
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Post-polymerization functionalized sulfonium nanogels for gene delivery†
Disraëli N. M.
Kusmus
 ,
Thijs
van Veldhuisen‡
,
Sandra
Michel-Souzy
,
Jeroen J. L. M.
Cornelissen
* and
Jos M. J.
Paulusse
*
,
Thijs
van Veldhuisen‡
,
Sandra
Michel-Souzy
,
Jeroen J. L. M.
Cornelissen
* and
Jos M. J.
Paulusse
*
MESA+ Institute and TechMed Institute for Health and Biomedical Technologies, Department of Molecules & Materials, University of Twente, Drienerlolaan 5, 7522 NB, Enschede, Netherlands. E-mail: j.m.j.paulusse@utwente.nl; j.j.l.m.cornelissen@utwente.nl
First published on 15th April 2024
Abstract
Gene therapy is widely recognized as a promising method in combating diseases caused by gene abnormalities or deletions. The effects of these deletions and mutations are ameliorated through gene therapy by means of transfection vectors. These delivery vehicles are tasked with protecting the gene and transporting it to the cell nucleus when necessary. Nano-sized hydrogel particles, also known as nanogels, are crosslinked polymeric nanoparticles that are promising materials for such biomedical applications. Whereas most cationic carriers for gene delivery are nitrogen-based, we are interested in utilizing a sulfonium moiety to this end. Diversifying the available gene vectors not only satisfies scientific curiosity, it could also offer improved gene delivery efficiencies. Here we describe the synthesis of glycidyl methacrylate (GMA) nanogels as a platform for subsequent functionalization. Ring-opening reactions with diethyl sulfide were carried out to install permanent cationic sulfonium groups on the nanogels, yielding readily water-soluble nanogels with a zeta potential of ζ = +40 ± 0.5 mV at neutral pH and a mean diameter of D = 29 ± 10 nm as determined by transmission electron microscopy (TEM). The degree of functionalization with sulfonium groups was found to be tunable. These nanogels were subjected to post-synthesis modifications resulting in biocompatible sulfonium nanogels containing a thioglycerol moiety. Polyplexes were formed by successful incubation with plasmid DNA encoding for green fluorescent protein (pCMV-GFP), at various ratios. In a next step, nucleic acid delivery by sulfonium nanogels was probed for various cell lines for the first time, showing poor delivery properties.
Introduction
In recent decades, the use of nanotechnology for biomedical applications has risen rapidly; for example, with drug and gene delivery systems, imaging and diagnostic agents, and implantable materials.1,2 This field, known as nanomedicine, exploits the unique properties (e.g. optical, electronic) of nanoscale materials. Gene therapy is a branch of nanomedicine in which foreign oligonucleotides are introduced into the human body with the aim to cure inherited or acquired disorders, such as AIDS,3,4 type 2 diabetes5–13 and cancer,14,15 by stimulating or downregulating certain gene expressions.16–19 There are three general approaches to gene therapy, the first of which involves replacing a dysfunctional gene by a healthy copy. Another approach is introducing new or missing genes for the expression of a therapeutic protein. The third approach involves silencing of a mutated gene by interfering with its expression after transcription.20 Unfortunately, therapeutic use of DNA or RNA is not without challenges; simply injecting nucleic acids intravenously leads to limited cellular uptake as both nucleic acids and cell membranes are negatively charged. Other challenges include enzymatic degradation by nucleases, renal clearance and expulsion by the mononuclear phagocyte system.21–25 Therefore, delivery systems, also known as vectors, are needed. Nanoscale delivery vehicles are used to protect, transport, mediate cellular uptake and endosomal release of genetic material such as nucleic acids.26 An ideal nanocarrier should protect the genetic material from enzymatic degradation in physiological fluids and removal by macrophages and the kidneys. As cellular membranes are negatively charged, the nanocarrier should also neutralize or shield the negative charges of the nucleic acids to prevent electrostatic repulsion. Ideally, the nanocarrier should allow easy functionalization, precise control over the amount of gene release and expression, be biocompatible, easy to produce and cost-effective.27,28 Satisfying these criteria requires the nanocarrier to have tunable properties such as size, shape, surface charge and hydrophilicity.29Gene delivery vehicles can generally be classified into three groups: viral, physical, and synthetic (chemical) vectors. Synthetic vectors can be inorganic, lipid-based, and polymeric. Lipid-based vectors employ amphiphilic lipids to encapsulate nucleic acids in liposomes or micelles for therapeutic use.30–33 Although several limitations are associated with lipid-based vectors, such as toxicity, premature payload release34–37 and poor stability,38–40 these vectors are among the most widely used non-viral vectors – most recently employed in the SARS-CoV-2 mRNA vaccine.11,24,41–51 Polymeric vectors are based on a polymer/nucleic acid complex (polyplex), formed by electrostatic interactions between a positively charged polymer and negatively charged nucleic acids.52,53 These polyplexes stimulate cellular uptake through endocytosis.54
Until recently, most polymeric gene delivery systems were nitrogen-based cationic systems such as poly-L-lysine, chitosan, Superfect and PEI.55–63 Although these nitrogen-based systems can achieve high transfection efficiency, many also exhibit unwanted high cytotoxicity. In 2012, Hemp et al. first showed nucleic acid delivery using phosphonium-based cationic macromolecules as an alternative to ammonium macromolecules.64 These materials have proven to be suitable alternatives, with reports of lower cytotoxicity, higher transfection efficiency and higher binding affinities with nucleic acids.64–66 Similarly, sulfonium has a stronger binding affinity for nucleic acids,67 a more centered positive charge68 and a larger ionic radius than ammonium69 – making it a great, potentially less toxic alternative. The group of Long at Virginia Tech were the first to utilize the sulfonium functionality in polymeric macrostructures to complex nucleic acids after their initial success with phosphonium macromolecules (Fig. 1a).64,70 Long and coworkers demonstrated successful luciferase expression in HeLa cells utilizing a sulfonium homopolymer and a sulfonium diblock copolymer as delivery vehicles. Others synthesized sulfonium cell penetrating peptides that have low cytotoxicity.71 Mackenzie et al. studied biocompatible sulfonium block copolymers that are stable towards dealkylation, as siRNA delivery vehicles in murine clavarial preosteoblasts (MC3T3s), achieving successful Gapdh knockdown (Fig. 1b).72 Zhu et al. designed intracellularly disintegrable polysulfonium compounds, which were able to effectively condense DNA into polyplexes and achieve high transfection efficiency (Fig. 1c).73
 | ||
| Fig. 1 Overview of polysulfonium structures for gene delivery, as reported by (a) Hemp et al.,70 (b) Mackenzie et al.,72 and (c) Zhu et al.73 | ||
Although the field of sulfonium-based polymers for gene delivery is still in its infancy, the results are quite promising. So far, exclusively linear polysulfonium vectors have been explored as gene carriers. In addition to linear or branched polymers, many nano-sized polymer architectures have been utilized for gene delivery, such as dendrimers,63,74 polymersomes75 and nanogels. Nanogels are highly biocompatible crosslinked three-dimensional polymer networks with high (therapeutic) cargo loading.76 They typically have a network-like structure with inter- as well as intramolecular crosslinks.77,78 Nanogels can be synthesized by controlled crosslinking polymerization (CCP) of monomers with multi-functional co-monomers, relying on simultaneous polymer growth and crosslinking in a homogenous reaction mixture.79 Synthesis via living polymerization endow nanogels with a very similar degree of polymerization across all individual polymer chains, leading to a low polydispersity index. This allows for the synthesis of precisely designed polymer nanostructures with a high degree of control over molecular mass.76,80,81 Nanogels have been shown to be high functioning delivery vehicles for many biomedical applications including gene delivery.82,83 Incorporating sulfonium groups into their three-dimensional architecture might offer new insights into their polyplex formation and transfection efficiency. Positive charge can be installed on polymer pendent groups by alkylation of a thioether with an alkyl halide, forming a sulfonium group.84,85 Likewise, polymers employing thioether moieties were shown to react with epoxides under acidic conditions, forming charged sulfonium groups in a single step.86,87 Bearing this in mind, combined with great success achieved by others transfecting cells with amine derivatives of poly(glycidyl methacrylate) (pGMA),88–91 we synthesized sulfonium GMA nanogels and evaluated their gene transfection efficiency.
Results and discussion
GMA-EGDMA functionalizations
GMA-EGDMA nanogel was synthesized as described in the Experimental section and in previous work.92 Once a desirable monomer conversion was achieved (31%), a large excess of AIBN was added to the reaction mixture to cap the remaining methacrylates with AIBN fragments. Dynamic Light Scattering (DLS) measurements gave a Z-average hydrodynamic diameter of Dh = 60 ± 0.1 nm (in DCM) and analysis by Transmission Electron Microscopy (TEM) resulted in a similar particle diameter of 56 ± 14 nm (Fig. 2). | ||
| Fig. 2 GMA-EGDMA nanogel at 31% monomer conversion was analyzed by DLS and TEM displaying unimodal distribution for both techniques. | ||
The next step towards the formation of a gene carrier is the installation of charge. Plasmid DNA that is utilized to mimic foreign genes trafficked into the human body has a negative charge. As such it is desired to have the gene vector be positively charged to facilitate interaction and ensure proper conjugation. Utilizing the method first published by Park86 – GMA-EGDMA nanogel was functionalized with diethyl sulfide in a ring-opening reaction to yield a sulfonium-functionalized nanogel (Scheme 1).
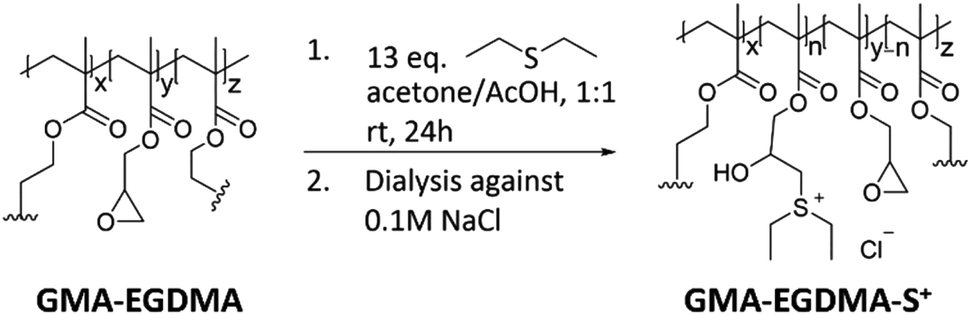 | ||
| Scheme 1 The synthesis of sulfonium-functionalized GMA-EGDMA-S+ was followed by dialysis against 0.1 M NaCl to exchange the acetate counter ion for a chlorine counter ion. | ||
Fig. 3 displays the 1H NMR spectrum of GMA-EGDMA-S+ at full epoxide conversion. Characteristic signals of the sulfonium group are labelled at δ = 1.4 ppm and δ = 3.5 ppm. These signals are shifted upfield compared to uncharged thioether, as expected. Full conversion of epoxides is assessed by the absence of epoxide signals at δ = 2.6 ppm, δ = 2.8 ppm and δ = 3.2 ppm.
 | ||
| Fig. 3 1H NMR spectrum in DMSO-D6 (residual solvent signals are labelled with red crosses) of GMA-EGDMA-S+, with characteristic signals of the sulfonium group at δ = 1.4 ppm labelled blue and at δ = 3.5 ppm labelled green. | ||
The zeta potential of GMA-EGDMA-S+ was determined to be ζ = +40 ± 0.5 mV at neutral pH (in 5 mM KCl), meaning a high surface charge. This is reflected in the hydrodynamic diameter of GMA-EGDMA-S+ compared to GMA-EGDMA. DLS measurements indicated a size increase from Dh = 60 ± 0.1 nm to Dh = 95 ± 1.6 nm, likely the result of the positive charges repelling each other (Fig. 4).
 | ||
| Fig. 4 The hydrodynamic diameters of GMA-EGDMA-S+ (in 5 mM KCl) and GMA-EGDMA (in DCM) were measured by means of DLS indicating a semi bimodal size distribution. This increase in size is likely the result of same charges repelling each other as well as a difference in solvents. | ||
The dynamic nature of these sulfonium nanogels was further illustrated by TEM analysis. A mean particle diameter of 29 ± 10 nm was measured, which is significantly lower than described above. This is expected as the nanogel is soft and well-solvated during DLS measurements as opposed to high vacuum during TEM measurements (Fig. 5).
 | ||
| Fig. 5 TEM micrographs of GMA-EGDMA-S+ give a mean particle diameter of 29 ± 10 nm. The scale bar represents 50 nm. | ||
Controlled epoxide conversion
Particles with high charge density are known to exhibit cytotoxicity.93 Therefore, control over the degree of functionalization with charged moieties is desired. By investigating the kinetics of GMA-EGDMA-S+ synthesis, a certain degree of functionalization and thus positive charge can be obtained by quenching the reaction at a certain time point. The evolution of characteristic 1H NMR signals for GMA-EGDMA-S+ during the diethyl sulfide induced ring-opening reaction is shown in Fig. 6. The characteristic signals of the sulfonium group at δ = 1.4 ppm and δ = 3.5 ppm increase with time. | ||
Fig. 6 Conversion of epoxides during a ring-opening reaction of GMA-EGDMA-S+ with 5 equiv. of diethyl sulfide in AcOH/acetone 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as analyzed by 1H NMR spectroscopy in acetone-d6. Conversion was calculated utilizing 1,3,5-trioxane as an internal standard. Its singlet peak can be seen at 5.1 ppm. :1, as analyzed by 1H NMR spectroscopy in acetone-d6. Conversion was calculated utilizing 1,3,5-trioxane as an internal standard. Its singlet peak can be seen at 5.1 ppm. | ||
Solubility of sulfonium-functionalized nanogels
Having demonstrated the control over conversion and thus charge density, a series of GMA-EGDMA-S+ nanogels were made at various epoxide conversions. Surprisingly, relatively high conversions were needed to achieve water-solubility. At 50% epoxide conversion solubility was achieved at a concentration of 0.5 mg mL−1 in water after 30 min of sonication.As these nanogels are to be used in the human body for transfection purposes, water-solubility is a strict requirement. The remaining epoxides of GMA-EGDMA-S+-x provide a convenient handle for further nanogel functionalizations to enhance water-solubility. Coincidentally this strategy also increases the biocompatibility of these nanogels as epoxides have been proven to be toxic.94 Having previously demonstrated the versatility and ease of epoxide ring opening reactions, it was decided to apply these strategies on GMA-EGDMA-S+-x.
Hydrolysis of epoxides using TFA
As a first strategy, hydrolysis of the remaining epoxides was carried out in an attempt to solubilize GMA-EGDMA-S+-x nanogels at low charge densities (Scheme 2). The nanogel was reacted with TFA in 9:1 THF/water. During this step the acetate counterion is replaced by a trifluoroacetate counterion as excess TFA (12 equivalents to epoxide groups) is used. However, this counterion has been reported as toxic in cell culture experiments by inhibition of proliferation.95 Therefore, dialysis against 0.1 M NaCl was done to facilitate counterion exchange to Cl−. The disappearance of the AcO− 1H NMR signal at δ = 1.9 ppm indicated a successful exchange. Nanogels GMA-EGDMA-S+-10, GMA-EGDMA-S+-15 and GMA-EGDMA-S+-25 (with 10, 15, 25 denoting the epoxide conversion) all formed insoluble (likely crosslinked) products. GMA-EGDMA-S+-45 was successfully hydrolyzed to GMA-EGDMA-S+-45-OH.
 | ||
| Scheme 2 A schematic overview of the attempts made to achieve water-soluble GMA-EGDMA-S+-x. Hydrolysis of the remaining epoxides with TFA to form GMA-EGDMA-S+-OH was attempted as well as ring opening reactions with hydrophilic thiols to GMA-EGDMA-S+-THG and GMA-EGDMA-S+-mPEG. | ||
IR measurements showed no characteristic epoxide band at 907 cm−1, indicating complete hydrolysis of epoxides. Although the exact mechanism behind crosslinking during acidic hydrolysis with a low charge density is not known, it is suspected that particles with higher charge density experience more electrostatic repulsion, preventing crosslinking due to decreased proximity. GMA-EGDMA-S+-45-OH was water-soluble. DLS and zeta potential measurements gave a Dh = 121 ± 1.8 nm and ζ = +34 ± 0.7 mV at neutral pH. As hydrolysis of the epoxides is troublesome for nanogels with low degrees of charge density, other reactions were investigated.
Thioglycerol-epoxy ring-opening
Thiol-epoxy ring-opening reactions are known as click chemistry reactions, proceeding with high efficiency at ambient temperatures.96 Therefore, thiol-epoxy reactions were investigated for the solubilization of GMA-EGDMA-S+-x. To enhance hydrophilicity, 1-thioglycerol was used during a ring-opening reaction under basic conditions. Unfortunately, these nanogels were also not water-soluble at a concentration of 0.5 mg mL−1. Cell transfection experiments can be carried out at lower nanogel concentrations – however, 0.5 mg mL−1 is the minimum required for accurate characterization.mPEG thiol-epoxy ring opening
PEGylation is a well-known strategy to improve water-solubility of particles.97 PEGylated particles are also known to have an increased blood circulation half-life. Therefore, a thiol-functionalized PEG oligomer (O-(2-mercaptoethyl)-O′-methyl-hexa(ethylene glycol)) was employed in efforts to solubilize GMA-EGDMA-S+-x. To prevent crosslinking, a thiol-functionalized PEG oligomer with six repeating ethylene glycol units and methoxy end-groups (mPEG) was chosen. Longer mPEG units are expected to experience a greater degree of steric hindrance when attacking epoxides within the nanogel matrix. Conversely, shorter mPEG units might not yield water-soluble nanogels due to their relatively low contribution to overall hydrophilicity of the nanogels. The degree of conversion could not be determined by 1H NMR spectroscopy due to overlapping signals in the 2.5–3.5 ppm region. These NMR spectra however are in agreement with the one obtained in the work of De et al.,96 who used a similar approach to attach thiolated mPEG chains to polymers with epoxide moieties. Sulfonium-mPEG functionalized nanogels GMA-EGDMA-S+-27-mPEG and GMA-EGDMA-S+-47-mPEG were synthesized. These compounds, however, exhibited poor water-solubility at 0.5 mg mL−1. DLS indicated the presence of large aggregates.Probing nanogel solubility by tuning its crosslinker
The experiments described above irrefutably indicate that proper solvation of GMA-EGDMA-S+-x nanogels at low positive charge density is not achievable solely by functionalization of the remaining epoxides with hydrophilic reactants. The remainder of the nanogel matrix is probably too hydrophobic. An alternative strategy would be reducing this hydrophobicity, for example by using a more hydrophilic crosslinker during nanogel synthesis. Test reactions were carried out with N,N′-methylenebisacrylamide (MBA), diethylene glycol dimethacrylate (DEGDMA) and tetraethylene glycol dimethacrylate (TTEGDMA) as crosslinker at varying conditions (Fig. 7). MBA did not form well-defined nanogels as it is not compatible with GMA – most likely due to differing reactivity ratios. DEGDMA and TTEGDMA were successfully crosslinked with GMA to form nanogels. For these nanogels, the remaining methacrylate groups were not quenched as they do not hinder the reaction with diethyl sulfide or cause crosslinking. | ||
| Fig. 7 In an attempt to achieve more hydrophilic nanogels, GMA was crosslinked with MBA, DEGDMA and TTEGDMA. | ||
In a next step, GMA-DEGDMA and GMA-TTEGDMA were subjected to epoxide ring opening reaction with diethyl sulfide to form GMA-DEGDMA-S+-x and GMA-TTEGDMA-S+-x at low epoxide conversions (low value for x) and evaluate their water-solubility. Both reactions were successful, however, only GMA-TTEGDMA-S+-15 was water-soluble at a desirable concentration (1.3 mg mL−1). 1H NMR spectroscopy indicated an epoxide conversion of 15% (x = 15) as seen in Fig. 8.
