
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring uracil derivatives: synthesis, crystal structure insights, and antibacterial activity†
Susital
Mal
a,
Chris H. J.
Franco
 *b,
Binay
Kumar
a,
Alexander M.
Kirillov
b and
Subrata
Das
*a
*b,
Binay
Kumar
a,
Alexander M.
Kirillov
b and
Subrata
Das
*a
aDepartment of Chemistry, National Institute of Technology Patna, Bihar 800005, India. E-mail: subrataorgchem@gmail.com
bMINDlab: Molecular Design & Innovation Laboratory, Centro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Lisboa, Portugal. E-mail: chris.franco@tecnico.ulisboa.pt
First published on 14th May 2025
Abstract
As one of four nucleobases of RNA, uracil and its analogues represent an important class of bioactive pyrimidine derivatives. Their molecular arrangements in the solid state can be explored from the crystal engineering approach to obtain an understanding of structure–bioactivity correlations. In the present study, a series of uracil derivatives (compounds 1–4) was synthesized and fully characterized. The effect of the functionalization of the uracil core with different –NCHN(CH3)2, –CH3, –Cl, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, –NH2, and –CH2–COOH groups on stability, solubility, and antibacterial activity was investigated. The single-crystal structures of these compounds show that the hydrogen bonds formed by distinct synthons (R22(8), R44(12), C11(6)) contribute to framework stability. The presence of water molecules in the lattice is an important feature, as they provide additional H-bonding interactions that influence lattice energy and solubility. Lattice energy minimization, Hirshfeld surface analysis, and 2D fingerprint plots were employed to investigate intermolecular interactions and the stability of the obtained uracil derivatives, particularly the effect of functional groups. Although all compounds exhibit antibacterial behavior, the derivatives with small polar functional groups revealed an enhanced activity against Gram-negative bacteria, while the compounds with moderately polar substituents are more active against Gram-positive bacteria. The established discussions expand the comprehension of uracil chemistry and highlight the relationship between crystal structure and the resulting properties of the compounds, thus contributing to the rational development of new antibacterial agents.
S, –NH2, and –CH2–COOH groups on stability, solubility, and antibacterial activity was investigated. The single-crystal structures of these compounds show that the hydrogen bonds formed by distinct synthons (R22(8), R44(12), C11(6)) contribute to framework stability. The presence of water molecules in the lattice is an important feature, as they provide additional H-bonding interactions that influence lattice energy and solubility. Lattice energy minimization, Hirshfeld surface analysis, and 2D fingerprint plots were employed to investigate intermolecular interactions and the stability of the obtained uracil derivatives, particularly the effect of functional groups. Although all compounds exhibit antibacterial behavior, the derivatives with small polar functional groups revealed an enhanced activity against Gram-negative bacteria, while the compounds with moderately polar substituents are more active against Gram-positive bacteria. The established discussions expand the comprehension of uracil chemistry and highlight the relationship between crystal structure and the resulting properties of the compounds, thus contributing to the rational development of new antibacterial agents.
Introduction
As one of the four nucleobases in RNA, uracil and its analogues form a crucial group of bioactive pyrimidine derivatives.1,2 These are often functionalized at different positions of the pyrimidine ring, resulting in a variety of molecular arrangements (Scheme 1). Investigating these arrangements in the solid state can give insights into the relationship between structure and bioactivity.3–8 In particular, uracil derivatives feature a broad range of bioactivity1,9–15 that makes them appealing for exploring chemical and biological properties, aiming at the development of potential antiviral,16 antitumor,17 antiplasmodial,18 antidiabetic,19 and antibacterial20,21 molecules. However, the therapeutic application of uracil derivatives is still limited because of toxicity levels in several human body systems,10 thus inspiring the synthesis of new derivatives with improved pharmacological and pharmacokinetic properties.1,10 | ||
| Scheme 1 Functionalization of uracil to derivatives 1–4. 1: (E)-N,N-dimethyl-N′-(3-methyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)formimidamide, C8H12N4O2. 2: 6-chloro-3-methylpyrimidine-2,4(1H,3H)-dione, C5H5N2O2Cl. 3: 2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetic acid, C7H8N2O4. 4: 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one, C4H5N3OS. | ||
Furthermore, the physicochemical and biological significance of these compounds is profoundly influenced by their capacity to establish hydrogen bonds and other non-covalent interactions, such as halogen bonding and π-stacking interactions.22–31 Among the various pyrimidine bases, uracil derivatives exhibit both hydrogen bond donor and acceptor sites, thus acting as robust H-bonded synthons.22 Despite being weaker than most covalent bonds,32,33 hydrogen bonds are essential to consider in crystal engineering, given their prevalence in biological systems. Many aspects of biochemistry are a result of these weak interactions in an aqueous environment,32 where H-bonding often determines the self-organization of organic molecules such as proteins, ribonucleic acid (RNA), and deoxyribonucleic acid (DNA).31
The elucidation of molecular arrangements in the solid state via crystal engineering has become a valuable tool, particularly for pyrimidine and uracil derivatives.34–36 Distinct supramolecular synthons can stabilize crystal packing through diverse intermolecular interactions, enabling the design of materials with desirable properties.37–40 Given the presence of uracil derivatives in some drugs, further diversification of uracil skeletons and synthesis of new molecules with prospective bioactivity is a current focus of synthetic chemistry, particularly with regard to fine-tuning of the physicochemical properties including solubility and stability.41–43
Inspired by our interest in uracil chemistry44–46 and development of new antimicrobial compounds,47 in the present work we focused on the synthesis, detailed structural characterization and antibacterial activity of several uracil derivatives (Scheme 1). The single-crystal X-ray structures of two new products, C8H12N4O2·H2O (1) and C7H8N2O4·2H2O (3), were determined and compared to related compounds such as uracil,48 C5H5N2O2Cl (2),36 and C4H5N3OS·H2O (4).49 The molecular Hirshfeld surface analysis and energy frameworks were used to verify the differences in the crystal structures and establish possible correlations between structure and bioactivity. This work widens the family of bioactive uracil derivatives.
Experimental
Synthesis and crystallization
 | ||
| Scheme 2 Synthesis of compound 1. | ||
 | ||
| Scheme 3 Synthesis of compound 3. | ||
Single-crystal X-ray diffraction
X-ray data for compounds 1 and 3 were acquired at room temperature (298 K) using a Rigaku Oxford Diffraction SuperNova diffractometer equipped with a high sensitivity EosS2 CCD detector and a Mo-Kα radiation (λ = 0.71073 Å). CrysAlisPro (ver. 1.171.40.69a, 2020)52 program package was used for data collection, data reduction, and absorption correction. For compound 2, the X-ray data were collected on a Bruker AXS APEXIII diffractometer with PHOTON II CCD detector using Cu-Kα radiation (λ = 1.54178 Å). Data collection and reduction were performed using APEXIII53 and SAINT.54 SADABS was used for absorption corrections.55 X-ray data for compound 4 were collected on a Bruker D8 Venture Diffractometer with PHOTON II detector using a Mo-Kα radiation (λ = 0.71073 Å) at room temperature (298 K). CrysAlisPro (ver. 1.171.40.69a, 2020) program package was used for data treatment and correction.52 With Olex256 as the graphical interface, the crystal structures of all compounds were solved with the SHELXS structure solution program using direct methods (SHELXS). The structures were refined with the SHELXL57 refinement package using the least squares minimization on F2. All non-hydrogen atoms were refined anisotropically, while hydrogen atom positions were calculated geometrically and placed in idealized positions. Methyl hydrogen atoms were constrained by C–H = 0.96 Å and Uiso(H) = 1.5Ueq(C); however, H atoms connected to nitrogen centers were constrained by N–H = 0.86 Å and Uiso(H) = 1.2Ueq(N). In the crystal structure of 1, a water molecule was found to be disordered over two positions. The disorder was modeled by splitting the oxygen atom into two sites, O3 and O3A, with fixed occupancies of 0.50 each, corresponding to a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio. The hydrogen atoms associated with each water position (H3A, H3B for O3; H3AA, H3AB for O3A) were placed in idealized positions and refined with distance restraints (O–H ≈ 0.84 Å, H⋯H ≈ 1.36 Å) and angle constraints (H–O–H ≈ 104.5°). The Mercury program58 was used to draw the molecules, crystal packing, and motifs. Crystal data and refinement parameters for compounds 1–4 are summarized in Table S1, ESI.†36,49 CCDC codes: 2306291 (1) and 2306292 (3).
:50 ratio. The hydrogen atoms associated with each water position (H3A, H3B for O3; H3AA, H3AB for O3A) were placed in idealized positions and refined with distance restraints (O–H ≈ 0.84 Å, H⋯H ≈ 1.36 Å) and angle constraints (H–O–H ≈ 104.5°). The Mercury program58 was used to draw the molecules, crystal packing, and motifs. Crystal data and refinement parameters for compounds 1–4 are summarized in Table S1, ESI.†36,49 CCDC codes: 2306291 (1) and 2306292 (3).
Hirshfeld surface analysis and energy frameworks
For initial geometry optimization, the experimental crystal structures of uracil (CSD code URACIL)48 and its derivatives were obtained from crystallographic information files (cif files). The positional disorder in compound 1 was preserved as modeled throughout the single-crystal X-ray refinement before the structure was loaded into CrystalExplorer.59 All model energies were calculated using molecular electron densities derived from wavefunctions computed by the Gaussian09 suite.60 The intermolecular interactions were investigated using Hirshfeld surface61,62 and 2D fingerprint plots,63,64 in addition to all molecular clusters' calculations, with a radius ranging from 3.8 to 22 Å. These calculations were performed at the B3LYP/6-31G(d,p) level of theory using the Crystal Explorer package.59 The energy frameworks and lattice energy were also calculated with the B3LYP/6-31G(d,p) level of theory, and lattice energies65 were considered convergent when they did not present consistent changes of less than 1 kJ mol−1, with the variation of the cut-off radius of interactions between pairs of molecules (convergence data can be found in the ESI,† Table S2, Fig. S1 and S2).Solubility studies
The solubility of compounds 1–4 was evaluated following a previously described procedure.35 Distinct concentrations of each compound were prepared to establish a calibration curve (concentration–absorbance graph based on a λmax value) employing UV-visible spectroscopy (Shimadzu UV-1780 spectrophotometer). Simultaneously, by adding an excess of each component to 2 mL of distilled water (pH = 6.8) and stirring for 24 h at room temperature, saturated solutions of each compound were prepared. Following this step, the resulting solution underwent centrifugation for 15 min. The clear solution obtained was subsequently diluted by a factor of 1000. The absorbance of the supernatant solution was evaluated, and then the concentration of each compound was calculated from the calibration curve. The solubility was determined by multiplying the concentration of the resulting solution by a factor of 1000. (detailed data can be found in Fig. S15 and S16, ESI†).Antibacterial Activity Investigation: compounds 1–4, each at a consistent concentration, were dissolved in MilliQ-grade water. Prior to experimentation, all bacterial cells were subjected to purification through the streaking method to obtain single, isolated colonies. These isolated colonies were cultured in Luria Bertani (LB) broth for 7–8 h, depending on the microorganism under investigation. A known quantity of each compound was introduced into a fresh LB broth, along with a fixed number of bacterial cells, followed by incubation at 37 °C. Concurrently, a fixed concentration of cells was introduced into LB agar media to facilitate visual inspection of bacterial growth. Optical density (OD) measurements were taken at 600 nm after 8–9 h of incubation.
Results and discussion
Structural features and solid-state studies
In this work, all uracil analogues are pyrimidine derivatives having a heterocyclic aromatic ring and a ketone group along with different substituents (–NCHN(CH3)2, –CH3, –Cl,S, –NH2, and –CH2–COOH groups), which represent the core pharmacophore of several drugs.19 Four uracil derivatives were synthesized (1–4, Scheme 1) and their molecular structures (Fig. 1a–d) were investigated using single-crystal X-ray diffraction and theoretical approaches (Hirsfeld surface, 2D-finger plots, electrostatic potential isosurface, energy frameworks, and lattice energy) to identify and compare the effects of different functional groups.
 | ||
| Fig. 1 Molecular structures of compounds (a) 1, (b) 2, (c) 3, and (d) 4, including atom labelling scheme. Thermal ellipsoids are drawn at the 50% probability level. H atoms were omitted for clarity. Molecular electrostatic potential isosurfaces for crystal structures of (e) 1, (f) 2, (g) 3, and (h) 4. The electron density surface drawn at ±0.05 a.u. contour. The quantum mechanical proprieties obtained using the B3LYP/6-31G(d,p) wave function. | ||
Compound 1 (C8H20N4O2·H2O), a mono-methyl-amino substituted uracil, crystallizes in a triclinic system and space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . There is some disorder that affects only one water molecule across the inversion center with a ratio of 50:50 at 0.7 Å distance. Thus, it is expected that the local energy minima are not affected by significant differences in total lattice energy. The mono-methyl-acid substituted uracil derivative, compound 3 (C7H16N2O4·2H2O), crystallizes in the same crystallographic system (Fig. 1). Two water molecules present in the crystal structure of 3 are responsible for extending the H-bonding network. The presence of –COOH group is evident from the difference between the C–O distance of carboxylate group, being also in agreement with the FTIR data (1703 cm−1). The protected amino group in 1 is practically planar to the uracil ring orientation, with the torsion C3–C4–N3–C5 angle of −8.64°. However, this angle in 3 is 110.78° (C4–N2–C6–C7) for the substituted acid group, which is potentially a result of intermolecular H-bonds. The bonding parameters for compounds 1–4 are listed in Tables S5 and S7 (ESI†). The mono-methyl-choro derivative 2, (C5H5N2O2Cl), and the amino-thiooxo derivative 4, (C4H5N3OS·H2O) crystallize in a system of high symmetry (monoclinic) with the space groups P21/c and C2/c, respectively (Fig. 1, Table S1†). The substitution of functional groups produces minor changes in the main uracil ring. The CC double bond lengths in 1–4 range between 1.340 and 1.378 Å, and agree with the distance in uracil (1.340 Å).48
. There is some disorder that affects only one water molecule across the inversion center with a ratio of 50:50 at 0.7 Å distance. Thus, it is expected that the local energy minima are not affected by significant differences in total lattice energy. The mono-methyl-acid substituted uracil derivative, compound 3 (C7H16N2O4·2H2O), crystallizes in the same crystallographic system (Fig. 1). Two water molecules present in the crystal structure of 3 are responsible for extending the H-bonding network. The presence of –COOH group is evident from the difference between the C–O distance of carboxylate group, being also in agreement with the FTIR data (1703 cm−1). The protected amino group in 1 is practically planar to the uracil ring orientation, with the torsion C3–C4–N3–C5 angle of −8.64°. However, this angle in 3 is 110.78° (C4–N2–C6–C7) for the substituted acid group, which is potentially a result of intermolecular H-bonds. The bonding parameters for compounds 1–4 are listed in Tables S5 and S7 (ESI†). The mono-methyl-choro derivative 2, (C5H5N2O2Cl), and the amino-thiooxo derivative 4, (C4H5N3OS·H2O) crystallize in a system of high symmetry (monoclinic) with the space groups P21/c and C2/c, respectively (Fig. 1, Table S1†). The substitution of functional groups produces minor changes in the main uracil ring. The CC double bond lengths in 1–4 range between 1.340 and 1.378 Å, and agree with the distance in uracil (1.340 Å).48
The molecular electrostatic potential isosurface map for the compounds 1–4 can be utilized for predicting non-covalent interactions.22 The isosurface was calculated at the B3LYP/6-31G(d,p) level of theory using the Crystal Explorer package.59,66 From the surface data in Fig. 1e–h, it is possible to note for 1 that the N1 atom exhibits an increased tendency to donate a proton (as indicated by the blue color). In contrast, the ketone oxygen atoms, shown in red, tend to accept a proton, according to the electrostatic potential representation. In compound 3, the same tendency is observed. However, the –COOH group tends to donate and accept protons from water molecules in the crystal lattice. For compounds 2 and 4, the acceptor atoms show a negative electrostatic potential with Cl atoms involved in donor sites with methyl-substituted groups. Intriguingly, the sulfur atom demonstrates a small partial region to accept protons (red color). Based on these data, the pyrimidinone ring protons show positive electrostatic potential and probably contribute to the non-covalent interactions with the available sites. This trend is similar in all analyzed crystal structures.
Because of differences in space group configurations, water molecules content, and unit cell features, each compound has a distinct crystal packing that is influenced by the electrostatic potential of the molecules and H-bonds.27,67 Considering only short-range contact D–H⋯A type hydrogen bonds [H⋯A ≤ 2.75 Å, D⋯A ≤ 3.50 Å, and ∠(D–H⋯A) > 120°], the main structural motif in 1 is a R22(8) synthon (Fig. 2a), formed by molecular dimers with the N–H⋯O type hydrogen bonds [dN1⋯O2 = 2.8658(19) Å]. There is a 1D zig-zag chain along the [201] direction through long-range contact H-bonds derived from the disordered water molecule [dO⋯O = 2.956(5) and 2.637(11) Å]. As a result, the independent 1D chains are arranged into column motifs stabilized by H-bonding interactions (Fig. S10, ESI†). Compound 3 shows two main structural motifs – the synthons R22(8) and R44(12) produced by molecular dimers [dN1⋯O2 = 2.860(3) Å] and water molecules [dO6⋯O1 = 2.858(3) Å and dO6⋯O2 = 2.917(3) Å] (Fig. 3a). These discrete units are connected by three types of H-bonds, resulting in the development of 2D sheets that are stacked by non-classic long-range H-bonds (C–H⋯O); all observations are consistent with electrostatic isosurfaces. Consequently, this compound tends to be more soluble in polar solvents, since these larger distances result in weaker and more dispersed interactions in the lattice. Differently, the structure of 2 has no crystallization solvent molecules and propagates in a 1D chain parallel to the b-axis by C11(6) synthons [dN1⋯O2 = 2.785(5) Å] as shown in Fig. 2b. In contrast, in the compound 4, the molecules are arranged in parallel 1D column motifs along the c-axis and connected by the N–H⋯O hydrogen bonds [dN2⋯O1 = 2.7364(17) Å and dN3⋯O1 = 3.136(2) Å] in bifurcated mode with C11(6) synthons (Fig. 3b). The interactions involving the NH2 group are not significant. The water molecules (trifurcated mode) connect the perpendicular 1D column motifs and establish a 3D network via the O–H⋯O and N–H⋯O hydrogen bonding [dN1⋯O2 = 2.8528(19) Å, dN3⋯O2 = 3.109(2) Å and dO2⋯O1 = 2.7632(19) Å] (Fig. S11, ESI†). Although the crystallographic data suggest a weak O–H⋯S interaction due to the long contact distance [dO2⋯S1 = 3.4889(15) Å], the electrostatic potential surface reveals that such interaction is important for stabilization of the crystal structure.
 | ||
| Fig. 2 Crystal packing diagrams of (a) 1 and (b) 2 showing the supramolecular R22(8) and C11(6) synthons (highlighted in light blue) and the arrangement of molecules. | ||
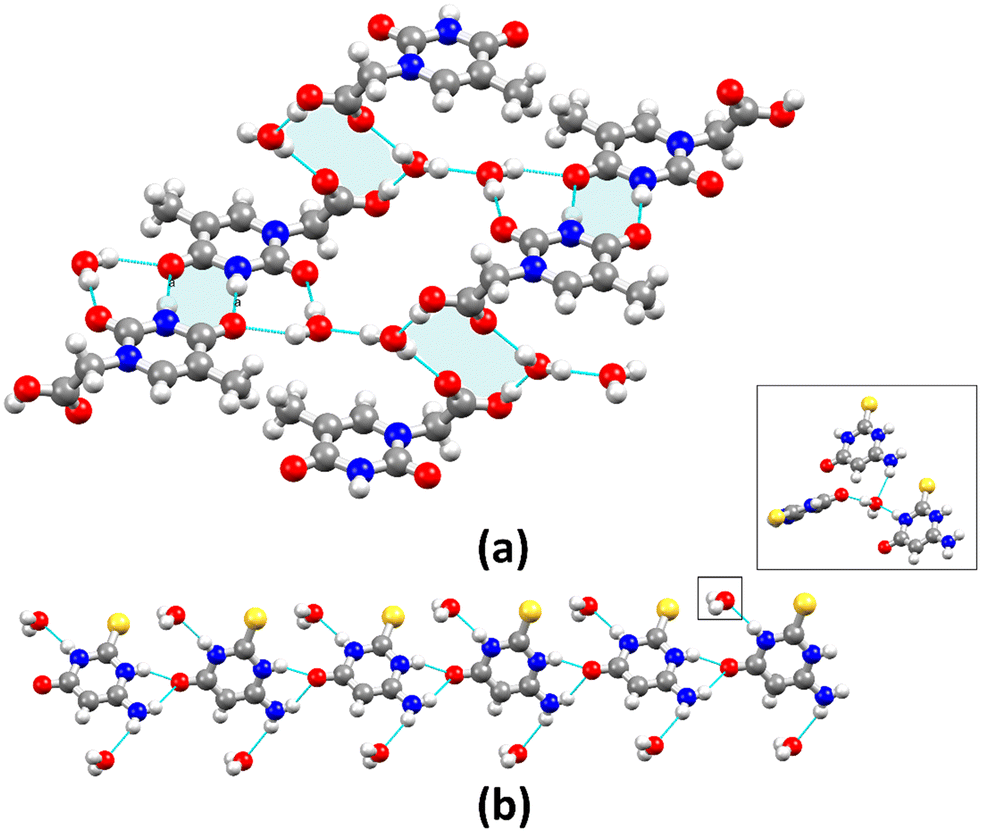 | ||
| Fig. 3 Crystal packing diagrams of (a) 3 and (b) 4 showing the supramolecular R22(8) and R44(12) synthons in 3 and the C11(6) synthon in 4 (highlighted in light blue), as well as the arrangement of molecules. Inset represents water molecules in trifurcate mode. | ||
The Hirshfeld surface (HS) analysis and 2D-fingerprint plots (2D-FP) were used to assess how the presence of different functional groups can affect the physicochemical properties and biological activity of compounds in the present study. The results were compared to the uracil precursor. In addition, energy framework and lattice energy parameters were used to get further insight on the molecular interactions and to predict some properties. Fig. 4 displays the HS mapped over dnorm, shape index, and 2D-fingerprint plot for the uracil molecule. The HS reveals the electron density accumulation and depletion regions around each atom, providing information on the nature of intermolecular interactions in the crystal structure.68,69 The intense red spots over the dnorm surface indicate the short intermolecular contacts, which are less than the sum of the vdW (van der Waals) radii of interacting atoms.37,69 The blue regions relate to longer contacts, and the white regions refer to the vdW contacts. Uracil shows three H-bonds of the N–H⋯O type with intense red areas (dnorm), which represent a main R66(32) synthon generated by molecular dimers (Fig. S12, ESI†), leading to the growth of 2D sheets stacked by weak interactions from pyrimidinone ring (Fig. 4b, red and blue triangular shapes above the rings) as expected to electrostatic potentials isosurface (Fig. S13†).
 | ||
| Fig. 4 Hirshfeld surface analysis of uracil. (a) Perpendicular view showing three short intermolecular contacts (red spots). (b) Shape index plots surface, proving the stacking of uracil molecules by weak pyrimidinone ring interactions. (c) 2D-fingerprint plot of N–H⋯O contacts. | ||
By examining the 2D-FP, it is possible to obtain the contribution of each type of interaction in stabilizing the crystal packing.63,69,70 Most interactions in uracil involve symmetric N–H⋯O contacts (55%). In contrast, van der Waals interactions represented by H⋯H contacts account for 15.9% of total interactions. These two types of interactions represent 70.9% of total interactions. The functionalization of the pyrimidinone ring via the incorporation of –CH3 and –NCHN(CH3)2 groups in 1 lead to a reduction of crystallographic symmetry and a change of the distribution of electron density in the main ring and, consequently, in all interactions. The 2D-FP of 1 reveals that the predominant interactions are H⋯H (46.2%), followed by the N–H⋯O and C–H⋯O hydrogen bonds (28.2%); these three interactions correspond to 70.4% of total interactions similar those of uracil (Fig. 5a). Disordered water molecules are present in the lattice and slightly influence on the stabilization of crystal packing (long-range D–H⋯A distance, Table S7†). The C⋯H and C⋯C contacts originating from methyl group show a secondary role in stabilizing the supramolecular structure of 1 (10.1% and 2.2%, respectively). In the methyl-chloro analogue (compound 2), two N–H⋯O and C–H⋯O types of H-bonds display intense red spots with 29.4% of interactions (Fig. 5b). Nonetheless, a much longer intermolecular C–H⋯O intermolecular contact shows pale red spots. Contacts involving the Cl atom contribute to 20.9% of all contacts. In contrast to 1, the water molecules in 3 are associated into a 2D sheet by the O–H⋯O and N–H⋯O hydrogen bonding along with C–H⋯O interactions (44.6% of contribution), thus directly affecting the stability of network (bright red spots, Fig. 5c). The contribution of the H⋯H contacts (vdW interactions) from methyl group is 34.9%. Particularly, the contribution of O⋯H contacts is increased (∼20%) due to the presence of different substituents in 3 if compared to 1. In 4, the S⋯H contacts account for 19.5% of interactions. They derive from the –NH2 functional group (Fig. 5d). The N–H⋯O interactions are more significant (26.4%) and responsible for the extension of crystal structure along the c-axis. The H⋯H contacts present due to the incorporation of methyl group in 4 account for 24.3% (more details for 2D-FP see Fig. S14†).
 | ||
| Fig. 5 Hirshfeld surfaces with short intermolecular contacts (red spots) in (a) 1, (b) 2, (c) 3, and (d) 4 (two perpendicular views). In the right column, 2D fingerprint plots of main contacts are represented. | ||
Intermolecular interactions37 can affect various properties of molecular compounds. The stability of 1–4 can be evaluated via lattice energy minimization (Elattice).65,71,72 The lattice energies can be estimated by the direct sum of the total interaction energies between pairs i and j ( ), until the lattice energy converted to a limit of 1 kJ mol−1. All these calculations were performed in Crystal Explorer,59 using a radius range of 3.8 to 22 Å (details about minimization process can be found in ESI†), and the calculated total energy values for all crystal structures are displayed in Scheme 4. The obtained values are consistent with those available in the literature for comparable compounds.43,72 The presence of water in the lattice is seen as an essential feature, since H-bonds involving H2O molecules provide additional intermolecular interactions, which can increase the lattice energy (Table S2, ESI†). According to Scheme 4, the stability order (kJ mol−1) of compounds is as follows: 4 > 3 > 1 > uracil > 2.
), until the lattice energy converted to a limit of 1 kJ mol−1. All these calculations were performed in Crystal Explorer,59 using a radius range of 3.8 to 22 Å (details about minimization process can be found in ESI†), and the calculated total energy values for all crystal structures are displayed in Scheme 4. The obtained values are consistent with those available in the literature for comparable compounds.43,72 The presence of water in the lattice is seen as an essential feature, since H-bonds involving H2O molecules provide additional intermolecular interactions, which can increase the lattice energy (Table S2, ESI†). According to Scheme 4, the stability order (kJ mol−1) of compounds is as follows: 4 > 3 > 1 > uracil > 2.
 | ||
| Scheme 4 Lattice energy profile and simplified energy frameworks for uracil and derivatives 1–4. Total framework interaction energies (blue) are shown as cylindrical tube size with 150 u.a. and cut-off 15 kJ mol−1. | ||
The analysis of the results reveals that the type of functional group affects the stability of uracil derivatives 1–4, influencing on how the electrostatic potential on the isosurface is distributed. While in uracil the major contribution to the lattice energy is derived from the energies of H-bond interactions (N–H⋯O motifs), in compound 1 the dispersion energy assumes a greater contribution, directly affecting the solubility values. The presence of solvated water facilitates the organization of organic molecules and their stabilization by long-range H-bonds, which decreases the lattice energy and reduces solubility. As for 2, the modification of uracil structure reduces the dimensionality and concentrates on the greatest energy contribution in a single direction along the crystallographic c-axis. The interactions caused by the presence of –Cl substituent (29%) present only a small contribution to total stability. Compound 3 features carboxylic acid group that affects the charge distribution, with Coulomb energy donating the most. It is worth noting that in 4, despite the formation of infinite 1D chains via intermolecular interactions, crystallization water molecules form H-bonds in a trifurcated mode, which provides 30% to the overall value of the lattice energy (−57.7 kJ mol−1) and favors the formation of stable lattice. Despite the N–H⋯O interactions are responsible for the formation of dimers between molecules of all the analyzed compounds (preferential binding mode is the double CO⋯H–N hydrogen bond). Consequently, in energy framework distribution, it is possible to notice that H-bonds from water molecules significantly influence the crystal lattice energy by contributing to the overall stability of the lattice and affecting the packing arrangement of molecules.
Antibacterial properties
Antibacterial activity of 1–4 was investigated using the pre-grown colonies of E. coli73 (Gram-negative) and S. aureus74 (Gram-positive), following the previously described protocol.75 The percentage of bacterial growth inhibition by 1–4 as well as solubility data are summarized in Table 1. Additional details on the solubility tests (Fig. S15 and S16) and antibacterial screening (Tables S8 and S9, Fig. S17–S24) are given in ESI.† The inhibitory activity (in %) of compounds 1–4 was determined at maximum concentrations of 5.6, 4.7, 12.5, and 2.2 μg mL−1 relative to negative control (water), and results are shown in Fig. 6. A comparison of the inhibitory activity of 1–4 suggests that the inhibition of bacterial activity is not proportional to the concentration of uracil derivatives in solution (Table 1). The compounds show a varying degree of bacterial growth inhibition. The presence of different functional groups influences the nature of non-covalent interactions, which may affect the antibacterial activity. Gram-positive and Gram-negative bacteria respond differently to uracil derivatives, depending on the size and polarity of the incorporated functional groups. It reveals a correlation between the incorporation of functional groups and the electrostatic potential of the pyrimidine ring. Small polar functional groups enhance the activity against Gram-negative bacteria (compound 4), suggesting a favorable interaction with the bacterial cell surfaces. In contrast, the introduction of both larger and smaller polar groups with greater density dispersion favors antibacterial activity against Gram-positive bacteria (compound 1) in the C4 position.| Compound | λmax | Saturation solubility (μg mL−1) | Inhibition (%) | |
|---|---|---|---|---|
| (E. coli) | (S. aureus) | |||
| 1 | 305 | 5.6 | 17.8 | 12.9 |
| 2 | 263 | 4.7 | 15.9 | 3.3 |
| 3 | 272 | 12.5 | 31.1 | 7.1 |
| 4 | 275 | 2.2 | 12.9 | 2.1 |
 | ||
| Fig. 6 Images of Petri dishes revealing the antibacterial activity of compounds 1 (a and b), 2 (c and d), 3 (e and f), and 4 (g and h) against E. coli (a–g) and S. aureus (b–h). Maximum concentrations (saturation solubility) of 1–4 were used. Conditions are those of Table 1. | ||
Although compounds 1 and 4 exhibit only a small difference in lattice energy (10.57 kJ mol−1), they display opposite trends in the wavenumber of their main absorption bands. This suggests that hydrogen bonding may influence the electronic structures of the compounds and contribute to the observed bathochromic effect (Table S4, Fig. S9†). The bathochromic shift observed in compound 1 can be attributed to enhanced charge delocalization (Fig. 7), resulting in a partial positive charge on nitrogen atom (N4). The resulting resonance-stabilized system promotes coplanarity between the formimidamide moiety and the adjacent heterocycle, stabilizing the electronic structure and shifting the absorption band to lower energy. A similar delocalization pattern is observed in compound 4, which is consistent with the X-ray crystallographic data. The resembling structural features were observed in both compounds 1 and 4—specifically, the elongation of the CC bond and the shortening of the C–C(O) bond if compared to 2 and 3. Moreover, the variation in spectroscopic and structural properties observed among the compounds can also be further clarified by differences in hydrogen bonding and van der Waals interactions.
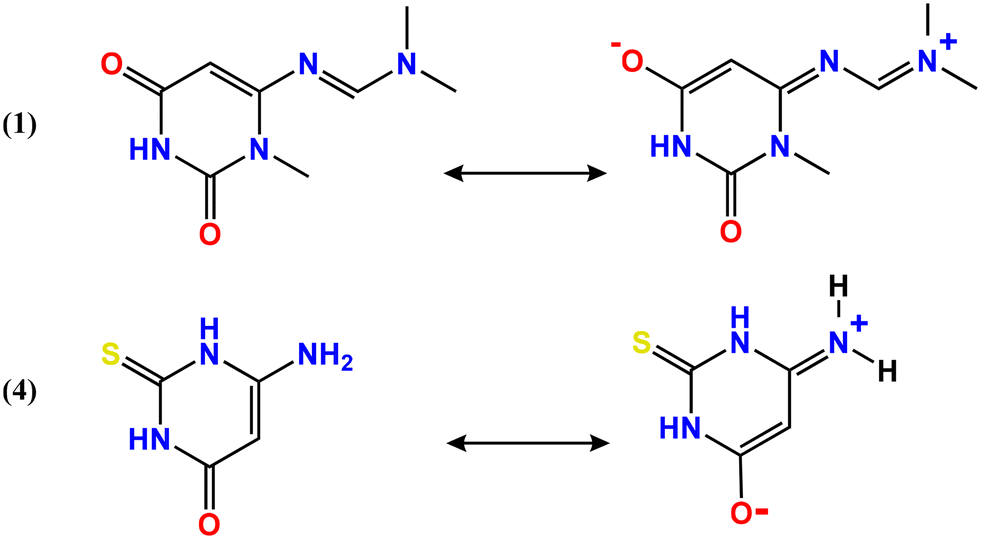 | ||
| Fig. 7 Resonance and charge delocalization in compounds 1 and 4. | ||
Notably, while the N–H⋯O and C–H⋯O interactions contribute similarly in both compounds, the H⋯H interactions exhibit a ratio of approximately 2:1, proportionally influencing the solubility and antibacterial activities of the compounds. In the case of 3, the introduction of carboxylic acid functionality, which promotes the formation of H-bonds within the crystal lattice, results in a reduction of the lattice energy compared to compound 2 and uracil. However, the antibacterial activity of compound 3 is negatively influenced by these modifications. When comparing the solubility, 3 is the most soluble, as expected since it presents a 2D dispersion of H-bond interactions as previously described. Nonetheless, it displays a lower inhibition in contrast to the concentration used. While lattice energy of 2 exhibits values of −112.7 kJ mol−1, it is essential to note that this compound exists in an anhydrous form. The organization of H-bond interactions [C11(6) synthons] plays an important role in stabilizing this structure, resulting in reduced solubility. The primary contributor to bioactivity remains the functional group rather than the solvation effects. These observations provide an elucidation of the key role of functional groups in customizing uracil derivatives for antibacterial applications.
Conclusions
In the current work, we reported a simple synthetic procedure for two new uracil derivatives, 1 and 3, as well as succeeded in the crystallization of two other uracil analogs, 2 and 4. All compounds were analyzed using standard techniques (single crystal X-ray diffraction, FTIR, UV-vis and NMR spectroscopy). The structural details were further emphasized using Hirshfeld surface analysis, energy frameworks and 2D-fingerprint plots.Despite the structural differences and space groups, the structures of compounds 1, 2 and 4 reveal the formation of 1D H-bonded motifs, while compound 3 presents a 2D supramolecular arrangement with a greater dispersion of interactions. The presence of H-bonds increases the lattice energy, especially for compounds with crystallization water molecules (1, 2, and 4), where the arrangement of H-bonds can also affect the solubility. Furthermore, the antibacterial activity of these compounds against Gram-negative (E. coli) and Gram-positive (S. aureus) bacteria were evaluated, and the presence of different functional groups within the uracil core appeared to be closely associated with antibacterial activity. Small polar functional groups tend to increase activity against Gram-negative bacteria (compound 4), while the introduction of larger and smaller polar groups with greater density dispersion (compound 1) favors antibacterial activity against Gram-positive bacteria. Despite having greater solubility in aqueous medium, compound 3 presents a lower antibacterial activity.
The interaction of these compounds with bacterial cell surfaces is influenced by variations in the electrostatic potential of the pyrimidine ring driven by changes in functional groups and the distribution of non-covalent interactions in the structure. Hence, this work not only expands our knowledge about pyrimidine and uracil derivatives but also contributes to the rational design of new bioactive uracil derivatives.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and included in the ESI.† Crystallographic data for the compounds have been deposited at the CCDC under CCDC numbers 2306291 and 2306292 and can be freely obtained from https://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. and S. D. are grateful to NIT Patna for providing research facilities and acknowledging the DST grant (EEQ/2019/000294) for financial assistance. We express our gratitude to IIT(ISM) Dhanbad, and SAIF IIT Patna for instrumental facilities. C. H. J. F. acknowledges the research contracts within the PTDC/QUI-QIN/29697/2017 and PTDC/QUI-QIN/3898/2020 projects (2021-2024). The authors also thank the Foundation for Science and Technology (FCT, Portugal: PTDC/QUI-QIN/3898/2020, PTDC/QUI-QIN/29697/2017, UIDB/00100/2020, LA/P/0056/2020, and UID/00100/2023).References
- H. U. Rashid, M. A. U. Martines, A. P. Duarte, J. Jorge, S. Rasool, R. Muhammad, N. Ahmad and M. N. Umar, RSC Adv., 2021, 11, 6060–6098 RSC.
- O. Michalak, P. Cmoch, P. Krzeczyński, M. Cybulski and A. Leś, Org. Biomol. Chem., 2019, 17, 354–362 RSC.
- K. Kajal, R. Shakya, M. Rashid, V. Nigam, B. D. Kurmi, G. D. Gupta and P. Patel, Sustainable Chem. Pharm., 2024, 37, 101374 CrossRef CAS.
- J. Ye, H. Pei, D. Liu, F. Wang, Y. Yu, J. Zhang and L. Zhang, Tetrahedron Lett., 2024, 142, 155101 Search PubMed.
- K. N. Jarzembska, M. Kubsik, R. Kamiński, K. Wozniak and P. M. Dominiak, Cryst. Growth Des., 2012, 12, 2508–2524 CrossRef CAS.
- B. Cui, B. Yan, K. Wang, L. Li, S. Chen and Z. Zhang, J. Med. Chem., 2022, 65, 12747–12780 CrossRef CAS PubMed.
- P. Chakraborty and P. Dastidar, Chem. Commun., 2019, 55, 7683–7686 Search PubMed.
- M. Katkevics, J. A. MacKay and E. Rozners, Chem. Commun., 2024, 60, 1999–2008 RSC.
- L. Baziar, L. Emami, Z. Rezaei, A. Solhjoo, A. Sakhteman and S. Khabnadideh, Sci. Rep., 2024, 14, 26538 CrossRef CAS PubMed.
- A. Pałasz and D. Cież, Eur. J. Med. Chem., 2015, 97, 582–611 CrossRef PubMed.
- K. Jain, S. Sharma and D. Utreja, ChemistrySelect, 2024, 9, e202303179 CrossRef CAS.
- T. Das, P. Kumar, S. Kumar, S. Mal, S. Kumar, V. K. Rajana, S. Singh, A. Dasgupta, D. Mondal and S. Das, ACS Infect. Dis., 2024, 10, 4314–4326 CrossRef CAS PubMed.
- C. Zhi, Z. Y. Long, A. Manikowski, N. C. Brown, P. M. Tarantino, K. Holm, E. J. Dix, G. E. Wright, K. A. Foster, M. M. Butler, W. A. LaMarr, D. J. Skow, I. Motorina, S. Lamothe and R. Storer, J. Med. Chem., 2005, 48, 7063–7074 CrossRef CAS PubMed.
- V. A. Kezin, E. S. Matyugina, M. S. Novikov, A. O. Chizhov, R. Snoeck, G. Andrei, S. N. Kochetkov and A. L. Khandazhinskaya, Molecules, 2022, 27, 2866 CrossRef CAS PubMed.
- S. Djaković, L. Glavaš-Obrovac, J. Lapić, S. Maračić, J. Kirchofer, M. Knežević, M. Jukić and S. Raić-Malić, Appl. Organomet. Chem., 2021, 35, e6052 CrossRef.
- M. S. Novikov, M. P. Paramonova, I. M. Kirillov, O. A. Smirnova, A. L. Khandazhinskaya, A. O. Chizhov, S. N. Kochetkov, A. V. Ivanov and I. F. Fedyakina, Mendeleev Commun., 2023, 33, 537–538 CrossRef CAS.
- G. Q. Lu, X. Y. Li, O. K. Mohamed, D. Wang and F. H. Meng, Eur. J. Med. Chem., 2019, 171, 282–296 CrossRef CAS PubMed.
- H. Cui, G. F. Ruda, J. Carrero-Lérida, L. M. Ruiz-Pérez, I. H. Gilbert and D. González-Pacanowska, Eur. J. Med. Chem., 2010, 45, 5140–5149 CrossRef CAS PubMed.
- D. Ramesh, B. G. Vijayakumar and T. Kannan, Eur. J. Med. Chem., 2020, 207, 112801 CrossRef CAS PubMed.
- Shivangi, S. Das, S. Mal and S. Naskar, ChemistrySelect, 2024, 9, e202400715 CrossRef CAS.
- S. Meshcheryakova, A. Shumadalova, O. Beylerli, I. Gareev, S. Zhao and J. Wu, Drug Dev. Res., 2022, 83, 578–585 CAS.
- L. H. Al-Wahaibi, S. R. S. Bysani, S. S. Tawfik, M. S. M. Abdelbaky, S. Garcia-Granda, A. A. El-Emam, M. J. Percino and S. Thamotharan, Cryst. Growth Des., 2021, 21, 3234–3250 CrossRef CAS.
- G. Portalone, J. O. Moilanen, H. M. Tuononen and K. Rissanen, Cryst. Growth Des., 2016, 16, 2631–2639 CrossRef CAS.
- A. O. Surov, A. P. Voronin, K. V. Drozd, T. V. Volkova, N. Vasilev, D. Batov, A. V. Churakov and G. L. Perlovich, Cryst. Growth Des., 2022, 22, 2569–2586 CrossRef CAS.
- Y. Roselló, M. Benito, M. Barceló-Oliver, A. Frontera and E. Molins, Cryst. Growth Des., 2021, 21, 4857–4870 CrossRef.
- S. J. Zamisa, N. P. Ngubane, A. A. Adeleke, S. B. Jonnalagadda and B. Omondi, Cryst. Growth Des., 2022, 22, 5814–5834 CrossRef CAS.
- M. Barceló-Oliver, C. Estarellas, A. García-Raso, A. Terrón, A. Frontera, D. Quiñonero, I. Mata, E. Molins and P. M. Deyà, CrystEngComm, 2010, 12, 3758–3767 RSC.
- B. Makiabadi, M. Zakarianezhad and E. Zeydabadi, Struct. Chem., 2023, 34, 755–767 CrossRef CAS.
- S. Jena, J. Dutta, K. D. Tulsiyan, A. K. Sahu, S. S. Choudhury and H. S. Biswal, Chem. Soc. Rev., 2022, 51, 4261–4286 RSC.
- P. W. Kenny, J. Med. Chem., 2022, 65, 14261–14275 CrossRef CAS PubMed.
- M. Zhang, Z. Li, Y. Zhu, L. Yan, X. Zhong, Y. Zhang, Z. Li, Y. Bai and H. Li, CrystEngComm, 2024, 26, 261–267 RSC.
- E. D. Głowacki, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, J. Mater. Chem. B, 2013, 1, 3742–3753 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- V. Kumar, P. K. Goswami, R. Thaimattam and A. Ramanan, CrystEngComm, 2018, 20, 3490–3504 RSC.
- M. Kumar, G. Jaiswar, M. Afzal, M. Muddassir, A. Alarifi, A. Fatima, N. Siddiqui, R. Ayub, N. A. Y. Abduh, W. S. Saeed and S. Javed, Molecules, 2023, 28, 2116 CrossRef CAS PubMed.
- V. Gerhardt and E. Egert, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 209–220 CrossRef CAS PubMed.
- L. F. Diniz, C. H. J. Franco, D. F. Silva, L. S. Martins, P. S. Carvalho Jr, M. A. C. Souza, N. F. A. Reis, C. Fernandes and R. Diniz, Int. J. Pharm., 2021, 605, 120790 CrossRef CAS PubMed.
- A. E. Krajewski and J. K. Lee, Gas-phase experimental and computational studies of 5-halouracils: intrinsic properties and biological implications, J. Org. Chem., 2021, 86, 6361–6370 CrossRef CAS PubMed.
- F. B. Ilhami, Y. S. Birhan and C. C. Cheng, ACS Biomater. Sci. Eng., 2023, 10, 234–254 CrossRef PubMed.
- F. Jubeen, A. Liaqat, F. Amjad, M. Sultan, S. Z. Iqbal, I. Sajid, M. B. K. Niazi and F. Sher, Cryst. Growth Des., 2020, 20, 2406–2414 CrossRef CAS.
- R. K. Shayakhmetova, E. M. Khamitov, A. G. Mustafin, S. P. Ivanov and S. L. Khursan, J. Chin. Chem. Soc., 2017, 64, 143–151 CrossRef CAS.
- J. L. Sessler and J. Jayawickramarajah, Chem. Commun., 2005, 1939–1949 Search PubMed.
- S. A. Barnett, A. T. Hulme, N. Issa, T. C. Lewis, L. S. Price, D. A. Tocher and S. L. Price, New J. Chem., 2008, 32, 1761–1775 RSC.
- D. Ramesh, A. K. Mohanty, A. De, B. G. Vijayakumar, A. Sethumadhavan, S. K. Muthuvel, M. Mani and T. Kannan, RSC Adv., 2022, 12, 17466–17480 RSC.
- E. A. Basiony, A. A. Hassan, M. Elsawalhy, A. H. Adel, H. Mansour, R. K. Arafa and N. A. Hassan, Bioorg. Chem., 2025, 154, 108066 CrossRef CAS PubMed.
- T. H. Abdalla, A. G. Ibrahim, S. Elabbady, E. Nassar, A. A. Hamed and A. Aboelnaga, Int. J. Biol. Macromol., 2025, 300, 140327 CrossRef CAS PubMed.
- T. A. Fernandes, F. Macedo, R. G. Cabral, T. Guiu, C. H. Franco, P. Jorge, A. C. Sousa, V. Andre, N. Cerca and A. M. Kirillov, RSC Appl. Interfaces, 2024, 1, 98–109 RSC.
- R. F. Stewart and L. H. Jensen, Acta Crystallogr., 1967, 23, 1102–1105 CrossRef CAS.
- S. Swaminathan and K. K. Chacko, Acta Crystallogr., Sect. B, 1978, 34, 3108–3110 CrossRef.
- K. Dzieszkowski, I. Barańska and Z. Rafiński, J. Org. Chem., 2020, 85, 6645–6662 CrossRef CAS PubMed.
- G. Marafon, D. Mosconi, D. Mazzier, B. Biondi, M. De Zotti and A. Moretto, RSC Adv., 2016, 6, 73650–73659 RSC.
- Rigaku, CrysAlisPro Software System, 1.171.40.69a, 2020 Search PubMed.
- APEXIII, APEXIII crystallography software suite, 2016 Search PubMed.
- Bruker, Bruker AXS: SAINT+, 6.22, 2016 Search PubMed.
- Bruker, Bruker AXS: SADABS, 2005 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.2., 2009 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- P. A. Wood, J. J. McKinnon, S. Parsons, E. Pidcock and M. A. Spackman, CrystEngComm, 2008, 10, 368–376 CAS.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS PubMed.
- M. A. Spackman, Z. Kristallogr. - Cryst. Mater., 2018, 233, 641–648 CrossRef CAS.
- D. Ž. Veljković, V. B. Medaković, J. M. Andrić and S. D. Zarić, CrystEngComm, 2014, 16, 10089–10096 RSC.
- M. Ziemniak, S. Pawlędzio, A. Zawadzka-Kaźmierczuk, P. M. Dominiak, D. Trzybiński, W. Koźmiński, R. Zieliński, I. Fokt, W. Priebe, K. Woźniak and B. Pająk, RSC Adv., 2022, 12, 8345–8360 RSC.
- M. S. Cunha, C. E. P. Ribeiro, C. C. Corrêa and R. Diniz, J. Mol. Struct., 2017, 1150, 586–594 CrossRef CAS.
- P. K. Deb, A. M. Menon, F. N. PSR, I. Shruti, S. Nidal, K. N. Venugopala and D. Chopra, CrystEngComm, 2025, 27, 2070–2085 RSC.
- M. K. Singh, J. Cryst. Growth, 2014, 396, 14–23 CrossRef CAS.
- S. L. Price and K. S. Wibley, J. Phys. Chem. A, 1997, 101, 2198–2206 CrossRef CAS.
- J. B. Kaper, J. P. Nataro and H. L. Mobley, Nat. Rev. Microbiol., 2004, 2, 123–140 CrossRef CAS PubMed.
- N. A. Turner, B. K. Sharma-Kuinkel, S. A. Maskarinec, E. M. Eichenberger, P. P. Shah, M. Carugati, T. L. Holland and V. G. Fowler Jr, Nat. Rev. Microbiol., 2019, 17, 203–218 CrossRef CAS PubMed.
- J. Campbell, Curr. Protoc. Chem. Biol., 2010, 2, 195–208 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional discussion, experimental and structural details for compounds 1–4; FT-IR (Fig. S3–S6, Table S3), UV-visible (Fig. S9, Table S4) and NMR spectra (Fig. S7 and S8), solubility data (Fig. S15 and S16) and antibacterial activity (Fig. S17–S24. Tables S8 and S9). CCDC 2306291 and 2306292 (PDF). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00362h |
| This journal is © The Royal Society of Chemistry 2025 |