
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the stability of photocatalytic systems for hydrogen evolution in water by using a tris-phenyl-phenanthroline sulfonate ruthenium photosensitizer†
Fakourou
Camara
ab,
Juan S.
Aguirre-Araque
a,
Jérôme
Fortage
 *a and
Marie-Noëlle
Collomb
*a
*a and
Marie-Noëlle
Collomb
*a
aUniv. Grenoble Alpes, CNRS, DCM, 38000 Grenoble, France. E-mail: jerome.fortage@univ-grenoble-alpes.fr; marie-noelle.collomb@univ-grenoble-alpes.fr
bUniv. Grenoble Alpes, CNRS, CEA, IRIG, SyMMES, 38000 Grenoble, France
First published on 23rd February 2024
Abstract
The family of ruthenium tris-bipyridine complexes remains among the most widely used molecular photosensitizers (PSs) to drive catalytic reactions using the energy of light. However, the main drawback of such PSs is the poor stability of their oxidized and reduced forms subject to ligand dissociation, especially in water that causes relatively fast deactivation of the photocatalytic systems. We were able to improve the stability and efficiency of a Ru based photocatalytic system for hydrogen production in water by using the water-soluble Na4[Ru((SO3Ph)2phen)3] derivative (RuSPhphen, (SO3Ph)2phen = disodium (1,10-phenanthroline-4,7-diyl)bis(benzenesulfonate)) in place of regular [RuII(bpy)]3Cl2 (Rubpy, bpy = 2,2′-bipyridine) PS. RuSPhphen was tested with the [CoIII(CR14)Cl2]Cl (Co) catalyst and ascorbate (HA−) as a sacrificial electron donor under visible-light irradiation. The RuSPhphen absorption coefficient being twice as high compared to Rubpy and the excited-state lifetime being much longer, while keeping almost similar potentials, more efficient intermolecular electron transfers have been observed allowing the concentration of Ru PSs to decrease by 5-fold, i.e. to 100 μM, compared to previous studies with the Rubpy/Co/HA−/H2A photocatalytic system. A substantially enhanced H2-evolving photocatalytic activity was obtained with RuSPhphen directly correlated to its better stability over Rubpy. With 1.1 M H2A/HA−, at catalyst concentrations of 10 an 5 μM, the H2 production is two times higher compared to that obtained with Rubpy. When the H2A/HA− concentration is decreased to 0.1 M, the stability of both Ru PSs is further improved although RuSPhphen still systematically outperforms Rubpy whatever the catalyst concentration, with TONs reaching up to 4770 at 5 μM catalyst. A faster electron transfer of the RuSPhphen excited state to HA− has been observed by time-resolved luminescence compared to that of Rubpy, that could be ascribed to its much longer lifetime. In addition, a much higher rate constant for back electron transfer from the RuSPhphen˙− reduced state to HA˙ was determined by nanosecond transient absorption spectroscopy that could contribute to the higher stability of RuSPhphen during photocatalysis. The greater stability of RuSPhphen over Rubpy could also be correlated to the geometry of the SPhphen ligand that makes it less prone to dissociation in water in its reduced state.
Introduction
The unique spectroscopic, photophysical and electrochemical properties of ruthenium(II) complexes of the type [RuII(L)]32+ with 2,2′-bipyridyl and 1,10-phenanthroline ligands and their derivatives have made them extensively used for decades as molecular photosensitizers (PSs) for the activation of catalytic reactions using visible light energy.1–9 Indeed, their long lifetime in their triplet excited state, their intense absorption in the visible range, and their strong reducing and oxidizing power both in the excited and ground states as well as their solubility in water and organic solvents, make them very interesting PSs able to activate a wide range of catalysts for both oxidation and reduction reactions. Various reactions have been successfully carried out with such Ru derivatives, both in homogeneous solution and on surfaces (photoelectrodes), including the activation of organic substrates,10–14 the oxidation of water to O2 (oxygen-evolution-reaction (OER)),15–18 the reduction of water (protons) to H2 (hydrogen-evolution-reaction (HER))19–22 and the reduction of CO2.23–30 However, besides the fact that they are based on rare and expensive metals, the main drawback of Ru derivatives is the instability of their reduced [RuII(L)2(L˙−)]+ and oxidized [RuIII(L)3]3+ forms, that easily undergo a bidentate ligand substitution by solvent molecules or anions present in the medium, especially in aqueous solvent.7,31–36 Molecular PSs based on Ir, Re and Pt noble metals have demonstrated good performances in photocatalysis for H2 production or CO2 reduction. However, Ir and Re PSs are generally limited by their poor absorption in the visible region, while Pt PSs commonly photobleach under prolonged irradiation.37 Regarding organic dyes,38,39 they are generally restrained by very short singlet excited state lifetimes and less negative reduction potentials than noble-metal based PSs, together with their fast photobleaching.40In this context, given the quite unique properties of Ru complexes as PSs, progress should be made to increase their stability. Among the existing Ru derivatives, the tris-diphenyl-phenanthroline complex, [Ru(Ph2phen)3]2+ (Ph2phen = 4,7-diphenyl-1,10-phenanthroline or bathophenanthroline) (denoted as RuPhphen),2 and the water-soluble sulfonate analog Na4[Ru((SO3Ph)2phen)3] ((SO3Ph)2phen = disodium (1,10-phenanthroline-4,7-diyl)bis(benzenesulfonate)) (denoted as RuSPhphen)41 (Scheme 1) have particularly retained our attention. In fact, such PSs exhibit absorption coefficients twice as high compared to regular [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) (denoted as Rubpy) and much longer lifetime of their excited-state, while keeping almost similar potentials. In addition, the group of Castellano42 has shown that RuPhphen is more stable than Rubpy allowing enhanced H2 production when used in a homogeneous photocatalytic system with a cobalt glyoxime catalyst (Cat), [Co(dmgH)2pyCl] (dmgH = dimethylglyoxime, py = pyridine) and N,N′-dimethyl-p-toluidine (DMT) as a sacrificial electron donor (SD) in a CH3CN/H2O mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). It has been proposed that the better stability of the photocatalytic system with RuPhphen over Rubpy may result from the higher rigidity of the RuPhphen complex induced by the large diphenyl-phenanthroline ligand, which makes it less prone to ligand loss/substitution. In addition, a high rate constant for reductive quenching between the excited state of RuPhphen and DMT was reported which was attributed to the long lifetime of the PS's excited state (5.86 μs) that facilitates bimolecular electron-transfer reactions.42 The group of Brewer also reported substantially enhanced H2-evolving photocatalytic activity with supramolecular RuRh photocatalysts of the triad and diad type through the use of diphenyl-phenanthroline terminal ligands on the Ru moieties in place of regular phen or bpy ligands (Scheme 1).43–48 Since the observed excited state rate constants do not vary greatly, the steric protection of the Rh catalyst by the bulkier terminal ligand was suggested as a possible factor in the enhanced catalytic performances, decreasing unfavorable side reactions such as dimerization of the Rh catalytic site in the case of the RuRh diad. In addition, it has been proposed that the larger size of the diphenyl-phenanthroline ligand may provide for a lower rate of back electron transfer to the photooxidized N,N-dimethylaniline (DMA) donor. Finally, the same group found that insertion of a hydrophilic sulfonate substituent on the phenyl units49 increase the quantum yield of emission and the lifetime of the 3MLCT excited state20 and led to a water soluble H2-evolution photocatalyst (Scheme 1). These interesting results prompt us to investigate the water-soluble RuSPhphen in a three-component photocatalytic system for H2 production in fully aqueous solution.
:1). It has been proposed that the better stability of the photocatalytic system with RuPhphen over Rubpy may result from the higher rigidity of the RuPhphen complex induced by the large diphenyl-phenanthroline ligand, which makes it less prone to ligand loss/substitution. In addition, a high rate constant for reductive quenching between the excited state of RuPhphen and DMT was reported which was attributed to the long lifetime of the PS's excited state (5.86 μs) that facilitates bimolecular electron-transfer reactions.42 The group of Brewer also reported substantially enhanced H2-evolving photocatalytic activity with supramolecular RuRh photocatalysts of the triad and diad type through the use of diphenyl-phenanthroline terminal ligands on the Ru moieties in place of regular phen or bpy ligands (Scheme 1).43–48 Since the observed excited state rate constants do not vary greatly, the steric protection of the Rh catalyst by the bulkier terminal ligand was suggested as a possible factor in the enhanced catalytic performances, decreasing unfavorable side reactions such as dimerization of the Rh catalytic site in the case of the RuRh diad. In addition, it has been proposed that the larger size of the diphenyl-phenanthroline ligand may provide for a lower rate of back electron transfer to the photooxidized N,N-dimethylaniline (DMA) donor. Finally, the same group found that insertion of a hydrophilic sulfonate substituent on the phenyl units49 increase the quantum yield of emission and the lifetime of the 3MLCT excited state20 and led to a water soluble H2-evolution photocatalyst (Scheme 1). These interesting results prompt us to investigate the water-soluble RuSPhphen in a three-component photocatalytic system for H2 production in fully aqueous solution.
 | ||
| Scheme 1 Ruthenium photosensitizers and cobalt catalysts studied in this work and ruthenium–rhodium photocatalysts of the triad and diad type developed by the group of Brewer.43–49 | ||
In this contribution, we have tested the performance as a PS of the water-soluble RuSPhphen PS in association with one of the most efficient H2-evolving catalysts in acidic water,40,50–53 the cobalt tetraazamacrocyclic complex, [CoIII(CR14)Cl2]+ (denoted, Co) (CR14 = 2,12-dimethyl-3,7,11,17-tetraazabicyclo[11.3.1]heptadeca-1(17),2,11,13,15-pentaene) and ascorbate (HA−) as a SD (Scheme 1) under visible-light irradiation. We also performed comparative studies under similar conditions to the regular Rubpy PS. Various PSs and catalyst concentrations were investigated in two different aqueous media: 1.1 M HA2/HA− at pH 4.0 and 0.1 M HA2/HA− at pH 4.5 with 1 M acetate buffer. A substantially enhanced H2-evolving photocatalytic activity was observed in both media with RuSPhphen compared to benchmark Rubpy, especially for lower catalyst concentrations, directly correlated to the better stability of RuSPhphen over Rubpy.
We have also investigated the redox properties of RuSPhphen in DMF to determine the stability of the reduced states of RuSPhphen and their UV-visible absorption signature. Finally, time-resolved luminescence and nanosecond transient absorption spectroscopy were employed to investigate the photocatalytic mechanism owing to the detection of key intermediates such as the excited and reduced states of the Ru PS as well as the reduced state of the catalyst (CoI). The extracted kinetics parameters of the two PSs were compared in an effort to rationalize the greater stability of RuSPhphen.
Results and discussion
Electrochemical properties of RuSPhphen: generation and UV-visible characterization of the reduced states
The redox properties of RuSPhphen have been investigated in a degassed solution of DMF, 0.1 M [Bu4N](ClO4) and compared to those of Rubpy under the same conditions. The cyclic voltammograms are characteristic of such derivatives2,41 with a one-electron reversible metal-centered oxidation process (RuIII/RuII) and three reversible ligand-centered reduction processes (eqn (1) for L = bpy and eqn (2) for L = (SO3Ph)2phen) (Fig. 1(A)). Potentials are quite similar for both complexes (Table S1†). For RuSPhphen, the reversible RuIII/RuII oxidation system is located at +0.80 V vs. Ag/AgNO3 (with this reference, the reversible ferrocene/ferrocenium redox couple was measured at E1/2 = 60 mV) and the three reversible ligand-centered reduction processes at E1/2 = −1.61, −1.78 and −2.00 V (Fig. 1). For Rubpy, the corresponding systems are observed at +0.88, −1.65, −1.82 and −2.09 V. The reducing power of RuSPhphen is thus nearly similar to that of Rubpy in organic solvent, with the first reduction process of RuSPhphen (L/L˙−) being less negative than that of Rubpy by 40 mV.| [RuII(L)3]2+ ⇌ [RuII(L)2(L˙−)]+ ⇌ [RuII(L)(L˙−)2]0 ⇌ [RuII(L˙−)3]− | (1) |
| [RuII(L)3]4− ⇌ [RuII(L)2(L˙−)]5− ⇌ [RuII(L)(L˙−)2]6− ⇌ [RuII(L˙−)3]7− | (2) |
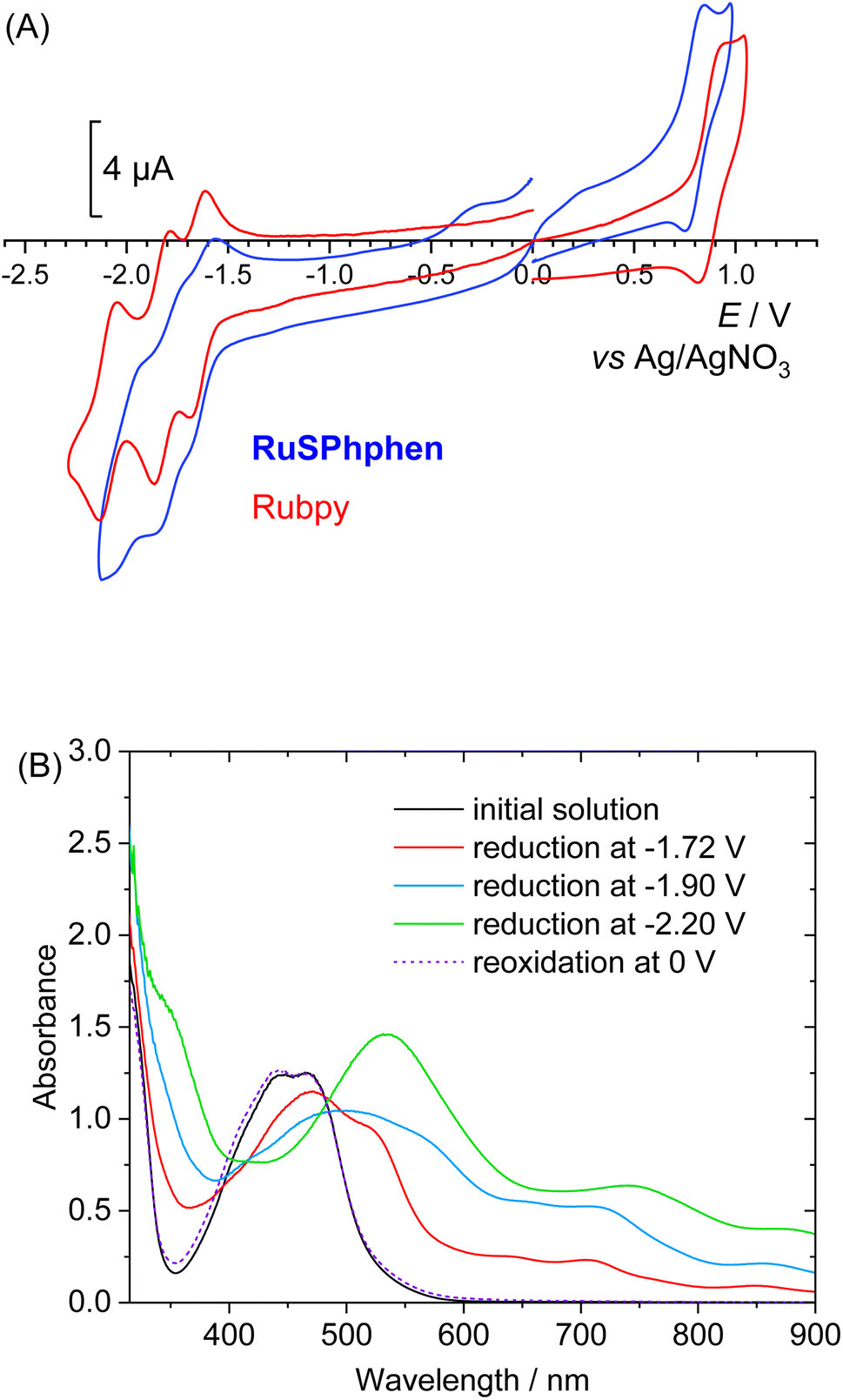 | ||
| Fig. 1 (A) Cyclic voltammograms in DMF, 0.1 M [Bu4N]ClO4 at a glassy carbon electrode (Ø = 3 mm, ν = 100 mV s−1) of solutions of RuSPhphen (0.44 mM, blue line) and Rubpy (0.3 mM, red line). (B) UV-visible absorption spectral changes of the solution of RuSPhphen (0.44 mM) in DMF, 0.1 M [Bu4N]ClO4, after three successive exhaustive reductions at (a) E = −1.72 V (formation of RuSPhphen5− also denoted as RuSPhphen˙−), (b) −1.90 V (formation of RuSPhphen6−), and (c) −2.20 V (formation of RuSPhphen7−) and (d) after a reoxidation at 0.0 V (reformation of RuSPhphen). The optical path is 1 mm. | ||
To evaluate the stability in DMF of the RuSPhphen reduced species with the respective charges 5−, 6− and 7− (eqn (2)) and assess their UV-vis signature, three successive electrolyses at −1.72, −1.90 and −2.20 V have been performed. The knowledge of the spectroscopic signature of the one electron reduced species of RuSPhphen (denoted as RuSPhphen˙−) will allow this species to be easily identified by transient absorption spectroscopy and thus to ascertain the reductive quenching mechanism (see below).
Fig. 1(B) displays the UV-visible spectra of the initial solution and of the stepwise reduction species. As for Rubpy,54 the spectra show the progressive decrease in intensity of the initial MLCT bands at 445 and 466 nm upon reduction, along with the corresponding increase of new bands progressively shifted at lower energy. An isobestic point is also observed at 485 nm. The maxima of these bands after the third reduction corresponding to the fully reduced species [RuII(L˙−)3]7− are located at 355 (sh), 553, 773 (sh) and 870 nm. The reduced solutions are perfectly stable showing the excellent stability of the three reduced states of the complex in organic solvent. A similar stability for the three reduced states of Rubpy is obtained in DMF, with the resulting spectra being similar to those previously obtained in CH3CN (Fig. S3†).54 So, from this experiment in organic solvent, it is not possible to conclude the better stability of the one-electron reduced state of RuSPhphen to explain the higher stability of the photocatalytic system with this PS (see below). All reduction steps were found to be reversible as further reoxidation of the solution at a potential of 0 V fully restores the initial solution for both RuSPhphen and Rubpy (Fig. 1(B) and S3†).
Photocatalytic activities for hydrogen production with the RuSPhphen and Rubpy/Co/HA−/H2A systems
Our previous studies with the Rubpy/[CoIII(CR14)Cl2]+ (Co)/HA−/H2A system for photo-induced H2 production were conducted in deaerated aqueous media under visible light irradiation (400–700 nm) with Rubpy at a concentration of 500 μM and various concentrations of Co catalyst, between 100 and 1 μM in the 1.1 M HA−/H2A buffer50,51,53 and between 50 and 5 μM in 1 M acetate buffer with 0.1 M of HA−/H2A.40 The best activities for H2 production in these media were obtained at pH 4.0 and 4.5, respectively. The visible absorption (29300 M−1 cm−1 at 462 nm) of RuSPhphen in water41 is twice as high as that of the reference PS Rubpy (14600 M−1 cm−1 at 452 nm) and the excited state lifetime of RuSPhphen (3.8 μs) is much longer than that of Rubpy (0.61 μs) in water (Table S2†). Electron transfers between the PS excited state and the SD and Cat should thus be favored and the photo-induced H2 production is more efficient even at lower PS concentrations. We therefore decrease the concentration of RuSPhphen by 5-fold compared to previous studies, i.e. to 100 μM. Comparative experiments with the Rubpy/Co/HA−/H2A system at a Rubpy concentration of 100 μM were also performed under the same experimental conditions for a more reliable comparison of the performance of the two PSs. Table S3† summarizes the photocatalytic activities of all studied systems.
The photocatalytic performances were first investigated in 1.1 M H2A/HA− at pH 4.0 with 100, 50, 10 and 5 μM of Co catalyst. Fig. 2 displays the comparison of both PSs activities as a function of time in terms of turnover number vs. catalyst (TONCat). The corresponding activities in terms of the number of moles and volume of H2 produced (nH2, vH2) are shown in Fig. S4.† For both PSs, H2 production is effective and increasing the ratio PS/Cat notably increases the activity of the catalyst, as indicated by steadily higher TONCat and TOFCat values. Such behavior has been previously observed with the Rubpy (500 μM)/Co/H2A/HA− systems50,51,53 and in many other H2-evolving photocatalytic systems using Co33,35,40,52,55–61 and Rh20,34,62 based-molecular catalysts. Increasing the PS/Cat ratio indeed promotes the reduction of the catalyst. The increase in catalytic activity (in terms of TONCat and TOFCat) induced by an increase in the PS/Cat ratio was well rationalized by a kinetic model recently reported by our group with the Rubpy/Co/HA−/H2A photocatalytic system.63 We showed that, in the case where PS degradation occurs, the limited TONCat and TOFcat values are directly proportional to the PS/Cat ratio, even if other factors can also strongly impact the catalytic activity such as the kinetics of the PS degradation and all kinetics of electron transfer between SD, PS and Cat. In any case, lowering the catalyst concentration often has a deleterious impact on the stability of the photocatalytic system, as it also accelerates Ru PS decomposition (see below). At higher catalyst concentrations of 100 and 50 μM, thus a PS/Cat ratio of 1/1 and 2/1, the amount of H2 produced is quite similar for both PSs after 28 h of irradiation with TONCat values of about 410 and 870, although the initial rate of H2 production is higher for Rubpy (see TOFCat values in Table S3†). By contrast, a significant difference between the two PSs appears at lower catalyst concentrations of 10 and 5 μM, with the catalytic activity of RuSPhphen being much more effective with a production of H2 multiplied by two (TONCat of about 2600 and 3170 for RuSPhphen at 10 and 5 μM, respectively versus 1380 and 1630 for Rubpy). However, the initial TOFCat values remain lower for RuSPhphen compared to Rubpy. The higher H2 production obtained with RuSPhphen can be directly correlated to the higher stability of the photocatalytic system under prolonged irradiation. Indeed, at catalyst concentrations of 10 and 5 μM, H2 is produced for more than 10 h with RuSPhphen while H2 evolution is fully stopped after only 3–4 h for Rubpy.
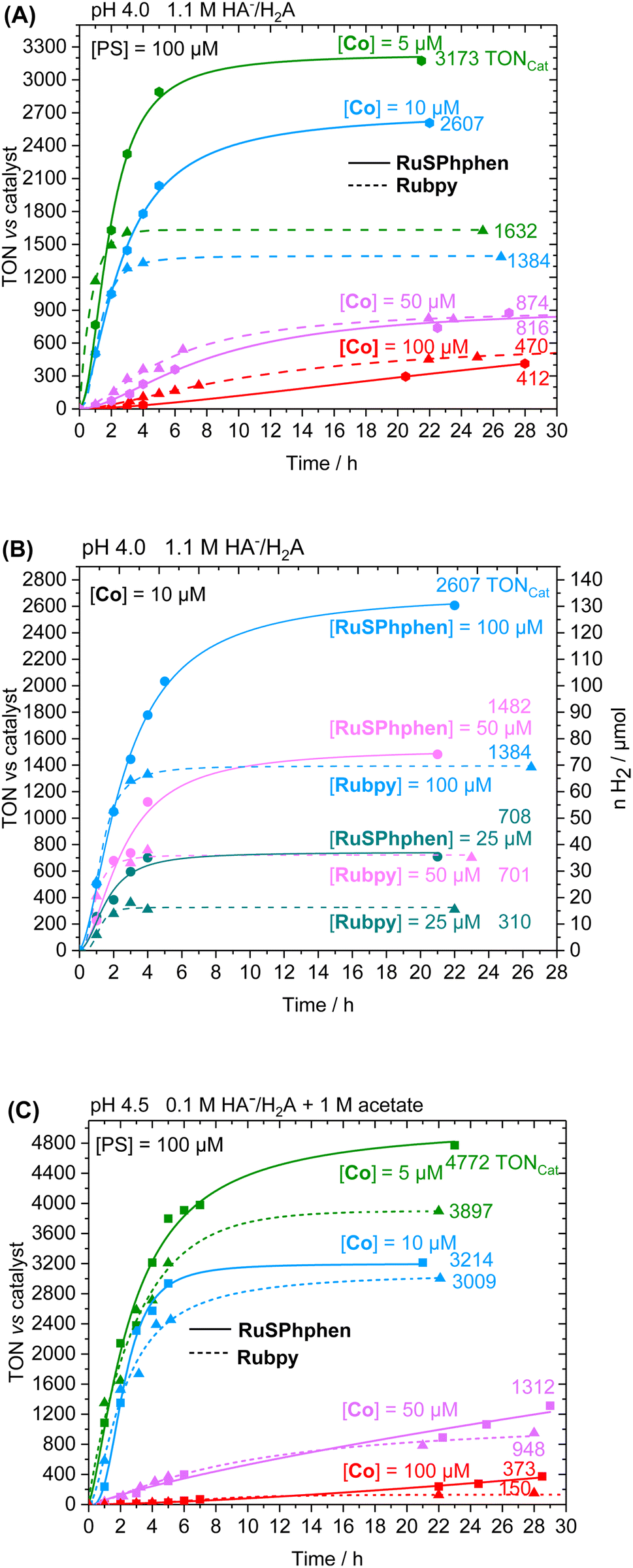 | ||
| Fig. 2 Photocatalytic hydrogen production as a function of time in TONCat under visible light irradiation (400–700 nm) from deaerated aqueous solutions (5 mL) of (A and B) NaHA (0.55 M) and H2A (0.55 M) at pH 4.0 and (C) of NaHA (0.0714 M), H2A (0.0286 M) and 1 M acetate buffer at pH 4.5, in the presence of a PS, RuSPhphen or Rubpy and the Co catalyst at various concentrations. | ||
By monitoring the evolution of UV-vis absorption spectra under prolonged irradiation of solutions of Rubpy and RuSPhphen (100 μM), in the absence and in the presence of Co catalyst at 10 μM, it appears that RuSPhphen is more stable than Rubpy. Indeed, in the absence of catalyst, Rubpy is almost fully transformed after only 1 h (Fig. 3(A)). At this stage, the intensity of the initial visible band at 452 nm has decreased by more than 70% and is shifted to slightly higher wavelength, with the appearance of a new band at 462 nm associated with another one at about 350 nm (shoulder). These changes are characteristic of the formation of Ru-bis-bipyridine species by substitution of a bipyridine ligand by solvent molecules or anions, owing to the well-known poor stability of Rubpy˙− in acidic water.33,36,40,61 Early work by Sutin et al. showed that Rubpy˙−, generated by photolysis of an acidic aqueous solution of Rubpy and HA−, undergoes a ligand substitution leading to the formation of [Ru(bpy)2(H2O)2]+ and the release of the protonated bipyridine ligand in solution.36 Castellano et al. also demonstrated that an aqueous mixture of Rubpy and HA− at pH 4, under an irradiation at 452 nm, mainly results in the formation of [Ru(bpy)2(HA)]+ along with side-products as [Ru(bpy)2(H2O)2]2+.33
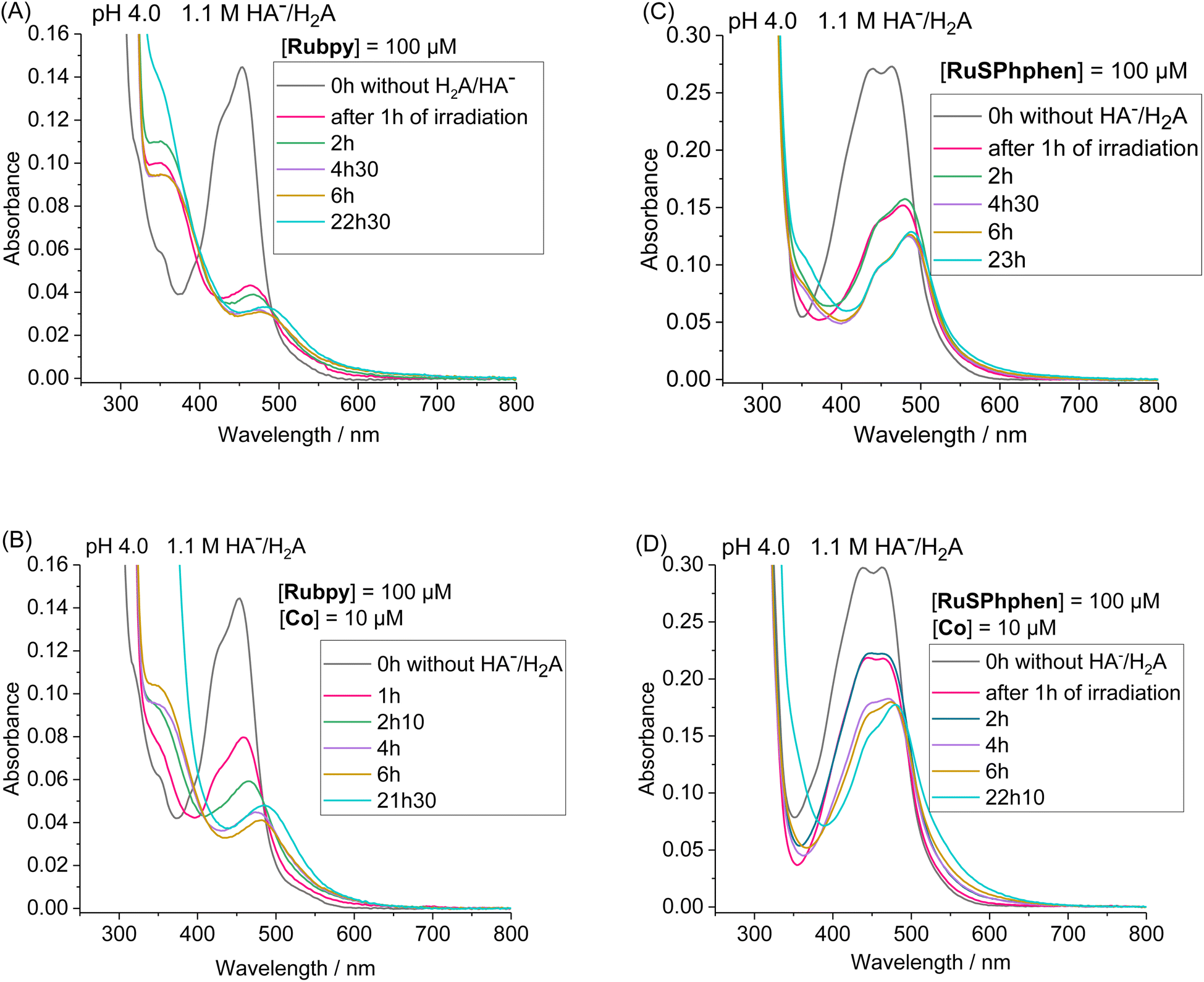 | ||
| Fig. 3 Evolution of the UV/vis absorption spectra under continuous visible light irradiation (λ = 400–700 nm) of a deaerated 1.1 M HA−/H2A aqueous solution at pH 4.0 containing Rubpy (100 μM) in the absence (A) or in the presence of the Co catalyst (10 μM) (B) and RuSPhphen (100 μM) in the absence (C) and in the presence of the Co catalyst (10 μM) (D). UV/vis absorption spectra of initial solutions before irradiation containing a mixture of Ru PS/Co or Ru PS alone without HA−/H2A are shown in black lines and for (A) after 1 h, 2 h, 4 h 30 min, 6 h and 22 h 30 min of irradiation, for (B) 1 h, 2 h 10, 4 h, 6 h and 21 h 30 min, for (C) 1 h, 2 h, 4 h 30, 6 h and 23 h, and for (D) 1 h, 2 h, 4 h, 6 h and 22 h 10 min. Cell path length: l = 1 mm. | ||
This degradation can only be partially limited in the presence of 10 μM of catalyst, with the catalyst acting as a quencher of Rubpy˙−. Indeed, Rubpy is still present at about 50% of its initial amount after 1 h of irradiation but fully transformed after 4 h (Fig. 3(B)). This is consistent with the fact that the H2 production has fully stopped after 3–4 h for 10 μM of catalyst (Fig. 2(A)).
Under similar conditions, RuSPhphen is much more stable in water in the presence of the catalyst (Fig. 3(D)). At least 75% of the initial PS is still present after 2 h of irradiation, and about 50–60% after 4–6 h (Fig. 3(D)) in agreement with H2 production maintained with the same efficiency for at least 6 h (Fig. 2(A)). However, RuSPhphen doesn't seem much more stable than Rubpy in the absence of the catalyst, with degradation appearing almost complete after just 1 hour, since at this stage, the intensity of the band has decreased by half and the two maxima have shifted to the longer wavelengths (Fig. 3(C)).
We also performed control experiments. In the absence of the Ru PS or HA−/H2A, no H2 is produced while the production of a small quantity of H2 was detected for solutions containing only the Ru PS and HA−/H2A (Table S3†). The amount of H2 produced, resulting from the direct reduction of protons by the reduced state of the Ru PS, is much lower than that in our previous experiments with a PS concentration of 500 μM.40,50,51,53 It can be considered negligible at all catalyst concentrations as it represents a maximum of approximately 1% of the H2 produced at a lower catalyst concentration of 5 μM (Table S3†). Also, addition of Hg in an experiment carried out with 100 μM RuSPhphen and 10 μM Co does not significantly change the photocatalytic activity (Fig. S5 and ESI†) ruling out the involvement of cobalt metallic colloids in H2 production that would result from the decomposition of the cobalt complex.64–66
The good efficiency of the different systems at 100 μM prompts us to further decrease the concentration of PS to 50 and 25 μM. The concentration of the Co catalyst was fixed at 10 μM (Fig. 2(B)). Overall, decreasing the concentration of the PS, i.e. the PS/Cat ratio, notably decreases the activity of the catalyst and thus the H2 production. Anyway, at this catalyst concentration, the H2 production is always two times higher with RuSPhphen compared to Rubpy (TONCat of 2600, 1480 and 708 for RuSPhphen at 100, 50 and 25 μM, respectively versus 1380, 700 and 310 for Rubpy), confirming once again the higher stability of photocatalytic systems with RuSPhphen. Indeed, it is interesting to see that even with these low concentrations of Ru, the production of H2 is still effective after 6–12 h for RuSPhphen while that with Rubpy is stopped after 3–4 h.
Another set of experiments was performed with a concentration of sacrificial electron donor of 0.1 M of H2A/HA− instead of 1.1 M. Under such conditions, the aqueous solution was buffered with 1 M acetate at pH 4.5 in order to keep the pH constant during the photocatalytic experiment. With a concentration of sacrificial electron donor divided by ten, a better stability of both Ru PSs is expected as the rate of formation of the reduced state, Ru˙−, subject to degradation by bidentate ligand dissociation, will be slower (see above). The photocatalytic performances of both Ru PSs at 100 μM in 1 M acetate buffer and 0.1 M HA−/H2A at pH 4.5 with 100, 50, 10 and 5 μM of Co catalyst are displayed in Fig. 2(C) and S4(B).† As anticipated, under such conditions, the photocatalytic systems are more stable, whatever the PS, with significantly higher TONCat obtained at low Co concentrations compared to those obtained in 1.1 M HA−/H2A at pH 4.0. This is even more visible for Rubpy at low Co concentrations of 10 and 5 μM, with TONsCat reaching up to 3010 and 3900, respectively after 22 h of irradiation versus 1380 and 1630 in 1.1 M H2A/HA− (Fig. 2(A and C)). The stability of the photocatalytic system with Rubpy in 0.1 M H2A/HA−, 1 M acetate buffer is now approaching that of RuSPhphen with H2 production still effective after 12 h leading to a less important difference in terms of TONCat between the two PSs. Nevertheless, higher TONCat values are obtained for RuSPhphen (4770, 3210, 1310, and 370 at 5, 10, 50, and 100 μM, respectively, versus 3900, 3010, 950, and 150 for Rubpy) with initial TOFs still higher for Rubpy. As observed in 1.1 M HA−/H2A at pH 4.0 (see above), the higher H2 production in the case of RuSPhphen comes from the better stability of this PS versusRubpy. From UV-visible absorption spectroscopy it is clear that the degradation of both PSs is slowed down either in the absence or in the presence of catalyst (10 μM) in 1 M acetate buffer with 0.1 M HA−/H2A (Fig. 4) compared to experiments performed in 1.1 M HA−/H2A (Fig. 3). Indeed, in the presence of catalyst, still about 50% of Rubpy and 75% of RuSPhphen are saved after about 6–8 h (Fig. 4(B and D)). The degradation of the PS is most probably the main limiting factor for H2 production after long term irradiation. However, the accumulation of dehydroascorbic acid (DHA) in the course of the photocatalysis formed by the oxidation reaction of ascorbate is also known to progressively short-circuit the catalysis.20,31,33,57,58,67,68 Dehydroascorbic acid is well-known to be a good electron acceptor able to prevent any electron transfer to the catalyst by trapping the electron from the reduced PS (see below).31,57,58,67,68 Finally, it should be noticed that, in comparison with our previous photocatalytic systems using the Co catalyst, the TONCat values measured here are very high, especially with the RuSPhphen (TONCat of 3200 and 4770 at 10 and 5 μM of Co catalyst), and almost reach those obtained with the organic dye TATA+ (TONCat of 4080 and 5910 at 10 and 5 μM of Co catalyst), although the TATA+ concentration was 500 μM.40 It should also be noted that the TONCat values obtained previously with the Rubpy/Co/HA−/H2A system (1.1 M or 0.1 M) for a Rubpy concentration of 500 μM never exceed 2000.40,50,51,53 These results show that, in acetate buffer with 0.1 M HA−/H2A, it is possible to divide by five the quantity of the Ru PS, while significantly improving the activity and thus saving precious metals.
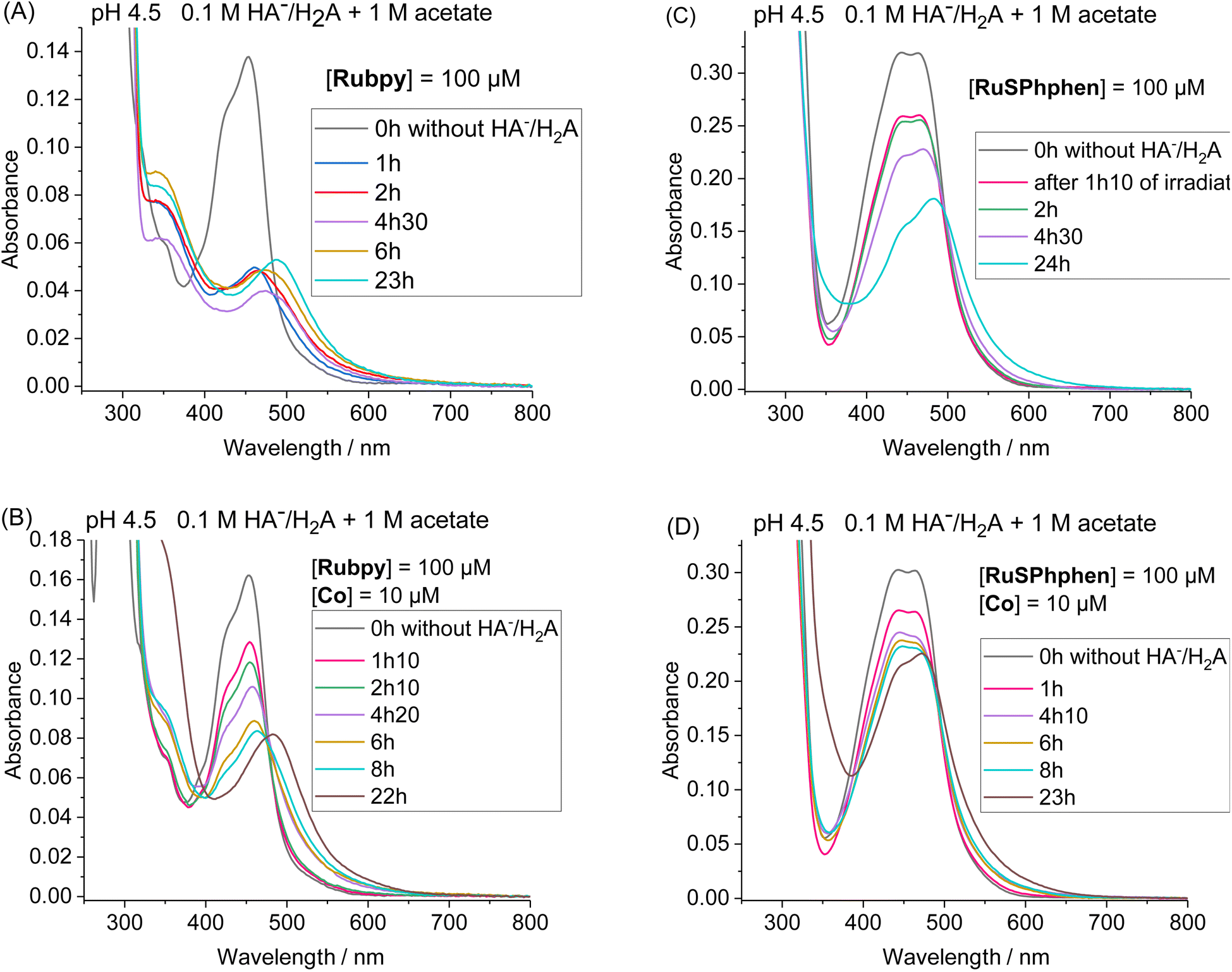 | ||
| Fig. 4 Evolution of the UV/vis absorption spectra under continuous visible light irradiation (λ = 400–700 nm) of a deaerated 1 M acetate buffer containing 0.1 M HA−/H2A at pH 4.5 containing Rubpy (100 μM) in the absence (A) or in the presence of Co catalyst (10 μM) (B) and RuSPhphen (100 μM) in the absence (C) and in the presence of Co catalyst (10 μM) (D). UV/vis absorption spectra of initial solutions containing a mixture of Ru PS/Co or Ru PS alone without HA−/H2A are shown in black lines and for (A) after 1 h, 2 h, 4 h 30, 6 h and 23 h of irradiation, for (B) 1 h 10, 2 h 10, 4 h 20, 6 h, 8 h and 22 h, for (C) 1 h 10, 2 h, 4 h 30 and 24 h, and for (D) 1 h, 4 h 10, 6 h, 8 h and 23 h. Cell path length: l = 1 mm. | ||
Mechanistic insight for the RuSPhphen/Co/HA−/H2A system from photophysical measurements
The possible steps for the three-component photocatalytic system Ru/Co/HA−/H2A are depicted in Scheme 2(A). It should be mentioned that the Co catalyst, initially in the +III oxidation state is immediately reduced by ascorbate into its +II state (denoted CoII), as previously shown.50 Thus, the cobalt species that enters the photocatalytic cycle is CoII. The photocatalytic cycle is initiated by the absorption of one photon by the Ru PS under visible light irradiation generating the excited state of the PS, denoted as *RuII. Due to the large excess of HA−versus the catalyst, *RuII is generally quenched by an electron transfer from HA− (reductive quenching), generating Ru˙− and the oxidized form of ascorbate, HA˙. Ru˙− is a good reductant (Table S1†) that can in turn reduce the CoII catalyst into CoI, reforming the initial state of the PS, RuII. HA˙ can easily release a proton to form A˙−, an unstable species which easily disproportionates to generate DHA (Scheme 2(C)). DHA is a very good electron acceptor able to withdraw electrons from Ru˙− and/or the reduced catalyst CoI (Scheme 2(A)). Therefore, when DHA accumulates in solution in the course of the catalysis, the latter is short-circuited by back electron transfers that occur from Ru˙− to HA˙ or DHA (BET process), restoring RuII and HA−, and from CoI to HA˙ or DHA (BETC process), reforming CoII. In other words, if the BET and BETC processes dominate over the catalytic process due to the accumulation of DHA, H2 generation is stopped. The other possibility is the quenching of *RuII by an electron transfer to the CoII catalyst (oxidative quenching), generating RuIII and the one-electron reduced catalyst, CoI, but this process is usually less favourable (see below). Then the CoI species can react with protons to form the elusive CoIII hydride species (denoted as CoIII-H) from which H2 can be released following different mechanisms that could proceed in parallel and that are difficult to demonstrate experimentally.50 Reduction of CoIII-H into CoII-H by another CoI or by Ru˙− is the most probable mechanism.50CoII-H can then produce H2 by reacting with a proton or another CoII-H present in solution.69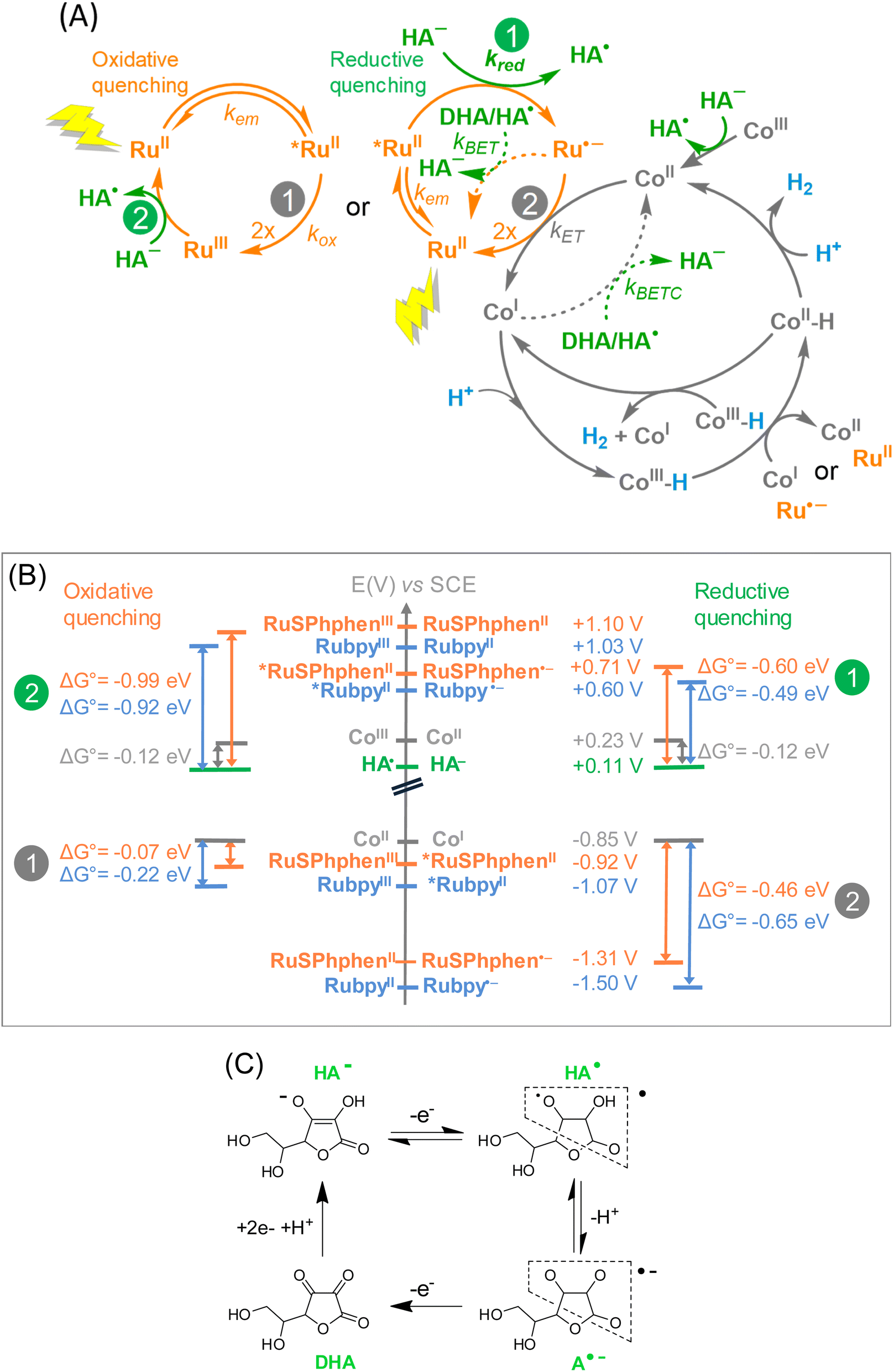 | ||
| Scheme 2 (A) Possible mechanisms for the light-driven H2 production with the system Ru/Co/HA−/H2A (Ru = Rubpy or RuSPhphen). (B) Diagrams of potentials referred versus SCE for the redox couples of RuSPhphen and Rubpy PSs at the ground and excited states, the [Co(CR14)Cl2]+ (Co) catalyst, and HA−. (C) Oxidation processes of ascorbate salt (HA−) to dehydroascorbic acid (DHA) and back reduction of DHA to HA−. | ||
Experimentally, we observed that, whatever the concentration of HA−/H2A, the systems with RuSPhphen are significantly more efficient than the corresponding systems with Rubpy. The enhanced activity can be attributed to the higher stability of RuSphen in acidic water compared to the regular Rubpy that could be the result of several parameters:
(i) a higher intrinsic stability of the reduced state, RuSPhphen˙−, in acidic water, compared to Rubpy˙−, due to the steric hindrance of the SPhphen ligand,70 and/or a greater delocalization of the radical anion over the reduced SPhphen ligand making it less prone to decoordination,
(ii) a faster electron transfer from RuSPhphen˙− to the catalyst (ET process), and/or
(iii) a faster back electron transfer from RuSPhphen˙− to HA˙ or DHA (BET process).
UV-visible absorption spectra recorded in the course of the photocatalysis (Fig. 3 and 4) do not give any information at this stage because only the ground state of the PSs is observed. The two last possibilities (ii) and (iii) do not necessarily require a better intrinsic stability of RuSPhphen˙− compared to that of Rubpy˙−. Further information can be obtained by the determination of the rate constants of the intermolecular electron transfers by time-resolved luminescence and nanosecond transient absorption spectroscopy thanks to the detection of the key intermediates *RuSPhphen and RuSPhphen˙− and the reduced form of the catalyst, CoI.
We can first estimate the driving force for the oxidative and reductive quenching processes of *Ru from the standard potentials at the ground and excited states of HA−, Rubpy, RuSPhphen and Co (Table S1,† and Scheme 2(B)). The standard potentials of RuSPhphen at the ground and excited states have only been obtained in DMF. We hypothesized that these values are close to those in water and used these values to calculate the driving force in water converted versus the SCE reference electrode by adding 298 mV (Table S1†). The reduction of the CoII catalyst by RuSPhphen˙− (reductive quenching mechanism) and by *RuSPhphen (oxidative quenching mechanism) are both thermodynamically favorable reactions (Scheme 2(B)). Indeed, considering the reduction potential of Co (E1/2 (CoII/CoI) = −0.85 V vs. SCE) in water and the redox potential of RuSPhphen at reduced and excited states (E1/2 (RuII/Ru˙−) = −1.31 V and E1/2 (RuIII/*Ru) = −0.92 V vs. SCE, Scheme 2), the driving force (ΔG0) is exergonic with values of −0.46 and −0.07 eV, respectively for the CoII reduction by Ru˙− and *Ru. The activation of the Co H2-evolving catalyst by RuSPhphen is thus thermodynamically possible.
In order to identify the most favourable first step of the photocatalytic mechanism (reductive vs. oxidative quenching of *RuSPhphen), Stern–Volmer plots were plotted between RuSPhphen and HA−/H2A (Fig. 5(A and C)) and between RuSPhphen and Co (Fig. 5(B and D)), in aqueous solution at pH 4.0 (Fig. 5(A and B)) and 4.5 (Fig. 5(C and D)), respectively. Stern–Volmer plots were obtained via stationary (I0/I) and time-resolved (τ0/τ) luminescence spectroscopies in order to survey the dynamic quenching of *Ru by HA− and Co and also to characterize a possible static quenching that could occur between *Ru, HA− and Co, during photocatalysis.71 The rate constants for the reductive quenching (kred) of *RuSPhphen by HA− were determined from τ0/τ values to be 5.04 × 107 at pH 4.0 and 9.41 × 107 at pH 4.5. These rate constants are higher than those reported with the reference PS Rubpy under similar conditions (Table 1; 2 × 107 M−1 s−1 at pH 4.0 (ref. 72 and 73) and 1 × 107 M−1 s−1 at pH 4.5 (ref. 72)). Of note, kred constants between *Ru and HA− determined from I0/I values (4.86 × 107 at pH 4.0 and 9.64 × 107 at pH 4.5) are very close to those estimated from τ0/τ values (Fig. 5(A and C)), which indicates that only dynamic quenching occurs between these two species, thus ruling out a static quenching and an interaction even weaker between *RuSPhphen and HA−. It is interesting to note that a higher rate constant was observed by the Brewer's group for reductive quenching (2.9 × 109 M−1 s−1 by HA− in water) of a Ru PS within the triad [{(SO3Ph)2phenRu(dpp)}RhX2]5+, having two 4,7-diphenyl-1,10-phenanthroline 4′,4′′-disulfonate ligands on the Ru units (Scheme 1).49 Castellano's group also reported a high rate constant (4.9 × 109 M−1 s−1) for reductive quenching between RuPhphen and DMT as a SD (see above), which was attributed to the long lifetime of the PS's excited state (5.86 μs) that facilitates bimolecular electron-transfer reactions.42
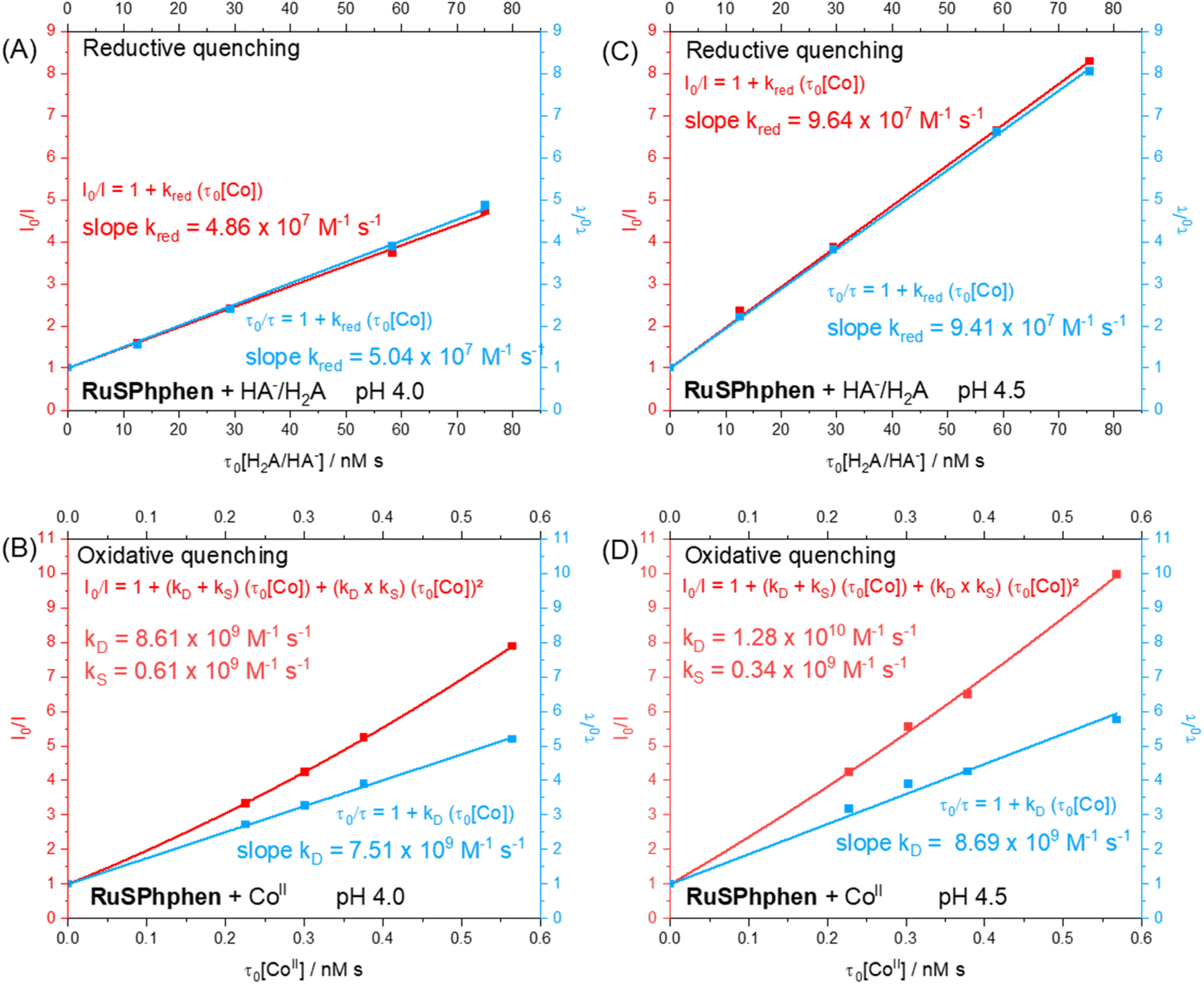 | ||
| Fig. 5 Stern–Volmer plot obtained by stationary (I0/I, red trace) and time-resolved (τ0/τ, blue trace) luminescence spectroscopies in deaerated aqueous solution at pH 4.0 (A and B) and 4.5 (C and D) (0.1 M acetate buffer) for the reductive quenching of RuSPhphen (3.4 μM) at the excited state by the NaHA/H2A couple by varying their total concentration (0, 3.3, 7.7, 15.4 and 19.8 mM) (A and C) and for the oxidative quenching of RuSPhphen (3.4 μM) at the excited state by CoII (i.e. [CoII(CR14)(H2O)2]2+) by varying its concentration (0, 0.06, 0.08, 0.1 and 0.15 mM) (B and D). I and I0 are respectively the emission intensities of *RuSPhphen registered at 614 nm with an excitation at 462 nm, with or without NaHA/H2A or CoII. τ and τ0 are respectively the lifetimes of *RuSPhphen registered at 614 nm with an excitation at 409 nm, with or without NaHA/H2A or CoII. The lifetime of *RuSPhphen without NaHA/H2A or CoII (τ0) was determined to be 3.79 μs (A), 3.76 μs (B), 3.82 μs (C) and 3.79 μs (D). | ||
| PS | RuSPhphen (3.4 μM) | Rubpy (10 μM) | RuSPhphen (3.4 μM) | Rubpy (83 or 10 μM) |
|---|---|---|---|---|
| 0.1 M acetate buffer at pH 4.0 | 0.1 M acetate buffer at pH 4.5 | |||
| a Measured with [Rubpy] = 83 μM. b Measured with [Rubpy] = 10 μM. c The CoII species (i.e. [CoII(CR14)(H2O)2]2+) used for the Stern–Volmer plot was obtained by exhaustive electrolysis at −0.20 V vs. Ag/AgCl of an aqueous solution (0.1 M NaClO4, pH 4.5) of 0.5 mM [CoIII(CR14)Cl2]+. d Global rate constant of oxidative quenching (kox) as the sum of static and dynamic quenching (kS + kD). e k ox was only estimated by the authors from τ0/τ, i.e. from the rate constant of dynamic quenching (kD). f k diss is the kinetic constant related to the disappearance of CoI in solution and can be expressed as k1[H+] + kbetc[HA˙]. n.d.: not determined. | ||||
| τ(*Ru) without HA−/CoIIc | 4 μs | 0.6 μs | 3.8 μs | 0.6 μs |
| k red with HA− | 5.04 × 107 M−1 s−1 | 2 × 107 M−1 s−1 (ref. 72 and 73) | 9.41 × 107 M−1 s−1 | 1 × 107a M−1 s−1 (ref. 72) |
| k ox with CoIIc | 9.22 × 109d M−1 s−1 | 5.1 × 109e M−1 s−1 (ref. 50) | 1.34 × 1010d M−1 s−1 | 7.07 × 109b,d M−1 s−1 |
| PS | RuSPhphen (50 μM) | Rubpy (130 μM) | RuSPhphen (50 μM) | Rubpy (100 μM) |
|---|---|---|---|---|
| 1.1 M HA−/H2A pH 4.0 | 0.1 M HA−/H2A, 1 M acetate pH 4.5 | |||
| k BET with HA− | 2.10 × 1010 M−1 s−1 | 3.5 × 109 M−1 s−1 (ref. 51) | 1.47 × 1010 M−1 s−1 | 7.4 × 109 M−1 s−1 (ref. 77) |
| k ET with HA−/Co | 2 × 109 M−1 s−1 [Co] = 200 μM | 1.4 × 109 M−1 s−1 (ref. 51) [Co] = 240 μM | 3.3 × 109 M−1 s−1 [Co] = 200 μM | n.d. |
| k diss with HA−/Co | 4.68 × 103 s−1 [Co] = 200 μM | 2.6 × 103 s−1 (ref. 51) [Co] = 240 μM | 7.11 × 103 s−1 [Co] = 200 μM | n.d. |
For the oxidative quenching of *RuSPhphen by CoII (Table 1), rate constants (kox) of 7.51 × 109 M−1 s−1 at pH 4.0 and 8.69 × 109 M−1 s−1 at pH 4.5 were estimated from τ0/τ values, the latter being higher than those reported by our group with Rubpy at pH 4.0 (5.1 × 109 M−1 s−1) and measured herein at pH 4.5 (1.27 × 109 M−1 s−1, Fig. S7†). In addition, upward-curving Stern–Volmer plots were obtained from I0/I values for the oxidative quenching of *RuSPhphen by CoII at pHs 4.0 and 4.5 (Fig. 5(B and D)), which deviates from the linear plots obtained from τ0/τ values. This behaviour clearly indicates that both static and dynamic quenching are involved in the disappearance of *RuSPhphen.71,74–76 The static quenching can be ascribed to a potential coulombic interaction between the negatively charged *RuSPhphen (beared on the sulfonate groups) and the positively charged CoII catalyst that favors the formation of an ion pair and then significantly increases the kinetics of electron transfer from *RuSPhphen to CoII. Classically, the upward curvature of the Stern–Volmer plot (i.e. I0/I = f(τ0[Q]), where [Q] is the quencher concentration) is fitted with a polynomial function of the second order, from which the static and dynamic quenching rate constants (kS and kD) are extracted by solving a quadratic equation (see Fig. 5 and the section devoted to the Stern–Volmer plot in the ESI†).71 So, from the polynomial fitting, kS and kD values for the oxidative quenching of *RuSPhphen by CoII were determined to be respectively 0.61 × 109 and 8.61 × 109 M−1 s−1 at pH 4.0, and respectively 0.34 × 109 and 1.28 × 1010 M−1 s−1 at pH 4.5. If we consider that the global rate constant of the oxidative quenching (kox) is the sum of kS and kD, the kox values are estimated to be 9.22 × 109 M−1 s−1 at pH 4.0 and 1.32 × 1010 M−1 s−1 at pH 4.5 (Table 1). The small contribution of static quenching (kS) at both pHs (6.6% at pH 4.0 and 2.6% at pH 4.5) to the global rate constant kox suggests that the phenomenon of ion pairing between *RuSPhphen and CoII is weak. Importantly, a deviation is also observed from the linear Stern–Volmer plot obtained between *Rubpy and CoII at pH 4.5, also suggesting a mixture of static and dynamic quenching, even if the global rate constant (kox = 7.07 × 109 M−1 s−1) remains lower than those between *RuSPhphen and CoII (Fig. S7†).
Considering the concentrations of Co catalyst (5–100 μM) and HA−/H2A (0.1 and 1.1 M) used during photocatalysis and the second order rate constants estimated above from τ0/τ, the reductive quenching of *RuSPhphen dominates over the oxidative one at both pHs with respective pseudo-first order rate constants of 5.54 × 107 s−1vs. 4.6–92.2 × 104 s−1 at pH 4.0, and of 9.47 × 106 s−1vs. 6.7–134 × 104 s−1 at pH 4.5. Regarding our study, the difference in the kinetic constants observed between the two PSs cannot be explained by the driving forces of the two quenching processes (Scheme 2(B)). Indeed, the driving force ΔG° for the oxidative quenching is lower for RuSPhphen (−0.07 eV) compared to Rubpy (−0.22 eV), while the driving force for the reductive quenching for both PSs is of the same order of magnitude (−0.60 eV for RuSPhphen and −0.49 eV for Rubpy). Thus, the higher rate constants of both reductive and oxidative quenching processes for *RuSPhphen could then be ascribed to its much longer lifetime (3.8 μs) compared to that of *Rubpy (0.6 μs) (Table S2†),62 promoting bimolecular electron transfer both with HA− and CoII.
The photocatalytic mechanism of the system RuSPhphen/Co/HA−/H2A was further investigated via transient absorption spectroscopy. In the absence of the Co catalyst, the transient absorption spectra of aqueous solutions of RuSPhphen (50 μM) and HA−/H2A (1.1 M for pH 4.0 and 0.1 M for pH 4.5), after a flash excitation at 462 nm, exhibit the characteristic absorption band of RuSPhphen˙− at 510–520 nm (MLCT transition) and ca. 350 nm (π–π* transition) (Fig. 6). An additional transition related to RuSPhphen˙− is observed at ca. 700 nm which could be ascribed to a d–d transition. This transition at 700 nm is also observed in the absorption spectrum of RuSPhphen˙− generated by an exhaustive reduction in DMF (Fig. 1(B)).
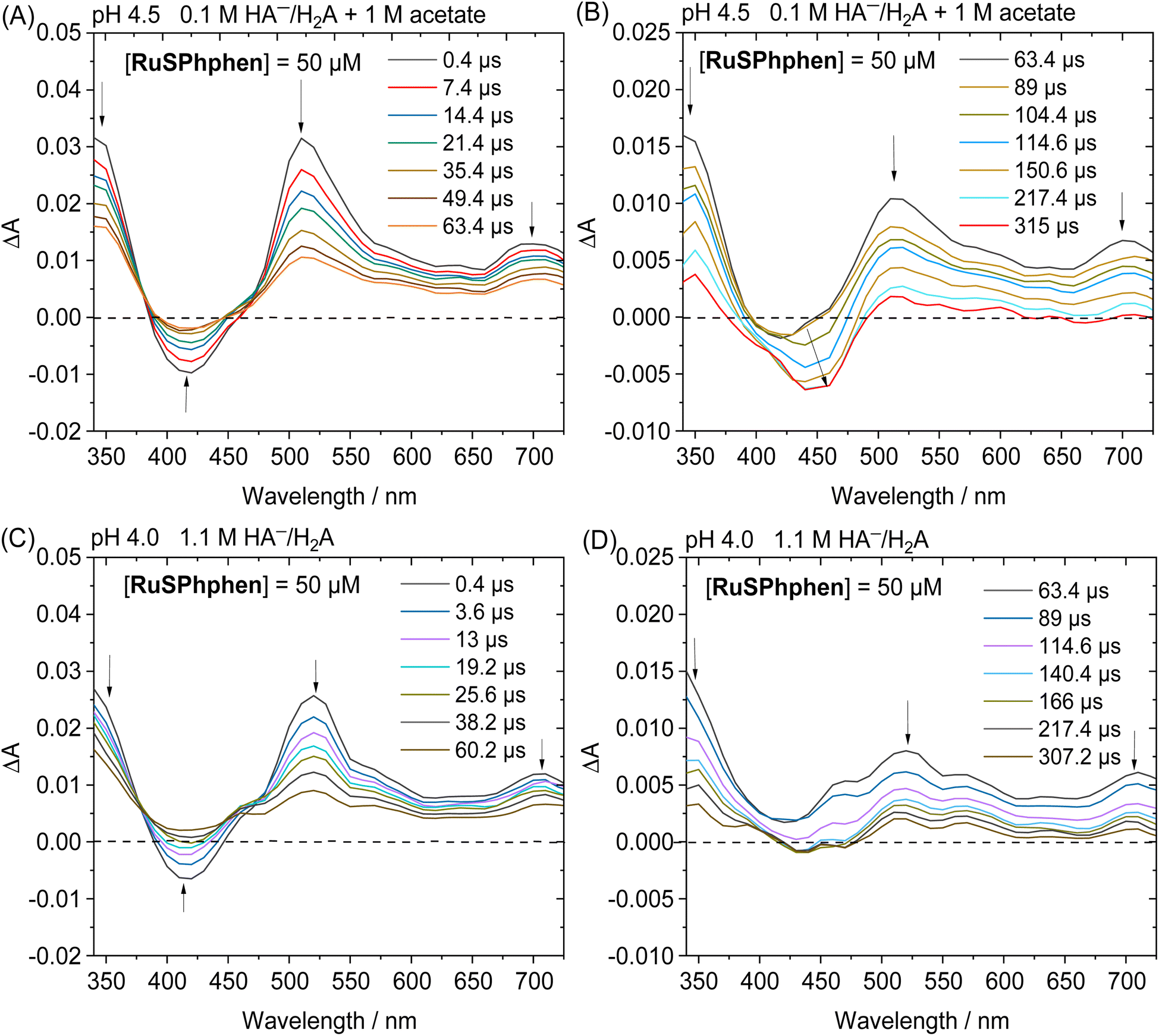 | ||
| Fig. 6 Transient absorption spectra after laser excitation (λ = 462 nm) of (A and B) a deaerated aqueous solution of 1 M acetate buffer and NaHA/H2A (0.1 M) at pH 4.5 containing RuSPhphen (50 μM) within the time ranges 0.4–63.4 μs (A) and 63.4–315 μs (B), and (C and D) a deaerated aqueous solution at pH 4.0 containing NaHA/H2A (1.1 M) and RuSPhphen (50 μM) within the time ranges 0.4–60.2 μs (C) and 63.4–307.2 μs (D); optical pathlength = 1 cm. | ||
The generation of RuSPhphen˙− through a photo-induced electron transfer from HA− to *RuSPhphen is complete within 0.4 μs following the laser excitation (Fig. 6). The decay of RuSPhphen˙−, monitored via the transient trace at 510 nm (Fig. S8†), can be fitted with a second-order kinetic law (eqn (3)) and its concentration tends to zero after almost 300 μs. A second-order kinetics can be employed to analyse the RuSPhphen˙− decay since the latter stems from the back-electron transfer between RuSPhphen˙− and one of the oxidized forms of ascorbate (i.e. HA˙ or DHA), and the concentrations of RuSPhphen˙− and HA˙ (or DHA) are assumed to be equal:
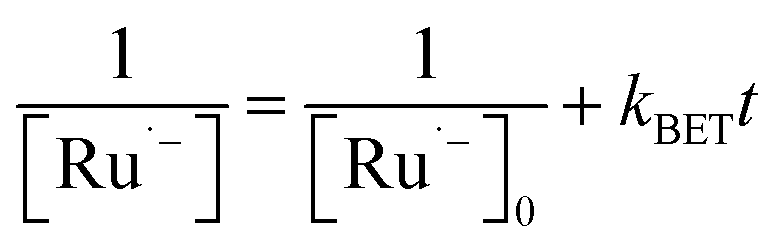 | (3) |
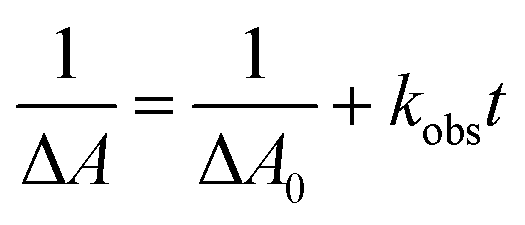 | (4) |
In the presence of a catalyst, the transient absorption spectra of the aqueous solutions containing RuSPhphen (50 μM), Co (200 μM) and HA−/H2A (1.1 M for pH 4.0 and 0.1 M for pH 4.5) also present the characteristic signature of RuSPhphen˙− at 350, 510 and 700 nm (Fig. 7), confirming a reductive quenching of *RuSPhphen by HA−. As observed above with the simple mixture of RuSPhphen and HA−/H2A, the growth of the RuSPhphen˙− absorption bands at 510 nm in the presence of Cat and SD still occurs within the first 0.4 μs after the laser flash. The decay of RuSPhphen˙− in the presence of Co and HA−, observed in the transient absorption spectra (Fig. 7) and the transient absorption trace at 510 nm (Fig. S9(A and B)†), is complete after ca. 8 μs, which is much faster than that observed without catalyst (after ca. 300 μs, Fig. 6 and S8†). Considering that the decay of RuSPhphen˙− is mainly due to an electron transfer towards the Co catalyst present in large excess in solution (200 μM), the latter can be fitted with a pseudo first order kinetic law following eqn (5):
| ln[Ru˙−] = ln[R˙−]0 − kobst | (5) |
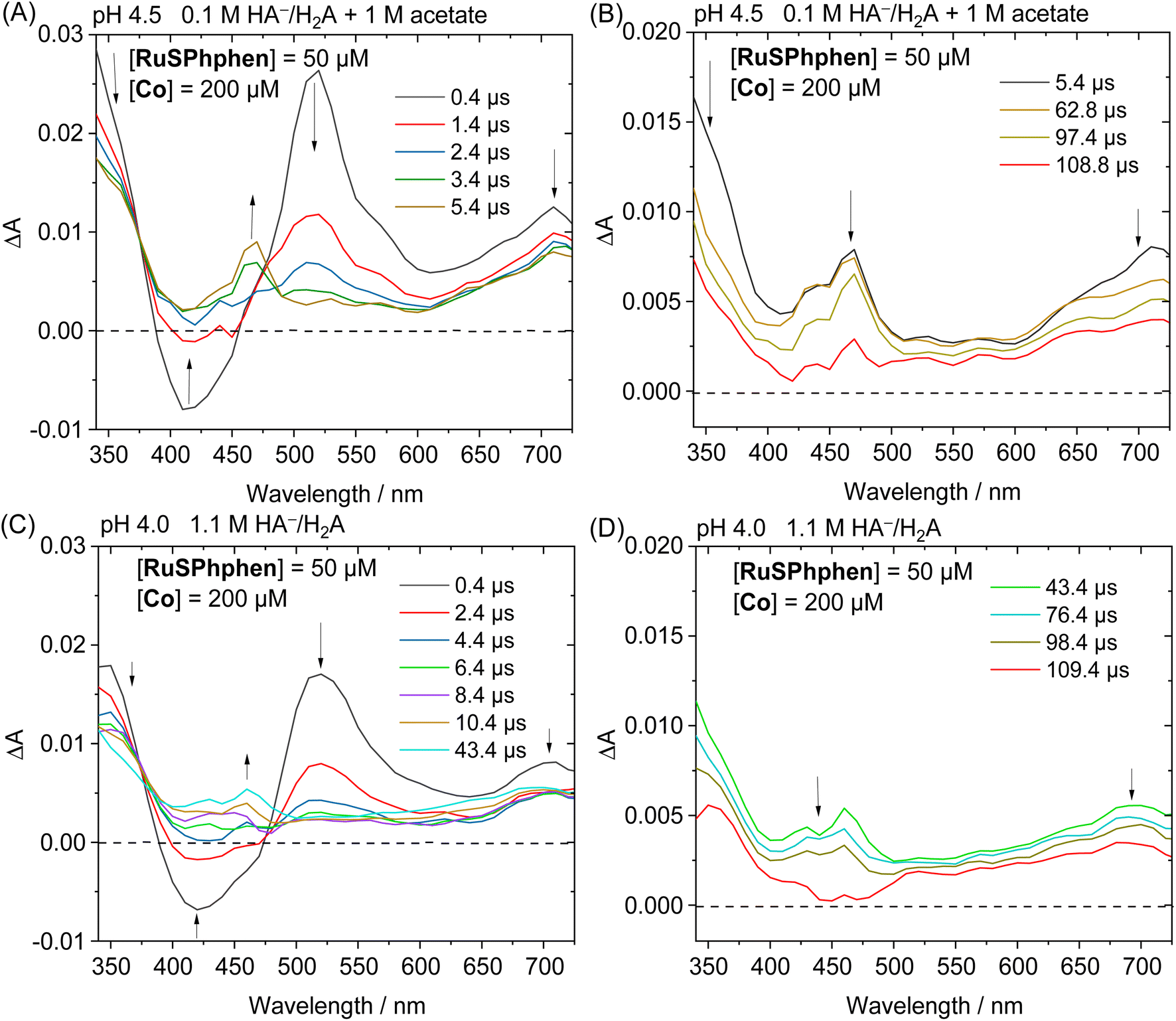 | ||
| Fig. 7 Transient absorption spectra after laser excitation (λ = 462 nm) of (A and B) a deaerated aqueous solution of 1 M acetate buffer and NaHA/H2A (0.1 M) at pH 4.5 containing RuSPhphen (50 μM) and Co (200 μM) within the time ranges 0.4–5.4 μs (A) and 5.4–108.8 μs (B) and (C and D) a deaerated 1.1 M NaHA/H2A aqueous solution at pH 4.0 containing RuSPhphen (50 μM) and Co (200 μM) within the time ranges 0.4–43.4 μs (C) and 43.4–109.4 μs (D); optical pathlength = 1 cm. | ||
Considering the Beer–Lambert law, the RuSPhphen˙− concentration (i.e. [Ru˙−]) could be converted to the variation of absorption at 510 nm in the transient absorption spectra following eqn (6):
| lnΔA = lnΔA0 − kobst | (6) |
In parallel with the disappearance of RuSPhphen˙− observed at 510 and 700 nm in the transient absorption spectra, the appearance of CoI, which is the result of an electron transfer from RuSPhphen˙− to CoII, is observed through the growth of two bands at about 430 and 460 nm and a large band at above 550 nm along with a maximum at 700 nm (Fig. 7). The same spectroscopic signature for the reduced form of the catalyst, CoI, was previously observed by transient absorption spectroscopy in the presence of the organic dye TATA+ and 0.1 M HA−/H2A at pH 4.5.40 The formation and the disappearance of the CoI species can be observed by monitoring the transient trace at 700 nm, but the changes in this band are quite complex (Fig. S10(A and D)†). First, a very fast decrease is observed at 700 nm with time constants of 89.9 ns with 0.1 M HA−/H2A and 14.1 ns with 1.1 M HA−/H2A (Fig. S10(B and E),† respectively), the latter being mainly ascribed to the disappearance of Ru˙− which exhibits an absorption band at this wavelength. Following this fast decay, new growth of the band at 700 nm occurs within a time scale of 20 μs (Fig. S10(A and D)†), along with a change of the shape of the absorption band between 550 and 725 nm observed in the transient absorption spectra (Fig. 7(A and C)). This band growth is related to the formation of CoI. Then, this band at 700 nm undergoes a decrease due to an oxidative protonation of CoI generating the CoIII-H hydride species, the key intermediate for H2 evolution (Scheme 2(A)).
This decay at 700 nm, 20 μs after the flash excitation, can be analysed via a pseudo-first order kinetic law (eqn (6)), since the concentration of the proton source (i.e. 0.55 > [H2A] > 0.03 M) is in large excess compared to that of the CoI species. The kobs rate constant for the formation of the CoIII-H hydride species can be determined as the slope of the linear function ln(ΔA) plotted as a function of time (vide supra), which gives values of 7.11 × 103 s−1 with 0.1 M HA−/H2A (pH 4.5) and 4.68 × 103 s−1 with 1.1 M HA−/H2A (pH 4.0) (Fig. S10(C and F)†). As expected, these rate constants for the generation of CoIII-H from the Co catalyst are consistent with those previously reported by our group of 7.1 × 103 s−1 with TATA+ and 0.1 M HA−/H2A (pH 4.5)40 and of 2.6 × 103 s−1 with Rubpy and 1.1 M HA−/H2A (pH 4.0).51 Castellano's group has also reported a similar kinetic value of 16 × 103 s−1 for the oxidative protonation of a polypyridine cobalt(I) catalyst, to form a CoIII-H hydride species, in the presence of Rubpy and 0.3 M HA−/H2A (pH 4.0).33
Conclusion
In view of increasing the stability of the ruthenium tris-bipyridine photosensitizer (Rubpy) which is the main limitation of such molecular PSs, the water soluble Na4[Ru((SO3Ph)2phen)3] analogue (RuSPhphen) was employed. This derivative was tested in combination with the cobalt tetraazamacrocyclic complex [CoIII(CR14)Cl2]+ (Co), one of the most efficient H2-evolving catalysts in acidic water, and ascorbate (HA−) as a SD, under visible-light irradiation. Comparative studies under similar experimental conditions with the PS reference Rubpy were also performed. Regardless of the medium, 1.1 M of HA−/H2A at pH 4.0 or 0.1 M HA−/H2A at pH 4.5 with 1 M acetate buffer, higher photocatalytic performances for H2 production were obtained with RuSPhphen. In 1.1 M HA−/H2A at pH 4.0, the production of H2 with RuSPhphen is twice at the lower catalyst concentrations of 10 and 5 μM with TONCat values of up to 2600 and 3170 for RuSPhphen, respectively versus 1380 and 1630 for Rubpy. The H2 production is always two times higher with RuSPhphen when the PS concentration further decreases to 50 and 25 μM for a catalyst concentration of 10 μM. The higher H2 production obtained with RuSPhphen can be directly correlated to the higher stability of the photocatalytic system under prolonged irradiation since at catalyst concentrations of 10 and 5 μM, H2 is produced for more than 10 h with RuSPhphen, while the H2 production is fully stopped after only 3–4 h for Rubpy. By monitoring the evolution of UV-vis absorption spectra of both PSs during photocatalysis, it was evidenced that RuSPhphen is more stable as a PS than Rubpy, although the initial rate of H2 production is higher for Rubpy. The stability of both Ru PSs is further improved when the concentration of the sacrificial electron donor is decreased to 0.1 M. Under such conditions, RuSPhphen still systematically outperforms Rubpy whatever the catalyst concentration, with TONs reaching up to 4770 at 5 μM in the catalyst. This study shows that it is possible to divide by five the quantity of ruthenium PS, while maintaining a high efficiency for H2 production.Time-resolved luminescence and nanosecond transient absorption spectroscopy were also employed to study the quenching kinetics of the Ru PS excited state, as well as the associated photocatalytic mechanism, through the detection of key intermediates such as the excited and reduced states of the PSs as well as the reduced form of the catalyst. This photophysical study clearly showed that the first step of the photocatalytic cycle is the reductive quenching of *Ru by HA−, generating the reduced state Ru˙−, and that this reductive quenching is faster (kred between 5 and 9 × 107 M−1 s−1) for RuSPhphen than for Rubpy (between 1 and 2 × 107 M−1 s−1). The RuSPhphen˙− species is thus generated very rapidly during photocatalysis, but transient absorption spectroscopy experiments also show that this species disappears very rapidly in the absence of the catalyst (kBET between 1.5 and 2.1 × 1010 M−1 s−1), through a back-electron transfer between RuSPhphen˙− and the oxidized state of the DS, i.e. HA˙ or DHA. This rapid charge recombination could contribute to the higher stability of RuSPhphen during photocatalysis, compared to Rubpy, that exhibits a much slower back electron transfer (between 3.5 and 7.4 × 109 M−1 s−1). Transient absorption spectroscopy also shows that the reduction of the CoII catalyst to CoI is just as fast and thermodynamically favorable by RuSPhphen˙− (kET between 2.0 and 3.3 × 109 M−1 s−1 and ΔG° of −0.46 eV) as by Rubpy˙− (kET of 1.4 × 109 M−1 s−1 and ΔG° of −0.65 eV). This tends to show that the RuSPhphen PS, in addition to a greater stability, presents a similar reducing power to Rubpy, which should allow it to photo-trigger the activation of a wide panel of molecular catalysts for the reduction of protons to H2 and also for the reduction of CO2. Finally, the steric hindrance and rigidity of the SPhphen ligand and the possible delocalization of the radical anion on the conjugated and extended ligand in the reduced state RuSPhphen˙−, which would then be less prone to losing a ligand in an acidic aqueous medium, could also contribute to the greater stability of RuSPhphen over Rubpy. DFT calculations are underway to evaluate the delocalization of the radical ion on the bpy and SPhphen bidentate ligands in view of rationalizing the difference in stability observed for the two PSs.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. C. thanks the University Grenoble Alpes for his PhD grant and J. A. A. thanks the French National Research Agency through the project TATADyes (ANR-20-CE05-0041) for his post-doctoral grant. We are grateful to the University Grenoble Alpes, CNRS and the French National Research Agency through the project TATADyes (ANR-20-CE05-0041), Labex ARCANE (ANR-11-LABX-0003-01) and CBH-EUR-GS (ANR-17-EURE-0003) for their financial support. This work was also supported by the ICMG Chemistry Nanobio Platform (FR2067).References
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- Y.-J. Yuan, Z.-T. Yu, D.-Q. Chen and Z.-G. Zou, Chem. Soc. Rev., 2017, 46, 603–631 RSC.
- D. Kim and T. S. Teets, Chem. Phys. Rev., 2022, 3, 021302 CrossRef CAS.
- R. S. Khnayzer, C. E. McCusker, B. S. Olaiya and F. N. Castellano, J. Am. Chem. Soc., 2013, 135, 14068–14070 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- B. Konig, Eur. J. Org Chem., 2017, 1979–1981, DOI:10.1002/ejoc.201700420.
- Y. Wu, D. Kim and T. S. Teets, Synlett, 2021, 33, 1154–1179 Search PubMed.
- V. Srivastava, P. K. Singh and P. P. Singh, J. Photochem. Photobiol., C, 2022, 50, 100488 CrossRef CAS.
- Y. Lattach, J. Fortage, A. Deronzier and J. C. Moutet, ACS Appl. Mater. Interfaces, 2015, 7, 4476–4480 CrossRef CAS PubMed.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS PubMed.
- S. Fukuzumi, J. Jung, Y. Yamada, T. Kojima and W. Nam, Chem.– Asian J., 2016, 11, 1138–1150 CrossRef CAS PubMed.
- S. Ye, C. Ding, M. Liu, A. Wang, Q. Huang and C. Li, Adv. Mater., 2019, 31, 1902069 CrossRef CAS PubMed.
- M.-N. Collomb, D. V. Morales, C. N. Astudillo, B. Dautreppe and J. Fortage, Sustainable Energy Fuels, 2020, 4, 31–49 RSC.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304, 3–19 CrossRef PubMed.
- T. Stoll, C. E. Castillo, M. Kayanuma, M. Sandroni, C. Daniel, F. Odobel, J. Fortage and M.-N. Collomb, Coord. Chem. Rev., 2015, 304–305, 20–37 CrossRef CAS.
- W. T. Eckenhoff, Coord. Chem. Rev., 2018, 373, 295–316 CrossRef CAS.
- L. Tong, L. Duan, A. Zhou and R. P. Thummel, Coord. Chem. Rev., 2020, 402, 213079 CrossRef CAS.
- R. J. Lim, M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- M. Stanbury, J.-D. Compain and S. Chardon-Noblat, Coord. Chem. Rev., 2018, 361, 120–137 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Chem. Soc. Rev., 2020, 49, 5772–5809 RSC.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed.
- J. R. Fisher and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1984, 809–813, 10.1039/dt9840000809.
- D. R. Whang, K. Sakai and S. Y. Park, Angew. Chem., Int. Ed., 2013, 52, 11612–11615 CrossRef CAS PubMed.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477–1488 RSC.
- T. Stoll, M. Gennari, J. Fortage, C. E. Castillo, M. Rebarz, M. Sliwa, O. Poizat, F. Odobel, A. Deronzier and M.-N. Collomb, Angew. Chem., Int. Ed., 2014, 53, 1654–1658 CrossRef CAS PubMed.
- M. Vennampalli, G. Liang, L. Katta, C. E. Webster and X. Zhao, Inorg. Chem., 2014, 53, 10094–10100 CrossRef CAS PubMed.
- C. V. Krishnan, C. Creutz, D. Mahajan, H. A. Schwarz and N. Sutin, Isr. J. Chem., 1982, 22, 98–106 CrossRef CAS.
- S. Guo, K.-K. Chen, R. Dong, Z.-M. Zhang, J. Zhao and T.-B. Lu, ACS Catal., 2018, 8, 8659–8670 CrossRef CAS.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC.
- B. Cecconi, N. Manfredi, T. Montini, P. Fornasiero and A. Abbotto, Eur. J. Org Chem., 2016, 2016, 5194–5215 CrossRef CAS.
- R. Gueret, L. Poulard, M. Oshinowo, J. Chauvin, M. Dahmane, G. Dupeyre, P. P. Lainé, J. Fortage and M.-N. Collomb, ACS Catal., 2018, 8, 3792–3802 CrossRef CAS.
- D. García-Fresnadillo and G. Orellana, Helv. Chim. Acta, 2001, 84, 2708–2730 CrossRef.
- R. S. Khnayzer, B. S. Olaiya, K. A. El Roz and F. N. Castellano, ChemPlusChem, 2016, 81, 1090–1097 CrossRef CAS PubMed.
- T. A. White, S. L. H. Higgins, S. M. Arachchige and K. J. Brewer, Angew. Chem., Int. Ed., 2011, 50, 12209–12213 CrossRef CAS PubMed.
- T. A. White, J. D. Knoll, S. M. Arachchige and K. J. Brewer, Materials, 2012, 5, 27–46 CrossRef CAS PubMed.
- G. F. Manbeck and K. J. Brewer, Coord. Chem. Rev., 2013, 257, 1660–1675 CrossRef CAS.
- T. A. White, H. E. Mallalieu, J. Wang and K. J. Brewer, Chem.– Eur. J., 2014, 20, 8265–8268 CrossRef CAS PubMed.
- G. F. Manbeck, T. Canterbury, R. Zhou, S. King, G. Nam and K. J. Brewer, Inorg. Chem., 2015, 54, 8148–8157 CrossRef CAS PubMed.
- H. M. Rogers, T. A. White, B. N. Stone, S. M. Arachchige and K. J. Brewer, Inorg. Chem., 2015, 54, 3545–3551 CrossRef CAS PubMed.
- T. R. Canterbury, S. M. Arachchige, K. J. Brewer and R. B. Moore, Chem. Commun., 2016, 52, 8663–8666 RSC.
- S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M.-N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544–17552 RSC.
- R. Gueret, C. E. Castillo, M. Rebarz, F. Thomas, A.-A. Hargrove, J. Pécaut, M. Sliwa, J. Fortage and M.-N. Collomb, J. Photochem. Photobiol., B, 2015, 152, 82–94 CrossRef CAS PubMed.
- M. Sandroni, R. Gueret, K. D. Wegner, P. Reiss, J. Fortage, D. Aldakov and M. N. Collomb, Energy Environ. Sci., 2018, 11, 1752–1761 RSC.
- R. Gueret, C. E. Castillo, M. Rebarz, F. Thomas, M. Sliwa, J. Chauvin, B. Dautreppe, J. Pécaut, J. Fortage and M.-N. Collomb, Inorg. Chem., 2019, 58, 9043–9056 CrossRef CAS PubMed.
- P. Bugnon and R. E. Hester, Chem. Phys. Lett., 1983, 102, 537–543 CrossRef CAS.
- W. K. C. Lo, C. E. Castillo, R. Gueret, J. Fortage, M. Rebarz, M. Sliwa, F. Thomas, C. J. McAdam, G. B. Jameson, D. A. McMorran, J. D. Crowley, M.-N. Collomb and A. G. Blackman, Inorg. Chem., 2016, 55, 4564–4581 CrossRef CAS PubMed.
- C. Bachmann, M. Guttentag, B. Spingler and R. Alberto, Inorg. Chem., 2013, 52, 6055–6061 CrossRef CAS PubMed.
- M. Guttentag, A. Rodenberg, C. Bachmann, A. Senn, P. Hamm and R. Alberto, Dalton Trans., 2013, 42, 334–337 RSC.
- E. Deponti, A. Luisa, M. Natali, E. Iengo and F. Scandola, Dalton Trans., 2014, 43, 16345–16353 RSC.
- M. Natali, A. Luisa, E. lengo and F. Scandola, Chem. Commun., 2014, 50, 1842–1844 RSC.
- L. P. Tong, R. F. Zong and R. P. Thummel, J. Am. Chem. Soc., 2014, 136, 4881–4884 CrossRef CAS PubMed.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed.
- T. Stoll, M. Gennari, I. Serrano, J. Fortage, J. Chauvin, F. Odobel, M. Rebarz, O. Poizat, M. Sliwa, A. Deronzier and M.-N. Collomb, Chem.– Eur. J., 2013, 19, 782–792 CrossRef CAS PubMed.
- C. Costentin, F. Camara, J. Fortage and M.-N. Collomb, ACS Catal., 2022, 6246–6254 CrossRef CAS.
- E. Amouyal and P. Koffi, J. Photochem., 1985, 29, 227–242 CrossRef CAS.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855–859 CrossRef CAS.
- B. R. Galan, M. L. Reback, A. Jain, A. M. Appel and W. J. Shaw, Eur. J. Inorg. Chem., 2013, 2013, 5366–5371 CrossRef CAS.
- G. M. Brown, B. S. Brunschwig, C. Creutz, J. F. Endicott and N. Sutin, J. Am. Chem. Soc., 1979, 101, 1298–1300 CrossRef CAS.
- M. Guttentag, A. Rodenberg, R. Kopelent, B. Probst, C. Buchwalder, M. Brandstätter, P. Hamm and R. Alberto, Eur. J. Inorg. Chem., 2012, 2012, 59–64 CrossRef CAS.
- C.-B. Li, A. J. Bagnall, D. Sun, J. Rendon, M. Koepf, S. Gambarelli, J.-M. Mouesca, M. Chavarot-Kerlidou and V. Artero, Sustainable Energy Fuels, 2022, 6, 143–149 RSC.
- M. L. A. Hakkennes, M. S. Meijer, J. P. Menzel, A.-C. Goetz, R. Van Duijn, M. A. Siegler, F. Buda and S. Bonnet, J. Am. Chem. Soc., 2023, 145, 13420–13434 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, NY, 2017 Search PubMed.
- B. Shan, T. Baine, X. A. N. Ma, X. Zhao and R. H. Schmehl, Inorg. Chem., 2013, 52, 4853–4859 CrossRef CAS PubMed.
- C. Creutz, N. Sutin and B. S. Brunschwig, J. Am. Chem. Soc., 1979, 101, 1297–1298 CrossRef CAS.
- X. Zhang, K. Yamauchi and K. Sakai, ACS Catal., 2021, 11, 10436–10449 CrossRef CAS.
- K. Kitamoto and K. Sakai, Chem. Commun., 2016, 52, 1385–1388 RSC.
- C. V. Suneesh, B. Balan, H. Ozawa, Y. Nakamura, T. Katayama, M. Muramatsu, Y. Nagasawa, H. Miyasaka and K. Sakai, Phys. Chem. Chem. Phys., 2014, 16, 1607–1616 RSC.
- F. Camara, T. Gavaggio, B. Dautreppe, J. Chauvin, J. Pécaut, D. Aldakov, M.-N. Collomb and J. Fortage, Molecules, 2022, 27, 6614 CrossRef CAS PubMed.
- The Δε value was determined from the difference in absorption at 510 nm between the spectra of RuSPhphen at ground and mono-reduced states presented in Fig. 2.
- W. M. Singh, M. Mirmohades, R. T. Jane, T. A. White, L. Hammarström, A. Thapper, R. Lomoth and S. Ott, Chem. Commun., 2013, 49, 8638–8640 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, general experimental details, preparation and characterisation (1H NMR, ESI-MS) of RuSPhphen, and additional data for electrochemistry, UV-visible absorption spectroscopy, photocatalytic hydrogen production, emission spectroscopy, Stern–Volmer plot and nanosecond transient absorption spectroscopy (Tables S1–S3 and Fig. S1–S10). See DOI: https://doi.org/10.1039/d3se01556d |
| This journal is © The Royal Society of Chemistry 2024 |