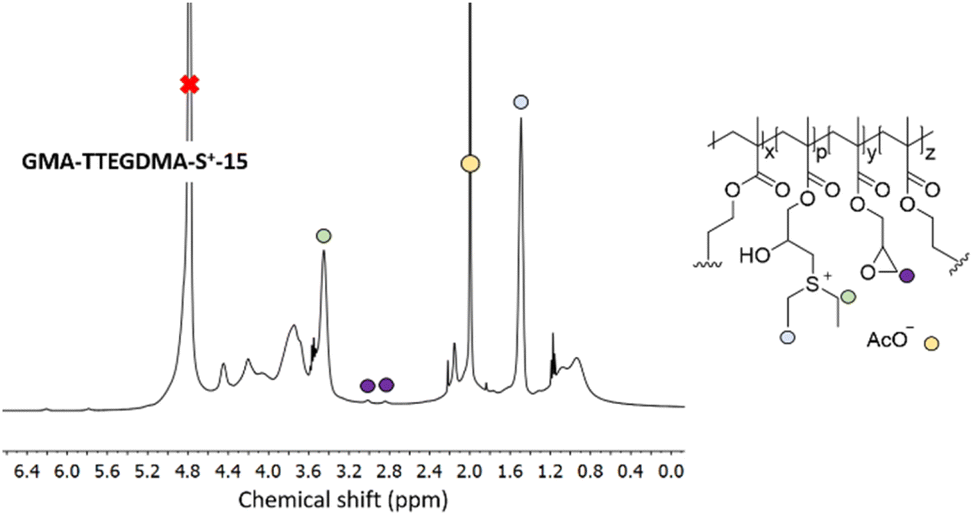 | ||
| Fig. 8 1H NMR spectrum of GMA-TTEGDMA-S+-15 in D2O displays the characteristic sulfonium peaks at δ = 1.4 ppm and δ = 3.5 ppm along with the remaining epoxide peaks at δ = 2.6 ppm and δ = 2.8 ppm (the epoxide peak at δ = 3.2 ppm is hidden under the methylene peaks of the sulfonium group). The acetate counter ion can also be seen at δ = 1.9 ppm. The structure of the nanogel was simplified to enhance clarity. | ||
Utilizing TTEGDMA as crosslinker during nanogel formation with GMA increases the hydrophilicity of the nanogel and its water-solubility at low surface charge density. GMA-TTEGDMA-S+-40 and GMA-TTEGDMA-S+-50 were synthesized. The hydrodynamic diameter and surface charge was measured for both nanogels in 10 mM HEPES at pH = 7. GMA-TTEGDMA-S+-50 was 27 ± 0.7 nm in diameter with ζ = +31 ± 1 mV. GMA-TTEGDMA-S+-40 was 67 ± 2.2 nm in diameter and ζ = +29 ± 0.4 mV. To enhance the biocompatibility of these nanogels they were reacted with 1-thioglycerol to GMA-TTEGDMA-S+-40-THG (40-THG) and GMA-TTEGDMA-S+-50-THG (50-THG), quenching the remaining epoxides. This reaction was carried out under basic conditions in HEPES solution. DLS measurements gave a Dh = 39 ± 0.2 nm in 10 mM HEPES buffer (2 mg mL−1) and ζ = +10 ± 0.2 mV at 0.5 mg mL−1 for 40-THG. 50-THG had a diameter of 24 ± 0.2 nm and ζ = +14 ± 1.2 mV. The decrease in surface charge in comparison to their precursors is likely an indication of the stability of the sulfonium charge on these nanogels. NMR analysis of 40-THG and 50-THG display a decrease in the intensity of the acetate counter ion peak compared to GMA-TTEGDMA-S+-40 and GMA-TTEGDMA-S+-50 – likely indicating hydrolysis or nucleophilic substitution of the sulfonium moiety. Surprisingly others observed minimal dealkylation of polysulfonium polymers in aqueous environment.72
To further analyze the properties of these nanogels their infrared (IR) absorptions were measured. The epoxide stretch at ν = 907 cm−1 is no longer present and new peaks arise for S+–C vibrational stretches at ν = 585 cm−1 and ν = 632 cm−1 as seen in Fig. 9. Analysis of the nanogels by TEM demonstrated spherical particles with a high polydispersity. Due to low quantities of 40-THG TEM micrographs had to be made at highly dilute concentrations. As such it was not possible to accurately calculate the mean particle diameter (Fig. 10).
 | ||
| Fig. 9 IR spectrum of 50-THG and 40-THG. The green dotted lines over peaks at ν = 585 cm−1 and ν = 632 cm−1 highlight the emergence of S+–C vibrational stretches. The green dotted line (and arrow in the inset) at ν = 907 cm−1 specifies the disappearance of epoxide moieties demonstrating a successful reaction. | ||
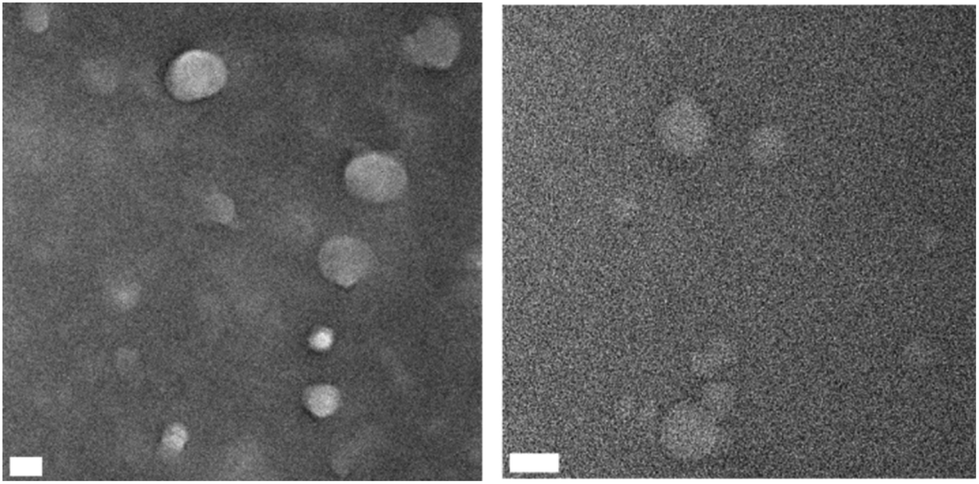 | ||
| Fig. 10 TEM images of 50-THG (left) and 40-THG (right) indicate the formation of spherical particles with high polydispersity. The scale bars represent 50 nm. 40-THG had a particle diameter of 79 ± 56 nm and 50-THG had a particle diameter of 61 ± 17 nm. | ||
Polyplex formation with sulfonium nanogels
The complexation abilities of the nanogels with plasmid DNA were studied utilizing plasmid DNA encoding for green fluorescent protein (pCMV-GFP). Polyplexes were formed at various S/P ratios and their size and surface charge were measured. S/P ratio refers to the ratio between the estimated S+ atoms in the nanogel and the negatively charged phosphate units in the plasmid DNA. As the DNA and cationic nanogel bind tighter to each other due to electrostatic interactions, the size of the polyplex decreases at higher S/P ratios until maximum polyplex density is achieved. 50-THG reached a maximum density and minimum hydrodynamic diameter at S/P = 50 as can be seen in Fig. 11. This plateau indicates the formation of stable polyplexes. A similar trend is seen for the surface charge of the polyplexes. The positively charged nanogel will complex and shield the DNA and its negative charge resulting in an increase in polyplex surface charge, which eventually stagnates.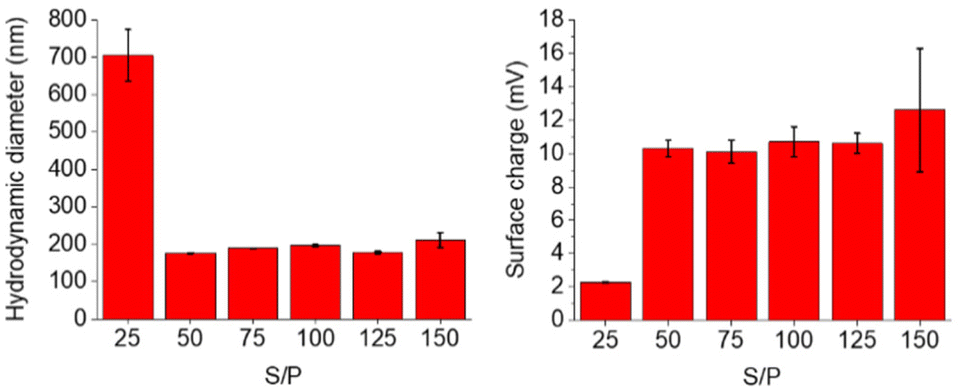 | ||
| Fig. 11 DLS and zeta potential measurements of 50-THG polyplexes at various S/P ratios indicate proper polyplex formation starting from S/P = 50, in 10 mM HEPES. | ||
The complexation abilities of 40-THG were studied in a similar manner. Polyplexes were formed at various S/P ratios and their hydrodynamic diameter and surface charge were measured. 40-THG required higher S/P ratios to form tight polyplexes. The eventual plateau in size was less pronounced for 40-THG than for 50-THG, and not at all present for the surface charge. Most likely 40-THG forms polyplexes with lower binding affinity between nanogel and pDNA, leading to weaker electrostatic interactions than 50-THG. As a result, the polyplexes formed are less stable and not always quantifiable. As is exemplified by the S/P = 125 sample of 40-THG that gave repetitive inconclusive zeta potential data (Fig. 12). Since further experiments were carried out with 50-THG based polyplexes, no attempts were made to stabilize the 40-THG samples.
 | ||
| Fig. 12 DLS and zeta potential measurements of 40-THG suggesting that stable polyplexes do not form. For S/P = 125 due to inconclusive measurements, no value is reported. | ||
The differences between the two sets of polyplexes are likely due to the different properties of the nanogels. 40-THG has a lower surface charge (ζ = +10 mV compared to ζ = +14 mV for 50-THG), requiring higher amounts of cationic nanogels (and thus higher S/P ratios) to form adequate, stable polyplexes with the negatively charged pDNA. Higher numbers of 39 nm nanogels directly translate to larger polyplexes (as witnessed in Fig. 12) as well as decreased stability – one negative pDNA charge must interact with increasingly bulkier positive nanogel charges.
