
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembly of biomimetic microreactors using caged-coacervate droplets†
Arjaree
Jobdeedamrong‡
ab,
Shoupeng
Cao‡
 b,
Iain
Harley
b,
Daniel
Crespy
a,
Katharina
Landfester
*b and
Lucas
Caire da Silva
*b
b,
Iain
Harley
b,
Daniel
Crespy
a,
Katharina
Landfester
*b and
Lucas
Caire da Silva
*b
aDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand
bDepartment of Physical Chemistry of Polymers, Max Planck Institute for Polymer Research, 55128 Mainz, Germany. E-mail: silva@mpip-mainz.mpg.de
First published on 21st December 2022
Abstract
Complex coacervates are liquid-like droplets that can be used to create adaptive cell-like compartments. These compartments offer a versatile platform for the construction of bioreactors inspired by living cells. However, the lack of a membrane significantly reduces the colloidal stability of coacervates in terms of fusion and surface wetting, which limits their suitability as compartments. Here, we describe the formation of caged-coacervates surrounded by a semipermeable shell of silica nanocapsules. We demonstrate that the silica nanocapsules create a protective shell that also regulates the molecular transport of water-soluble compounds as a function of nanocapasule size. The adjustable semipermeability and intrinsic affinity of enzymes for the interior of the caged-coacervates allowed us to assemble biomimetic microreactors with enhanced colloidal stability.
 Lucas Caire da Silva | Lucas Caire da Silva studied chemistry at the University of Campinas in Brazil and received his PhD in polymer chemistry in 2015 under the supervision of Professor Kenneth Wagener at the University of Florida. In 2016, he joined Professor Katharina Landfester's group at the Max Planck Institute for Polymer Research (MPIP) as a postdoctoral researcher to work on the development of synthetic cells from synthetic building blocks. He is currently a group leader in the Department of Physical Chemistry of Polymers at MPIP. The Silva Lab develops biomimetic supramolecular systems for applications in synthetic biology, catalysis, and soft matter. |
Introduction
Biomimetic microreactors are chemical systems that mimic the properties of natural cells. A typical design consists of a micron-sized compartment that encapsulates an active core which may contain catalysts (e.g., enzymes), biomolecules (e.g., DNA/RNA) and even entire cellular structures and other nano objects (e.g., chloroplasts, nanoparticles).1–4 Like in a simple cell, the nature of the encapsulated active materials and the properties of the compartment determine the function of the microreactors. Due to their synthetic nature, biomimetic microreactors can be designed to perform diverse chemical tasks such as protein expression, DNA replication and biocatalysis.5–8 In addition to their application as standalone (bio)chemical factories, biomimetic microreactors have the potential to interact with living cells, creating the possibility to directly control their behaviour or induce desired synergistic properties.9–11Biomimetic microreactors have been designed using different types of compartments. For instance, polymer and lipid vesicles provide membrane-bound compartmentalization that shares some characteristics of the phospholipid compartments in cells such as semi-permeability and membrane fluidity.12–14 Polymeric capsules formed by layer-by-layer assembly offer another approach, where the permeability of the compartment can be tailored by the properties of the polymeric material.11
Recently, coacervates have been introduced as a versatile micron-sized compartment for engineering of biomimetic microreactors.15–18 In contrast to membrane-bound compartments, coacervates are not surrounded by a membrane.19 The membrane-free architecture allows molecules to freely diffuse across the interface. The use of coacervate as compartments in the design of microreactors have many advantages over traditional approaches that use vesicles and capsules. For example, coacervates formed from stimuli-responsive components can be dynamically assembled/disassembled by changes in conditions such as pH.20 This property allows the development of microreactors that can be modulated by their environment. Another advantage comes from the microenvironment created by the coacervate droplet, as it can selectively sequester different molecules based on intrinsic properties such as charge and molecular weight.
Coacervates are formed via liquid–liquid phase separation (LLPS) of colloidal solutions.21 They differ by the mechanism of phase separation, which can be dominated by attractive electrostatic interactions and solvent interactions. Complex coacervates, in particular, are formed via the condensation of oppositely charged polyelectrolytes (e.g., polymers, proteins, nucleic acids) in solution. The interior of coacervate droplets is characterized by a dynamic and molecularly crowded environment that simulates the conditions of the cytosol, rendering these materials ideal for development of cell-like systems.5,22,23 As compartments, coacervates can control biochemical processes via the selective partitioning of molecules and also via the localization of substrates and enzymes to enhance selectivity and kinetics.20,21,24,25
The absence of a physical membrane in complex coacervates significantly reduces their colloidal stability. Compared to membrane-bound compartments, such as liposomes and polymersomes, coacervates are prone to coalescence and wetting effects.26 The colloidal stability of coacervates can be improved via the formation of a surrounding membrane obtained from fatty acids,27 lipids,28 and polymers.29 In addition to improving colloidal stability, the introduction of semi-permeable membranes surrounding the coacervates allows a control on the molecular transport between coacervates and the surrounding medium.
An alternative to “membranization” is the use of Pickering stabilization by solid particles. Here, nanoparticles present at the interface between the two liquid phases provide colloidal stabilization. However, this method has been mostly applied to the formation of colloidosomes, which involves a crosslinking of the particles to avoid disassembly of the nanoparticle-cage before transferring them to the aqueous medium.30–32
We here describe the construction of a coacervate-based biomimetic microreactor containing a non-crosslinked silica nanocapsule (SiO2NC) shell (Fig. 1). The presence of a shell of SiO2NC enhanced the stability of the coacervate compartment and allowed the adjustment of its membrane permeability depending on the size of the SiO2NCs. The resulting caged-coacervates were able to sequestrate and confined active enzymes, which was further developed into a functional controlled bioreactor with selective molecular transport.
 | ||
Fig. 1 Schematic illustration and characterization of coacervates stabilized with large and small SiO2NCs. (a) Bright field images of uncaged coacervates (C-Am![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Q-Am 1:2 weight ratio). (b) TEM images of ultrasmall PEG-SiO2CNs (10 nm). (c) TEM images of large PEG-SiO2CNs (100 nm). (d–f) Bright field and fluorescence microscopy images of caged-coacervates stabilized by 10 nm PEG-SiO2CNs. (g–i) Bright field and fluorescence microscopy images of caged-coacervates stabilized with 100 nm PEG-SiO2CNs. :Q-Am 1:2 weight ratio). (b) TEM images of ultrasmall PEG-SiO2CNs (10 nm). (c) TEM images of large PEG-SiO2CNs (100 nm). (d–f) Bright field and fluorescence microscopy images of caged-coacervates stabilized by 10 nm PEG-SiO2CNs. (g–i) Bright field and fluorescence microscopy images of caged-coacervates stabilized with 100 nm PEG-SiO2CNs. | ||
Results and discussion
Pickering stabilization of coacervate compartments with SiO2NC
Coacervate microcompartments were obtained by mixing carboxy-functionalized amylose (C-Am) and quaternized amylose (Q-Am) in phosphate buffered saline (PBS). Droplet formation was driven by electrostatic interactions between C-Am (negatively charged) and Q-Am (positively charged), resulting in the formation of dense liquid-like droplets. Charged derivatives of amylose were synthesized using a modular approach described in the literature.29 Coacervates with a positive surface charge (ζ = +20 mV) were obtained by setting the Q-Am:C-Am ratio to 2:1 during coacervation (Fig. S1†). The resulting coacervates were largely unstable, as indicated by the rapid coalescence (<1 min) of coacervate droplets and the wetting of the glass microscope slides by the droplets (Fig. 1a). These effects are a consequence of the lack of a protective membrane around the droplets, which favours Ostwald ripening and wetting of hydrophilic surfaces.33
Silica nanocapsules (SiO2NCs) were synthesized via a miniemulsion approach based on the hydrolysis and condensation of tetraethyl orthosilicate (TEOS).34 The silica shell was formed at the interface of miniemulsion droplets stabilized with a reactive cationic surfactant, dimethyloctadecyl[3-(trimethoxysilyl)propyl] ammonium chloride (TPOAc), which can be hydrolyzed and co-condensed with TEOS to chemically bond to silica surfaces.35 The diameters of SiO2NCs were controlled by varying the compositions of hydrophobic substances in the miniemulsion droplets. A volume ratio of 2:1:10:0 in cyclohexane (Cy), chloroform (CHCl3), TEOS, and hexadecane (HD), respectively, was used to regulate the Ostwald ripening in the miniemulsion system, which yielded ultrasmall silica nanocapsules (ul-SiO2NCs) with a diameter about 10 nm (Fig. S2a†).36 Large SiO2NCs (L-SiO2NCs), with a diameter about 100 nm, were obtained using a 0:1:1:0.08 volume ratio of Cy, CHCl3, TEOS, and HD, respectively (Fig. S2b†). The surface of SiO2NCs were functionalized with polyethylene glycol (PEG, Mw = 5000 g mol−1) to prevent aggregation and precipitation through steric stabilization.37 The PEG content of SiO2NCs were determined by 1H NMR spectroscopy. A calibration curve for the determination of PEG content by NMR is shown in Fig. S3.† The NMR spectra of the SiO2NCs confirmed the presence of PEG (3.52 ppm). Integration of this signal showed that the PEG chain density on the surface of the SiO2NCs was 0.037 and 1.07 chains per nm2 for ul-SiO2NCs and L-SiO2NCs, respectively. Transmission electron microscopy (TEM) images confirmed the core–shell structure of the SiO2NCs (Fig. 1b and c). The diameters of PEG-SiO2NCs were similar to the diameters of pristine SiO2NCs. Zeta potentials of PEG-ul-SiO2NCs and PEG-L-SiO2NCs were negative (−8 and −6 mV, respectively).
Caged-coacervates were prepared by mixing of all components (C-Am:Q-Am:PEG-SiO2NC) in a buffer solution at a weight ratio of 1:2:0.75. Stable coacervates were obtained as a result of the interfacial assembly of PEG-SiO2NCs around the coacervates, creating a caged structure that enhanced the colloidal stability of the droplets. In contrast with non-stabilized coacervates, caged-coacervates did not coalesce or fuse, and were stable for at least 24 h (Fig. 1d and g). The driving force for the shell formation can be attributed to the decreasing of total free energy associated with the presence of the SiO2NCs at the liquid–liquid interface.38 Caged-coacervates were also stable in aqueous solutions containing up to 100 mM of NaCl (Fig. S4 and S5†).
Formation of the caged coacervates and the interfacial stabilization are affected by the ratio of C-AM and Q-AM used. Stable coacervates stabilized with PEG-L-SiO2NCs in spherical shape are obtained with a 1:1 or 1:2 ratio of C-AM and Q-AM, while excess C-AM hinders the interfacial assembly attributed to the low absorption between the negatively charged SiO2 on the negative coacervate interface (Fig. S6†). After preparation, the caged coacervates displayed sensitivity to pH, which breakdown into very small droplets or fully dissolve upon subjecting to ∼pH 12 or ∼pH 3, respectively (Fig. S7†). Interestingly, the caged coacervates are relatively stable upon ultrasonication even up to 5 min, highlighting their stability against mechanical forces (Fig. S8†).
The shell formed by the SiO2NCs was visualized by confocal microscopy imaging. As shown in Fig. 1e, f and h, i, Nile red labelled PEG-ul-SiO2NCs and PEG-L-SiO2NCs successfully coated the coacervate droplets. A non-homogenous distribution of nanocapsules was obtained with PEG-ul-SiO2NCs. Here, the NCs were also internalized by the coacervate droplet. We attributed this result to the small diameter of the SiO2NCs (10 nm), which could penetrate the coacervate core more easily than the larger SiO2NCs. In contrast, PEG-L-SiO2NCs were not internalized, showing a high accumulation at the interface. Cryo-TEM was used to characterize the structure of the caged coacervates. As shown in Fig. S9,† the PEG-ul-SiO2NCs are scattered at the interface of coacervates in a multilayer mode.
Selective permeability of caged-coacervates
The presence of the SiO2NCs shell was expected to limit the transport of molecules across the coacervate interface, allowing the molecular transport to be controlled. Molecular transport of caged-coacervates was investigated in aqueous media containing fluorescent probes of increasing molecular weight: calcein (Mw = 623 Da), FITC-PEG (Mw = 1 kDa), and FITC-Dextran (Mw = 10 kDa or 70 kDa). The permeability of the caged-coacervates to the different fluorescent molecules was evaluated by confocal microscopy measurements (Fig. 2 and S10†). The data indicates that caged-coacervates formed with the large SiO2NCs were more permeable than those formed with the small SiO2NCs. As shown in Fig. 2a, caged-coacervates with PEG-ul-SiO2NCs were only permeable to the 0.6 kDa probe. Larger molecules could not cross the shell.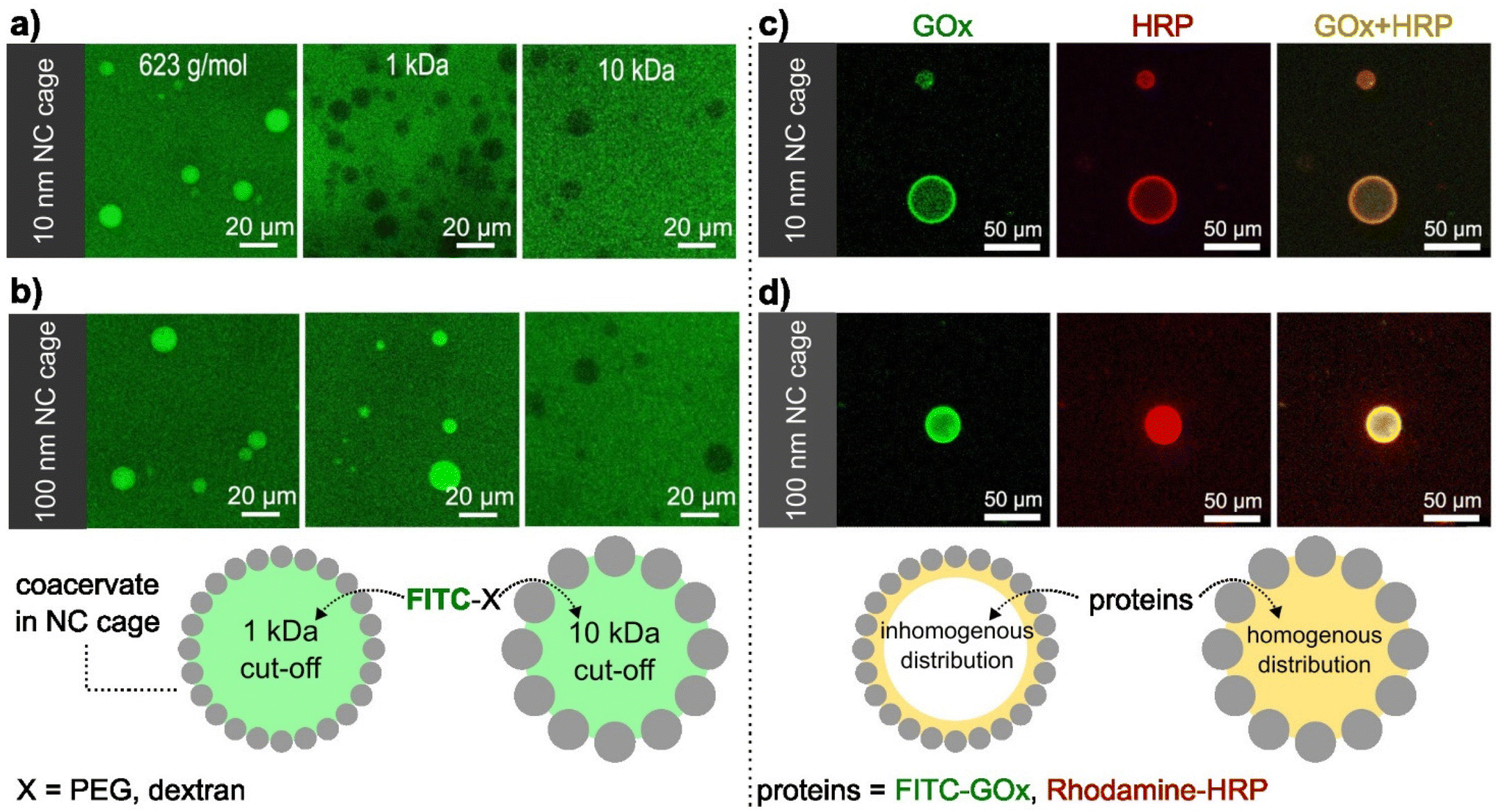 | ||
| Fig. 2 Determination of semi-permeability and enzyme encapsulation properties of caged-coacervates. (a and b) Confocal microscopy images of caged-coacervates dispersed in PBS buffer containing calcein (623 g mol−1), FITC-PEG-NH2 (1 kDa), and FITC-dextran (10 kDa). (c and d) Confocal microscopy images of caged-coacervates loaded with FITC-GOx and Rhodamine-HRP. | ||
In contrast, caged-coacervates with PEG-L-SiO2NCs were permeable to both 0.6 and 1 kDa probes, indicating a higher cut-off value. Plots of the fluorescent intensity relative to the background are shown in Fig. S11.† These results demonstrate that the permeability of the caged-coacervates can be controlled by the size of the SiO2NCs. The effect can be attributed to the differences in the packing density of the shell, leading to different pore sizes. Smaller nanocapsules closely packed on the surface resulted in smaller pores and in a less permeable shell.
Biomimetic microreactor
The highly charged microenvironment of the caged-coacervates allows them to be utilized as selective compartments for different molecules. For instance, the caged coacervates displayed effective sequestration and partitioning towards negative charged sulfo-rhodamine b and zwitterionic rhodamine b, with partitioning coefficients ∼52.7 ± 7.6 and 9.9 ± 1.3, respectively. While upon incubation with positively charged rhodamine 6G, its emission inside and outside the coacervates are nearly equal, with a partitioning coefficient ∼1.16 ± 0.4 (Fig. S12†).The semipermeability and colloidal stability of the caged-coacervates allow these materials to be used as compartments for biomimetic microreactors. To demonstrate that, we designed a caged-microreactor containing two active enzymes, glucose oxidase (GOx) and horseradish peroxidase (HRP). First, GOx and HRP were mixed with the solution of negatively charged amylose (C-Am). This solution was then mixed with positively charged amylose (Q-Am) to form the enzyme-carrying coacervates. The caged-microreactors were obtained after the formation of the SiO2NC shells by mixing the coacervates with PEG-ul-SiO2NCs or PEG-L-SiO2NCs. The loading efficiency was measured after centrifuge-based purification process. Determined via a calibration curve, the loading efficiency of GOx and HRP were calculated to be 78.3% and 83.2%, respectively (Fig. S13 and S14†). Fig. 2c and d shows the distribution of GOx and HRP inside the caged-coacervates. The enzymes were labelled with FITC (GOx) and rhodamine b (HRP) for visualization by confocal microscopy. The images show that both enzymes were encapsulated into the caged-coacervates. The distribution of the enzymes inside the compartments depended on the type of SiO2NCs used.
The core of caged-coacervates stabilized with PEG-SiO2NCs 10 nm showed lower fluorescent intensities of FITC-GOx and Rhodamine-HRP compared with the fluorescent intensity at the interface (Fig. S15†). The localization of the enzymes at the interface of coacervates stabilized with PEG-SiO2NCs (10 nm) is attributed to the larger surface area produced by 10 nm NCs compared to that of 100 nm SiO2NCs. The larger surface area lead to an enhanced enzyme adsorption at the interface.39
The partition coefficients of GOx and HRP were determined via confocal imaging (Fig. S16†). The values were 42.4 ± 11.4 for GOx, and 50.2 ± 10.1 for HRP.
Caged-microreactors were tested for their ability to carry out an enzymatic cascade reaction involving GOx and HRP. The reaction was started by adding glucose and Amplex Red to a dispersion of caged-microreactors. Once internalized by the microreactors, glucose is consumed by GOx to produce hydrogen peroxide. The peroxide is then used internally by HRP to convert a profluorescent substrate, Amplex Red, into the fluorescent product resorufin (Fig. 3b). The speed of resorufin production by the microreactors depended on the type of SiO2NC employed. Resorufin production was slower in microreactors stabilized by PEG-ul-SiO2NC than in microreactors stabilized with PEG-L-SiO2NCs (Fig. 3a). These results imply that the diffusion of glucose depended on the permeability of the SiO2NC shell. Measurements of the loading efficiency of GOx and HRP showed that the concentration of these enzymes was slightly higher in caged-coacervates stabilized by 100 nm NCs (Fig. S13 and S14†). However, the difference in reaction rate, which is more than twice as large, cannot be explained by the different loading efficiency alone. The silica cage has a strong influence on the reaction rate of the microreactor. The more permeable PEG-L-SiO2NC shell allowed glucose to rapidly reach the interior of the microreactors. The higher permeability of the PEG-L-SiO2NC microreactors also allowed the resorufin product to be released into the outer medium. Fig. 3c shows plots of the fluorescence intensity of resorufin production by caged-microreactors over time as a function of SiO2NC type. The plots highlight the restricting effect of the less permeable SiO2NC shell on the reaction kinetics. These results illustrate how the SiO2NC coating can be utilized to regulate the substrate diffusion and release in caged-microreactors.
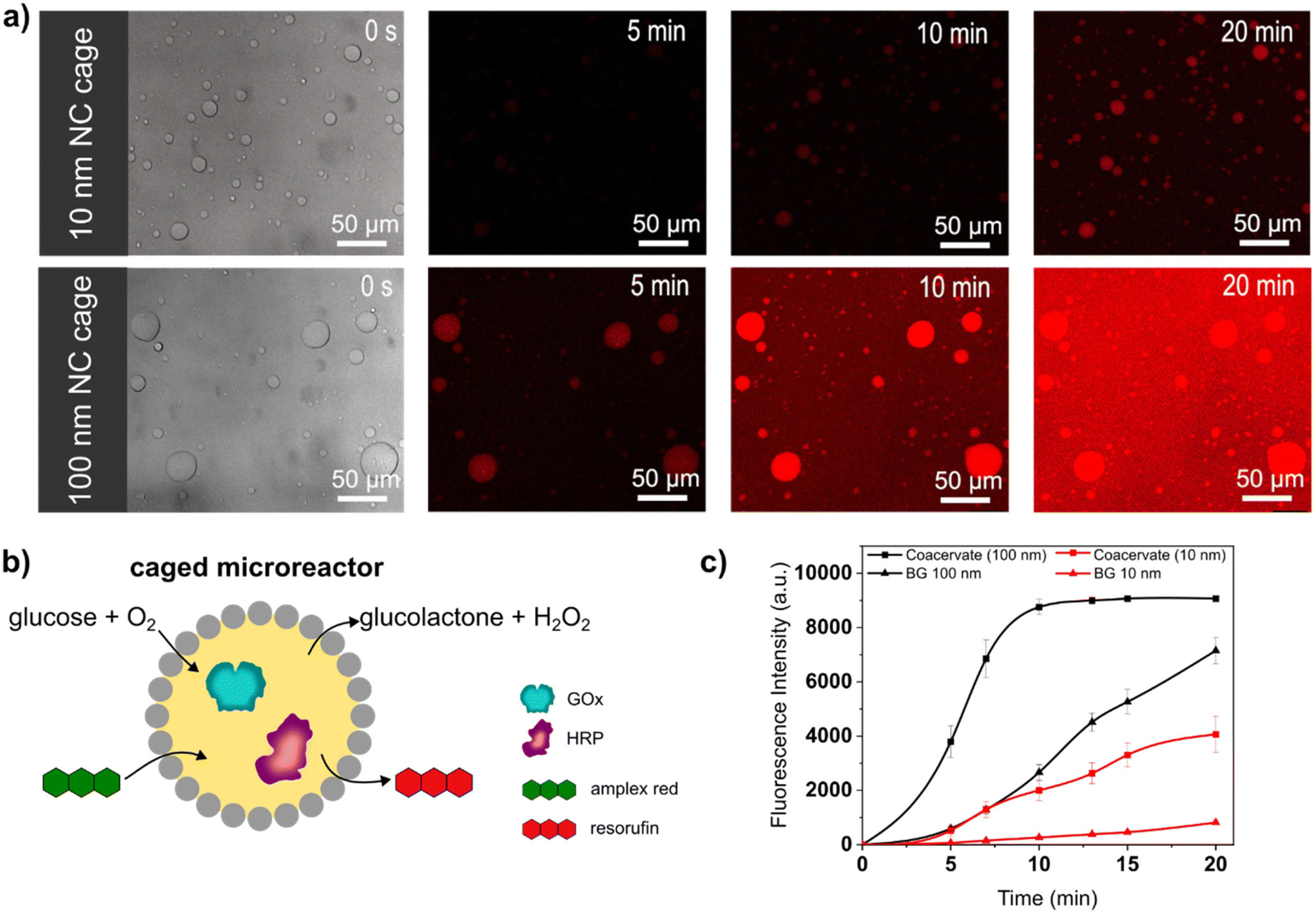 | ||
| Fig. 3 Caged-microreactors from caged-coacervates (a) bright field and time-dependent confocal microscopy images of caged microreactors stabilized with 10 nm (top) and 100 nm (bottom) PEG-SiO2NCs. The reaction started with the addition of glucose (50 mM, 5 μL) and Amplex red (1 mM, 3 μL) to the external medium. The enzymes GOx and HRP were encapsulated in the core of the caged-coacervates. (b) Schematic representation of caged microreactors. (c) The time evolution of the resorufin produced by the oxidation of Amplex Red by GOx/HRP. Determined by changes in the fluorescence intensity inside the coacervates (squares) and in the dispersing medium (triangles). 10 nm PEG-SiO2NCs: black lines, 100 nm PEG-SiO2NCs: red lines. BG: background intensity from external medium. | ||
Conclusions
Caged-coacervates stabilized by silica nanocapsules were fabricated in a one-step process that allowed straightforward assembly of biomimetic microreactors. The silica cage enabled regulation of substrate diffusion into the microreactors, with a molecular weight cut-off determined by the size of the silica nanocapsules. The non-covalent interaction of the silica shell with the coacervate core opens new possibilities for dynamic control of the interfacial properties in microreactors. For example, the assembly/disassembly of the shell could be used to dynamically control the permeability to a wider range of substrates and nano-objects. The modification of the interfacial properties could also be used to induce fusion of different microreactors, allowing the contents of different populations to merge, allowing hierarchical interactions between compartmentalized chemical systems to be explored in synthetic cell-like systems. In addition, the large capacity and high loading of silica nanocapsules could facilitate the encapsulation of various substances in the core of silica nanocapsules, such as enzymes, proteins, or photocatalysts, which could be used as novel multifunctional protocells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the Vidyasirimedhi Institute of Science and Technology. S. C. thanks the Alexander von Humboldt Foundation for a fellowship and financial support (No. 3.5-CHN-1222717-HFST-P). This work is part of research within the Max Planck Consortium for Synthetic Biology (MaxSynBio), which is jointly funded by the German Federal Ministry of Education and Research (BMBF) and the Max Planck Society. I. H. gratefully acknowledges the H2020 Marie Curie Actions Fellowship of the European Commission (ITN SUPERCOL, Grant Agreement 860914). Open Access funding provided by the Max Planck Society.References
- S. Koga, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2011, 3, 720–724 CrossRef CAS PubMed.
- B. V. V. S. P. Kumar, J. Fothergill, J. Bretherton, L. F. Tian, A. J. Patil, S. A. Davis and S. Mann, Chem. Commun., 2018, 54, 3594–3597 RSC.
- M. Matsuo and K. Kurihara, Nat. Commun., 2021, 12, 5487 CrossRef CAS PubMed.
- K. K. Nakashima, J. F. Baaij and E. Spruijt, Soft Matter, 2018, 14, 361–367 RSC.
- C. Guindani, L. Caire da Silva, S. Cao, T. Ivanov and K. Landfester, Angew. Chem., Int. Ed., 2022, 61, e202110855 CrossRef CAS PubMed.
- D. T. Gonzales, N. Yandrapalli, T. Robinson, C. Zechner and T. Y. D. Tang, ACS Synth. Biol., 2022, 11, 205–215 CrossRef CAS PubMed.
- K. Kurihara, M. Tamura, K. Shohda, T. Toyota, K. Suzuki and T. Sugawara, Nat. Chem., 2011, 3, 775–781 CrossRef CAS.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 CrossRef CAS PubMed.
- Y. Zhang, P. S. Schattling, F. Itel and B. Stadler, ACS Omega, 2017, 2, 7085–7095 CrossRef.
- Y. Elani, Angew. Chem., Int. Ed., 2021, 60, 5602–5611 CrossRef CAS PubMed.
- M. Godoy-Gallardo, C. Labay, V. D. Trikalitis, P. J. Kempen, J. B. Larsen, T. L. Andresen and L. Hosta-Rigau, ACS Appl. Mater. Interfaces, 2017, 9, 15907–15921 CrossRef CAS.
- S. Cao, L. Caire da Silva and K. Landfester, Angew. Chem., Int. Ed., 2022, 61, e202205266 CAS.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- C. E. Sing and S. L. Perry, Soft Matter, 2020, 16, 2885–2914 RSC.
- A. Samanta, V. Sabatino, T. R. Ward and A. Walther, Nat. Nanotechnol., 2020, 15, 914–921 CrossRef CAS PubMed.
- W. J. Mu, Z. Ji, M. S. Zhou, J. Z. Wu, Y. Y. Lin and Y. Qiao, Sci. Adv., 2021, 7, eabf9000 CrossRef CAS PubMed.
- N. A. Yewdall, A. A. M. Andre, T. M. Lu and E. Spruijt, Curr. Opin. Colloid Interface Sci., 2021, 52, 101416 CrossRef CAS.
- M. Abbas, J. O. Law, S. N. Grellscheid, W. T. S. Huck and E. Spruijt, Adv. Mater., 2022, 34, 2202913 CrossRef CAS.
- C. Love, J. Steinkuhler, D. T. Gonzales, N. Yandrapalli, T. Robinson, R. Dimova and T. Y. D. Tang, Angew. Chem., Int. Ed., 2020, 59, 5950–5957 CrossRef CAS.
- N. Martin, ChemBioChem, 2019, 20, 2553–2568 CrossRef CAS PubMed.
- C. D. Crowe and C. D. Keating, Interface Focus, 2018, 8, 20180032 CrossRef PubMed.
- W. M. Aumiller, F. P. Cakmak, B. W. Davis and C. D. Keating, Langmuir, 2016, 32, 10042–10053 CrossRef CAS.
- W. J. Altenburg, N. A. Yewdall, D. F. M. Vervoort, M. H. M. E. van Stevendaal, A. F. Mason and J. C. M. van Hest, Nat. Commun., 2020, 11, 6282 CrossRef CAS PubMed.
- B. Drobot, J. M. Iglesias-Artola, K. Le Vay, V. Mayr, M. Kar, M. Kreysing, H. Mutschler and T. Y. D. Tang, Nat. Commun., 2018, 9, 3643 CrossRef PubMed.
- J. T. Cirulis, C. M. Bellingham, E. C. Davis, D. Hubmacher, D. P. Reinhardt, R. P. Mecham and F. W. Keeley, Biochemistry, 2008, 47, 12601–12613 CrossRef CAS PubMed.
- L. L. Zhou, J. J. Koh, J. Wu, X. T. Fan, H. M. Chen, X. A. Hou, L. Jiang, X. H. Lu, Z. B. A. Li and C. B. He, Bioconjugate Chem., 2022, 33, 444–451 CrossRef CAS.
- S. Deshpande, F. Brandenburg, A. Lau, M. G. F. Last, W. K. Spoelstra, L. Reese, S. Wunnava, M. Dogterom and C. Dekker, Nat. Commun., 2019, 10, 1800 CrossRef.
- A. F. Mason, B. C. Buddingh, D. S. Williams and J. C. M. van Hest, J. Am. Chem. Soc., 2017, 139, 17309–17312 CrossRef CAS.
- J. P. Douliez, N. Martin, T. Beneyton, J. C. Eloi, J. P. Chapel, L. Navailles, J. C. Baret, S. Mann and L. Beven, Angew. Chem., Int. Ed., 2018, 57, 7780–7784 CrossRef CAS PubMed.
- J. Fothergill, M. Li, S. A. Davis, J. A. Cunningham and S. Mann, Langmuir, 2014, 30, 14591–14596 CrossRef CAS.
- M. Li, R. L. Harbron, J. V. M. Weaver, B. P. Binks and S. Mann, Nat. Chem., 2013, 5, 529–536 CrossRef CAS.
- S. Mondal and Q. Cui, Chem. Sci., 2022, 13, 7933–7946 RSC.
- J. Fickert, P. Rupper, R. Graf, K. Landfester and D. Crespy, J. Mater. Chem., 2012, 22, 2286–2291 RSC.
- A. Jobdeedamrong, M. Theerasilp, N. Nasongkla and D. Crespy, Biomater. Sci., 2021, 9, 5781–5784 RSC.
- T. P. Doan-Nguyen, S. Jiang, K. Koynov, K. Landfester and D. Crespy, Angew. Chem., Int. Ed., 2021, 60, 18094–18102 CrossRef CAS PubMed.
- V. Cauda, C. Argyo and T. Bein, J. Mater. Chem., 2010, 20, 8693–8699 RSC.
- T. Y. D. Tang, D. van Swaay, A. deMello, J. L. R. Anderson and S. Mann, Chem. Commun., 2015, 51, 11429–11432 RSC.
- G. T. Rengarajan, D. Enke, M. Steinhart and M. Beiner, J. Mater. Chem., 2008, 18, 2537–2539 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05101j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |