
Improving the bioactivity and mechanical properties of poly(ethylene glycol)-based hydrogels through a supramolecular support network†
Yuzhu
Liu
a,
Md Shariful
Islam
a,
Anna
Bakker
b,
Zihao
Li
 c,
Alaa
Ajam
b,
Jamie J.
Kruzic
b and
Kristopher A.
Kilian
*ac
c,
Alaa
Ajam
b,
Jamie J.
Kruzic
b and
Kristopher A.
Kilian
*ac
aSchool of Materials Science and Engineering, University of New South Wales (UNSW Sydney), Sydney, NSW 2052, Australia. E-mail: k.kilian@unsw.edu.au
bSchool of Mechanical and Manufacturing Engineering, University of New South Wales (UNSW Sydney), Sydney, NSW 2052, Australia
cAustralian Centre for NanoMedicine, School of Chemistry, University of New South Wales (UNSW Sydney), Sydney, NSW 2052, Australia
First published on 27th December 2024
Abstract
Most synthetic hydrogels are formed through radical polymerization to yield a homogenous covalent meshwork. In contrast, natural hydrogels form through mechanisms involving both covalent assembly and supramolecular interactions. In this communication, we expand the capabilities of covalent poly(ethylene glycol) (PEG) networks through co-assembly of supramolecular peptide nanofibers. Using a peptide hydrogelator derived from the tryptophan zipper (Trpzip) motif, we demonstrate how in situ formation of nanofiber networks can tune the stiffness of PEG-based hydrogels, while also imparting shear thinning, stress relaxation, and self-healing properties. The hybrid networks show enhanced toughness and durability under tension, providing scope for use in load bearing applications. A small quantity of Trpzip peptide renders the non-adhesive PEG network adhesive, supporting adipose derived stromal cell adhesion, elongation, and growth. The integration of supramolecular networks into covalent meshworks expands the versatility of these materials, opening up new avenues for applications in biotechnology and medicine.
Introduction
Hydrogels are water–swollen polymer networks that are found in nature and can be prepared synthetically, which have proved useful for a broad spectrum of commercial and technological applications.1–3 With their diverse characteristics, hydrogels have demonstrated significant potential in applications such as drug delivery systems, scaffolds, stents, wound dressings, and sensors.4 Their ability to mimic biological structures and incorporate functional elements enables them to adapt to different tissue environments, making them invaluable for biomedical applications.5–7Hydrogel systems have been designed using both natural and synthetic materials for soft tissue engineering. Natural materials like alginate, hyaluronic acid, collagen, gelatin, chitosan, and cellulose are commonly used because they are biocompatible, degradable, and promote cell growth.8–13 On the other hand, synthetic materials such as poly(vinyl alcohol) (PVA), poly(ethylene glycol) (PEG), and their derivatives offer advantages such as reproducibility, tunability, and the ability to modify functional groups for diverse applications.14–18 However, traditional natural polymers may lack the mechanical strength needed to accurately replicate tissue environments, while synthetic polymers often lack the biological signals necessary for effective cell and tissue level processes. Furthermore, the most widely used covalently crosslinked hydrogels display purely elastic properties and are notoriously brittle, making them unsuitable for applications demanding both viscous character and toughness.
Biologically inspired hydrogels, particularly self-assembling peptide and protein-based materials, have shown promise as innovative building blocks in biomaterials design.19 Research has increasingly focused on developing supramolecular systems that mimic the nano- and micro-architecture of natural soft structures, while providing bioactivity to support cell viability, proliferation, and differentiation.20–24 Additionally, hydrogels containing bioinspired protein assemblies using recombinant DNA techniques also hold significant potential for biomedical applications. For instance, elastin-like proteins have been incorporated with synthetic polymers to create shear-thinning and self-healing hydrogels, which are being explored as injectable tools for minimally invasive surgeries.25 Similarly, bacterial collagen-like proteins have been integrated into synthetically modified hyaluronic acid hydrogels to establish bioactive matrices that mimic aspects of the tissue microenvironment.26 These hydrid synthetic and protein-based materials expand the possibilities for the tunable design of novel biomaterials that mimic natural matrices.27
Short peptide-based supramolecular systems, with their controllable amino acid sequences and inherent cytocompatibility, address many functional needs in hydrogel design, including the incorporation of adhesive and bioactive sequences.28,29 Classic examples include peptide amphiphiles,30 amyloid-derived peptides,31 collagen-imitated peptides32 and surfactant-like peptides.33 Recently we demonstrated a self-assembling short peptide hydrogel based on the tryptophan zipper motif, so-called ‘Trpzip’34 that exhibits a range of desirable properties, such as stress-relaxation, self-healing, antibacterial activity, and the ability to promote 3D cell culture and organoid propagation.34 However, despite its potential, Trpzip alone remains fragile and unstable under external forces, obviating its use in many applications.
In this communication, we describe a solution to the limitations of natural and synthetic hydrogel systems, by integrating short peptide hydrogelators with covalent PEG-based materials. Poly(ethylene glycol)dimethacrylate (PEGDM) yields a strong covalent network that provides elasticity and mechanical stability, while the Trpzip network contributes to bioactivity and facilitates stress relaxation and self-healing. This unique hybrid material supports cell adhesion and spreading, with both networks playing a role, indicating that cells detect PEGDM elasticity through engagement of the entangled nanofibrous Trpzip. These characteristics position PEGDM/Trpzip hydrogels as promising new materials for cell culture, tissue engineering and regenerative medicine applications.