The ideal size of particles for in vitro studies are smaller than 200 nm to promote cellular uptake through endocytosis.98–100 As such it can be concluded that 40-THG is not suitable for in vitro transfection experiments. Optimizing the sulfonium nanogel properties, such as increasing the surface charge could result in stable polyplexes at lower S/P ratios. However, a higher charge density is expected to cause a size increase as sulfonium moieties will repel each other, leading to polyplexes larger than the desired 200 nm. This trade-off can be eased by starting with smaller GMA-TTEGDMA epoxide nanogels. It is worth noting however, that this strategy cannot be utilized to its full extent as high positive charge has been proven to be toxic to cells,93 meaning there is an upper limit to the cationic charge of the nanogels/polyplexes.
Polyplex stability
In order to quantify the stability of 50-THG polyplexes their hydrodynamic diameter and surface charge were studied for an extended period of time. Polyplexes were formed at S/P = 75 and their properties were measured every 60 min. The electrostatic interactions that form the polyplexes start dissociating, releasing the negatively charged DNA that is inside. As a result these polyplexes become less dense and increase in size while decreasing in overall surface charge. Starting at 3 h significant changes are seen, likely due to the dynamic nature of the system that apparently leads to larger entities (Fig. 13). | ||
| Fig. 13 DLS and zeta potential measurements of 50-THG polyplexes with pCMV-GFP indicate that the polyplexes formed are stable for 3 h, after which their bonds start dissociating. This is mirrored in the size increase and charge decrease. Measurements were done once. DLS and zeta potential measurements were conducted separately, each with freshly made polyplex solutions. Inset: TEM micrograph at S/P = 75, t = 0; mean diameter = 127 nm. Complete image in ESI (Fig. S5†). | ||
Gene transfection
To assess the transfection efficiency of sulfonium nanogel 50-THG, experiments were conducted in human cervical cancer cells (HeLa) with pCMV-GFP. Branched polyethyleneimine at N/P = 25 (bPEI25k) was used as a positive control along with commercial lipid vector Lipofectamine™ 3000. Various ratios between 50-THG and plasmid DNA were evaluated in a 96-well plate under serum-free (in DMEM−) conditions. Transfection experiments were carried out by incubating HeLa cells (seeded at 15000 cells per well) with plasmid DNA encoding for green fluorescent protein (pCMV-GFP, 0.25 μg per well) for 6 h in DMEM− followed by an additional 40 h in DMEM+ to allow GFP expression. The transfection efficiency was qualitatively analyzed by fluorescence microscopy as seen in Fig. 14. The cell viability was quantitatively analyzed by alamarBlue™ assay and compared to cells that were not exposed to the transfection medium (Fig. S6 in ESI†). Transfection and cytotoxicity experiments were conducted in parallel meaning microscopy photos and metabolic activity were measured at the same time point. GFP expression was only observed in HeLa cells at an S/P ratio of 100.
 | ||
| Fig. 14 Fluorescence microscope images of HeLa cells transfected with pCMV-GFP at 0.25 μg per well via50-THG. Top row depicts the controls: untreated HeLa cells, Lipofectamine™ 3000, plasmid DNA and bPEI25k at N/P = 25. 100× magnification. Scale bar represents 200 μm (both top and bottom images). | ||
As seen in Fig. 14, minimal to no GFP expression was observed for 50-THG. Fig. 14 also illustrates the high toxicity of Lipofectamine™ 3000 and bPEI25k in comparison to the 50-THG polyplexes. A high positive charge is toxic to cells resulting in bPEI25k and its polyplexes being intrinsically toxic.56 This was further confirmed during the cytotoxicity studies as depicted in Fig. S6,† resulting in low cell viability. The 50-THG polyplexes contrarily are non-toxic, displaying cell viability similar to cells that were not exposed to polyplexes.
To allow higher uptake and transfection efficiency the experiment was repeated with a longer incubation period of 20 h (Fig. S7 and S8†) as well as at a higher DNA/well concentration of 1 μg (Fig. S8†). Transfection and cytotoxicity experiments were carried out in parallel meaning microscopy photos and metabolic activity were measured at the same time point. Once the polyplexes were removed the cells were incubated for an additional 24 h to allow GFP expression. Cells were seeded at 15000 cells per well. Both Fig. S8 and S9† exhibit no GFP expression for the 50-THG polyplexes. A longer incubation period did not increase transfection efficiency. The incubation time also had a negative impact on the HeLa cells as significant reduction in living cells is observed. This is most likely due to the fact that serum-free medium (DMEM−) is used during this incubation period. The cells do not tolerate the absence of FBS for such an extended period of time. An increased incubation period with the 50-THG polyplexes and a higher plasmid DNA concentration had no effect on the cell viability of HeLa cells. On the contrary, longer incubation with bPEI25k polyplexes and a higher plasmid DNA concentration seem to significantly impact the viability of the HeLa cells (Fig. S9†).
Transfection efficiency varies between different cell lines for the same vector.101 This feature is attractive as it can be exploited to achieve selectivity and enhance targeting. Bearing this in mind various cell lines were subjected to transfection experiments to evaluate the transfection efficiency and potential selectivity of 50-THG polyplexes. HeLa (human cervical cancer), bEnd.3 (mouse endothelial brain), HepG2 (human epithelial liver cancer), C2C12 (mouse muscle myoblasts), RAW264 (mouse macrophages), HEK293 (human embryonic kidney cells) and HFF (human fibroblast) cells were incubated with polyplexes for 5 h in serum-free medium (DMEM−) followed by 44 h of incubation to allow GFP expression. All experiments were carried out at 1 μg pCMV-GFP and 15000 cells per well in a 96-well plate. The number of polyplex ratios was increased from four to five and the S/P ratio was increased to include 125 and 150. Micrographs and metabolic activity are from the same time point. The polyplexes did not exhibit GFP expression (Fig. S10–S16†) or any significant cytotoxicity (Fig. S17†) for the cell lines mentioned above. Lipofectamine™ 3000 and especially bPEI25k showed high levels of cytotoxicity, resulting in a minimum of 40% cell death (data not shown). Fig. S10–S16† indicate reduced tolerance for a concentration of 1 μg pCMV-GFP per well compared to 0.25 μg pCMV-GFP per well (Fig. S7 and S8†). Notably, cells transfected by bPEI25k polyplexes exhibited significant cell death and reduced transfection. As such all following experiments were done at 0.25 μg pCMV-GFP per well.
Having tested 7 different cell lines and observing no transfection, the conclusion can be drawn that 50-THG does not function as a vector for gene transfection. Successful transfection, resulting in GFP gene expression, requires a series of events to occur successfully, from polyplex formation to protein expression.102 Previous work done in our group confirms that polyplexes are formed successfully and can be imaged (TEM) and their properties measured (DLS, zeta potential). Protein expression, in the studied systems, was also successful as seen for Lipofectamine™ 3000 and bPEI25k in the figures above. The reason for the unsuccessful transfection experiments thus lies with the intermediate steps. As stated previously, the ideal gene vector has a positive charge that interacts with the negative charge of DNA forming a polyplex. The ideal gene vector enhances uptake of the plasmid DNA,103,104 protects it from interacting with proteins in the cytosol or from degradation by intracellular nucleases105,106 and transports it to the cell nucleus and across the nucleus membrane.107 As such the vector is expected to barricade the plasmid in some form, for example through encapsulation. Considering that the polyplexes are formed by mixing spherical nanogels with relatively large plasmid DNA, it is possible that the DNA does not enter the nanogel matrix but rather sits on top of it, interacting with the positive charges on its surface. As a result, the plasmid DNA is not adequately protected. Most likely the plasmid DNA blankets a grand majority, if not all, of the positive surface charges. Having few remaining available positive moieties left on its surface, uptake of the nanogel is significantly hindered as it is no longer capable of properly interacting with the negatively charged cell membrane. Another possible cause is the stability of the sulfonium moiety and the formed polyplexes.108 DLS measurements indicate that the polyplexes are not stable and start disassembling within 3 h (data not shown). Both of these factors would lead to minimal uptake, and consequently no protein expression.
To test these hypotheses transfection experiments were conducted with DTAF-labelled 50-THG sulfonium nanogels, in order to visualize the nanogels and monitor their uptake. Polyplexes were formed with pCMV-lacZ and DTAF-labeled 50-THG at S/P = 125 to evaluate the influence of bound DNA on polyplex uptake without the interference of the green fluorescence of GFP. Linear sulfonium polymers (synthesized from pGMA), equivalent to 50-THG, were synthesized to evaluate their nucleic acid condensing properties, in comparison to their crosslinked nanogel counterparts. Sulfonium polymers carried either an epoxide moiety (S/P = 50, Dh = 259 ± 3.9 nm, ζ = +37 ± 0.9 mV) or a thioglycerol moiety (S/P = 94, Dh = 90 ± 2.9 nm, ζ = +16 ± 1.7 mV). The choice was made for HeLa cells, C2C12 cells and HEK293 cells (at a seed density of 10000 cells per well) as they showed most promise. The cells were incubated with polyplexes (Fig. S18†) or 50-THG for 2 h in serum-free medium (DMEM−), followed by 48 h of incubation to allow GFP expression for the polyplexes carrying pCMV-GFP. All experiments were carried out in duplo and at 0.25 μg pCMV-GFP per well (or its equivalent) in a 96-well plate. Polymer polyplexes were incubated for 45 min at rt prior to use and 50-THG polyplexes for 10 min at rt prior to use. Microscopy photos and metabolic activity are from the same time point.
Fig. 15, S19 and S20† show no improvement in the transfection efficiency of 50-THG polyplexes conducted at lower plasmid DNA concentration. They do however indicate less cell death and thus a higher tolerance at 0.25 μg pCMV-GFP per well. Although slightly better than before (Fig. S15†), HEK293 cells (Fig. S20†) still exhibited poor adherence to the well plate. Fig. 15 and S19† highlight the cytotoxicity of epoxides.94 Interestingly, transfection was only witnessed in HEK293 cells for polyplexes formed from linear sulfonium polymers bearing 8% epoxides (Fig. S20†). It was hypothesized that linear polymers have higher transfection efficiencies than nanogels as they have more freedom of movement and can thus better encapsulate pDNA. If this were truly the case and linear polymers were indeed far better at transfection, both sulfonium polymers would exhibit much higher GFP expression than what is seen in Fig. 15, S19 and S20.† However, the two polysulfoniums differed significantly from each other in size and surface charge. The epoxide-containing polymer that achieved transfection for HEK293 cells may be hindered in properly transfecting other cell lines as it is >200 nm and thus not the ideal size for endocytosis. Contrarily, the thioglycerol polymer has a more favorable size, but a surface charge that is potentially too low to transfect.
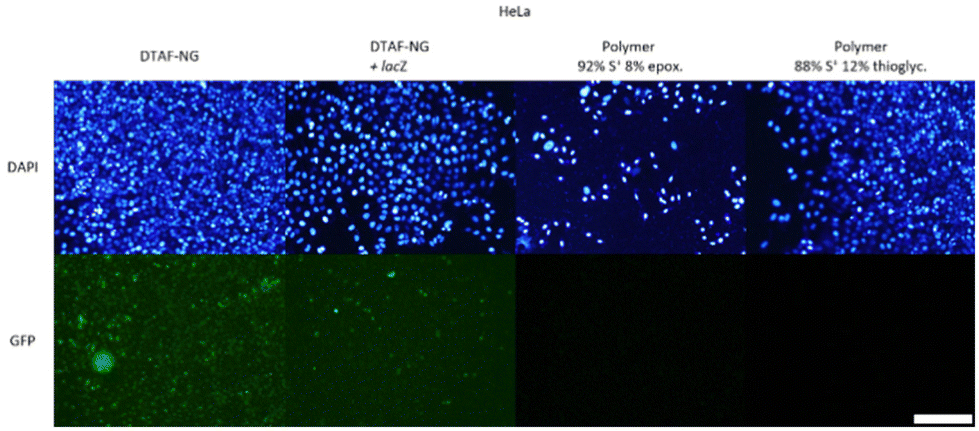 | ||
| Fig. 15 Transfection with 50-THG at S/P = 75, 100 and 125 on HeLa cells was repeated, along with Lipofectamine™ 3000 and bPEI25k as positive controls (Fig. S18†). Uptake was evaluated for empty DTAF-dyed nanogels (41 ± 26.3 nm, ζ = −11 ± 2.8 mV) and lacZ polyplexes in dyed nanogel. Transfection efficiency was monitored for GFP polyplexes formed from sulfonium polymers carrying either an epoxide moiety (Dh = 259 ± 4 nm, ζ = +37 ± 0.9 mV) or a thioglycerol moiety (Dh = 89 ± 3 nm, ζ = +16 ± 1.7 mV). Scale bar represents 200 μm. | ||
As seen in Fig. 15, uptake is witnessed for free/empty dyed 50-THG and pCMV-lacZ polyplexes (top row). Examining these two at a higher magnification (Fig. S22–24†) reveals that empty 50-THG is taken up much more than its pCMV-lacZ polyplex. This observation confirms our hypothesis that plasmid DNA actually hinders the uptake of 50-THG polyplexes – most likely by concealing the positive surface charge. Nanogels containing no pCMV-lacZ exhibited higher uptake than the polyplexes, revealing endocytosis to be the obstacle hindering gene transfection. A vast majority of the polyplexes added to the cells during the experiments do not enter the cells. The few that do most likely are not enough to achieve gene transfection and express GFP fluorescence. These experiments show that for 50-THG endocytosis is the (first) major obstacle in the transfection pathway. This, however, does not exclude the possibility of subsequent steps also requiring optimization.
Confocal images were taken to ensure that the green fluorescence seen in the images above arise from nanogels endocytosed by the cells and not from nanogels on the cell surface. This enables us to calculate the amount of nanogels taken up by the various cell lines more accurately. The confocal images revealed that the grand majority of the nanogels present were located inside the cells with a negligible amount residing on the cell surface. The intensity of the green fluorescence in the confocal images in Fig. S25† was quantified, revealing that for HeLa cells the endocytosed empty 50-THG had a 2.4 times higher fluorescence intensity than the endocytosed polyplexes, meaning that 2.4 times more 50-THG was taken up. For C2C12 cells (Fig. S26†) and HEK293 cells (Fig. S27†) the green fluorescence intensity and uptake was respectively 12 times and 3.4 times higher for empty 50-THG than for polyplexes.
Conclusions
The objective of this work was to synthesize sulfonium-functionalized nanogels for gene delivery. GMA-EGDMA nanogels were synthesized by a RAFT CCP approach. Sulfonium groups were installed by applying a ring-opening reaction of epoxide moieties with diethyl sulfide under acidic conditions. The degree of functionalization is controllable by monitoring the conversion with NMR spectroscopy. The solubility of sulfonium functionalized nanogels in water was achieved by substituting the crosslinker EGDMA for its longer and more hydrophilic equivalent TTEGDMA. GMA-TTEGDMA nanogels that are appreciably water-soluble start from 15% epoxide conversion (i.e.GMA-TTEGDMA-S+-15).The efficiency of sulfonium nanogels at forming polyplexes with negatively charged plasmid DNA was evaluated. While 40-THG did not form stable polyplexes, 50-THG formed polyplexes starting from S/P = 50, with an average size of 150 nm and an average surface charge of ζ = +10 mV. These polyplexes were stable for up to 3 h.
The transfection efficiency of 50-THG was evaluated for various cell lines at varying plasmid DNA concentrations and incubation periods. After trailing multiple cell lines with no success, the hypothesis that endocytosis is the bottleneck was confirmed. As such it is concluded that 50-THG is not suitable as plasmid delivery vehicle for gene therapy. A linear polysulfonium with high surface charge however, did exhibit some transfection for HEK293 cells. This suggests that both shape and surface charge should be probed in future studies, when evaluating the transfection efficiency of sulfonium nanogels. Cytotoxicity experiments indicated that 50-THG is biocompatible and non-toxic. We believe that sulfonium nanogels can find use as vectors for smaller, negatively charged nucleotides such as ssDNA and mRNA.
Experimental
Materials
Glycidyl methacrylate (GMA, 97%), ethylene glycol dimethacrylate (EGDMA, 98%), diethylene glycol dimethacrylate (DEGDMA, 95%) and tetraethylene glycol dimethacrylate (TTEGDMA, >90%) were purchased from Sigma-Aldrich and passed through a column of neutral Al2O3 prior to use. Potassium chloride (KCl), N,N-dimethylformamide (DMF, anhydrous, 99.8%), trifluoroacetic acid (TFA, HPLC grade, >99.0%), 1,4-dioxane (HPLC grade, >99.5%), azobisisobutyronitrile (AIBN, 98%), lithium hydroxide (LiOH, reagent grade, 98%), HEPES sodium salt (99.5%), sodium hydroxide pellets (NaOH), branched polyethylene imine (bPEI25k, 25 kDa), HEPES (99.5%) sodium salt, L-glutamine solution, Penicillin–Streptomycin solution, Dulbecco's Modified Eagle's medium, high glucose (DMEM), Dulbecco's Phosphate-Buffered Saline (DPBS), Trypsin – EDTA solution, Fetal Bovine Serum (FBS), Lipofectamine™ 3000 Transfection Reagent, 4′,6-diamidino-2-phenylindole (98%, DAPI) and Resazurin Sodium Salt were also purchased from Sigma-Aldrich and used without further purification. Acetic acid glacial (AcOH) was obtained from VWR chemicals. Plasmid DNA (pCMV-GFP) was purchased from the Plasmid Factory. Dichloromethane (DCM, 99.7%), acetone (99.5%) and ethylacetate (EtOAc, 99.5%) were obtained from Ossum Chemicals. The RAFT agent 2-[[(butylthio)thioxomethyl] thio] propanoic acid was synthesized following a literature procedure.109 Deuterated solvents were purchased from Sigma-Aldrich and used as received. Demi-water was used unless stated otherwise. Dialysis was carried out using either SnakeSkin dialysis tubing with a molecular weight cut-off of 10 kDa or with Spectra/Por 6 Dialysis Membrane Pre-wetted RC Tubing with a molecular weight cut-off of 1 kDa or 3.5 kDa. pGMA was gifted by Dr Anzar Khan. Lipofectamine™ 3000 Transfection Reagent was prepared according to provided protocol. Wheat Germ Agglutinin CF®405S Conjugate (WGA) was obtained from Biotium. 5-(4,6-Dichlorotriazinyl) aminofluorescein single isomer (5-DTAF) was purchased from Thermo Fisher. pCMV-GFP and pCMV-lacZ were purchased from the Plasmid Factory. HeLa, bEnd.3, HepG2, C2C12, RAW264, HEK293 and HFF cells were purchased from ECACC. Super folder green fluorescent protein (sfGFP) was synthesized according to literature.110Analysis
Lyophilization was performed using a Labconco FreeZone 4.5 Liter Benchtop Freeze Dry System. NMR spectra were recorded on a Bruker Ascend 400 MHz NMR spectrometer at 298 K. Residual solvent signals were used as internal reference according to the literature.111 Multiplicities are abbreviated as follows: singlet (s), doublet (d), and multiplet (m). IR spectra were recorded on a Nicolet 6700 FT-IR spectrometer from Thermo Scientific, equipped with a Smart Orbit Diamond Attenuated Total Reflectance accessory. Infrared spectroscopy for 40-THG and 50-THG were performed on a Bruker Alpha and analyzed with OPUS software. DLS and zeta potential measurements were carried out on a Malvern Zetasizer 4000, performed in triplicate. As a reference material for DLS, poly(ethylene glycol)-co-poly(isobutyl methacrylate) with a refractive index (RI) of 1.465 was used. DLS measurements were carried with concentrations in the range of 0.5–2.0 mg mL−1. Zeta potential measurements were carried out at similar concentrations in Milli-Q in the presence of 5 mM KCl or in 10 mM HEPES buffer solution (HEPES-NaOH, pH = 7). TEM was carried out on a Philips CM300ST-FEG Transmission Electron Microscope. Samples were prepared on Electron Microscopy Sciences FCF200-Cu Formvar/copper support grids (200 mesh). Solutions of nanogels were prepared with concentrations of 0.5 mg mL−1 in water or 1,4-dioxane, or 1 mg mL−1 in 10 mM HEPES. They were subsequently sonicated for 30 min. Of this solution 5 μL was deposited on the grid and dried after 15 min with paper filter. After drying the grid in the fume hood, 5 μL of an aqueous 1% w/v uranyl acetate solution was deposited on the grid and dried after 30 seconds, using a paper filter. Particle size analysis using TEM micrographs was carried out using ImageJ software by calibrating to the scale given in the micrographs and a minimum of 200 particles. Fluorescence microscope images were taken on an Olympus IX2-ILL 100 with 10x and 20x objective, operating at ex 350 nm/em 420 nm for DAPI and WGA and ex 460–490 nm/em 525 nm for GFP and DTAF. Fluorescence was measured on an EnSight™ multimode plate reader exciting at 560 nm and emitting at 590 nm. Confocal Fluorescence Microscopy (CFM) images were taken on a Zeiss Confocal LSM880 with 20x objective, exciting at 405 nm and emitting at 454 nm for DAPI and WGA. GFP and DTAF images were excited at 488 nm and emission was recorded at 548 nm. The intensities were quantified in ImageJ and corrected for background signals.Synthetic procedures
GMA-EGDMA nanogel formation. In a typical procedure, GMA (2.1 mL, 16 mmol, 3.0 equiv.), EGDMA (1.0 mL, 5.3 mmol, 1.0 equiv.), AIBN (1.4 mL, 61 mM in 1,4-dioxane, 8.4 μmol, 1.6 mol%), and 2-(((butylthio)carbonothioyl)thio)propanoic acid (1.0 mL, 0.42 M in 1,4-dioxane, 0.42 mmol, 8.0 mol%) were added to a flask with 50 mL of 1,4-dioxane, giving a monomer concentration of 6% w/w. The flask was sealed with a septum and degassed by nitrogen bubbling for 30 min. Then, the reaction mixture was heated to 70 °C and stirred at 200 rpm for 90 min. An excess of AIBN (4.6 gram, 28 mmol, 5.3 equiv.) was dissolved in 30 mL of 1,4-dioxane. This AIBN solution was degassed by nitrogen bubbling for 30 min and added to the reaction mixture. After 22 h of stirring at 70 °C, the reaction mixture was precipitated three times in hexane. The solids were dried under a stream of nitrogen yielding white solids (1.33 gram, quantitative yield). Conversion of monomers was determined by 1H NMR spectroscopy using 1,4-dioxane as an internal standard for relative signal intensity. 1H NMR spectroscopy at 90 min gave a monomer conversion of 30%.1H NMR (GMA-EGDMA, 400 MHz, CDCl3): δ (ppm): 4.67–4.06 (m, CH2), 4.05–3.55 (m, CH2), 3.54–3.07 (m, CH, epoxide), 3.05–2.76 (m, CH2, epoxide), 2.76–2.50 (m, CH2, epoxide), 2.29–1.81 (s, CH2), 2.21–1.67 (s, CH2), 1.47–0.65 (m, CH3).
IR (GMA-EGDMA, neat) λmax: 2990, 2952, 1724, 1450, 1387, 1257, 1146, 993, 906, 848, 759 cm−1.
GMA-DEGDMA nanogel formation. RAFT agent 2-[[(butylthio)thioxomethyl] thio] propanoic acid (22.1 mg, 0.1 mmol, 1 equiv.), AIBN (3 mg, 0.02 mmol, 0.2 equiv.), GMA (0.46 mL, 3.5 mmol, 37.5 equiv.), DEGDMA (0.26 mL, 1.2 mmol, 12.5 equiv.) and 1,4-dioxane (10 mL, 93 w/w%) were added to a 25 mL round bottom flask equipped with a stir bar, sealed with a septum and purged with nitrogen for 30 min. The flask was subsequently placed in an oil bath at 70 °C and allowed to react for 1.5 h after which it was taken out of the oil bath and quenched with 3 mL DCM. The nanogel was precipitated three times in hexane yielding a white solid (235.9 mg, 35% conversion, 8.73 mmol g−1 epoxides).
GMA-TTEGDMA nanogel formation. RAFT agent 2-[[(butylthio)thioxomethyl] thio] propanoic acid (19.5 mg, 0.08 mmol, 1 equiv.), AIBN (2.7 mg, 0.02 mmol, 0.2 equiv.), GMA (0.41 mL, 3.1 mmol, 37.5 equiv.), TTEGDMA (0.31 mL, 1 mmol, 12.5 equiv.), DMF (0.24 mL, 3 mmol, 37.5 equiv.) as internal standard and 1,4-dioxane (10 mL, 93 w/w%) were added to a 25 mL round bottom flask equipped with a stir bar, sealed with a septum and purged with nitrogen for 30 min. The flask was subsequently placed in an oil bath at 70 °C and allowed to react for 1.5 h after which it was taken out of the oil bath and quenched with 6 mL DCM. The nanogel was precipitated twice in hexane yielding a white solid (187.3 mg, 27% conversion, 4.38 mmol g−1 epoxides).
GMA-EGDMA-S+-x nanogel formation. In a typical procedure, GMA-EGDMA (68 mg, 0.44 mmol of epoxides) was dissolved in a mixture of AcOH/acetone (1:1, 3.2 mL). To the flask, diethyl sulfide (250 μL, 2.3 mmol, 7.0 equiv.) was added. After stirring for 230 min, the product was purified by precipitation into ice-cold diethyl ether twice. After drying under a stream of nitrogen, GMA-EGDMA-S+-x was obtained as a glassy colorless solid; x indicating the epoxide conversion to sulfonium.
1H NMR (GMA-EGDMA-S+, 400 MHz, DMSO-d6): δ (ppm): 6.95–6.13 (s, OH), 4.68–2.79 (m, CH and CH2), 2.28–1.83 (s, CH2), 1.79–0.44 (m, CH3).
GMA-EGDMA-S+-45-OH nanogel formation. In a typical procedure, GMA-EGDMA-S+-45 (119 mg, 0.31 mmol of epoxides) was dissolved in a mixture of tetrahydrofuran (THF) and water (9:1, 10 mL). To the mixture, TFA (280 μL, 3.6 mmol, 12 equiv. to epoxides) was added. The reaction mixture was stirred at room temperature for 7 days. Next, the reaction mixture was dialyzed against 0.1 M NaCl for 2 days and against Milli-Q for 4 days. After lyophilization, GMA-EGDMA-S+-45-OH was obtained as white solids (84 mg, 67% yield).
1H NMR (GMA-EGDMA-S+-45-OH, 400 MHz, DMSO-d6): δ (ppm): 6.72–6.03 (s, OH), 5.75–4.50 (m, OH), 4.50–3.37 (m, CH2 and CH), 2.26–1.80 (CH2), 1.65–1.24 (s, CH3), 1.24–0.41 (m, CH3).
IR (GMA-EGDMA-S+-45-OH, neat): λmax: 3368 (broad), 2944, 1723, 1456, 1387, 1263, 1154, 1051, 978, 938, 752 cm−1.
GMA-EGDMA-S+-39-THG nanogel formation. In a typical procedure, GMA-EGDMA-S+-39 (98 mg, 0.47 mmol of epoxides, 1.0 equiv.) and LiOH (14 mg, 0.58 mmol, 1.2 equiv.) were dissolved in a mixture of dimethylformamide (DMF) and water (9:1, 9 mL). To the mixture, 1-thioglycerol (100 μL, 1.15 mmol, 2.4 equiv.) was added. The reaction mixture was stirred at room temperature for 4 days. Next, the reaction mixture was dialyzed against 0.1 M NaCl for 24 h and against Milli-Q for 48 h. After lyophilization, GMA-EGDMA-S+-39-THG was obtained as white solids (121 mg, 88% yield).
GMA-EGDMA-S+-47-mPEG nanogel formation. In a typical procedure, GMA-EGDMA-S+-47 (30 mg, 76 μmol of epoxides, 1.0 equiv.) and LiOH (5.6 mg, 0.23 mmol, 3.1 equiv.) were dissolved in a mixture of DMF and water (9:1, 10 mL). To the mixture, O-(2-mercaptoethyl)-O′-methyl-hexa(ethylene glycol) (33 μL, 99 μmol, 1.3 equiv.) was added. The reaction mixture was stirred at room temperature for 2 days. Next, the reaction mixture was dialyzed against 0.1 M NaCl for 2 days and against Milli-Q for 5 days. After lyophilization, GMA-EGDMA-S+-47-mPEG was obtained as white solids (34 mg, 52% yield).
GMA-DEGDMA-S+-x nanogel formation. GMA-DEGDMA (91.9 mg, 0.802 mmol of epoxides, 1 equiv.) was dissolved in a mixture of AcOH/acetone (1:1, 6 mL), followed by addition of diethyl sulfide (0.26 mL, 2.4 mmol, 3 equiv.). After stirring overnight at room temperature, the nanogel was precipitated in diethyl ether obtaining GMA-DEGDMA-S+-x.
GMA-TTEGDMA-S+-x nanogel formation. In a typical procedure, GMA-TTEGDMA (245.5 mg, 1.13 mmol of epoxides, 1 equiv.) was dissolved in a mixture of AcOH/acetone (1:1, 25 mL), followed by addition of diethyl sulfide (1.2 mL, 11.3 mmol, 10 equiv.). After stirring for 2 h at room temperature, the nanogel was precipitated three times in diethyl ether and dissolved in 10 mM HEPES (HEPES-NaOH, pH = 7) obtaining a solution of GMA-TTEGDMA-S+-x.
x-THG nanogel formation. In a typical procedure, to a snap cap vial containing GMA-TTEGDMA-S+-x (81 mg, 0.37 mmol of epoxides, 1.0 equiv.) in 10 mM HEPES (HEPES-NaOH, pH = 7) solution was added, LiOH (26.5 mg, 1.1 mmol, 3 equiv.) and 1-thioglycerol (0.1 mL, 1.1 mmol, 3 equiv.) and was allowed to stir overnight at room temperature. The reaction mixture was then dialyzed (MWCO = 1 kDa) for 2d against water. HEPES salt and NaOH were added to the aqueous solution to obtain x-THG 10 mM HEPES (3.3 or 2 mg mL−1, HEPES-NaOH, pH = 7).
pGMA (88%S+ 12% thioglycerol) formation. pGMA (80 repeating units, 100 mg, 0.68 mmol epoxides) was dissolved in a mixture of AcOH/acetone (1:1, 3 mL). To the flask, diethyl sulfide (0.22 mL, 2.04 mmol, 3.0 equiv.) was added. After stirring for 24 h, the product was precipitated twice into diethyl ether yielding a water-soluble polymer with 88% conversion. The polymer (0.056 mmol epoxides) was subsequently dissolved in 10 mL of water and added to a snap cap vial. A stir bar, LiOH (38 mg, 1.59 mmol, 3 equiv.) and 1-thioglycerol (0.14 mL, 1.59 mmol, 3 equiv.) were added to the vial and the reaction mixture stirred for 24 h. The polymer was then dialyzed (MWCO = 3.5 kDa) against water for 3 d, concentrated and set to pH 7. The addition of HEPES salt gave a polymer solution of 3.77 mg mL−1 in 10 mM HEPES buffer (HEPES·NaOH, pH = 7). At 2 mg mL−1 in 10 mM HEPES buffer, the polymer exhibited a hydrodynamic diameter of Dh = 89 ± 3 nm and a surface charge of ζ = +16 ± 1.7 mV.
1H NMR (400 MHz, D2O): δ (ppm): 3.46 (s, sulfonium CH2), 2.80 (s, thioglycerol CH), 2.68 (s, thioglycerol CH2), 1.90 (s, AcO−), 1.50 (s, sulfonium CH3).
pGMA (92% S+ 8% epoxides) formation. pGMA (80 repeating units, 100 mg, 0.68 mmol epoxides) was dissolved in a mixture of AcOH/acetone (1:1, 3 mL). To the flask, diethyl sulfide (0.59 mL, 5.43 mmol, 8.0 equiv.) was added. After stirring for 24 h, the product was precipitated twice into diethyl ether followed by dialysis (MWCO = 3.5 kDa) against water for 3d. Freeze drying yielded 130 mg of a water-soluble polymer with 92% conversion. The sulfonium polymer was stored in 10 mM HEPES buffer (HEPES·NaOH, pH = 7) at 2 mg mL−1, Dh = 259 ± 4 nm and ζ = + 37 ± 0.9 mV.
1H NMR (400 MHz, D2O): δ (ppm): 3.44 (s, sulfonium CH2), 2.78 (s, epoxide CH), 2.66 (s, epoxide CH), 1.90 (s, AcO−), 1.48 (s, sulfonium CH3).
Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Michel Klein Gunnewiek is acknowledged for his help with AFM imaging. Dr Rico Keim is acknowledged for his help with TEM imaging. Dr Naomi Hamelmann is acknowledged for helping guide the experiments and giving great feedback. Dr Anzar Khan is thanked for providing us with pGMA.References
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed.
- M. L. Etheridge, S. A. Campbell, A. G. Erdman, C. L. Haynes, S. M. Wolf and J. McCullough, Nanomedicine, 2013, 9, 1–14 CrossRef CAS PubMed.
- J. J. Rossi, C. H. June and D. B. Kohn, Nat. Biotechnol., 2007, 25, 1444–1454 CrossRef CAS PubMed.
- X.-F. Qin, D. S. An, I. S. Y. Chen and D. Baltimore, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 183–188 CrossRef CAS PubMed.
- F. Mingozzi and K. A. High, Nat. Rev. Genet., 2011, 12, 341–355 CrossRef CAS PubMed.
- M. Jean, M. Alameh, M. D. Buschmann and A. Merzouki, Gene Ther., 2011, 18, 807–816 CrossRef CAS PubMed.
- J. Han, B. McLane, E.-H. Kim, J.-W. Yoon and H.-S. Jun, Mol. Ther., 2011, 19, 470–478 CrossRef CAS PubMed.
- Y.-W. Won, P. P. Adhikary, K. S. Lim, H. J. Kim, J. K. Kim and Y.-H. Kim, Nat. Mater., 2014, 13, 1157–1164 CrossRef CAS PubMed.
- M. Izquierdo, Cancer Gene Ther., 2005, 12, 217–227 CrossRef CAS PubMed.
- J. Park, K. Singha, S. Son, J. Kim, R. Namgung, C. O. Yun and W. J. Kim, Cancer Gene Ther., 2012, 19, 741–748 CrossRef CAS PubMed.
- C. J. Cheng, R. Bahal, I. A. Babar, Z. Pincus, F. Barrera, C. Liu, A. Svoronos, D. T. Braddock, P. M. Glazer and D. M. Engelman, Nature, 2015, 518, 107–110 CrossRef CAS PubMed.
- X. Yu, L. Gong, J. Zhang, Z. Zhao, X. Zhang and W. Tan, Sci. China: Chem., 2017, 60, 1318–1323 CrossRef CAS.
- Z.-Y. He, Z.-H. Jin, M. Zhan, Z. Qin, Z. Li and T. Xu, Chin. Chem. Lett., 2017, 28, 1851–1856 CrossRef CAS.
- M. H. Amer, Mol. Cell. Ther., 2014, 2, 1–19 CrossRef PubMed.
- D. Cross and J. K. Burmester, Clin. Med. Res., 2006, 4, 218–227 CrossRef CAS PubMed.
- D. Luo and W. M. Saltzman, Nat. Biotechnol., 2000, 18, 33–37 CrossRef CAS PubMed.
- K. Tatiparti, S. Sau, S. K. Kashaw and A. K. Iyer, Nanomaterials, 2017, 7, 77 CrossRef PubMed.
- R. Goswami, G. Subramanian, L. Silayeva, I. Newkirk, D. Doctor, K. Chawla, S. Chattopadhyay, D. Chandra, N. Chilukuri and V. Betapudi, Front. Oncol., 2019, 9, 297 CrossRef PubMed.
- D. Ibraheem, A. Elaissari and H. Fessi, Int. J. Pharm., 2014, 459, 70–83 CrossRef CAS PubMed.
- J. C. Burnett, J. J. Rossi and K. Tiemann, Biotechnol. J., 2011, 6, 1130–1146 CrossRef CAS PubMed.
- D. Lechardeur, K. J. Sohn, M. Haardt, P. B. Joshi, M. Monck, R. W. Graham, B. Beatty, J. Squire, H. O′brodovich and G. L. Lukacs, Gene Ther., 1999, 6, 482–497 CrossRef CAS PubMed.
- A. Singh, P. Trivedi and N. K. Jain, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 274–283 CrossRef CAS PubMed.
- H. Herweijer and J. A. Wolff, Gene Ther., 2003, 10, 453–458 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- N. S. Petrova, I. V. Chernikov, M. I. Meschaninova, Ii. S. Dovydenko, A. G. Venyaminova, M. A. Zenkova, V. V. Vlassov and E. L. Chernolovskaya, Nucleic Acids Res., 2012, 40, 2330–2344 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- N. Somia and I. M. Verma, Nat. Rev. Genet., 2000, 1, 91–99 CrossRef CAS PubMed.
- S. Y. Wong, J. M. Pelet and D. Putnam, Prog. Polym. Sci., 2007, 32, 799–837 CrossRef CAS.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS PubMed.
- L. Wasungu and D. Hoekstra, J. Controlled Release, 2006, 116, 255–264 CrossRef CAS PubMed.
- I. Koltover, T. Salditt, J. O. Rädler and C. R. Safinya, Science, 1998, 281, 78–81 CrossRef CAS PubMed.
- F. Liu, H. Qi, L. Huang and D. Liu, Gene Ther., 1997, 4, 517–523 CrossRef CAS PubMed.
- Y. H. Bae and H. Yin, J. Controlled Release, 2008, 131, 2–4 CrossRef CAS PubMed.
- H. Chen, S. Kim, L. Li, S. Wang, K. Park and J.-X. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6596–6601 CrossRef CAS PubMed.
- H. Chen, S. Kim, W. He, H. Wang, P. S. Low, K. Park and J.-X. Cheng, Langmuir, 2008, 24, 5213–5217 CrossRef CAS PubMed.
- P. Xu, E. Gullotti, L. Tong, C. B. Highley, D. R. Errabelli, T. Hasan, J.-X. Cheng, D. S. Kohane and Y. Yeo, Mol. Pharm., 2009, 6, 190–201 CrossRef CAS PubMed.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS PubMed.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- L. A. Jackson, E. J. Anderson, N. G. Rouphael, P. C. Roberts, M. Makhene, R. N. Coler, M. P. McCullough, J. D. Chappell, M. R. Denison, L. J. Stevens, A. J. Pruijssers, A. McDermott, B. Flach, N. A. Doria-Rose, K. S. Corbett, K. M. Morabito, S. O'Dell, S. D. Schmidt, P. A. Swanson, M. Padilla, J. R. Mascola, K. M. Neuzil, H. Bennett, W. Sun, E. Peters, M. Makowski, J. Albert, K. Cross, W. Buchanan, R. Pikaart-Tautges, J. E. Ledgerwood, B. S. Graham and J. H. Beigel, N. Engl. J. Med., 2020, 383, 1920–1931 CrossRef CAS PubMed.
- M. R. Molla and P. A. Levkin, Adv. Mater., 2016, 28, 1159–1175 CrossRef CAS PubMed.
- Y. Zhao, W. Wang, S. Guo, Y. Wang, L. Miao, Y. Xiong and L. Huang, Nat. Commun., 2016, 7, 11822 CrossRef PubMed.
- M. A. Kay, Nat. Rev. Genet., 2011, 12, 316–328 CrossRef CAS PubMed.
- C. H. Jones, C.-K. Chen, A. Ravikrishnan, S. Rane and B. A. Pfeifer, Mol. Pharm., 2013, 10, 4082–4098 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- A. S. A. Lila, T. Ishida and H. Kiwada, Expert Opin. Drug Delivery, 2009, 6, 1297–1309 CrossRef PubMed.
- R. E. Eliaz, S. Nir, C. Marty and F. C. Szoka Jr., Cancer Res., 2004, 64, 711–718 CrossRef CAS PubMed.
- T. L. Andresen, S. S. Jensen and K. Jørgensen, Prog. Lipid Res., 2005, 44, 68–97 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- M. Mammen, S. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- J. Zhou, J. Liu, C. J. Cheng, T. R. Patel, C. E. Weller, J. M. Piepmeier, Z. Jiang and W. M. Saltzman, Nat. Mater., 2012, 11, 82–90 CrossRef CAS PubMed.
- H. H. Ahn, J. H. Lee, K. S. Kim, J. Y. Lee, M. S. Kim, G. Khang, I. W. Lee and H. B. Lee, Biomaterials, 2008, 29, 2415–2422 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualc’h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J.-P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS PubMed.
- U. Lungwitz, M. Breunig, T. Blunk and A. Göpferich, Eur. J. Pharm. Biopharm., 2005, 60, 247–266 CrossRef CAS PubMed.
- L. Parhamifar, A. K. Larsen, A. C. Hunter, T. L. Andresen and S. M. Moghimi, Soft Matter, 2010, 6, 4001–4009 RSC.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CrossRef CAS PubMed.
- M. Köping-Höggård, I. Tubulekas, H. Guan, K. Edwards, M. Nilsson, K. M. Vårum and P. Artursson, Gene Ther., 2001, 8, 1108–1121 CrossRef PubMed.
- S. P. Strand, S. Lelu, N. K. Reitan, C. de Lange Davies, P. Artursson and K. M. Vårum, Biomaterials, 2010, 31, 975–987 CrossRef CAS PubMed.
- D. Luo, K. Haverstick, N. Belcheva, E. Han and W. M. Saltzman, Macromolecules, 2002, 35, 3456–3462 CrossRef CAS.
- C. Dufès, I. F. Uchegbu and A. G. Schätzlein, Adv. Drug Delivery Rev., 2005, 57, 2177–2202 CrossRef PubMed.
- S. T. Hemp, M. H. Allen, M. D. Green and T. E. Long, Biomacromolecules, 2012, 13, 231–238 CrossRef CAS PubMed.
- C. Ornelas-Megiatto, P. R. Wich and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 1902–1905 CrossRef CAS PubMed.
- V. L. Rose, F. Mastrotto and G. Mantovani, Polym. Chem., 2017, 8, 353–360 RSC.
- X. Nie, Z. Zhang, C.-H. Wang, Y.-S. Fan, Q.-Y. Meng and Y.-Z. You, Bioconjugate Chem., 2018, 30, 284–292 CrossRef PubMed.
- K. A. Kurnia, M. V. Quental, L. M. N. B. F. Santos, M. G. Freire and J. A. P. Coutinho, Phys. Chem. Chem. Phys., 2015, 17, 4569–4577 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. T. Hemp, M. H. Allen, A. E. Smith and T. E. Long, ACS Macro Lett., 2013, 2, 731–735 CrossRef CAS PubMed.
- J. R. Kramer, N. W. Schmidt, K. M. Mayle, D. T. Kamei, G. C. L. Wong and T. J. Deming, ACS Cent. Sci., 2015, 1, 83–88 CrossRef CAS PubMed.
- M. C. Mackenzie, A. R. Shrivats, D. Konkolewicz, S. E. Averick, M. C. McDermott, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2015, 16, 236–245 CrossRef CAS PubMed.
- D. Zhu, H. Yan, X. Liu, J. Xiang, Z. Zhou, J. Tang, X. Liu and Y. Shen, Adv. Funct. Mater., 2017, 27, 1606826 CrossRef.
- M. Yamagata, T. Kawano, K. Shiba, T. Mori, Y. Katayama and T. Niidome, Bioorg. Med. Chem., 2007, 15, 526–532 CrossRef CAS PubMed.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- A. E. Ekkelenkamp, M. R. Elzes, J. F. J. Engbersen and J. M. J. Paulusse, J. Mater. Chem. B, 2018, 6, 210–235 RSC.
- R. W. Graff, Y. Shi, X. Wang and H. Gao, Macromol. Rapid Commun., 2015, 36, 2076–2082 CrossRef CAS PubMed.
- X. Wang and H. Gao, Polymer, 2017, 9, 188 Search PubMed.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- J. Spanswick and K. Matyjaszewski, Mater. Today, 2005, 8, 26–33 Search PubMed.
- K. S. Soni, S. S. Desale and T. K. Bronich, J. Controlled Release, 2016, 240, 109–126 CrossRef CAS PubMed.
- F. Sabir, M. I. Asad, M. Qindeel, I. Afzal, M. J. Dar, K. U. Shah, A. Zeb, G. M. Khan, N. Ahmed and F. Din, J. Nanomater., 2019, 2019, 1 CrossRef.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2014, 136, 5547–5550 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, Biomacromolecules, 2012, 13, 1719–1723 CrossRef CAS PubMed.
- N. H. Park, M. Fevre, Z. X. Voo, R. J. Ono, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2016, 5, 1247–1252 CrossRef CAS PubMed.
- E. G. Gharakhanian and T. J. Deming, Biomacromolecules, 2015, 16, 1802–1806 CrossRef CAS PubMed.
- N. Zhao, X. Lin, Q. Zhang, Z. Ji and F. Xu, Small, 2015, 11, 6467–6479 CrossRef CAS PubMed.
- F. J. Xu, Y. Zhu, M. Y. Chai and F. S. Liu, Acta Biomater., 2011, 7, 3131–3140 CrossRef CAS PubMed.
- F. J. Xu, M. Y. Chai, W. B. Li, Y. Ping, G. P. Tang, W. T. Yang, J. Ma and F. S. Liu, Biomacromolecules, 2010, 11, 1437–1442 CrossRef CAS PubMed.
- X. B. Dou, M. Y. Chai, Y. Zhu, W. T. Yang and F. J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 3212–3218 CrossRef CAS PubMed.
- D. N. M. Kusmus, T. W. van Veldhuisen, A. Khan, J. J. L. M. Cornelissen and J. M. J. Paulusse, RSC Adv., 2022, 12, 29423–29432 RSC.
- E. Fröhlich, Int. J. Nanomed., 2012, 5577–5591 CrossRef PubMed.
- L. Ehrenberg and S. Hussain, Mutat. Res., 1981, 86, 1–113 CAS.
- J. Cornish, K. E. Callon, C.-X. Lin, C. L. Xiao, T. B. Mulvey, G. J. S. Cooper and I. R. Reid, Am. J. Physiol. Endocrinol. Metab., 1999, 277, E779–E783 CrossRef CAS PubMed.
- S. De and A. Khan, ChemComm, 2012, 48, 3130–3132 RSC.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- D. Venturoli and B. Rippe, Am. J. Physiol.: Renal Physiol., 2005, 288, 605–613 CrossRef PubMed.
- S. A. Kulkarni and S.-S. Feng, Pharm. Res., 2013, 30, 2512–2522 CrossRef CAS.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed.
- S. Yamano, J. Dai and A. M. Moursi, Mol. Biotechnol., 2010, 46, 287–300 CrossRef CAS PubMed.
- M. R. Elzes, G. Si, J. F. J. Engbersen and J. M. J. Paulusse, ACS Symp. Ser., 2019, 93–117 CrossRef CAS.
- J. M. McMahon, E. Signori, K. E. Wells, V. M. Fazio and D. J. Wells, Gene Ther., 2001, 8, 1264–1270 CrossRef CAS PubMed.
- D. J. Stephens and R. Pepperkok, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4295–4298 CrossRef CAS PubMed.
- M. F. Bureau, S. Naimi, R. T. Ibad, J. Seguin, C. Georger, E. Arnould, L. Maton, F. Blanche, P. Delaere and D. Scherman, Biochim. Biophys. Acta, Gene Struct. Expression, 2004, 1676, 138–148 CrossRef CAS PubMed.
- G. L. Lukacs, P. Haggie, O. Seksek, D. Lechardeur, N. Freedman and A. S. Verkman, J. Biol. Chem., 2000, 275, 1625–1629 CrossRef CAS PubMed.
- E. E. Vaughan, J. V. DeGiulio and D. A. Dean, Curr. Gene Ther., 2006, 6, 671–681 CrossRef CAS PubMed.
- M. J. Hatch, F. J. Meyer and W. D. Lloyd, J. Appl. Polym. Sci., 1969, 13, 721–744 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00011k |
| ‡ Present address: Department of Biomedical Engineering and Institute for Complex Molecular Systems, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, Netherlands. |
| This journal is © The Royal Society of Chemistry 2024 |