
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flow bioprocessing of citrus glycosides for high-value aglycone preparation†
Agostina
Colacicco
 a,
Giorgia
Catinella
a,
Cecilia
Pinna
a,
Alessandro
Pellis
b,
Stefano
Farris
a,
Lucia
Tamborini
c,
Sabrina
Dallavalle
a,
Francesco
Molinari
a,
Martina Letizia
Contente
*a and
Andrea
Pinto
a
a,
Giorgia
Catinella
a,
Cecilia
Pinna
a,
Alessandro
Pellis
b,
Stefano
Farris
a,
Lucia
Tamborini
c,
Sabrina
Dallavalle
a,
Francesco
Molinari
a,
Martina Letizia
Contente
*a and
Andrea
Pinto
a
aDepartment of Food, Environmental and Nutritional Sciences (DeFENS), University of Milan, via Celoria 2, 20133, Milan, Italy. E-mail: martina.contente@unimi.it
bDepartment of Chemistry and Industrial Chemistry, University of Genoa, via Dodecaneso 31, 16146, Genoa, Italy
cDepartment of Pharmaceutical Sciences (DISFARM), University of Milan, via Mangiagalli 25, 20133, Milan, Italy
First published on 28th June 2023
Abstract
An efficient one-pot, 2-step flow bioprocess for the hydrolysis of hesperidin (HES) and rutin (RT), citrus rutinosyl flavonoids, has been developed for the obtainment of the corresponding aglycones. A commercially available α-rhamnosidase (RN) and an extremophilic home-made β-glycosidase (HOR) have been co-immobilized on glyoxyl-agarose beads to prepare a high-performing multi-active biocatalyst (imm-RN–HOR). After the optimization of the reaction conditions in batch mode, a “flow switch” was applied, increasing the productivity (>99% m.c., 5 min), cost-efficiency and sustainability of the overall process. Due to the recovery and reuse of all the materials involved in the flow biotransformation, this strategy is effectively a zero-waste process.
Citrus agriculture and citrus-related processing industries produce tonnes of waste and by-products every year, mainly consisting in seeds, peels, leaves and branches which need to be managed and disposed of.1–3 In terms of sustainability, the maximization of residue reuse and exploitation has become a key issue since these waste materials are rich in natural valuable compounds with a variety of different properties.4 Among them, flavonoids present a huge potential in the pharmaceutical, food and cosmetic sectors, due to their wide range of biological activities such as anti-inflammatory, antioxidant, antiviral, anticancer and neuroprotective properties.5–8 In particular, citrus flavonoids can be divided into three categories: rutinosyl flavonoids, their mono-glycosides and aglycones. Although these compounds are characterized by similar in vitro activities, higher bioavailability, stability and membrane permeability are generally observed for aglycones.9–11 In this context, hesperidin (HES) and rutin (RT) (Scheme 1) represent widespread rutinosyl flavonoids in citrus species, both in fruits and their by-products. Peels and leaves possessing a high content of HES and RT (260–670 mg g−1, 7–15 mg g−1 respectively)12–14 are nowadays considered as alternative natural sources for their recovery. Chemical attempts to obtain the corresponding aglycones hesperetin (HP) and quercetin (Q) (Scheme 1) from their rutinosyl counterparts mainly rely on hydrolysis approaches. However, the conditions for chemical hydrolysis of flavonoid glycosides are typically too harsh to preserve the aglycone structure (e.g., polymerization), thus affecting their bioactivities and/or generating side-products.15–17
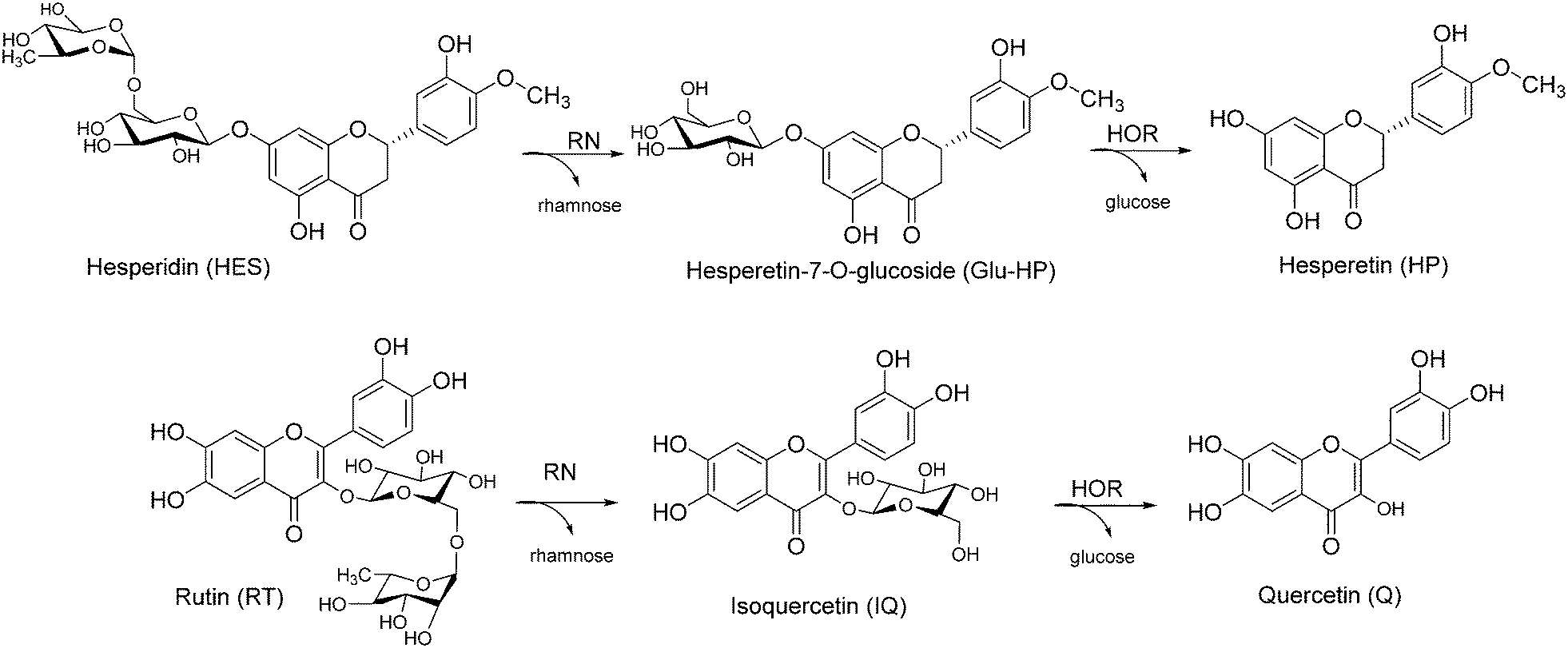 | ||
| Scheme 1 Obtainment of quercetin and hesperetin as aglycones from the corresponding natural rutinosides. | ||
Recent advances in biocatalysis have confirmed its advantages over conventional chemistry, especially in terms of higher selectivity and milder operational conditions.18–20 Although whole-cell biocatalysis has been the leading technique for the preparation of flavonoid aglycones, these processes are typically characterized by low substrate loading, high reaction volumes (e.g., fermentation technology) and poor yields,21–24 while just a few examples employing commercial hydrolytic enzymatic preparations have been reported.25–27 On the other hand, the home-made production of proteins is generally considered a costly and time-consuming technique. Moreover, the application of pure enzymes is sometimes limited by their low operational stability and reusability. Thus, enzyme immobilization is considered as a key strategy to overcome these drawbacks.28–30 Enzyme immobilization not only enhances the catalyst stability, and its facile isolation and reuse, but also allows for biocatalyst incorporation in flow chemistry reactors.31,32 The combination of biocatalysis with flow facilities, namely flow biocatalysis, is nowadays recognized as a greener way to operate in chemistry, improving biotransformation productivity and shortening reaction times, while minimizing waste generation and energy consumption.33–35 Flow (bio)systems are characterized by a smaller equipment footprint and better control of biotransformations, rendering scale-up more predictable: process productivity can be improved by simply allowing the system to work over time without the need for proportionally increasing the reactor size and the biocatalyst amount.
Moreover, multi-step biotransformations can be performed by sequentially connecting bioreactors filled with different enzymes, or via enzymatic co-immobilization techniques.36–38 During the last 20 years, although separately immobilized enzymes were the most described, co-immobilization of several proteins on the same carrier was demonstrated to enhance the catalytic performance, reducing the number of reaction steps, minimizing by-product formation, decreasing the accumulation of unstable/toxic intermediates, shifting the thermodynamic equilibrium toward the target product, in situ recycling enzyme cofactors (when necessary), and finally increasing bioprocess productivity and cost-efficiency.39,40
In this work, we developed highly-productive and rapid one-pot, 2-step flow biotransformations to have direct access to HP and Q starting from the natural rutinosides HES and RT (Scheme 1). A commercially available α-rhamnosidase (RN) and an “in-house” prepared extremophilic β-glycosidase from Halotermotrix orenii (HOR), so far employed for food applications (e.g., bitterness removal from juice containing naringin, wine aroma enhancement after the release of volatile aglycones from non-volatile glucosides)41,42 have been selected as catalysts to be co-immobilized on the same matrix.
Free enzyme-mediated batch biotransformations for the obtainment of glucoside-intermediates (Glu-HP, IQ) and subsequently of HP and Q as aglycones (Scheme 1) have been firstly investigated. To avoid any solubility issue, typical of flavonoid glycosides, biphasic reactions employing a water-immiscible green solvent 2,2,5,5-tetramethyloxolane (TMO)43 have been set up (i.e., 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water/TMO, 1 mg mL−1 catalyst concentration, 5 g L−1 substrate loading). Glu-HP and IQ were obtained with 15% and 30% m.c. in 14 h and 3 h, while HP and Q with 18% and 32% m.c. in 2 and 0.5 h, respectively (see the ESI†). To enhance the catalyst stability under operational conditions (i.e., biphasic media, 40 °C, 5 g L−1 rutinoside concentration), a highly tailored immobilization of the two single proteins (imm-RN and imm-HOR) was developed. A covalent bond between the matrix and the enzymes has been selected to specifically obtain robust and durable catalysts to be used at high flow rates for scalable productivity.35,44,45 Agarose previously functionalized with aldehyde groups (i.e., glyoxyl-agarose) has been chosen as a support for its better performance in terms of retained activity when compared with other popular immobilization carriers (e.g., methacrylate resins).45 Different concentrations of the two enzymes (RN and HOR) have been tested (1, 2, 5 mg gresin−1); the highest recovered activity (35%, 53% respectively) was obtained employing 1 mg gresin−1 pure proteins (see the ESI†). With the idea of speeding up the first reaction (i.e., rhamnose cleavage), characterized by long reaction times when the free catalyst was employed (14 h HES → Glu-HP; 2 h RT → IQ), a higher enzymatic loading was selected for imm-RN (5 mgprotein gresin−1), allowing us to obtain a final catalyst with improved total activity (U gresin−1). Subsequently, simultaneous co-immobilization of the two biocatalysts (imm-RN–HOR, RN: 5 mg gresin−1; HOR: 1 mg gresin−1) has been carried out with the final aim of running enzymatic cascades into one-pot reaction systems. To further characterize the carrier before and after enzyme (co)-immobilization, imaging analysis was carried out via scanning electron microscopy (SEM) (see the ESI†). Particles supported with 5 mg gresin−1 RN, 1 mg gresin−1 HOR, and a combination of both enzymes (5 mgRN gresin−1, 1 mgHOR gresin−1) showed neither surface changes nor aggregation phenomena, thus maximizing the contact area during the reactions (Fig. 1).
:50 water/TMO, 1 mg mL−1 catalyst concentration, 5 g L−1 substrate loading). Glu-HP and IQ were obtained with 15% and 30% m.c. in 14 h and 3 h, while HP and Q with 18% and 32% m.c. in 2 and 0.5 h, respectively (see the ESI†). To enhance the catalyst stability under operational conditions (i.e., biphasic media, 40 °C, 5 g L−1 rutinoside concentration), a highly tailored immobilization of the two single proteins (imm-RN and imm-HOR) was developed. A covalent bond between the matrix and the enzymes has been selected to specifically obtain robust and durable catalysts to be used at high flow rates for scalable productivity.35,44,45 Agarose previously functionalized with aldehyde groups (i.e., glyoxyl-agarose) has been chosen as a support for its better performance in terms of retained activity when compared with other popular immobilization carriers (e.g., methacrylate resins).45 Different concentrations of the two enzymes (RN and HOR) have been tested (1, 2, 5 mg gresin−1); the highest recovered activity (35%, 53% respectively) was obtained employing 1 mg gresin−1 pure proteins (see the ESI†). With the idea of speeding up the first reaction (i.e., rhamnose cleavage), characterized by long reaction times when the free catalyst was employed (14 h HES → Glu-HP; 2 h RT → IQ), a higher enzymatic loading was selected for imm-RN (5 mgprotein gresin−1), allowing us to obtain a final catalyst with improved total activity (U gresin−1). Subsequently, simultaneous co-immobilization of the two biocatalysts (imm-RN–HOR, RN: 5 mg gresin−1; HOR: 1 mg gresin−1) has been carried out with the final aim of running enzymatic cascades into one-pot reaction systems. To further characterize the carrier before and after enzyme (co)-immobilization, imaging analysis was carried out via scanning electron microscopy (SEM) (see the ESI†). Particles supported with 5 mg gresin−1 RN, 1 mg gresin−1 HOR, and a combination of both enzymes (5 mgRN gresin−1, 1 mgHOR gresin−1) showed neither surface changes nor aggregation phenomena, thus maximizing the contact area during the reactions (Fig. 1).
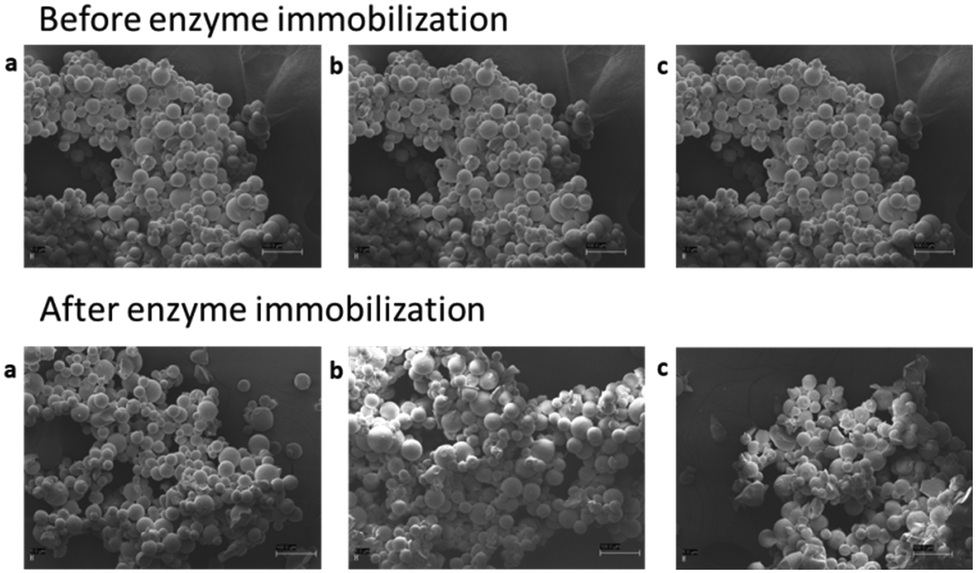 | ||
| Fig. 1 SEM images (500× magnification) of the agarose matrix before and after enzyme (co)-immobilization of a: RN (5 mg gresin−1); b: HOR (1 mg gresin−1); c: RN–HOR (5 mgRN gresin−1, 1 mgHOR gresin−1). | ||
Additionally, the spatial distribution of fluorophore-labelled proteins was investigated (see the ESI†). In the single-enzyme preparations (Fig. 2a and b), the biocatalysts were localized across the porous surface of glyoxyl-agarose beads, thus favouring close contact with substrates. In the combined formulation, a spatial organization with HOR more internally localized surrounded by RN was observed (Fig. 2c). This can be explained considering the 1:5 HOR/RN loading ratio.
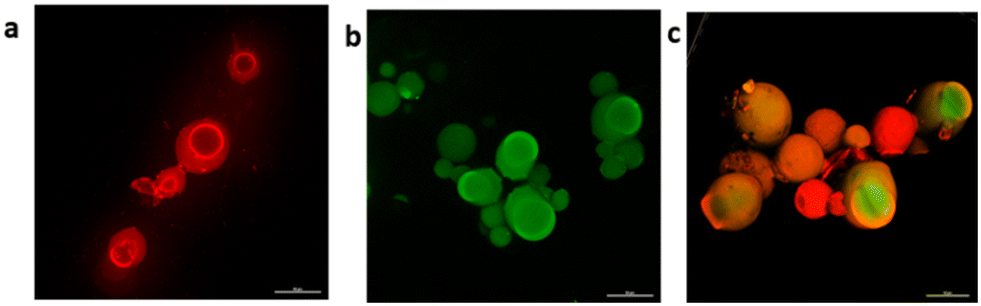 | ||
| Fig. 2 Confocal microscopy of glyoxyl-agarose particles immobilizing a: rhodamine-labelled RN (5 mg gresin−1); b: fluorescein-labelled HOR (1 mg gresin−1); c: rhodamine-labelled RN and fluorescein-labelled HOR (5 mgRN gresin−1, 1 mgHOR gresin−1). Magnification 20×. | ||
To test the catalytic performance of the agarose (co)-immobilized enzymes (imm-RN, imm-HOR, imm-RN–HOR), batch reactions as previously described have been set up, fixing the substrate concentration at 5 g L−1. Employing RT as a substrate for rhamnose hydrolysis the higher conversion (50% m.c.) was reached after 1 h, whereas, starting from IQ, the higher molar conversion for glucose cleavage was 80% after 1.5 h (see the ESI†). Direct obtainment of the HP and Q aglycones using imm-RN–HOR was achieved with 60% and 70% molar conversion in 1 and 0.5 h, respectively (see the ESI†). It is worth noting that the immobilized system afforded much higher activity than the free enzymes since the catalyst concentrations (RN: 50 mg mL−1, enzyme loading: 5 mg gresin−1; HOR: 50 mg mL−1, enzyme loading: 1 mg gresin−1) required for these reactions were between 4- and 20-times lower than that of their free counterparts (1 mg mL−1).
Although better results have been observed after protein immobilization, to further accelerate biotransformations while also enhancing the productivity, flow bioprocesses have been developed. Single step reactions were firstly optimized flowing a segmented liquid–liquid stream composed of HEPES buffer 0.05 M, pH 7.5/TMO 50:50 into a packed-bed reactor containing imm-RN or imm-HOR (Scheme 2). Keeping constant the starting concentration of rutinosides (5 g L−1), the flow was varied till the obtainment of the best results in terms of molar conversion (see the ESI†). Imm-RN gave Glu-HP with 90% m.c. in 30 min of residence time, while IQ was obtained with complete conversion in 5 min. Imm-HOR-mediated biotransformation delivered HP and Q with >99% and 89% conversion in 5 and 1 min, respectively (see the ESI†). A further leap forward has been made with the employment of the co-immobilized RN–HOR preparation, obtaining directly the desired aglycones HP and Q from the corresponding rutinosides with complete conversion in just 5 min of residence time (reactor volume: 1.2 mL). The biphasic stream exiting from the flow bioreactor (20 mL, 16 reactor volumes) was collected. After phase separation, the aqueous phase containing the sugars was recovered as the mixture could be potentially reutilized for cell culture/cell feeding operations.46 The evaporation of the organic phase allowed for product isolation (95–98 mg) without any further purification. TMO was also recovered and used again for other biotransformation cycles. Since all the materials involved in the developed procedure can be recovered and reused, the designed strategy can be considered an ultra-efficient, zero-waste process.35,37
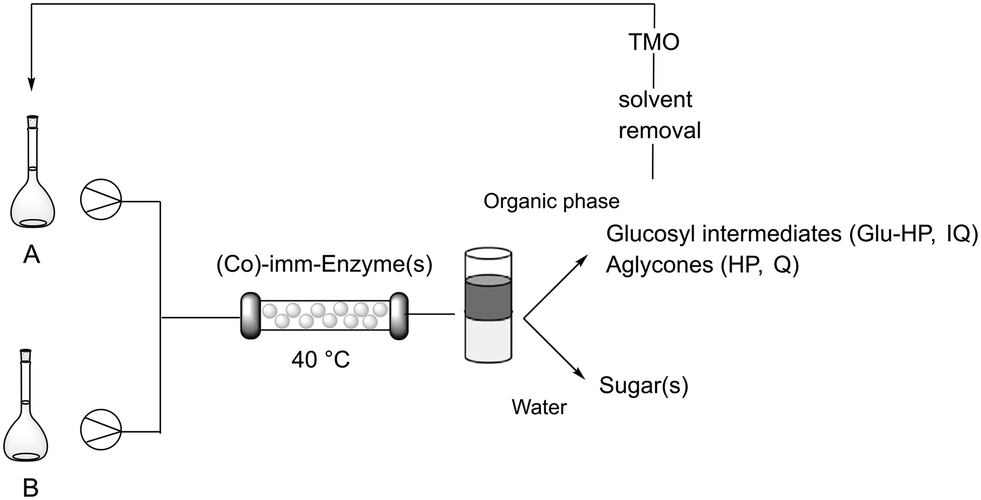 | ||
| Scheme 2 Flow bioprocessing of HES and RT. Solution A: 10 g L−1 HES or RT in TMO. Solution B: HEPES buffer, 0.05 M, pH 7.5. Co-imm-enzyme(s): imm-RN (5 mg gresin−1) or HOR (1 mg gresin−1) or imm-RN–HOR (5 mgRN gresin−1, 1 mgHOR gresin−1). T = 40 °C. | ||
In conclusion, hesperidin and rutin, among the most abundant rutinosyl flavonoids recoverable from citrus residues, have been successfully transformed into the corresponding aglycones (hesperetin and quercetin), characterized by better pharmacokinetic profiles. The combination of biocatalytic approaches and flow facilities allowed for higher yields and rapid reaction times (>99% m.c., 5 min) with respect to batch reactions (60–70%, 0.5–1 h). Enzyme co-immobilization was demonstrated to be a smart strategy to solve some drawbacks typical of step-by-step cascade reactions (e.g., multiple reaction steps, sequential pots, intermediate accumulation, unfavourable equilibrium, and difficult work-up procedures, among others). By providing a compartmentalized microenvironment where enzymes are spatially organized at the right density, the imm-RN–HOR preparation allowed for one-pot, 2-step flow bioprocesses characterized by enhanced catalytic performance. Due to the robustness of the imm-RN–HOR system (16 reactor volumes with no activity loss), together with the flow methodology here developed, by simply leaving the bioreactors working overtime, large scale production of natural products can be achieved without any further optimization. The recovery and the potential reuse of all the materials involved in the biotransformations, including solvents, make this process appealing for its overall sustainability.
Author contributions
Conceptualization: M. L. C., and A. Pi; methodology: M. L. C., A. C., G. C., C. P., A. Pe., L. T., and S. F.; investigation: M. L. C., A. C., G. C., C. P., L. T. and S. F.; resources: M. L. C., S. F., L. T., S. D., F. M., and A. Pi.; data curation: M. L. C., A. C., G. C., C. P., and S. F.; writing—review and editing: all authors. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was carried out within the Agritech National Research Center and received funding from the European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) – MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4 – D.D. 1032 17/06/2022, CN00000022). This manuscript reflects only the author views and opinions. Neither the European Union nor the European Commission can be considered responsible for them. M. L. C. acknowledges funding from the University of Milan (Piano di Sostegno UNIMI Linea 2-2021) (Italy) through the project W-BioFlow “From agro-food waste to valuable bioactives through flow biocatalysis”. S. D. acknowledges “ON Foods – Research and innovation network on food and nutrition Sustainability, Safety and Security – Working ON Foods”, PE00000003, Concession Decree No. 1550 of 11 October 2022 adopted by the Italian Ministry of University and Research (National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 – Call for tender no. 341 of 15 March 2022 of the Italian Ministry of University and Research funded by the European Union – Next-GenerationEU). The authors wish to thank Prof. Paradisi (University of Bern) for the kind donation of the plasmid containing HOR gene and UNITECH NO LIMITS for the SEM and confocal microscopy experiments.Notes and references
- U. M. Khan, A. Sameen, R. M. Aadil, M. Shahid, S. Sezen, A. Zarrabi, B. Ozdemir, M. Sevindik, D. N. Kaplan, Z. Selamoglu, A. Ydyrys, T. Anitha, M. Kumar, J. Sharifi-Rad and M. Butnariu, J. Evidence-Based Complementary Altern. Med., 2021, 2021, 2488804 Search PubMed.
- S. Suri, A. Singh and P. K. Nema, Appl. Food Res., 2022, 2, 100050 CrossRef CAS.
- D. Tonini, P. F. Albizzati and T. F. Astrup, Waste Manage., 2018, 76, 744–766 CrossRef PubMed.
- M. L. Contente, F. Annunziata, P. Cannazza, S. Donzella, C. Pinna, D. Romano, L. Tamborini, F. G. Barbosa, F. Molinari and A. Pinto, J. Agric. Food Chem., 2021, 69, 13669–13681 CrossRef CAS PubMed.
- G. Oboh and A. O. Ademosun, J. Food Sci. Technol., 2012, 49, 729–736 CrossRef CAS PubMed.
- K. Pyrzynska, Nutrients, 2022, 2387–2398 CrossRef CAS.
- A. Roohbakhsh, H. Parhiz, F. Soltani, R. Rezaee and M. Iranshahi, Life Sci., 2015, 124, 64–74 CrossRef CAS PubMed.
- J. Cho, Arch. Pharmacal Res., 2006, 29, 699–706 CrossRef CAS PubMed.
- J. Xiao, Crit. Rev. Food Sci. Nutr., 2017, 57, 1874–1905 CAS.
- M. Sasaki, H. T. Manalu, R. Kamogawa, C. S. C. Issasi, A. T. Quitain and T. Kida, Food Chem., 2023, 405, 134808 CrossRef CAS PubMed.
- N. Guo, Y. W. Zhu, Y. W. Jiang, H. K. Li, Z. M. Liu, W. Wang, C. H. Shan and Y. J. Fu, Ind. Crops Prod., 2020, 148, 112287 CrossRef CAS.
- A. Gupta, H. A. Al-Aubaidy, C. K. Narkowicz, H. F. Jelinek, D. S. Nichols, J. R. Burgess and G. A. Jacobson, Molecules, 2022, 27, 4741 CrossRef CAS PubMed.
- P. Li, X. Yao, Q. Zhou, X. Meng, T. Zhou and Q. Gu, Front. Nutr., 2022, 9, 1–13 Search PubMed.
- E. Gómez-Mejía, N. Rosales-Conrado, M. E. León-González and Y. Madrid, Food Chem., 2019, 295, 289–299 CrossRef PubMed.
- J. M. Carceller, J. P. Martínez Galán, R. Monti, J. C. Bassan, M. Filice, S. Iborra, J. Yu and A. Corma, Green Chem., 2019, 21, 839–849 RSC.
- A. M. Nuutila, K. Kammiovirta and K. M. Oksman-Caldentey, Food Chem., 2002, 76, 519–525 CrossRef CAS.
- K. Grohmann, J. A. Manthey and R. G. Cameron, Carbohydr. Res., 2000, 328, 141–146 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- P. Cannazza, S. Donzella, A. Pellis and M. L. Contente, Biotechnol. Adv., 2022, 59, 107985 CrossRef CAS PubMed.
- C. M. Park, G. M. Kim and G. S. Cha, Microorganisms, 2021, 9, 1–8 Search PubMed.
- J. Kapešová, L. Petrásková, K. Markošová, M. Rebroš, M. Kotik, P. Bojarová and V. Křen, Int. J. Mol. Sci., 2019, 20, 1112 CrossRef PubMed.
- Y. Zou, X. Xin, H. Xu, H. Yuan, X. Li, Y. Yu and G. Zhao, Green Chem., 2020, 22, 3196–3207 RSC.
- S. Yu, X. Shan, Y. Lyv and J. Zhou, Bioresour. Bioprocess., 2022, 9, 48 CrossRef.
- A. Gong, D. Zhu, Y. Y. Mei, X. H. Xu, F. A. Wu and J. Wang, Bioresour. Technol., 2016, 205, 48–57 CrossRef CAS PubMed.
- Y. S. Lee, J. Y. Huh, S. H. Nam, D. Kim and S. B. Lee, J. Food Sci., 2013, 78, C411–C415 CrossRef CAS PubMed.
- J. Wang, J. Med. Plants Res., 2012, 6, 1130–1139 CAS.
- J. M. Bolivar, J. M. Woodley and R. Fernandez-Lafuente, Chem. Soc. Rev., 2022, 51, 6251–6290 RSC.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- R. A. Sheldon and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- M. Romero-Fernández and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef PubMed.
- J. M. Bolivar and F. López-Gallego, Curr. Opin. Green Sustainable Chem., 2020, 25, 100349 CrossRef.
- A. I. Benítez-Mateos, M. L. Contente, D. Roura Padrosa and F. Paradisi, React. Chem. Eng., 2021, 6, 599–611 RSC.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- M. L. Contente, S. Farris, L. Tamborini, F. Molinari and F. Paradisi, Green Chem., 2019, 21, 3263–3266 RSC.
- S. Donzella, A. Colacicco, L. Nespoli and M. L. Contente, ChemBioChem, 2022, 23, e202200462 CrossRef CAS PubMed.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- S. Velasco-Lozano, J. Santiago-Arcos, J. A. Mayoral and F. López-Gallego, ChemCatChem, 2020, 12, 3030–3041 CrossRef CAS.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Nat. Chem., 2016, 8, 299–309 CrossRef CAS PubMed.
- S. Schoffelen and J. C. M. Van Hest, Curr. Opin. Struct. Biol., 2013, 23, 613–621 CrossRef CAS PubMed.
- A. E. Alvarenga, C. M. Romero and G. R. Castro, Eur. Food Res. Technol., 2013, 237, 977–985 CrossRef CAS.
- L. Delgado, M. Parker, I. Fisk and F. Paradisi, Food Chem., 2020, 323, 126825 CrossRef CAS PubMed.
- A. Pellis, F. P. Byrne, J. Sherwood, M. Vastano, J. W. Comerford and T. J. Farmer, Green Chem., 2019, 21, 1686–1694 RSC.
- M. L. Contente, L. Tamborini, F. Molinari and F. Paradisi, J. Flow Chem., 2020, 10, 235–240 CrossRef CAS.
- A. I. Benitez-Mateos and M. L. Contente, Catalysts, 2021, 11, 814–824 CrossRef CAS.
- S. Donzella, I. Serra, A. Fumagalli, L. Pellegrino, G. Mosconi, R. Lo Scalzo and C. Compagno, Biotechnol. Biofuels Bioprod., 2022, 15, 1–13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00603d |
| This journal is © The Royal Society of Chemistry 2023 |