The structures of PEGDM and Trpzip are shown in Fig. 1(a). Covalent PEGDM networks were formed via the reaction of methacrylate groups on PEGDM through radical polymerization (Fig. 1(b)). This process is initiated by the addition of APS and TEMED, leads to gelation within 15–20 min.35 PEGDM hydrogels can be formulated to display varying stiffness by changing the weight fraction of the monomer. Despite their popularity as synthetic biomaterials, PEG-based hydrogels are notoriously brittle, lack the desirable viscoelastic properties of natural materials, and do not support cellular processes unless modified with peptides or protein motifs. We hypothized that incorporation of Trpzip hydrogelators into the PEGDM pre-gel solution would result in a hybrid hydrogel, where PEGDM forms a covalent skeleton that provides mechanical stability, while Trpzip introduces a self-healing nanofiber network to provide bioactivity and viscous characteristics. To explore the potential of PEGDM/Trpzip hydrogels as multifunctional biomaterials, 0.5% (w/v) Trpzip was incorporated into various ratios of PEGDM (5%, 7.5%, and 10% w/v).
 | ||
| Fig. 1 Schematic and morphology of PEGDM/Trpzip hydrogels. (a) The chemical structures and components of Trpzip and PEGDM used to form the hydrogels. (b) Schematic representation of the crosslinking process of PEGDM/Trpzip hydrogels. APS and TEMED act as crosslinkers to initiate the reaction, resulting in a double network structure where PEGDM provides a covalent skeleton and Trpzip forms a self-healing network. (c) Visual comparison of gelation in glass vials for Trpzip alone (0.5%T), and PEGDM/Trpzip at different ratios (5%P + 0.5%T, 7.5%P + 0.5%T, and 10%P + 0.5%T). (d)–(i) Scanning electron microscope (SEM) images of the internal structure of hydrogels, showing porous architectures at different magnifications: (d) 0.5% (w/v) Trpzip (scale bar = 200 μm), (e), (f) 7.5% (w/v) PEGDM and 10% (w/v) PEGDM, (g) PEGDM/Trpzip 5%P + 0.5%T (w/v), (h) PEGDM/Trpzip 7.5%P + 0.5%T (w/v), (i) PEGDM/Trpzip 10%P + 0.5%T (w/v) (scale bar = 20 μm). The network structure changes with increasing PEGDM concentrations, resulting in more densely packed and interconnected pores. | ||
As shown in Fig. 1(c), all hydrogel samples containing Trpzip maintained their integrity during the inversion test after polymerization. In contrast to the 7.5% and 10% (w/v) PEGDM-only solutions, which form hydrogels within 30 minutes, the 5% (w/v) PEGDM-only solution does not form a hydrogel under the conditions tested, requiring the addition of Trpzip nanofibres to support full gelation. This is an interesting finding, suggesting that Trpzip can be used as an additive to assist gelation. Trpzip hydrogels appeared slightly cloudy due to microscale ordering; while the introduction of PEGDM improved their transparency, an important attribute for imaging and for applications in ophthalmology, such as delivery of cells36 or vitreous body replacements.37 To investigate how combining PEGDM with Trpzip might impact the network morphology, we performed scanning electron microscopy (SEM). The SEM images in Fig. 1(d)–(i) provide insights into the nano- and micro-structural properties of the hydrogels. The pure Trpzip hydrogel exhibited a sponge-like porous structure (Fig. 1(d)) while the PEGDM network displayed a loose and porous architecture (Fig. 1(e) and (f)). When Trpzip was added to PEGDM at increasing concentrations (5%, 7.5%, and 10% w/v), the resulting PEGDM/Trpzip hydrogels displayed an interconnected porous network, resembling a honeycomb configuration (Fig. 1(g)–(i)), with the PEGDM providing a uniform network with Trpzip adding nanofibrous character. This configuration became denser as PEGDM concentration increased, providing an indication of a change in architecture aligning with improved transparency. The Trpzip network appears to infiltrate the PEGDM pores, creating small threadlike structures that have the potential to provide bioactivity cues to nurture cell attachment and proliferation. This appearance is consistent with other hydrogel blends,38–41 where each network is contributing to the final architecture.
Hydrogel gelation kinetics were analyzed using shear rheology to compare the native hydrogels with the hybrid networks. Hydrogel precursors were loaded onto the rheometer, and storage (G′) and loss (G′′) modulus data were collected under varying shear forces and frequencies, as shown in Fig. 2(a). These measurements provided insights into the gelation dynamics and mechanical behavior of the hydrogels. According to the time sweep results, the pure Trpzip hydrogel required over 12 hours to reach a storage modulus of 1 kPa (Fig. 2(b)), indicating a slow gelation process. However, after introducing the PEGDM network, the gelation time for all PEGDM/Trpzip hydrogels was dramatically reduced to 15–20 min, demonstrating the dominance of radical polymerization. The storage modulus of the PEGDM/Trpzip hydrogels were tunable between 0.1 and 9 kPa depending on PEGDM concentration, with the addition of 0.5 wt% Trpzip into each formulation further increasing the modulus (Fig. 2(c)).
 | ||
| Fig. 2 Rheological analysis and gelation kinetics of PEGDM/Trpzip hydrogels. (a) Schematic representation of the rheological testing setup used to evaluate the mechanical properties of the hydrogels. (b) Time sweep showing the evolution of the storage modulus (G′) and loss modulus (G′′) of 0.5% (w/v) Trpzip over 12 h, indicating the stability and gelation dynamics of the peptide network. (c) Comparison of the storage modulus (G′) of hydrogels containing varying concentrations of PEGDM (5%, 7.5%, and 10% w/v) with and without 0.5% (w/v) Trpzip over a 30-min gelation period. (d)–(f) Strain sweep measurements of the storage (G′) and loss modulus (G′′) for hydrogels composed of 0.5% (w/v) Trpzip, 7.5% (w/v) PEGDM, and 10% (w/v) PEGDM, showing the response to increasing strain. (g)–(i) Strain sweep measurements of the storage (G′) and loss modulus (G′′) for hybrid hydrogels with 5%, 7.5%, and 10% (w/v) PEGDM combined with 0.5% (w/v) Trpzip, indicating the influence of Trpzip on the mechanical behavior and strain tolerance of the PEGDM network. All tests were performed in triplicate (n = 3). | ||
In addition to gelation time, the hydrogels’ ability to withstand shear deformation was also examined through strain sweep tests. Pure Trpzip began to deform plastically at a low strain of 0.2% (Fig. 2(d)), indicating its yield-stress fluid-like properties. In contrast, the addition of 0.5% Trpzip to 5% PEGDM significantly enhanced resistance to deformation, raising the strain threshold to approximately 1% (Fig. 2(g)). This indicates that the hybrid networks exhibit improved mechanical stability compared to Trpzip alone. Moreover, when comparing the PEGDM-only hydrogels (Fig. 2(e) and (f)) from 7.5% to 10% resulted in a 3.3-fold increase in storage modulus (0.94 kPa for 7.5% PEGDM and 3.1 kPa for 10% PEGDM). The addition of Trpzip increased the storage modulus by 2.6-fold for both 7.5% and 10% formulations, with the hybrid PEGDM/Trpzip hydrogels (Fig. 2(h) and (i)), showing the same 3.3-fold increase in storage modulus when going from 7.5% to 10% (2.43 kPa for 7.5% PEGDM/0.5% Trpzip to 8.01 kPa for 10% PEGDM/0.5% Trpzip). This result shows how Trpzip addition can increase the storage modulus within a formulation but with the same increase in modulus when changing the PEGDM content. Trpzip addition also increased the loss modulus for each formulation, indicating increased viscous character of the hybrid materials. The 5% hybrid network displayed a lower storage modulus (0.14 kPa) compared to Trpzip alone (Fig. 2(d) and (g)), suggesting formation of a softer hybrid network. Tuning both PEGDM and Trpzip content at these lower concentrations could lead to new interesting viscoelastic properties. Compared to Trpzip alone, the PEGDM/Trpzip system showed a greater ability to withstand shear deformation due to the covalent PEGDM network, enduring up to 100% strain before yielding. Frequency sweep tests showed that the PEGDM/Trpzip gels exhibited higher modulus values and a more stable change in both storage and loss modulus across a range of frequencies (ESI,† Fig. S1a–f).
