-life of the Z-isomer was 22 days at 0 °C, which made the system effective for long-term energy storage. The STF material could be charged and discharged at −1 °C with bidirectional visible light with a storage energy density of 250 J g−1. The sample was coated on a glass as a miniatured energy storage window, exhibiting a temperature increase from −1 °C to 21.7 °C upon discharging. The chemical design of the PCM-STF materials from the structure–property relationship provides a blueprint for designing PCM-STF materials functional at below 0 °C.
 | ||
| Fig. 11 (a) Molecular structures of bidirectional visible light-switchable PCM-STF materials rechargeable at low temperature (55–74). (b) and (c) Melting temperatures (Tm) of the E and Z isomers of 55–74. (d) Photograph of the optical microscope image and polarisation microscope image of 66 on an ice surface. The crystal E-isomer (top), liquid Z-isomer after 400 nm light irradiation (middle) and crystal E-isomer (top) upon 532 light irradiation (bottom). (e) The X-ray crystal structure of the E-isomer of 66. Reproduced from ref. 68 with permission from the Royal Society of Chemistry. | ||
Presently, a maximum energy density of ∼400 J g−1 is obtained using PCM-STF materials (Table 1). Current research mainly focuses on achieving a large difference in the melting temperatures of the E and Z-isomers and the photo-liquefaction of the E-isomer at room temperature or below room temperature, along with a high storage density. With novel structural design and tuned intermolecular interactions, these materials may be exploited as promising, highly efficient STF materials.
| Photoswitch | Entry no. | Material type | Energy density (J g−1) | t 1/2 (h) | Ref. |
|---|---|---|---|---|---|
| p-Methoxy azobenzene on fabric | 13 | Phase changing | 201 | 40 | 37 |
| p-Pentoxy azobenzene | 14 | Phase changing | 343 | 20.5 | 58 |
| p-Methoxy azobenzene | 18 | Phase changing | 292 | 48 | 47 |
| p-Ethoxy azobenzene | 18 | Phase changing | 349 | 45.6 | 47 |
| Tetra-o-fluoro azobenzene | 26 | Phase changing | 150 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 352 352 |
61 |
| Pyrazolylazo phenyl ether | 30–50 | Phase changing | 320–370 | ∼2160 | 65 |
| Arylazo pyrazole ether | 52 | Phase changing | 231 | — | 67 |
| Pyrazolylazo phenyl thioether | 70 | Phase changing | 250 | 528 | 66 |
| Azo bispyrazole with small alkyl terminals | — | Phase changing | 331 | 168–144 | 48 |
| Alkoxy azobenzene grafted on acrylate polymer | 78 | Azo-derivative templated polymer | 143 | 125 | 69 |
| Azobenzene-anchored polymer with intrinsic microporosity | 86 | Azo-derivative templated polymer | 180 | 47 | 38 |
| p-cyanoazobenzene grafted on a polycarbazole backbone | 88 | Azo-derivative templated polymer | 145 | 1 | 70 |
| Syndiotactic fabrication of azobenzene on the methyl acrylate polymer | 89 | Azo-derivative templated polymer | 698 | 75.5 | 71 |
| Azobenzene grafted on a polynorbornene backbone | 90 | Azo-derivative templated polymer | 176 | 0.33 | 72 |
| Azobenzene grafted on a poly(amido amine) dendrimer | 93 | Azo-derivative templated polymer | 214 | 32.6 | 73 |
| Azobenzene-linked dendrimers on a polydopamine-coated fabric | 94 | Azo-derivative templated polymer | 460 | 28.5 | 74 |
| Azobenzene containing a liquid crystalline block copolymer blended with poly(ethylene oxide) (1 kDa) | 96 | Azo-derivative templated polymer | 210 | 9.3 | 75 |
| Metal-coordinated p-azobenzenesulfonic acid grafted on polyacrylic acid | 97 | Azo-derivative templated polymer | 409 | 108 | 76 |
| Azobenzene grafted on a single-walled carbon nanotube | 98 | Azo-derivative grafted carbon material | 200 | — | 97 |
| 4-((3,5-Dimethoxyaniline)diazenyl)-benzene grafted RGO | 99 | Azo-derivative grafted carbon material | 245 | 6 | 77 |
| 4-((3,5-Dimethoxyaniline)diazenyl)-4-benzoic acid grafted RGO | 100 | Azo-derivative grafted carbon material | 403 | 792 | 77 |
| (4-((3,5-Dimethoxyaniline)-diazenyl)-4-isophthalic acid grafted RGO | 101 | Azo-derivative grafted carbon material | 497 | 1250 | 78 |
| 2-Sulfo-2′-hydroxy-4′-amino-azobenzene grafted on RGO | 102 | Azo-derivative grafted carbon material | 150 | 5400 | 79 |
| Ortho-tetrafluoro azobenzene grafted RGO | 103 | Azo-derivative grafted carbon material | 346 | 80 | 80 |
| Triazobenzene grafted on reduced graphene oxide | 104 | Azo-derivative grafted carbon material | 541 | 1250 | 81 |
| Phenylazene grafted on graphene oxide | 105 | Azo-derivative grafted carbon material | 864 | 10 | 82 |
| Dendritic hyperbranched azobenzene grafted on graphene | 106 | Azo-derivative grafted carbon material | 374 | 80 | 83 |
| Metal-coordinated p-azobenzenesulfonic acid grafted on reduced graphene oxide | 107 | Azo-derivative grafted carbon material | 720 | 96 | 84 |
3. Polymer templated STF materials
Azobenzene functionalized polymer-based STF materials are a promising class of STF materials with the advantage of controlling the various STF properties through the template-assisted assembly strategy.85 Contrary to the randomly oriented small molecule-based STF materials, highly ordered azobenzene moieties in the STF polymer may significantly increase the energy density with improved storage duration. These materials can offer the bulk properties of solid materials while providing molecular level free volume to enable the structural change due to the photoisomerization.38 Therefore, the intermolecular interaction inside the polymer-based STF material can be tuned with an appropriate photoisomerizable moiety and a suitable backbone structure to obtain a high-energy density STF material. Other advantages of these materials are ease of synthesis, moldability and high stability. Importantly, these materials may be deposited or developed with the desired thickness using various techniques like spin coating, electrodeposition, electrospinning polymerization, and dynamic stretching to optimize the heat-releasing behavior of the STF materials. Herein, we have discussed a few important polymer-based Azo-STF materials focusing on the role of intermolecular interactions in achieving high fuel efficiency.The photoisomerization yield of azobenzene-based polymers decreases significantly in the solid state due to limited isomerization volume inside the polymer matrixes. This reduces the storage efficiency of the polymer-based STF materials.38,72,86,87 With a rational choice of the polymer backbone and linked azo-derivative, Griffin, Görtz and co-workers significantly improved the photoisomerization yield in solid polymer-based STF materials (Fig. 12).69 The STF material was developed by functionalizing methacrylate and acrylate-based polymers with an azobenzene side group. The alkoxy linker between the polymer backbone and the azobenzene side group played an important role in enhancing the photoisomerization yield. The alkoxy linker not only separated the π–π* absorbance band between the stable and metastable state but also increased the free volume of isomerization in the solid sample to achieve ∼90% E-to-Z photoisomerization at PSS365 (Fig. 12(b)). The glass temperature (Tg) of the sample gradually decreased with increasing linker chain length (Fig. 12(b)).69,88 An increased local mobility and free isomerization volume in the alkoxy-linked azobenzene polymers are attributed to lower Tg of the polymers. The enhanced photoconversion in alkoxy-linked polymer caused an increase in the energy density by 44% as compared to acrylate polymers directly attached to the azobenzene moieties. A maximum of 143 J g−1 energy density was achieved using these STF polymers in the solid state. All the STF materials (75–80) showed a storage half-life of more than four days. It was indicated that rather than the mobility of the backbone chain, the local mobility and the free isomerization volume of the pendant azobenzene are more important for fast Z-to-E thermal back isomerization.
