
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenase-mimicking selective chlorination of unactivated C–H bonds by a Fe-complex†
Himangshu Kuiry,
Anish Gangopadhyay,
Bittu Chandra and
Sayam Sen Gupta*
and
Sayam Sen Gupta*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, West Bengal 741246, India. E-mail: sayam.sengupta@iiserkol.ac.in
First published on 11th June 2025
Abstract
In this study, we report a selective aliphatic C–H bond chlorination mediated by Fe-bTAML (bTAML: biuret-modified tetraamido macrocyclic ligand) using sodium hypochlorite as the chlorine source. The reaction predominantly yields chlorinated products over hydroxylated ones across various unactivated C–H bonds in acetonitrile–water medium. The formation of rearranged chlorinated products in norcarane and the absence of stereo-retention in cis-dimethylcyclohexane suggests the involvement of a long-lived, cage-escaped carbon radical intermediate. UV-Vis and EPR spectroscopic analyses confirm the presence of [FeV(O)–(NO2)bTAML]− (2) as the reactive intermediate. The 3°:2° selectivity in hydrocarbons, kinetic isotope effect (KIE), and detailed kinetic studies indicate hydrogen atom abstraction (HAA) by [FeV(O)–(NO2)bTAML]− (2) as the rate-determining step.
Halogenated organic compounds are essential to chemistry because they provide significant building blocks for pharmacologically and physiologically active molecules.1 Nature has deployed many enzymes that perform halogenation of bioactive molecules. For instance, halogenation of organic compounds has been accomplished by the halogenase enzymes CytC3 and SyrB2.2 The α-ketoglutarate motif associated with these enzymes includes an iron(II) co-factor facially coordinated with two histidine moieties, which in the presence of O2 forms a high-spin (S = 2) cis-iron-oxo-halide intermediate that subsequently abstracts a hydrogen atom from the C–H bond. The active site of this enzyme arranges itself in such a way towards the newly formed alkyl radical that the radical selectively rebounds with the halide instead of the hydroxyl group to form a halogenated product. However, mimicking the natural halogenase in vitro has been one of the biggest challenges. Although a wide variety of peroxidase-mimicking iron and manganese oxo-complexes are known to be excellent hydroxylating agents for organic substrates,3 only a handful of halogenase-mimicking synthetic complexes have been reported. In the quest to replicate the halogenase active site, a few biomimetic model complexes such as [FeV(TPA)(O)(Cl)]2+, [FeIV(O)(TQA)(X)]+, [FeIV(O)(X)(Pytacn)]+, and [TpPh2FeIV(O)(X)] (X = Cl and Br) have been prepared and their reactivity has been studied over several decades to aid in understanding these halogenation processes.4–9 Paine and co-workers also developed two non-heme iron(II)-α-keto acid model complexes with [(phdpa)Fe(BF)Cl] and [(1,4-tpbd)Fe2(BF)2Cl2] frameworks, which, in the presence of O2, activated C–H bonds of aliphatic substrates to yield the corresponding halogenated products.10 In 2018, Maiti and co-workers reported an aliphatic and benzylic C–H bond halogenation protocol where an iron(IV)-oxo intermediate ([FeIV(2PyN2Q)(O)]2+) abstracts a hydrogen from the C–H bond and another iron(III) halide complex provides a halide atom to the alkyl radical.11 Recently, Costas and co-workers demonstrated similar halogenation reactivity using the [FeIV(O)(R,R′Pytacn)(S)]2+ (R = Np, Me; R′ = H, Me) intermediate, employing NBu4Cl as the chloride source in the presence of a strong acid.12 Herein, we show the use of a single [Et4N]2[FeIII(Cl)–(NO2)bTAML] (1) complex, which performs both hydrogen atom abstraction in its high-valent iron(V)-oxo state and a chloride radical source using a transient iron(IV)-hypochlorite intermediate under the reaction conditions, similar to the strategy developed by Groves and co-workers, which includes C–H halogenation reaction by the manganese(V)-oxo complex.13
Our group has earlier reported the bTAML framework, [Et4N]2[FeIII(Cl)–(NO2)bTAML] (1), known to catalyse the oxidation of 3° C–H bonds with unprecedented selectivity using NaOCl or mCPBA as the terminal oxidant.14,15 This peroxygenase-mimicking [FeIII(Cl)–(NO2)bTAML]2− complex is known to abstract a H-atom in its high-valent [FeV(O)–(NO2)bTAML]− (2) state, followed by a rebound to give the hydroxylated product. While performing the selective hydroxylation with NaOCl as a terminal oxidant, the system always ends up in trace amounts of chlorinated product. Despite being insignificant, the yield prompted us to investigate the halogenase enzymatic route of a peroxygenase-mimicking enzyme. In this work, we assessed the feasibility of tuning this 100% hydroxylating catalytic system to function as a halogenase mimic. The strategy adopted for generating the chlorinated products was to alter the catalyst:oxidant ratio. The addition of excess NaOCl to the catalyst system led to significantly higher yields of the chlorinated product. Thereafter, we investigated a broad range of substrates for chlorination reactions and obtained superior selectivity and yield. Intrigued by these excellent results, we also investigated the mechanism of the halogenation reaction. It was observed that the abstraction of a hydrogen from the C–H bond by the [FeV(O)–(NO2)bTAML]− (2) intermediate was followed by the escape of the alkyl radical from the solvent cage and rebound with the FeIV(OCl) intermediate instead of FeIV(OH). In this work, we have reported the first instance of this reactivity in a non-heme catalyst framework where an unprecedented selectivity is achieved by a high-valent oxo-metal intermediate (Scheme 1).
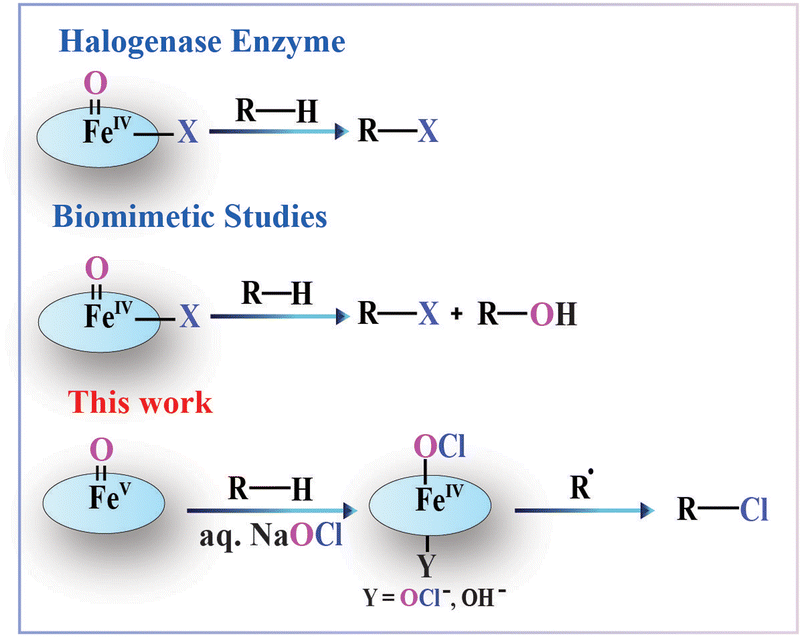 | ||
| Scheme 1 C(sp3)–H halogenation by natural enzymes and biomimetic complexes. | ||
In the hydroxylation reaction of adamantane, NaOCl functions as a stoichiometric terminal oxidant. The reaction mechanism involves hydrogen atom abstraction by the in situ generated high-valent [FeV(O)–(NO2)bTAML]− (2) intermediate.15 Herein, for the chlorination reaction, excess NaOCl was added (10–15 eq. of the substrate) (Fig. 1A). As we gradually increased the concentration of NaOCl (from 2 to 12 eq. of adamantane), we noticed a decrease in the yield of 1-adamantanol, along with a corresponding increase of 1-chloroadamantane (Fig. 1B).
 | ||
| Fig. 1 (A) General scheme of the chlorination reaction. (B) Yield vs. eq. of NaOCl plot for adamantane oxidation by 1. (C) Substrate scope for the chlorination reaction (all reactions were performed under an N2 atmosphere at rt for 1–2 h; yield and selectivity were calculated in GC-MS with standard substrate and product, OH = alcohol, O = ketone, Cl = chloro product). | ||
After several optimization studies (Table S1, ESI†), the maximum yield of the product(s) (e.g. 1-chloroadamantane for adamantane chlorination) was obtained in acetonitrile–water medium with 5 mol% of 1 (0.2 mM) and iterative addition of 60 mM of NaOCl. All reactions were performed under an N2 atmosphere at room temperature for 1–2 h with constant stirring. GC-MS analysis of the reaction mixture showed 1-chloroadamantane as the major product with 86% yield and 3°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2° = 52:1 regioselectivity (Fig. S9, ESI†).
:2° = 52:1 regioselectivity (Fig. S9, ESI†).
This excellent yield and regioselectivity in the halogenation reactivity profile of 1 in the presence of elevated NaOCl concentration prompted us to investigate the reaction mechanism in detail. Kinetic analysis was done at 298 K by monitoring the decay of the peak at 547 nm corresponding to 2, which showed an exponential decay with time in presence of substrate (Fig. S22, ESI†). The rate of this exponential decay (kobs) further increased linearly with increase in concentration of cyclooctane (0.075–0.150 M), which indicates that the rate of the reaction is first-order with respect to both the catalyst and substrate concentration (Fig. S23, ESI†). From the linear fitting of the plot of kobs with cyclooctane concentration, we obtained a second-order rate constant value (k2) of 2.6 × 10−1 M−1 s−1 (Fig. 2A).
 | ||
| Fig. 2 (A) Plot of kobs vs. cyclooctane concentration (0,075–0.150 M). (B) Kinetic isotope effect of the chlorination reaction with adamantane and adamantane-D16 (competitive reaction) at 298 K. | ||
Furthermore, we studied the kinetic isotopic effect (KIE) of this chlorination reaction by conducting intermolecular competition experiments with adamantane and adamantane-D16 at 298 K (ESI,† Section S3.6). The kinetic isotope effect (KIE) value was 4.2, which suggests that H-atom abstraction is the rate-determining step (Fig. 2B). The KIE value and remarkable regioselectivity in adamantane chlorination reaction are in agreement with those measured from C(sp3)–H oxidation reactions catalysed by metal-based oxidants, including metal-oxo species. This data excludes a radical-mediated pathway for the chlorination reaction of adamantane.16 Costas and co-workers have reported that chlorine radical-mediated halogenation reactions, using the metal complex [(Me,HPyTACN)NiII(CH3CN)2]2+ in combination with NaOCl as the chlorine source, exhibit a kinetic isotope effect (KIE) of 2 and a 3°:2° selectivity ratio of 7:1.17 In contrast, our chlorination reaction of adamantane using complex 2 yielded a significantly higher KIE value of 4.2 and a 3°:2° selectivity of 52:1. These values far exceed those observed for typical chlorine radical-mediated reactions or hydroxylation reactions by OH radicals.18 Notably, the KIE and regioselectivity observed in our system closely match those reported for the hydroxylation of adamantane by complex 2.15 Based on these observations, we ruled out the involvement of free chlorine radicals in our reaction. Additionally, no product formation was observed in the absence of the catalyst or when FeCl3 was used in place of complex 1, further supporting the essential role of complex 1 in the chlorination process. The involvement of a high-valent oxo-metal intermediate in the hydrogen atom abstraction step was therefore postulated akin to what has been shown by Maiti (FeIVO), Costas (FeIVO), and Groves (MnVO).11–13 The addition of NaOCl to 1 was accompanied by a colour change from orange to violet. The intermediate formed was analysed by UV-Vis spectroscopy, EPR spectroscopy, and mass spectrometry. The UV-Vis spectrum of the violet-coloured species showed the absence of spectral features at 372 nm (characteristic of the starting Fe(III)-complex) and the concomitant appearance of new spectral features at 547 nm (Fig. 3A), indicative of the [FeV(O)–(NO2)bTAML]− (2) intermediate that has been reported earlier.14,15 Furthermore, ESI-MS of the violet species in the reaction mixture revealed a prominent ion peak at the mass-to-charge ratio/m/z (negative mode) 474.0569 (calculated 474.0586). The X-band EPR spectrum at 80 K showed g values of 2.01, 1.98, and 1.79 (Fig. 3B), corresponding to an S = 1/2 species, confirming [FeV(O)–(NO2)bTAML]− (2) to be the violet-coloured intermediate.
 | ||
| Fig. 3 (A) UV-Vis spectral change after the addition of 60 mM aq. NaOCl to 1 (0.2 mM) in CH3CN–H2O (5:1) at 298 K (half diluted). (B) EPR spectrum measured at 85 K after 5 s of mixing of 60 mM aqueous NaOCl with 1 (0.2 mM) in CH3CN–H2O (5:1) at 298 K. | ||
Based on these observations, we propose that the high-valent [FeV(O)–(NO2)bTAML]− (2) complex formed upon the addition of NaOCl abstracts a hydrogen atom from the C–H bond, generating a carbon-centred radical and a [FeIV(OH)–(NO2)bTAML]− intermediate. However, the rebound “reaction” from the [FeIV(OH)–(NO2)bTAML]− intermediate to the carbon-centred radical leading to the formation of alcohol does not take place in the presence of excess quantities of NaOCl. We hypothesize that in the presence of a high concentration of sodium hypochlorite, the excess OCl−/OH− present in the solution possibly coordinates in the sixth position of the five-coordinate [FeIV(OH)–(NO2)bTAML]−.
Due to this electron-donating ligand in the sixth position, the rate of rebound of the –OH group in [FeIV(OH)–(NO2)bTAML]− to the carbon radical decreases,13 giving enough time for this carbon-centred radical to escape from the solvent cage. The proposed cage escape mechanism was tested using the strained cycloalkane norcarane as a diagnostic radical clock in the chlorination reaction by 1. Under the chlorination reaction conditions, norcarane afforded a significant amount of rearranged product, 3-chloromethylcyclohexene (4.0%), along with unrearranged product 2-chloronorcarane (4.3%), which supports the cage-escape phenomenon of the organic radical (Fig. 4A).19 Furthermore, a reaction between a pure stereoisomer of cis-1,2-dimethylcyclohexane and 1 in the chlorination condition showed the formation of a mixture of cis/trans-1-chloro-1,2-dimethylcyclohexane (60% yield) with a 1.1:1 ratio. This high degree of epimerization in the products arises from a long-lived radical, which could be generated due to the dissociation of the radical from the solvent cage after HAA (Fig. 4B).11
 | ||
| Fig. 4 Chlorination reaction of (A) norcarane, (B) cis-DMCH, and (C) adamantane in the presence of 1 eq. imidazole. | ||
Finally, to support the hypothesis of the formation of a six-coordinated [FeIV(OH)(Y)–(NO2)bTAML]2− (Y = OCl−, OH−), we performed the chlorination reaction of adamantane in the presence of imidazole (1 eq.). A change in product selectivity was observed, during which 1-adamantanol was obtained as the major product instead of 1-chloroadamantane (Fig. 4C). This suggests the role of the sixth-coordinated ligand in determining product selectivity. Imidazole competitively coordinates to the sixth position of [FeIV(OH)–(NO2)bTAML]−, replacing OCl−/OH−. This change in coordination leads to an increase in the rebound rate between the alkyl radical and iron(IV)hydroxide intermediate. Thus, the OCl−/OH− ligation in the sixth position plays a role in decreasing the OH− rebound rate of the alkyl radical in the reaction medium and supports our hypothesis of the formation of a six-coordinated [FeIV(OH)(Y)–(NO2)bTAML]2− (Y = OCl−, OH−). Subsequently, –OH in the [FeIV(OH)(Y)–(NO2)bTAML]2− was substituted by –OCl, and [FeIV(OCl)(Y)–(NO2)bTAML]2− was formed. Attempts to trap this intermediate were unsuccessful; however, the formation of the [FeIV(OCl)(Y)–(NO2)bTAML]2− intermediate under the reaction conditions was indirectly probed by using 1,3,5-trimethoxybenzene as the chlorine radical trap (Fig. S24, ESI†). After the formation of [FeIV(OCl)(Y)–(NO2)bTAML]2−, the cage-escaped alkyl radical rebounds with the Cl atom of the [FeIV(OCl)(Y)–(NO2)bTAML]2− intermediate, leading to the formation of the chlorinated product.
We attempted the chlorination reaction with substrates with moderate to high C–H bond energies (BDE > 90 kcal mol−1), such as 2,3-dimethylbutane, cyclopentane, cyclohexane, cycloheptane, cyclooctane, and N-acetyl-3,5-dimethyl-1-adamantanamine (Fig. 1C). The formation of the chlorinated hydrocarbon was the major product for all these substrates. However, for substrates with weak C–H bonds (BDE < 84 kcal mol−1), such as xanthene, fluorene, and ambroxide, the hydroxylated products were obtained in 100% yield, and no chlorinated product was obtained. This suggests that these substrates do not follow the chlorination pathway described above. The alkyl radicals generated from these weak C–H bonds have significantly lower oxidation potentials compared to those derived from stronger C–H bonds. As a result, the rebound rates of these radicals with the hydroxyl group of the [FeIV(OH)(Y)–(NO2)bTAML]2− complex are very fast during the reaction.20 We believe that due to this fast rebound, the [FeIV(OCl)(Y)–(NO2)bTAML]2− species, which is responsible for the chlorination reaction, is not formed. As a result, the reaction proceeds with the exclusive formation of the hydroxylated product.
Mechanism: Based on spectroscopic studies (UV-Vis, EPR), product analysis via mass spectrometry, trapping of reactive intermediates and analysis of the reaction kinetics, we propose the following catalytic cycle for the chlorination of alkanes by 1 and NaOCl. In the reaction, the in situ generated [FeV(O)–(NO2)bTAML]− (2) intermediate abstracts a hydrogen atom from the C–H bond, which is the rate-determining step of the chlorination reaction. Then, in the presence of excess hypochlorite, a six-coordinated [FeIV(OH)(Y)–(NO2)bTAML]2− (Y = OCl−, OH−) forms, and the rebound rate of the alkyl radical decreases, allowing it to escape from the solvent cage. Subsequently, [FeIV(OCl)(Y)–(NO2)bTAML]2− forms and the cage-escaped alkyl radical rebounds with the Cl-atom of this [FeIV(OCl)(Y)–(NO2)bTAML]2− intermediate to yield alkyl chloride, regenerating the [FeV(O)–(NO2)bTAML]− (2) for the next catalytic cycle (Fig. 5).
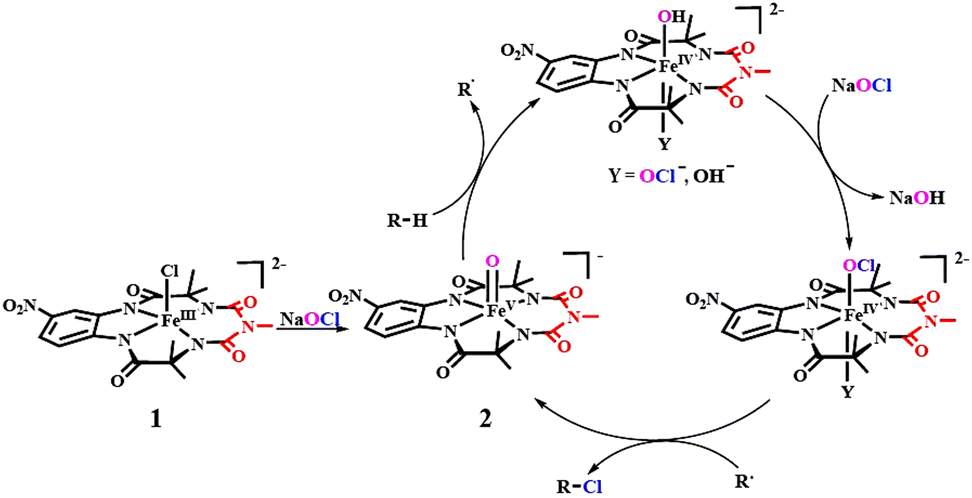 | ||
| Fig. 5 Plausible mechanism of the chlorination reaction by complex 1. | ||
In summary, we explored the selective catalytic oxidative chlorination over hydroxylation of unactivated C–H bonds for a series of substrates using iron complex 1 in the presence of NaOCl. The overall reaction consists of four major steps: (i) formation of the [FeV(O)–(NO2)bTAML]− (2), (ii) hydrogen atom abstraction, (iii) exchange of the axial-OH group of [FeIV(OH)–(NO2)bTAML]− by OCl− and (iv) rebound of the cage-escaped alkyl radical to the chlorine atom of the [FeIV(OCl)(Y)–(NO2)bTAML]2− intermediate. To the best of our knowledge, complex 1 serves as an excellent halogenase-mimicking system.
H. K. thanks CSIR for the fellowship. A. G. thanks INSPIRE for the fellowship. S. S. G. acknowledges the SERB core research grant, New Delhi (grant no. CRG/2022/007285), MoE Stars (grant no. MoE-STARS/STARS-2/2023-0689) for funding.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- (a) A. Podgoršek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef PubMed; (b) S. Stavber, M. Jereb and M. Zupan, Synthesis, 2008, 1487–1513 CrossRef CAS.
- T. Borowski, H. Noack, M. Radoń, K. Zych and P. E. M. Siegbahn, J. Am. Chem. Soc., 2010, 132, 12887–12898 CrossRef CAS PubMed.
- D. Chatterjee and S. S. Gupta, Handbook of CH-Functionalization, 2022, pp. 1–35 Search PubMed.
- M. Puri, A. N. Biswas, R. Fan, Y. Guo and L. Que Jr, J. Am. Chem. Soc., 2016, 138, 2484–2487 CrossRef CAS PubMed.
- P. Comba and S. Wunderlich, Chem. – Eur. J., 2010, 16, 7293–7299 CrossRef CAS PubMed.
- O. Planas, M. Clemancey, J.-M. Latour, A. Company and M. Costas, Chem. Commun., 2014, 50, 10887–10890 RSC.
- S. Chatterjee and T. K. Paine, Angew. Chem., Int. Ed., 2016, 55, 7717–7722 CrossRef CAS PubMed.
- V. Yadav, R. J. Rodriguez, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2020, 142, 7259–7264 CrossRef CAS PubMed.
- E. F. Gerard, V. Yadav, D. P. Goldberg and S. P. de Visser, J. Am. Chem. Soc., 2022, 144, 10752–10767 CrossRef CAS PubMed.
- R. D. Jana, D. Sheet, S. Chatterjee and T. K. Paine, Inorg. Chem., 2018, 57, 8769–8777 CrossRef CAS PubMed.
- S. Rana, J. P. Biswas, A. Sen, M. Clémancey, G. Blondin, J.-M. Latour, G. Rajaraman and D. Maiti, Chem. Sci., 2018, 9, 7843–7858 RSC.
- N. Pagès-Vilà, I. Gamba, M. Clémancey, J. M. Latour, A. Company and M. Costas, J. Inorg. Biochem., 2024, 259, 112643 CrossRef PubMed.
- (a) W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef CAS PubMed; (b) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- S. Jana, M. Ghosh, M. Ambule and S. S. Gupta, Org. Lett., 2017, 19, 746–749 CrossRef CAS PubMed.
- M. Ghosh, S. Pattanayak, B. B. Dhar, K. K. Singh, C. Panda and S. S. Gupta, Inorg. Chem., 2017, 56, 10852–10860 CrossRef CAS PubMed.
- J. T. Groves and T. E. Nemo, J. Am. Chem. Soc., 1983, 105, 6243–6248 CrossRef CAS.
- A. Draksharapu, Z. Codola, L. Gomez, J. Lloret-Fillol, W. R. Browne and M. Costas, Inorg. Chem., 2015, 54, 10656–10666 CrossRef CAS PubMed.
- C. Walling, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- W. Liu, X. Y. Huang, M. J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- D. C. Cummins, J. G. Alvarado, J. P. T. Zaragoza, M. Q. E. Mubarak, Y. Lin, S. P. de Visser and D. P. Goldberg, Inorg. Chem., 2020, 59(21), 16053–16064 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02614h |
| This journal is © The Royal Society of Chemistry 2025 |