The improved mechanical properties of the PEGDM/Trpzip hydrogels can be attributed to the formation of a mixed-network structure.38 PEGDM establishes a covalent network through rapid free-radical polymerization, which creates strong carbon–carbon bonds between PEGDM monomers.35 This high-density crosslinking reinforces the hydrogel, resulting in a higher storage modulus. Meanwhile, the Trpzip network forms through non-covalent interactions, such as hydrogen bonding and hydrophobic interactions, contributing to secondary network interactions.30,42 Though slower to assemble on its own, the Trpzip's network is stabilized and supported by the covalent PEGDM network, leading to faster gelation and enhanced overall mechanical properties. Together, these networks distribute and absorb mechanical stress in a complementary fashion, leading to a hybrid hydrogel that is both stiffer and more resistant to deformation than the individual components.
One of the exciting attributes of supramolecular systems like the Trpzip network is their tunable viscoelastic properties.34 Remarkably, the addition of Trpzip to PEGDM imparts shear-thinning properties that are comparable to pure Trpzip, with a linear decrease in viscosity with increasing shear rate (Fig. 3(a)–(d)). However, the hybrid PEGDM/Trpzip hydrogels maintained significantly higher viscosity compared to pure Trpzip across all shear rates. This indicates that the hybrid network preserves Trpzip's inherent shear thinning properties while enhancing the mechanical stability provided by the PEGDM network.
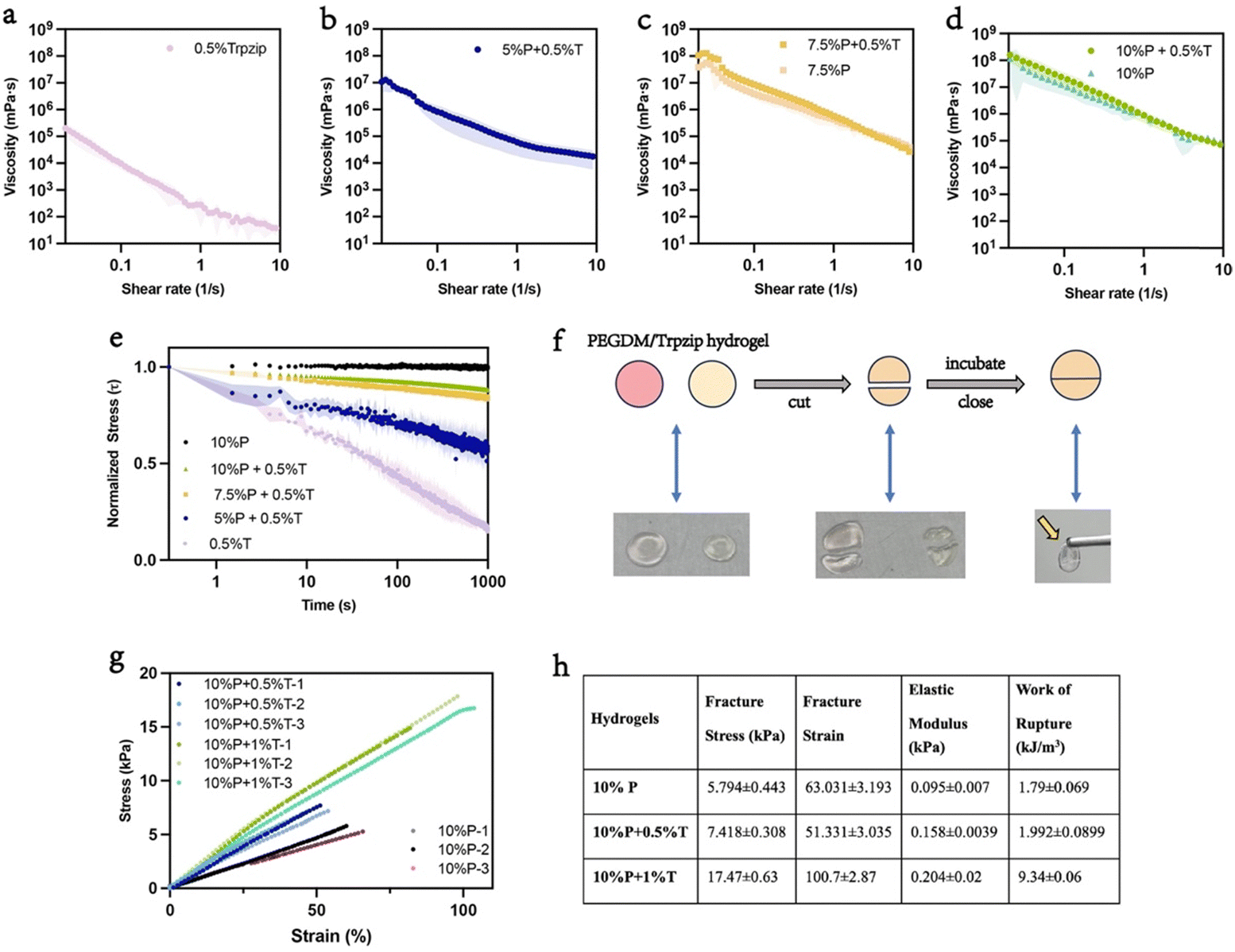 | ||
| Fig. 3 Functional mechanical properties and self-healing ability of PEGDM/Trpzip hydrogels. (a)–(d) Viscosity profiles of hydrogels composed of 0.5% (w/v) Trpzip, 5% (w/v), 7.5% (w/v), and 10% (w/v) PEGDM, with and without 0.5% (w/v) Trpzip, over a range of shear rates (0.01–10 s−1). These profiles highlight the shear-thinning behavior of the hydrogels, indicating their ability to flow under stress and return to a solid-like state when at rest. (e) Normalized stress relaxation curves for PEGDM/Trpzip hydrogels, showing the ability of different formulations to dissipate applied stress over time, with varying degrees of relaxation depending on PEGDM content. (f) Self-healing capabilities of PEGDM/Trpzip hydrogels demonstrated by cutting the hydrogel and allowing it to reassemble upon incubation, regaining its original form. (g) Stress–strain curves illustrating the mechanical properties under tension for 10% PEGDM hydrogels with and without 0.5% (w/v) and 1% (w/v) Trpzip, demonstrating enhanced mechanical performance in the presence of Trpzip. (h) Table summarizing the mechanical properties of the hydrogels, including fracture stress, fracture strain, elastic modulus, and work of rupture, comparing 10% PEGDM hydrogels with and without 0.5% (w/v) and 1% (w/v) Trpzip. Results show significantly improved mechanical toughness and elasticity with the addition of Trpzip. (n = 3). | ||
An important attribute of viscoelastic materials found in nature is stress-relaxation, which provides important signals to cells during morphogenesis and homeostasis.43,44 To assess the stress relaxation behavior of the PEGDM/Trpzip hydrogels, a constant shear force was applied to the samples. Pure PEGDM showed no stress relaxation due to the rigidity of its covalent network, while PEGDM/Trpzip hydrogels demonstrated tunable stress relaxation depending on their composition (Fig. 3(e)). For example, the half-life of stress relaxation increased from 200 seconds in pure Trpzip to over 1000 seconds in the PEGDM/Trpzip hybrids. This tunability is a key advantage, enabling the mechanical properties of the hydrogel to be tailored for different applications; such as mimicking the stiffness and stress relaxation characteristics of specific tissues. While previous studies on hydrogels composed of PEG polymers and short peptides have shown some tunable stress relaxation, their values were much closer to that of Trpzip alone and significantly lower than the PEGDM/Trpzip hybrids.45
In contrast to covalent networks that often fail irreversibly under mechanical stress, supramolecular systems such as the Trpzip peptide network showcase remarkable self-healing capabilities due to their dynamic and reversible non-covalent interactions. These interactions, which include hydrogen bonding, electrostatic forces, and hydrophobic effects, drive the self-assembly of the Trpzip peptide into a robust yet tunable network. Within the PEGDM/Trpzip hydrogel, the Trpzip network significantly enhances self-healing, as demonstrated by the hydrogel's ability to recover its structural integrity after being cut and incubated at 37 °C overnight (Fig. 3(f)). This inherent self-assembly mechanism distinguishes the hybrid hydrogel as a dynamic system capable of restoring functionality after mechanical damage.
Thixotropic tests further illustrate the pivotal role of the Trpzip peptide in endowing the double network with self-healing properties. Hydrogels containing Trpzip (Fig. S3c, ESI†) exhibited greater stiffness compared to 10% PEGDM hydrogels alone (Fig. S3a, ESI†), and the normalized storage modulus (G′) of the PEGDM/Trpzip hydrogel displayed reversible behavior under repeated cycles of high shear strain (Fig. S3d, ESI†). Conversely, the storage modulus of the PEGDM-only hydrogel showed minimal recovery (Fig. S3b, ESI†), emphasizing the unique contribution of the supramolecular Trpzip network. Over successive shear cycles, the normalized G′ changes in the PEGDM/Trpzip hydrogel stabilized, reflecting the dynamic and reversible nature of the non-covalent bonds within the Trpzip network. Meanwhile, the covalent PEGDM network experienced some degree of permanent fracture (Fig. S3e, ESI†). These findings underscore the complementarity between the Trpzip and PEGDM networks in creating a robust, self-healing hydrogel.
The ability of the Trpzip peptide to self-assemble into a tunable supramolecular network also enhances the viscoelastic and mechanical properties of the double-network hydrogel. By integrating the dynamic Trpzip peptide, the PEGDM/Trpzip system achieves a balance between rigidity and adaptability, closely mimicking some aspects of natural tissue matrices. The interplay of peptide self-assembly and covalent polymer crosslinking offers a promising approach for the design of multifunctional hydrogels with tailored mechanical and biological properties, making the material relevant to both tissue engineering and regenerative medicine.46
In addition to their viscoelastic properties, mixed-network hydrogels have attracted considerable interest for their propensity to increase toughness and durability.47 To evaluate the fracture resistance of the hydrogels after swelling, we formed “dog bone” specimens for tensile testing, in order to simulate the mechanical stresses encountered in load-bearing biomaterial applications. PEGDM hydrogels underwent brittle failure at low stress in most cases. In contrast, the PEGDM/Trpzip hydrogel displayed excellent repeatability and uniformity during stress–strain testing (Fig. 3(g)). The inclusion of Trpzip significantly improved the mechanical properties of the double networks, with fracture stress increasing more than threefold, from 5.794 kPa in pure PEGDM to 7.418 kPa of 10%PEGDM/0.5%Trpzip and 17.47 kPa of 10%PEGDM/1%Trpzip hydrogel (Fig. 3(h)). Moreover, the 10%PEGDM/1%Trpzip hydrogel was capable of withstanding 100% deformation before failure, compared to only 66% deformation for the 10% PEGDM hydrogel (Fig. 3(h)). This indicates that the addition of Trpzip not only improves the strength of the hydrogel but also enhances its elasticity and deformation tolerance. The elastic modulus of the hybrid hydrogel also increased twofold compared to PEGDM alone, further demonstrating the improved mechanical resilience provided by the mixed network structure (Fig. 3(h)). The work of rupture, which measures the toughness of the material, revealed a substantial improvement in the PEGDM/Trpzip material, with the value of 1.994 kJ m−3 for 10%PEGDM/0.5%Trpzip hydrogel and 9.34 kJ m−3 for 10%PEGDM/1%Trpzip hydrogel compared to 1.85 kJ m−3 for PEGDM alone (Fig. 3(h)). This level of toughness makes the hybrid hydrogel suitable for replicating the mechanical behavior of many soft tissues.48
PEGDM hydrogels are inherently non-adhesive to mammalian cells, requiring integration of protein or peptide-based cell binding motifs.36 In contrast, Trpzip shows innate bioactivity, presumably due to the net positive charge at physiological pH promoting sequestration of matrix. To assess the biological performance of our hybrid networks, we seeded adipose derived stromal cells (ADSC) on the PEGDM/Trpzip hydrogels to evaluate cell morphology, adhesion and growth. ADSCs were seeded at a concentration of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per gel on top of 5%, 7.5%, and 10% PEGDM hydrogels, both with and without 0.5% Trpzip, and cultured for 48 h. The cells were then stained for actin filaments (phalloidin) and nuclei (DAPI) to evaluate cell growth and morphology. As shown in Fig. 4(a), cells on the 0.5% Trpzip hydrogel displayed the most elongated morphology, confirming the excellent bioactivity of Trpzip, consistent with previous studies.34 In contrast, cells failed to adhere to all of the pure PEGDM networks (ESI,† Fig. S2). When Trpzip was introduced into the PEGDM matrix, it effectively mitigated the bioinert nature of PEGDM, promoting cell adhesion and spreading on the hybrid hydrogels (Fig. 4(a)). The incorporation of Trpzip enhanced the bioactivity of the otherwise inert PEGDM matrix, demonstrating its potential to improve cell compatibility in hybrid networks.
000 cells per gel on top of 5%, 7.5%, and 10% PEGDM hydrogels, both with and without 0.5% Trpzip, and cultured for 48 h. The cells were then stained for actin filaments (phalloidin) and nuclei (DAPI) to evaluate cell growth and morphology. As shown in Fig. 4(a), cells on the 0.5% Trpzip hydrogel displayed the most elongated morphology, confirming the excellent bioactivity of Trpzip, consistent with previous studies.34 In contrast, cells failed to adhere to all of the pure PEGDM networks (ESI,† Fig. S2). When Trpzip was introduced into the PEGDM matrix, it effectively mitigated the bioinert nature of PEGDM, promoting cell adhesion and spreading on the hybrid hydrogels (Fig. 4(a)). The incorporation of Trpzip enhanced the bioactivity of the otherwise inert PEGDM matrix, demonstrating its potential to improve cell compatibility in hybrid networks.
 | ||
| Fig. 4 Morphology and quantitative analysis of ADSCs cultured on PEGDM/Trpzip hydrogels after 2 days. (a) Confocal microscopy images of adipose-derived stem cells (ADSCs) cultured on various hydrogel formulations stained for nuclei (blue, DAPI) and actin (green, phalloidin) after 2 days. The different hydrogel formulations include 0.5% (w/v) Trpzip, 5% PEGDM + 0.5% Trpzip, 7.5% PEGDM + 0.5% Trpzip, and 10% PEGDM + 0.5% Trpzip. Scale bar: 100 μm. (b) Box plot showing the average cell area (μm2) of ADSCs across different hydrogel formulations. (c) Box plot depicting the aspect ratio of ADSCs on various hydrogels, providing insights into cell elongation. (d) Box plot representing cell roundness on different hydrogel formulations, indicating the degree of circularity of ADSCs. Data were collected from 100 cells per condition, with whiskers showing the 5th to 95th percentile. Statistical significance was determined using one-way ANOVA with p-values indicated by ‘*’p < 0.1, ‘**’p < 0.01, ‘***’p < 0.001, ‘****’p < 0.0001. | ||
Despite variations in hydrogel stiffness across the hybrid formulations, stiffness did not significantly influence cell morphology, as indicated by the relatively consistent aspect ratio across the different hybrid hydrogels (Fig. 4(c)). This result contrasts with previous studies that demonstrated a positive correlation between hydrogel stiffness and increased cell elongation,49 This suggests that the cells are “feeling” the matrix via the underlying Trpzip nanofiber network. The self-assembled Trpzip network provides a fibrous, bioactive network that mimics extracellular matrix features, enabling cell adhesion and spreading. This secondary supramolecular network offers nanoscale topographical cues and dynamic mechanical interactions, which may complement or override the stiffness differences of the PEGDM matrix, depending on the interfacial architecture. Notably, among the hybrid hydrogels, the 7.5% PEGDM/0.5% Trpzip formulation performed best, showing the largest average cell area and the lowest cell roundness, suggesting optimal conditions for nurturing cell adhesion and spreading (Fig. 4(b) and (d)). This balance likely arises from sufficient PEGDM structural support combined with accessible Trpzip nanofibers, maximizing cell–matrix interactions.
The observed cell behaviour can be rationalised to the hybrid network's architecture and mechanics, with stress relaxation influencing the stiffness perceived by the cells. The 5% PEGDM/0.5% Trpzip hydrogel, having the lowest stiffness among the hybrids, supported good cell adhesion but had relatively fewer elongated cells compared to the other PEGDM hybrids, likely due to the softer mechanical environment, i.e., lower ability for cells to exert traction stress. On the other extreme, the 10% PEGDM/0.5% Trpzip hydrogel showed the highest stiffness due to increased PEG density, leading to a higher proportion of rounded cells, likely due to the dense non-adhesive meshwork masking the underlying adhesive Trpzip network. Between these extremes, we propose that the 7.5% PEGDM/0.5% Trpzip hydrogel shows a good balance of stiffness and accessible bioactive motifs to provide effective cell spreading and elongation.
To further explore the role of Trpzip content in guiding cell morphology, we prepared hydrogels with varying Trpzip concentrations (0.125%, 0.25%, 0.5%, and 1%) in the presence of 7.5% PEGDM. The results revealed a positive correlation between Trpzip concentration and cell spreading, with the 0.5% and 1% Trpzip hydrogels supporting the highest levels of cell elongation and growth (Fig. S5, ESI†). At concentrations below 0.5%, cell elongation was limited, although cell adhesion remained higher than on PEG-only hydrogels. These findings demonstrate that inclusion of trpzip peptides at different concentrations can be used to tune adhesion and spreading characteristics.
In addition to cell morphology, the proliferation rate of ADSCs was examined to evaluate the hydrogels’ ability to support cell growth. The number of cells adherent to the 7.5% PEGDM/0.5% Trpzip hydrogel increased significantly over three days, with a 66% rise in cell numbers from day 1 to day 3 (Fig. S6, ESI†). This marked improvement contrasts with the 7.5% PEGDM hydrogel without Trpzip, where cell numbers decreased over the same period due to absence of adhesion cues. These results highlight the Trpzip network's role in supporting cell viability and growth, positioning the PEGDM/Trpzip hydrogels as promising candidates for biomedical applications where cell proliferation is desirable.
These findings highlight the complementary properties of PEGDM and Trpzip in the design of mixed covalent self-assembling peptide hydrogels. The optimal balance of polymers and peptides plays a crucial role in achieving the desired mechanical and biological properties. The ability of the PEGDM/Trpzip hydrogels to support ADSC adhesion and growth indicates their potential for various biomedical applications. For instance, ADSCs have been widely studied for their role in promoting tissue regeneration,50 and these hydrogels could be particularly useful in tissue engineering and regenerative medicine, where controlling cell behaviour through material properties is essential. Furthermore, the tunable viscoelastic properties of these PEGDM/Trpzip hybrids enable them to mimic the mechanical characteristics of various soft tissues, such as brain, lungs, kidneys, and skin,49 suggesting future utility in biomedical applications where mechanical properties need to be tailored to mimic specific tissue types and disease states.
Conclusion
Hybrid networks combining covalent hydrogels and supramolecular nanofiber assemblies like the PEGDM/Trpzip system presented here demonstrate a promising approach to access a wide array of mechanical properties and bioactivity. The introduction of PEGDM into the Trpzip network significantly improves the hydrogel's mechanical properties, including faster gelation time, enhanced storage modulus, tunable stress relaxation, and increased fracture resistance. The combination of covalent and non-covalent interactions in the hybrid network system mimics the non-linear viscoelastic properties of native tissues, making the PEGDM/Trpzip hydrogels a promising candidate for a wide range of biomedical applications. The hybrid hydrogel's ability to mimic the mechanical behavior of various soft tissues and its suitability for sustained cell adhesion and growth make it a viable candidate for regenerative medicine and other biomedical applications, where mechanical and biological properties must be finely tuned to meet specific therapeutic needs.Materials and methods
Synthesis of poly(ethylene glycol)dimethacrylate (PEGDM)
Poly(ethylene glycol)dimethacrylate (PEGDM, 10 kDa) was synthesized based on a previously established method.51 PEG (10 kDa, Sigma-Aldrich, 81280) powder was dissolved in toluene (Sigma-Aldrich, 108883) and dehydrated through evaporation. Dehydrated PEG was then dissolved in a mixture of toluene, dichloromethane (Chem-Supply, 75092), and triethylamine (ThermoFisher, 121448), followed by reaction with 2.2 equivalents of methacrylic anhydride (Sigma-Aldrich, 276685). The reaction was stirred on ice for 48 h to produce PEGDM. The reaction was quenched using potassium carbonate, and PEGDM was filtered and precipitated by adding diethyl ether (Chem-Supply, EA036). The product was dried and stored at −20 °C for future use.Synthesis and morphological characterization of PEGDM/Trpzip hydrogel
A 1% (w/v) Trpzip (purchased from GeneScript Biotech, purity >98%) stock solution was prepared by dissolving Trpzip powder in deionized (DI) water at 37 °C. A 20% (w/v) PEGDM stock solution was prepared by dissolving PEGDM powder in DMEM. Hydrogels with different PEGDM concentrations (5%, 7.5%, and 10% w/v) were prepared with 0.5% (w/v) Trpzip by mixing DMEM, PEGDM, and Trpzip stock solutions at specific ratios: 0.5:0.5:1, 0.25:0.75:1, and 0:1:1, respectively. Crosslinking was initiated by adding ammonium persulfate (APS, Chem-Supply, AL019) and N,N,N′,N′-tetramethylethylenediamine (TEMED, Sigma-Aldrich, T22500) to the PEGDM/Trpzip mixtures at final concentrations of 0.14% (w/v) and 0.28% (w/v), respectively. The hydrogels formed within 15–20 min.
The morphology of the hydrogel samples was characterized using scanning electron microscopy (SEM). After lyophilization, the hydrogel samples were mounted onto SEM plates and coated with platinum using an Emitech K575x sputter coater. The samples were then imaged at 10 kV using a Hitachi S230 scanning electron microscope.
Evaluating the mechanical properties of the hydrogels
The mechanical properties of the hydrogel samples were assessed using both a rheometer and a tensile tester.Rheological tests were performed using an Anton Paar MCR 302e Rheometer equipped with a 25 mm disc plate. Each test involved loading a 600 μL sample with a 1 mm gap between the plates. The time sweep test was conducted at 0.2% strain and 1 Hz frequency at 37 °C for 1 h. The following tests were all tested after hydrogel gelation in time sweep. Strain sweep tests were carried out at a 1 Hz frequency, with shear strain increasing from 0% to 1000% over 10 min. Frequency sweep tests were performed with 0.2% shear strain, ranging from 0 to 100 Hz, over 10 min. Viscosity flow curves were measured using a shear rate ramp from 0.01 to 10 s−1 over 10 min. Stress relaxation tests were conducted at 1% shear strain for 30 min, with the data normalized to the initial value. The thixotropic tests were exposed to 600% shear strain for 3 min, followed by 0.2% shear strain for hydrogel recovery, repeated in 3 cycles.
