
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvation rules: aromatic interactions outcompete cation–π interactions in synthetic host–guest complexes in water†
Gloria
Tobajas-Curiel
 a,
Qingqing
Sun
bc,
Jeremy K. M.
Sanders
a,
Pablo
Ballester
*bd and
Christopher A.
Hunter
*a
a,
Qingqing
Sun
bc,
Jeremy K. M.
Sanders
a,
Pablo
Ballester
*bd and
Christopher A.
Hunter
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: herchelsmith.orgchem@ch.cam.ac.uk
bInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology (BIST), Av. Països Catalans, 16, 43007, Tarragona, Spain. E-mail: pballester@iciq.es
cYangzhou University, School of Chemistry and Chemical Engineering, Yangzhou, Jiangsu 225002, China. E-mail: sunqingqing@snnu.edu.cn
dICREA, Passeig Lluís Companys 23, Barcelona 08010, Spain
First published on 13th November 2023
Abstract
Chemical double mutant cycles were used to measure the interaction of a N-methyl pyridinium cation with a π-box in a calix[4]pyrrole receptor. Although the cation–π interaction is attractive (−11 kJ mol−1), it is 7 kJ mol−1 less favourable than the corresponding aromatic interaction with the isosteric but uncharged tolyl group.
The main driving force for molecular recognition in water is usually provided by hydrophobic contacts between non-polar surfaces, and polar interactions between H-bonding groups control the specificity and geometrical properties of the interaction.1,2 The cation–π interaction between a quaternary ammonium ion and an aromatic ring lies somewhere between these two extremes: both moieties have non-polar hydrocarbon surfaces, but the positive charge and π-electron density make attractive electrostatic interactions.3,4 The importance of the cation–π interaction in biology has been highlighted in a number of studies of ion channels and protein–ligand interactions,4–6 but the relative contributions of polar interactions and desolvation to the cation–π interaction in water are difficult to disentangle.
One experiment that has offered some insight is to measure the difference between the non-covalent interaction of an aromatic ring with a quaternary ammonium ion and the corresponding interaction with an isosteric hydrocarbon. For the hydrocarbon, electrostatic interactions are minimal, and the interaction free energy is entirely due to desolvation, whereas for the quaternary ammonium ion, polar interactions with the positive charge make a significant contribution. For example, experiments carried out on biomolecules suggest that interaction with a trimethyl ammonium group is preferred over a t-butyl group.7,8 However, complexes of dumbbell guests with synthetic cavitand receptors consistently showed that the t-butyl group outcompetes the trimethyl ammonium group for the aromatic binding pocket in water (Fig. 1a).9
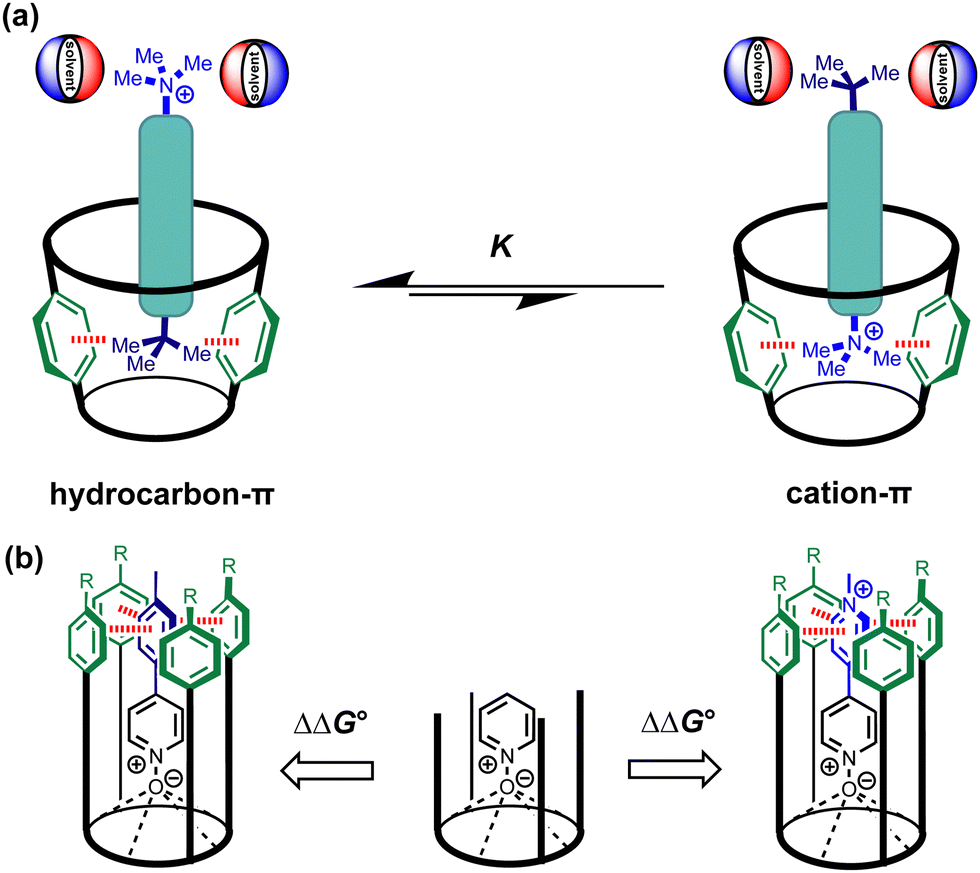 | ||
| Fig. 1 (a) Dumbbell guests previously used to measure the competition between solvation and interactions with a π-box at the base of a cavitand receptor. In water, the receptor binds the uncharged t-butyl group in preference to the cation–π interaction. (b) Comparison of binding affinities for closely related complexes allows direct measurement of the free energy contributions of the functional group interactions highlighted in red (aromatic interactions on the left versus cation–π interactions on the right) to the stabilities of calix[4]pyrrole·pyridine N-oxide complexes. | ||
Here, we use chemical double mutant cycles (DMC) to directly measure the free energy contributions due to non-covalent interactions with the aromatic pocket of a synthetic calix[4]pyrrole receptor in water. The interaction with an uncharged aromatic hydrocarbon is 7 kJ mol−1 more favourable than the interaction with an isosteric N-methyl pyridinium cation, which suggests that desolvation of the polar cation strongly disfavours the cation–π interaction in water (Fig. 1b).
We recently described a supramolecular approach for measuring aromatic interactions using complexes of calix[4]pyrrole receptors and pyridine N-oxide guests.10Fig. 2 shows that H-bonding interactions between N-oxide group of the guest and the four pyrrole NH protons at the base of the receptor cavity lock the complex into a well-defined conformation. When the side-walls of the receptor are extended with aromatic groups (green), a π-box is formed, which makes geometrically well-defined interactions with substituents attached to the para-position of the guest (blue). Here, the free energy contribution of cation–π interactions to the stability of the host–guest complex formed by a super-aryl-extended calix[4]pyrrole and a pyridine N-oxide guest equipped with a pyridinium substituent was measured in aqueous solution using the DMC approach (Fig. 3). By comparing closely-related complexes, the DMC dissects out the free energy contribution due to the non-covalent interaction highlighted in red in Fig. 3 from all other contributions to the stability of complex A, including for example changes in the strength of the H-bonding interactions with the N-oxide group.
 | ||
| Fig. 2 Energy-minimized structure of a truncated version of the 1·5 complex with the receptor solubilising groups replaced by methyl groups: (a) side view and (b) top view showing the two stacking and two edge-to-face cation–π interactions. Geometry optimizations were performed in the gas phase at the RI-BP86-D3BJ-def2-TZVP level of theory as implemented in TURBOMOLE v7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.com.13–22 | ||
 | ||
| Fig. 3 Chemical double mutant cycle for evaluating the free energy contribution (ΔΔG°) due to cation–π interactions to the overall stability of complex A. Only one of the four calix[4]pyrrole side-arms is shown for clarity, but the DMC measures the sum of interactions with four green aromatic rings on the host (see Fig. 1b). | ||
The chemical structures of the compounds used to construct DMCs are shown in Fig. 4. The two water-soluble calix[4]pyrrole receptors were synthesised according to previously reported procedures.11,12 Guest 5 was synthesised by alkylation of the corresponding pyridine (see ESI†), and the other guests were commercially available. The pyridinium solubilising groups used on the receptor have chloride counterions, and pyridinium guest has an iodide counterion. The extent of ion-pairing of halide anions with N-methyl pyridinium cations is negligible in aqueous solution, so we assume that the counterions have no influence on the DMC measurements.23 Long range electrostatic interactions with the charged solubilising groups on the receptors could affect the DMC measurements for cationic guest 5. Receptors 1 and 2 have four pyridinium solubilising groups in common, so any contribution from these charges cancels out in the DMC, but electrostatic contributions associated with the additional four pyridinium solubilising groups on the top of receptor 1 do not. Previous studies indicate four remote positive charges could destabilise the cation–π interaction by up to 4 kJ mol−1.9
 | ||
| Fig. 4 Chemical structures of calix[4]pyrroles 1 and 2 and pyridine N-oxide guests 3–6. | ||
The thermodynamic properties of complexes B, C and D of the DMC shown in Fig. 3 were determined using isothermal titration calorimetry (ITC). The stabilities of complex A, which are significantly more strongly bound in water, were measured using 1H NMR competition experiments. The results are reported in Table 1. The geometries of the aromatic interactions in these systems were analysed using complexation-induced changes in 1H NMR chemical shift. The results show that structures of the complexes do not vary significantly from one guest to another, and that the geometrical arrangement of the aromatic interactions is similar in all cases (see ESI† for details).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes in water at 298 Ka
:1 complexes in water at 298 Ka
| Guest | Host | |||
|---|---|---|---|---|
| 1 | 2 | |||
| K /M−1 | ΔG°b/kJ mol−1 | K/M−1c | ΔG°c/kJ mol−1 | |
| a Errors are quoted as twice the standard deviation of at least three measurements. b Determined by pairwise 1H NMR competition experiments. c Determined by ITC experiments. d Values reported previously in ref. 10. | ||||
| 3 | 3.5 ± 0.1 × 106 | −37.4 ± 0.1 | 3.3 ± 0.6 × 105 | −31.5 ± 0.4 |
| 4 | 2.3 ± 0.9 × 1010 | −59.1 ± 1.0 | 1.4 ± 0.1 × 106 | −35.0 ± 0.2 |
| 5 | 7.2 ± 3.4 × 107 | −44.8 ± 1.2 | 7.6 ± 0.2 × 104 | −27.9 ± 0.1 |
| 6 | 1.8 ± 0.5 × 1010 | −58.6 ± 0.6 | 1.1 ± 0.1 × 106 | −34.6 ± 0.1 |
The association constants for all of complexes formed with receptor 2 and with guest 3, i.e. complexes B, C and D of the DMC, are all in the range 105–106 M−1. The other complexes, i.e. complex A of the DMC (1–4, 1–5 and 1–6), which have the additional aromatic interactions highlighted in red in Fig. 1b, are all significantly more stable with association constants in the range 108–1010 M−1. The total free energy contribution (ΔΔG°/kJ mol−1) due to these interactions was determined using the data in Table 1 and eqn (1).
 | (1) |
It is important to note that the cation–π interaction is favourable in water.4 In this case, the N-methyl pyridinium group stabilises the complex by −11 kJ mol−1. As pointed out in seminal contributions by Dougherty,3,4 it is remarkable that the cation binds to the hydrophobic pocket at all, given the very large desolvation penalty in water. The fact that there is a net favourable contribution to the free energy of binding must reflect a highly attractive cation–π interaction that overcomes the desolvation penalty. Aromatic groups that lack the positive charge make weaker interactions with the receptor, but desolvation of these groups is favourable in water, and as a result the two aromatic interactions measured here both stabilise the complex by an additional −7 kJ mol−1 compared with the cation–π interaction.
These double mutant cycle results are consistent with the previous dumbbell guest experiments on the trimethyl ammonium cation–π interaction and confirm that the binding of an uncharged hydrocarbon to a π-box in these synthetic receptors is favoured over the binding of an isosteric quaternary ammonium cation. Although the cation–π interaction is favourable in water, interactions between uncharged hydrocarbons are significantly more favourable, due to the differences in the free energy contributions associated with the desolvation of these different types of functional group.
The manuscript was written through contributions of all authors.
We thank the Herchel Smith fund, Gobierno de España MICINN/AEI/FEDER (PID2020-114020GB-I00 and CEX2019-000925-S), the CERCA Programme/Generalitat de Catalunya, and AGAUR (2017 SGR 1123) for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS.
- J. Dong and A. P. Davis, Angew. Chem., Int. Ed., 2021, 60, 8035–8048 CrossRef CAS PubMed.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- R. M. Hughes, K. R. Wiggins, S. Khorasanizadeh and M. L. Waters, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11184–11188 CrossRef CAS.
- K. Schärer, M. Morgenthaler, R. Paulini, U. Obst-Sander, D. W. Banner, D. Schlatter, J. Benz, M. Stihle and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 4400–4404 CrossRef.
- Y. Zhu, M. Tang, H. Zhang, F. U. Rahman, P. Ballester, J. Rebek, C. A. Hunter and Y. Yu, J. Am. Chem. Soc., 2021, 143, 12397–12403 CrossRef CAS PubMed.
- G. Tobajas-Curiel, Q. Sun, J. K. M. Sanders, P. Ballester and C. A. Hunter, Chem. Sci., 2023, 14, 6226–6236 RSC.
- L. Escobar, A. Díaz-Moscoso and P. Ballester, Chem. Sci., 2018, 9, 7186–7192 RSC.
- L. Escobar and P. Ballester, Org. Chem. Front., 2019, 6, 1738–1748 RSC.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc. Theory, Comput. Model., 1997, 97, 119–124 CrossRef CAS.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- G. Saielli, J. Phys. Chem. A, 2008, 112(35), 7987–7995 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure and characterization including 1H and 13C NMR spectra, details of ITC and pairwise NMR competition experiments to characterize binding affinities. See DOI: https://doi.org/10.1039/d3cc04399a |
| This journal is © The Royal Society of Chemistry 2023 |