
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in tunable metal–support interactions for enhancing the photocatalytic nitrogen reduction reaction
Bing-Hao
Wang†
a,
Guang-Hui
Chen†
a,
Biao
Hu†
a,
Lang
Chen
*ab,
Xiong
Wang
a,
Sheng
Tian
a,
Xing-Sheng
Hu
a,
Yang
Li
a,
Chao
Peng
a and
Shuang-Feng
Yin
 *abc
*abc
aAdvanced Catalytic Engineering Research Center of the Ministry of Education, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, P. R. China. E-mail: huagong042cl@163.com; sf_yin@hnu.edu.cn
bGreater Bay Area Institute for Innovation, Hunan University, Guangzhou, 511300, P. R. China
cCollege of Science, Central South University of Forestry and Technology, Changsha 410004, P. R. China
First published on 15th September 2023
Abstract
Ammonia (NH3), as an important foundational chemical and green hydrogen energy carrier, plays an indispensable role in the development of human society. However, it is evident that the traditional process for NH3 synthesis is no longer in line with the times due to its drawbacks, such as high energy consumption and high carbon emission. In recent years, the photocatalytic nitrogen reduction reaction (PNRR), which reduces N2 to NH3 under mild conditions using inexhaustible solar energy, has been considered as a promising alternative. Nevertheless, the catalytic efficiency of the PNRR is low and far from realizing practical applications owing to the weak N2 adsorption, hard dissociation of inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, and competing reactions of hydrogen precipitation. Metal–support interactions (MSIs) provide an efficient way to adjust the performance of both the active metal and support in the photocatalytic process through geometric, electronic and bifunctional effects. The design of heterogeneous photocatalysts with tunable MSIs has been proved to be a feasible way to enhance their catalytic performance for the PNRR. In this review, we summarize the recent developments in MSI photocatalysts involved in nitrogen fixation. Firstly, the mechanism of MSIs and their characterization as well as the synthesis strategies for photocatalysts with MSIs are briefly outlined. Subsequently, the electronic and bifunctional effects of MSI photocatalysts and the corresponding PNRR mechanism are focused on, from the aspects of supports such as metal oxides, bismuth oxyhalides, metal sulfides, metal–organic frameworks (MOFs) and carbon nitrides. Finally, the future developments in this area such as creating state-of-the-art materials with MSIs and synthesis strategies and developing advanced techniques to investigate reaction mechanisms for N2 fixation are discussed. It is expected that this review can provide some guidance for understanding and rationally designing MSI photocatalysts, especially for boosting the PNRR.
N, and competing reactions of hydrogen precipitation. Metal–support interactions (MSIs) provide an efficient way to adjust the performance of both the active metal and support in the photocatalytic process through geometric, electronic and bifunctional effects. The design of heterogeneous photocatalysts with tunable MSIs has been proved to be a feasible way to enhance their catalytic performance for the PNRR. In this review, we summarize the recent developments in MSI photocatalysts involved in nitrogen fixation. Firstly, the mechanism of MSIs and their characterization as well as the synthesis strategies for photocatalysts with MSIs are briefly outlined. Subsequently, the electronic and bifunctional effects of MSI photocatalysts and the corresponding PNRR mechanism are focused on, from the aspects of supports such as metal oxides, bismuth oxyhalides, metal sulfides, metal–organic frameworks (MOFs) and carbon nitrides. Finally, the future developments in this area such as creating state-of-the-art materials with MSIs and synthesis strategies and developing advanced techniques to investigate reaction mechanisms for N2 fixation are discussed. It is expected that this review can provide some guidance for understanding and rationally designing MSI photocatalysts, especially for boosting the PNRR.
 Bing-Hao Wang | Bing-Hao Wang received his B.S. degree from Hunan University of Science and Technology in 2019. He is currently a PhD student under the supervision of Professor Shuang-Feng Yin in the Department of Chemical Engineering of Hunan University. His current research focuses on photocatalytic nitrogen fixation, plasmonic photocatalysis and first-principles calculations. |
 Guang-Hui Chen | Guang-Hui Chen received his B.S. degree in chemical engineering from Guangxi University in 2021. He is currently a master's student under the supervision of Associate Professor Lang Chen in the Department of Chemical Engineering, Hunan University. His current research interests include the design and synthesis of photocatalysts and their applications in nitrogen fixation. |
 Biao Hu | Biao Hu obtained his B.S. degree in metallurgical engineering from Hunan University of Technology in 2016 and his M.S. degree in Metallurgical Engineering from Central South University in 2019. He is currently a PhD student under the supervision of Professor Shuang-Feng Yin in the Department of Chemical Engineering of Hunan University. His current research interests include the design and synthesis of nanostructured semiconductors and application in photocatalytic nitrogen fixation. |
 Lang Chen | Lang Chen received his B.S. degree from Henan University of Science and Technology in 2008 and then obtained his PhD in Hunan University under the supervision of Professor Shuang-Feng Yin. He is currently an associate professor in Hunan University. His research interests focus on the design and synthesis of efficient heterogeneous photocatalysts/photo-electro-catalyst and application in conversion of inert small molecules. |
 Shuang-Feng Yin | Shuang-Feng Yin obtained his B.S. degree in 1996 from Beijing University of Chemical Technology. He subsequently received his PhD from Tsinghua University in 2003. He was promoted to full Professor in Hunan University in 2006. He worked as a senior visiting scholar in the Hong Kong Baptist University and Japan Institute of Integrated Industrial Technology from 2008 to 2009. His research interests focus on photo/electrocatalytic energy conversion and C–H bond activation. |
Broader contextAmmonia (NH3) is an important chemical raw material, and is indispensable for the sustainable development of human society and agriculture. In general, we rely mainly on the artificial nitrogen fixation process, i.e. the Haber–Bosch process, to obtain NH3. However, this traditional method is not in line with modern society due to its high energy consumption and high carbon emissions. The use of photocatalytic technology for nitrogen fixation is expected to replace the conventional process due to its environmentally-friendly nature and no fossil energy consumption. Unfortunately, the low activity of photocatalysts has greatly limited the economic feasibility of the photocatalytic nitrogen reduction reaction (PNRR), so there is still a long way to go to realize its practical application. Metal–support interactions (MSIs) can play an important role in influencing the activity, selectivity, and stability of supported catalysts through geometric, electronic, and bifunctional effects. Therefore, the design of heterogeneous photocatalysts with MSIs is likely to improve the efficiency of the photocatalytic nitrogen fixation system. This paper summarizes the most recent developments in tunable MSI photocatalysts mainly from the perspective of supports, even the metals which were commonly regarded as active sites played an indispensable role in the PNRR. It is hoped to provide some guidance for the design of efficient heterogeneous photocatalysts using tunable MSIs, especially for the PNRR process. |
1. Introduction
Supported catalysts, where nano/subnano-structured metals are uniformly dispersed and loaded on specially selected supports, are widely used in the area of thermal and photo as well as electro-catalyses.1–4 At the beginning of the development of catalytic science, it was generally believed that supports only played the role of supporting and dispersing the active components.5 However, as understanding deepened, it was found that the catalytic performances are highly dependent on both the active component and the support rather than individually.6 Attention has been paid to the interrelated systems of supports and active components (taking a metal as an example) for designing efficient catalysts, such as metal–support interactions (MSIs), which were originally recognized by the sudden loss of adsorption capacity for CO and H2 after high-temperature reduction of group VIII noble metal (Pt, Pd, Ir, Ru, and Rh) loaded TiO2 due to the covering of noble metals by TiO2.7 Nowadays, MSIs are commonly divided into different types regarding the main interactions between metals and supports. For example, a metal being partially or fully encapsulated by a support to form a physical cladding layer is widely considered a strong metal–support interaction (SMSI).7–10 The other type is an electronic metal–support interaction (EMSI) with the charge transfer between a metal and a support.11–14 In addition, MSI phenomena can also be distinguished based on the interfacial perimeter, nanoparticle morphology, and chemical composition.15 These phenomena often do not exist alone and can exhibit different intensities depending on the catalyst and reaction system.In heterogeneous catalytic reaction systems, MSIs play an indispensable role in tuning the activity, selectivity, and stability of catalysts through geometric, electronic, and bifunctional effects.16–18 Diverse influences of MSIs have been revealed on catalytic reactions, as they can enhance catalytic activity by transferring the inert support into active centers, tuning the selectivity of outcomes by promoting or inhibiting the occurrence of a particular reaction and increasing the stability of the catalyst by forming covalent or ionic bonds between the metal and the support.19–22 Moreover, the heterogeneous catalysts with MSIs can adjust the electronic structure of both active metals and supports to regulate their performances. Nowadays, MSIs have been widely used in photocatalytic reactions such as hydrogen evolution,23–25 ethylene oxidation,26 environmental remediation,27,28 and nitrogen fixation.
Since the pioneering report of light-driven nitrogen fixation in 1977,29 this pathway has attracted the interest of a large number of researchers, because the photocatalytic nitrogen reduction reaction (PNRR) can be performed at ambient temperature and pressure, with H2O as the source of H atoms and almost zero carbon emission, and it has great potential to be used as an alternative to traditional methods (Table 1).30–34 The mechanism of the PNRR is as follows: (1) a semiconductor matrix composite is excited by light to produce photogenerated electron–hole pairs, (2) some of the electron–hole pairs rapidly recombine and the others migrate to the surface, which then undergo an oxidation–reduction reaction, and (3) H2O reacts with holes in an oxidation reaction to produce protons (H+), while at the same time, N2 adsorbed at the catalyst active site is activated by electron excitation, and subsequently, H atoms are continuously added to the N atoms.35–37 Attributing to the inert nature of N2, both its adsorption and activation on photocatalysts are limited, and the production rate of NH3 in the PNRR is extremely low and far from the requirements for industrial applications. To be specific, the conversion of N2 to NH3 is extremely difficult because the two N atoms of the N2 molecule share three pairs of electrons from the σ and π orbitals thus forming an extremely strong NN bond with strong bond energy, weak polarization, and a lack of a dipole moment.38–40
| Project | Haber–Bosch process | Photocatalytic nitrogen fixation |
|---|---|---|
| Hydrogen source | H2 | Water |
| Catalyst | Iron-based (commonly) | Semiconductors |
| Temperature | 400–500 °C | Room temperature |
| Pressure | 20–50 MPa | 0.1 MPa |
| Energy source | Fossil fuel | Solar energy |
| CO2 emissions per year | 1.6% of the global total | Zero |
Based on the above facts, we can conclude that from a thermodynamic point of view, the solar-driven nitrogen fixation process needs to satisfy the energy criterion of oxidation and reduction potential. However, the nitrogen fixation efficiency of unmodified catalysts is generally low due to kinetic limitations. Hence, designing active centers to promote N2 adsorption and activation, and thus improve the catalytic efficiency, is a great challenge for photocatalytic nitrogen fixation at present. In order to overcome these unsolved problems, a series of advanced strategies have been proposed, such as structural engineering, defect engineering, constructing heterojunctions, and metal modification as well as interfacial/hydrophilic modulation. Among them, MSIs are considered to be a promising method because they can modulate the geometrical and electronic structures of a catalyst, optimize the nature of the active center, and improve the adsorption of N2. In addition, the hydrogen reduction reaction (HER) as a competitive reaction is a key scientific issue limiting the development of the PNRR. The method of stabilizing the defect sites of a support by a metal, which in turn transforms the active center into a metal site unfavourable for hydrogen precipitation reactions, has great potential. Furthermore, low light absorption and light utilization, which are common in photocatalytic systems, can be improved and optimized by loading metal nanoparticles with plasma effects. They can act synergistically with supports, inducing a bifunctional effect of MSIs to enhance the catalytic efficiency for the PNRR.
In general, transition metals (Fe, Mn, Cu, Ru, Mo, etc.) were widely used in nitrogen fixation reactions because they have unfilled valence d-orbitals that can accept unpaired electrons from N2 and feed electrons back to the antibonding orbitals of NN.1,41,42 The essence of this approach is the electronic “acceptance–donation” between a transition metal and N2.43 It is well acknowledged that adjusting the electronic structure of photocatalysts is an efficient way to enhance the PNRR. Surprisingly, MSIs not only disperse and stabilize transition metals, but also allow for charge transfer, thereby optimizing the electronic structure of catalysts.44–47 Specifically, the charge transfer will lead to the redistribution of electrons on the active metals, adjusting the occupation state of their d-orbitals. For transition metals, changes in the electron distribution of the d-orbitals can alter their d-band center, thus affecting the N2 adsorption and activation capacity, the reaction pathway, and the desorption of intermediates and NH3.19
Among the reported MSI photocatalysts, metals have attracted much attention due to their available active sites for N2.41,42 However, it has been shown that a support has a strong influence on the PNRR by optimizing the geometry and electronic structure of a catalyst, which is often overlooked. In particular, the activation of N2 in a photocatalytic system essentially relies on the photogenerated electrons of semiconductors. In addition, the mechanism of metal–support interactions in the PNRR is still not clear enough. Therefore, it is necessary to summarize the application of MSIs in the PNRR from the perspective of support classification.
In this review, we will summarize and understand the application of MSIs in photocatalytic nitrogen fixation from the following aspects: (1) the mechanism of MSIs and their characterization, (2) methods for the fabrication of heterogeneous photocatalysts with MSIs, (3) photocatalytic performance and mechanism under MSI action, and (4) challenges and prospects for MSI photocatalysts in the future. The electronic effect, bifunctional effect and reaction mechanisms of supported metal catalysts will be focused. We expect that this review will be of interest to researchers and provide guidance for the design of heterogeneous photocatalysts with tunable MSIs to enhance the PNRR.