For tensile testing, 2 mm thick dog-bone shaped samples were used and prepared as shown in ESI,† Fig. S4. The samples were sprayed with black speckle paint to facilitate the tracking of two displacement points within the gauge length by recording a video for each sample at 2 fps. The frames were then imported into GOM software to obtain the axial displacement data. The Mark-10 Tensile Tester was carefully aligned to ensure proper positioning of the grips. Tensile load was applied at a rate of 0.5 mm min−1, and load vs. time record was captured. A stress–strain graph was generated from the correlation of the axial displacement data and the load vs. time record, with maximum stress corresponding to the peak of the curve. The engineering strain and stress were calculated as follows:52

Assessment of self-healing capabilities of hydrogel samples
Hydrogel samples were prepared in a columnar shape and cut in half. The halves were brought back together and incubated at 37 °C overnight to assess self-healing properties.Preparation of the hydrogel samples on glass cover slips to seed adipose-derived stem cells (ADSC)
Hydrogel samples were fabricated on 1 cm-diameter glass coverslips. To chemically bind the hydrogels to the glass, the coverslips were first functionalized by treating them with a 3-(trimethoxysilyl)propyl methacrylate (TMSPMA) solution (92% ethanol, 6% glacial acetic acid, and 2% TMSPMA (Sigma, 440159)) at room temperature for 30 min. After the coverslips were air-dried, 50 μL of the hydrogel precursor solution (prepared as described in Section 2.2) was pipetted onto the functionalized coverslips. A second hydrophobic glass coverslip was carefully placed on top to create a uniform thin layer of hydrogel. Once gelation was complete, the top coverslip was gently peeled off, and the hydrogels were washed with PBS. The hydrogel-coated coverslips were then transferred to a 24-well plate and sterilized under UV light for 30 min.Adipose-derived stem cells (ADSCs, ATCC, PCS-500-011) were seeded onto the hydrogel samples at a density of 100000 cells per mL. The cells were cultured in low-glucose Dulbecco's Modified Eagle's Medium (DMEM, ThermoFisher Scientific, 11885084), which was supplemented with 10% (v/v) fetal bovine serum (FBS, BOVO-GEN, Australia, SFBS-AU) and 1% (v/v) penicillin/streptomycin (Sigma-Aldrich, P4333).
Immunostaining of ADSC seeded hydrogels
ADSCs were cultured on hydrogels with the following formulations for 2 days (w/v): 0.5% Trpzip, 5% PEGDM with 0.5% Trpzip, 7.5% PEGDM with 0.5% Trpzip, 10% PEGDM with 0.5% Trpzip, and 10% PEGDM. To evaluate the proliferation rate, ADSCs were also cultured on the same hydrogel formulations in 96-well plates for 1 and 3 days.Additionally, ADSCs were seeded on hydrogels with varying Trpzip concentrations (1% Trpzip, 7.5% PEGDM with 0.125%, 0.25%, 0.5%, and 1% Trpzip) for 2 days to determine the upper and lower limits of Trpzip concentration that influence cell morphological spread.
The cells were fixed with 4% paraformaldehyde (PFA, Sigma-Aldrich, P6148) at room temperature for 30 min and washed with PBS overnight at 4 °C. The samples were blocked with 0.5% Triton X-100 (Sigma-Aldrich, 562380) and 3% Bovine Serum Albumin (BSA, Sigma-Aldrich, A3858) in DI water, followed by washing twice with PBS. The cells were stained with DAPI (1:500) and phalloidin (1:200) in 1% BSA overnight and washed again with PBS at 4 °C. Finally, the samples were stored at 4 °C overnight before imaging with a Zeiss LSM 800 confocal microscope using a 10× objective.
Data availability
This study generated data including shear rheology and spectroscopy files which are stored in instrument specific software formats and in ASCiI file format, and microscopy files and images. The datasets generated during the current study are not publicly available due to further analysis related to future publications but are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported through funding from the Australian Research Council grant DP230101804 (K. A. K.) and DP210103654 (K. A. K. and J. K.), the National Health and Medical Research Council grant APP1185021 (K. A. K.), and the National Cancer Institute of the National Institutes of Health grant R01CA251443 (K. A. K.).References
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- J. Kopecek, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5929–5946 CrossRef CAS PubMed.
- E. A. Kamoun, E.-R. S. Kenawy and X. Chen, J. Adv. Res., 2017, 8, 217–233 CrossRef CAS PubMed.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- J. S. Boateng, K. H. Matthews, H. N. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- T. Yuan, L. Zhang, K. Li, H. Fan, Y. Fan, J. Liang and X. Zhang, J. Biomed. Mater. Res., Part B, 2014, 102, 337–344 CrossRef PubMed.
- C. Vinatier, O. Gauthier, A. Fatimi, C. Merceron, M. Masson, A. Moreau, F. Moreau, B. Fellah, P. Weiss and J. Guicheux, Biotechnol. Bioeng., 2009, 102, 1259–1267 CrossRef CAS PubMed.
- W. S. Toh, E. H. Lee, X.-M. Guo, J. K. Chan, C. H. Yeow, A. B. Choo and T. Cao, Biomaterials, 2010, 31, 6968–6980 CrossRef CAS PubMed.
- P. Sajkiewicz and D. Kołbuk, J. Biomater. Sci., Polym. Ed., 2014, 25, 2009–2022 CrossRef CAS PubMed.
- S. W. Kang, B. H. Cha, H. Park, K. S. Park, K. Y. Lee and S. H. Lee, Macromol. Biosci., 2011, 11, 673–679 CrossRef CAS PubMed.
- Z.-K. Cui, S. Kim, J. J. Baljon, B. M. Wu, T. Aghaloo and M. Lee, Nat. Commun., 2019, 10, 3523 CrossRef PubMed.
- J. Liu, S. Lin, X. Liu, Z. Qin, Y. Yang, J. Zang and X. Zhao, Nat. Commun., 2020, 11, 1071 CrossRef CAS PubMed.
- H. Kamata, Y. Akagi, Y. Kayasuga-Kariya, U.-I. Chung and T. Sakai, Science, 2014, 343, 873–875 CrossRef CAS PubMed.
- H. Yuk, C. E. Varela, C. S. Nabzdyk, X. Mao, R. F. Padera, E. T. Roche and X. Zhao, Nature, 2019, 575, 169–174 CrossRef CAS PubMed.
- T. V. Chirila, Biomaterials, 2001, 22, 3311–3317 CrossRef CAS PubMed.
- H. Kim, H. Witt, T. A. Oswald and M. Tarantola, ACS Appl. Mater. Interfaces, 2020, 12, 33516–33529 CrossRef CAS PubMed.
- S. Peressotti, G. E. Koehl, J. A. Goding and R. A. Green, ACS Biomater. Sci. Eng., 2021, 7, 4136–4163 CrossRef CAS PubMed.
