
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmmonia synthesis by the reductive N–N bond cleavage of hydrazine using an air-stable, phosphine-free ruthenium catalyst†
Aisa
Mohanty
a,
Smruti Rekha
Rout
b,
Rambabu
Dandela
b and
Prosenjit
Daw
 *a
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Berhampur, Transit Campus, (Govt. ITI Building), Engg. School Junction, Berhampur 760010, Odisha, India. E-mail: pdaw@iiserbpr.ac.in
bDepartment of Industrial and Engineering Chemistry, Institute of Chemical Technology, Bhubaneswar 751013, Odisha, India
First published on 1st December 2023
Abstract
The development of an effective molecular catalyst to reduce hydrazine efficiently to ammonia using a suitable reductant and proton source is demanding. Herein, an unprecedented air-stable, phosphine-free ruthenium complex is used as a potent catalyst for hydrazine hydrate reduction to generate ammonia using SmI2 and water under ambient reaction conditions. Maximizing the flow of electrons from the reductant to the hydrazine hydrate via the metal centre results in a greater yield of ammonia while minimizing the evolution of H2 gas as a competing product.
In the biological nitrogen fixation reaction, the MoFe-protein-containing nitrogenase enzyme plays a crucial role in ammonia formation from the dinitrogen reduction under milder reaction conditions.1–3 During this transformation, hydrazine is proposed to act as an active intermediate, which may be released as a byproduct along with the ammonia via an alternative pathway (Scheme 1a).4 The exploration of hydrazine as a potential substrate and its 2e−/2H+ reduction process to generate ammonia is highly essential to determine the mechanism involved in both enzymatic sites and other molecular catalyst sites.5 However, while the process of the reduction of hydrazine to ammonia has been thoroughly investigated using early transition metals such as Mo, W, and V, studies involving Ru as a metal centre for the process lie far behind, demanding further exploration at the current state.
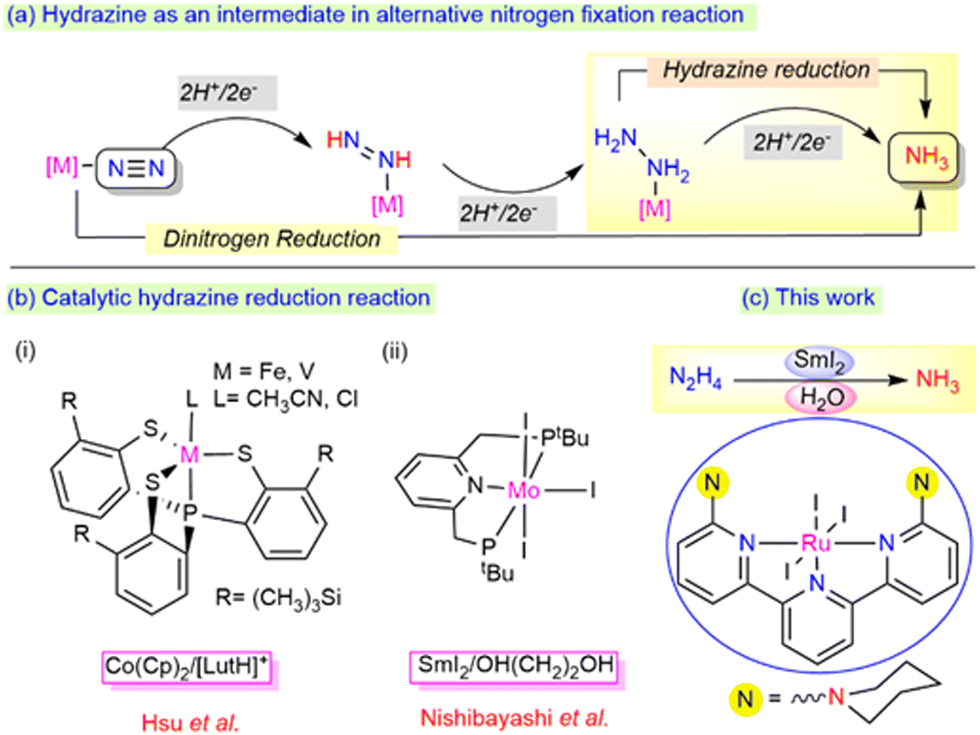 | ||
| Scheme 1 (a) Hydrazine as an intermediate in the nitrogen fixation reaction. (b) Monometallic system for the hydrazine reduction reaction (c) This work. | ||
The disproportionation and reduction reaction processes with the aid of suitable metal catalysts are the two alternative mechanisms involved in the formation of ammonia from hydrazine. Notably, Ikariya et al.6 and Peter et al.7 reported the catalytic disproportionation of hydrazine to ammonia and dinitrogen under milder conditions. In parallel, reduction reactions have also gained significant attention towards the generation of single reduced product, ammonia (Scheme 1b). A few bimetallic complexes mimicking the bimetallic cooperativity at the Fe-Mo protein sites of the nitrogenase enzyme have been described for the catalytic hydrazine reduction reaction.8–10 In addition to bimetallic cooperativity, recent catalytic reduction at mononuclear metal centres has also drawn a lot of attention. A mononuclear vanadium or iron core, upon ligation with the thiolate ligands designed by the Hsu group, efficiently reduced hydrazine to ammonia using CoCp2 and lutidinium salts (LutH+) as the reductant and proton source (e−/H+), respectively [Scheme 1b(i)].11,12 The reduction of dinitrogen to ammonia was achieved by the Nishibayashi group using a Mo complex employing SmI2/ROH [R = H, OH–(CH2)2] as an e−/H+ pair, which was also found to reduce hydrazine to ammonia at room temperature [Scheme 1b(ii)].13
The major drawbacks associated with these systems are that using a strong reductant and proton source together in the reaction system can evolve dihydrogen gas as a side product.14 Thus, it is highly necessary to tune the selectivity between the ammonia production (AP) and hydrogen evolution reaction (HER). To the best of our knowledge, the use of SmI2/H2O as a reductant and proton pair using ruthenium metal centres has never been employed before for the reduction of hydrazine to ammonia. In this work, we highlight the use of the SmI2/H2O pair to promote the efficient production of ammonia from hydrazine hydrate using ambient reaction conditions with an air-stable, phosphine-free ruthenium complex. This process revealed a possible strategy to deliver electron transfer from SmI2 to the substrate via the metal centre by minimizing the evolution of H2 as the side product (Scheme 1c).
A piperidine-functionalised terpyridine ligand scaffold L1 was synthesized and characterized using single crystal X-ray analysis (see ESI,† Fig. S28). L1 was treated with RuCl3·3H2O to afford Ru complex 1, a paramagnetic complex that showed an HRMS peak at m/z = 571.08 corresponding to the [1-Cl]+ unit (see ESI,† Fig. S34). This complex was further used for halogen exchange reaction with sodium iodide in dry THF to afford the triiodo-Ru complex 2, which showed an HRMS peak at m/z = 754.95 for the [2-I]+ unit (see ESI,† Fig. S37) (Scheme 2).