2. Mechanism of MSIs and their characterization
Since the early 1980s, researchers have tried to use various techniques to investigate the mechanism of metal–support interactions in catalysis, but unfortunately, progress in this area has been slow owing to the limitations of characterization and analytical tools available at the time.7 After decades of development, benefiting from advanced characterization techniques and theoretical computational analysis, the atomic and electronic structure at the interface has been studied in depth, which in turn reveals the nature of MSIs.48,49 In general, the process of metal–support interface formation is accompanied by charge redistribution and chemical reactions, which are considered to be the cause of MSIs.9 In this section, we will illustrate the mechanism of MSI action mainly in terms of geometric, electronic, and bifunctional effects (Fig. 1), and briefly describe its characteristics.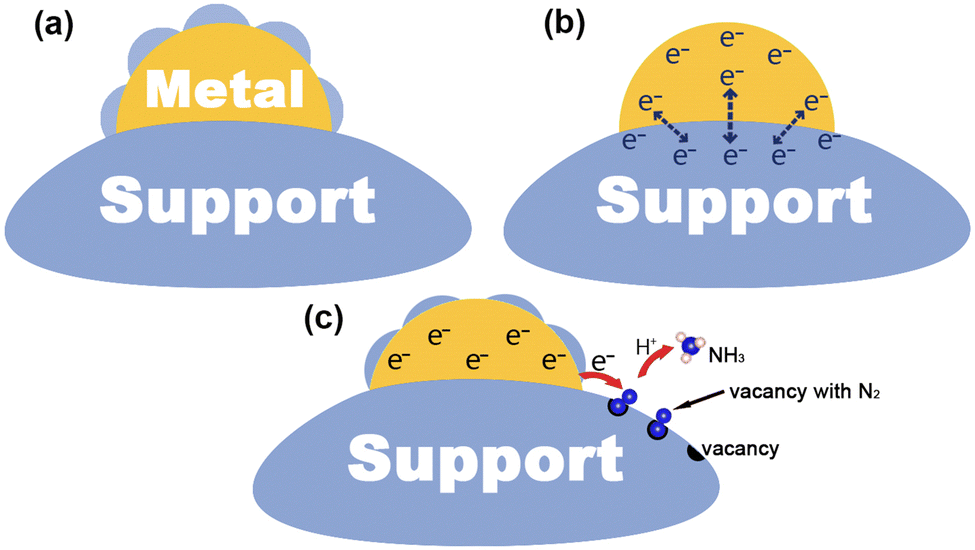 | ||
| Fig. 1 (a)–(c) Geometric, electronic, and bifunctional effects of MSIs. | ||
2.1. Geometric effect
The geometric effect of MSIs is considered to be due to the partial or total coating of a metal surface by a support, thus forming an encapsulation layer. The geometric effect of MSIs is attributed to the surface energy minimization, which can be divided into three steps:8–10,50 (1) the continuous transfer of off-domain electrons through the oxide to the metal to reach the Fermi level (EF) equilibrium, (2) the diffusion of positive metal ions (Mn+) towards the metal surface through the local electric field formed by the positive charge on the oxide and the negative charge on the metal, and (3) the subsequently reduced positive charge on the oxide, leading to the diffusion of negative oxygen ions (On−) to the metal surface. Commonly, the strength of the MSI geometry effect can be determined by the surface energy magnitude of a metal. Generally, when the surface energy of a metal is greater than 2 J cm−2 it can drive the encapsulation of the metal by a support.51 In addition, geometric effects can usually be observed directly using a transmission electron microscope (TEM) and a high-resolution transmission electron microscope (HRTEM). Electron energy loss spectroscopy (EELS) and energy dispersive spectroscopy (EDS) can be used to identify the chemical composition of the overlay to confirm whether the support diffusion occurs.8,52 The MSI geometry effect is commonly beneficial for the stabilization and dispersion of a metal, however, this comes at the expense of the metal active sites, which may hinder the metal active sites and prevent the adsorption of reactants and ultimately may lead to a reduction in reactivity. From another point of view, yet, the total encapsulation of a metal by a support may favor electron transport, leading to an increase in the activity of some catalytic reactions. Thus, we need to control the geometrical effect of MSIs and utilize this “double-edged sword” to achieve a dynamic balance between the catalyst stability and activity.2.2. Electronic effect
Based on the theory of energy minimization and solid potential continuity, when contact occurs between a metal and a support, a redistribution of the charge at the interface will happen.53,54 The local charge transfer that occurs within a few atomic layers at the metal–support interface is often accompanied by the breaking of old chemical bonds and the formation of new ones between the metal and the support in the form of covalent or ionic bonds.55,56 In particular, for photocatalytic nitrogen fixation systems, semiconductors are often chosen as supports. At this point, the occurrence of long-range charge transfer is attributed to the EF of the metal and the support to be aligned, which may lead to space charge generation on the surface of the semiconductor.42 Herrmann et al. found that the direction and intensity of charge transfer were determined by the magnitude of the surface work function (ϕ).57 Specifically, if ϕ of a metal is larger than that of a semiconductor, electrons will transfer from the semiconductor to the metal. In addition, charge transfer between the metal and the support can be detected using two powerful characterization techniques: X-ray absorption near edge structure (XANES) and X-ray photoelectron spectroscopy (XPS). To be specific, for XANES, the intensity change of a peak is related to the unoccupied density states of the metal orbitals. When the d-electrons on the metal are transferred to the support, the unoccupied density states of the metal d-orbitals increase, and thus, the peak intensity increases.19 Moreover, in XPS, a negative shift of the metal binding energy implies an enrichment of electrons on the metal, which may be the result of charge transfer from the support to the metal. The electronic effect of MSIs tends to optimize the catalyst's electronic structure, which may lead to changes in the EF and d-band center, thus promoting the catalytic activity for the PNRR.58 Hence, reasonable modulation of electronic effects in heterogeneous catalysts is an important way to regulate their catalytic performance.2.3. Bifunctional effect
It is commonly believed that in MSI systems, metals act as the active sites for the reaction, however, more and more research studies have shown that supports can also provide active sites to participate in the catalytic reaction.75,143 The so-called MSI bifunctional effect is that the metal and the support provide independent and synergistic reactive sites at the metal–support interface to enhance the activity for the catalytic reaction. For example, oxygen vacancies on metal oxides can often act as adsorption and activation sites for N2, at which time the metal can act synergistically with the support through plasma effects,75,151 electron storage,143etc.,85 and thus promote the catalytic activity for the PNRR. In situ characterization techniques (e.g., in situ TEM and in situ extended X-ray absorption fine structure) enable dynamic, in situ real-time monitoring of the corresponding behavior of the catalyst's microstructure in the external field, while providing its fine structural, chemical, and electronic information at the atomic scale, which is very helpful for our understanding of the bifunctional effects of MSIs.12,14,16 Hence, the accurate understanding and regulation of the bifunctional effect of the MSIs is also an important strategy to improve the catalytic activity.3. Synthesis
So far, many methods have been developed for the preparation of catalysts with MSIs. However, these methods are still not universal but have their characteristics. For example, when preparing single-atom catalysts by impregnation, weak van der Waals forces may form between the metal and the support, which is not conducive to the anchoring and dispersion of single-atom metals.12,16,17 Heat treatment methods avoid these problems, but higher temperatures can lead to metal agglomeration.18,59,60 Thus, the selection of appropriate preparation methods and optimization of conditions are crucial. In this section, we will briefly introduce the principles and specifics of some common preparation methods, such as thermal treatment,95 photoreduction,77,96,98 impregnation,146 wet chemical synthesis,91,104 and others99 (Fig. 2).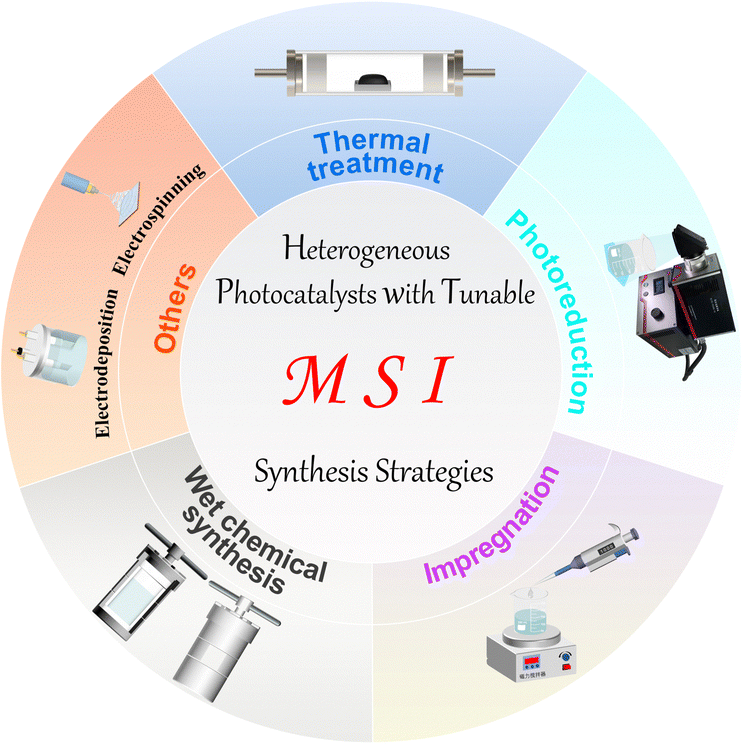 | ||
| Fig. 2 Summary diagram of the typical preparation methods for heterogeneous photocatalysts with MSIs. | ||
High-temperature reduction is one of the most classical methods in thermal treatment and is widely used for the synthesis of heterogeneous catalysts with MSIs. It is a method of reducing a high-valent compound to a low-valent compound or a simple substance using a reducing agent. In general, at low temperatures, the obtained metals have a large surface area and strong reaction activity. Increasing the reduction temperature causes the metal particles to polymerize, thus reducing their surface area and active site exposure, since the high-temperature results in a more stable structure.21 The heat treatment method is attracting much attention for its large treatment capacity and easy operation, yet, it often consumes a lot of energy. In contrast, photoreduction is becoming a popular method for the synthesis of metal/support photocatalysts due to its mild preparation conditions. The mechanism of photoreduction is based on the photoelectric effect, where a metal precursor and a semiconductor are first mixed in a solution and then excited by light irradiation, at which point the photogenerated electrons over the semiconductor will reduce the metal precursor to metal and simultaneously deposit on the semiconductor surface.8 However, photoreduction can only happen when the reduction ability of the photogenerated electrons from the semiconductor is higher than the reduction potential of the metal oxidant/metal.
Surprisingly, the impregnation method as an old and simple method will compensate for the lack of photoreduction. It is based on the principle of mixing the support with the soluble compound containing the active component in the liquid phase, and after sufficient contact, the desired catalyst is obtained by separation and drying.18 Nevertheless, catalysts synthesized by impregnation usually result in the production of nonuniform metal species owing to the weak interaction between the metal precursor and the support, which may hinder their use in MSIs. Wet chemical synthesis methods are also of interest, which usually require a series of sequential steps such as impregnation, co-precipitation or deposition precipitation, followed by calcination or reduction.17 The first step is to stabilize the metal atoms by modulating the metal–support interaction, while the second step often enhances the MSI effect by improving the electronic properties at the active site. Wet chemical methods provide a simpler and more feasible approach for the large-scale synthesis of MSI catalysts. While the most commonly used method is the solvothermal method, in which a mixed solution of a precursor and an organic solvent is kept in a high temperature and high-pressure resistant reactor to obtain nanomaterials by separation and drying after a certain period of reaction. The solvothermal method has been widely used in the synthesis of supported catalysts because of the advantages of universal application and good product crystallinity. Nevertheless, there are often multiple product types and potentially toxic substances that have to be considered. Besides, electrochemical deposition and electrostatic spinning have also been applied to prepare supported metal catalysts due to their ability to obtain nanomaterials of various grain sizes and prepare fibers with adjustable slenderness, respectively.16 In general, the various preparation methods have their own advantages and disadvantages, so how to synthesize MSI catalysts with sufficiently high metal coverage, high structural homogeneity and stability in a simpler, low-toxicity and inexpensive way remains an open question.
4. Support classification
In general, metals (e.g., Fe,95,104,124 Mo,91,115 Co,147 Ru,83,84,97,100 and Au110,127) often act as adsorption and activity centers in PNRR systems. However, more studies have shown that supports can not only act as active sites (e.g., oxygen vacancies,75,76,111 and nitrogen vacancies143,151), but also tune the electronic and geometric structures of metals, thus enhancing the catalytic activity for the PNRR.61 For example, metal oxides tend to easily induce geometrical effects in MSIs, while metal–organic frameworks are prone to electronic interactions with metals.9,16 In addition, different synthesis strategies can affect the support–metal interaction relationship. It is worth mentioning that in a photocatalytic system, the activation of N2 molecules is still essentially dependent on the photogenerated electrons generated by semiconductors (commonly supports).62,63 Therefore, it is essential to summarize the recent advances in different supports for the PNRR. So far, the catalysts with MSIs for PNRR systems are mainly based on the supports of metal oxides,75–78,82–89,91–100 bismuth oxyhalides,102–111 metal sulfides,113–115,117 metal–organic frameworks (MOFs),123–128 carbon nitrides138–153 and so on.4.1. Metal oxides
Metal oxide semiconductors are the most commonly used supports for the PNRR due to their property of being stable, easily available, and rich in oxygen species.64 Since the first report of N2 fixation over a TiO2 photocatalyst, a series of strategies were developed to enhance the performance of TiO2 on one hand. On the other hand, other kinds of metal oxides such as W18O49, In2O3, etc. were used in this reaction, and great progress was made (Table 2). In this part, the progress in the MSI photocatalysts with metal oxide-based supports will be discussed.| No. | Catalyst | Scavenger | Light source | NH3 yield (μmol g−1 h−1) | AQEa | Ref. |
|---|---|---|---|---|---|---|
| a The apparent quantum efficiency (AQE) is calculated. b nmol h−1 cm−1. Optical image of a representative as-printed TiO2 pillar array, featuring twenty-by-twenty TiO2 pillars in 10 × 10 mm area.N.A.: not addressed | ||||||
| 1 | Au/TiO2-OV | 10 vol% | 300 W Xenon lamp | 130.5 | 0.82% | 75 |
| Methanol | λ > 420 nm | 550 nm | ||||
| 2 | Au/P25-K+ | No | 300 W Xenon lamp | 428.7 | 0.34% | 76 |
| Full spectrum | 400 nm | |||||
| 3 | Au@TiO2 NT | No | 500 W Mercury lamp | 36.3 | 0.101% | 77 |
| Full spectrum | 350 nm | |||||
| 4 | Au/TiO2 | N.A. | 300 W Xenon lamp | 40.3b | N.A. | 78 |
| 5 | TR-1.0 | 20 vol% | 300 W Xenon lamp | 3.3 | N.A. | 82 |
| Ethanol | ||||||
| 6 | Ru-SAs/Def-TNs | No | 300 W Xenon lamp | 125.2 | N.A. | 83 |
| Full spectrum | ||||||
| 7 | Ru/TiO2-Ov | No | 300 W Xenon lamp | 18.9 | N.A. | 84 |
| Full spectrum | ||||||
| 8 | Fe-T-S | No | 300 W Xenon lamp | 32 | N.A. | 85 |
| Full spectrum | ||||||
| 9 | Ru/TiO2(P25) | No | 300 W Xenon lamp | 6.1 | N.A. | 86 |
| Full spectrum | ||||||
| 10 | Sb/TiO2 | 20 vol% | 300 W Xenon lamp | 20.8 | N.A. | 87 |
| Methanol | λ > 420 nm | |||||
| 11 | Cu/TiO2 | 5 vol% | 300 W Xenon lamp | 6780 | N.A. | 88 |
| Glycerin | ||||||
| 12 | 5-FTNFs | No | 300 W Xenon lamp | 64.2 | N.A. | 89 |
| Full spectrum | ||||||
| 13 | MWO | 1 mM | 300 W Xenon lamp | 195.5 | 0.33% | 91 |
| Na2SO3 | Full spectrum | 400 nm | ||||
| 14 | P-Fe/W18O49 | 1 mM | Simulated solar light | 187.6 | 0.54% | 92 |
| Na2SO3 | 300 nm | |||||
| 15 | Au-MWO | 20 vol% | 300 W Xenon lamp | 399.2 | 0.611% | 93 |
| Methanol | Full spectrum | 540 nm | ||||
| 16 | Ru-W18O49 | No | 300 W Xenon lamp | 44.3 | 0.33% | 94 |
| Visible light | 365 nm | |||||
| 17 | Fe-SA/WO2.72−x | No | 300 W Xenon lamp | 186.5 | 0.41% | 95 |
| Full spectrum | 350 nm | |||||
| 18 | Au@In2O3 HS | No | 500 W Mercury lamp | 118.4 | N.A. | 96 |
| Full spectrum | ||||||
| 19 | Ru-In2O3 HPNs | 8 vol% | N.A. | 44.5 | N.A. | 97 |
| Methanol | ||||||
| 20 | La/MoO3−x | No | 300 W Xenon lamp | 209 | N.A. | 98 |
| λ > 420 nm | ||||||
| 21 | RuO+Co/CoO | No | 300 W Xenon lamp | 306 | N.A. | 99 |
| λ > 400 nm | ||||||
| 22 | Ru1/2DAF | No | 300 W Xenon lamp | 213 | 0.34% | 100 |
| Full spectrum | 385 nm | |||||
Localized surface plasmon resonance (LSPR) refers to the collective oscillation of free charges confined in a nanocrystal induced by photoexcitation.68,69 Some metal nanocrystals, especially Au and Ag, have plasmonic properties in the visible and near-infrared regions.70–72 In the PNRR system, surface plasmons in metal nanostructures can activate the NN bond by two mechanisms. On the one hand, the metal plasma is excited by light to generate hot electrons, which are subsequently injected into the antibonding orbitals of N2. On the other hand, the carriers are excited by the hybridized metal–adsorbate interface to convert N2 directly into vibrational or electronically excited states.73,74 By loading plasma Au on TiO2, it not only broadens the light absorption range of the catalyst, but also enhances the electronic effect of MSI, thus improving the catalytic activity for the PNRR. Yu et al. reported a TiO2 nanosheet catalyst with abundant oxygen vacancies to support Au nanospheres (Au/TiO2-OV).75 By XPS analysis of Au 4f7/2 (Fig. 3a), the Au/TiO2-OV sample showed a significant negative shift concerning the bulk Au, which indicates the transfer of electrons from the TiO2-OV nanosheets to the Au nanospheres due to the equilibrium of the EF. This result is also evidenced by the positive shift of Ti 2p3/2 (Fig. 3b). Such a MSI-induced electron transfer process decreased the local electron density at oxygen vacancies (OVs) and therefore weakened their chemisorption capacity for N2. Nevertheless, further studies confirm that the activation of N2 relies on hot electrons from Au nanospheres under visible light excitation. Thus, Au nanospheres have the highest nitrogen fixation yield when their loading is around 2.53 wt%. Finally, the mechanism of nitrogen fixation induced by the bifunctional effect of MSIs is proposed as follows (Fig. 3c): OVs act as N2 adsorption and activation centers, which collect hot electrons generated by plasma Au nanospheres and finally N2 is reduced at the OVs through the electronic “acceptance–donation” process. A similar electron transfer from TiO2 to Au and the presence of OVs facilitating the chemisorption of N2 were found on Au/P25-K+, Au@TiO2 NT, and Au/TiO2.76–78 Surprisingly, alkali metal cations (e.g. Li+, Na+, and K+) are found to promote the activity for the PNRR, and the nitrogen fixation rate of Au/P25-K+ reaches 428.7 μmol g−1 h−1 under the conditions of 1 mol L−1 potassium ion solution and full spectrum, which is 7 times higher than the one without K+.76 The promoting effect of K+ can be attributed to both electronic and electric field effects (Fig. 3d): (1) K+ can change the electronic structure at the active sites of OVs, enhancing its ability to adsorb N2, and (2) K+ also provides a strong local electric field, which weakens the NN bond and stabilizes key intermediates. However, this work requires further characterization analysis to prove the above points. In contrast to previous studies, Jiang et al. prepared Au nanoparticle-loaded TiO2 catalysts (Au/TiO2) inspired by the concept of forest structure design and the principle of vertical 3D printing (Fig. 4a).78 MSI induced by this special structure has enhanced the close contact between Au nanoparticles and TiO2 (Fig. 4b–d), promoting the charge transfer as well as overcoming the easy agglomeration of Au nanoparticles after the reaction, thus significantly improving the stability of the catalyst. Moreover, researchers also discovered a nitrogen fixation mechanism similar to that induced by the bifunctional effect of MSIs on Au/TiO2-OV.75 Thus, compared with planar TiO2 films decorated with Au nanoparticles, the nitrogen fixation rate of Au/TiO2 is 15 times higher.