- L. Rijns, J. W. Peeters, S. I. Hendrikse, M. E. Vleugels, X. Lou, H. M. Janssen, E. Meijer and P. Y. Dankers, Chem. Mater., 2023, 35, 8203–8217 CrossRef CAS.
- A. F. Vrehen, J. F. van Sprang, M. J. Schotman and P. Y. Dankers, Mater. Today Bio, 2024, 26, 101021 CrossRef CAS PubMed.
- N. Al Balushi, M. Boyd-Moss, R. M. Samarasinghe, A. Rifai, S. J. Franks, K. Firipis, B. M. Long, I. A. Darby, D. R. Nisbet and D. Pouniotis, Gels, 2022, 8, 332 CrossRef CAS PubMed.
- B. J. Cossette, S. Shetty, L. A. Issah and J. H. Collier, ACS Biomater. Sci. Eng., 2024, 10, 1819–1829 CrossRef CAS PubMed.
- K. M. Wong, A. S. Robang, A. H. Lint, Y. Wang, X. Dong, X. Xiao, D. T. Seroski, R. Liu, Q. Shao and G. A. Hudalla, J. Phys. Chem. B, 2021, 125, 13599–13609 CrossRef CAS PubMed.
- H. Wang, D. Zhu, A. Paul, L. Cai, A. Enejder, F. Yang and S. C. Heilshorn, Adv. Funct. Mater., 2017, 27, 1605609 CrossRef PubMed.
- S. Nemec, S. Ganda, K. Al Taief, C. Kopecky, R. Kuchel, H. N. Lebhar, C. P. Marquis, P. Thordarson and K. A. Kilian, ACS Appl. Bio Mater., 2022, 5, 4581–4588 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- M. J. Webber, J. Tongers, M.-A. Renault, J. G. Roncalli, D. W. Losordo and S. I. Stupp, Acta Biomater., 2010, 6, 3–11 CrossRef CAS PubMed.
- K. Koss and L. Unsworth, Acta Biomater., 2016, 44, 2–15 CrossRef CAS PubMed.
- M. Sedighi, N. Shrestha, Z. Mahmoudi, Z. Khademi, A. Ghasempour, H. Dehghan, S. F. Talebi, M. Toolabi, V. Préat, B. Chen, X. Guo and M.-A. Shahbazi, Polymers, 2023, 15, 1160 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- S. A. Hulgan and J. D. Hartgerink, Biomacromolecules, 2022, 23, 1475–1489 CrossRef CAS PubMed.
- X. Zhao, Curr. Opin. Colloid Interface Sci., 2009, 14, 340–348 CrossRef CAS.
- A. K. Nguyen, T. G. Molley, E. Kardia, S. Ganda, S. Chakraborty, S. L. Wong, J. Ruan, B. E. Yee, J. Mata, A. Vijayan, N. Kumar, R. D. Tilley, S. A. Waters and K. A. Kilian, Nat. Commun., 2023, 14, 6604 CrossRef CAS PubMed.
- I. Mironi-Harpaz, D. Y. Wang, S. Venkatraman and D. Seliktar, Acta Biomater., 2012, 8, 1838–1848 CrossRef CAS PubMed.
- S. Johannsmeier, M. T. T. Nguyen, R. Hohndorf, G. Dräger, D. Heinemann, T. Ripken and A. Heisterkamp, ACS Appl. Bio Mater., 2020, 3, 7011–7020 CrossRef CAS PubMed.
- M. Chen, J. Hu, H. Gao, J. Shen, T. Wei, J. Yao, Y. Zhang, P. Gu, Z. Liu and Q. Chen, Sci. Adv., 2023, 9, eadh1582 CrossRef CAS PubMed.
- H. Asokan-Sheeja, K. Awad, J. Xu, M. Le, J. N. Nguyen, N. Nguyen, T. P. Nguyen, K. T. Nguyen, Y. Hong, V. G. Varanasi, X. Liu and H. Dong, Biomacromolecules, 2024, 25, 2814–2822 CrossRef CAS PubMed.
- B. Shagdarova, M. Konovalova, Y. Zhuikova, A. Lunkov, V. Zhuikov, D. Khaydapova, A. Il’ina, E. Svirshchevskaya and V. Varlamov, Materials, 2021, 15, 15 CrossRef PubMed.
- L. L. Fernandes, C. X. Resende, D. S. Tavares, G. A. Soares, L. O. Castro and J. M. Granjeiro, Polímeros, 2011, 21, 1–6 CrossRef CAS.
- F. Zhang, C. Hu, Q. Kong, R. Luo and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 37147–37155 CrossRef CAS PubMed.
- P. K. Gavel, N. Kumar, H. S. Parmar and A. K. Das, ACS Appl. Bio Mater., 2020, 3, 3326–3336 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H.-P. Lee, E. Lippens and G. N. Duda, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, M. Darnell, D. Klumpers, S. A. Bencherif, J. C. Weaver, N. Huebsch and D. J. Mooney, Nat. Commun., 2015, 6, 6365 CrossRef CAS PubMed.
- R. Pugliese and F. Gelain, J. Appl. Polym. Sci., 2022, 139, 51759 CrossRef CAS.
- F. Gelain, Z. Luo and S. Zhang, Chem. Rev., 2020, 120, 13434–13460 CrossRef CAS PubMed.
- X. Huang, J. Li, J. Luo, Q. Gao, A. Mao and J. Li, Mater. Today Commun., 2021, 29, 102757 CrossRef CAS.
- D. Taylor, N. O’Mara, E. Ryan, M. Takaza and C. Simms, J. Mech. Behav. Biomed. Mater., 2012, 6, 139–147 CrossRef PubMed.
- C. F. Guimarães, L. Gasperini, A. P. Marques and R. L. Reis, Nat. Rev. Mater., 2020, 5, 351–370 CrossRef.
- A. Krawczenko and A. Klimczak, Int. J. Mol. Sci., 2022, 23, 2425 CrossRef CAS PubMed.
- M. S. Islam, T. G. Molley, J. Ireland, J. J. Kruzic and K. A. Kilian, Adv. NanoBiomed Res., 2021, 1, 2000072 CrossRef CAS.
- A. Ajam, Y. Huang, M. S. Islam, K. A. Kilian and J. J. Kruzic, J. Mech. Behav. Biomed. Mater., 2024, 157, 106642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02002b |
| This journal is © The Royal Society of Chemistry 2025 |