
Construction of an Sc-NiFe-LDH electrocatalyst for highly efficient electrooxidation of 5-hydroxymethylfurfural at industrial current density†
Yufeng
Wu
,
Zhiyan
Hou
and
Changlong
Wang
 *
*
Institute of Circular Economy, College of Materials Science and Engineering, Beijing University of Technology, Beijing 100124, P. R. China. E-mail: clwang1987@126.com
First published on 9th December 2024
Abstract
Renewable electricity-powered electrooxidation of 5-hydroxymethylfurfural (HMFOR) to FDCA offers a green and sustainable approach to producing an essential monomer for bio-polymers, provided that a highly efficient electrocatalyst is present. Herein, we show that the doping of scandium (Sc) into an NiFe-LDH electrocatalyst (Sc-NiFe-LDH) considerably promotes HMFOR by enhancing the formation of high-valence NiIII–O active sites, facilitating electron transport and HMF adsorption and suppressing the oxygen evolution reaction. In the presence of the Sc-NiFe-LDH electrocatalyst, an FDCA faradaic efficiency and selectivity of 96.5% and 99.5%, respectively, were achieved at a current density of >600 mA cm−2. The high performance of the Sc-NiFe-LDH electrocatalyst is the best among those of other reported electrocatalysts for this reaction, showing great promise in upgrading biomass to valuable chemicals. This work would inspire further studies on the rational design of novel and efficient electrocatalysts for green and sustainable energy transformations.
 Changlong Wang | Changlong Wang was born in Shandong Province, China. He carried out his graduate studies with Prof. Dezhi Wang at the Central South University in Changsha, China, and completed his PhD at the University of Toulouse III under the supervision of Prof. Didier Astruc. He later joined Prof. Ferdi Schüth's group at the Max-Planck-Institut für Kohlenforschung, Germany, for postdoctoral studies. He is presently Professor at the Beijing University of Technology. His research interests include the design of transition-metal nanomaterials and nanocatalysis for waste upcycling and for a circular economy. |
1. Introduction
Polyethylene terephthalate (PET) has versatile applications and is an essential component in our daily life. However, it is very difficult to degrade it under natural conditions, which results in white pollution and therefore calls for biodegradable alternatives.1–3 Polyethylene furanoate (PEF), synthesized through the polymerization of 2,5-furandicarboxylic acid (FDCA) and ethylene glycol, has superior biodegradability and mechanical properties and is considered a promising bio-substitute to PET.4,5 However, the high cost of FDCA has become an essential problem that limits large-scale PEF production, posing challenges to its green and cost-effective synthesis. Renewable energy-powered electrooxidation of 5-hydroxymethylfurfural (HMFOR) offers an eco-friendly method for generating the bio-monomer FDCA.6 The green and carbon-free electro-synthesis of FDCA not only addresses environmental concerns raised by fossil fuels but also provides a sustainable pathway for bio-plastic production.Ni-based materials, including composited catalysts such as NiCo,7 NiCu,8 NiMn,9 and NiFe,10 have been extensively investigated as non-noble metal-based electrocatalysts for the electrooxidation of HMF to FDCA. Although Ni–Fe layered double hydroxide (LDH) exhibits good reactivity for HMF oxidation, it is presented with low faradaic efficiencies (FEs) for FDCA at elevated potentials.11 Owing to its high efficiency for the oxygen evolution reaction, O2 bubbles would block active sites, leading to poor catalyst stability. Moreover, bulk LDH exhibits inherent low conductivity, limiting the number of accessible active sites that are essential for high efficiency. Doping LDHs with foreign metals has been shown to effectively regulate the electronic structure and electrocatalytic performance of LDHs without causing unexpected phase separation.12–14 Recent research has highlighted that rare-earth metal doping is an effective method to modify electronic structures. It regulates intermediate adsorption and generates new active sites, improving the activity and performance of electrocatalysts.15,16 For instance, Fang's group suggested that the electron-promoting effect of Pr additives would substantially improve the electrocatalytic properties of Co(OH)2 toward HMF electrooxidation.17 Wang's group found that Ce doping in NiFe-LDH considerably facilitated electron transport and optimized the adsorption of key intermediates, lowering the activation energy of the rate-determining step—the transformation of 5-formyl-2-furancarboxylic acid (FFCA).12
Scandium (Sc) is considered as the omnipotent “industrial ajinomoto” in electrochemistry, and indeed, Sc-based electrocatalysts show high promise in applications towards fuel cells, lithium–sulfur batteries, water oxidation, etc.18–21 However, Sc doping has rarely been used to enhance the electrocatalytic properties of NiFe-LDH. Thus, in this study, we first demonstrate that introducing Sc into the crystal structure of NiFe-LDH (Sc-NiFe-LDH) leads to lattice expansion and alterations in the electronic structure, resulting in improved performance towards the HMF oxidation. Raman spectroscopic characterization confirms that the active species are the high-valence NiIII–O. In situ electrochemical impedance spectroscopy (EIS) further validates the suppression of OER by Sc doping in the HMFOR. Additionally, Sc doping enhances the ability of NiOOH species to capture protons from HMF, facilitating electron transfer and promoting the conversion of HMF to FDCA. As a result, a current density of 802.8 mA cm−2 was achieved at 1.50 V vs. RHE, surpassing the performance of most previously reported HMF oxidation catalysts. Introducing Sc suppressed the competitive OER process considerably, showing high performance toward HMF electrooxidation. Furthermore, the best performance of Sc-NiFe-LDH was achieved with the FDCA yield of 96.7% and FE of 96.5% at 1.477 V vs. RHE with excellent catalyst stability.
2. Experimental section
2.1. Materials
5-Hydroxymethylfurfural (HMF, Aladdin, 99%), 2,5-furandicarboxylic acid (FDCA, Aladdin, 98%), diformylfuran (DFF; Aladdin, 99%), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA; Aladdin, 98%), 5-formyl-2-furancarboxylic acid (FFCA; Aladdin, 98%), Ni(NO3)2·6H2O (Aladdin, 99.99%), Fe(NO3)3·9H2O (Aladdin, 99.99%), Sc(NO3)3·xH2O (Aladdin, 99.99%), anion-exchange membrane (Fumasep FAB-PK-130), potassium hydroxide (KOH, Sigma-Aldrich, ≥85%), and Milli-Q water (18.25 MΩ) were used for all the synthesis and catalysis experiments. All solvents and chemicals were used as purchased without further purifications.2.2. Preparation of Sc-NiFe-LDH and NiFe-LDH
Prior to synthesis, nickel foam (NF) was washed with 3 M HCl to remove any metal residues. Subsequently, it was rinsed with acetone, ethanol, and water, respectively, for 15 min. Sc-NiFe-LDH was synthesized using a one-step hydrothermal method. Typically, a solution of Ni(NO3)2·6H2O (3 mmol), Fe(NO3)3·9H2O (0.95 mmol), Sc(NO3)3·xH2O (0.05 mmol), and urea (5 mmol) were dissolved in 30 mL H2O. Pre-treated NF is placed in the above mixed solution and transferred to a sealed Teflon-lined stainless-steel autoclave (50 mL), sealed and maintained at 120 °C for 10 h. After the autoclave was cooled to room temperature, the resulting Sc-NiFe-LDH was washed three times with deionized water and ethanol and then dried at 60 °C overnight.The preparations of NiFe-LDH, 2.5% Sc-NiFe-LDH, and 10% Sc-NiFe-LDH were similar to that of 5% Sc-NiFe-LDH, except that the amounts of the Sc precursor were adjusted from 0 to 0.025 and 0.1 mmol, while the amount of Fe(NO3)3·9H2O was varied from 1 to 0.975 and 0.9 mmol.
2.3. Physical characterizations
The scanning electron microscopy test was conducted on ZEISS GeminiSEM 300. The X-ray powder diffraction (XRD) characterization was conducted using a Bruker AXS D8 Advance diffractometer with a Cu-Kα source (1.54056 Å). Diffraction data were collected for 2θ angles from 5° to 80°. Transmission electron microscopy was performed using a JEOL-JEM-2100F transmission electron microscope at an accelerating voltage of 200 kV. The FT-IR measurements were conducted in a Bruker INVENIO-S equipped with a DLaTGS detector. The in situ Raman spectra were recorded on the aforementioned Raman microscope equipped with an Ar laser (wavelength = 532 nm) under controlled potentials using the Chenhua CHI760 electrochemical workstation. The spectral range was 200–1000 cm−1. The laser power was set at 1 and the exposure time was 10 seconds. The electrolytic cell, purchased from Shanghai Chu Xi Industrial Co., Ltd, was made of polytetrafluoroethylene (PTFE), and a thin round quartz glass plate was used as a protective cover for the objective. A Pt wire was used as the counter electrode, and an Ag/AgCl electrode was used as the reference electrode. The working electrode was inserted through the wall of the electrolytic cell to ensure that its plane was perpendicular to the incident laser. Electrochemical in situ ATR-IR spectroscopy was conducted on the Bruker-INVENIO instrument in 1 M KOH solution with or without 10 mM HMF. The scanned potential range was from 1.20 to 1.55 V vs. RHE with a step of 0.05 V. The electrolysis experiment was conducted at 1.45 V vs. RHE for 10 min, and the infrared spectra were recorded at different times. During the experiment, the infrared optical table was purged with clean nitrogen to remove internal water vapor and gas-phase CO2.2.4. Electrochemical measurements
The OER and electrocatalytic oxidation of HMF were tested using an Ivium-n-Stat (Ivium Technologies B.V., Netherlands) electrochemical workstation with the standard three-electrode system in an H-type electrochemical cell separated by an anion-exchange membrane (PK-130) at room temperature. The Sc-NiFe-LDH (∼1 cm × 1 cm) was directly used as the working electrode (WE), a carbon rod was used as the counter electrode (CE), and a standard Hg/HgO electrode was used as the reference electrode (RE). The Hg/HgO electrode is available from ALS Co., Ltd. In a typical electrochemical experiment sequence, cyclic voltammetry (CVs) at a rate of 100 mV s−1 from 0.00–0.70 V vs. Hg/HgO for 20 cycles was measured to guarantee a stable electrode performance during the experiments. The potentials were converted to the reversible hydrogen electrode (RHE) through the Nernst equation: (E(RHE) = E(Hg/HgO) + 0.059 pH + 0.098 V). The scan rate for LSV was kept at 5 mV s−1. The electrochemical surface area (ECSA) was evaluated in terms of the double-layer capacitance (Cdl). Cyclic voltammetry (CV) was performed in 1 M KOH at different scan rates of 20–60 mV s−1 in a potential window where no faradaic process occurs (1.087–1.187 V vs. RHE). In situ EIS measurements were conducted with a three-electrode system. The frequency ranged from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 0.01 Hz with an amplitude of 5 mV, and the applied potential ranged from 1.15 to 1.60 V (vs. RHE) with a 0.05 V interval.
000 to 0.01 Hz with an amplitude of 5 mV, and the applied potential ranged from 1.15 to 1.60 V (vs. RHE) with a 0.05 V interval.
2.5. Product analysis
HPLC (Agilent 1260 Infinity), with an ultraviolet-visible detector, was used to analyze the HMF oxidation products. Specifically, during potentiostatic electrolysis, 100 μL of the electrolyte was taken out and diluted with ultrapure water 100 times and analyzed by HPLC. The specific parameters are set as follows. The wavelength of the UV detector is set to 265 nm, mobile phase A is methanol and phase B is 5 mM ammonium formate aqueous solution, the ratio of A:B is 15:85, flow rate is 0.5 ml min−1, and column temperature is 30 °C. Using a Polaris 5 C18-A column (4.6 mm × 150 mm), 20 μL of the liquid is injected into the HPLC and separation lasted for 10 min. The FDCA yield (%), FE and selectivity were calculated using eqn (1)–(3): | (1) |
 | (2) |
 | (3) |
F is the Faraday constant (96485 C mol−1), and n is the mol of the reactant calculated from the concentration measured by HPLC.
The theoretic total charge of HMF electrooxidation is as follows:

3. Results and discussion
3.1. Electrocatalyst synthesis and characterizations
The Sc-NiFe-LDH and the counterpart electrocatalysts are grown in NF using a hydrothermal method (Experimental section). The resulting electrodes are characterized as follows. In the X-ray powder diffraction (XRD) spectra shown in Fig. 1a, both NiFe-LDH and Sc-NiFe-LDH are well indexed, and the positions of the diffraction peaks are in good agreement with the lattice parameters characteristic of NiFe-LDH (PDF#40-0215).22 In comparison to NiFe-LDH, Sc-NiFe-LDH exhibits a narrower reflection, indicating a higher crystallinity and larger grain size, which benefit the electrical conductivity.23Fig. 1b is the scanning electron microscope (SEM) image of Sc-NiFe-LDH, showing numerous ultrathin nanosheets that self-assembled into a nanoflower structure. Compared to NiFe-LDH (Fig. S1, ESI†), the thickness of the nanosheets is relatively reduced. Because the ionic radius of Sc3+ (74 pm) is larger than that of Fe3+ (64 pm), the doping of Sc further promotes lattice expansion. It enhances the interactions between metal ions and facilitates crystal growth, resulting in structurally intact ultrathin nanoflower spheres.24 Detailed observations using transmission electron microscopy (TEM) revealed the layered crystalline structure of nanoflower spheres (Fig. 1c). Moreover, the elemental mapping images (Fig. 1d) show that Ni, Fe, Sc, C and O elements are homogeneously distributed in the nanoflower spheres of Sc-NiFe-LDH. This unique three-dimensional (3D) nanoflower architecture would facilitate the rapid substrate mass transfer during the electrooxidation of HMF reactions, thereby enhancing the reaction kinetics (vide infra).25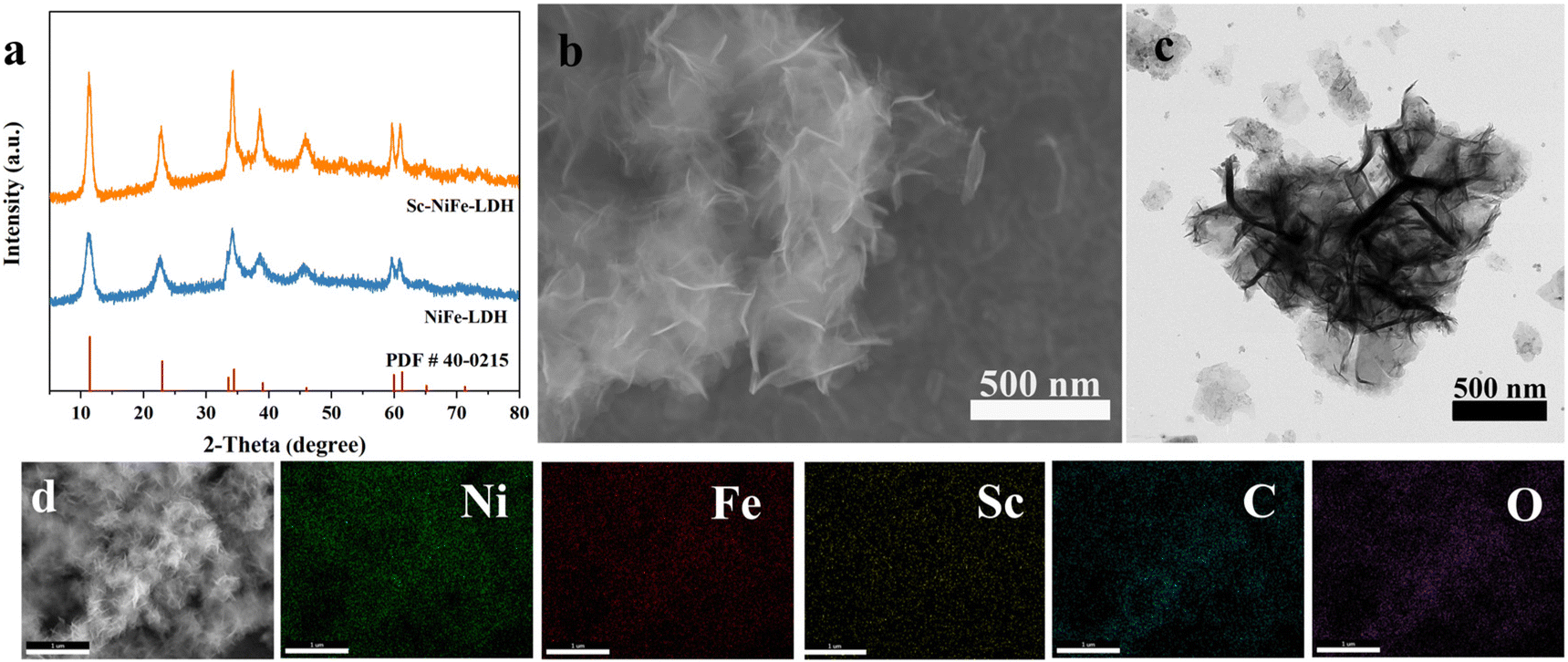 | ||
| Fig. 1 (a) XRD patterns Sc-NiFe-LDH and NiFe-LDH; (b) SEM image of Sc-NiFe-LDH; (c) TEM image of Sc-NiFe-LDH; (d) TEM image and the elemental mappings of Sc-NiFe-LDH. | ||
3.2. Electrocatalytic HMF oxidation over Sc-NiFe-LDH
We then evaluated the performances of Sc-NiFe-LDH and other control samples toward HMF electrooxidation in a three-electrode setup in the H-type cell. First, linear sweep voltammetry (LSV) was performed using the catalysts with different amounts of Sc, allowing the identification of the best catalyst, 5% Sc-NiFe-LDH, in terms of the current responses to HMF (Fig. S2, ESI†). Fig. 2a shows the LSV curves of this Sc-NiFe-LDH and NiFe-LDH in 1 M KOH, with and without 10 mM HMF. In the presence of HMF, the onset potential shifts to much lower potentials, indicating the kinetically more favorable HMFOR than OER. Compared to NiFe-LDH for HMFOR, Sc-NiFe-LDH exhibits a lower onset potential of approximately 1.35 V vs. RHE. Upon introducing HMF, the current density increases from 100, 200, 500 to 1000 mA cm−2 at the potentials of 1.39, 1.42, 1.46 V to 1.52 V vs. RHE, giving the first hint of the high efficiency of Sc-NiFe-LDH. To optimize the reaction parameters, HMF electrooxidations were conducted at different potentials. The optimal potential was identified to be 1.477 V vs. RHE in terms of FDCA yield, faradaic efficiency (FE), and selectivity (Fig. S3, ESI†). Consequently, chronoamperometry experiments at this potential were conducted to identify and quantify the products, as well as to calculate the faradaic efficiency (FE) based on the substrate concentration changes monitored by high-performance liquid chromatography (HPLC) (Fig. 2b, and Fig. S4, S5, ESI†). HMF progressively decreased and efficiently converted to FDCA with a carbon balance close to 100%. Fig. 2c shows the evolution of yield, FE, and selectivity for FDCA at 1.477 V vs. RHE. For Sc-NiFe-LDH, the yield is high (95.3%), with a faradaic efficiency (FE) of 92.5% and selectivity of 99.5%. In contrast, at the same potential, the NiFe-LDH catalyzed HMF electrooxidation proceeded along with OER, leading to a considerable decrease in FDCA yield and FE.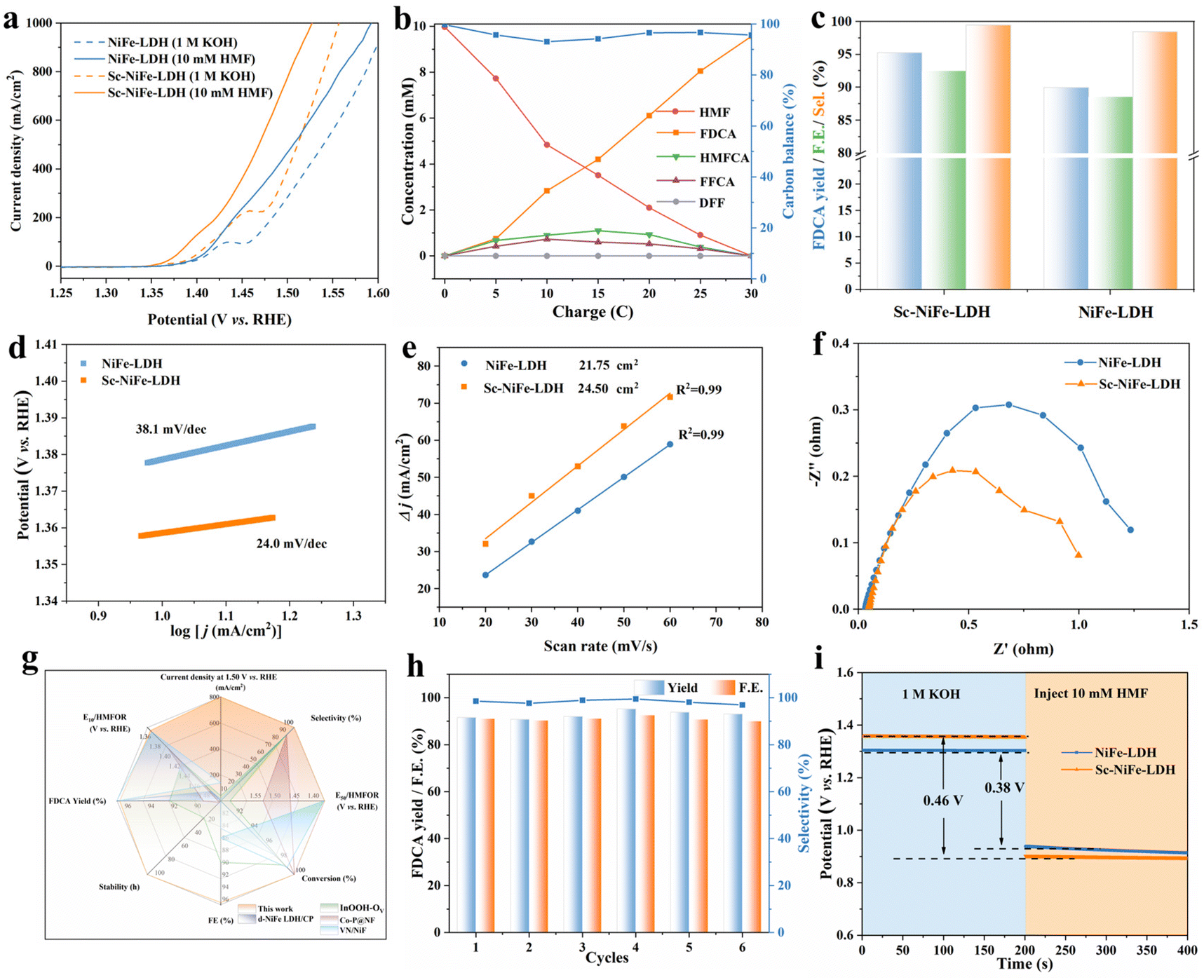 | ||
| Fig. 2 (a) LSV curves of Sc-NiFe-LDH and NiFe-LDH in 1 M KOH with 10 mM HMF at 5 mV s−1. (b) Concentration changes of each substance of Sc-NiFe-LDH during the HMFOR. (c) Comparison of FDCA yield, FE and Sel. (d) Tafel slopes. (e) ECSA. (f) Nyquist plots of samples in the presence of 10 mM HMF at 1.50 V vs. RHE. (g) Comprehensive comparison of the catalytic performance of known catalysts reported in the literature and the catalyst in this work. (h) FDCA yield, FE and selectivity after six successive electrolysis cycles. (i) OCP of Sc-NiFe-LDH and NiFe-LDH in 1 M KOH solution before and after 10 mM HMF was injected. | ||
To provide insights into the high efficiency of Sc-NiFe-LDH, the Tafel slopes were calculated (Fig. 2d). It shows that the Tafel slope of Sc-NiFe-LDH is 24.0 mV dec−1, significantly lower than that of NiFe-LDH (38.1 mV dec−1). This suggests the fast electron transfer at the interface between HMF and the Sc-NiFe-LDH electrocatalyst. Second, the electrochemical surface area (ECSA) was compared based on the calculated electrochemical double layer capacitance (Cdl) from the non-faradaic regions by cyclic voltammetry measurements (Fig. S6, ESI†). As shown in Fig. 2e, the ECSA of Sc-NiFe-LDH is 24.50 cm2; the value is significantly higher than that of NiFe-LDH (21.75 cm2). This suggests that the doping of Sc into NiFe-LDH increases the active electrochemical area for HMFOR. Furthermore, the Nyquist plot of Sc-NiFe-LDH (Fig. 2f) shows a semicircle with a smaller diameter than that of NiFe-LDH, indicating that charge transfer occurs more rapidly on the surface of the electrocatalysts. Thus, all those results confirm that the 3D nanoflower-structured Sc-NiFe-LDH provides numerous active sites for enhanced properties by promoting charge transfer and facilitating the rapid mass transfer of HMF molecules to the electrocatalyst.
Compared with the previously reported catalysts, the Sc-NiFe-LDH catalyst exhibits significant advantages for HMFOR and HER electrolysis, yielding a current density at 1.50 V vs. RHE, voltages at current densities of 10 and 50 mA cm−2 (E10 and E50), and FDCA yield and FE (Fig. 2g and Table S1, ESI†).25–28 This strongly suggests its promising application for simultaneous biomass upgradation and sustainable H2 generation. The high stability of Sc-NiFe-LDH was also revealed by conducting six successive electrolysis cycles, showing no obvious decays in FDCA yields, FEs, and selectivity (Fig. 2h). After the HMFOR electrolysis for six runs, TEM was further employed to track the structural evolution of Sc-NiFe-LDH (Fig. S7, ESI†). The catalyst retains the original nanosheet structure of Sc-NiFe-LDH, indicating its high stability during HMFOR. Furthermore, the introduction of various substrates (including glucose, furfural, benzyl alcohol, urea, ethanol, and methanol) into the Sc-NiFe-LDH system showed significant increases in current densities at lower potentials in sharp comparison to those required for the OER. This indicates its broad potential applications toward electrochemical energy transformations (Fig. S8–S13, ESI†).
To understand the adsorption behavior of HMF over Sc-NiFe-LDH, the open circuit potential (OCP) measurements were conducted, providing information on the changes in HMF adsorption on the Helmholtz layer.29 As can be seen, a stronger interaction between the catalyst surface and HMF results in a pronounced change in OCP upon the introduction of HMF. The significant drop in OCP, shown in Fig. 2i, indicates the stronger adsorption of HMF on Sc-NiFe-LDH compared to NiFe-LDH. This strong HMF adsorption would facilitate the HMF oxidation even further.
3.3. Kinetic analysis and catalytic mechanism
The HMF molecule contains two functional groups, and its oxidation can be categorized into two pathways. The first pathway involves the oxidation of the formyl group, forming 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) as the intermediate. The second pathway involves the oxidation of the hydroxymethyl group, resulting in a diformylfuran (DFF) intermediate (Fig. S14, ESI†).30 As indicated in Fig. 2b, almost no DFF is detected by HPLC, suggesting that the formation of FDCA primarily proceeds through the HMFCA pathway (Fig. 2b).31 To gain insight into the reaction pathway, the potential-dependent in situ ATR-IR measurements were conducted (Fig. 3a). At potentials higher than 1.35 V vs. RHE, new bands are observed, indicating the progress of the HMF electrocatalytic oxidation; the result is consistent with the LSV results (vide supra). When the potential increases, the progressively downward band at 1668 cm−1 suggests the consumption of HMF.32 On the other hand, the bands at 1529 and 1569 cm−1 are assigned to the intermediate HMFCA,33 and the peak at 1366 cm−1 is attributed to the stretching vibration of the carboxyl group in either FFCA or FDCA.7 Moreover, the band at 1389 cm−1 corresponds to FDCA formation. The significant downward vibration peak around 1680 cm−1 corresponds to the bending vibration of water molecules (δ(H2O)) or the bending vibration of hydroxyl groups (O–H bending). Next, we conducted the time-dependent in situ ATR-IR spectra at 1.477 V vs. RHE to investigate the reaction pathway. Fig. 3b shows that HMF is gradually consumed while FDCA is formed. During this transformation, the peak intensities at 1529 and 1569 cm−1, which are assigned to the formation of the HMFCA intermediate, were enhanced. However, we were unable to detect any bands for DFF in the IR spectrum. The IR absorption peak of DFF resembles that of HMF, and it is rapidly oxidized to FFCA under base-catalyzed conditions, making it difficult to detect. This result agrees well with the HPLC analysis, confirming that the Sc-NiFe-LDH-catalyzed HMF electrooxidation to FDCA follows the HMFCA pathway. | ||
| Fig. 3 (a) Potential-dependent in situ ATR-IR spectrum of Sc-NiFe-LDH in 1 M KOH with 10 mM HMF. (b) Time-dependent in situ ATR-IR spectrum of Sc-NiFe-LDH at the potential of 1.477 V vs. RHE in 1 M KOH with 10 mM HMF. (c) Potential-dependent in situ Raman spectrum of Sc-NiFe-LDH in 1 M KOH and (d) 1 M KOH with 10 mM HMF. | ||
We further employed in situ potential-dependent Raman spectroscopy to investigate the reaction mechanism of HMF oxidation over Sc-NiFe-LDH. For both HMFOR and OER processes, no significant signals were detected until the applied potential was up to 1.35 V vs. RHE. When the applied potential reached 1.35 V vs. RHE, the OER process showed two peaks at 473 and 557 cm−1, corresponding to NiIII–O vibrations,33 and the intensities were gradually increased with the increasing potential (Fig. 3c). However, the NiIII–O vibration peaks weakened when the applied potential reached 1.60 V vs. RHE, which is tentatively attributed to the generation of large amounts of bubbles during the OER process.34 Upon the addition of HMF, the NiIII–O vibrational signal was delayed until the applied potential reached 1.45 V vs. RHE (Fig. 3d). This suggests that the oxidation of HMF is accompanied by the reduction of Ni3+ to Ni2+ in NiOOH.35 When the applied potential exceeds 1.45 V vs. RHE, OER starts with the generation of an oxygen-containing intermediate that promotes the formation of NiOOH again.34
To explore the correlation between the electrochemical potential and interfacial reactions, in situ EIS measurements were conducted (Fig. 4a–d). The peak observed in the high-frequency range (101 to 105 Hz) is the signal of the electrode oxidation, while the peak in the low-frequency range (10−1–101 Hz) is typically attributed to the non-homogeneous charge distribution caused by the oxidized species forming on the electrode surface.36,37 Notably, an apparent transition peak was identified at 1.45 V vs. RHE for Sc-NiFe-LDH during OER in the absence of HMF, indicating the start of oxygen evolution; the result is in agreement with the LSV-measured OER onset potential38 (Fig. 4a and 2a).
 | ||
| Fig. 4 Bode phase plots of in situ EIS in 1 M KOH with and without 10 mM HMF: (a and b) Sc-NiFe-LDH; (c and d) NiFe-LDH. | ||
Upon adding HMF, an additional peak at 1.35 V vs. RHE, which is lower than that for OER, appeared for Sc-NiFe-LDH. This suggests a more favorable HMF oxidation compared to OER. It should be noted that the phase angle value of Sc-NiFe-LDH in the high-frequency region is significantly smaller than that of NiFe-LDH, indicating its faster reaction kinetics (Fig. 4c).39 Furthermore, in the potential range of 1.1–1.4 V vs. RHE, the phase angle value for Sc-NiFe-LDH remains smaller than that of NiFe-LDH, further confirming the enhanced reaction rate of HMF. When HMF was introduced, a low-frequency peak, indicative of OER, was observed for NiFe-LDH (Fig. 4d) at 1.45 V vs. RHE, while Sc-NiFe-LDH showed a peak at 1.50 V vs. RHE (Fig. 4b). This indicates that the OER begins at a lower potential for NiFe-LDH, confirming that Sc-NiFe-LDH catalysts can effectively lower the potential of the OER. The related equivalent circuit and Nyquist plots, as well as their fitted results, are shown in ESI Fig. S15 and Tables S3–S6.† Moreover, Sc-NiFe-LDH exhibits much lower resistances than NiFe-LDH in all the measured potentials, indicating more favorable kinetics for the oxidation of both the electrode and the HMF.40
The electrocatalytic HER performance of Sc-NiFe-LDH was evaluated in 1 M KOH with and without 10 mM HMF. The two LSV curves almost overlap with each other (Fig. 5a), and there is almost no change in the calculated Tafel slope (Fig. 5a inset) from 192.4 mV dec−1 to 195.3 mV dec−1. This indicates that the impact of HMF on the HER kinetics of Sc-NiFe-LDH is negligible.36 Moreover, there are also no noticeable voltage changes in the chronoamperometry experiments at −10 mA cm−2 for 110 h (Fig. 5b), showing the high stability of the Sc-NiFe-LDH electrode for HER. Furthermore, based on the outstanding electrocatalytic performance of Sc-NiFe-LDH for both HMFOR and HER, a dual-electrode cell employing Sc-NiFe-LDH as both the anode and cathode was assembled (Fig. 5c and d). The oxidation of HMF at the anode required a potential of 1.74 V to achieve a current density of 50 mA cm−2, which is significantly lower than the potential needed for water electrolysis (1.82 V). This performance is comparable to that of novel metal catalysts for overall water splitting, suggesting a promising dual-functional electrocatalyst for the simultaneous production of FDCA and H2. Moreover, the potential at a current density of 100 mA cm−2 is also better than the state-of-the-art electrocatalysts for HMFOR (Fig. 5e and Table S2, ESI†), highlighting the potential of the Sc-NiFe-LDH electrocatalyst for efficient and selective HMF oxidation to FDCA.32,41–53
 | ||
| Fig. 5 (a) Electrochemical system for the oxidation of HMF. (b) LSV curves of the HER of the Sc-NiFe-LDH electrode in 1 M KOH with and without 10 mM HMF. (c) LSV curves of Sc-NiFe-LDH || Sc-NiFe-LDH couple in 1 M KOH with and without 10 mM HMF. (d) Chronopotentiometry curve of the Sc-NiFe-LDH electrode at a current density of −10 mA cm−2 in 1 M KOH with 10 mM HMF. (e) Comparison of the potential at a current density of 100 mA cm−2 in this work and state-of-the-art works. | ||
4. Conclusion
We have shown here for the first time the rare-earth metal Sc doping in enhancing the electrooxidation efficiency of HMF. The Sc doping considerably improved the adsorption of HMF and promoted the formation of high-valence NiIII–O active species for the enhanced electrooxidation of HMF to FDCA. Using in situ EIS, we further showed that the Sc doping of NiFe-LDH effectively suppressed the OER for enhanced HMF electrooxidation. As a result, the Sc-NiFe-LDH-catalyzed HMF electrooxidation to FDCA proceeded with remarkable FDCA yield (96.7%), FE (96.5%), and 99.5% selectivity for FDCA at a current density of >600 mA cm−2 at a relatively low potential of 1.477 V vs. RHE. The findings in this work would contribute to the rational design of highly efficient electrocatalysts and understanding of the chemical behaviors of the dopants for enhanced electrochemistry. On the other hand, this work also paves the way for green, efficient and sustainable biomass upgradation to valuable bio-chemicals, promoting the synergy of pollution and carbon reduction in the polymer industry.Author contributions
Yufeng Wu: conceptualization, writing – original draft, writing – review & editing, supervision. Zhiyan Hou: writing – original draft. Changlong Wang: writing – review & editing, conceptualization, supervision.Data availability
Data are available on reasonable request from the authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
Financial support from the National Key R&D Program of China (2021YFC2902505), National Natural Science Foundation of China (52425004), the start-up funding and urban carbon neutrality technology innovation funding by Beijing University of Technology (C. W.) is gratefully acknowledgedReferences
- E. Barnard, J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- R. Cao, M.-Q. Zhang, Y. Jiao, Y. Li, B. Sun, D. Xiao, M. Wang and D. Ma, Nat. Sustainability, 2023, 6, 1685–1692 CrossRef.
- M. Li and S. Zhang, ACS Catal., 2024, 14, 2949–2958 CrossRef CAS.
- M. Song, Y. Wu, Z. Zhao, M. Zheng, C. Wang and J. Lu, Adv. Mater., 2024, 36, 2403234 CrossRef CAS PubMed.
- T. Fang, W. Jiang, T. Zheng, X. Yao and W. Zhu, Adv. Mater., 2024, 4, 2403728 CrossRef PubMed.
- Z. Jiang, Y. Zeng, D. Hu, R. Guo, K. Yan and R. Luque, Green Chem., 2023, 25, 871–892 RSC.
- Y. Song, S. Jiang, Y. He, Y. Wu, X. Wan, W. Xie, J. Wang, Z. Li, H. Duan and M. Shao, Fundam. Res., 2024, 4, 69–76 CrossRef CAS PubMed.
- D. Chen, Y. Ding, X. Cao, L. Wang, H. Lee, G. Lin, W. Li, G. Ding and L. Sun, Angew. Chem., Int. Ed., 2023, 62, e202309478 CrossRef CAS PubMed.
- Y. Yang, W. H. Lie, R. R. Unocic, J. A. Yuwono, M. Klingenhof, T. Merzdorf, P. W. Buchheister, M. Kroschel, A. Walker, L. C. Gallington, L. Thomsen, P. V. Kumar, P. Strasser, J. A. Scott and N. M. Bedford, Adv. Mater., 2023, 35, 2305573 CrossRef CAS PubMed.
- M. Cai, Q. Zhu, X. Wang, Z. Shao, L. Yao, H. Zeng, X. Wu, J. Chen, K. Huang and S. Feng, Adv. Mater., 2023, 35, 2209338 CrossRef CAS PubMed.
- H. Xu, G. Xin, W. Hu, Z. Zhang, C. Si, J. Chen, L. Lu, Y. Peng and X. Li, Appl. Catal., B, 2023, 339, 123157 CrossRef CAS.
- Y. Zhang, G. Hai, Z. Huang, Z. Liu, X. Huang and G. Wang, Adv. Energy Mater., 2024, 14, 2401449 CrossRef CAS.
- Y. Tang, Q. Liu, L. Dong, H. B. Wu and X.-Y. Yu, Appl. Catal., B, 2020, 266, 118627 CrossRef CAS.
- D. Li, W. Wan, Z. Wang, H. Wu, S. Wu, T. Jiang, G. Cai, C. Jiang and F. Ren, Adv. Energy Mater., 2022, 12, 2201913 CrossRef CAS.
- A. Pei, P. Wang, S. Zhang, Q. Zhang, X. Jiang, Z. Chen, W. Zhou, Q. Qin, R. Liu, R. Du, Z. Li, Y. Qiu, K. Yan, L. Gu, J. Ye, G. I. N. Waterhouse, W.-H. Huang, C.-L. Chen, Y. Zhao and G. Chen, Nat. Commun., 2024, 15, 5899 CrossRef CAS PubMed.
- B. Zhang, Y. Zheng, Z. Xing, Z. Wu, C. Cheng, T. Ma and S. Li, J. Mater. Chem. A, 2024, 12, 4484–4491 RSC.
- B. Fan, H. Zhang, B. Gu, F. Qiu, Q. Cao and W. Fang, J. Energy Chem., 2025, 100, 234–244 CrossRef CAS.
- W. Sun, N. Zhang, Y. Mao and K. Sun, J. Power Sources, 2012, 218, 352–356 CrossRef CAS.
- S. Hosseini, M. Shaterian, M. Vahedpour and M. A. Rezvani, Int. J. Hydrogen Energy, 2018, 43, 4961–4966 CrossRef CAS.
- J. Xu, Q. Zhang, X. Liang, J. Yan, J. Liu and Y. Wu, Nanoscale, 2020, 12, 6832–6843 RSC.
- S. Tamilarasi, R. S. Kumar, A. R. Kim, H. J. Kim and D. J. Yoo, Small, 2024, 2405939 CrossRef CAS PubMed.
- L. Tan, J. Yu, C. Wang, H. Wang, X. Liu, H. Gao, L. Xin, D. Liu, W. Hou and T. Zhan, Adv. Funct. Mater., 2022, 32, 2200951 CrossRef CAS.
- H. Ding, C. Su, J. Wu, H. Lv, Y. Tan, X. Tai, W. Wang, T. Zhou, Y. Lin, W. Chu, X. Wu, Y. Xie and C. Wu, J. Am. Chem. Soc., 2024, 146, 7858–7867 CrossRef CAS PubMed.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- C. Yang, C. Wang, L. Zhou, W. Duan, Y. Song, F. Zhang, Y. Zhen, J. Zhang, W. Bao, Y. Lu, D. Wang and F. Fu, Chem. Eng. J., 2021, 422, 130125 CrossRef CAS.
- F. Ye, S. Zhang, Q. Cheng, Y. Long, D. Liu, R. Paul, Y. Fang, Y. Su, L. Qu, L. Dai and C. Hu, Nat. Commun., 2023, 14, 2040 CrossRef CAS PubMed.
- Y. Qiang, D. Ouyang, L. You, D. Liu and X. Zhao, Chem. Eng. J., 2023, 469, 143832 CrossRef CAS.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS.
- X. Liu, X. Wang, C. Mao, J. Qiu, R. Wang, Y. Liu, Y. Chen and D. Wang, Angew. Chem., Int. Ed., 2024, 63, e202408109 CrossRef CAS PubMed.
- C. Wang, H.-J. Bongard, M. Yu and F. Schüth, ChemSusChem, 2021, 14, 5199–5206 CrossRef CAS PubMed.
- C. Wang, H.-J. Bongard, C. Weidenthaler, Y. Wu and F. Schüth, Chem. Mater., 2022, 34, 3123–3132 CrossRef CAS.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- Y. Xie, Z. Zhou, N. Yang and G. Zhao, Adv. Funct. Mater., 2021, 31, 2102886 CrossRef CAS.
- Y. Song, W. Xie, Y. Song, H. Li, S. Li, S. Jiang, J. Y. Lee and M. Shao, Appl. Catal., B, 2022, 312, 121400 CrossRef CAS.
- B. Zhou, Y. Li, Y. Zou, W. Chen, W. Zhou, M. Song, Y. Wu, Y. Lu, J. Liu, Y. Wang and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 22908–22914 CrossRef CAS PubMed.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., 2019, 131, 16042–16050 CrossRef.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- Y. Lu, C.-L. Dong, Y.-C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- P. Zhou, X. Lv, S. Tao, J. Wu, H. Wang, X. Wei, T. Wang, B. Zhou, Y. Lu, T. Frauenheim, X. Fu, S. Wang and Y. Zou, Adv. Mater., 2022, 34, 2204089 CrossRef CAS PubMed.
- G. Zhao, G. Hai, P. Zhou, Z. Liu, Y. Zhang, B. Peng, W. Xia, X. Huang and G. Wang, Adv. Funct. Mater., 2023, 33, 2213170 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- Z. Zhou, C. Chen, M. Gao, B. Xia and J. Zhang, Green Chem., 2019, 21, 6699–6706 RSC.
- X. Huang, J. Song, M. Hua, Z. Xie, S. Liu, T. Wu, G. Yang and B. Han, Green Chem., 2020, 22, 843–849 RSC.
- Y. Song, Z. Li, K. Fan, Z. Ren, W. Xie, Y. Yang, M. Shao and M. Wei, Appl. Catal., B, 2021, 299, 120669 CrossRef CAS.
- C. Chen, Z. Zhou, J. Liu, B. Zhu, H. Hu, Y. Yang, G. Chen, M. Gao and J. Zhang, Appl. Catal., B, 2022, 307, 121209 CrossRef CAS.
- R. Luo, Y. Li, L. Xing, N. Wang, R. Zhong, Z. Qian, C. Du, G. Yin, Y. Wang and L. Du, Appl. Catal., B, 2022, 311, 121357 CrossRef CAS.
- M. Xing, D. Zhang, D. Liu, C. Song and D. Wang, J. Colloid Interface Sci., 2023, 629, 451–460 CrossRef CAS PubMed.
- Y. Feng, R. Lee Smith, J. Fu and X. Qi, Green Chem., 2023, 25, 8698–8705 RSC.
- G. Ren, B. Liu, L. Liu, M. Hu, J. Zhu, X. Xu, P. Jing, J. Wu and J. Zhang, Inorg. Chem., 2023, 62, 12534–12547 CrossRef CAS PubMed.
- T. Wu, Z. Xu, X. Wang, M. Luo, Y. Xia, X. Zhang, J. Li, J. Liu, J. Wang, H.-L. Wang and F. Huang, Appl. Catal., B, 2023, 323, 122126 CrossRef CAS.
- R. Zhang, F. Gao, C. Yang, Y. Bian, G. Wang, K. Xue, J. Zhang, C. Wang and X. Gao, Mater. Today Nano, 2023, 23, 100373 CrossRef CAS.
- L. Zhao, S. Du, R. Gong, W. Jia, Z. Chen and Z. Ren, Molecules, 2023, 28, 3040 CrossRef CAS PubMed.
- D. Liu, Y. Li, C. Wang, H. Yang, R. Wang, S. Li and X. Yang, Appl. Catal., A, 2024, 669, 119497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04389h |
| This journal is © The Royal Society of Chemistry 2025 |