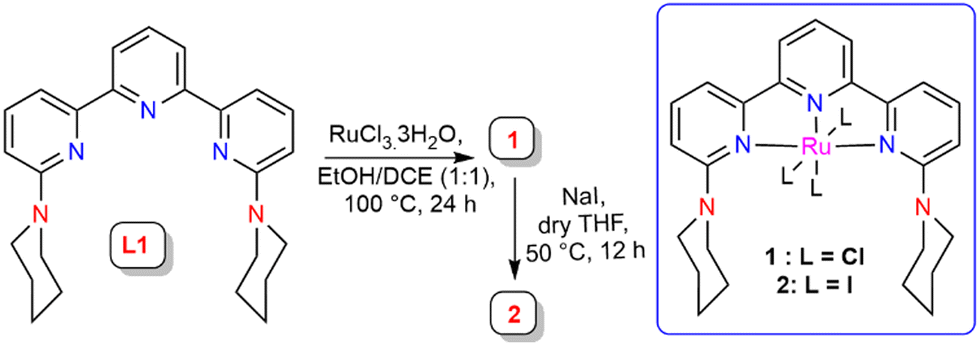 | ||
| Scheme 2 Synthesis of complexes 1 and 2. | ||
The synthesized Ru-complexes were utilized for the reduction reaction in the presence of an external reductant and proton pair, SmI2 and H2O, respectively (Table 1). Triiodo Ru-complex 2 was investigated for the reduction reaction under the operating reaction conditions [N2H4·H2O (0.09 mmol, 30 equiv. with respect to the catalyst), 2 (0.003 mmol, 3.3 mol%), SmI2(thf)2 (0.1 M in THF, 0.18 mmol), H2O (0.18 mmol) for 4 h] at room temperature, resulting in only a 17% yield of ammonium (0.030 mmol) by capturing the ammonia with acid (HCl in 2 M Et2O) treatment following the equation mentioned in Table 1 (entry 1, see ESI,† Section S6; Table S2). When the reaction temperature was elevated to 55 °C under the same reaction conditions, the colour of the reaction mixture changed from deep blue to yellow after 20 minutes, representing the consumption of SmI2, and resulting in a 51% yield of ammonia (entry 2) and a 36% yield of hydrogen gas (analyzed using GC-TCD; see ESI,† Section S7; Fig. S6). The corresponding amount of unreacted hydrazine was also detected using the p-(dimethylamino)benzaldehyde (p-DMAB) method (see ESI,† Section S8).15 Upon increasing the reaction time from 4 h to 12 h under the optimized reaction conditions at 55 °C, the formation of ammonia was similar (57%), which implied that the initial hours were quite impactful for the catalysis reaction (entry 3, see ESI,† Section S9.1; Fig. S14). In the absence of an additional proton source (H2O), a 23% yield of ammonia was afforded, indicating that hydrazine monohydrate affects the catalysis reaction (entry 4). However, without any catalyst loading, the combination of SmI2 and water resulted in a 30% yield of ammonia with 10% H2 gas formation (entry 5, see ESI,† Section S7; Fig. S7). The trichloro-derivative complex 1 was found to be less active as compared to 2, resulting in only a 32% yield of ammonia (entry 6) (vide infra). The [(terpyridine)RuCl3] complex (3, see ESI,† Section S3.3) and the RuCl3·3H2O were found to result in 31% and 26% yields, respectively, which were similar to that of the blank reaction without any catalyst (see ESI,† Table S2). Replacing hydrazine hydrate with N2H4 (1 M in THF) in the presence of SmI2 and H2O and complex 2 yielded only 21% ammonia under the optimized reaction conditions (entry 7). The role of water concentration was investigated using anhydrous hydrazine, and the results indicated a requirement for a certain water loading concentration for the effective catalysis reaction (see ESI,† Section S9.4). The reaction in the absence of a reductant showed a drastic decrease in ammonia production to 9%, via the equation presented in Table S1 (ESI†), which indicated the need for the participation of both a reductant and a proton source (entry 8). In order to validate this, a disproportionation reaction was performed with hydrazine hydrate in the absence of any reductant/proton pair, and only a 7% yield of ammonia was detected along with the corresponding unreacted hydrazine (entry 9, see ESI,† Sections S5 and S8; Table S1). No H2 gas was detected using GC-TCD for the disproportionation reaction (see ESI,† Fig. S5), which supported the inefficiency of complex 2 towards the transfer hydrogenation reaction of substrates such as nitrobenzene and benzophenone in the presence of hydrazine hydrate as the hydrogen source (see ESI,† Section S12). The role of SmI2 as a reductant (E1/2 = −1.22 V in acetonitrile)16 was evaluated by comparison with other strong reducing agents such as CoCp2 (E1/2 = −1.3 in acetonitrile) and CoCp2* (E1/2 = −1.85 V in THF)17 using [LutH]OTf as the proton source, and the yields of ammonia were 48% and 8%, respectively (entry 10, 11). Around 14% and 62% yields of H2 gas were detected using CoCp2 and CoCp2*, respectively (see ESI,† Fig. S9 and S10). To validate this observation with the complex 2 redox potential, a cyclic voltammetry study was carried out, and 2 showed an active redox property, unlike complex 1 (see ESI,† Section S4). Complex 2 bearing the +3 oxidation state on the Ru centre displayed redox activity in the range of 0.9 V to −1.2 V, excluding the possibility of redox changes at the terpyridine backbone, which would likely occur at higher negative or positive potential.18 A reduction potential at −0.753 V was observed and assigned to the Ru(III) → Ru(II) process (see ESI,† Section S4.3; Fig. S3A, S3B).19 To investigate the involvement of atmospheric dinitrogen in ammonia generation in the presence of SmI2/H2O, a reaction was performed with a 60 equiv. of SmI2 loading and the same scale of catalyst. This reaction resulted in no yield of ammonia under the optimized reaction conditions (entry 12), indicating that the ammonia is not formed from atmospheric dinitrogen reduction.
|
|
||||
|---|---|---|---|---|
| Entrya | Cat | Electron source | Proton source | NH4+ (% yield) |
| a Reaction conditions: Catalyst (0.003 mmol, 3.3 mol%), N2H4·H2O (0.09 mmol, 30 equiv. with respect to the catalyst), SmI2 (0.18 mmol), H2O (0.18 mmol) at 55 °C for 4 h. b Reaction performed at room temperature. c Reaction performed for 12 h. d N2H4 (1 M in THF) was used. e Reaction performed without hydrazine. f Reaction performed with 1.5 mol% catalyst loading. g Reaction performed with 5.5 mol% catalyst loading. h Reaction performed with 13 mol% catalyst loading. | ||||
| 1b | 2 | SmI2 | H2O | 17 |
| 2 | 2 | SmI2 | H2O | 51 |
| 3c | 2 | SmI2 | H2O | 57 |
| 4 | 2 | SmI2 | — | 23 |
| 5 | — | SmI2 | H2O | 30 |
| 6 | 1 | SmI2 | H2O | 32 |
| 7d | 2 | SmI2 | H2O | 21 |
| 8 | 2 | — | H2O | 9 |
| 9 | 2 | — | — | 7 |
| 10 | 2 | CoCp2 | [LutH]OTf | 48 |
| 11 | 2 | CoCp2* | [LutH]OTf | 8 |
| 12e | 2 | SmI2 | H2O | — |
| 13f | 2 | SmI2 | H2O | 42 |
| 14g | 2 | SmI2 | H2O | 62 |
| 15h | 2 | SmI2 | H2O | 92.5 ± 3 |

In the hydrazine hydrate reduction, the reductant and catalyst concentration play a definite role in maximizing the production of ammonia by increasing the flow of electrons from the reductant to the substrate through the metal centre. Enhancing the reductant (SmI2) loading two-fold did not result in a noticeable increase in ammonia production (66%) (see ESI,† Section S9.3; Table S7). Further, a series of catalyst loading optimizations with complex 2 was performed by progressively increasing the catalyst loading to achieve a superior yield of ammonia while controlling the H2 production (see ESI,† Section S9.2; Fig. S15). As the catalyst 2 loading was varied to 1.5 mol%, 5.5 mol%, and 13 mol%, the ammonium production progressively increased to 42%, 62%, and 92.5% yield, respectively, indicating the role of the catalyst under the optimized reaction conditions (entry 13–15). A relatively minor amount of H2 gas was produced (analyzed using GC-TCD, see ESI,† Fig. S8) when the catalyst 2 intake exceeded 13 mol% (entry 15), which was qualitatively less than the amount of H2 gas produced when the catalyst 2 loading was set to 3.3 mol% (entry 2), which indicates the effective transfer of electrons to the N–N bond of the hydrazine.
To analyze the broader scope of this reduction process, the reactions with other N–N bonded analogue substrates were also examined. In the presence of catalyst 2 under the optimized reaction conditions (hydrazine (0.09 mmol), 2 (0.003 mmol, 3.3 mol%), SmI2(thf)2 (0.1 M in THF, 0.18 mmol), and H2O (0.18 mmol) for 4 h at 55 °C), phenylhydrazine produced an 82% yield of ammonia and 74% yield of aniline, and the bulkier analogue 1,2-diphenyl hydrazine showed a good response, generating an 86% yield of aniline through significant N–N bond cleavage (see ESI,† Section S11).
To detect the possible intermediate involved in the mechanistic studies during the reaction, complex 2 was treated with hydrazine hydrate or hydrazine (1 M in THF) for 2 h. Hydrazine-coordinated species A was afforded, which exhibited an ESI-MS peak at 660.11 corresponding to [A-I]+ units, where a reduction of the metal center [Ru(III) to Ru(II)] was observed (Scheme 3a, see ESI,† Section S10.3; Fig. S20). It was predicted that the iodide ion may behave as a one-electron reductant for the source of reduction at the Ru centre, which was verified using UV-vis spectroscopy of the liberation of iodine during the transformation (see ESI,† Section S10.8; Fig. S24).20,21 Treatment of complex 2 with gaseous ammonia in dry THF resulted in an ammonia-bound Ru complex (B), as confirmed by mass spectrometry, with a m/z value of 771.98 (Scheme 3b, see ESI,† Section S10.4; Fig. S21). The existence of both the in situ generated intermediates A and B in the catalytic cycle was investigated by utilizing both of them as the catalyst for the reduction reaction process. Intermediates A and B gave a 55.8% and 60.5% yield of ammonia, respectively, under the optimized reaction conditions (hydrazine (0.1 mmol), SmI2(thf)2 (0.1 M in THF, 0.2 mmol), and H2O (0.2 mmol) for 4 h at 55 °C) (see ESI,† Sections S10.6 and S10.7).
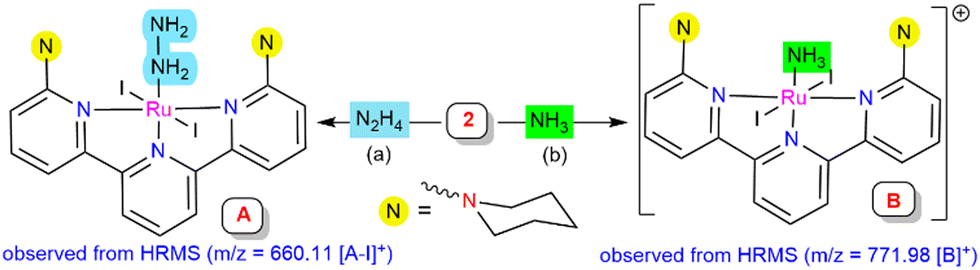 | ||
| Scheme 3 (a) Reactivity of complex 2 with hydrazine. (b) Reactivity of complex 2 with ammonia. | ||
Based on the experimental studies and the literature support,22 a plausible mechanistic pathway is proposed (Scheme 4). A series of control experiments were carried out with the observed intermediates in order to obtain a clear insight into the mechanism associated with this process. At the initial stage, complex 2 may generate intermediate A with the coordination of hydrazine to the Ru centre. Intermediate A was suggested to be stable, since treatment with an excess of ammonia gas did not cause any changes in the HRMS spectra (see ESI,† Section S10.5). This suggested that for the successive reduction of hydrazine to occur, the catalytically active species A requires external electrons and protons to facilitate substantial cleavage of the N–N bond. Intermediate A might protonate in the presence of the proton source to give intermediate A′, where the hydrazinium ion is predicted to stabilize with a hydrogen bonding interaction with the substituted N-arm of piperidine in the secondary coordination sphere.23 To verify the protonation at the piperidine N-arm, the ligand L1 was treated with formic acid, which resulted in a doubly protonated species distinguished from the HRMS and 1H NMR spectra (see ESI,† Section S10.10). This proposed H-bonding interaction may assist the release of one equivalent of ammonia to afford the amido-coordinated intermediate A′′ through N–N bond cleavage. The nitrogen atom (Nα) coordinated to the Ru centre of intermediate A′′ might protonate to generate ammonia-coordinated intermediate species B, which was independently observed upon treatment of complex 2 with NH3, as illustrated in Scheme 3b. The reactivity of intermediate B was determined by treatment with hydrazine (1 M in THF, 50 equiv.), resulting in an instantaneous shift in the peak from m/z = 771.98 (representing [B]+) to m/z = 660.09 (representing [A-I]+) in the HRMS spectra, suggesting the generation of intermediate A (see ESI,† Section S10.5), which was also supported by the UV-Visible spectrometry studies (see ESI,† Section S10.9). From the experimental finding, it was envisaged that upon further electron and hydrazine addition, intermediate B could eliminate the second equivalent of ammonia to regenerate intermediate A, which leads to further continuation of the catalytic cycle.
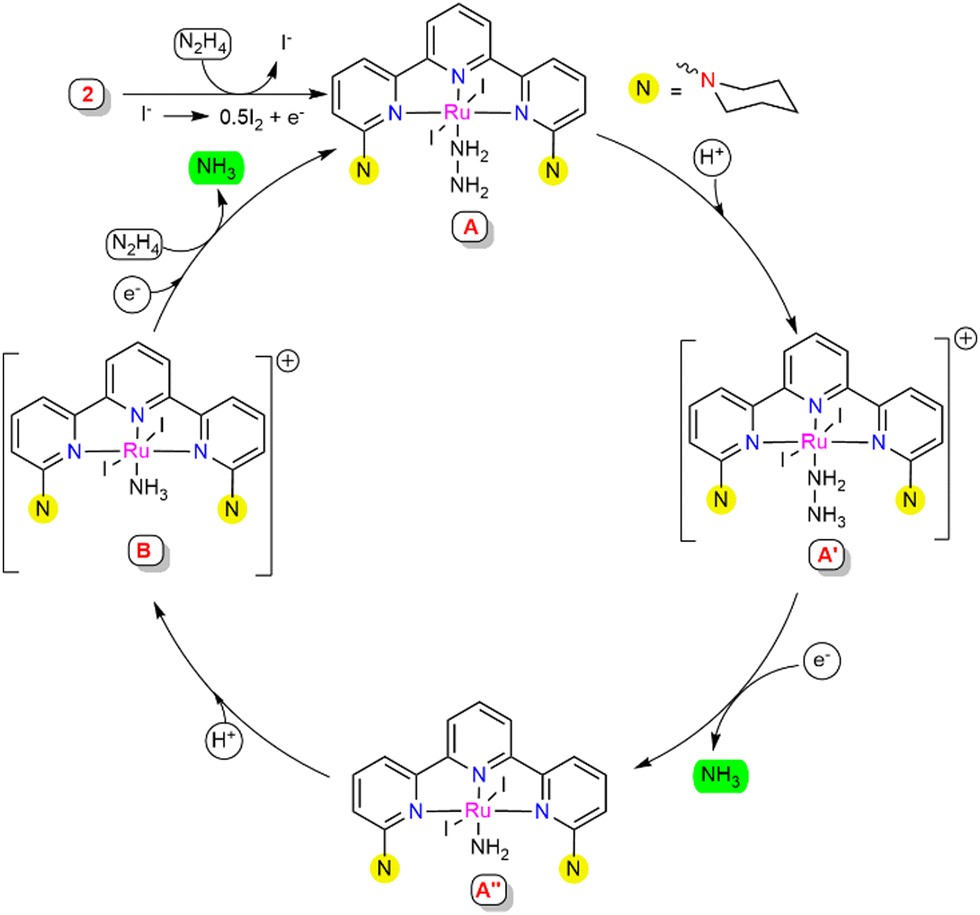 | ||
| Scheme 4 Proposed catalytic cycle. | ||
In summary, the synthesized triiodo-Ru complex 2 successfully catalyses the formation of ammonia from hydrazine in up to 92% yield through N–N bond scission using SmI2/H2O under ambient reaction conditions, thereby suppressing the generation of H2. The developed reaction systems are also compatible with the N–N bond cleavage of substituted hydrazines. The substitution of the piperidine arm in the C2 position of the terpyridine ring of L1 in the secondary coordination sphere is suggested to assist in the H-bonding interaction for facile catalytic efficiency towards the reduction reaction. Further investigation of the absolute arrangement and function of the piperidine arms with modification of the catalyst is currently in progress.
P. D. thanks SERB, DST, India (SRG/2020/000424) and IISER Berhampur for the financial support. We are thankful to IISER Berhampur for the Central Advance Instrument Facility (CAIF). We acknowledge Dr Santanu Bhattacharyya for the gas chromatography measurements. A. M. thanks IISER Berhampur for the fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. R. Eady, Chem. Rev., 1996, 96, 3013–3030 CrossRef CAS PubMed.
- J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS PubMed.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3011 CrossRef CAS PubMed.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- B. M. Barney, T. C. Yang, R. Y. Igarashi, P. C. Dos Santos, M. Laryukhin, H. I. Lee, B. M. Hoffman, D. R. Dean and L. C. Seefeldt, J. Am. Chem. Soc., 2005, 127, 14960–14961 CrossRef CAS PubMed.
- K. Umehara, S. Kuwata and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 6754–6757 CrossRef CAS PubMed.
- N. X. Gu, G. Ung and J. C. Peters, Chem. Commun., 2019, 55, 5363–5366 RSC.
- Y. Chen, Y. Zhou, P. Chen, Y. Tao, Y. Li and J. Qu, J. Am. Chem. Soc., 2008, 130, 15250–15251 CrossRef CAS PubMed.
- Y. Li, L. Su, D. Yang, K. Di, B. Wang and J. Qu, Dalton Trans., 2022, 51, 10866–10870 RSC.
- M. Yuki, Y. Miyake and Y. Nishibayashi, Organometallics, 2012, 31, 2953–2956 CrossRef CAS.
- W. C. Chu, C. C. Wu and H. F. Hsu, Inorg. Chem., 2006, 45, 3164–3166 CrossRef CAS PubMed.
- Y. H. Chang, P. M. Chan, Y. F. Tsai, G. H. Lee and H. F. Hsu, Inorg. Chem., 2014, 53, 664–666 CrossRef CAS PubMed.
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- J. Junge, T. A. Engesser and F. Tuczek, Chem. – Eur. J., 2023, 29, e202202629 CrossRef CAS PubMed.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- M. L. Kuhlman and R. A. Flowers, Tetrahedron Lett., 2000, 41, 8049–8052 CrossRef CAS.
- J. L. Robbins, N. Edelstein, B. Spencer and J. C. Smart, J. Am. Chem. Soc., 1982, 104, 1882–1893 CrossRef CAS.
- J. Lombard, D. A. Jose, C. E. Castillo, R. Pansu, J. Chauvin, A. Deronzier and M. N. Collomb, J. Mater. Chem. C, 2014, 2, 9824–9835 RSC.
- A. Breivogel, K. Hempel and K. Heinze, Inorg. Chim. Acta, 2011, 374, 152–162 CrossRef CAS.
- A. Reynal and E. Palomares, Eur. J. Inorg. Chem., 2011, 4509–4526 CrossRef CAS.
- G. Kerenskaya, I. U. Goldschleger, V. A. Apkarian, E. Fleischer and K. C. Janda, J. Phys. Chem. A, 2007, 111, 10969–10976 CrossRef CAS PubMed.
- A. K. Guha and A. K. Phukan, Inorg. Chim. Acta, 2010, 363, 3270–3273 CrossRef CAS.
- P. Bhattacharya, D. E. Prokopchuk and M. T. Mock, Coord. Chem. Rev., 2017, 334, 67–83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Detailed experimental techniques, spectral information, and crystallographic information for ligand L1 (CCDC 2290962) are provided. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04490d |
| This journal is © The Royal Society of Chemistry 2024 |