 | ||
| Fig. 12 (a) Molecular structures of azobenzene linked polyacrylate based STF materials (75–80). (b) Various physical properties of the STF materials in the solid state. Reproduced from ref. 69 with permission from the American Chemical Society. | ||
Grossman and co-workers functionalized azobenzene on a rigid conjugated polydiacetylene backbone to obtain a high-energy density STF material (Fig. 13).89 The polymer materials exhibited 89–173 kJ mol−1 energy density, which was more than twice the pristine azobenzene storage density (41 kJ mol−1). The X-ray diffraction study interpreted that the polymer's molecular packing occurred along the long molecular axis with a herringbone arrangement of the neighbouring azobenzene groups. Interestingly, the crystalline behavior was observed only with the E-isomer. The liquid Z-isomer turned into a solid E-isomer crystal during the discharging process. Based on the differential scanning calorimetry, X-ray diffraction and theoretical results, it was concluded that the enhanced storage energy density was attributed to the combined release of isomerization enthalpy (ΔHZ–E) and crystallization energy (ΔHl). They synthesized different polydiacetylene–azobenzene materials (Fig. 13) to obtain various energy storage densities. With increasing spacer alkyl chain length, the intermolecular interaction, the self-assembly nature and the crystallization behavior of the E-isomer were also altered, which led to a higher energy storage density. A maximum storage energy density of 239 J g−1 was reported for this hybrid-type polymer-based STF. However, one major drawback was that the system could not undergo photoisomerization in a solid state and required solvent-assisted charging. The author suggested that the photoisomerization of azobenzene in a solid state might be possible by attaching a bulky functional group to provide sufficient free volume for the photoisomerization.
 | ||
| Fig. 13 An azobenzene functionalized diacetylene-based (a) monomer and (b) polymer for STF applications. Reproduced from ref. 89 with permission from the Royal Society of Chemistry. | ||
To enhance the isomerization-free volume inside the STF polymer, Tang and coworkers developed an azobenzene–anchored polymer with intrinsic microporosity (Fig. 14).38 Polymers with intrinsic microporosity are a new class of amorphous materials that possess nano-dimensional void due to rigid and contorted polymer chains. The microporous material was developed by polymerising Tröger's base and appending azobenzene derivatives by simple quaternization of the tertiary amines (86). The quaternization process also generated cation–π interactions inside the polymer material, which was reported to enhance the energy density of STF materials.90 The microporous volume of the material was 0.02 cc g−1. The thermal back half-life of the Z-isomer inside the polymer matrix was 47 h, slightly higher than that of its monomer form (45 h). The reverse thermal isomerization was performed at 100 °C. The material showed a maximum energy density of 180 J g−1. Thermally-induced discharging achieved a 6.6 °C temperature difference using the STF material with intrinsic microporosity. It was demonstrated that the cation–π interactions, intrinsic microporosity, and template-enforced steric strain synergistically enhanced the STF energy density.
 | ||
| Fig. 14 (a) Molecular structure of an azobenzene–anchored polymer with intrinsic microporosity (86). (b) The enhanced storage density and charging/discharging process of the material are shown with a schematic diagram. Reproduced from ref. 38 with permission from the American Chemical Society. | ||
Electrodeposition of STF polymers is an excellent technique for conformal coating of large surface area with a desired polymer layer thickness.91 The electrodeposition technique usually generates a smooth surface with a uniform layer structure. This technique provides an enormous opportunity for developing a thin layer coating with nano- to micrometre thickness. Zhang, Liu and co-workers fabricated multiple layers of crosslinked 1,2-bis(4-(4-(9H-carbazol-9-yl)butoxy)phenyl)-diazene (BCz-BO-Azo) (87) on an indium tin oxide (ITO) glass surface by the electrodeposition (ED) technique to achieve a longer storage duration (Fig. 15).92 The E-to-Z photoisomerization ratio decreased in the polymer film (40%) as compared to the dissolved monomer (64%). However, the ED film showed a 7.5-fold enlarged thermal back half-life (20 h) compared to the monomer half-life in the solid state. The heat release from the charged ED film caused a ∼12 °C increase of the surface temperature. The energy density of the charged monomer was 101 J g−1. However, the energy density was not reported for the ED film. Since the crosslinked polymer film was insoluble in water, the author proposed its potential application in severe environments like rainy and snowy environments. This example indicates that a suitably crosslinked ED film may be exploited to generate STF materials with prolonged storage duration.
 | ||
| Fig. 15 Schematic diagram of electrochemical polymerization with a BCz-BO-azo monomer. Reproduced from ref. 92 with permission from John Wiley and Sons. | ||
Metin and co-workers reported the electro-polymerization of a carbazole containing cyanobenzene (Cz-Azo-CN) derivative to demonstrate 128% enhancement of storage density in polymer-STF (88, Fig. 16) compared to its monomer.70 With this rational choice of the conjugated carbazole backbone, they obtained 72% E-to-Z isomerization yield as compared to 64% isomerization yield in monomer solution in toluene. The thermal back half-life of the polymer film was 64 min, which was higher compared to the monomer. The energy density of the polymer film was 145 J g−1. The author interpreted that the polycarbazole matrix obtained by the electrochemical method provided a closely packed arrangement between the cyano-azobenzene moieties, leaving sufficient free volume for isomerization and ultimately increasing the storage capacity.
 | ||
| Fig. 16 Representation of E and Z- forms of the p(Cz-Azo-CN) polymer (88) film and their corresponding color. Reproduced from ref. 70 with permission from Elsevier. | ||
Venkataraman and co-workers developed poly(methacrylate) polymer scaffolds with pendent azobenzene to achieve photothermal batteries with a very high energy density of 510 ± 115 J g−1 (Fig. 17).71 The syndiotactic fabrication of the high density of functional azobenzene group on poly(methacrylate) polymers (89) critically controlled the intermolecular interactions between the neighbouring azobenzene groups and within the neighbouring polymer chains. Since it was difficult to charge the material in the solid state, the polymer STF material was charged in tetrahydrofuran (THF) or dichloromethane (DCM) medium. The dried charged-sample developed from THF solvent had a higher energy density (510 ± 115 J g−1) than the sample dried from DCM (110 ± 25 J g−1) (Fig. 17(b)). The charged sample prepared from various THF/DCM ratios showed that the energy density increased with increasing THF percentage (Fig. 17(c)). The thermal back half-life of the charged AzoPM film fabricated from THF and DMC was 75.5 and 48.9 h, respectively. These physical behaviours indicated structural differences between the films fabricated from THF and DCM. Wide-angle X-ray scattering (WAXS) indicated an interpolymer packing distance of ∼16–21 Å, and a d-spacing of ∼5 Å between two neighbouring syn-azobenzene moieties in the syndiotactic triad (Fig. 17(d) and (e)). A more compact intermolecular packing and intercalation of azobenzene units were observed using polymer films fabricated from DCM than those fabricated from THF. It was interpreted from the polymer–solvent interaction parameter that DCM preferentially interacted with the polymer backbone over the pendent azobenzene moieties, which led to the aggregation of the pendent azobenzene unit. On the other hand, THF solvated the Z-isomer better, and it did not have a strong interaction with the polymer backbone. Significant π–π interactions between the neighbouring E-isomers occurred upon discharging because of the particular packing of Z-rich 89 in THF. It led to nearly three times larger heat release than isolated azobenzene. Their work revealed the important role of specific molecular orientation in modulating intermolecular interaction and developing high-energy density STF materials.
 | ||
| Fig. 17 (a) Chemical structure of AzoPMA polymer 89. (b) DSC exotherm of the first heating cycle with Z-89 dried from THF and DCM. (c) Full width at half maximum (FWHM, unit °C) and energy density obtained for the exotherm peak of Z-89 samples dried from various THF/DCM solvent mixtures. (d) WAXS of solid Z-89 films dried from DCM vs. THF. (e) The interpolymer packing distance ∼15–21 Å, and a d-spacing of ∼5 Å between two neighbouring syn-azobenzene moieties in the syndiotactic triad. Reproduced from ref. 71 with permission from Springer Nature. | ||
Azobenzene was grafted to the polynorbornene template via a flexible spacer link to increase the free volume of isomerization inside the polymer matrix and to develop a stretchable STF film (90, Fig. 18).72 The material showed a high degree of photo-charging (72%) with 365 nm light irradiation. The fast-relaxing azo dye exhibited a comparatively slower and controlled heat release in the polymer matrix due to enhanced intermolecular interaction and steric hindrance. Stretching of the polymer film allowed a higher degree of photoisomerization (85%) than the normal film. It was suggested that stretching of the polymer film not only enhanced the film transparency and the possibility of charging at the back surface of the film due to the partial orientation of the random entangled chain along the stretching direction but also increased free volume between the chains. The rate of thermal back isomerization increased in the stretched film compared to that in the normal film. A maximum energy density of 176 J g−1 was obtained with this STF material. The photo-discharging process caused a temperature increase of 1.5 °C. The result demonstrated that intermolecular interactions and steric hindrance inside the polymer film play an important role in balancing the isomerization yield and fast discharging of the polymer-based STF film.
 | ||
| Fig. 18 (a) Molecular structure of azobenzne grafted polynorbornene (90) and the optical image of the film before stretching, rolling into a cylinder and after stretching. (b) Heat-release performance under blue-light irradiation at room temperature of (b) the charged stretched film, (c) charged film without stretching and (d) uncharged film without stretching. Reproduced from ref. 72 with permission from the American Chemical Society. | ||
Compared to linear polymers, dendrimers are highly integrated branched macromolecular nanostructures with the ability to undergo surface functionalization. Integrating azo-derivatives on the dendrimer surface is an efficient technique for developing STF materials with high storage density. Yu, Wang and co-workers developed photochromic dendrimers by grafting azobenzene acrylate on a poly-(amidoamine) dendrimer (91–93, Fig. 19).73 These materials showed solid-to-liquid phase transition upon E-to-Z photoisomerization. The phase-changing behaviour originated from the different intermolecular interactions between the azo-isomers on the dendrimer surfaces. The energy density corresponding to the dendrimer-based STF materials was higher than that of the pristine azo-derivative. The isomerization enthalpy and the phase-changing energy were released together during the Z-to-E isomerization to yield a higher energy density. The energy density was gradually increased in higher-generation dendrimers. A maximum energy density of 214 J g−1 was reported with STF-93. The storage half-life of the polymer film was increased from 18.3 h in STF-91 to 32.6 h in STF-93. This indicates higher-order intermolecular interactions between the photoisomers with increasing dendrimer generations. Later, the same group developed an arylazopyrazole-based dendrimer for STF applications to obtain longer storage durations.93 However, no photochemical phase change was observed for arylazopyrazole-based dendrimers. The novel strategy may be applied to develop high-energy STF materials with a rational choice of photochromic molecules.
 | ||
| Fig. 19 Schematic diagram of synthesizing photochromic dendrimers (91–93) for STF applications. Reproduced from ref. 73 with permission from the American Chemical Society. | ||
To achieve better STF efficiency, azobenzene-linked dendrimers were further deposited on a polydopamine-coated cotton fabric.74,94ortho-Substituted tetrafluoro and fluorochloro azobenzene were grafted on a poly-(amidoamine) dendrimer and deposited on poly-dopamine coated cotton fabric to obtain wearable STF materials 94 and 95, respectively (Fig. 20). The materials showed only 27% E-to-Z isomerization in the film state at PSS520. However, in solution, the azobenzene-linked dendrimer materials exhibited approximately 70% E-to-Z isomerization at PSS520. An efficient back isomerization (95%) could be achieved with 420 nm light irradiation. The dendrimer-based STF materials showed a long storage half-life (t1/2), 28.5 and 20.6 days, respectively, for STF-94 and STF-95. The Z-rich STF material generated with 520 nm light irradiation showed a storage density of 460 J g−1. The azo-derivative grafted dendrimer on polydopamine-coated fabric materials showed a higher storage energy density than the conventional deposition of the free-photoswitches on the polydopamine-coated fabric. The strong interaction and steric hindrance between the azo-derivatives on the dendrimer resulted in higher storage density of the STF materials. The polydopamine coating on cotton fabric also enhanced the storage efficiency by rendering intermolecular interactions (van der Waals interaction) between the polydopamine amine and the azo-derivatives. The glass transition temperature (Tg) of the Z-rich state of STF-94 and STF-95 was −3 and 0 °C, respectively. The low Tg of the Z-rich state made these materials suitable for low-temperature STF applications in solvent-free environments. STF-94 showed an approximately 12 °C temperature increase with 420 nm light irradiation. The STF materials were successfully used for low-temperature warming and de-icing with blue light irradiation.
 | ||
| Fig. 20 Bidirectional visible light switchable, wearable STF materials composed of a tetra-o-halo substituted azobenzene-containing poly(amidoamine) dendrimer (PAMAM), polydopamine (PDA) and cotton fabric (94 and 95). The materials efficiently work as solar thermal fuels at low temperatures and room temperature. Reproduced from ref. 74 and 94 with permission from the Royal Society of Chemistry and Elsevier, respectively. | ||
Yu and coworkers reported a microphase separation and nanoconfinement technique to enhance the STF efficiency of an azobenzene-containing liquid crystalline block copolymer (PEO-b-PAzo).75 The self-assembly of PEO-b-PAzo generated a continuous mesogenic phase with a liquid-crystalline block copolymer containing azobenzene and a separated phase with poly(ethylene oxide) (5 kD) (Fig. 21). The liquid-crystalline block copolymer containing azobenzene showed the energy release by Z-to-E isomerization and liquid crystalline to isotropic phase transition. A blending of PEO-b-PAzo with a small molecular weight of poly(ethylene oxide) (s-PEO) generated a composite (96) of s-PEO and PEO-b-PAzo where s-PEO selectively dispersed into the PEO nano-domain without alteration of the micro-phase separated morphology. The crystalline temperature of the composite material was reduced to −38 °C compared to the crystalline temperature of s-PEO, 24 °C. The composite material (90% s-PEO) displayed 95% E-to-Z photoisomerization and a concomitant liquid crystalline to isotropic phase transition with 365 nm light irradiation. A subsequent 460 nm light irradiation induced 69% Z-to-E back isomerization. The composite material stored the light energy and the ambient heat energy in the form of isomerization enthalpy (ΔHIso), phase-changing enthalpy (ΔHAzo) of the liquid-crystalline block copolymer containing azobenzene and crystallization enthalpy of s-PEO (ΔHs-PEO) and PEO-block (ΔHPEO). Also, the s-PEO material in the composite facilitated more azo moieties to assemble in the liquid crystalline phase. A maximum energy density of 210.25 J g−1 was achieved with the composite material. The composite was applied to the fabric to obtain a flexible STF material, which showed a temperature increase from 32.2 °C to 43.6 °C upon blue light irradiation.
 | ||
| Fig. 21 (a) Molecular structure of a liquid crystalline block copolymer (PEO-b-PAzo). (b) Illustration of UV irradiation, microphase separation and assembled structure with and without sPEO. (c) Solar thermal energy storage using a composite (96). Contribution of various parameters to the total storage energy density (ΔHTotal). (d) IR thermal images of sPEO and PEO-b-PAzo composites (90:10) within cloth on the wrist under blue light irradiation. Reproduced from ref. 75 with permission from the American Chemical Society. | ||
Polymer-based STF materials with metal ion-controlled dynamic bonds were reported to harvest the isomerization energy and the bond enthalpy for high energy output.76 Sulfonate azobenzene grafted on polyacrylic acid was treated with various metal ions (Mg2+, Ca2+, Ni2+, Zn2+, Cu2+, and Fe3+) to generate dynamic metal–ligand bonds (97, Fig. 22). The photoisomerization of the azobenzene moiety reversibly controlled the formation and dissociation of the dynamic bond. An efficient dynamic bond with the E-isomer enhanced the energy gap between the E and Z-isomers. UV irradiation dissociated the dynamic bond and caused E-to-Z photoisomerization. Despite dynamic bonding, E-97(Mg2+) showed 86% E-to-Z photoisomerization with UV light irradiation. The metastable Z-97(Mg2+) simultaneously stored the isomerization enthalpy and the bond enthalpy. Blue light irradiation caused Z-to-E photoisomerization and released high isomerization energy. The charged materials exhibited a thermal back half-life of 108 h. The energy density of the STF material was 408.6 J g−1, which was two times higher than the polymer without dynamic bond cross-linking. The charged STF material on nylon fabric efficiently functioned as a low-temperature heat-releasing material and showed a temperature increase of 7.7–12 °C at cold temperatures upon blue light irradiation.
 | ||
| Fig. 22 (a) Chemical structure and schematic illustration of polymer-based STF materials with metal ion-controlled dynamic bonds (97) reversibly switch with UV and blue light irradiation. The formation of the M–O dynamic bond with various metal ions is shown at the bottom right. Reproduced from ref. 76 with permission from John Wiley and Sons. | ||
An increased functional density is a major advantage of polymer-based STF materials. However, due to the reduced isomerization volume inside the polymer matrix, the photoisomerization yield decreases. This mainly limits the energy storage density of polymer-based STF materials. Suitable polymerization techniques with a rational choice of monomer and crosslinker may be employed to achieve an orderly assembled polymer structure with sufficient isomerization volume to yield efficient STF materials (Table 1).
4. Nanocarbon material templated STF materials
Light-weight carbon-based nano-templates provide a high surface-to-volume ratio for functionalizing photoactive molecules with a high packing density.95 The surface functionalization also allows an ordered arrangement of the azo-derivatives to facilitate the intra- and intermolecular interaction to tune the STF efficiency.96 Various nano-templates like carbon nanotubes and graphene oxide were covalently linked with different azo-derivatives with suitable linker moieties to develop efficient STF materials by controlling the molecular packing interactions.95 Kolpak, Grossman and co-workers demonstrated the templated assembly of photoswitches to obtain high energy density STF materials.96,97 With in silico experiments, it was established that the templating of the photoactive materials facilitates a crystalline, closed-pack ordered arrangement of the photoactive material.96 The Z-to-E isomerization activation energy (Ea) and the isomerization enthalpy (ΔHZ–E) were enhanced simultaneously by controlling the orientation, symmetry and intermolecular spacing of neighbouring azo-derivatives on the nano-template (Fig. 23(a)). A very high volumetric energy density (2484 J g−1) along with an enhanced storage duration was predicted with azobenzene-derivative functionalized carbon nanotubes.96 Later, the group developed azobenzene-functionalized single-walled carbon nanotubes (Azo-SWCNTs, 98) to demonstrate the inter- and intra-molecular interactions between the azobenzene molecules in the templated system (Fig. 23(b)–(d)).97 The rigid system in the solid state allowed bundling between the azobenzene moieties of neighbouring Azo-SWCNTs. This bundling nature was absent in diluted suspension in acetonitrile. The assembly nature increased the activation energy for the thermal back isomerization in the solid state. The energy storage efficiency of the STF system was 200 J g−1. The stored energy per molecule increased from 58 kJ mol−1 to 120 kJ mol−1 using templated azobenzene assembly. Though the Azo-SWCNT absorbs light in the UV region, it was proposed that the effect could be minimized by using a high azobenzene functional density or by disrupting the conjugation corresponding to the electronic absorption. Later, different azo-derivatives were templated on carbon nanotubes to obtain more efficient STF materials.98,99 | ||
| Fig. 23 (a) A space-filling model of 2,2′,5′-trihydroxy azobenzene linked carbon nanotubes. White, grey, blue, and red spheres represent H, C, N, and O atoms, respectively; nanotube carbons are shown with lighter grey. Various physical parameters like Z-to-E isomerization activation energy (Ea) and isomerization enthalpy (ΔH) were calculated for 2,2′,5′-trihydroxy azobenzene linked carbon nanotubes. The inset shows the corresponding parameters for untethered azobenzene molecules. (b) Photochemical/thermal isomerization in Azo-SWCNT-98. (c) Non-interacting Azo-SWCNT-98 in suspension. (d) Interacting Azo-SWCNT-98 bundle in the solid state. Reproduced from ref. 96,97 with permission from American Chemical Society and Springer Nature, respectively. | ||
Feng et al. reported a covalently attached azobenzene derivative on reduced graphene oxide (RGO) to demonstrate the role of molecular packing and intermolecular hydrogen bonding in designing high energy density STF materials (Fig. 24).77,79 Using a triple iterating functionalization technique, one azo-derivative was covalently supported with ∼16–19 carbon atoms in Azo-RGO STF material. The orderly distributed microstructure of the azobenzene immobilized reduced graphene sheet was confirmed using the transmission electron microscope image and powder X-ray diffraction pattern. The high packing density not only allowed intermolecular interaction between two azo-moieties attached to the same graphene oxide sheet but also favoured interlayer interactions (Fig. 24(b)). The charged Azo-RGO STF-100 showed a thermal back half-life of 33 days, which was several folds higher than the parent azo-compounds. The intermolecular hydrogen bond between the Z-isomers inside the densely packed structure is attributed to the extended storage duration of the STF material. The energy density of Azo-RGO STF-100 was 403 J g−1, which was almost twofold higher than that of the parent molecule.56 The intermolecular hydrogen bond and proximity-induced interactions in the bundled Azo-RGO STF material resulted in a significant difference in the isomerization enthalpy (ΔHZ–E). The same group also grafted substituted azo-derivatives to increase the number of intermolecular hydrogen bonds in Azo-RGO STF material.78,81 (4-((3,5-dimethoxyaniline)-diazenyl)-4-isophthalic acid (101) grafted RGO showed an enhanced gravimetric energy density of 497 J g−1 and a long storage half-life of 52 days.56,78 The intermolecular hydrogen bond and steric configuration between the photoisomers were favoured due to the high grafting density (azo-to-carbon ratio: 1:17) on the nanosheet. ortho-Substituted azo-derivatives (102) capable of forming intermolecular hydrogen bonds were reported to enhance the Z-isomer's stability and to increase the storage half-life up to 225 days.79Ortho-tetrafluoro azobenzene (103) was grafted onto reduced graphene oxide to obtain a visible light-driven STF material using a similar strategy.80 A similar correlation between the high grafting density and energy density was demonstrated based on enhanced intermolecular interactions. The STF material exhibited a storage density of 346 J g−1 and a power density of 2401 W kg−1.
 | ||
| Fig. 24 (a) Chemical structure of a covalently linked azo-derivative onto a reduced graphene oxide surface. (b) Bundling of 100 in the solid state. Reproduced from ref. 77 with permission from the Royal Society of Chemistry. (c) Molecular structures of different azo-derivatives grafted on RGO to efficiently tune the intermolecular interactions. | ||
A tri-azobenzene grafted RGO material was developed as a highly efficient STF material to demonstrate thermochromic display applications (104, Fig. 25).81 The degree of isomerization and the rate of isomerization, two crucial factors for high energy density and power density, were altered by controlling the grafting density. This approach modulated the intermolecular interactions between the azo moieties. Rather than having a maximum grafting density, a high isomerization degree and good cycling ability were achieved by losing a certain degree of grafting density (Fig. 25(b)). A lower grafting density than the maximum also lowered the heat loss. The heat release rate measured under different stimuli revealed that a high heating rate led to fast heat release over a narrow temperature range. Also, an efficient heat release with blue light irradiation was observed even at 5 °C. The material displayed a high energy density (541 J g−1), a long storage duration (t1/2 = 1250 h) and a high power density (3036.9 W kg−1). This high energy density was generated from the high ΔH (stabilizing E-tri-Azo on RGO by H-bonds) and a relatively high isomerization degree (72.6% for E-to-Z and 87.9% for Z-to-E). The film was applied to induce a reversible color change of a thermochromic-pattern display by releasing heat to increase the temperature by 6–7 °C.
 | ||
| Fig. 25 Representative hybrid structure of triazobenzene grafted RGO (104). (b) DSC first heating traces of tri-Azo/rGO with different grafting densities (tri-azo unit to carbon ratio 1/60, 1/68, 1/90, and 1/120). Maximum energy density was obtained with 1:68 grafting density. Reproduced from ref. 81 with permission from the Royal Society of Chemistry. | ||
The Pang group reported a novel azobenzene grafted graphene oxide-based STF material in which graphene was part of the photochrome as well as the template material (Fig. 26).82 This technique simultaneously offered reduced molecular weight and high grafting density. A high functional density (1/16, one azo-moiety for every sixteen carbon atoms of graphene oxide) with an average neighbouring distance between two azo-moieties ∼3.76 Å was achieved by direct addition of the diazonium salt onto the graphene oxide surface (105). The STF material exhibited ∼69% E-to-Z conversion in a UV-photostationary state in the dispersed medium. The STF material showed a very high storage energy density of 864 J g−1 with a storage half-life of 10 h.56 The dense grafting of the azo-material facilitated the intermolecular interactions between the azo-derivative to yield a very high energy density. The author indicated that an enhanced CH–π interaction in the Z-rich photo-stationary state increased the storage half-life of the material.
 | ||
| Fig. 26 Representative chromophore/graphene oxide hybrid structure (105). Graphene oxide is both part of the chromophore and the template. Reproduced from ref. 82 with permission from Springer Nature. | ||
Yu, Wang and co-workers grafted azobenzene on graphene with layer-by-layer dendritic hyperbranched structures (106) to achieve STF material with high storage performance (Fig. 27).83 The azobenzene grafting density was gradually increased with dendritic amplification. The energy density of the STF material with one, two and three-layer hyperbranched structures was 234, 291 and 374 J g−1. The storage half-life of the azobenzene–graphene hybrid was increased to 80 h due to the dendritic amplification. The increased energy density and the storage half-life of the STF materials with increasing hyperbranched layer structure were attributed to the increased intermolecular interactions and steric hindrance between the neighbouring azobenzenes on the graphene surface. These materials showed a moderate power density of 186–390 W kg−1 with good cycling performance.
 | ||
| Fig. 27 Azobenzene–graphene hybrids with hyperbranched structures (106). | ||
Sulphonate-substituted azobenzene covalently grafted on reduced graphene oxide (G-Azo) was coordinated with metal ions to harvest the isomerization enthalpy and bond energy for high energy density STF materials (G-Azo-M, Fig. 28).84 In the presence of magnesium ions, the Mg–O coordinate bond was formed between two E-isomers, which resulted in the crosslinked self-assembled layered structure. The coordination bond stabilized the E-structure. The coordination bond was dissociated upon E-to-Z photoisomerization. The optically induced formation and dissociation of coordination bonds enlarged the molecular energy gap by lowering the energy level of the E-isomer and increasing that of the Z-isomer. The total enthalpy (ΔE) is the sum of photothermal enthalpy (ΔEp, the original energy gap) and the bond enthalpy (ΔEp) due to the metal coordination (Fig. 28(b)). The coordination bond also altered the arrangement of steric interaction. The E-to-Z photoisomerization yield decreased slightly from 75% to 73%. The thermal back isomerization rate was increased in the coordinated system. The thermal back half-life of STF-107 was 96 h. The reversible photoisomerization in the solid state was effective in both directions. The maximum energy density obtained with this STF material was 720 J g−1, 85% higher than that of the no-metal coordinated STF material. The temperature of the G-Azo-M film was increased from 25 to 40 °C upon UV-light irradiation (Fig. 28(c)).
 | ||
| Fig. 28 (a) Schematic illustration of reversible photo-isomerization of the G-Azo-Mg (107) film in the solid state with the formation and dissociation of coordination bonds. (b) Schematic energy diagram of G-Azo tuned by coordination bonds. (c) IR-thermal image of time-evolved temperature release at the centre of the G-Azo-M film upon blue light irradiation. Reproduced from ref. 84 with permission from Elsevier. | ||
Nanocarbon-based STF materials yield increased storage density and long storage half-life (Table 1). Therefore, these STF materials are highly suitable for large-scale applications. The high functional density of the photochromic material and an ordered grafting technology contribute to the high efficiency of these STF materials. A further improvement in the STF efficiency may be possible with increasing noncovalent interactions between the photochromic materials and the tuned spacer length between the carbon surface and the isomerizing unit.
5. Conclusions and perspectives
In summary, we discussed the recent progress in azo-based STF materials developed using photochemical phase-changing materials and template-based azo-derivatives. The molecular structure of the photochromic materials and their assembly nature play an important role in tuning the energy density, storage half-life and photoisomerization yield of the STF materials. The assembly of the azo-molecules can be effectively tuned with a suitable molecular design to modulate the efficiency of the STF material. Regarding the structural design of PCM STF materials, long alkyl chains are mostly used to control the phase behaviours. However, a suitable chemical design of the photochromic material along with a small to moderate terminal alkyl chain for controlling the assembled structure is highly beneficial because of low molecular mass, which may significantly improve the gravimetric energy density. Additionally, these materials allow intense light penetration to achieve multi-layer illumination. Template-based STF materials usually have a higher energy density than single-molecule-based STF materials due to the suitable orientation of the azo-derivatives in space to accomplish effective intermolecular interaction. The grafting density is also highly tunable with various chemical methodologies. The low isomerization ratio inside the polymer matrix strongly limits polymer-based STF materials’ efficiency. With a suitable choice of monomer unit, crosslinker and, most importantly, an appropriate polymerization technique, a photochromic material can be grafted densely within the polymer matrix without compromising the required isomerization volume. The energy density and storage half-life of carbon-nano-material-based STF materials are much better than those of other techniques because of the crystalline ordered structure and high grafting density in these materials. However, a significant improvement can be made with a suitable structural design of the photochromic compound and an effective molecular assembly.Although significant progress has been made in developing efficient STF materials, large-scale industrial implementation of STF materials is inadequate. Simultaneously achieving high energy density, long storage half-life, and good cyclability with high efficiency in absorbing the solar spectrum is still challenging in the STF field and is not cost-effective in the present scenario. Parallelly, solvent-free STF materials with highly flexible behaviour are desirable for practical applications. The light penetration depth of the STF materials is another critical challenge for diverse applications. Low light penetration results in slow and inefficient charging of STF materials in the solid state. Generally, the solid-state STF materials release a small amount of the heat induced by a high temperature or reveal a slow heat release upon light irradiation. Suitable techniques should be developed to achieve a fast, on-demand heat release with minimum leakage to the surroundings. The STF materials also suffer from inefficient functioning at low or ultralow temperatures. The heat-releasing efficiency of the STF material is significantly truncated under ultralow temperature conditions. All these factors limit the implementation of large-scale STF devices. New structural design of the photochromic materials, advanced grafting technology, and suitable coating technology are necessary for developing next-generation STF materials without the present shortcomings.
Data availability
The data can be assessed directly from the references cited in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SS acknowledges Prof. Dr Nobuyuki Tamaoki, Hokkaido University, for his valuable suggestions and comments. SKB thanks Dr Manas Ranjan Satapathy, Principal Model Degree College, Nayagarh, for his valuable suggestions.Notes and references
- T. Z. Ang, M. Salem, M. Kamarol, H. S. Das, M. A. Nazari and N. Prabaharan, Energy Strateg. Rev., 2022, 43, 100939 CrossRef
.
- World Energy Council (2013) World Energy Resources: Solar, https://www.worldenergy.org/wp-content/uploads/2013/10/WER_2013_8_Solar_revised.pdf.
- W. Feng, S. W. Feng, L. Dong, Y. Feng, L. Wang, X. Duan and P. Jingkun, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC
- Z. Wang, H. Hölzel and K. Moth-Poulsen, Chem. Soc. Rev., 2022, 51, 7313–7326 RSC
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS
- B. Zhang, Y. Feng and W. Feng, Nano-Micro Lett., 2022, 14, 1–37 CrossRef PubMed
- R. Luther and F. Weigert, Z. Phys. Chem., 1905, 51U, 297–328 CrossRef
- N. Abas, A. Kalair and N. Khan, Futures, 2015, 69, 31–49 CrossRef
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef
- C. Bastianelli, V. Caia, G. Cum, R. Gallo and V. Mancini, J. Chem. Soc., Perkin Trans. 2, 1991, 679–683 RSC
- V. Caia, G. Cum, R. Gallo, V. Mancini and E. Pitoni, Tetrahedron Lett., 1983, 24, 3903–3904 CrossRef CAS
- M. Brøndsted Nielsen, N. Ree, K. V. Mikkelsen and M. Cacciarini, Russ. Chem. Rev., 2020, 89, 573–586 CrossRef
- M. Cacciarini, A. B. Skov, M. Jevric, A. S. Hansen, J. Elm, H. G. Kjaergaard, K. V. Mikkelsen and M. Brøndsted Nielsen, Chem. – Eur. J., 2015, 21, 7454–7461 CrossRef CAS PubMed
- E. M. Arpa and B. Durbeej, Chem.: Methods, 2023, 3, e202200060 CAS
- Z. Wang, J. Udmark, K. Börjesson, R. Rodrigues, A. Roffey, M. Abrahamsson, M. B. Nielsen and K. Moth-Poulsen, ChemSusChem, 2017, 10, 3049–3055 CrossRef CAS PubMed
- Z. i Yoshida, J. Photochem., 1985, 29, 27–40 CrossRef CAS
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed
- M. Mansø, A. U. Petersen, Z. Wang, P. Erhart, M. B. Nielsen and K. Moth-Poulsen, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed
- R. Schulte, S. Afflerbach, T. Paululat and H. Ihmels, Angew. Chem., Int. Ed., 2023, 62, e202309544 CrossRef CAS PubMed
- R. J. Salthouse and K. Moth-Poulsen, J. Mater. Chem. A, 2024, 12, 3180–3208 RSC
- Z. Wang, P. Erhart, T. Li, Z. Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Joule, 2021, 5, 3116–3136 CrossRef CAS
- I. I. O. F. Walls, J. Padilla, P. Joseph-Nathan, F. Gird, J. Romo and B. Hermann Rau, Angew. Chem., Int. Ed. Engl., 1973, 12, 224–235 CrossRef
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 CrossRef CAS PubMed
- S. Axelrod, E. Shakhnovich and R. Gómez-Bombarelli, ACS Cent. Sci., 2022, 31, 41 Search PubMed
- G. L. Hallett-Tapley, C. D’alfonso, N. L. Pacioni, C. D. McTiernan, M. González-Béjar, O. Lanzalunga, E. I. Alarcon and J. C. Scaiano, Chem. Commun., 2013, 49, 10073–10075 RSC
- S. Simoncelli and P. F. Aramendía, Catal. Sci. Technol., 2015, 5, 2110–2116 RSC
- E. Titov, L. Lysyakova, N. Lomadze, A. V. Kabashin, P. Saalfrank and S. Santer, J. Phys. Chem. C, 2015, 119, 17369–17377 CrossRef CAS
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS PubMed
- R. J. Corruccini and E. C. Gilbert, J. Am. Chem. Soc., 1939, 61, 2925–2927 CrossRef CAS
- M. J. Hansen, M. M. Lerch, W. Szymanski, B. L. Feringa, M. J. Hansen, M. M. Lerch, W. Szymanski and B. L. F. Eringa, Angew. Chem., Int. Ed., 2016, 55, 13514–13518 CrossRef CAS PubMed
- T. Dang, D. Dong, J. Zhang, Y. He, Z. Y. Zhang and T. Li, Angew. Chem., Int. Ed., 2023, 62, e202301992 CrossRef CAS PubMed
- R. Lin, P. K. Hashim, S. Sahu, A. S. Amrutha, N. M. Cheruthu, S. Thazhathethil, K. Takahashi, T. Nakamura, T. Kikukawa and N. Tamaoki, J. Am. Chem. Soc., 2023, 145, 9072–9080 CrossRef CAS PubMed
- S. A. M. Steinmüller, M. Odaybat, G. Galli, D. Prischich, M. J. Fuchter and M. Decker, Chem. Sci., 2024, 15, 5360–5367 RSC
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed
- P. K. Hashim, S. Sahu, K. Takahashi, S. Thazhathethil, T. Nakamura and N. Tamaoki, Chem. – Eur. J., 2024, 30, e202400047 CrossRef CAS PubMed
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed
- J. Hu, S. Huang, M. Yu and H. Yu, Adv. Energy Mater., 2019, 9, 1901363 CrossRef CAS
- H. Ren, H. Chen, M. Lu, X. Cui, H. Wang, L. Wang and Q. Tang, ACS Appl. Polym. Mater., 2023, 5, 5592–5599 CrossRef CAS
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Nat. Rev. Chem., 2021, 6, 51–69 CrossRef PubMed
- A. Y. Hao, X. Q. Wang, Y. Z. Mei, J. F. Nie, Y. Q. Yang and C. C. Dai, Spectrochim. Acta, Part A, 2021, 249, 119310 CrossRef CAS PubMed
- A. Kunz, A. H. Heindl, A. Dreos, Z. Wang, K. Moth-Poulsen, J. Becker, H. A. Wegner, A. H. Heindl, H. A. Wegner, A. Dreos, Z. Wang, K. Moth-Poulsen and J. Becker, ChemPlusChem, 2019, 84, 1145–1148 CrossRef CAS PubMed
- W. C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS PubMed
- Q. Qiu, Y. Shi and G. G. D. Han, J. Mater. Chem. C, 2021, 9, 11444–11463 RSC
- K. Wang, H. Yu, J. Gao, Y. Feng and W. Feng, J. Mater. Chem. C, 2024, 12, 3811–3837 RSC
- L. Schweighauser, M. A. Strauss, S. Bellotto and H. A. Wegner, Angew. Chem., Int. Ed., 2015, 54, 13436–13439 CrossRef CAS PubMed
- Q. Qiu, M. A. Gerkman, Y. Shi and G. G. D. Han, Chem. Commun., 2021, 57, 9458–9461 RSC
- A. Gonzalez, M. Odaybat, M. Le, J. L. Greenfield, A. J. P. White, X. Li, M. J. Fuchter and G. G. D. Han, J. Am. Chem. Soc., 2022, 144, 19430–19436 CrossRef CAS PubMed
- H. Akiyama, M. Yoshida, H. Akiyama and M. Yoshida, Adv. Mater., 2012, 24, 2353–2356 CrossRef CAS PubMed
- Y. Norikane, E. Uchida, S. Tanaka, K. Fujiwara, E. Koyama, R. Azumi, H. Akiyama, H. Kihara and M. Yoshida, Org. Lett., 2014, 16, 5012–5015 CrossRef CAS PubMed
- Y. Okui and M. Han, Chem. Commun., 2012, 48, 11763–11765 RSC
- E. Uchida, K. Sakaki, Y. Nakamura, R. Azumi, Y. Hirai, H. Akiyama, M. Yoshida and Y. Norikane, Chem. – Eur. J., 2013, 19, 17391–17397 CrossRef CAS PubMed
- M. Hoshino, E. Uchida, Y. Norikane, R. Azumi, S. Nozawa, A. Tomita, T. Sato, S. I. Adachi and S. Y. Koshihara, J. Am. Chem. Soc., 2014, 136, 9158–9164 CrossRef CAS PubMed
- Y. Kikkawa, M. Nagasaki and Y. Norikane, Phys. Chem. Chem. Phys., 2022, 24, 29757–29764 RSC
- K. Ishiba, M. A. Morikawa, C. Chikara, T. Yamada, K. Iwase, M. Kawakita and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 1532–1536 CrossRef CAS PubMed
- Unit conversion was performed by using either of the following equations. a) 1 kJ mol−1 = 1000/molecular − weight(g) J g−1, and b) 1 W h kg−1 = 3.6 J g−1.
- J. L. Greenfield, M. A. Gerkman, R. S. L. Gibson, G. G. D. Han and M. J. Fuchter, J. Am. Chem. Soc., 2021, 143, 15250–15257 CrossRef CAS PubMed
- Q. Yang, J. Ge, M. Qin, H. Wang, X. Yang, X. Zhou, B. Zhang, Y. Feng and W. Feng, Sci. China Mater., 2023, 66, 3609–3620 CrossRef CAS
- H. Liu, Y. Feng and W. Feng, Compos. Commun., 2020, 21, 100402 CrossRef
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS PubMed
- Y. Shi, M. A. Gerkman, Q. Qiu, S. Zhang and G. G. D. Han, J. Mater. Chem. A, 2021, 9, 9798–9808 RSC
- G. G. D. Han, H. Li and J. C. Grossman, Nat. Commun., 2017, 8, 1–10 CrossRef CAS PubMed
- D. Kwaria, K. McGehee, S. Liu, Y. Kikkawa, S. Ito and Y. Norikane, ACS Appl. Opt. Mater., 2023, 1, 633–639 CrossRef CAS
- M. Gupta and Ashy, Adv. Energy Mater., 2024, 14, 2303845 CrossRef CAS
- Z. Y. Zhang, Y. He, Z. Wang, J. Xu, M. Xie, P. Tao, D. Ji, K. Moth-Poulsen and T. Li, J. Am. Chem. Soc., 2020, 142, 12256–12264 CrossRef CAS PubMed
- S. Wu, Z. Y. Zhang, T. Li, R. Wang and T. Li, ACS Mater. Lett., 2023, 5, 2019–2027 CrossRef CAS
- M. A. Gerkman, R. S. L. Gibson, J. Calbo, Y. Shi, M. J. Fuchter and G. G. D. Han, J. Am. Chem. Soc., 2020, 142, 8688–8695 CrossRef PubMed
- Z. Shangguan, W. Sun, Z. Y. Zhang, D. Fang, Z. Wang, S. Wu, C. Deng, X. Huang, Y. He, R. Wang, T. T. Li, K. Moth-Poulsen and T. T. Li, Chem. Sci., 2022, 13, 6950–6958 RSC
- C. Wallace, K. Griffiths, B. L. Dale, S. Roberts, J. Parsons, J. M. Griffin and V. Görtz, ACS Appl. Mater. Interfaces, 2023, 15, 31787–31794 CrossRef CAS PubMed
- S. Sert, R. Ayranci, G. K. Çılgı and M. Ak, J. Energy Storage, 2023, 72, 108551 CrossRef
- S. P. Jeong, L. A. Renna, C. J. Boyle, H. S. Kwak, E. Harder, W. Damm and D. Venkataraman, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed
- L. Fu, J. Yang, L. Dong, H. Yu, Q. Yan, F. Zhao, F. Zhai, Y. Xu, Y. Dang, W. Hu, Y. Feng and W. Feng, Macromolecules, 2019, 52, 4222–4231 CrossRef CAS
- X. Xu, P. Zhang, B. Wu, Y. Xing, K. Shi, W. Fang, H. Yu and G. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 50135–50142 CrossRef CAS PubMed
- X. Xu, C. Li, W. Chen, J. Feng, W. Y. Li, G. Wang and H. Yu, J. Mater. Chem. A, 2024, 12, 23723–23731 RSC
- F. Cai, T. Song, B. Yang, X. Lv, L. Zhang and H. Yu, Chem. Mater., 2021, 33, 9750–9759 CrossRef CAS
- H. Wang, Y. Feng, J. Gao, W. Fang, J. Ge, X. Yang, F. Zhai, Y. Yu, W. Feng, H. Wang, Y. Feng, J. Gao, W. Fang, J. Ge, X. Yang, F. Zhai, Y. Yu and W. Feng, Adv. Sci., 2022, 9, 2201657 CrossRef CAS PubMed
- W. Luo, Y. Feng, C. Cao, M. Li, E. Liu, S. Li, C. Qin, W. Hu and W. Feng, J. Mater. Chem. A, 2015, 3, 11787–11795 RSC
- W. Luo, Y. Feng, C. Qin, M. Li, S. Li, C. Cao, P. Long, E. Liu, W. Hu, K. Yoshino and W. Feng, Nanoscale, 2015, 7, 16214–16221 RSC
- Y. Feng, H. Liu, W. Luo, E. Liu, N. Zhao, K. Yoshino and W. Feng, Sci. Rep., 2013, 3, 1–8 Search PubMed
- X. Yang, S. Li, J. Zhao, X. Wang, H. Huang and Y. Wang, Sol. Energy Mater. Sol. Cells, 2021, 231, 111330 CrossRef CAS
- W. Yang, Y. Feng, Q. Si, Q. Yan, P. Long, L. Dong, L. Fu and W. Feng, J. Mater. Chem. A, 2019, 7, 97–106 RSC
- W. Pang, J. Xue and H. Pang, Sci. Rep., 2019, 9, 1–12 CrossRef CAS PubMed
- X. Xu, B. Wu, P. Zhang, H. Yu and G. Wang, Sol. RRL, 2020, 4, 1900422 CrossRef CAS
- H. Wang, Y. Feng, H. Yu, L. Dong, F. Zhai, J. Tang, J. Ge and W. Feng, Nano Energy, 2021, 89, 106401 CrossRef CAS
- X. Xu, J. Feng, W. Y. Li, G. Wang, W. Feng and H. Yu, Prog. Polym. Sci., 2024, 149, 101782 CrossRef CAS
- D. Zhitomirsky, E. Cho and J. C. Grossman, Adv. Energy Mater., 2016, 6, 1502006 CrossRef
- C. Yang, M. Lu, H. Chen, X. Wang, H. Ren, X. Cui, H. Wang, L. Wang and Q. Tang, React. Funct. Polym., 2024, 194, 105790 CrossRef CAS
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H. J. Butt and S. Wu, Nat. Chem., 2016, 9, 145–151 CrossRef PubMed
- G. D. Han, S. S. Park, Y. Liu, D. Zhitomirsky, E. Cho, M. Dincǎ and J. C. Grossman, J. Mater. Chem. A, 2016, 4, 16157–16165 RSC
- T. Song, H. Lei, F. Cai, Y. Kang, H. Yu and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 1940–1949 CrossRef CAS PubMed
- D. Zhitomirsky and J. C. Grossman, ACS Appl. Mater. Interfaces, 2016, 8, 26319–26325 CrossRef CAS PubMed
- R. Zhao, Y. Zhang, J. Mu, C. Yu, Y. Pan, J. Zhao, Q. Wang, S. Zhang, B. Yang, F. Liu, R. Zhao, Y. Zhang, J. Mu, C. Yu, J. Zhao, Q. Wang, F. Liu, Y. Pan, S. Zhang and B. Yang, Adv. Mater. Interfaces, 2020, 7, 2001079 CrossRef CAS
- X. Xu, B. Wu, P. Zhang, Y. Xing, K. Shi, W. Fang, H. Yu and G. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 22655–22663 CrossRef CAS PubMed
- X. Xu, Y. Xing, Y. Yin, W. Fang, B. Wu, P. Bei, J. Feng, H. Yu, G. Wang and W. Y. Li, Chem. Eng. J., 2023, 466, 143175 CrossRef CAS
- G. F. Martins, B. de, P. Cardoso, N. Galamba and B. J. C. Cabral, Energy Fuels, 2023, 37, 1731–1756 CrossRef CAS
- A. M. Kolpak and J. C. Grossman, Nano Lett., 2011, 11, 3156–3162 CrossRef CAS PubMed
- T. J. Kucharski, N. Ferralis, A. M. Kolpak, J. O. Zheng, D. G. Nocera and J. C. Grossman, Nat. Chem., 2014, 6, 441–447 CrossRef CAS PubMed
- Y. Jiang, J. Huang, W. Feng, X. Zhao, T. Wang, C. Li and W. Luo, Sol. Energy Mater. Sol. Cells, 2019, 193, 198–205 CrossRef CAS
- J. Huang, Y. Jiang, J. Wang, C. Li and W. Luo, Thermochim. Acta, 2017, 657, 163–169 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |