
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExfoliation of a metal–organic framework enabled by post-synthetic cleavage of a dipyridyl dianthracene ligand†
Madison E.
Logelin
 ,
Eric
Schreiber
,
Brandon Q.
Mercado
,
Michael J.
Burke
,
Caitlin M.
Davis
and
Amymarie K.
Bartholomew
*
,
Eric
Schreiber
,
Brandon Q.
Mercado
,
Michael J.
Burke
,
Caitlin M.
Davis
and
Amymarie K.
Bartholomew
*
Department of Chemistry, Yale University, New Haven, Connecticut 06520, USA. E-mail: amymarie.bartholomew@yale.edu
First published on 20th August 2024
Abstract
The synthetic tunability and porosity of two-dimensional (2D) metal–organic frameworks (MOFs) renders them a promising class of materials for ultrathin and nanoscale applications. Conductive 2D MOFs are of particular interest for applications in nanoelectronics, chemo-sensing, and memory storage. However, the lack of covalency along the stacking axis typically leads to poor crystallinity in 2D MOFs, limiting structural analysis and precluding exfoliation. One strategy to improve crystal growth is to increase order along the stacking direction. Here, we demonstrate the synthesis of mechanically exfoliatable macroscopic crystals of a 2D zinc MOF by selective dimensional reduction of a 3D zinc MOF bearing a dianthracene (diAn) ligand along the stacking axis. The diAn ligand, a thermally cleavable analogue of 4,4′-bipyridine, is synthesized by the direct functionalization of dianthraldehyde in a novel “dianthracene-first” approach. This work presents a new strategy for the growth of macroscopic crystals of 2D materials while introducing the functionalization of dianthraldehyde as a means to access new stimuli-responsive ligands.
Introduction
Since the isolation and characterization of monolayer graphene as a two-dimensional (2D) semi-metal, the search for other 2D materials that can be exfoliated, studied, and eventually used at the ultrathin limit has captured the attention of chemists, materials scientists, and engineers alike.1–12 2D metal–organic frameworks (MOFs) have distinct properties from traditional 2D materials, particularly in terms of their high porosity and synthetic tunability.10–18Synthesizing large, exfoliatable single crystals of 2D MOFs remains a major challenge, limiting our understanding of their structure and properties, as well as their use at the 2D limit.11,17–20 Lack of covalent bonding between the 2D sheets often leads to stacking faults and misalignment of layers along the stacking axis, producing needle-like crystalline morphologies or microcrystalline powders.11,18–21 As the crystallinity of these materials decreases, grain boundaries between the crystallites increase, reducing the electrical and thermal conductivity of the bulk material.11,18,19,21–23 Highly crystalline material is therefore crucial in understanding the intrinsic properties, trends, and structure–function relationships of 2D MOFs.11,18,19
There have been numerous attempts to improve the growth of large, exfoliatable, single crystals of 2D MOFs.18,19,24–27 Besides optimizing synthetic conditions (i.e. temperature, solvent, metal source, etc.), different methods, including biphasic19,27 approaches and the addition of modulators,28,29 have been developed to increase control over the kinetics of crystal growth. Modifying the MOF itself by increasing the strength of interlayer covalent interactions has also been shown to increase crystal size by improving order along the stacking axis.18,19,24–26 Additionally, dimensional reduction by post-synthetic ligand insertion or removal30–33 has resulted in some successful examples of exfoliatable MOFs, leading us to hypothesize that the introduction of a post-synthetically cleavable, covalent link could be effective.
Herein, we introduce a new approach for preparing large, exfoliatable crystals of 2D MOFs by exploiting the reversible photodimerization reaction of anthracene (An) to dianthracene (diAn). Linking 2D MOF sheets with diAn ligands creates strong covalent bonds in the stacking direction, enabling the synthesis of large, single crystals. Once the dianthracene-linked 3D MOF has been synthesized, the diAn ligands can be selectively undimerized through post-synthetic annealing (Fig. 1). After annealing, the resulting 2D MOF can be mechanically exfoliated into thin sheets.
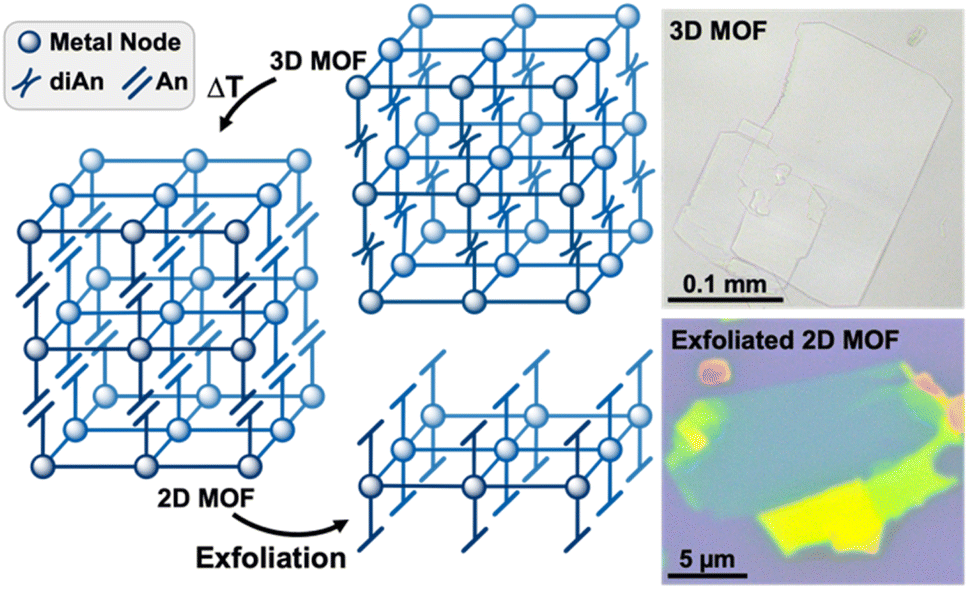 | ||
| Fig. 1 Generalized pathway from a diAn-linked 3D MOF to an exfoliated, 2D monolayer (left). Optical images of the 3D (top right) and exfoliated 2D MOF (bottom right) produced in this work. The teal region represents 5–6 layers, as measured by atomic force microscopy (AFM). | ||
Results and discussion
The reversible photodimerization of anthracene has only recently been exploited for stimuli-responsive inorganic and materials applications.34–37 Although prevalent within polymer chemistry,38–42 few 9,9′-functionalized diAn compounds exist as linking ligands within coordination networks (CNs).43–52 These examples have exhibited changes in magnetism, luminescence, and photomechanical response upon undimerization and feature diAn as a multi-dimensional ligand (Fig. 2a).43–52 Thus, upon undimerization, these frameworks break into molecular species or chains.43–52 In order to use diAn to prepare a 2D MOF by dimensional reduction, the diAn ligand must selectively bind along the stacking axis.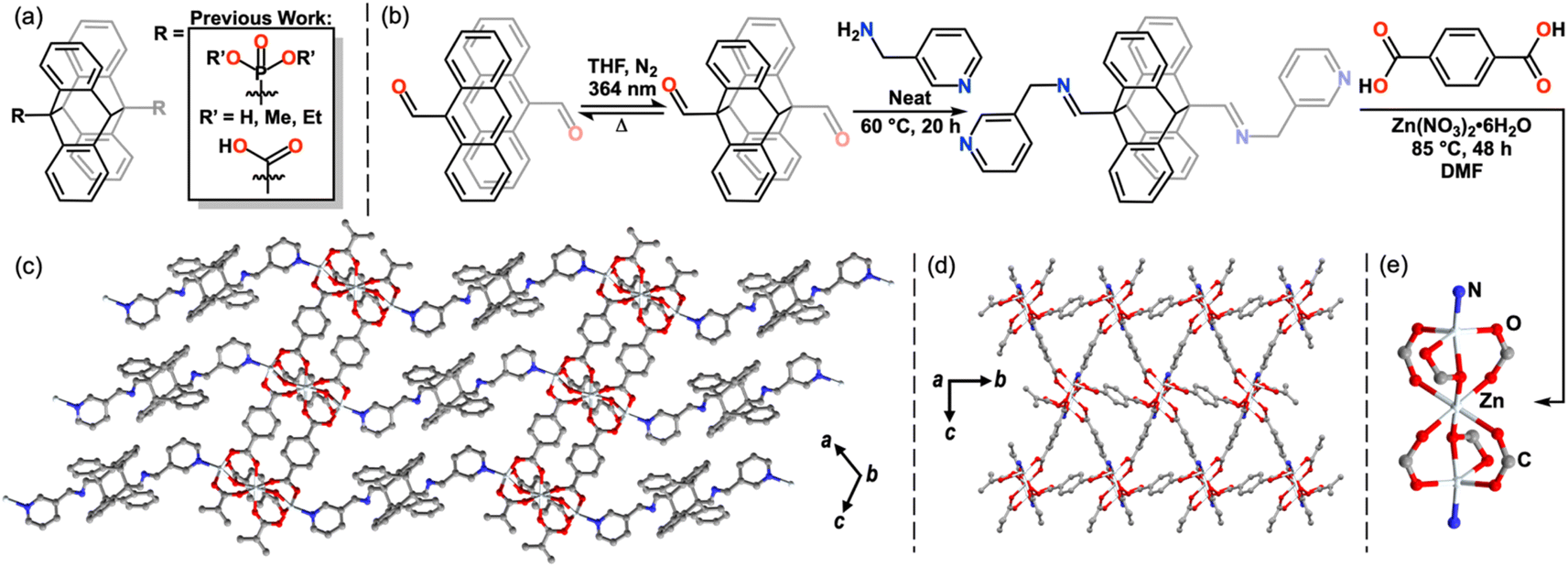 | ||
| Fig. 2 Previously known 9,9′-functionalized diAn ligands for MOFs and CNs (a). “Dianthracene-first” synthesis of the diAnCNC-3Py ligand (b). The crystal structure of 1, depicting the view along the stacking direction (c), across the 2D Zn3BDC3 sheets within the 3D structure, where the diAn ligand has been reduced to a single nitrogen atom for clarity (d), and the tri-zinc node (e). All hydrogen atoms and DMF molecules have been omitted for clarity. | ||
4,4′-Bipyridine (bipy) is a commonly used linker in coordination polymers and 3D MOFs,13,14 frequently forming networks in which bipy links metal sites along one axis, while anionic ligands link in the other two dimensions.53–55 We hypothesized that a 9,9′-functionalized diAn analogue of bipy could enable the desired orientation of the cleavable diAn group in the stacking direction. In most known examples of 9,9′-functionalized diAn ligands, the diAn group presents considerable steric bulk close to the donor atom(s), limiting the synthesis of MOFs to those that use large cations, such as lanthanides or precious metals, and, rarely, first-row transition metals (Fig. 2a).43–52 We targeted the synthesis of a diAn ligand analogue of bipy as a selective linker for 2D sheets with an imine link and methyl spacer to provide more distance between the pyridyl ligands and the sterically bulky diAn core (Fig. 2b).
The preparation of diAn ligands presents a unique synthetic challenge. While syntheses of functionalized anthracene monomers are often straightforward, dimerization must then be achieved either by irradiation of crystalline material or in a highly concentrated solution. Solid state photodimerization requires that the anthracenes crystal pack serendipitously within 4.2 Å and in the correct alignment, with the anthracenes fully overlapped.46,56 In solution, the concentration must be high enough so that an excited anthracene can react with a nearby anthracene to form an excimer before decaying.57 Functionalized monomers such as those required to produce a diAn bipy analogue are typically much less soluble than more simply substituted anthracenes, precluding solution-phase dimerization.57
To bypass this dilemma, we employed a “dianthracene-first” approach by direct functionalization of 9,9′-dianthraldehyde (diAnCHO), which can be prepared from inexpensive 9-anthraldehyde on gram scale (Fig. 2b).58 Functionalization of diAn derivatives has been scarcely reported,38,39,59,60 with no previous literature reports of diAnCHO derivatization. Reaction conditions must account for the solution phase thermal undimerization of diAnCHO, which occurs within hours above 65 °C, as well as its marginal solubility in most solvents. Therefore, we elected to functionalize diAnCHO with a liquid primary amine, affording high reagent concentrations to overcome the thermal and solubility challenges inherent to diAnCHO. Condensation in neat 3-picolylamine at 60 °C produced the desired pyridine-terminated dimer, di-(N-(9-anthracenylmethylene)-3-pyridinemethanamine) (diAnCNC-3Py), in 94.4% yield.
With the pyridyl-terminated diAn in hand, we sought to generate a layered MOF incorporating this ditopic, neutral linker. One such platform is the Zn3BDC3L2 lattice, where BDC = 1,4-benzenedicarboxylic acid, first reported by Hubberstey, Schröder and coworkers in 2005.61 This general motif features 2D Zn-carboxylate sheets capped or linked by neutral mono- and ditopic ligands to produce 2- and 3D structures.62–65 An example of a Zn3BDC3L2 framework from Yu and coworkers65 features an apical pyridine, while a 3D analogue from Zheng and coworkers features a bridging bis(imidazole) spacer.63 The ability of the Zn3BDC3L2 motif to bind neutral, nitrogen-based capping ligands and the tolerance of the overall structure toward different dimensionalities make this an attractive platform in which to incorporate diAnCNC-3Py. Indeed, solvothermal synthesis in N,N-dimethylformamide (DMF) at 85 °C using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4 ratio of Zn(NO3)2·6H2O:diAnCNC-3Py:BDC yields a crystalline 3D MOF, Zn3BDC3diAnCNC-3Py (1), with two lattice DMF molecules per formula unit in the P21/c space group (Fig. 2c–e). The clear, colorless plates were characterized by single crystal X-ray diffraction (SCXRD) and the bulk material was confirmed to be uniform by powder X-ray diffraction (PXRD) and optical microscopy (Fig. 3g, S19, S26 and S27†). The secondary building unit (SBU) of 1 consists of three zinc atoms and six BDC molecules (Fig. 2e). The central zinc atom is pseudo-octahedral, bound to two monoatomic and four syn–syn bidentate bridging oxygen atoms of six discrete BDC carboxylates. The outer zinc atoms, related by symmetry, are distorted square pyramidal in coordination geometry. Each is bound to a terminal pyridine moiety of diAnCNC-3Py and four oxygen atoms from three BDC carboxylates. The BDC ligands connect individual SBUs, forming a 2D sheet (Fig. 2d). These 2D Zn3BDC3 sheets are linked by diAnCNC-3Py, resulting in a 3D MOF with similar connectivity to the Zn3BDC3(1,2-bis(2-(1H-imidazol-1-yl)ethoxy)ethane)2 framework, which also bears a ditopic, N-donor ligand in the stacking dimension (Fig. 2c).63
:1:4 ratio of Zn(NO3)2·6H2O:diAnCNC-3Py:BDC yields a crystalline 3D MOF, Zn3BDC3diAnCNC-3Py (1), with two lattice DMF molecules per formula unit in the P21/c space group (Fig. 2c–e). The clear, colorless plates were characterized by single crystal X-ray diffraction (SCXRD) and the bulk material was confirmed to be uniform by powder X-ray diffraction (PXRD) and optical microscopy (Fig. 3g, S19, S26 and S27†). The secondary building unit (SBU) of 1 consists of three zinc atoms and six BDC molecules (Fig. 2e). The central zinc atom is pseudo-octahedral, bound to two monoatomic and four syn–syn bidentate bridging oxygen atoms of six discrete BDC carboxylates. The outer zinc atoms, related by symmetry, are distorted square pyramidal in coordination geometry. Each is bound to a terminal pyridine moiety of diAnCNC-3Py and four oxygen atoms from three BDC carboxylates. The BDC ligands connect individual SBUs, forming a 2D sheet (Fig. 2d). These 2D Zn3BDC3 sheets are linked by diAnCNC-3Py, resulting in a 3D MOF with similar connectivity to the Zn3BDC3(1,2-bis(2-(1H-imidazol-1-yl)ethoxy)ethane)2 framework, which also bears a ditopic, N-donor ligand in the stacking dimension (Fig. 2c).63
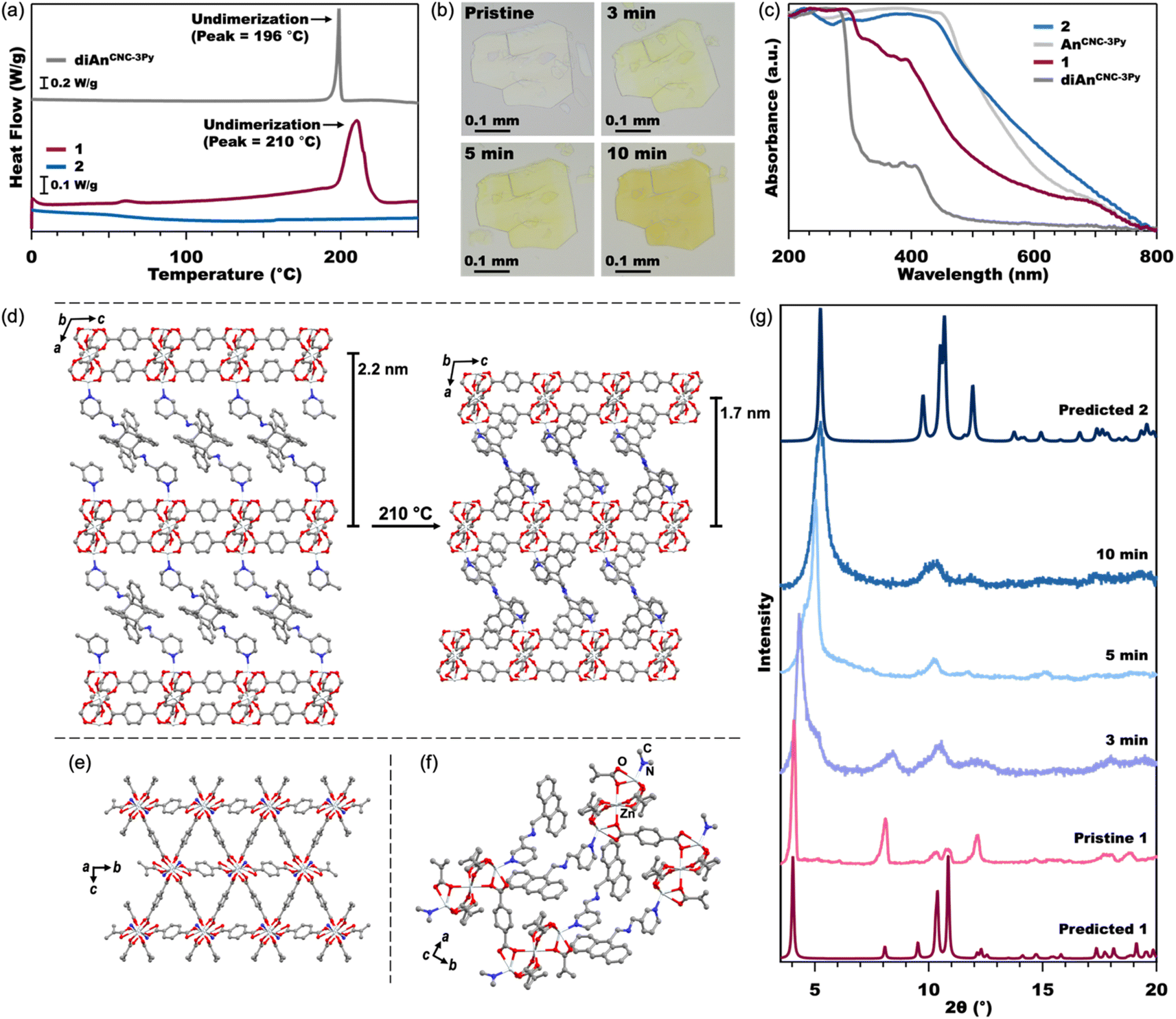 | ||
| Fig. 3 DSC (a) of the diAnCNC-3Py ligand and MOFs 1 and 2 from 0–250 °C (see Fig. S7† for full data plot). Optical microscope images (b) of pristine crystals of 1 and crystals annealed at 210 °C. Normalized solid-state UV-vis spectra (c) of the diAnCNC-3Py and AnCNC-3Py ligands and MOFs 1 and 2. Crystal structures (d) of 1 (left) and 2 (right) showing compression of the 2D Zn3BDC3 sheets upon undimerization of diAn. A view across the 2D Zn3BDC3 sheets of 2, (e) where the An ligand has been reduced to a single nitrogen atom for clarity. A close-up view of the stacking between An ligands in 2 (f). All hydrogen atoms have been omitted for clarity. PXRD (g) of pristine crystals of 1, crystals annealed at 210 °C, and predicted spectra from the SCXRD and MicroED structures of 1 and 2, respectively. | ||
The connection of 2D Zn3BDC3 sheets by diAnCNC-3Py within the 3D lattice makes 1 an excellent candidate for dimensional reduction into an exfoliatable 2D material. Photochemical and thermal cleavage of diAn is well established, with several diAn-bridged MOFs known to undimerize into lower-dimensional compounds (3D to 1D or 3D to 0D molecules).43,46 To determine suitable annealing conditions for 1, we turned to thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The TGA curve for 1 revealed a mass loss of 14.3% upon heating to 190 °C, broadly consistent with the loss of the two solvent molecules (calculated 112.51 g mol−1, 1.54 equiv. DMF) from within the lattice (Fig. S6†). No further mass loss was observed until the onset of material degradation at 300 °C. DSC analysis of 1 revealed an exothermic peak centered at 210 °C, consistent with undimerization of the diAn core (Fig. 3a).45 The DSC curve for the free diAnCNC-3Py ligand features a sharp peak at 196 °C; the shift in this feature to higher temperature in 1 is likely a consequence of structural confinement within the 3D lattice as has been observed for other diAn-linked MOFs.45–47,49,51
Based on the DSC of 1, pristine crystals were annealed at 210 °C. After 2 min at 210 °C, the once-colorless crystals began to turn yellow while retaining their overall size and shape (Fig. 3b). The crystals continued to turn a darker yellow-orange color until annealed for about 10 minutes, after which the color remained constant. The yellow color is consistent with conversion of diAnCNC-3Py ligands to their monomeric form, and thus the generation of the 2D MOF, Zn3BDC3AnCNC-3Py2 (2). This is supported by an analogous annealing experiment, where free diAnCNC-3Py was heated to 196 °C for 10 min, revealing complete transformation of diAnCNC-3Py to the corresponding monomer, AnCNC-3Py, by 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S4†).
Solid-state UV-vis spectroscopy was also used to compare the dimer and monomer ligands to MOFs 1 and 2 (Fig. 3c). The MOF spectra have similar features to their ligand counterparts. The normalized spectra of diAnCNC-3Py and 1 show strong absorbance features in the UV region at approximately 300 nm and 400 nm, while the normalized spectra of AnCNC-3Py and 2 show strong absorbance features at 460 nm, corresponding to their yellow-orange color. The DSC data obtained for a sample of 1 annealed for 10 min at 210 °C lacks any exothermic feature, further confirming complete undimerization of the diAnCNC-3Py ligand (Fig. 3a). This relationship between DSC peak temperature, annealing time, and complete undimerization is consistent with that observed for other diAn-linked MOFs.45–47,50,51 The TGA curve for 2 reveals little mass loss until the onset of material degradation at 310 °C, indicating that the DMF molecules trapped in the lattice of 1 are liberated upon annealing (Fig. S6†).
Characterization of 2 by SCXRD was attempted, however, annealing decreases the crystal domain size, prohibiting collection of an SCXRD structure. This is also observed through the line broadening of PXRD traces as the sample is annealed (Fig. 3g, S26, and S27†). We instead turned to microcrystal electron diffraction (MicroED), which allows for structural determination of significantly smaller crystal domains. MicroED analysis of crystals of 1 annealed for 3 min at 210 °C yields the fully undimerized, 2D MOF crystal structure in the P21/c space group with no solvent in the lattice (Fig. 3d–f). The only substantive difference between the structures of 1 and 2 is in the stacking direction. In 2, the AnCNC-3Py ligands are stacked so that the anthracene of one layer is next to the pyridine of the adjacent layer, resulting in a stacking distance approximately 0.5 nm shorter between the 2D layers (Fig. 3d). The 2D Zn3BDC3 sheets maintain the same structural connectivity as in the pristine 3D MOF, 1, but the SBUs of 2 are now slightly offset from the SBUs of adjacent layers (Fig. 2e and S30†). Attempts to independently synthesize 2 from the monomeric ligand, AnCNC-3Py, were unsuccessful and consistently produced the water-capped analogue.62 This highlights the importance of diAnCNC-3Py in generating 2.
While diAn-linked MOFs that undergo 3D to 1D or 3D to 0D dimensional reduction often crack upon thermal annealing, we note that annealed samples of 1 are identical in appearance to their pristine state aside from the yellow color arising from the anthracene monomers, with no new cracks or features observable by optical microscopy (Fig. 3b). We propose that this greater morphological fidelity arises from the decreased strain induced by undimerization along one axis compared to that induced by undimerization along two or three axes. The thermal annealing of 1 produces a rapid phase change in the PXRD pattern of the material (Fig. 3g). The features at 4.2° and 8.2° in 1, which both correspond to reflections along the stacking axis, a, increase to 5.3° and 10.4° as the MOF is annealed. Although the MicroED structure of 2 was collected on crystals annealed for 3 min, we note that this structure does not feature any disorder corresponding to a mixture of phases between 1 and 2 as is demonstrated in the measured PXRD at 3 min (Fig. 3g). Therefore, although bulk conversion of 1 to 2 does not occur until 10 min at 210 °C, we conclude that the domains analyzed by MicroED were fully undimerized in the short annealing time.
The layered nature of both 1 and 2 is clear from scanning electron microscopy (SEM) and indicates that 2 is a suitable material for exfoliation (Fig. 4a, b, S13, and S14†). We note that, in our hands, the Py-capped analogue65 is similarly produced as plate-like crystals which can be mechanically exfoliated (Fig. S24†). Therefore, annealing conditions were optimized for mechanical exfoliation by the Scotch tape method (Fig. S22†). Exfoliation of crystals annealed for longer times at lower temperatures produced larger exfoliated flakes than those annealed at 210 °C. Thus, crystals of 1 were annealed at 160 °C for 18 h, which shows complete conversion to 2 by PXRD (Fig. S26†). Mechanical exfoliation of 2 onto Si/SiO2 substrates produced atomically flat flakes as thin as 4 nm (determined by AFM, Fig. 4, S17, and S18†). A representative 10 nm flake is shown in Fig. 4c–e. The height of a single layer of 2 is expected to be ∼1.7 nm according to the MicroED crystal structure of 2, with a 10 nm sample representing roughly five or six layers. The average root mean squared (RMS) roughness of a single layer sampled from 16 flakes is 0.471 nm (standard deviation = 0.179 nm) (Fig. S18 and Table S1†).
 | ||
| Fig. 4 SEM of MOFs 1 (a) and 2 (b) with progressively magnified images (left to right) showing the layers of the crystals' edges. Optical image (c) of an exfoliated flake of 2 on a Si/SiO2 wafer, its AFM topography map (d), and height profile (e). | ||
Confirmation that exfoliated flakes were indeed crystals of 2 was gained from optical photothermal infrared (O-PTIR) spectroscopy, which allows for the acquisition of spatially-resolved vibrational spectra of thin samples. The O-PTIR spectra of exfoliated crystals were found to be in excellent agreement with those of bulk annealed 2, indicating that the material remained unchanged after exfoliation (Fig. S11 and S12†). Additional evidence of material integrity upon exfoliation was found by X-ray Photoelectron Spectroscopy (XPS), which revealed the expected elemental composition of the exfoliated MOF (Fig. S16†). While pristine crystals of 1 cannot be exfoliated at all, 2 can be mechanically exfoliated to few-layer samples, demonstrating the first example of the conversion of a dianthracene-based 3D framework into exfoliatable 2D sheets.
Conclusions
The “dianthracene-first” method of direct functionalization of diAnCHO allowed us to prepare a new, neutral, minimally sterically encumbered diAn analogue of 4,4′-bipyridine (bipy), which in turn enabled the preparation of a 3D Zn MOF with cleavable diAn links along only one axis. 3D MOFs and CNs commonly feature bipy as a linker between 2D sheets, making our approach promising for application to diverse molecular materials. MOF 1 can be thought of as a masked 2D material that can be grown with complete control over structure in the stacking direction, giving rise to large, single crystals with a favorable aspect ratio for exfoliation. Annealing releases the diAn linkage, achieving dimensional reduction to the 2D, mechanically exfoliatable form without loss of sample integrity or size. Additionally, in magnetic and/or conductive MOFs, the compression of 2D sheets from the undimerization of diAn would be expected to modify the coupling or electron transport between the layers. Ongoing work in our group seeks to harness this strategy to both engineer specific interlayer alignments in 2D molecular materials, as well as to access large crystals of magnetic and/or electrically conductive 2D MOFs.Data availability
Data for this article are available at https://doi.org/10.1039/D4SC03524K. The data supporting this article (detailed experimental and synthetic methods and characterization, including NMR, TGA, DSC, ATR-FTIR, O-PTIR, SEM, XPS, AFM, optical images, exfoliation techniques, XRD, MicroED data, and crystallographic tables) have been included as part of the ESI.† Crystallographic data for 1 and 2 has been deposited at the CCDC under 2342280 (1) and 2356228 (2) and can be obtained from https://www.ccdc.cam.ac.uk/.Author contributions
M. E. L. and A. K. B. conceived and planned the experiments. M. E. L. synthesized all complexes and collected all data except for SEM, XPS, DSC, TGA, MicroED, and O-PTIR. E. S. performed SEM, DSC and TGA experiments. B. Q. M. collected MicroED data and solved and refined the MicroED crystal structure. M. J. B. collected O-PTIR data and contributed O-PTIR-related text. C. M. D. conceived of O-PTIR experimental setup, interpreted O-PTIR, and contributed to manuscript editing. M. E. L., E. S., and A. K. B. contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the Yale University Provost's Office and the Yale University Chemistry Department. E. S. was supported by a Rudolph J. Anderson Postdoctoral Fellowship from Yale University. NMR, Single crystal and powder XRD measurements were performed in the Yale CBIC and AFM measurements were performed in the YINQE shared core facility. The authors acknowledge the use and research support provided by the Yale Mechanical and Thermal Analysis Instrumentation Core for TGA and DSC experiments. The authors acknowledge the use and research support provided by the Yale West Campus Materials Characterization Core and Dr Min Li for SEM and XPS experiments. The authors would like to thank Kassandra Castaldi, Prof. Robert H. Crabtree, Dr Andrew Mayfield, Cassie Rodrigues, Prof. Xavier Roy, Annie Tyler, and Sadie Wolfarth for helpful discussions.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, A. Mishchenko, A. Carvalho and A. H. Castro Neto, Science, 2016, 353, aac9439 CrossRef CAS.
- A. J. Clough, N. M. Orchanian, J. M. Skelton, A. J. Neer, S. A. Howard, C. A. Downes, L. F. J. Piper, A. Walsh, B. C. Melot and S. C. Marinescu, J. Am. Chem. Soc., 2019, 141, 16323–16330 CrossRef CAS PubMed.
- J. Kwon, H. Ma, A. Giri, P. E. Hopkins, N. B. Shustova and Z. Tian, ACS Nano, 2023, 17, 15222–15230 CrossRef CAS PubMed.
- Q. Jiang, P. Xiong, J. Liu, Z. Xie, Q. Wang, X. Q. Yang, E. Hu, Y. Cao, J. Sun, Y. Xu and L. Chen, Angew Chem. Int. Ed. Engl., 2020, 59, 5273–5277 CrossRef CAS PubMed.
- M. Zeng, Y. Xiao, J. Liu, K. Yang and L. Fu, Chem. Rev., 2018, 118, 6236–6296 CrossRef CAS PubMed.
- X. Wang, T. Fellowes, M. Bahri, H. Qu, B. Li, H. Niu, N. D. Browning, W. Zhang, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2024, 146, 14128–14135 CrossRef CAS PubMed.
- E. C. Ahn, npj 2D Mater. Appl., 2020, 4, 17 CrossRef CAS.
- D. Jayachandran, R. Pendurthi, M. U. K. Sadaf, N. U. Sakib, A. Pannone, C. Chen, Y. Han, N. Trainor, S. Kumari, T. V. Mc Knight, J. M. Redwing, Y. Yang and S. Das, Nature, 2024, 625, 276–281 CrossRef CAS PubMed.
- A. Devarakonda, H. Inoue, S. Fang, C. Ozsoy-Keskinbora, T. Suzuki, M. Kriener, L. Fu, E. Kaxiras, D. C. Bell and J. G. Checkelsky, Science, 2020, 370, 231–236 CrossRef CAS.
- J. H. Dou, M. Q. Arguilla, Y. Luo, J. Li, W. Zhang, L. Sun, J. L. Mancuso, L. Yang, T. Chen, L. R. Parent, G. Skorupskii, N. J. Libretto, C. Sun, M. C. Yang, P. V. Dip, E. J. Brignole, J. T. Miller, J. Kong, C. H. Hendon and M. Dinca, Nat. Mater., 2021, 20, 222–228 CrossRef CAS.
- J. H. Dou, L. Sun, Y. Ge, W. Li, C. H. Hendon, J. Li, S. Gul, J. Yano, E. A. Stach and M. Dinca, J. Am. Chem. Soc., 2017, 139, 13608–13611 CrossRef CAS PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS.
- L. S. Xie, G. Skorupskii and M. Dinca, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- J. Nicks, K. Sasitharan, R. R. R. Prasad, D. J. Ashworth and J. A. Foster, Adv. Funct. Mater., 2021, 31(42), 2103723 CrossRef CAS.
- P. Perlepe, I. Oyarzabal, A. Mailman, M. Yquel, M. Platunov, I. Dovgaliuk, M. Rouzieres, P. Negrier, D. Mondieig, E. A. Suturina, M. A. Dourges, S. Bonhommeau, R. A. Musgrave, K. S. Pedersen, D. Chernyshov, F. Wilhelm, A. Rogalev, C. Mathoniere and R. Clerac, Science, 2020, 370, 587–592 CrossRef CAS PubMed.
- L. Cao and C. Wang, ACS Cent. Sci., 2020, 6, 2149–2158 CrossRef CAS PubMed.
- M. Ko, L. Mendecki and K. A. Mirica, Chem. Commun., 2018, 54, 7873–7891 RSC.
- D. G. Ha, M. Rezaee, Y. Han, S. A. Siddiqui, R. W. Day, L. S. Xie, B. J. Modtland, D. A. Muller, J. Kong, P. Kim, M. Dinca and M. A. Baldo, ACS Cent. Sci., 2021, 7, 104–109 CrossRef CAS PubMed.
- E. M. Johnson, S. Ilic and A. J. Morris, ACS Cent. Sci., 2021, 7, 445–453 CrossRef CAS PubMed.
- Y. Lu, Z. Hu, P. Petkov, S. Fu, H. Qi, C. Huang, Y. Liu, X. Huang, M. Wang, P. Zhang, U. Kaiser, M. Bonn, H. I. Wang, P. Samori, E. Coronado, R. Dong and X. Feng, J. Am. Chem. Soc., 2024, 146, 2574–2582 CrossRef CAS PubMed.
- J. Y. Choi and J. Park, ACS Appl. Electron. Mater., 2021, 3, 4197–4202 CrossRef CAS.
- W. Kosaka, N. Eguchi, T. Kitayama, R. Sato, R. Nakao, Y. Sekine, S. Hayami, K. Taniguchi and H. Miyasaka, Chem. Mater., 2024, 36, 3563–3573 CrossRef CAS.
- M. A. Solomos, F. J. Claire and T. J. Kempa, J. Mater. Chem. A, 2019, 7, 23537–23562 RSC.
- Y. Song, X. Song, X. Wang, J. Bai, F. Cheng, C. Lin, X. Wang, H. Zhang, J. Sun, T. Zhao, H. Nara, Y. Sugahara, X. Li and Y. Yamauchi, J. Am. Chem. Soc., 2022, 144, 17457–17467 CrossRef CAS PubMed.
- X. Su, Z. Zhong, X. Yan, Y. Xu, T. Zhang, Y. Ma and L. Chen, J. Am. Chem. Soc., 2024, 146, 9036–9044 CrossRef CAS.
- Q. Jiang, C. Chen, N. Chai, Q. Guo, T. Chen, X. Ma and F. Y. Yi, Inorg. Chem., 2024, 63, 4636–4645 CrossRef CAS PubMed.
- F. E. Chen, T. A. Pitt, D. J. Okong'o, L. G. Wetherbee, J. J. Fuentes-Rivera and P. J. Milner, Chem. Mater., 2022, 34, 3383–3394 CrossRef CAS PubMed.
- L. Chen, J. Lu, X. Li, N. Luan, Y. Song, S. Yang, M. Yuan, H. Qin, H. Zhu, X. Dong, K. Li, D. Zhang, L. Chen, X. Dai, Y. Wang, Y. Wang, C. Xu, Z. Chai and S. Wang, J. Am. Chem. Soc., 2024, 146, 6697–6705 CrossRef CAS PubMed.
- Y. Ding, Y. P. Chen, X. Zhang, L. Chen, Z. Dong, H. L. Jiang, H. Xu and H. C. Zhou, J. Am. Chem. Soc., 2017, 139, 9136–9139 CrossRef CAS PubMed.
- J. H. Deng, Y. Q. Wen, J. Willman, W. J. Liu, Y. N. Gong, D. C. Zhong, T. B. Lu and H. C. Zhou, Inorg. Chem., 2019, 58, 11020–11027 CrossRef CAS PubMed.
- P. Wu, S. Geng, X. Wang, X. Zhang, H. Li, L. Zhang, Y. Shen, B. Zha, S. Zhang, F. Huo and W. Zhang, Angew Chem. Int. Ed. Engl., 2024, 63, e202402969 CrossRef CAS PubMed.
- Z. Y. Gu, J. M. Cao, K. Li, J. Z. Guo, X. T. Wang, S. H. Zheng, X. X. Zhao, B. Li, S. Y. Li, W. L. Li and X. L. Wu, Angew Chem. Int. Ed. Engl., 2024, 63, e202402371 CrossRef CAS PubMed.
- Y.-L. Chen, Y.-G. Wang and Q. Yu, Organometallics, 2023, 42, 2159–2170 CrossRef CAS.
- F. Spinelli, S. d'Agostino, P. Taddei, C. D. Jones, J. W. Steed and F. Grepioni, Dalton Trans., 2018, 47, 5725–5733 RSC.
- T. D. Trouts, D. S. Tyson, R. Pohl, D. V. Kozlov, A. G. Waldron and F. N. Castellano, Adv. Funct. Mater., 2003, 13, 398–402 CrossRef CAS.
- N. Huang, X. Ding, J. Kim, H. Ihee and D. Jiang, Angew Chem. Int. Ed. Engl., 2015, 54, 8704–8707 CrossRef CAS PubMed.
- T. Hughes, G. P. Simon and K. Saito, ACS Appl. Mater. Interfaces, 2019, 11, 19429–19443 CrossRef CAS PubMed.
- K. Tano and E. Sato, Chem. Lett., 2021, 50, 1787–1790 CrossRef CAS.
- Q. Yu, X. Yang, Y. Chen, K. Yu, J. Gao, Z. Liu, P. Cheng, Z. Zhang, B. Aguila and S. Ma, Angew Chem. Int. Ed. Engl., 2018, 57, 10192–10196 CrossRef CAS PubMed.
- Z.-J. Diao, G.-Z. Wu, T.-J. Zhang, H.-O. Qi, J.-J. Li, M. Lu, G. Liu, X.-Q. Liu and L.-B. Sun, J. Mater. Chem. A, 2023, 11, 16293–16302 RSC.
- N. Deneke, M. L. Rencheck and C. S. Davis, Soft Matter, 2020, 16, 6230–6252 RSC.
- G. Collet, T. Lathion, C. Besnard, C. Piguet and S. Petoud, J. Am. Chem. Soc., 2018, 140, 10820–10828 CrossRef CAS PubMed.
- J. C. Liu, Q. Zou, X. D. Huang, S. S. Bao and L. M. Zheng, Eur. J. Inorg. Chem., 2021, 2021, 1565–1570 CrossRef CAS.
- Q. Zou, T. Shang, X.-D. Huang, Q.-Q. Guo, J.-G. Jia, S.-S. Bao, Y.-Q. Zhang and L.-M. Zheng, J. Mater. Chem. C, 2021, 9, 10749–10758 RSC.
- Q. Zou, S. S. Bao, X. D. Huang, G. H. Wen, J. G. Jia, L. Q. Wu and L. M. Zheng, Chem.–Asian J., 2021, 16, 1456–1465 CrossRef CAS PubMed.
- X. D. Huang, G. H. Wen, S. S. Bao, J. G. Jia and L. M. Zheng, Chem. Sci., 2020, 12, 929–937 RSC.
- X. D. Huang, X. F. Ma, T. Shang, Y. Q. Zhang and L. M. Zheng, Inorg. Chem., 2023, 62, 1864–1874 CrossRef CAS PubMed.
- X. D. Huang, X. F. Ma and L. M. Zheng, Angew Chem. Int. Ed. Engl., 2023, 62, e202300088 CrossRef CAS PubMed.
- X. D. Huang, B. K. Hong, G. H. Wen, S. H. Li and L. M. Zheng, Chem. Sci., 2023, 14, 1852–1860 RSC.
- Q. Yu, M. Li, J. Gao, P. Xu, Q. Chen, D. Xing, J. Yan, M. J. Zaworotko, J. Xu, Y. Chen, P. Cheng and Z. Zhang, Angew Chem. Int. Ed. Engl., 2019, 58, 18634–18640 CrossRef CAS PubMed.
- X.-F. Ma, X.-D. Huang and L.-M. Zheng, Cryst. Growth Des., 2022, 23, 1095–1103 CrossRef.
- J. Feng, X. Wang, Y. Luo, J. Wang, Z. Wang, C. Wei and G. Cai, ACS Appl. Mater. Interfaces, 2024, 16, 1170–1178 CrossRef CAS PubMed.
- T. Yan, J. Yang, J. Lu, L. Zhou, Y. Zhang and G. He, ACS Appl. Mater. Interfaces, 2023, 15, 20571–20582 CrossRef CAS PubMed.
- J. Y. Choi, J. Flood, M. Stodolka, H. T. B. Pham and J. Park, ACS Nano, 2022, 16, 3145–3151 CrossRef CAS PubMed.
- C. Sun, J. J. Oppenheim, G. Skorupskii, L. Yang and M. Dincă, Chem, 2022, 8, 3215–3224 CAS.
- H. Bouas-Laurent, J.-P. Desvergne, A. Castellan and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- L. Yang, L. Qin, Y. Dou, D. Zhang, Z. Zhou and S. Wang, CrystEngComm, 2020, 22, 5411–5415 RSC.
- S. Nakatsuji, T. Ojima, H. Akutsu and J. Yamada, J. Org. Chem., 2002, 67, 916–921 CrossRef CAS PubMed.
- J. Van Damme, L. Vlaminck, G. Van Assche, B. Van Mele, O. van den Berg and F. Du Prez, Tetrahedron, 2016, 72, 4303–4311 CrossRef CAS.
- C. A. Williams, A. J. Blake, P. Hubberstey and M. Schröder, Chem. Commun., 2005, 5435–5437 RSC.
- S. M. Hawxwell, H. Adams and L. Brammer, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 808–814 CrossRef PubMed.
- Y. Qi, Y.-X. Che and J.-M. Zheng, CrystEngComm, 2008, 10, 1137–1139 RSC.
- K. Hirai, J. Reboul, N. Morone, J. E. Heuser, S. Furukawa and S. Kitagawa, J. Am. Chem. Soc., 2014, 136, 14966–14973 CrossRef CAS PubMed.
- J. He, Y. Zhang, J. Yu, Q. Pan and R. Xu, Mater. Res. Bull., 2006, 41, 925–933 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental and synthetic methods and characterization, including NMR, TGA, DSC, ATR-FTIR, O-PTIR, SEM, XPS, AFM, optical images, exfoliation techniques, XRD, MicroED data, and crystallographic tables. CCDC 2342280 (1) and 2356228 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03524k |
| This journal is © The Royal Society of Chemistry 2024 |