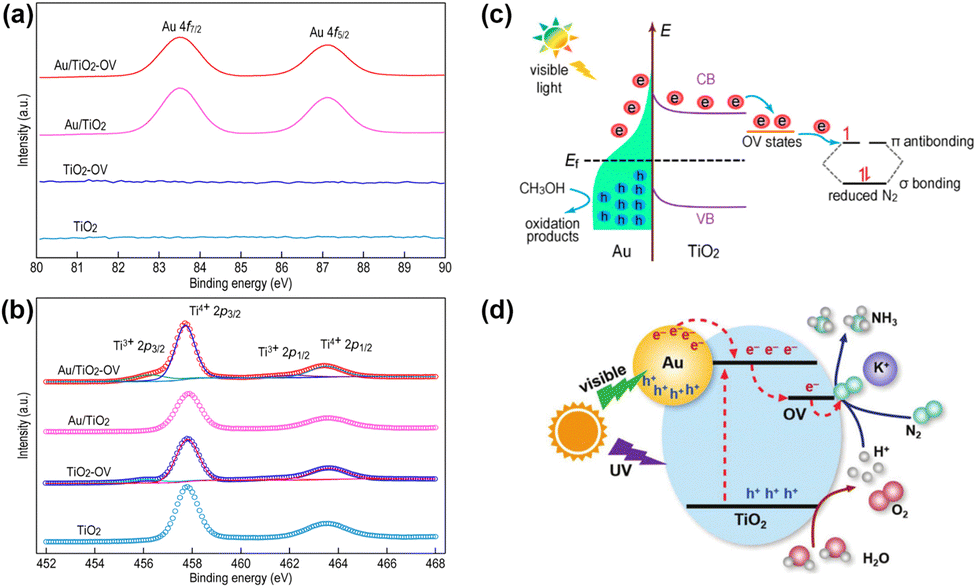 | ||
| Fig. 3 (a) and (b) Au 4f and Ti 2p High-resolution XPS spectra of Au/TiO2-OV. (c) Schematic illustration of the PNRR process involving plasma hot electrons in Au/TiO2-OV. Reprinted with permission from ref. 75. Copyright 2018 American Chemical Society. (d) Schematic diagram of the Au/P25-K+ PNRR mechanism. Reprinted with permission from ref. 76. Copyright the Royal Society of Chemistry 2019. | ||
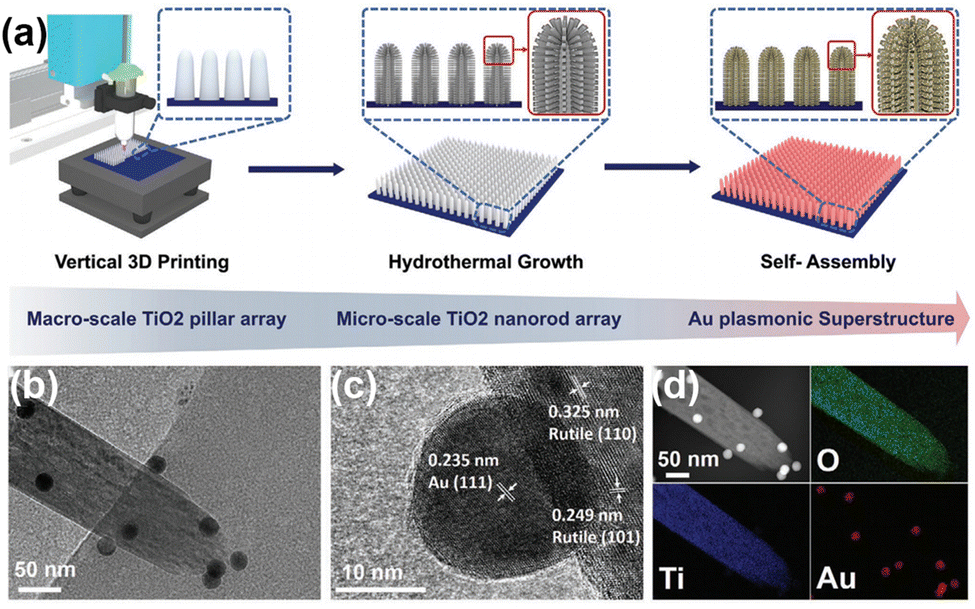 | ||
| Fig. 4 (a) Schematic demonstration of the fabrication of a forest-inspired hierarchical Au plasmonic superstructure. The vertically aligned TiO2 pillar arrays are first printed on a planar substrate (as tree trunks) (left). A layer of dense TiO2 nanorod arrays is then hydrothermally grown on the surface of the printed pillars (as branches) (middle). The forest-like plasmonic superstructure is finally constructed by self-assembling Au NPs (as leaves) on the hierarchical TiO2 structure (right). (b) TEM image of a TiO2 nanorod decorated with Au NPs. (c) High-resolution TEM images of the Au NPs and the TiO2 nanorod. The lattice spacing of 0.235 nm is corresponding to the (111) crystalline planes of Au. The lattice spacings of 0.249 and 0.324 nm are corresponding to the (101) and (110) crystalline planes of TiO2, respectively. (d) Elemental maps of O, Ti, and Au of the TiO2 nanorod decorated with Au NPs. Reprinted with permission from ref. 78. Copyright 2021 Wiley-VCH GmbH. | ||
Single-atom catalysts (SACs) are widely used in PNRR systems due to their high atomic utilization, well-defined local coordination environment, tunable electronic structure, and the advantages of both heterogeneous and homogeneous catalyses.79–81 In general, a unique coordination structure is formed between the single atom (SA) and the support, which will lead to the transfer of electrons, thus optimizing the active sites and enhancing the catalytic activity for the PNRR. Sun et al. found that compared with the electron densities of absorbed Ru4d and O2p over the perfect TiO2 (001) surface, the electron density of the TiO2 (001) surface with oxygen vacancies is lower.82 This charge transfer induced by the MSI electron effect enhances the Ru–O interaction and makes it more stable. In addition, further studies suggested that the Ru single atom stabilized on OVs might be converted into active centers, thereby inhibiting the hydrogen precipitation reaction and thus improving the selectivity of the catalyst. Electron transfer from the Ru single atom to TiO2 can be observed on Ru-SAs/Def-TNs83 and Ru/TiO2-Ov.84 Furthermore, a systematic study of photoexcited electron dynamics reveals a successive three-part transfer route of photogenerated electrons (Fig. 5a and b).83
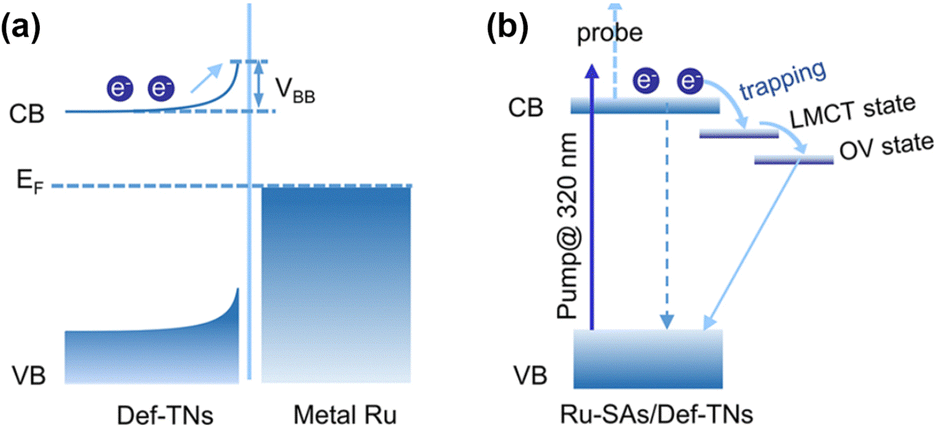 | ||
| Fig. 5 Mechanisms underlying the photoexcited electron dynamics involved in (a) Ru-NPs/Def-TNs and (b) Ru-SAs/Def-TNs. Reprinted with permission from ref. 83. Copyright 2020 American Chemical Society. | ||
Benefiting from the coordinated mode of Ru-SA covalent bonding in Ru-SAs/Def-TNs, there is no Schottky barrier in the catalyst system, which turns Ru into a powerful electron trap and thus promoted efficient electron transfer. Their work elucidates the key role of strong coordination of single atoms to support in facilitating photogenerated charge separation. Meng et al. discovered that a significant EMSI between Ru single atoms and TiO2 led to an unbalanced local charge distribution, which greatly reduced the energy barrier of the rate-limiting step and affected the local electron density of the adsorbed N2, promoting the activation of N2.84 Interestingly, a bifunctional effect of MSI is revealed on Fe single-atom doped TiO2–SiO2.85 First, the Fe sites are oxidized to Fe(IV) species by trapping holes, thereby promoting the separation of photogenerated carriers. Subsequently, the high-valent Fe(IV) species are responsible for the oxidation of water and promoted the N2 hydrogenation process on the adjacent oxygen vacancies by lowering the reaction energy barrier.
Loading Ru (Ru/TiO2(P25)),86 Sb (Sb/TiO2),87 or Cu (Cu/TiO2)88 metal on TiO2 also induced the electronic effect of MSIs and promoted the separation of photogenerated carriers, thus improving the catalytic activity for the PNRR. OVs have been shown to act as active sites for the PNRR, but it is worthwhile to ponder how to stabilize OVs and improve the local electronic structure of OVs to change the hydrogenation mechanism and, in turn, promote the activity for the PNRR. Xiong et al. elucidated how the Fe dopant affects the PNRR process through the electronic effect of MSIs on Fe-doped TiO2 (5-FTNFs).89 The local electron density of Fe decreases due to the formation of Fe–O bonds, while the Fe-induced formation of OVs increases the local electron density of Ti. This unbalanced charge distribution affected the electron density of the adsorbed N2 promoting its activation. In addition, since Fe atoms had similar radii to Ti atoms, the successful replacement of Ti(IV) by Fe(III) would stabilize the unsaturated coordination of Ti atoms through charge compensation, thus stabilizing the adjacent OVs. As found using in situ near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) plots (Fig. 6a and b), two peaks appeared at 395.3 and 397–399 eV corresponding to N and NHy (y = 1, 2), respectively. Hence, the observed N and NHy species during the reaction confirm the reduction of N2 by 5-FTNFs using a distal association pathway, unlike the alternating association on TiO2 (0-FTNFs-Ar) without the introduction of Fe dopants. Finally, theoretical calculations demonstrated (Fig. 6c) that this change in the hydrogenation mechanism caused by the MSI electron effect lowers the reaction energy barrier for the rate-limiting step, thus making the N2 hydrogenation pathway easier to occur.
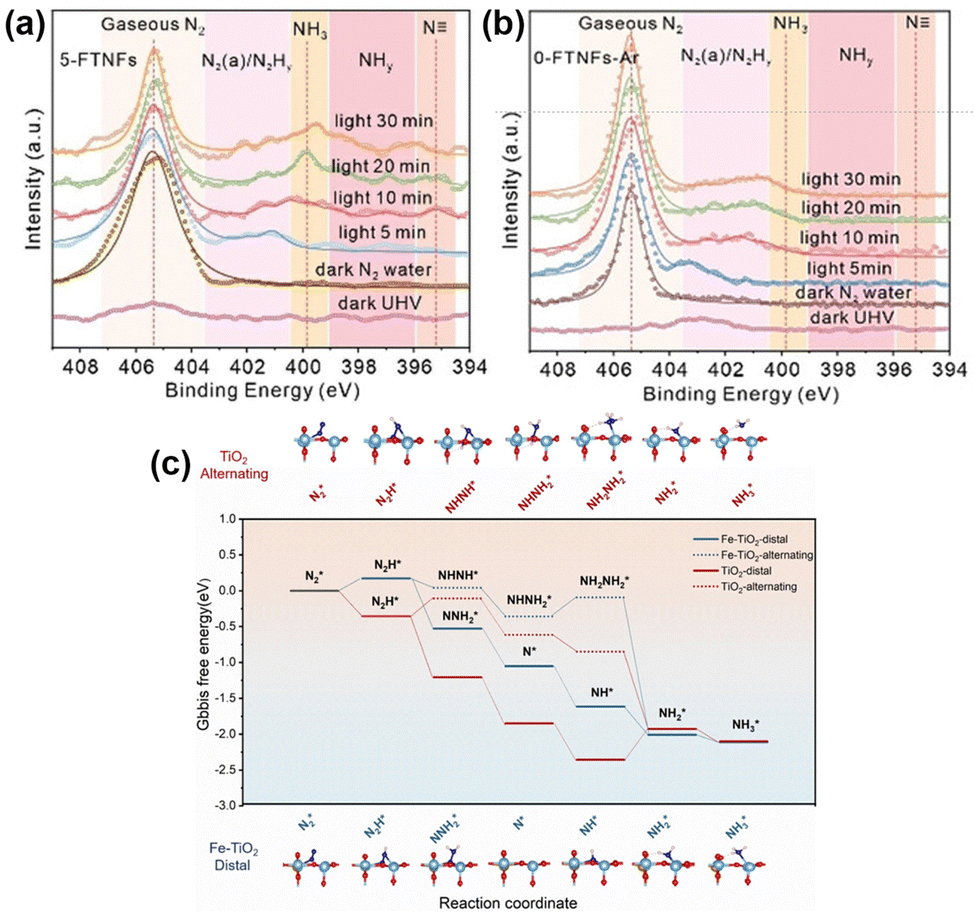 | ||
| Fig. 6 (a) and (b) In situ NAP-XPS spectra of 5-FTNFs and 0-FTNFs-Ar collected under the conditions of ultrahigh vacuum under dark, N2 atmosphere under dark, and N2 atmosphere under light irradiation, respectively. (c) Gibbs free energy spectra of PNRR processes under 0-FTNFs-Ar (red) and 5-FTNFs (blue) distal and alternate association pathways, and the optimized geometries of the corresponding intermediates adsorbed on the substrate. Reprinted with permission from ref. 89. Copyright 2021 Wiley-VCH GmbH. | ||
In contrast to previous reports, Fe single-atom doped WO2.72−x catalysts (Fe-SA/WO2.72−x) have been fabricated in our group.95 High-angle annular dark field scanning transmission (HAADF-STEM) and XPS combined with EXAFS (Fig. 7a–c) illustrate that Fe presents as Fe–O coordination on the surface of WO2.72−x rather than Fe particles or Fe clusters, which proves the successful loading of Fe single atoms. In general, OVs are susceptible and unstable as active sites due to the generation of O2 in the PNRR process commonly, which will lead to a decrease in catalyst stability. Nevertheless, based on experiments and DFT (Fig. 7d–f), we demonstrate that Fe is the adsorption and activation centre of N2, which is anchored to the support in the form of covalent bonding to O. This strong coordination induced by MSIs effectively enhances the stability of the catalyst. Also, the electronic interaction between WO2.72−x and Fe single atoms leads to the transfer of the d-band centre of the catalyst to the EF, which enhances N2 adsorption. Additionally, due to the MSI-induced electronic effects, electrons were enriched at the Fe single atom and subsequently transferred to the antibonding orbital of N2, ultimately activating the NN bond. Furthermore, benefiting from the strong interaction between Fe and WO2.72−x, the photocatalytic charge generation, separation, and transport are significantly enhanced, thus improving the photocatalytic nitrogen fixation efficiency.
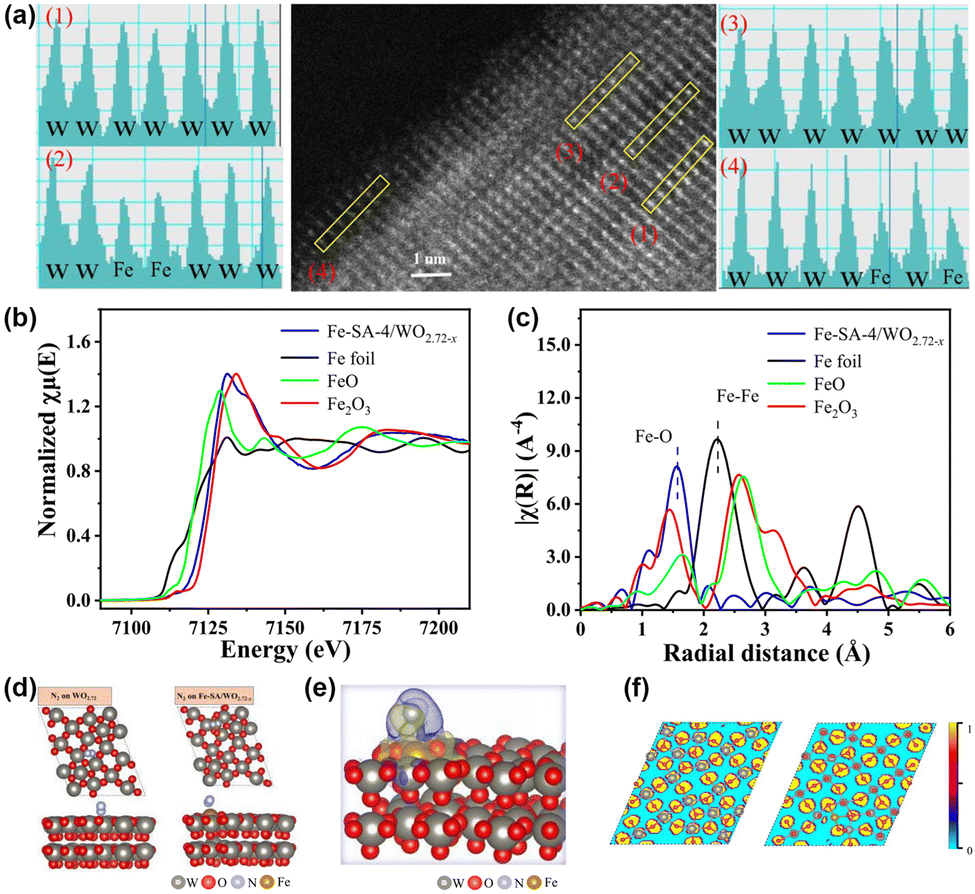 | ||
| Fig. 7 (a) HAADF-STEM and the element intensity profiles of Fe-SA-4/WO2.72−x. (b) and (c) Fe K-edge XANES spectra and Fourier-transform infrared spectra of Fe-SA-4/WO2.72−x. (d) Optimized structures of WO2.72 and Fe-SA/WO2.72−x with N2. (e) Differential charge densities of Fe-SA/WO2.72−x with adsorbed N2. (f) ELF of WO2.72 and Fe-SA/WO2.72−x. Reprinted with permission from ref. 95. Copyright 2022 American Chemical Society. | ||
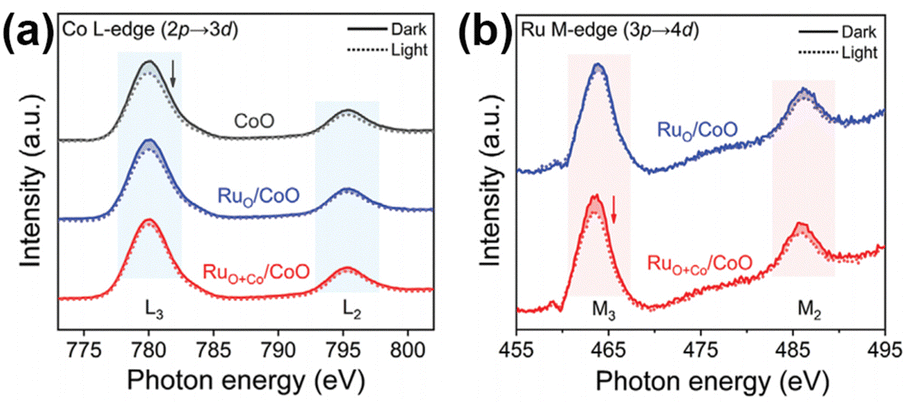 | ||
| Fig. 8 (a) In situ sXAS spectra of Co L-edge for CoO, RuO/CoO, and RuO+Co/CoO. (b) In situ sXAS spectra of Ru M-edge for RuO/CoO and RuO+Co/CoO. Reprinted with permission from ref. 99. Copyright 2022 Wiley-VCH GmbH. | ||
4.2. Bismuth oxyhalides
Bismuth oxyhalides (BiOX) (X = Cl, Br) have attracted interest in the PNRR because of their unique layered structure, large number of surface defects, and adjustable structure (Table 3). As important supports with MSIs, the rich surface defects of BiOX inducing a tunable EMSI between metals in the form of atoms, clusters, and nanoparticles have been demonstrated.| No. | Catalyst | Scavenger | Light source | NH3 yield (μmol g−1 h−1) | AQEa | Ref. |
|---|---|---|---|---|---|---|
| a The apparent quantum efficiency (AQE) is calculated. b The ammonia synthesis was tested at 270 °C. c μmol L−1 h−1: the thin film catalyst is not in direct contact with the water surface and provides a proton source for N2 through water vapor.N.A.: not addressed. | ||||||
| 1 | BiOCl NSs-Fe-5% | No | 300 W Xenon lamp | 1022 | 1.8% | 102 |
| Full spectrum | 420 nm | |||||
| 2 | PtO/Pt4+-BiOCl | 2 vol% | 300 W Xenon lamp | 7512 | N.A. | 103 |
| Methanol | ||||||
| 3 | Fe-BiOBr | No | 300 W Xenon lamp | 382.7 | N.A. | 104 |
| λ > 420 nm | ||||||
| 4 | BiOBr-Fe-S | No | 300 W Xenon lamp | 46.1 | 0.60% | 105 |
| λ > 400 nm | 365 nm | |||||
| 5 | Mo-Bi5O7Br | No | 300 W Xenon lamp | 122.9 | N.A. | 106 |
| λ > 400 nm | ||||||
| 6 | Pd-EG-BiOBr | 2 vol% | 300 W Xenon lamp | 249.3 | N.A. | 107 |
| Methanol | Full spectrum | |||||
| 7 | Cu-Bi24O31Br10 | No | 300 W Xenon lamp | 291.1 | 3.14% | 108 |
| Full spectrum | 400 nm | |||||
| 8 | Bi-BiOBr | 0.025 M | λ > 420 nm | 78.6 | 0.23% | 109 |
| Na2SO3 | 420 nm | |||||
| 9 | BiOBr/Au-CP | 10 vol% | 300 W Xenon lamp | 80.5 | N.A. | 110 |
| Methanol | λ > 400 nm | |||||
| 10 | CL@BiOBr-OV/Au | No | 300 W Xenon lamp | 2640 | N.A. | 111 |
| λ > 400 nm | ||||||
| 11 | Mo1−xWxS2-2 | 1 mM | 300 W Xenon lamp | 111 | N.A. | 113 |
| Na2SO3 | λ > 400 nm | |||||
| 12 | Fe-MoS2 | No | 70 W UV cut-off tungsten lamp | 2105b | N.A. | 114 |
| 13 | Mn-MoS2−x-4 | No | 300 W Xenon lamp | 213.2 | 0.26% | 115 |
| Full spectrum | 350 nm | |||||
| 14 | Ag/PM-CdS(e) | No | 350 W Xenon lamp | 20.2 | N.A. | 117 |
| λ > 420 nm | ||||||
| 15 | Ru@Mo-MOF-tri | No | 300 W Xenon lamp | 641 | N.A. | 123 |
| λ > 400 nm | ||||||
| 16 | Al-PMOF(Fe) | 20 vol% | λ > 420 nm | 7.5 | N.A. | 124 |
| Methanol | ||||||
| 17 | Cu-MIL-101(Fe) | N.A. | 300 W Xenon lamp | 315.2 | N.A. | 125 |
| Full spectrum | ||||||
| 18 | MIL-88A(Fe/Mo)/Fe | No | 350 W Xenon lamp | 20.7c | N.A. | 126 |
| Full spectrum | ||||||
| 19 | Au@UiO-66 | No | 300 W Xenon lamp | 360 | 1.54% | 127 |
| λ > 400 nm | 520 nm | |||||
| 20 | Ag@MIL-101(Cr) | No | 300 W Xenon lamp | 138.8 | N.A. | 128 |
| Full spectrum | ||||||
Metal–support interactions can also affect the performance of single-atom catalysts. Zhang et al. reported a single-atom Pd-modified BiOBr photocatalyst (Pd-EG-BiOBr) by using ethylene glycol (EG) as a molecular bridge to link Pd and BiOBr, which effectively avoids the agglomeration of Pd and simultaneously improves the catalytic activity for the PNRR.107 EG not only anchors the Pd single atom, but also enhances the N2 adsorption capacity of the Pd atom by modulating its electron density. The surface polarization engineering of nanomaterials can influence the electronic structure and the coordination mode of N2, favoring the activation of NN bonds.108 Studies showed that there is a significant potential difference at the Cu–Bi sites on the Cu-Bi24O31Br10 photocatalyst, which indicates the formation of local polarization, leading to a large local electric field. This local polarization facilitates the transfer of photogenerated electrons from Bi atoms to Cu atoms, promoting charge separation. In addition, the surface polarized Cu–Bi sites have increased the non-covalent bonding interactions between the catalyst surface and N2, then further weakened the covalent bond order in NN, enabling continuous nitrogen activation. Their study shows the special effect of MSIs in the PNRR from the perspective of surface polarization engineering.
Schottky junctions at the metal–semiconductor interface can establish unidirectional channels for photogenerated electrons, leading to efficient separation of photogenerated carriers, as demonstrated for Bi-loaded109 and Au-loaded110 BiOBr. In addition, the Bi nanoparticles with low hydrogen precipitation activity acted as a center for N2 adsorption and activation, which enhances the selectivity of the catalyst.109 Thus, the PNRR activity of Bi-BiOBr is 65 times higher than that of BiOBr under the conditions of Na2SO3 as a sacrificial agent and visible light. Sun et al. deposited BiOBr-OV/Au on a hydrophobic carbonization loofah (CL) sponge (CL@BiOBr-OV/Au) and constructed a gas–liquid–solid three-phase contact surface to enhance the diffusion rate of N2 and its ability to bind to protons.111 There are significant lattice distortions between Au nanoparticles and BiOBr-OV, confirming their close association, which facilitates the efficient transfer of electrons. Furthermore, based on XPS, electrons might be transferred from Au nanoparticles to BiOBr-OV, indicating the existence of electronic coupling between them. Finally, the mechanism of nitrogen fixation induced by the bifunctional effect of MSIs is as follows: OVs serve as the adsorption and activation centers of N2, capturing hot electrons generated from Au nanoparticles and photogenerated electrons from BiOBr-OV, which in turn promotes efficient activation of N2.
4.3. Metal sulfides
Transition metal sulfides are typical layered materials that also arouse great attention as PNRR photocatalysts (Table 3) due to their good visible light absorption. In which, the transition metal atoms have incompletely filled outer d-orbitals allowing the formation of compounds with a 3d valence electron layer structure.112 The ultra-thin layered structure and edge effect significantly enhance the MSI effect of metal sulfides.In our group, Mn as a dopant in MoS2 nanosheets has been found to change the electronic structure, and finally altering the inert MoS2 edge sites into active ones.115 At first, through theoretical calculations, the active sites of MoS2 are found to be at the Mo edge rather than the S edge. Moreover, the introduction of Mn could transform the inert S-edge sites into active Mo and Mn sites by forming S vacancies. Interestingly, the S-edge sites of MoS2 are more inclined to form single S vacancies and double S vacancies (Mn-MoS2−x-Sv and Mn-MoS2−x-2Sv), and the mechanism of N2 adsorption and activation on these two vacancies is different. Specifically, Mn doping forms S vacancies at the S edge, and N2 molecules preferentially adsorb on the Mn sites or Mn–Mo sites with lower adsorption energy (Fig. 9a–c). The creation of S vacancies not only reduces the adsorption energy of N2 molecules but also facilitates the activation of N2 by extending the NN bond. The hydrogenation of Mn-MoS2−x-Sv and Mn-MoS2−x-2Sv pathways are distinguished as shown in Fig. 9d. The difference between the two might be related to the exposed Mo site near the Mn site over Mn-MoS2−x-2Sv. Using the partial density of states calculations (Fig. 9e–g), we have found that the essence of the interaction between Mn-MoS2−x and N2 molecules follows the electron “acceptance–donation” rule. Specifically, the Mn-MoS2−x catalyst is inclined to accept unpaired electrons from N2 molecules and infused electrons into the NN antibonding orbital, thus activating N2 molecules. Secondly, to verify the theoretical concept, we prepare Mn-doped MoS2−x photocatalysts (Mn-MoS2−x-4) using a hydrothermal method. Based on the HAADF-STEM images (Fig. 9h–m), the successful doping of Mn atoms is demonstrated, and more edge sites are also generated by forming surface defects after Mn doping. Electron Paramagnetic Resonance (EPR) spectroscopy shows a significant enhancement of the signal intensity with the increase of Mn doping, which proves the results obtained for Mn doping to generate S vacancies using DFT. Furthermore, based on high-resolution XPS spectroscopy, electrons in the Mn-MoS2−x-4 material are transferred from Mn to Mo and S centers due to the electronic effect of MSIs, which favors the catalytic activity for the PNRR. As a result, the rate of NH3 production by the Mn-MoS2−x-4 photocatalyst reaches 213.2 μmol g−1 h−1 without any sacrificial agent, which is superior to those of most of the reported MoS2-based photocatalysts.
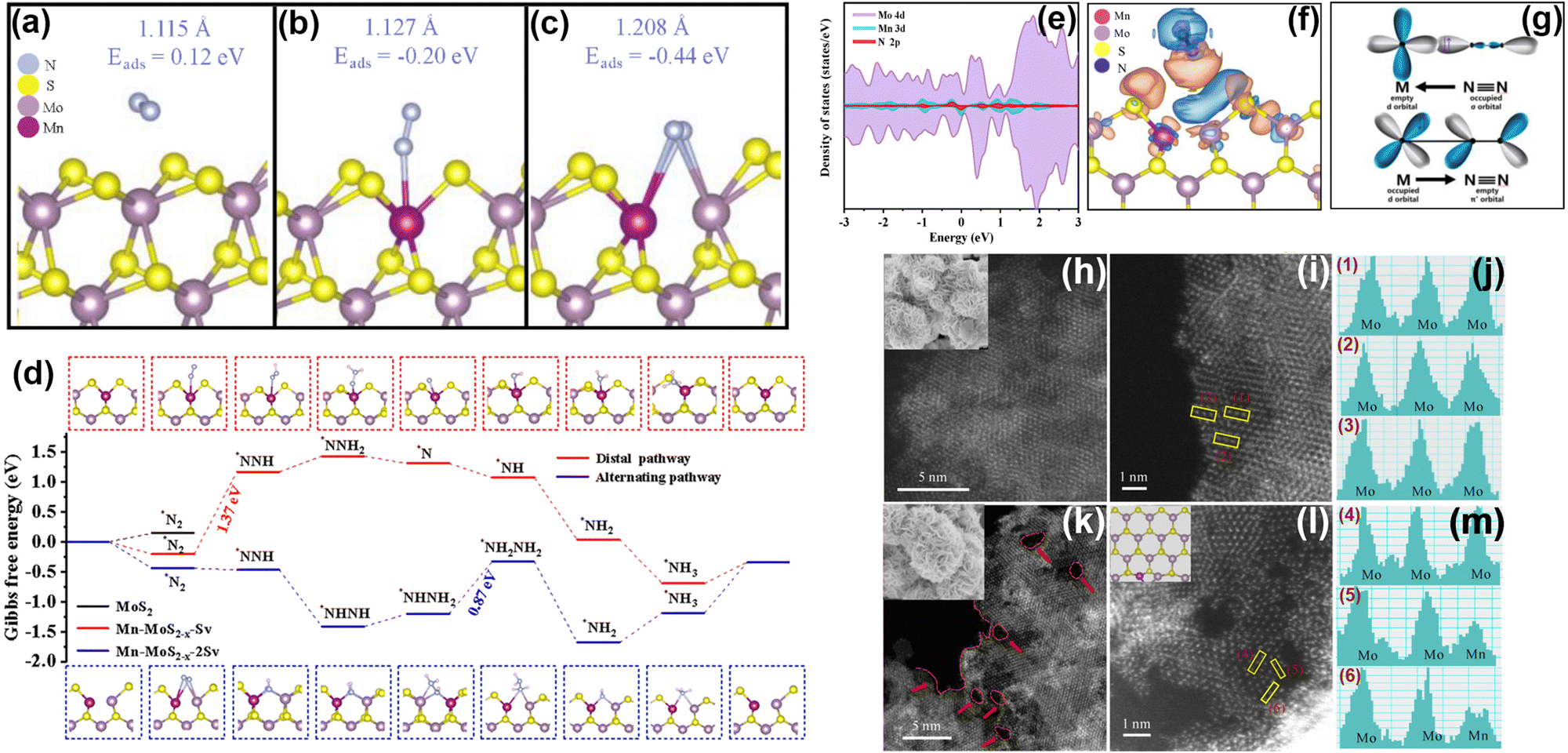 | ||
| Fig. 9 (a)–(c) DFT simulation of S edge sites interacting with a N2 molecule on MoS2, Mn-MoS2−x-Sv and Mn-MoS2−x-2 Sv. (d) Gibbs free energy diagram for N2 reduction over the basal plane and S edges of Mo-doped MoS2−x with single and continuous S vacancies. (e)–(g) PDOS calculations using the PBE method of (e) Mn-MoS2−x with adsorbed N2; (f) differential charge densities for Mn-MoS2−x with adsorbed N2 (orange is the electron accumulation area and blue is the electron dissipation area); and (g) simplified schematic of N2 bonding to transition metals. (h)–(m) HAADF-STEM and the corresponding high-resolution images as well as the element intensity profiles of (h), (j) and (m) pure MoS2; (i), (k) and (l) Mn-doped MoS2−x. Reprinted with permission from ref. 115. Copyright 2022 Elsevier B.V. | ||
4.4. MOFs
MOFs are crystalline materials constructed from metal ions and organic ligands, and their unique structure and properties make them an important class of porous materials.118–121 MOFs have the advantages of a large specific surface area, controllable structure, easy modification and high exposure of active sites, and have received much attention in PNRR systems recently (Table 3). Nevertheless, most of the MOFs themselves have insufficient visible light absorption ability. Moreover, problems such as poor electrical conductivity, insufficient stability, and rapid combination of photogenerated carriers limit the application of MOFs in PNRR systems.122 To address these problems, the MOF substrates are modified by metal doping and plasma loading to enhance their electronic effects as well as to optimize the electronic structure based on the EMSI, thus promoting the adsorption and activation of N2.Bu et al. prepared a Ru single-atom modified Mo-MOF with a [Mo8O26(im)2]4− structure (Ru@Mo-MOF-tri), thus forming Ruδ+–O3–Mo3 single-site heterojunctions.123 Based on the X-ray absorption fine structures (XAFs), the peak intensity of Ru in Ru@Mo-MOF-tri is significantly higher than that of Ru foil, which indicates the transfer of electrons from Ru monoatoms to O atoms. Such MSI electronic effects could simultaneously tune the energy band structure of the catalyst and extend the light absorption range to 700 nm. Further studies have showed that the increase in nitrogen fixation activity is attributed to an upward shift in the EF of the catalyst, which would lead to elevated electron energy and thus lower the reaction energy barrier of the rate-limiting step *NNH. Significantly enhanced nitrogen fixation activity is also confirmed on Fe single-atom modified porphyrin-based metal–organic frameworks (Al-PMOF(Fe))124 and Cu-doped MIL-101(Fe) (Cu-MIL-101(Fe)).125 Interestingly, on Mo-doped MIL-88A/Fe, Mo dopants not only leads to a long-range ordering distortion of the catalyst lattice to form a “MoFe cofactor”, resulting in catalyst electronic structure modification, but also induces a bifunctional effect of MSI.126 “MoFe cofactor” promotes photogenerated carrier transport and separation via a dual electron transport pathway. Among them, the Mo6+/Mo4+ redox pair acted as a carrier of photogenerated electrons, and Mo6+ is reduced to Mo4+ by capturing electrons. The generated Mo4+ intermediate easily reacts with N2 adsorbed on the catalyst surface to form NH3 due to the empty d-orbitals, and at the same time being oxidized back to its original stable state (Mo6+) by losing electrons. The Fe3+/Fe2+ redox pair functions similarly. The doping of Mo along with Fe can act as an adsorption and activation center for N2, resulting in a 9.6-fold increase in catalytic activity compared to the original MIL-88A/Fe.
Normally, it is difficult for Au and Ag to adsorb N2 as an active center owing to the absence of unfilled d-orbitals. Nevertheless, UiO-66 encapsulated gold nanoparticles (NPs) (Au@UiO-66)127 and Ag NPs in the cages of MIL-101(Cr) (Ag@MIL-101(Cr))128 in which Au and Ag acted as active sites to utilize the localized surface plasmon resonance (LSPR) enhancing the PNRR. It was demonstrated that the inner cavities of MOFs in both catalysts could disperse and stabilize the metal NPs as well as limit their size (Fig. 10c and d). Intriguingly, different MSI effects are observed on the two catalysts. Among them, Au@UiO-66 has no Schottky barriers, neither charge transfer, whose hot electrons generated under light could be directly transferred to the adsorbed N2 molecules. Furthermore, the electron transfer, energy transfer, and local electric field polarization effects induced by LSPR could also lower the reaction energy barriers of the intermediates, thus promoting the activation of N2 (Fig. 10a). Moreover, N2 could be directly diffused in high flux through the gas-permeable Au@UiO-66 membranes supported by porous PTFE films (Fig. 10b). On the other hand, there is a significant electron transfer process between MIL-101 (Cr) and Ag NPs and by forming Schottky junctions for facilitating photogenerated electron transfer from ligands to Ag NPs. Such a ligand–metal charge transfer (LMCT) mechanism could synergize with the LSPR of Ag NPs, resulting in a 12-fold increase in the nitrogen fixation rate of Ag@MIL-101(Cr) over that of pure MIL-101(Cr).
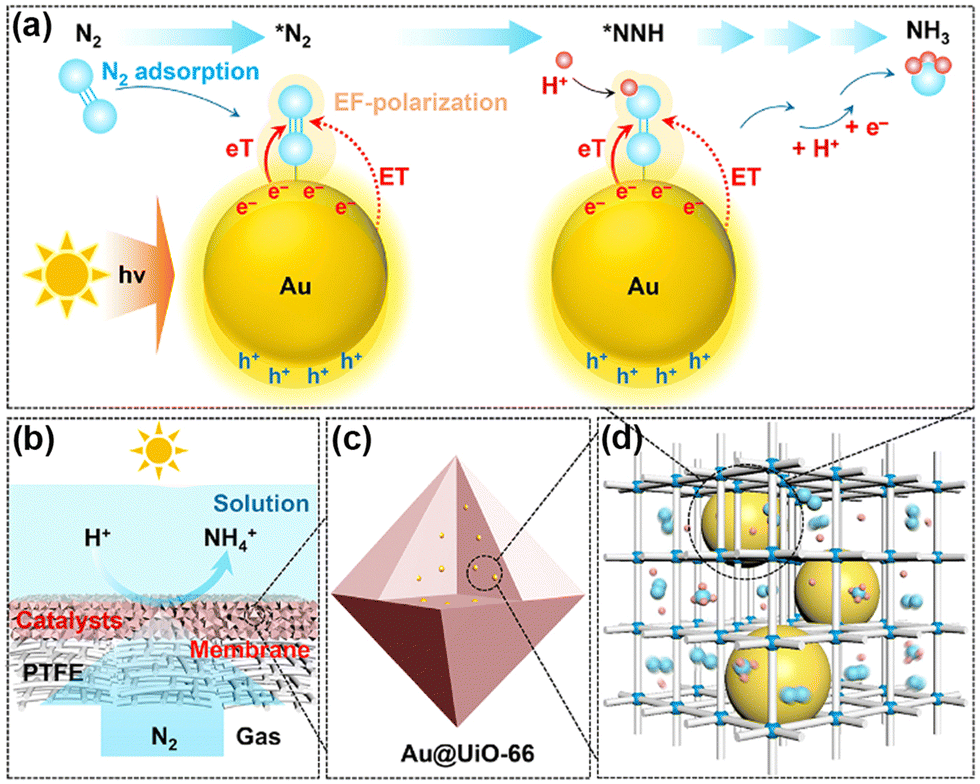 | ||
| Fig. 10 Schematic illustration for direct PNRR on Au@UiO-66. (a) Illustration of the direct PNRR on AuNPs. (b) Schematic of the GMS reaction interface design. (c) and (d) Schematic diagram showing the dispersion of plasmonic AuNPs within the cavities of the MOF matrix. Reprinted with permission from ref. 127. Copyright 2021 American Chemical Society. | ||
4.5. Carbon nitrides
Since 2006, graphitic phase carbon nitride (g-C3N4) has been applied in heterogeneous catalytic systems, and Wang et al. first reported its use as a semiconductor for photocatalytic hydrogen precipitation reactions in 2009.129,130 It has received a lot of attention from researchers in recent years and been broadly developed and applied to PNRR processes (Table 4). Compared with TiO2, g-C3N4 has the advantages of a suitable band gap (2.7–2.8 eV) and visible light excitation (450–460 nm).131–133 Moreover, g-C3N4 can be easily fabricated and is stable under environmental conditions. It is generally accepted that g-C3N4 has two basic construction units, the s-triazine (C3N3) and the homotriazine (C6N7) ring structures, respectively. The structure of g-C3N4 based on the homotriazine unit is highly recognized because it is the most stable phase under ambient conditions.134 However, g-C3N4 also suffers from the disadvantages of the rapid combination of photogenerated carriers, low electrical conductivity, and insufficient light absorption above 460 nm.135–137 In response to the above problems, researchers have also done a lot of work to modify g-C3N4. This subsection will summarize the recent progress of g-C3N4 in the PNRR from the perspective of MSIs.| No. | Catalyst | Scavenger | Light source | NH3 yield (μmol g−1 h−1) | AQEa | Ref. |
|---|---|---|---|---|---|---|
| a The apparent quantum efficiency (AQE) is calculated.N.A.: not addressed. | ||||||
| 1 | Cu-CN | 20 vol% | 300 W Xenon lamp | 186 | N.A. | 138 |
| Ethanol | 780 nm > λ > 420 nm | |||||
| 2 | Mo-PCN | No | 300 W Xenon lamp | 50.9 | N.A. | 139 |
| Full spectrum | ||||||
| 3 | Co@g-C3N4 | 4 wt% | 500 W Xenon lamp | 2510 | N.A. | 140 |
| Methanol | λ > 400 nm | |||||
| 4 | Ru-CCN-0.05 | No | 300 W Xenon lamp | 132.9 | N.A. | 141 |
| 5 | mCNN | Ethylene glycol | 300 W Xenon lamp | 3420 | N.A. | 142 |
| λ > 400 nm | ||||||
| 6 | KCCN-NVs | Methanol | 300 W Xenon lamp | 3510 | 2.05% | 143 |
| λ > 420 nm | 430 nm | |||||
| 7 | CNNR | 20 vol% | 300 W Xenon lamp | 23500 | N.A. | 144 |
| Methanol | Full spectrum | |||||
| 8 | Ru-K/B-g-C3N4 | 6.25 vol% | 300 W Xenon lamp | 85 | N.A. | 145 |
| Methanol | ||||||
| 9 | RuCN | No | 300 W Xenon lamp | 10.3 | N.A. | 146 |
| λ > 420 nm | ||||||
| 10 | Co-GCN | 0.2 mL | 250 W Sodium lamp | 341.2 | N.A. | 147 |
| Methanol | λ > 400 nm | |||||
| 11 | Fe-EDTA-CNNS | 20 vol% | 300 W Xenon lamp | 2500 | 0.86% | 148 |
| Methanol | λ > 420 nm | 420 nm | ||||
| 12 | Ag/BCN | 5 vol% | 300 W Xenon lamp | 305.3 | N.A. | 149 |
| Methanol | ||||||
| 13 | Ag/CN | 5 vol% | 300 W Xenon lamp | 1450 | N.A. | 150 |
| Ethanol | λ > 400 nm | |||||
| 14 | Au/HCNS-NV | 20 vol% | 300 W Xenon lamp | 783.4 | N.A. | 151 |
| Methanol | λ > 420 nm | |||||
| 15 | AuCNNVs | No | 300 W Xenon lamp | 122.7 | N.A. | 152 |
| Full spectrum | ||||||
| 16 | Ru-Vs-CoS/CN | 10 vol% | 300 W Xenon lamp | 438 | 1.28% | 153 |
| Methanol | Full spectrum | 400 nm | ||||
In the design of tunable MSI heterogeneous photocatalysts, single-atom catalysts anchored on g-C3N4 have been widely used for the PNRR due to their susceptibility to form strong coordination with N atoms.138–140 Xie et al. found that single-atom Cu is dispersed in g-C3N4 (Cu-CN) in the form of Cu–N bonds through a lone pair of electrons bonded to three N atoms.138 Such strong coordination induced by MSI deforms the conjugated π-electron cloud of g-C3N4, which in turn promotes the single valence electrons of the edge N atoms to leave the domains as well as to isolate even from the previous π-electron cloud. These separated valence electrons are easily photoactivated and then rapidly transferred to the π* antibonding orbitals of N2via the Cu–N bond, thus enhancing the separation of photogenerated carriers and inhibiting the complexation of photoexcited electron holes. Coordination of Mo single atoms with two N to form Mo–N bonds thereby lowering the electron density of N atoms has been demonstrated.139 Ultra-low coordination of Mo centers could enhance N2 adsorption because of the increased empty d orbitals. Intriguingly, electron transfer from g-C3N4 to Co single atoms was observed.140 Such an electronic effect of MSIs results in the d-band center being closer to the EF, leading to enhanced electron reduction and the creation of more active sites for the PNRR. Xu et al. showed that Ru single-atom modifications on g-C3N4 not only form cyano defects but also enhance the light absorption range.141
Metal–support interactions are reflected not only in geometric encapsulation and electronic modulation, but also by stabilizing key intermediates, thus maintaining the sustainability of the PNRR. Zhao et al. prepared cyano (–CN) and K+ intercalation-modified g-C3N4 nanoribbon photocatalysts (mCNN).142 K+ not only interacts with different g-C3N4 layers through N–K bonds, but also acts as an electronic promoter to enhance the PNRR. Besides, the involvement of K+ in the PNRR process could be divided into four steps as confirmed by DFT calculations (Fig. 11): (1) K+ stabilizes the C active site by coupling with the unsaturated C site, (2) followed by spontaneous adsorption of N2 by electron transfer, (3) subsequently, the rearrangement of the coordination states of K+, C and N2, and (4) finally, K+ is removed to form the C2N4 ring. The above DFT calculations have demonstrated that the involvement of K+ not only stabilizes the key intermediates but also contributes to the adsorption and activation of N2. Therefore, the NH3 production rate of the mCNN photocatalyst could reach 3420 μmol g−1 h−1 under the conditions of ethylene glycol as a sacrificial agent and visible light.
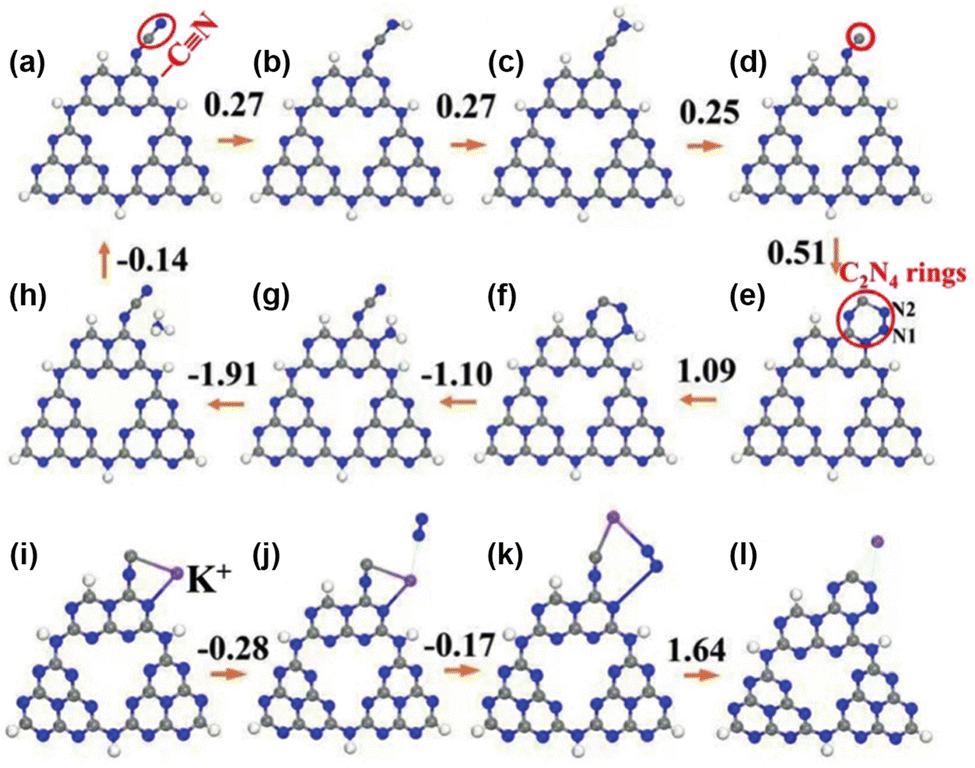 | ||
| Fig. 11 Free-energy diagrams of the PNRR pathway with the mCNN photocatalyst. (a)–(h) NRR pathway without K+. (i)–(l) K+-assisted –CN regeneration process. Blue sphere: N, grey sphere: C, white sphere: H, and purple sphere: K+. Reprinted with permission from ref. 142. Copyright 2019 Wiley-VCH. | ||
The potassium ions have a unique electron storage capacity, and can also act as an electron trapping agent, in turn inducing a bifunctional effect of MSIs with g-C3N4, promoting N2 activation.143,144 Among them, Zhao et al. found that N2 is adsorbed onto nitrogen vacancies (NVs) on potassium ion-intercalated carbon nitride (KCCN-NVs) and subsequently reduced by electrons stored in K+ (Fig. 12).143 Importantly, N2 could undergo a six-electron reduction process, thus being reduced directly to NH3 without producing intermediate species, which kinetically increases the PNRR activity. Interestingly, a record ammonia production rate of 23,500 μmol g−1 h−1 can be achieved by the K+ and –CN modified g-C3N4 catalyst with nanorod arrays (CNNR) prepared by Wang et al. (Fig. 13a and b).144 Under artificial light, photogenerated electrons are easily captured by K+ and –CN, which promotes effective charge separation; subsequently, –CN, as an adsorption center of N2, is able to transfer electrons to N2 thus activating them. Whereas, K+, as an electron-capture center, effectively inhibits charge recombination.
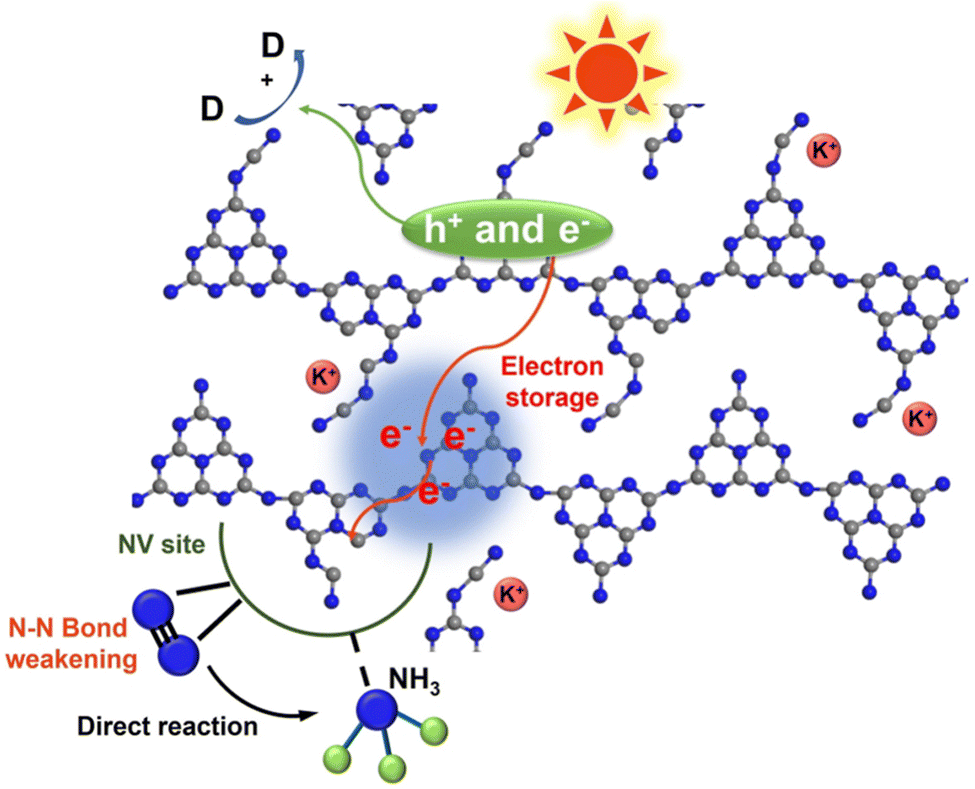 | ||
| Fig. 12 Schematic diagram of the N2 reduction reaction over KCCN-NVs. Reprinted with permission from ref. 143. Copyright 2021 Elsevier B.V. | ||
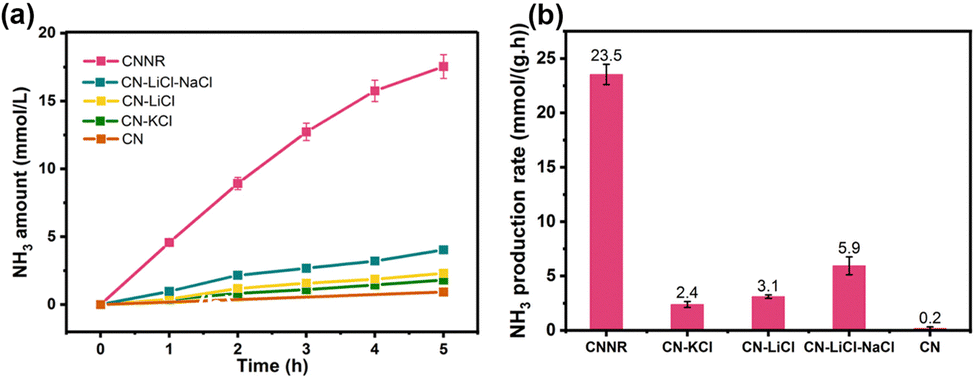 | ||
| Fig. 13 (a) NH3 production from N2 reduction with light irradiation time. (b) Photocatalytic NH3 production with different g-C3N4 samples. Reprinted with permission from ref. 144. Copyright 2022 Elsevier B.V. | ||
Electrons transfer from g-C3N4 to Ru clusters145 and Ru quantum dots146 and Co147 phenomena are confirmed. Such MSI-induced electronic effects lead to a tighter binding between Ru clusters and the edge sites of the bulk carbon nitride (B-g-C3N4), which in turn exhibits a lower N2 activation barrier.145 Thus, when Ru–K is loaded on bulk carbon nitride (B-g-C3N4), its nitrogen fixation activity is higher than that loaded on exfoliated carbon nitride (E-g-C3N4) and graphite (C). Their study reveals the subtle relationship between the PNRR activity and the support structure, which provides important guidance for the rational design of tunable MSI heterogeneous photocatalysts. He et al. covalently grafted ethylenediaminetetraacetic acid (EDTA) onto g-C3N4 nanosheets and then chelated Fe3+ (Fe-EDTA-CNNS).148 EDTA not only stabilizes the metal ions on the support surface to avoid their agglomeration or detachment, but also enhances the interaction between the metal and the support. Besides, the electron-absorbing properties of Fe-EDTA promote the separation and transfer of photogenerated carriers, making it a photogenerated electron-harvesting device itself. As a result, the nitrogen fixation rate of Fe-EDTA-CNNS is 13.5 times higher than that of pristine g-C3N4.
Similar to TiO2, MSI geometry effects are induced between Ag and g-C3N4, thus enhancing the stability of the catalyst.149,150 Wang et al. loaded Ag on the B-doped g-C3N4 composite (Ag/BCN).149 Ag was found to be partially encapsulated by g-C3N4 and the close contact can induce the MSI geometry effect. Thus, the activity of Ag/BCN has not been decreased after 5 cycles, proving its good stability. Interestingly, not only the geometric effect of MSIs but also the significant electronic effect of MSIs is found on the Ag-loaded g-C3N4 photocatalyst (Ag/CN).150 Based on the TEM and HRTEM images (Fig. 14a–f), Ag is partially coated by g-C3N4 and they are in close contact with each other. As mentioned earlier, the geometric cladding of MSIs often leads to a decrease in catalytic activity. However, the PNRR activity of Ag/CN is 1.4-fold and 15-fold higher than that of large-size Ag/g-C3N4 and pristine g-C3N4, respectively. Further analysis by theoretical calculations reveals that the highly dispersed Ag has a better and inhomogeneous electron distribution owing to the strong interfacial interaction between Ag and g-C3N4, which facilitates the transfer of electrons from Ag to the antibonding orbital of the N2 molecule to promote its activation.
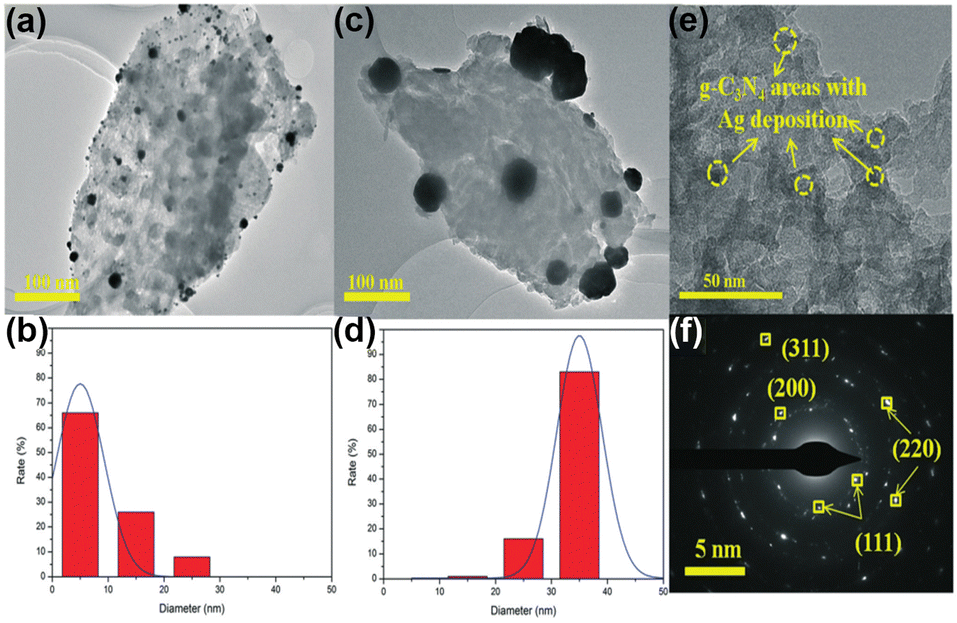 | ||
| Fig. 14 TEM images (a and c) and Ag nanoparticle size distribution (b and d) of Ag4%/CN (−60) and Ag4%/CN (room) and (e) HR-TEM and (f) SAED of Ag4%/CN (−60). Reprinted with permission from ref. 150. Copyright the Royal Society of Chemistry 2020. | ||
The LSPR effect of plasma nanoparticles has been widely used to modify the g-C3N4 supports because of their ability to increase the range of light absorption, generate high-energy hot electrons and local electric fields. Yang et al.151 and Zhang et al.152 found a bifunctional effect induced by MSIs on Au-loaded g-C3N4. Specifically, the NVs not only adsorb N2 molecules, but also collect photogenerated electrons generated by g-C3N4 and thermal electrons generated by Au nanoparticles, and subsequently, electrons entered the antibonding orbitals of N2 molecules to activate them. Liu et al. loaded a Ru/CoSx heterojunction with S vacancies as a co-catalyst on g-C3N4 (Ru-Vs-CoS/CN), constructing a Ru–Co bimetallic center to act as an active site for the side bridges of N2.153 The electron cloud density of N is reduced owing to the coordination of the lone pair of electrons of sp3-N on g-C3N4 with the unoccupied d orbitals of Co or Ru. Such MSI electronic effects optimize the electronic structure of the catalyst, and synergize with the plasma Ru/CoSx interface to promote the production of high-energy carriers as well as accelerated charge separation and transfer, which in turn contributes to the activation of N2.
4.6. Summary
Based on the above discussion, we found that the different supports applied to the PNRR process have their own characteristics, so it is necessary to make a brief summary of them. Firstly, there are advantages and disadvantages for unmodified supports, so we need to select appropriate supports to be explored as MSI model catalysts. Metal oxide supports (represented by TiO2 and W18O49) are widely used due to their good stability, ease of synthesis and abundance of oxygen vacancies. TiO2 has weak visible light absorption ability owing to its wide band gap, which may hinder its practical application and development. Interestingly, W18O49, in contrast, is visible light responsive. In addition, BiOX supports are also reported by researchers for their unique atomic structure, tunability and large number of defect sites. Among them, BiOBr has good visible light absorption ability, which makes it quite promising. It is worth noting that while OVs are favourable for N2 adsorption, they are also active sites for the HER, which may result in reduced selectivity. Moreover, the OVs are susceptible to be filled by the oxygen generated during the PNRR process leading to a decrease in stability. Metal sulfides, MOFs and carbon–nitrogen materials are expected to overcome the above problems. Of these, metal sulfides, as typical layered materials, have visible light absorption capabilities. But they are susceptible to photocorrosion, causing a decrease in stability. Surprisingly, MOFs have a unique pore structure that can effectively improve the diffusion of N2 and facilitate the mass transfer process. Besides, their inimitable ligand–metal charge transfer process is expected to endow the metal sites with distinctive electronic properties, thus optimizing the adsorption and activation of N2. Unfortunately, it is also vulnerable to effects such as photocorrosion in PNRR systems. Finally, we found that g-C3N4 attracted great attention owing to its visible light absorption ability, easy preparation and better stability. After a simple treatment, g-C3N4 containing NVs have the ability to adsorb and activate N2. Nevertheless, its own oxidizing capability is insufficient and often requires the addition of a sacrificial agent to drive the PNRR process. Additionally, because it contains a nitrogen source, this is likely to cause some interference with the detection of NH4+. Here, we also expect to introduce a variety of assays to prove the reliability of NH4+ (colorimetric, electrode, and ion chromatography) and, if necessary, to demonstrate the origin of the nitrogen using isotope labeling experiments.154What is more worth elaborating is that the activity, selectivity and stability of the above supports can be significantly enhanced after metal modification owing to the MSI effect. Intriguingly, we found that the forms, results, and strengths of the geometric, electronic, and bifunctional effects induced by different supports are distinctive, which necessitates a brief summary to guide the design of tunable MSI photocatalysts in a more rational way. The MSI electronic effect induced on TiO2 is prone to accelerate the electron transfer through stabilized metal–oxygen bonding and lower the reaction energy barrier of the rate-limiting step. In addition, the bifunctional effect is generally triggered by OVs synergizing with Au nanoparticles having plasma effects, which is beneficial for the activity enhancement. Moreover, single atoms stabilized at OVs can be converted into active sites, thereby enhancing selectivity. Differently, the MSI electronic effect on the W18O49 support tends to lead to a shift of the DB toward the EF, thus retaining more photogenerated electron energy for N2 activation. Furthermore, this electronic effect induces the raising of the d-band center to enhance N2 adsorption. We observed that the MSI electronic effect on bismuth halide supports tends to promote photogenerated carrier segregation, which retains more photogenerated electrons for the reduction of N2. Upon modification of a metal on the metal sulfide, the electrons are inclined to be transferred from the support to the active metal, and subsequently into the antibonding orbitals of N2, in order to activate it. Interestingly, MOFs can often synergize the unique LMCT and LSPR effects to enhance the catalytic activity for the PNRR. Finally, the modified single atoms on g-C3N4 are prone to form strong coordination with N atoms, which not only enhances the stability of the catalyst but also facilitates carrier separation. Besides, it is capable of inducing geometrical effects with some noble metal nanoparticles, which significantly improves the stability. Furthermore, g-C3N4 is able to induce MSI bifunctional effects on alkali metal ions with electron storage capacity, which is favourable for activity improvement.
5. Conclusions, challenges, and prospects
Photocatalytic nitrogen fixation, as a potential alternative to the traditional NH3 synthesis strategy, conducted under mild conditions without any fossil energy input and zero carbon emissions has aroused tremendous attention. However, there are still big challenges before realizing its practical application due to the lack of efficient photocatalysts. Owing to the inert nature of N2 molecules, weak adsorption and hard to activation are the main issues to be solved in developing photocatalysts. Among the reported photocatalysts, a class of photocatalysts with tunable MSIs showed potential for improving activity, stability as well as selectivity. In this review, recent advances in the supported metal photocatalysts in terms of the mechanism of MSIs have been summarized and discussed. From the analysis of the MSI mechanism, the synthesis strategy and the characterization of MSI photocatalysts, we focused on the discussion of supports which played an indispensable role in MSIs. Metal oxides, bismuth oxyhalides, metal sulphides, MOFs, and carbon nitrides as supports of MSI photocatalysts in the PNRR have been discussed, and how the supports affect the structural and catalytic behaviour has been emphasized. We have also highlighted how MSIs show geometric, electronic, and bifunctional effects influencing the catalytic efficiency for the PNRR. In addition, surface polarization engineering, stabilization of key intermediates, electron storage, strong coordination, and anchoring also have non-negligible effects on PNRR systems. Remarkable advancement for the PNRR is brought about through a tunable MSI system, using which the highest activity for NH3 generation obtained is 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 μmol g−1 h−1, which is 1655 times that of the first reported Fe–TiO2 (14.2 μmol g−1 h−1). However, there is still a long way to go before replacing Haber's process because of photocatalytic N2 fixation efficiency, problems in scale-up, and so on. Additionally, the detection of ammonia is also a key concern. In particular, for trace amounts of ammonia, the isotopic labelling method is a critical means of ruling out errors.155 The following aspects could be urgent problems to be solved in MSI photocatalysts to promote the progress of photocatalytic N2 fixation.
500 μmol g−1 h−1, which is 1655 times that of the first reported Fe–TiO2 (14.2 μmol g−1 h−1). However, there is still a long way to go before replacing Haber's process because of photocatalytic N2 fixation efficiency, problems in scale-up, and so on. Additionally, the detection of ammonia is also a key concern. In particular, for trace amounts of ammonia, the isotopic labelling method is a critical means of ruling out errors.155 The following aspects could be urgent problems to be solved in MSI photocatalysts to promote the progress of photocatalytic N2 fixation.
(I) Synthesizing photocatalysts with high metal loading and stability
According to the literature data, to prevent the aggregation of metal clusters during preparation and reaction, the metal loading of heterogeneous photocatalysts is generally low. Nevertheless, the insufficient number of active sites could be the main reason for low activity. Even supports with large specific surface areas (e.g., MOFs, 2D materials) and abundant anchor localization sites (e.g., surface defects and amorphous materials) have been paid more and more attention. Exposing the inner surface of porous materials is still a big challenge. Moreover, defects of supports are a limitation to maintaining their stability.156 Additionally, the generated O2 in the PNRR process may compete with metals on the anchor sites such as O-defects. A recent work effectively activated the TiO2 surface by high-temperature water vapor treatment, thereby inducing the migration of the support to the surface of the metal nanoparticles forming an oxygen-saturated encapsulated structure.157 Unlike classical SMSI, this type of wrapped structure does not contain oxygen defects and can be stabilized in oxidizing, reducing, and aqueous atmospheres. This oxygen-saturated SMSI (osSMSI) may open up new perspectives for us to construct MSI catalysts.(II) Optimizing the metal species and loading form
Composites represented by noble metals (Au, Ag, Ru, etc.) exhibit better PNRR catalytic activity and stability. However, the expensive price and limited resources of precious metals have hindered their wide application in PNRR systems. Surprisingly, inspired by the unique structure and superior performance of nitrogen-fixing enzymes, the design of photocatalysts with Fe or Mo metal as the active centre can also exhibit excellent performance, which opens a new path for non-precious metal composite catalysts. In addition, when the supported metal nanoparticles are shrunk to a single atom, they are directly coordinated with the support, thus exhibiting a special MSI effect. Single-atom catalysts also show good PNRR catalytic activity due to their ultra-high atomic utilization and unique electronic structure, thus they should be considered for their great potential.158 Recently, dual single-atom catalysts have attracted the attention of researchers because their two sets of single atoms can perform different functions in catalytic reactions, and the synergistic effect of the two can maximize the atom utilization and can improve the catalytic activity and selectivity. Therefore, we believe that the design of heterogeneous photocatalysts with dual single-atom sites is also a promising option.(III) Selecting the appropriate support
According to the discussion in the section of support classification and our viewpoint, the nature of the support plays a crucial role in the construction of MSIs, thus determining the activity for the PNRR and the selectivity of the product. For the PNRR system, visible light responsiveness, stability, and separation efficiency of the photogenerated carriers of the semiconductor are essential. TiO2 is widely used in the PNRR due to its properties such as high stability and easy preparation, yet, it does not have a visible light response, which hinders its practical applications. In contrast, g-C3N4, a common and stable semiconductor, has visible light (below 460 nm) responsiveness, but its valence band oxidation capability is insufficient and often requires the addition of a sacrificial agent to drive the PNRR. In addition, MOFs have a unique pore structure, which can effectively improve the diffusion of N2 molecules and promote the mass transfer process, but it is susceptible to photo corrosion, etc. in the PNRR system, leading to a decrease in stability. Surprisingly, the hydrogenated metal oxides (e.g., HyMoO3−x, HyWO3−x, and HyTiO2−x, etc.) obtained by hydrogen reduction treatment of metal oxides may serve as adsorption and activation centres for N2 molecules because of their large number of surface defect sites and plasma effect, which would effectively enhance the catalytic activity for the PNRR.(IV) Using theoretical calculations to guide experimental synthesis
It is an important strategy to use theoretical calculations to guide the synthesis of heterogeneous photocatalysts and to understand the reaction mechanism. Theoretical calculations can provide some unique insights into the electronic structure of photocatalysts, the adsorption and desorption of intermediates, and the optimal loading sites for metals. In addition, the behaviour of supported metal catalysts in catalysis is often complex and variable. Therefore, it is extremely challenging to use theoretical calculations to examine the dynamic changes in catalytic behaviour. Future work may have to combine operando characterization techniques on the atomic scale and theoretical simulations to quantitatively describe the support–metal nanoparticle interactions. Additionally, model catalyst systems can be focused on in order to simplify the study at the interface. The combination of advanced analytical techniques and computational methods in modeling studies is essential for a deeper understanding of the nature of interactions at interfaces.(V) Opening up new response systems
It is important to understand the nature of MSIs and the preparation of efficient photocatalysts using the mechanism of adsorption and activation of N2, the formation of intermediates, and the rate-determining steps. There are two main reaction pathways for the photoreduction of N2 molecules to ammonia (associative distal pathway and associative alternating pathway), which are closely dependent on the type of tunable MSI heterogeneous photocatalyst. If we can accurately obtain the information on active sites and MSI using in situ characterization and theoretical calculations, we can clarify the key intermediates in the PNRR process and the decisive steps, which is an important guideline for the design of tunable MSI heterogeneous photocatalysts. Furthermore, the photocatalytic synthesis of C–N bond-containing compounds (e.g., urea, amino acids, amides, amines, and their derivatives) from CO2, N2 and their derivatives is emerging as a promising and sustainable strategy.159–162 Among them, Zhang et al. reported a very innovative green pathway for the direct synthesis of urea from N2, CO2 and H2O by Cu single-atom modified TiO2 using solar energy, which will open up a new pathway for the PNRR.162 Besides, a new development pathway which used commercially available TiO2 as the catalyst to synthesise nitrate by N2 photochemical oxidation is useful in guiding the development of green nitrogen fixation pathways and is also worth focusing on.163However, the low solubility of N2 molecules and the high dissociation energy of NN bonds limit its application. Designing heterogeneous photocatalysts with MSI effects is expected to solve the above challenges and thus open up new reaction paths for photocatalytic N2 molecule immobilization. Besides fabricating efficient photocatalysts, the development of novel photocatalytic reactors should be advanced side by side. To our knowledge, there is limited work focused on this area. Indeed, the amplification effect in the photocatalytic process must be different from that of thermal catalysts. The transfer efficiency and utilization of incident light are the most important factors to be considered, but hard to investigate due to the wave-particle duality phenomenon.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (Grants 21975069, 21938002, 22278119 and 21725602), the Science and Technology Planning Project of Hunan Province (Grant 2019RS3010) and the Postgraduate Scientific Research Innovation Project of Hunan Province (Grant CX20220387).References
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed.
- P. G. Lustemberg, Z. Mao, A. Salcedo, B. Irigoyen, M. V. Ganduglia-Pirovano and C. T. Campbell, ACS Catal., 2021, 11, 10604–10613 CrossRef CAS.
- K. Stefkova, M. G. Guerzoni, Y. van Ingen, E. Richards and R. L. Melen, Org. Lett., 2023, 25, 500–505 CrossRef CAS.
- J. Feng, Y. He, Y. Liu, Y. Du and D. Li, Chem. Soc. Rev., 2015, 44, 5291–5319 RSC.
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 RSC.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- T. Pu, W. Zhang and M. Zhu, Angew. Chem., Int. Ed., 2022, 62, e202212278 CrossRef PubMed.
- C.-J. Pan, M.-C. Tsai, W.-N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S.-Y. Cheng and B.-J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154–186 CrossRef CAS.
- Z. Luo, G. Zhao, H. Pan and W. Sun, Adv. Energy Mater., 2022, 12, 2201395 CrossRef CAS.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS.
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y.-G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS.
- Y. Lou, J. Xu, Y. Zhang, C. Pan, Y. Dong and Y. Zhu, Mater. Today Nano, 2020, 12, 100093 CrossRef.
- T. W. Van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- K. Qi, M. Chhowalla and D. Voiry, Mater. Today, 2020, 40, 173–192 CrossRef CAS.
- Q.-Q. Yan, P. Yin and H.-W. Liang, ACS Mater. Lett., 2021, 3, 1197–1212 CrossRef CAS.
- Z. Li, R. Wu, L. Zhao, P. Li, X. Wei, J. Wang, J. S. Chen and T. Zhang, Nano Res., 2021, 14, 3795–3809 CrossRef CAS.
- S. Kim, J. Park, J. Hwang and J. Lee, Energy Chem., 2021, 3, 100054 CrossRef CAS.
- C. Wu, D. Cheng, M. Wang and D. Ma, Energy Fuels, 2021, 35, 19012–19023 CrossRef CAS.
- M. Ma, H. Wang and H. Liu, Chin. Chem. Lett., 2021, 32, 3613–3618 CrossRef CAS.
- G. D. Gesesse, C. Wang, B. K. Chang, S. H. Tai, P. Beaunier, R. Wojcieszak, H. Remita, C. Colbeau-Justin and M. N. Ghazzal, Nanoscale, 2020, 12, 7011–7023 RSC.
- G. M. Haselmann and D. Eder, ACS Catal., 2017, 7, 4668–4675 CrossRef CAS.
- W. Zhang, X. Pan, P. Long, X. Liu, X. Long, Y. Yu and Z. Yi, J. Mater. Chem. A, 2017, 5, 18998–19006 RSC.
- K. Fujiwara, K. Okuyama and S. E. Pratsinis, Environ. Sci.: Nano, 2017, 4, 2076–2092 RSC.
- F. Wu, C. Pu, M. Zhang, B. Liu and J. Yang, Surf. Interfaces, 2021, 25, 101298 CrossRef CAS.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- K. H. R. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, Energy Environ. Sci., 2021, 14, 2520–2534 RSC.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu, N. H. Deissler, J. B. V. Mygind, C. Wei, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef CAS.
- H.-L. Du, M. Chatti, R. Y. Hodgetts, P. V. Cherepanov, C. K. Nguyen, K. Matuszek, D. R. MacFarlane and A. N. Simonov, Nature, 2022, 609, 722–727 CrossRef CAS PubMed.
- G.-F. Han, F. Li, Z.-W. Chen, C. Coppex, S.-J. Kim, H.-J. Noh, Z. Fu, Y. Lu, C. V. Singh, S. Siahrostami, Q. Jiang and J.-B. Baek, Nat. Nanotechnol., 2021, 16, 325–330 CrossRef CAS.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- Y. Guo, Q. Zhou, B. Zhu, C. Y. Tang and Y. Zhu, EES. Catal., 2023, 1, 333–352 RSC.
- S. Xie, W. Ma, X. Wu, H. Zhang, Q. Zhang, Y. Wang and Y. Wang, Energy Environ. Sci., 2021, 14, 37–89 RSC.
- X. Zhang, S. Pollitt, G. Jung, W. Niu, P. Adams, J. Buhler, N. S. Grundmann, R. Erni, M. Nachtegaal, N. Ha, J. Jung, B. Shin, W. Yang and S. D. Tilley, Chem. Mater., 2023, 35, 2371–2380 CrossRef CAS PubMed.
- B. Yang, W. Ding, H. Zhang and S. Zhang, Energy Environ. Sci., 2021, 14, 672–687 RSC.
- Y. Fu, Y. Liao, P. Li, H. Li, S. Jiang, H. Huang, W. Sun, T. Li, H. Yu, K. Li, H. Li, B. Jia and T. Ma, Coord. Chem. Rev., 2022, 460, 214468 CrossRef CAS.
- Y. Xia, Y. Xu, X. Yu, K. Chang, H. Gong, X. Fan, X. Meng, X. Huang, T. Wang and J. He, J. Mater. Chem. A, 2022, 10, 17377–17394 RSC.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS PubMed.
- S. Chen, D. Liu and T. Peng, Sol. RRL, 2021, 5, 2000487 CrossRef CAS.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- K. U. D. Calvinho, A. W. Alherz, K. M. K. Yap, A. B. Laursen, S. Hwang, Z. J. L. Bare, Z. Clifford, C. B. Musgrave and G. C. Dismukes, J. Am. Chem. Soc., 2021, 143, 21275–21285 CrossRef CAS.
- C. Wei, Y. Sun, G. G. Scherer, A. C. Fisher, M. Sherburne, J. W. Ager and Z. J. Xu, J. Am. Chem. Soc., 2020, 142, 7765–7775 CrossRef CAS.
- X. Du, Y. Qin, B. Gao, J. H. Jang, C. Xiao, Y. Li, S. Ding, Z. Song, Y. Su and K. T. Nam, J. Mater. Chem. A, 2022, 10, 7082–7089 RSC.
- A. Caballero, J. P. Holgado, V. M. Gonzalez-delaCruz, S. E. Habas, T. Herranz and M. Salmeron, Chem. Commun., 2010, 46, 1097–1099 RSC.
- X. Han, T. Mou, S. Liu, M. Ji, Q. Gao, Q. He, H. Xin and H. Zhu, Nanoscale Horiz., 2022, 7, 508–514 RSC.
- H. Wang, L. Wang and F.-S. Xiao, Sci. China: Chem., 2022, 65, 2051–2057 CrossRef CAS.
- Q. Fu, T. Wagner, S. Olliges and H.-D. Carstanjen, J. Phys. Chem. B, 2005, 109, 944–951 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, K. Qian and W. Huang, ACS Catal., 2022, 12, 1268–1287 CrossRef CAS.
- J. Chen, Y. Zhang, Z. Zhang, D. Hou, F. Bai, Y. Han, C. Zhang, Y. Zhang and J. Hu, J. Mater. Chem. A, 2023, 11, 8540–8572 RSC.
- H. Xu, Y. Zhao, Q. Wang, G. He and H. Chen, Coord. Chem. Rev., 2022, 451, 214261 CrossRef CAS.
- Y. Sun, G. Chen, S. Xi and Z. J. Xu, ACS Catal., 2021, 11, 13947–13954 CrossRef CAS.
- T. Wu, M. Y. Han and Z. J. Xu, ACS Nano, 2022, 16, 8531–8539 CrossRef.
- J. M. Herrmann, J. Catal., 1984, 89, 404–412 CrossRef CAS.
- X. Tian, X. Zhao, Y.-Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS.
- L. Sun, V. Reddu and X. Wang, Chem. Soc. Rev., 2022, 51, 8923–8956 RSC.
- X. Xiao, Z. Ruan, Q. Li, L. Zhang, H. Meng, Q. Zhang, H. Bao, B. Jiang, J. Zhou, C. Guo, X. Wang and H. Fu, Adv. Mater., 2022, 34, 2200612 CrossRef CAS.
- X. Du, H. Tang and B. Qiao, Catalysts, 2021, 11, 896 CrossRef CAS.
- R. Shi, X. Zhang, G. I. N. Waterhouse, Y. Zhao and T. Zhang, Adv. Energy Mater., 2020, 10, 2000659 CrossRef CAS.
- X. Bian, Y. Zhao, G. I. N. Waterhouse, Y. Miao, C. Zhou, L.-Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304452 CrossRef CAS PubMed.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, Z. Wang, Y. Weng and T. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2554–2560 CrossRef CAS PubMed.
- X. Bian, Y. Zhao, S. Zhang, D. Li, R. Shi, C. Zhou, L.-Z. Wu and T. Zhang, ACS Mater. Lett., 2021, 3, 1521–1527 CrossRef CAS.
- K. Li, C. Sun, Z. Chen, H. Qu, H. Xie and Q. Zhong, Chem. Eng. J., 2022, 429, 132440 CrossRef CAS.
- L. Chen, J. Shou, Y. Chen, W. Han, X. Tu, L. Zhang, Q. Sun, J. Cao, Y. Chang and H. Zheng, Chem. Eng. J., 2023, 451, 138592 CrossRef CAS.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- G. Zheng, J. He, V. Kumar, S. Wang, I. Pastoriza-Santos, J. Perez-Juste, L. M. Liz-Marzan and K.-Y. Wong, Chem. Soc. Rev., 2021, 50, 3738–3754 RSC.
- P. Xing, S. Wu, Y. Chen, P. Chen, X. Hu, H. Lin, L. Zhao and Y. He, ACS Sustainable Chem. Eng., 2019, 7, 12408–12418 CAS.
- X. Gao, Y. Shang, L. Liu and F. Fu, J. Colloid Interface Sci., 2019, 533, 649–657 CrossRef CAS.
- R. Shi, Y. Cao, Y. Bao, Y. Zhao, G. I. N. Waterhouse, Z. Fang, L. Z. Wu, C. H. Tung, Y. Yin and T. Zhang, Adv. Mater., 2017, 29, 1700803 CrossRef.
- J. Wang, C. Hua, X. Dong, Y. Wang and N. Zheng, Sustainable Energy Fuels, 2020, 4, 1855–1862 RSC.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS.
- J. Yang, Y. Guo, R. Jiang, F. Qin, H. Zhang, W. Lu, J. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS.
- T.-A. Bu, Y.-C. Hao, W.-Y. Gao, X. Su, L.-W. Chen, N. Zhang and A.-X. Yin, Nanoscale, 2019, 11, 10072–10079 RSC.
- S. Chang and X. Xu, Inorg. Chem. Front., 2020, 7, 620–624 RSC.
- W. Guo, Y. Liu, Y. Sun, Y. Wang, W. Qin, B. Zhao, Z. Liang and L. Jiang, Adv. Funct. Mater., 2021, 31, 2100768 CrossRef CAS.
- P. Sharma, M. Sharma, M. Dearg, M. Wilding, T. J. A. Slater and C. R. A. Catlow, Angew. Chem., Int. Ed., 2023, 62, e202301239 CrossRef CAS PubMed.
- Y. Wang, Y. Qu, B. Qu, L. Bai, Y. Liu, Z. D. Yang, W. Zhang, L. Jing and H. Fu, Adv. Mater., 2021, 33, 2105482 CrossRef CAS.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- S. Liu, Y. Wang, S. Wang, M. You, S. Hong, T.-S. Wu, Y.-L. Soo, Z. Zhao, G. Jiang, Q. Jieshan, B. Wang and Z. Sun, ACS Sustainable Chem. Eng., 2019, 7, 6813–6820 CrossRef CAS.
- X. Niu, Q. Zhu, S. Jiang and Q. Zhang, J. Phys. Chem. Lett., 2020, 11, 9579–9586 CrossRef CAS PubMed.
- G. Ren, M. Shi, S. Liu, Z. Li, Z. Zhang and X. Meng, Chem. Eng. J., 2023, 454, 140158 CrossRef CAS.
- S. Wu, Z. Chen, W. Yue, S. Mine, T. Toyao, M. Matsuoka, X. Xi, L. Wang and J. Zhang, ACS Catal., 2021, 11, 4362–4371 CrossRef CAS.
- Y. Liao, J. Lin, B. Cui, G. Xie and S. Hu, J. Photochem. Photobiol., A, 2020, 387, 112100 CrossRef CAS.
- Z. Zhao, S. Hong, C. Yan, C. Choi, Y. Jung, Y. Liu, S. Liu, X. Li, J. Qiu and Z. Sun, Chem. Commun., 2019, 55, 7171–7174 RSC.
- Y. Liu, Z. Yu, S. Guo, L. Yao, R. Sun, X. Huang and W. Zhao, New J. Chem., 2020, 44, 19924–19932 RSC.
- Y. Bo, H. Wang, Y. Lin, T. Yang, R. Ye, Y. Li, C. Hu, P. Du, Y. Hu, Z. Liu, R. Long, C. Gao, B. Ye, L. Song, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 16085–16092 CrossRef CAS PubMed.
- Q. Han, X. Zhao, Y. Luo, L. Wu, S. Sun, J. Li, Y. Wang, G. Liu and Z. Chen, Adv. Sci., 2022, 9, 2104237 CrossRef CAS PubMed.
- N. Zhang, A. Jalil, D. Wu, S. Chen, Y. Liu, C. Gao, W. Ye, Z. Qi, H. Ju, C. Wang, X. Wu, L. Song, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 9434–9443 CrossRef CAS PubMed.
- G. Dong, X. Huang and Y. Bi, Angew. Chem., Int. Ed., 2022, 61, e202204271 CrossRef CAS PubMed.
- P. Qiu, C. Huang, G. Dong, F. Chen, F. Zhao, Y. Yu, X. Liu, Z. Li and Y. Wang, J. Mater. Chem. A, 2021, 9, 14459–14465 RSC.
- H. Shang, Y. Wang, H. Jia, M. Qu, X. Ye, Q. Zhu, D. Zhang, D. Wang, G. Li and H. Li, Catal. Sci. Technol., 2023, 13, 854–861 RSC.
- B. Hu, B.-H. Wang, L. Chen, Z.-J. Bai, W. Zhou, J.-K. Guo, S. Shen, T.-L. Xie, C.-T. Au, L.-L. Jiang and S.-F. Yin, ACS Catal., 2022, 12, 11860–11869 CrossRef CAS.
- R. Han, S. Chang and X. Xu, Inorg. Chem. Front., 2020, 7, 2778–2782 RSC.
- M.-H. Vu, T.-A. Quach and T.-O. Do, Sustainable Energy Fuels, 2021, 5, 2528–2536 RSC.
- X. Liu, Y. Luo, C. Ling, Y. Shi, G. Zhan, H. Li, H. Gu, K. Wei, F. Guo, Z. Ai and L. Zhang, Appl. Catal., B, 2022, 301, 120766 CrossRef CAS.
- Y. Zhang, Q. Wang, S. Yang, H. Wang, D. Rao, T. Chen, G. Wang, J. Lu, J. Zhu, S. Wei, X. Zheng and J. Zeng, Adv. Funct. Mater., 2022, 32, 2112452 CrossRef CAS.
- P. Liu, Z. Huang, S. Yang, J. Du, Y. Zhang, R. Cao, C. Chen, L. Li, T. Chen, G. Wang, D. Rao, X. Zheng and X. Hong, ACS Catal., 2022, 12, 8139–8146 CrossRef CAS.
- H. Han, H. Choi, S. Mhin, Y.-R. Hong, K. M. Kim, J. Kwon, G. Ali, K. Y. Chung, M. Je, H. N. Umh, D.-H. Lim, K. Davey, S.-Z. Qiao, U. Paik and T. Song, Energy Environ. Sci., 2019, 12, 2443–2454 RSC.
- N. Zhang, L. Li, Q. Shao, T. Zhu, X. Huang and X. Xiao, ACS Appl. Energy Mater., 2019, 2, 8394–8398 CrossRef CAS.
- J. Liu, D. Li, R. Li, Y. Wang, Y. Wang and C. Fan, Chem. Eng. J., 2020, 395, 123954 CrossRef CAS.
- Y. Liu, Z. Hu and J. C. Yu, Chem. Mater., 2020, 32, 1488–1494 CrossRef CAS.
- X. Chen, X. Zhang, Y.-H. Li, M.-Y. Qi, J.-Y. Li, Z.-R. Tang, Z. Zhou and Y.-J. Xu, Appl. Catal., B, 2021, 281, 119516 CrossRef CAS.
- X. Chen, M.-Y. Qi, Y.-H. Li, Z.-R. Tang and Y.-J. Xu, Chin. J. Catal., 2021, 42, 2020–2026 CrossRef CAS.
- J. Liu, F. Li, J. Lu, R. Li, Y. Wang, Y. Wang, X. Zhang, C. Fan and R. Zhang, J. Colloid Interface Sci., 2021, 603, 17–24 CrossRef CAS.
- J. Di, C. Chen, Y. Wu, Y. Zhao, C. Zhu, Y. Zhang, C. Wang, H. Chen, J. Xiong, M. Xu, J. Xia, J. Zhou, Y. Weng, L. Song, S. Li, W. Jiang and Z. Liu, Adv. Mater., 2022, 34, 2204959 CrossRef CAS.
- Y. Huang, Y. Zhu, S. Chen, X. Xie, Z. Wu and N. Zhang, Adv. Sci., 2021, 8, 2003626 CrossRef CAS.
- C. Wang, J. Wang and X. Yu, Colloids Surf., A, 2023, 662, 131055 CrossRef CAS.
- X. Wang, B. Wang, S. Yin, M. Xu, L. Yang and H. Sun, J. Cleaner Prod., 2022, 360, 132162 CrossRef CAS.
- J. He, L. Chen, F. Wang, Y. Liu, P. Chen, C.-T. Au and S.-F. Yin, ChemSusChem, 2016, 9, 624–630 CrossRef CAS.
- J. Qin, W. Zhao, X. Hu, J. Li, P. Ndokoye and B. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 7127–7134 CrossRef CAS PubMed.
- J. Zheng, L. Lu, K. Lebedev, S. Wu, P. Zhao, I. J. McPherson, T.-S. Wu, R. Kato, Y. Li, P.-L. Ho, G. Li, L. Bai, J. Sun, D. Prabhakaran, R. A. Taylor, Y.-L. Soo, K. Suenaga and S. C. E. Tsang, Chem. Catal., 2021, 1, 162–182 CrossRef CAS.
- B. Hu, B.-H. Wang, Z.-J. Bai, L. Chen, J.-K. Guo, S. Shen, T.-L. Xie, C.-T. Au, L.-L. Jiang and S.-F. Yin, Chem. Eng. J., 2022, 442, 136211 CrossRef CAS.
- P. Zhou, Q. Zhang, Z. Xu, Q. Shang, L. Wang, Y. Chao, Y. Li, H. Chen, F. Lv, Q. Zhang, L. Gu and S. Guo, Adv. Mater., 2020, 32, 1904249 CrossRef CAS.
- X. Guo, J. Duan, C. Li, Z. Zhang and W. Wang, Opt. Mater., 2022, 125, 112137 CrossRef CAS.
- J. Du, F. Li and L. Sun, Chem. Soc. Rev., 2021, 50, 2663–2695 RSC.
- J. Guo, Y. Qin, Y. Zhu, X. Zhang, C. Long, M. Zhao and Z. Tang, Chem. Soc. Rev., 2021, 50, 5366–5396 RSC.
- M. K. Aslam, K. Yang, S. Chen, Q. Li and J. Duan, EES Catal., 2023, 1, 179–229 RSC.
- L. Xiao, Q. Zhang, P. Chen, L. Chen, F. Ding, J. Tang, Y.-J. Li, C.-T. Au and S.-F. Yin, Appl. Catal., B, 2019, 248, 380–387 CrossRef CAS.
- J. B. Pan, B. H. Wang, J. B. Wang, H. Z. Ding, W. Zhou, X. Liu, J. R. Zhang, S. Shen, J. K. Guo, L. Chen, C. T. Au, L. L. Jiang and S. F. Yin, Angew. Chem., Int. Ed., 2021, 60, 1433–1440 CrossRef CAS PubMed.
- S. Wang, J.-J. Zhang, M.-Y. Zong, J. Xu, D.-H. Wang and X.-H. Bu, ACS Catal., 2022, 12, 7960–7974 CrossRef CAS.
- S. Shang, W. Xiong, C. Yang, B. Johannessen, R. Liu, H. Y. Hsu, Q. Gu, M. K. H. Leung and J. Shang, ACS Nano, 2021, 15, 9670–9678 CrossRef CAS PubMed.
- Z. Zhang, F. Li, G. Li, R. Li, Y. Wang, Y. Wang, X. Zhang, L. Zhang, F. Li, J. Liu and C. Fan, J. Solid State Chem., 2022, 310, 123041 CrossRef CAS.
- W. Wang, J. Qu, C. Li, L. Guo, X. Fang, G. Chen and J. Duan, Mol. Catal., 2022, 532, 112730 CrossRef CAS.
- L. W. Chen, Y. C. Hao, Y. Guo, Q. Zhang, J. Li, W. Y. Gao, L. Ren, X. Su, L. Hu, N. Zhang, S. Li, X. Feng, L. Gu, Y. W. Zhang, A. X. Yin and B. Wang, J. Am. Chem. Soc., 2021, 143, 5727–5736 CrossRef CAS.
- X. Zhang, X. Li, S. Su, M. Tan, G. Liu, Y. Wang and M. Luo, Catal. Sci. Technol., 2023, 13, 705–713 RSC.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- C. Xiao, L. Zhang, K. Wang, H. Wang, Y. Zhou and W. Wang, Appl. Catal., B, 2018, 239, 260–267 CrossRef CAS.
- J. Zander, J. Timm, M. Weiss and R. Marschall, Adv. Energy Mater., 2022, 12, 2202403 CrossRef CAS.
- G. Dong, W. Ho and C. Wang, J. Mater. Chem. A, 2015, 3, 23435–23441 RSC.
- J. Barrio, M. Volokh and M. Shalom, J. Mater. Chem. A, 2020, 8, 11075–11116 RSC.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- P. Chen, F. Liu, S. Chen, J.-K. Guo, S. Shen, L. Chen, C.-T. Au and S.-F. Yin, Chem. Eng. Sci., 2019, 207, 271–279 CrossRef CAS.
- J. He, L. Chen, Z.-Q. Yi, D. Ding, C.-T. Au and S.-F. Yin, Catal. Commun., 2017, 99, 79–82 CrossRef CAS.
- P. Huang, W. Liu, Z. He, C. Xiao, T. Yao, Y. Zou, C. Wang, Z. Qi, W. Tong, B. Pan, S. Wei and Y. Xie, Sci. China: Chem., 2018, 61, 1187–1196 CrossRef CAS.
- X.-W. Guo, S.-M. Chen, H.-J. Wang, Z.-M. Zhang, H. Lin, L. Song and T.-B. Lu, J. Mater. Chem. A, 2019, 7, 19831–19837 RSC.
- W. Zhang, Y. Fu, Q. Peng, Q. Yao, X. Wang, A. Yu and Z. Chen, Chem. Eng. J., 2020, 394, 124822 CrossRef CAS.
- L. Li, Y. Yu, S. Lin, W. Chu, D. Sun, Q. Su, S. Ma, G. Du and B. Xu, Catal. Commun., 2021, 153, 106294 CrossRef CAS.
- W. Wang, H. Zhang, S. Zhang, Y. Liu, G. Wang, C. Sun and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16644–16650 CrossRef CAS.
- G. Dong, P. Qiu, Q. Chen, C. Huang, F. Chen, X. Liu, Z. Li, Y. Wang and Y. Zhao, Chem. Eng. J., 2022, 433, 133573 CrossRef CAS.
- G. Liu, Z. Tang, X. Gu, N. Li, H. Lv, Y. Huang, Y. Zeng, M. Yuan, Q. Meng, Y. Zhou and C. Wang, Appl. Catal., B, 2022, 317, 121752 CrossRef CAS.
- H. Liu, P. Wu, H. Li, Z. Chen, L. Wang, X. Zeng, Y. Zhu, Y. Jiang, X. Liao, B. S. Haynes, J. Ye, C. Stampfl and J. Huang, Appl. Catal., B, 2019, 259, 118026 CrossRef CAS.
- S. Wang, D. Guo, M. Zong, C. Fan, X. Jun and D.-H. Wang, Appl. Catal., A, 2021, 617, 118112 CrossRef CAS.
- K. Wang, G. Gu, S. Hu, J. Zhang, X. Sun, F. Wang, P. Li, Y. Zhao, Z. Fan and X. Zou, Chem. Eng. J., 2019, 368, 896–904 CrossRef CAS.
- C. Yao, R. Wang, Z. Wang, H. Lei, X. Dong and C. He, J. Mater. Chem. A, 2019, 7, 27547–27559 RSC.
- X. Yao, W. Zhang, J. Huang, Z. Du, X. Hong, X. Chen, X. Hu and X. Wang, Appl. Catal., A, 2020, 601, 117647 CrossRef CAS.
- Y. Wang, R. Zhao, F. Wang, Y. Liu, X. Yu, L. Chen, Y. Yao, S. Lu and X. Liao, Catal. Sci. Technol., 2020, 10, 7652–7660 RSC.
- Y. Guo, J. Yang, D. Wu, H. Bai, Z. Yang, J. Wang and B. Yang, J. Mater. Chem. A, 2020, 8, 16218–16231 RSC.
- S. Wu, Z. Chen, K. Liu, W. Yue, L. Wang and J. Zhang, ChemSusChem, 2020, 13, 3455–3461 CrossRef CAS PubMed.
- J. Yuan, X. Yi, Y. Tang, M. Liu and C. Liu, Adv. Funct. Mater., 2020, 30, 1906983 CrossRef CAS.
- Y. Zhao, Y. Miao, C. Zhou and T. Zhang, Mol. Catal., 2022, 518, 112107 CrossRef CAS.
- Y. Zhao, F. Wu, Y. Miao, C. Zhou, N. Xu, R. Shi, L.-Z. Wu, J. Tang and T. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21728–21731 CrossRef CAS PubMed.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S. Z. Qiao, Adv. Mater., 2019, 31, 1902709 CrossRef.
- H. Wang, X. Dong, Y. Hui, Y. Niu, B. Zhang, L. Liu, J. Cao, M. Yabushita, Y. Nakagawa, K. Tomishige, Y. Qin, L. Song, J. Xiao, L. Wang and F.-S. Xiao, Adv. Funct. Mater., 2023, 2304303 CrossRef CAS.
- J. Zhao, J. Liu, Z. Li, K. Wang, R. Shi, P. Wang, Q. Wang, G. I. N. Waterhouse, X. Wen and T. Zhang, Nat. Commun., 2023, 14, 1909 CrossRef CAS PubMed.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed.
- D. Li, Y. Zhao, Y. Miao, C. Zhou, L.-P. Zhang, L.-Z. Wu and T. Zhang, Adv. Mater., 2022, 34, 2207793 CrossRef CAS.
- S. Zhang, Y. Zhao, Y. Miao, Y. Xu, J. Ran, Z. Wang, Y. Weng and T. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202211469 CrossRef CAS.
Footnote |
| † The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |