
Unprecedented urea oxidation on Zn@Ni-MOF with an ultra-high current density: understanding the competition between UOR and OER, catalytic activity limitation and reaction selectivity†
Nabeen K.
Shrestha
 *a,
Supriya A.
Patil
b,
Amol S.
Salunke
a,
Akbar I.
Inamdar
a and
Hyunsik
Im
*a
*a,
Supriya A.
Patil
b,
Amol S.
Salunke
a,
Akbar I.
Inamdar
a and
Hyunsik
Im
*a
aDivision of Physics and Semiconductor Science, Dongguk University, Seoul 04620, Republic of Korea. E-mail: nabeenkshrestha@hotmail.com; hyunsik7@dongguk.edu
bDepartment Nanotechnology & Advanced Materials Engineering, Sejong University, Seoul 05006, Republic of Korea
First published on 26th June 2023
Abstract
Urea oxidation reaction (UOR) has been extensively studied as an alternative to the sluggish oxygen evolution reaction (OER) for energy-efficient hydrogen generation. However, the detrimental competition between the UOR and OER limits the UOR current density to less than 500 mA cm−2 and ultimately switches the reaction toward the OER. In this study, we attempted to gain a fundamental understanding of the catalytic activity limitation for the UOR and the possible factors influencing the reaction selectivity employing Ni-MOF as an example. The study showed that upon doping the Ni-lattices of the framework with Zn, the factors influencing the detrimental competition, such as the mass and charge transport ability of the MOF catalyst could be enhanced and the formation of the catalytically active Ni3+-OOH phase could be accelerated. This populated Zn@Ni-MOF with Ni3+-OOH sites, and subsequently prevented the detrimental competition between the anodic reactions. Consequently, Zn@Ni-MOF demonstrated an outstanding ultra-high UOR current density of 1780 mA cm−2 at a low electrode potential of 1.52 V vs. RHE and the benchmark current density of 10 and 100 mA cm−2 at a lower electrode potential of 1.31 and 1.32 V vs. RHE, respectively, hence outperforming most of the high-performance UOR catalysts.
Introduction
Eco-friendly clean technologies for generating green fuels together with the simultaneous remediation of the current environmental issues are essential for the development of a sustainable society. Recently, the transformation of the conventional fossil to hydrogen economy has been identified as a great opportunity for enhancing the global energy security and mitigating greenhouse gases.1–3 The electrolysis route for splitting water into molecular hydrogen and oxygen is emerging as carbon-free clean-energy technology.4–6 However, the intrinsically sluggish kinetics of the oxygen evolution reaction (OER) at the anode is a bottleneck for the overall splitting of water.7–10 Owing to the lower theoretical overall urea electrolysis potential than that of water electrolysis and the huge difference in their cell voltage (+0.37 V for urea electrolysis versus +1.23 V for water electrolysis), the electrochemical urea oxidation reaction (UOR) is apparently a thermodynamically more feasible process than the OER.11–15 Consequently, recently extensive attention has been paid to the UOR as a promising alternative anodic reaction for energy-efficient hydrogen production in water electrolysis.16–26 In addition, the urea-rich wastewater from the fertilizer industry, agricultural residues, and human/animal wastes are abundantly available and easily transportable resources of hydrogen. Therefore, the UOR as an alternative anodic reaction to the OER can reduce the cost of hydrogen production and decontaminate urea waste. However, the UOR (i.e., CO(NH2)2 + 6OH− → CO2 + N2 + 5H2O + 6e−) involves a 6-electron transfer process, leading to a high energy consumption, and hence high operational cost is inevitable. In this case, a straightforward approach for mitigating the energy consumption is to develop efficient and low-cost UOR electrocatalysts to boost the UOR at lower overpotentials. Also, to substitute the expensive noble metal-based state-of-the art electrocatalysts, various materials derived from earth-abundant elements have been explored for the OER and UOR.27–40However, it is worth noting that regardless of the potential bias, the maximum UOR current density, which reflects the urea oxidation rate, is often reported to be less than 500 mA cm−2.11–26,32–40 This is because of the detrimental competition existing between the UOR and OER, resulting in a current trade-off region in the anodic polarization curves.17,41–45 Although many works have been reported on the catalytic performance of various catalysts for easily oxidizable organic molecules including urea, hardly any work can be found in the literature regarding the fundamental understanding of the catalytic activity limitation and reaction selectivity during urea electrolysis.46 Encouraged by this pioneer work, the present work attempted to achieve a basic understanding of the possible factors contributing to the detrimental competition between the UOR and OER, leading to a trade-off in the current during urea electrolysis. Accordingly, the design of a highly porous material such as metal–organic-frameworks (MOFs), offering abundant catalytic sites and facilitating charge/mass transfer,47–49 is highly desirable. In this case, a thin film of zinc-doped nickel-based metal–organic framework (Zn@Ni-MOF) was uniformly deposited on a nickel foam (NF) substrate via the solvothermal route. The highly ordered porous crystalline structure of the MOF uniformly possessed high densities of Ni-centers in its framework structure, which acted as the active catalytic centers. This enhanced the population of the surface-bound catalytic sites for the UOR. In addition, Zn-doping modulated the electronic structure of the framework,50 weakening the coordination bond between the nickel and organic ligand. This led to the delocalization of electrons, facilitating the charge transport, and hence enhancing the conductivity of the framework. Consequently, in contrast to the pristine Ni-MOF/NF, the Zn@Ni-MOF/NF inhibited the current trade-off at the UOR ↔ OER competition zone in the anodic polarization curve. Thus, compared with the pristine Ni-MOFs/NF, Zn@Ni-MOF/NF demonstrated a remarkably high UOR current density of 1500, 1000, and 500 mA cm−2 at a lower electrode potential of 1.50, 1.44, and 1.37 V vs. RHE, respectively. In addition, Zn@Ni-MOF/NF achieved a benchmark UOR current density of 100 and 10 mA cm−2 at a notably lower electrode potential of 1.32 and 1.31 V vs. RHE, respectively, thus outperforming the state-of-the-art IrO2/NF and pristine Ni-MOF/NF. This catalytic performance lies in the range of high-performance UOR electrocatalysts (Table S1†). Most importantly, the above-mentioned UOR potentials are lower by 310, 310, 300, 260, and 185 mV than that of the OER counterparts to deliver the UOR current density of 1500, 1000, 500, 100 and 10 mA cm−2, respectively. Thus, this work illustrates the UOR as a promising alternative to the OER on the Zn@Ni-MOF/NF anode for energy efficient green-hydrogen production at the cathodic side of a water-electrolyzer.
Results and discussion
Characterization of MOF film
Fig. 1 shows the scanning electron microscopy (SEM) images of the surface of the Ni-MOF and Zn@Ni-MOF films deposited on the nickel backbone of the NF-substrate. As is evident, the pristine Ni-MOF film consisted of a sheet-like structure, whereas the Zn@Ni-MOF film showed an assembly of overlapping nanosheets, forming 3D-microstructures. Upon the introduction of the Zn-dopant and increasing its concentration in the Ni-MOF precursor, the gradual transformation of the MOF film from 2D-nanosheets into 3D structures, and finally an assembly of tubular bulky structures was realized, as shown in Fig. S1.† All these MOF film samples were tested briefly in a control experiment for UOR activity via anodic LSV polarization in 0.33 M urea containing 1.0 M KOH aqueous electrolyte, as displayed in Fig. S2a.† Amongst the samples, the MOF film obtained by adding 0.5 mmol of ZnCl2 to the Ni-MOF precursor (i.e., 0.5-Zn@Ni-MOF/NF sample) exhibited the best UOR activity. For simplicity, this sample and the pristine Ni-MOF (0.0-Zn@Ni-MOF/NF) are named hereafter as Zn@Ni-MOF/NF and Ni-MOF/NF, respectively. Their detailed polarization curves recorded in urea electrolysis with and without iR loss compensation of 90% are displayed in Fig. S2b.† Based on the catalytic activity in the UOR, further characterization of the samples mainly focused on Ni-MOF/NF and Zn@Ni-MOF/NF. | ||
| Fig. 1 SEM surface views of (a) Ni-MOF and (b) Zn@Ni-MOF films deposited on a nickel foam. | ||
As a result of the strong X-ray diffraction (XRD) from the NF-substrate, Zn@Ni-MOF/NF only exhibited a few weak peaks from the MOF film, as shown in Fig. S3.† However, the majority of the peaks from the bulk MOF powder well-matched that of the Zn@Ni-MOF/NF sample, indicating the successful deposition of the MOF film on the NF-substrate. Further XRD analysis was conducted by collecting the powdery MOF from the loosely adhered deposits on the NF-substrate. Fig. 2a displays the powder-XRD patterns of the pristine and Zn-doped Ni-MOF. It should be noted that the XRD patterns are well-matched to that of the Ni(NH2-BDC) MOF-based frameworks reported previously, showing the major peaks at 2θ values of approximately 5.6°, 10.9°, 16.4°, 19.7°, 20.3°, 22.7° and 26.1°.51–54 The XRD pattern and the peak positions are very close to that of the MIL-88C(Fe) MOF system belonging to hexagonal symmetry with the space group of P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2c. The structure of Zn@Ni-MOF is schematically shown in Fig. S4.† However, it should be noted that the major XRD peaks after Zn-doping slightly shifted toward a lower diffraction angle. This can be ascribed to the insertion of the larger Zn atoms (atomic radius: Zn = 0.138 nm and Ni = 0.124 nm) into the crystal lattices of the frameworks. This finding is also supported by the existing electronic interaction between the Zn and Ni atoms, as indicated by the X-ray photoelectron spectroscopic (XPS) study of the MOF films. Fig. S5† displays the XPS elemental survey spectra, showing the presence of Zn in addition to the C, N, O and Ni species in the Zn@Ni-MOF/NF sample. Fig. 2b and c display the high-resolution XPS 2p spectra of Ni in the Ni-MOF/NF and Zn@Ni-MOF/NF samples, respectively. Evidently, a clear shift in the Ni 2p peaks toward a lower binding energy can be observed in the Zn@Ni-MOF/NF sample. This shift implies that electronic rearrangements in the coordination bond existing between the Ni-core and organic ligand took place after the introduction of Zn atoms. This shift can be explained based on the relative electronegativity of Zn and Ni. The electronegativity of Ni in the Pauling scale is 1.91, which indicates that it is more electronegative than Zn, having an electronegativity value of 1.65. Based on their difference in electronegativity, it can be assumed that electron transfer occurred from the Zn-dopant to the Ni-nodes in the framework. Consequently, the Ni 2p peaks of the Zn@Ni-MOF/NF sample shifted toward the lower binding energy region.
2c. The structure of Zn@Ni-MOF is schematically shown in Fig. S4.† However, it should be noted that the major XRD peaks after Zn-doping slightly shifted toward a lower diffraction angle. This can be ascribed to the insertion of the larger Zn atoms (atomic radius: Zn = 0.138 nm and Ni = 0.124 nm) into the crystal lattices of the frameworks. This finding is also supported by the existing electronic interaction between the Zn and Ni atoms, as indicated by the X-ray photoelectron spectroscopic (XPS) study of the MOF films. Fig. S5† displays the XPS elemental survey spectra, showing the presence of Zn in addition to the C, N, O and Ni species in the Zn@Ni-MOF/NF sample. Fig. 2b and c display the high-resolution XPS 2p spectra of Ni in the Ni-MOF/NF and Zn@Ni-MOF/NF samples, respectively. Evidently, a clear shift in the Ni 2p peaks toward a lower binding energy can be observed in the Zn@Ni-MOF/NF sample. This shift implies that electronic rearrangements in the coordination bond existing between the Ni-core and organic ligand took place after the introduction of Zn atoms. This shift can be explained based on the relative electronegativity of Zn and Ni. The electronegativity of Ni in the Pauling scale is 1.91, which indicates that it is more electronegative than Zn, having an electronegativity value of 1.65. Based on their difference in electronegativity, it can be assumed that electron transfer occurred from the Zn-dopant to the Ni-nodes in the framework. Consequently, the Ni 2p peaks of the Zn@Ni-MOF/NF sample shifted toward the lower binding energy region.
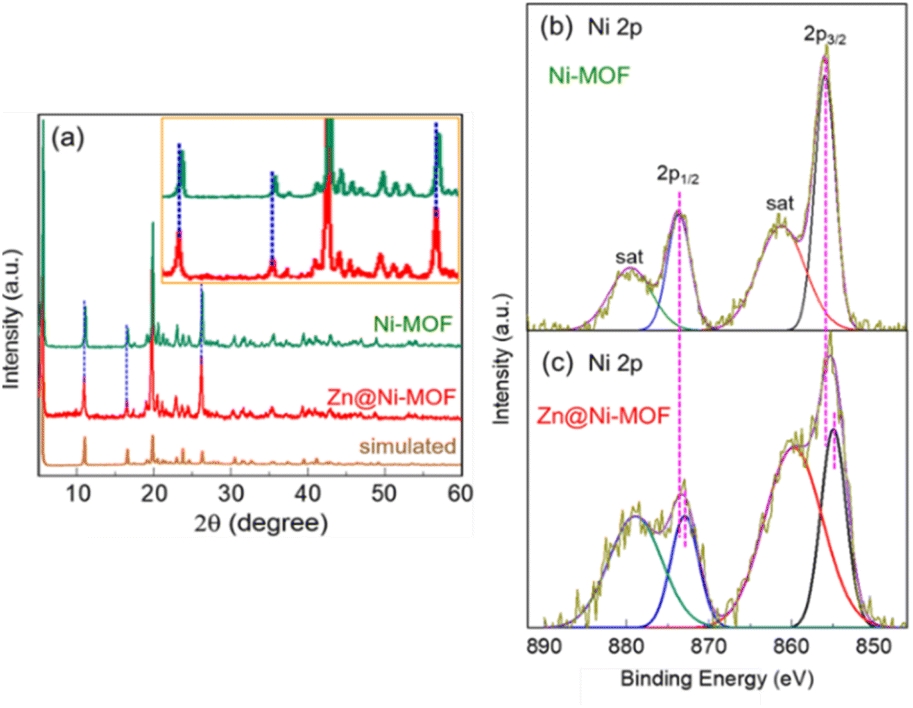 | ||
| Fig. 2 XRD pattens of Ni-MOF and Zn@Ni-MOF powders. XPS Ni 2p spectra of (b) Ni-MOF and (c) Zn@Ni-MOF films on nickel foam. Inset in figure (a) shows an enlarged view showing the shift in the XRD peaks after Zn-doping. | ||
The morphology and crystal structure of the Zn@Ni-MOF film was further studied using transmission electron microscopy (TEM). Consistent with the SEM result, the TEM image also showed the thin sheet-like structure of the Zn@Ni-MOF film (Fig. 3a). In the high-resolution mode, the film showed crystal lattice fringes, and one of them is highlighted as the (311) plane having the lattice spacing of 0.44 nm. To elucidate the distribution of the elements in the film, scanning transmission electron microscopy (STEM) was employed, which showed the uniform distribution of Zn, Ni and N throughout the film (Fig. 3d–f), respectively.
 | ||
| Fig. 3 (a) TEM and (b) HR-TEM images of the Zn@Ni-MOF film. (c) STEM-EDS elemental distribution mapping image and individual elemental distribution mapping images for (d) Zn, (e) Ni and (f) N. | ||
Influence of Zn-doping on the OER and UOR and reaction selectivity
To evaluate the catalytic activity, firstly the OER catalytic performance of the pristine Ni-MOF/NF and Zn@Ni-MOF/NF was studied by polarizing the electrodes anodically in 1.0 M KOH aqueous electrolyte. The resulting LSV polarization curves are displayed in Fig. S6a.† As can be noticed from the polarization curves, the bare NF-substrate has the poorest catalytic performance for the OER, while the pristine Ni-MOF/NF electrode exhibited a slightly better catalytic performance. More importantly, Zn@Ni-MOF/NF demonstrated comparable catalytic activity to that of the benchmark IrO2/NF electrode, particularly up to the current density of about 100 mA cm−2 with an OER overpotential of 340 mV. Beyond this point, Zn@Ni-MOF/NF demonstrated notably lower OER overpotentials of 385, 440, 520, 580 and 630 mV to attain the OER current density of 250, 500, 1000, 1500 and 2000 mA cm−2, respectively (Fig. S6b†). In addition, the chronopotentiometric stability test, as displayed in Fig. S6c,† revealed that the Zn@Ni-MOF/NF anode exhibited excellent long-term electrochemical durability against 100 mA cm−2 load, maintaining its OER catalytic activity for the 48 hours test.Similar to the OER, the catalytic performance of the pristine and doped MOF-based electrodes in the UOR was evaluated by polarizing the electrodes in 0.33 M urea containing 1.0 M KOH aqueous electrolyte. Fig. 4a depicts that the UOR occurred more readily than the OER. However, with a gradual increase in the polarization potential, a transition from the UOR to the OER took place at a certain potential depending on the catalyst material. Lin et al. identified that the detrimental competition between the UOR and OER in alkaline electrolyte is mainly due to the inadequate concentration of the catalytically active Ni3+-OOH phase.46 During the UOR, the Ni2+-centres existing on the surface of the frameworks are first hydrolysed to the corresponding hydroxide, followed by oxidation to the high-valence Ni3+-OOH phase. The UOR proceeds by consuming this catalytically active phase constantly, as shown by reaction (1).12,18,45
| 6Ni3+-OOH (s) + CO(NH2)2 (aq) + 6OH− → 6Ni2+-(OH)2 (s) + N2 (g) + 5H2O + CO2 (g) + 6e− | (1) |
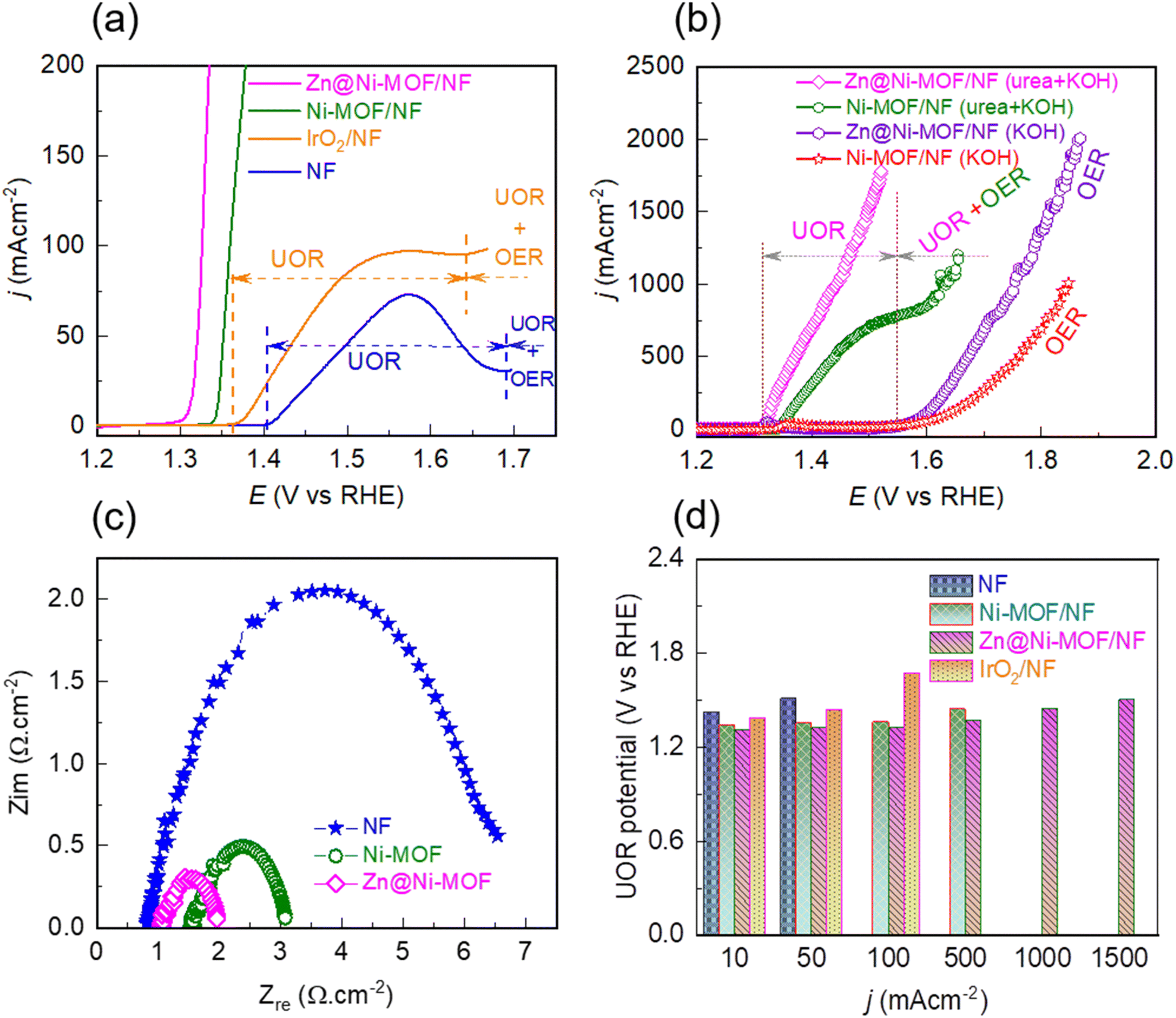 | ||
| Fig. 4 Linear sweep voltammetry curves of various catalytic electrodes in (a) 0.33 M urea containing 1.0 M KOH aqueous electrolyte, (b) 1.0 M KOH aqueous electrolyte indicated by the OER curve, and 0.33 M urea containing 1.0 M KOH aqueous electrolyte indicated by the UOR and UOR + OER curves. (c) Nyquist plots obtained by measuring electrochemical impedance in 0.33 M urea containing 1.0 M KOH aqueous electrolyte at 1.34 V vs. RHE. (d) UOR potential verses current density profile of various catalytic electrodes. | ||
With an increase in the polarization potential, more and more Ni3+-OOH sites developed, and subsequently consumed. At a certain bias potential when the formation of the Ni3+-OOH sites was slower than their consumption, the oxidation current became limited, leading to the formation of an UOR ↔ OER trade-off zone, as illustrated in Fig. 4a and b. Interestingly, when the Ni-MOF/NF and Zn@Ni-MOF/NF electrodes were polarized in 1.0 M KOH aqueous electrolyte, an oxidation peak corresponding to the formation of the Ni3+-OOH phase was observed (Fig. S7†). It should be noted that the peak appeared earlier in the case of Zn@Ni-MOF/NF. This can be ascribed to the electronic structure modulation caused by Zn-doping, thereby weakening the coordination bond between Ni and the organic ligand, and hence facilitating the formation of the Ni3+-OOH phase. In addition, the peak current was also higher, which based on the Randles–Sevcik equation indicates that Zn-doping populates the density of the Ni3+-OOH sites in Zn@Ni-MOF/NF. Another important observation in Fig. S7† is the onset potentials for the formation of the Ni3+-OOH phase and the UOR, which exhibit that the UOR takes place soon after the formation of the Ni3+-OOH active phase.11,16,17,39,42,44,55 The formation of the Ni3+-OOH phase on the anode surface is also supported by the XPS analysis (Fig. S8†). Guided by the oxidation peak for the formation of the Ni3+-OOH phase shown in Fig. S7,† when urea electrolysis was performed at 1.40 V vs. RHE, the Zn@Ni-MOF/NF anode exhibited additional peaks for the Ni3+-phase that were not initially present. Furthermore, direct evidence for the formation of the Ni3+-OOH phase is apparent when the MOF/NF anodes after urea electrolysis at 1.40 V vs. RHE were analysed through Raman spectroscopy. As is evident in Fig. S9,† when the urea electrolysis was performed at 1.40 V vs. RHE, a sharp characteristic doublet located at ca. 479 and 558 cm−1 corresponding to the existence of the Ni3+-OOH phase was observed in the Raman spectra.56 It should be noted that these characteristic Raman peaks did not appear when urea electrolysis was performed at 1.25 V vs. RHE. Consequently, when the polarization was continued beyond the onset potential for the formation of the Ni3+-OOH phase (i.e., ca. 1.30 V vs. RHE, Fig. S7†), the MOF/NF anodes demonstrated an abrupt rise in current due to urea oxidation. It is worth noting that Zn@Ni-MOF/NF exhibited a remarkably high UOR current density of 1780 mA cm−2 at the bias of 1.52 V vs. RHE (Fig. 4b). Most importantly, no UOR ↔ OER trade-off zone was observed in this case. To the best of our knowledge, this is the highest UOR current density reported to date without interfering with the UOR by OER. In contrast, Ni-MOF/NF exhibited a UOR ↔ OER trade-off zone at ca. 1.55 V vs. RHE with a maximum UOR current density of 779 mA cm−2.
The other possible factors that contribute to the UOR ↔ OER competition can be the charge and mass transfer limitation. The poor charge transfer can slow down the UOR, which involves a 6-electron process compared with the OER, proceeding only with a 4-electron process. Alternatively, the accumulation of gas bubbles at the electrode surface can hinder the diffusion of urea molecules to the active sites. Thus, the charge and mass transfer limitation inhibit the UOR, thereby diminishing the current density at a certain bias potential. The charge transfer property of the electrodes was studied via electrochemical impedance spectroscopy (EIS). The impedance was recorded at a bias of 1.34 V vs. RHE to ensure the existence of the UOR at the Ni-MOF/NF and Zn@Ni-MOF/NF electrodes. Fig. 4c depicts the Nyquist plot, wherein the semicircles represent the charge transfer resistance (Rct) between the electrode and electrolyte. Thus, Zn@Ni-MOF/NF showed the smallest semicircle with the lowest Rct of 0.87 Ω, while Ni-MOF/NF showed a higher Rct of 1.54 Ω. This reveals that the UOR kinetics at the Zn@Ni-MOF/NF electrode/electrolyte interface is relatively faster. In addition, the chronopotentiometric curves recorded at the bias of 100 mA cm−2 clearly demonstrate the impact of gas bubble build-up on the surface of the electrode, as shown in Fig. S10.† As a result of the continuous dissipation of gas bubbles at the Zn@Ni-MOF/NF electrode surface, the chronopotentiometric curve is smooth. In contrast, Ni-MOF/NF displayed a fluctuation in its chronopotentiometric curve due to the slower release of the gas bubbles. The bubbles residing on the electrode surface also hinder the access of the electrolyte, limiting the available active sites, which ultimately slows down the rate of the UOR. According to the above-mentioned findings, enhanced charge and mass transfer characteristics of the Zn@Ni-MOF/NF electrode can be realized.
Fig. 4d presents the UOR potential verses current density profile of the various catalytic electrodes under study. Notably, Zn@Ni-MOF/NF exhibited the relatively lower UOR potential of 1.31, 1.32 and 1.37 V vs. RHE to deliver the UOR current density of 10, 100 and 500 mA cm−2, outperforming Ni-MOF/NF, which showed the higher UOR potential of 1.34, 1.36 and 1.44 V vs. RHE to achieve the same UOR current density, respectively. The superior catalytic performance of the Zn@Ni-MOF/NF electrode can be attributed to its facile charge/mass transfer characteristics of. Moreover, the electrochemically active surface area (ECSA) of the electrodes was estimated by determining the double layer capacitance (Cdl) from cyclic voltammetry carried out at various potential scan rates in the non-faradic region. The ESCA was calculated using the relation ECSA = Cdl/Cs, where Cs is the specific capacitance of the electrode and is generally recommended to be the value of 0.04 mF cm−2 in 1.0 M KOH alkaline electrolyte.57 Thus, the ECSA of the Zn@Ni-MOF/NF electrode was found to be 173.50 cm−2, which is larger than that of Ni-MOF/NF (118.25 cm−2), as presented in Fig. S11.† This finding reveals that Zn@Ni-MOF/NF has a higher density of active catalytic sites. Hence, Zn@Ni-MOF/NF having relatively larger number of catalytic sites with facile charge and mass transfer characteristics is indisputably the superior UOR catalyst. The UOR performance of Zn@Ni-MOF/NF is among the top UOR electrocatalysts reported to date (see Table S1†). Most importantly, this electrode exhibited a remarkably high UOR current density of 1780 mA cm−2 at a small bias of only 1.52 V vs. RHE. To the best of our knowledge, previous studies have rarely presented a maximum UOR current density above 500 mA cm−2 (Table S1†). This is because of the existence of a UOR ↔ OER trade-off zone, switching the reaction toward the OER side. In some cases, a higher UOR current density without showing a clear UOR ↔ OER trade-off zone was found, which can be ascribed to the closer onset potential for the UOR and OER activating the electrode for both the anodic reactions simultaneously.25 The transition potential between the UOR and UOR + OER can be ascertained from the onset of the OER, as illustrated by the vertical dotted line in Fig. 4a and b.
In addition to the UOR, the catalytic activity of the pristine Ni-MOF/NF and Zn@Ni-MOF/NF electrodes was also evaluated toward the hydrogen evolution reaction (HER) by polarizing the electrodes cathodically in 1.0 M KOH solution containing 0.33 M urea. The LSV polarization curves are shown in Fig. S12,† which reveal the HER overpotential of 189 and 195 mV for the Ni-MOF/NF and Zn@Ni-MOF/NF cathodes, respectively. However, their HER activity was not that impressive. Hence, no further evaluation toward the HER was carried out.
Long-term electrochemical stability
To study the practicability of the electrodes in the UOR, their long-term electrochemical stability was tested chronopotentiometrically. The recorded chronopotentiometric traces are presented in Fig. 5. As is evident, the Ni-NOF/NF electrode showed a gradual increase in potential over the electrolysis time. However, the chronopotentiometric trace deviated aggressively when the bias load increased from 10 to 100 mA cm−2, indicating the failure of the electrode to maintain its catalytic activity. In contrast, the Zn@Ni-MOF/NF electrode showed an increment in potential from 1.37 V to 1.40 V vs. RHE in 24 h at 100 mA cm−2 bias, revealing that it has the ability to maintain its catalytic activity even at a high operational bias of 100 mA cm−2 for a long electrolysis time. It is worth noting that there are very few reports of the UOR catalysts showing long-term stability at 100 mA cm−2, while the majority of UOR catalysts have only been tested at 10 mA cm−2 due to their poor stability at a higher current bias (for example, see Table S1†).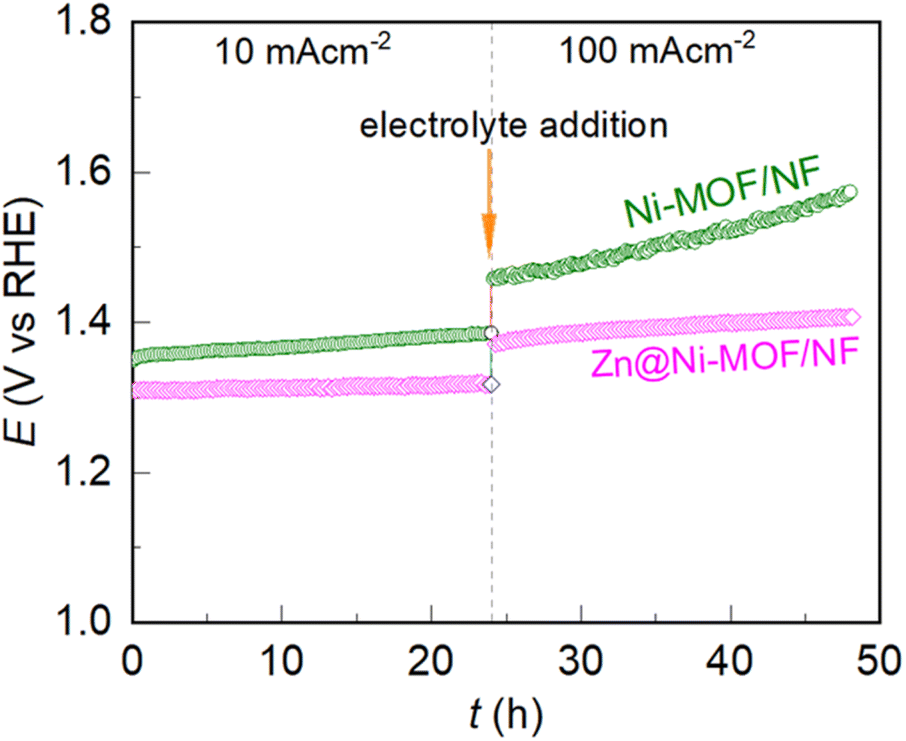 | ||
| Fig. 5 Chronopotentiometric responses recorded during the long-term electrochemical stability test at 10 and 100 mA cm−2 in 0.33 M urea containing 1.0 M KOH aqueous electrolyte. | ||
After the stability test, the electrode was examined with SEM and XRD. Fig. 6a and b display the SEM surface view of the Zn@Ni-MOF/NF before and after the long-term stability test, respectively. The electrode surface after the long-term stability test changed drastically. A smother surface was observed after the stability test, as shown in Fig. 6b, suggesting that electrochemical etching of the catalyst film took place during the long-term UOR.
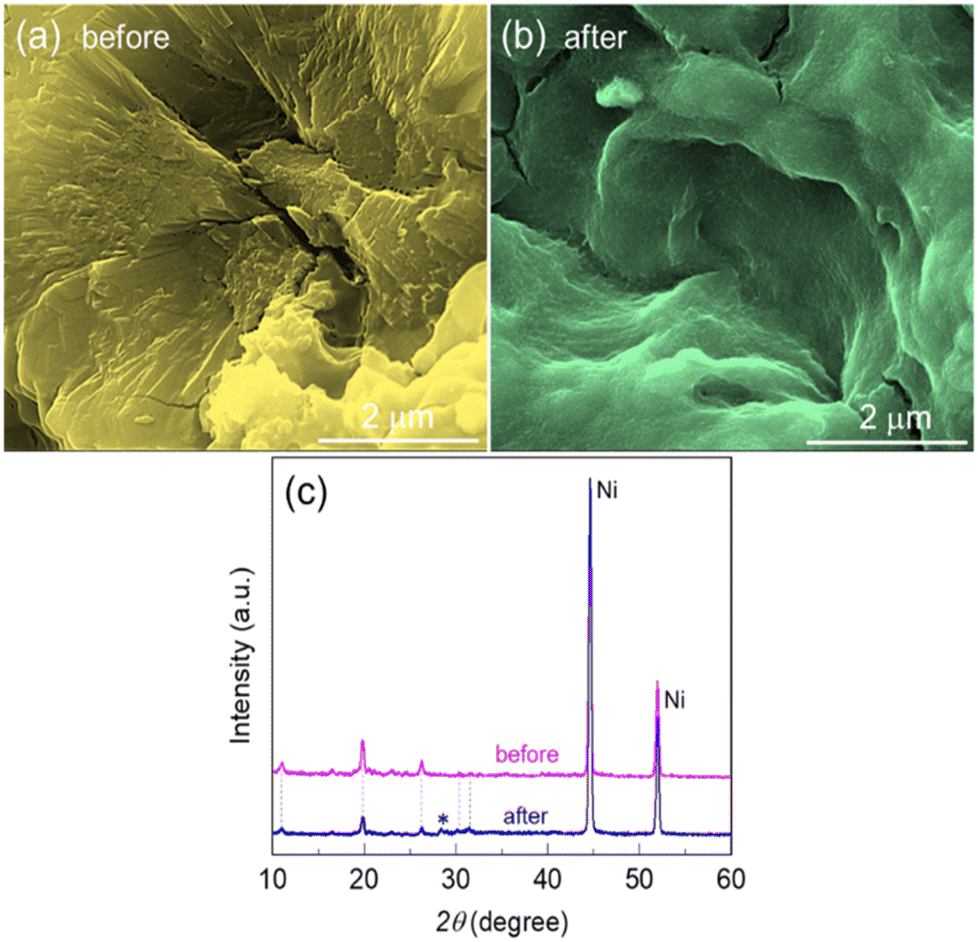 | ||
| Fig. 6 SEM images showing the surface of the Zn@Ni-MOF/NF electrode (a) before, and (b) after the long-term electrochemical stability test for 24 h at 10 and 100 mA cm−2 in 0.33 M urea containing 1.0 M KOH aqueous electrolyte. (c) XRD patterns of the Zn@Ni-MOF/NF electrode before and after the long-term stability test. The peak assigned to * is the unknown peak that appeared after the stability test. | ||
This observation is also supported by the XRD patterns, as shown in Fig. 6c, wherein the peaks from the MOF-deposits diminished, while that of the NF-substrate intensified. These findings suggest that there may be some leaching of the catalyst materials from the anode surface after the long-term stability test. However, it should be noted that the major XRD peaks from the MOF-deposits still remained in their positions even after the long-term electrolysis. A small unknown XRD peak (indicated by *) appeared after the stability test, which could be due to the partial oxidation of the electrode materials. Overall, the catalytic activity performance and the long-term electrochemical durability suggest the promising practicability of the Zn@Ni-MOF/NF electrode in UOR catalysis.
Conclusions
This work demonstrated that the consumption of the high-valence Ni3+-OOH phase acting as the active catalytic sites for the UOR is the key to inducing the detrimental competition between the UOR and OER, resulting in a current trade-off region in the anodic polarization curves. Alternatively, the limitation of the charge- and/or mass-transfer of the electrode materials, which gradually slows down the rate of the UOR at a high potential bias, ultimately switches the reaction toward the OER. Thus, the charge and mass transport abilities of the electrodes can possibly be the additional factors contributing equally to the detrimental competition. By doping the relatively electropositive Zn-atoms in the Ni-lattice in Ni-MOF, the electronic structure of the framework could be modulated, thereby weakening the coordination bond between the nickel and organic ligand. This led to the delocalization of electrons, thereby facilitating the charge transport and accelerating the formation of the Ni3+-OOH phase. Thus, compared to the pristine Ni-MOF, the doped Zn@Ni-MOF prevented the detrimental competition between the UOR and OER, thereby resulting an outstanding ultra-high UOR current density of 1780, 1500, and 1000 mA cm−2 at a lower electrode potential of 1.52, 1.50, and 1.44 V vs. RHE, respectively, hence outperforming the state-of-the-art IrO2 catalyst and the pristine Ni-MOF/NF.Experimental
Materials and methods
Reagent grade ZnCl2 ≥ 98%, NiCl2 98%, 2-aminoterephthalic acid 99%, triethylamine ≥ 99.5%, N,N-dimethylformamide 99.8%, KOH pellets ≥ 85% and urea 99.0–100.5% were obtained from Sigma-Aldrich, USA. 1.6 mm thick nickel foam (NF) sheets were obtained from Alantum Corporation, South Korea. Prior to use, the NF sheets were chopped into 1 × 5 cm2 sizes and washed ultrasonically for 10 min in 2 M HCl, deionized water, ethanol, and acetone sequentially, and then dried overnight at room temperature.Deposition of MOF films
MOF films were synthesized on the NF substrates following the conventional solvothermal route. For the deposition of the Ni-MOF film, 1.5 mmol of NiCl2 and 2-aminoterephthalic acid were dissolved in 50 mL dimethylformamide containing 20 μL of triethylamine via ultrasonication. The mixture was transferred to a Teflon-lined stainless-steel autoclave. Then, 2 to 3 pieces of NF substrate were immersed in the solution and reacted at 150 °C for 15 h. After deposition, the obtained films were washed thoroughly with dimethylformamide and dried in a desiccator. For Zn-doping, x mmol of ZnCl2 in the range of 0.25 to 1.0 mmol and (1.5 − x) mmol of NiCl2 were reacted with 1.5 mmol aminoterephthalic acid in 50 mL dimethylformamide containing 20 μL of triethylamine, as described above.Electrochemical measurements
In a home-made glass cell, an SCE (saturated calomel electrode), graphite and NF deposited with the MOF films were used as the reference, counter and working electrodes, respectively. For the electrocatalytic activity measurements, the working electrodes were first activated via cyclic voltammetry (CV) at a potential sweep of 100 mV s−1 continuously until stable voltammograms were obtained. Then, the catalytic performance was measured by polarizing the working electrode anodically at the scan rate of 5 mV s−1 in 1.0 M KOH or 0.33 M urea containing 1.0 M KOH solution using a BioLogic Science Instruments workstation. The obtained potentials were converted to the reversible hydrogen electrode (RHE) scale using the relation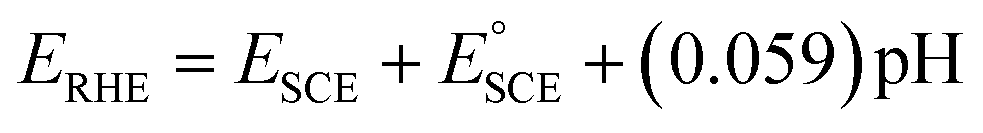 , where
, where 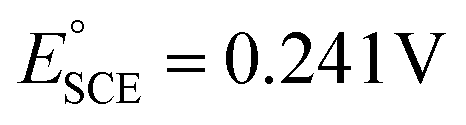 and pH is the measured pH of the electrolytic solution. All voltammograms were recorded with 90% iR loss compensation.
and pH is the measured pH of the electrolytic solution. All voltammograms were recorded with 90% iR loss compensation.
Author contributions
Nabeen K. Shrestha: conceptualization, investigation, experimental analysis, and writing original draft & editing. Supriya A. Patil: experimental analysis, data curation, and writing. Amol S. Salunke: experimental analysis. Akbar I. Inamdar: writing & editing. Hyunsik Im: supervision, project administration, and funding acquisition.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the National Research Foundation (NRF) of Korea (grant no. NRF-2022R1F1A070724).References
- J. O. Abe, A. P. I. Popoola, E. Ajenifuja and O. M. Popoola, Int. J. Hydrogen Energy, 2019, 44, 15072 CrossRef CAS.
- B. C. Tashie-Lewis and S. G. Nnabuife, Chem. Eng. J. Adv., 2021, 8, 100172 CrossRef CAS.
- A. M. Oliveira, R. R. Beswick and Y. Yan, Curr. Opin. Chem. Eng., 2021, 33, 100701 CrossRef.
- T. Terlouw, C. Bauer, R. McKenna and M. Mazzotti, Energy Environ. Sci., 2022, 15, 3583 RSC.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114 RSC.
- F. Y. Gao, P. C. Yu and M. R. Gao, Curr. Opin. Chem. Eng., 2022, 36, 100827 CrossRef.
- N. K. Shrestha, S. A. Patil, S. Cho, Y. Jo, H. Kim and H. Im, J. Mater. Chem. A, 2020, 8, 24408 RSC.
- S. A. Patil, S. Cho, Y. Jo, N. K. Shrestha, H. Kim and H. Im, Chem. Eng. J., 2021, 426, 130773 CrossRef CAS.
- N. K. Shrestha, S. A. Patil, J. Han, S. Cho, A. Inamdar, H. Kim and H. Im, J. Mater. Chem. A, 2022, 10, 8989 RSC.
- H. S. Chavan, C. H. Lee, A. I. Inamdar, J. Han, S. Park, S. Cho, N. K. Shreshta, S. U. Lee, B. Hou, H. Im and H. Kim, ACS Catal., 2022, 12, 3821 CrossRef CAS.
- J. Li, S. Wang, S. Sun, X. Wu, B. Zhang and L. Feng, J. Mater. Chem. A, 2022, 10, 9308 RSC.
- R. K. Singh, K. Rajavelu, M. Montag and A. Schechter, Energy Technol., 2021, 9, 2100017 CrossRef CAS.
- X. Sun and R. Ding, Catal. Sci. Technol., 2020, 10, 1567 RSC.
- Z. Chen, W. Wei and B. J. Ni, Curr. Opin. Electrochem., 2022, 31, 100888 CrossRef CAS.
- E. T. Sayed, T. Eisa, H. O. Mohamed, M. A. Abdelkareem, A. Allagui, H. Alawadhi and K. J. Chae, J. Power Sources, 2019, 417, 159 CrossRef CAS.
- Q. Li, S. Zheng, M. Du and H. Pang, Chem. Eng. J., 2021, 417, 129201 CrossRef CAS.
- Q. Zhang, F. M. Kazim, S. Ma, K. Qu, M. Li, Y. Wang, H. Hu, W. Cai and Z. Yang, Appl. Catal., B, 2021, 280, 119436 CrossRef CAS.
- L. Sha, K. Ye, J. Yin, K. Zhu, K. Cheng, J. Yan, G. Wang and D. Cao, Chem. Eng. J., 2020, 381, 122603 CrossRef CAS.
- W. Zhang, Q. Jia, H. Liang, L. Cui, D. Wei and J. Liu, Chem. Eng. J., 2020, 396, 125315 CrossRef CAS.
- X. Liu, H. Qin, G. Wang, Q. Li, Q. Huang, Z. Wen and S. Mao, J. Mater. Chem. A, 2022, 10, 16825 RSC.
- H. Xu, K. Ye, K. Zhu, Y. Gao, J. Yin, J. Yan, G. Wang and D. Cao, ACS Sustainable Chem. Eng., 2020, 8, 16037 CrossRef CAS.
- X. Xu, Q. Deng, H.-C. Chen, M. Humayun, D. Duan, X. Zhang, H. Sun, X. Ao, X. Xue, A. Nikiforov, K. Huo, C. Wang and Y. Xiong, Research, 2022, 2022, 9837109 CAS.
- B. Z. Desalegn, K. Hern and J. Gil Seo, ChemCatChem, 2022, 14, 202100969 Search PubMed.
- S. A. Patil, N. K. Shrestha, A. I. Inamdar, C. Bathula, J. Jung, H. Im and H. Kim, Int. J. Energy Res., 2022, 46, 15143 CrossRef CAS.
- S. A. Patil, N. K. Shrestha, A. I. Inamdar, C. Bathula and J. Jung, Nanomaterials, 2022, 12, 1916 CrossRef CAS PubMed.
- S. A. Patil, N. K. Shrestha, H. T. Bui, V. D. Chavan, D. Kee Kim, S. F. Shaikh, M. Ubaidullah, H. Kim and H. Im, Int. J. Energy Res., 2022, 46, 8413 CrossRef CAS.
- H. Im, A. Inamdar, H. S. Chavan, J. H. Seok, C. H. Lee, G. Shin, S. Park, S. Yeon, S. Cho, Y. Park, N. K. Shrestha, S. U. Lee and H. Kim, J. Mater. Chem. A, 2022, 10, 20497 RSC.
- J. Chen, Q. Long, K. Xiao, T. Ouyang, N. Li, S. Ye and Z. Q. Liu, Sci. Bull., 2021, 66, 1063 CrossRef CAS PubMed.
- J. Y. Wang, W. T. Liu, X. P. Li, T. Ouyang and Z. Q. Liu, Chem. Commun., 2020, 56, 1489 RSC.
- D. V. Shinde, S. A. Patil, K. Cho, D. Y. Ahn, N. K. Shrestha, R. S. Mane, J. K. Lee and S. H. Han, Adv. Funct. Mater., 2015, 25, 5739 CrossRef CAS.
- H. T. Bui, D. Y. Ahn, N. K. Shrestha, M. M. Sung, J. K. Lee and S. H. Han, J. Mater. Chem. A, 2016, 4, 9781 RSC.
- N. K. Shrestha, S. A. Patil, A. I. Inamdar, S. Park, S. Yeon, G. Shin, S. Cho, H. Kim and H. Im, Dalton Trans., 2022, 51, 8994 RSC.
- D. Zhu, C. Guo, J. Liu, L. Wang, Y. Du and S. Z. Qiao, Chem. Commun., 2017, 53, 10906 RSC.
- Y. Cheng, X. Xiao, X. Guo, H. Yao and H. Pang, ACS Sustainable Chem. Eng., 2020, 8, 8675 CrossRef CAS.
- X. Wang, S. Song and H. Zhang, Mater. Lab., 2022, 1, 220009 Search PubMed.
- M. Li, H. Sun, J. Yang, M. Humayun, L. Li, X. Xu, X. Xue, A. Habibi-Yangjeh, K. Temst and C. Wang, Chem. Eng. J., 2022, 430, 132733 CrossRef CAS.
- Z. Dai, X. Du and X. Zhang, Int. J. Hydrogen Energy, 2022, 47, 17252 CrossRef CAS.
- C. Chen, L. Jin, L. Hu, T. Zhang, J. He, P. Gu, Q. Xu and J. Lu, J. Colloid Interface Sci., 2022, 628, 1008 CrossRef CAS PubMed.
- H. Jiang, S. Bu, Q. Gao, J. Long, P. Wang, C. S. Lee and W. Zhang, Mater. Today Energy, 2022, 27, 101024 CrossRef CAS.
- J. L. Liu, X. Y. Zhou, J. L. An, Y. Q. Wang, M. D. Zhang and L. Qin, Energy Fuels, 2022, 36, 10346 CrossRef CAS.
- H. Sun, W. Zhang, J. G. Li, Z. Li, X. Ao, K. H. Xue, K. K. Ostrikov, J. Tang and C. Wang, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- Z.-Y. Yu, C.-C. Lang, M.-R. Gao, Y. Chen, Q.-Q. Fu, Y. Duan and S.-H. Yu, Energy Environ. Sci., 2018, 11, 1890 RSC.
- Z. Lv, Z. Li, X. Tan, Z. Li, R. Wang, M. Wen, X. Liu, G. Wang, G. Xie and L. Jiang, Appl. Surf. Sci., 2021, 552, 149514 CrossRef CAS.
- W. Shi, X. Sun, R. Ding, D. Ying, Y. Huang, Y. Huang, C. Tan, Z. Jia and E. Liu, Chem. Commun., 2020, 56, 6503 RSC.
- R. P. Forslund, J. T. Mefford, W. G. Hardin, C. T. Alexander, K. P. Johnston and K. J. Stevenson, ACS Catal., 2016, 6, 5044 CrossRef CAS.
- R. Lin, L. Kang, T. Zhao, J. Feng, V. Celorrio, G. Zhang, G. Cibin, A. Kucernak, D. J. L. Brett, F. Corà, I. P. Parkin and G. He, Energy Environ. Sci., 2022, 15, 2386 RSC.
- W. Li, X. Guo, P. Geng, M. Du, Q. Jing, X. Chen, G. Zhang, H. Li, Q. Xu, P. Braunstein and H. Pang, Adv. Mater., 2021, 33, 2105163 CrossRef CAS PubMed.
- G. Zhang, Y. Li, X. Xiao, Y. Shan, Y. Bai, H.-G. Xue, H. Pang, Z. Tian and Q. Xu, Nano Lett., 2021, 21, 3016 CrossRef CAS PubMed.
- Y. Bai, G. Zhang, S. Zheng, Q. Li, H. Pang and Q. Xu, Sci. China Mater., 2021, 64, 137 CrossRef CAS.
- S. Y. Lim, S. Park, S. W. Im, H. Ha, H. Seo and K. T. Nam, ACS Catal., 2020, 10, 235 CrossRef CAS.
- K. Huang and X. Zhang, Aust. J. Chem., 2020, 74, 282 CrossRef.
- S. Chen, G. Hai, H. Gao, X. Chen, A. Li, X. Zhang and W. Dong, Chem. Eng. J., 2021, 406, 1268862 Search PubMed.
- P. Qi, Y. Gu, H. Sun, Y. Lian, X. Yuan, J. Hu, Z. Deng, H.-C. Yao, J. Guo and Y. Peng, J. Catal., 2020, 389, 29 CrossRef CAS.
- H. Guo, Z. Zheng, Y. Zhang, H. Lin and Q. Xu, Sens. Actuators, B, 2017, 248, 430 CrossRef CAS.
- S. K. Geng, Y. Zheng, S. Q. Li, H. Su, X. Zhao, J. Hu, H. B. Shu, M. Jaroniec, P. Chen, Q. H. Liu and S. Z. Qiao, Nat. Energy, 2021, 6, 904 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01962d |
| This journal is © The Royal Society of Chemistry 2023 |