
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective access to dihydrophenanthridines and phenanthridinones via cyclisation of aryl amines onto N-tethered arynes†
Weitao
Sun‡
,
Maria
Uttendorfer‡
 ,
Fahima I. M.
Idiris
,
A. Yannic R.
Werling
,
Khushal
Siddiq
and
Christopher R.
Jones
*
,
Fahima I. M.
Idiris
,
A. Yannic R.
Werling
,
Khushal
Siddiq
and
Christopher R.
Jones
*
Department of Chemistry, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: c.jones@qmul.ac.uk
First published on 12th September 2023
Abstract
5,6-Dihydrophenanthridines are prepared from aryl amines via intramolecular addition to N-tethered arynes under mild conditions. A new o-silylaryl triflate precursor was developed to increase reactivity and enable electron-rich and electron-poor aryl amines to undergo cyclisation. A complete switch in product selectivity occurs when the reaction is conducted in air, affording the corresponding phenanthridin-6(5H)-one as the sole product under otherwise identical reaction conditions.
Phenanthridines and phenanthridinones are privileged heterocyclic scaffolds found in a range of natural products and therapeutically active compounds.1 They possess broad biological properties, including anticancer,2a antitumour,2b antiviral,2c antimicrobial,2d antifungal2e and antimalarial2f activity. In addition, a high charge mobility renders these frameworks versatile building blocks for functional materials.3 As a result, significant synthetic effort has been devoted to the synthesis of phenanthridines and their derivatives.4 This includes the Bischler–Napieralski reaction,5a photochemical,5b radical5c and microwave-assisted5d cyclisations, transition metal free C–H arylation,5e aza-Wittig,5f anionic ring-closure,5g hypervalent iodine5h and metal catalysed approaches.5i
Arynes have also been utilised in the synthesis of phenanthridine derivatives and related alkaloid natural products.6,7 These versatile reactive intermediates afford valuable benzenoid and heterocyclic frameworks8 and have experienced a recent resurgence in interest due to the advent of aryne precursors that act under mild conditions, namely the o-trimethylsilylaryl triflates (oSATs)9 and the hexadehydro-Diels–Alder reaction of polyalkynes.10 Cyclisation onto a pendant aryne has proven a valuable method to furnish phenanthridine derivatives, most notably in the synthesis of natural products.6a–e,g,h However, these approaches all generate arynes using organometallic reagents or strong bases at low temperatures which restricts the functional group tolerance. Methods that operate via electrophilic aromatic substitution (SEAr) have also been limited to electron-rich aryl nucleophiles such as 1 (Scheme 1a),6a–e which although common in the natural products, precludes preparation of more diverse substrate analogues. Alternatively, Barluenga and Sanz utilised anionic cyclisation onto pendant arynes (accessed from 3) to yield phenanthridine derivatives 4 and 5 by exploiting stronger in situ-formed aryllithium nucleophiles (Scheme 1b).6g,h However, this strategy also required organolithium reagents (in excess) to form both the aryne and the nucleophile, plus pre-functionalisation (o-bromination) of the pronucleophilic arene.
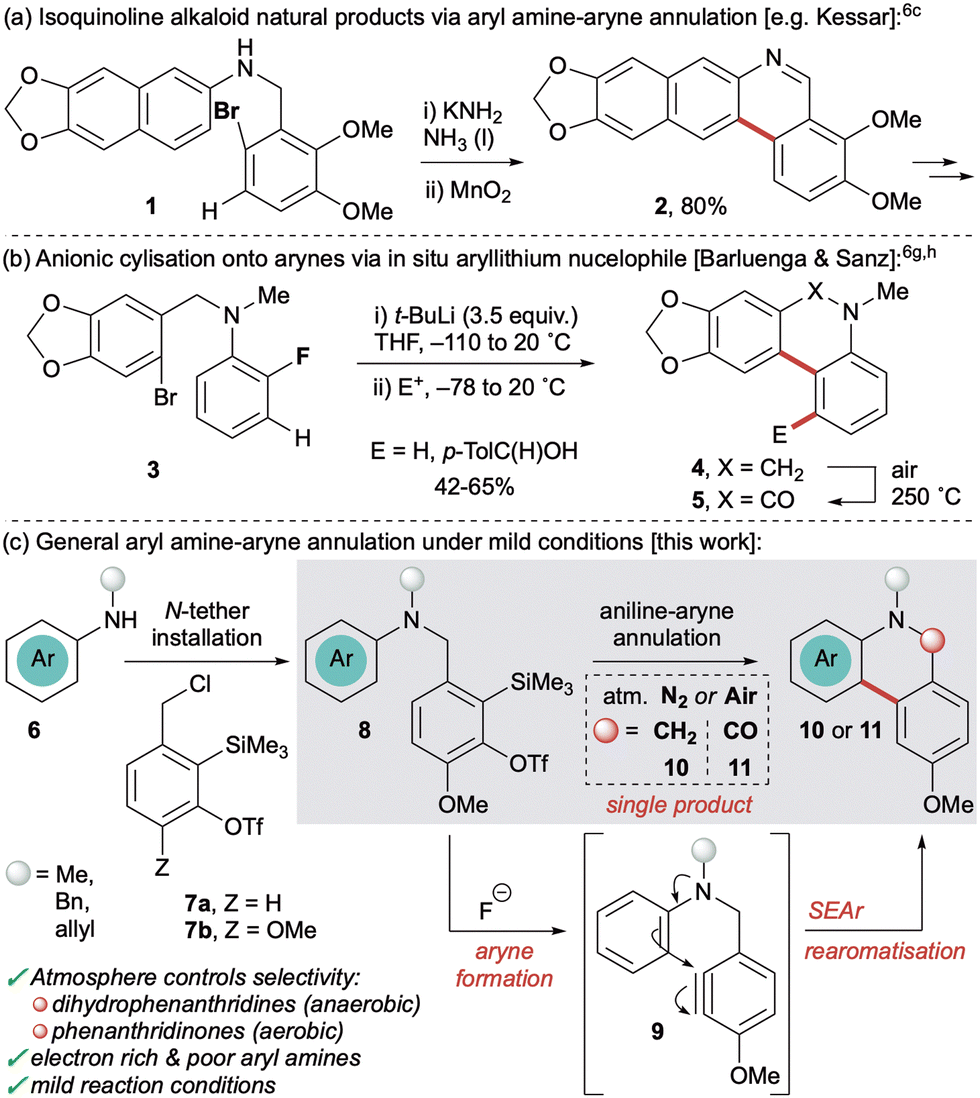 | ||
| Scheme 1 Cyclisation onto tethered arynes for the synthesis of phenanthridines and derivatives. | ||
Given our interests in the chemistry of arynes,11 we sought to develop a cyclisation approach to phenanthridine derivatives that exploited the benzylic oSAT precursor 7a previously used in our group (Scheme 1c).11b Inspired by the strategy of both Kessar and Stermitz in their syntheses of alkaloid natural products,6c,d it was proposed that aryl amines bearing an N-tethered aryne precursor (8) would undergo SEAr, upon generation of the aryne 9, to afford 5,6-dihydrophenanthridine derivatives 10. Given the propensity for such frameworks to undergo oxidation there would also be potential to access the corresponding phenanthridinones 11. Importantly, by using the oSAT precursor it removes the harsher reaction conditions of previous reports and should facilitate a wider range of aryl amine nucleophiles 6, especially those containing useful functional handles. Furthermore, tethering the aryne at nitrogen enables more facile synthesis of the starting materials, compared to linking via C–C bonds,6a,b thereby expediting the preparation of structural analogues.
To test this cyclisation hypothesis, benzylic oSAT precursor 7a was tethered to N-methyl aniline to afford precursor 12, which was then subjected to our standard aryne-forming conditions previously used with this tether (Scheme 2a).11b Unfortunately, this afforded a mixture of compounds and no clear evidence of cyclisation to dihydrophenanthridine 13 nor phenanthridinone 14. Intriguingly, a significant by-product was observed in the 1H NMR spectrum of the crude reaction mixture that suggested intermolecular attack at the aryne was taking precedence over the desired intramolecular process (although we were unable to isolate a clean sample for characterisation).12 With a goal to promote the cyclisation and suppress undesired intermolecular reactivity, a modified aryne precursor 17 was developed (Scheme 2b). It was rationalised that a p-methoxy substituent in 17 should increase reactivity by inductively polarising the aryne towards nucleophilic addition ortho to the tether – as supported by Garg and Houk's aryne distortion model13 – in addition to providing increased stabilisation of the resulting aryl anion.8a To this end, the novel p-methoxybenzyl aryne precursor 17 was prepared in four steps from commercially available 2-bromo-isovanillin 15 with no purification of the intermediates required.
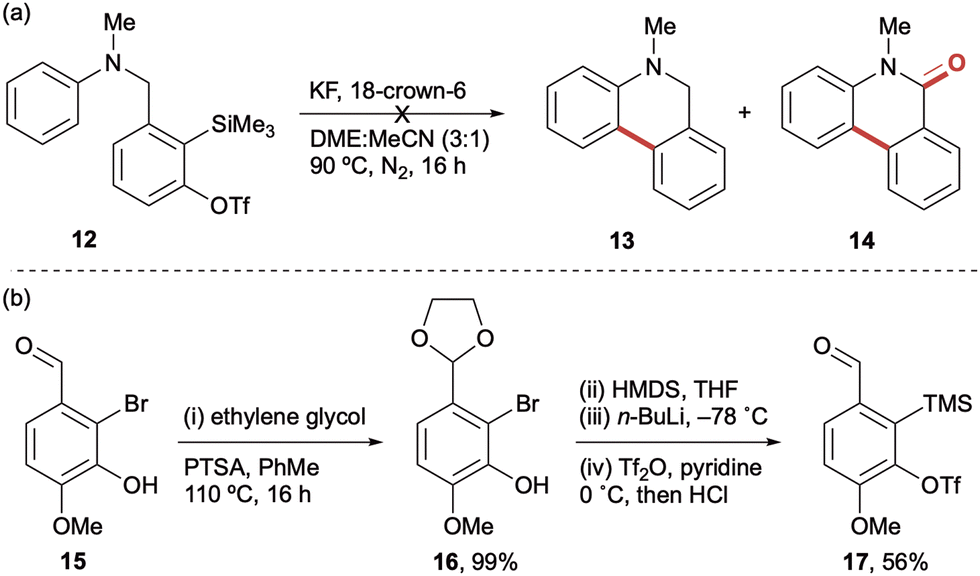 | ||
Scheme 2 Investigating cyclisation of aryl amine onto N-tethered aryne, (a) unsuccessful preliminary attempt to form 5,6-dihydrophenanthridine 13 or phenanthridinone 14. Reaction conditions: aryl amine (1.0 equiv.), KF (2.0 equiv.), 18-crown-6 (2.0 equiv.), DME![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN (3:1 by volume, 0.01 M), 90 °C, 24 h, N2 atmosphere. (b) Synthesis of a new p-methoxy benzylic aryne precursor 17. Reaction conditions: (i) ethylene glycol (5.0 equiv.), pyridinium p-toluenesulfonate (0.12 equiv.), PhMe, 110 °C, (ii) hexamethyldisilazane (0.8 equiv.), THF, 66 °C, 2 h, (iii) n-BuLi, THF, −78 °C, 30 min, (iv) trifluoromethanesulfonic anhydride (1.3 equiv.), pyridine (3.0 equiv.), CH2Cl2, 0 °C, 3 h, then HCl (4 M aq.), 16 h. Yields of isolated products throughout. :MeCN (3:1 by volume, 0.01 M), 90 °C, 24 h, N2 atmosphere. (b) Synthesis of a new p-methoxy benzylic aryne precursor 17. Reaction conditions: (i) ethylene glycol (5.0 equiv.), pyridinium p-toluenesulfonate (0.12 equiv.), PhMe, 110 °C, (ii) hexamethyldisilazane (0.8 equiv.), THF, 66 °C, 2 h, (iii) n-BuLi, THF, −78 °C, 30 min, (iv) trifluoromethanesulfonic anhydride (1.3 equiv.), pyridine (3.0 equiv.), CH2Cl2, 0 °C, 3 h, then HCl (4 M aq.), 16 h. Yields of isolated products throughout. | ||
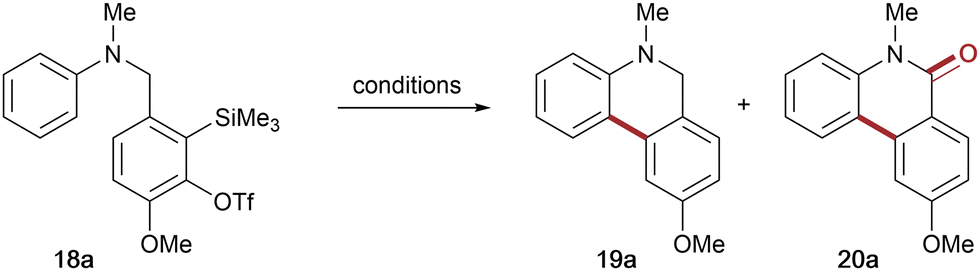
With the second generation aryne tether 17 in hand, the corresponding N-methyl aniline precursor 18a was then subjected to the conditions used for attempted cyclisation of 12. Gratifyingly, the new aryne tether promoted significant intramolecular reaction, affording dihydrophenanthridine 19a in 50% yield and the corresponding phenanthridinone 20a in 23% yield, with a marked decrease in deleterious N-arylation (8% by-product) (entry 1, Table 1). Having addressed the issue of overall reactivity, attention turned to optimising the reaction conditions to avoid mixtures of cyclisation products (see Table 1 for selected optimisation experiments). Evaluation of some common o-SAT activators (entries 2–5) identified CsF in acetonitrile and KF/18-crown-6 in THF as promoting cyclisation to the dihydrophenanthridine 19a in good yields, albeit with phenanthridinone and by-product still present. Encouragingly, replacing THF with DME led to an excellent yield of the desired dihydrophenanthridine 19a, with only trace amounts of the phenanthridinone 20a and no evidence of the competing intermolecular by-product (entries 5 & 6). Further investigations found that lowering the reaction temperature and concentration both decreased the overall yield (entries 7 and 8). Finally, increasing concentration led to more of the competing intermolecular N-arylation and subsequent erosion of the dihydrophenanthridine yield (entry 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Activator | Solvent | T (°C) | Yieldb (%) | ||
| 19a | 20a | By-product | ||||
| a Reaction conditions: aryl amine (1.0 equiv.), activator (2.0 equiv.), additive (2.0 equiv.), solvent [0.01 M], 16 h, N2 atmosphere. b 1H NMR yield vs. dibromomethane internal standard, isolated yield in parentheses, all reactions proceeded to full conversion after 16 h. c 1.0 M in THF. d 0.002 M. e 0.05 M. | ||||||
| 1 | KF 18-crown-6 | DME:MeCN (3:1) |
90 | 50 | 23 | 8 |
| 2 | CsF | MeCN | 90 | 69 | 2 | 7 |
| 3 | CsF | PhMe:MeCN |
90 | 64 | 10 | 11 |
| 4 | TBAFc | THF | 90 | 36 | — | — |
| 5 | KF 18-crown-6 | THF | 90 | 69 | 10 | 5 |
| 6 | KF 18-crown-6 | DME | 90 | 84(82) | 2 | — |
| 7 | KF 18-crown-6 | DME | 70 | 69 | 2 | 5 |
| 8 | KF 18-crown-6 | DMEd | 90 | 69 | — | 9 |
| 9 | KF 18-crown-6 | DMEe | 90 | 46 | — | 35 |
Having optimised the reaction conditions, attention turned to investigating substrate scope. Pleasingly, a range of substituted aniline derivatives were found to undergo cyclisation in generally good to excellent yields (Scheme 3). 4-Substituted anilines were evaluated first, with the electron-rich 4-methoxy derivative affording the corresponding dihydrophenanthridine 19b in 98% yield. The halogenated series showed an interesting trend, with the 4-iodo and 4-fluoro precursors undergoing efficient cyclisation to 19c (67%) and 19f (70%), respectively, however, the 4-bromo and 4-chloro derivatives furnished 19d and 19e in more moderate yields (52% and 48%). With the electron deficient 4-fluoro analogue proving highly amenable to the cyclisation, it was encouraging to observe that the 4-nitroaniline precursor also underwent the transformation to generate the corresponding electron-poor dihydrophenanthridine 19g in 34% yield. These are particularly noteworthy as most reports of intramolecular SEAr with arynes rely on markedly electron rich aromatic systems. A comparison of the propensity for cyclisation of the 3-methyl (18h) and 2-methylaniline (18i) precursors revealed a key role played by sterics. 3-Methylaniline 18h afforded the corresponding dihydrophenanthridine 19h in an excellent 83% yield, whereas only a trace of the 2-methyl analogue was observed. This suggested that 19i experiences significant 1,5-strain in the transition state required for cyclisation; instead favouring a mixture of the N-arylation by-product and an intramolecular dearomative aryne Diels–Alder cycloaddition.14 Interestingly, the more conformationally rigid indoline (18k) and tetrahydroquinoline (18l) precursors underwent efficient SEAr to furnish the corresponding tetracyclic products, 19k and 19l, respectively. It was noted that the THQ-derived framework afforded 19l in 76% yield by 1H NMR spectroscopic analysis; however, during attempted purification it proved susceptible to oxidation, leading to an isolated mixture (1:1.2) of 19l and the corresponding phenanthridinone.
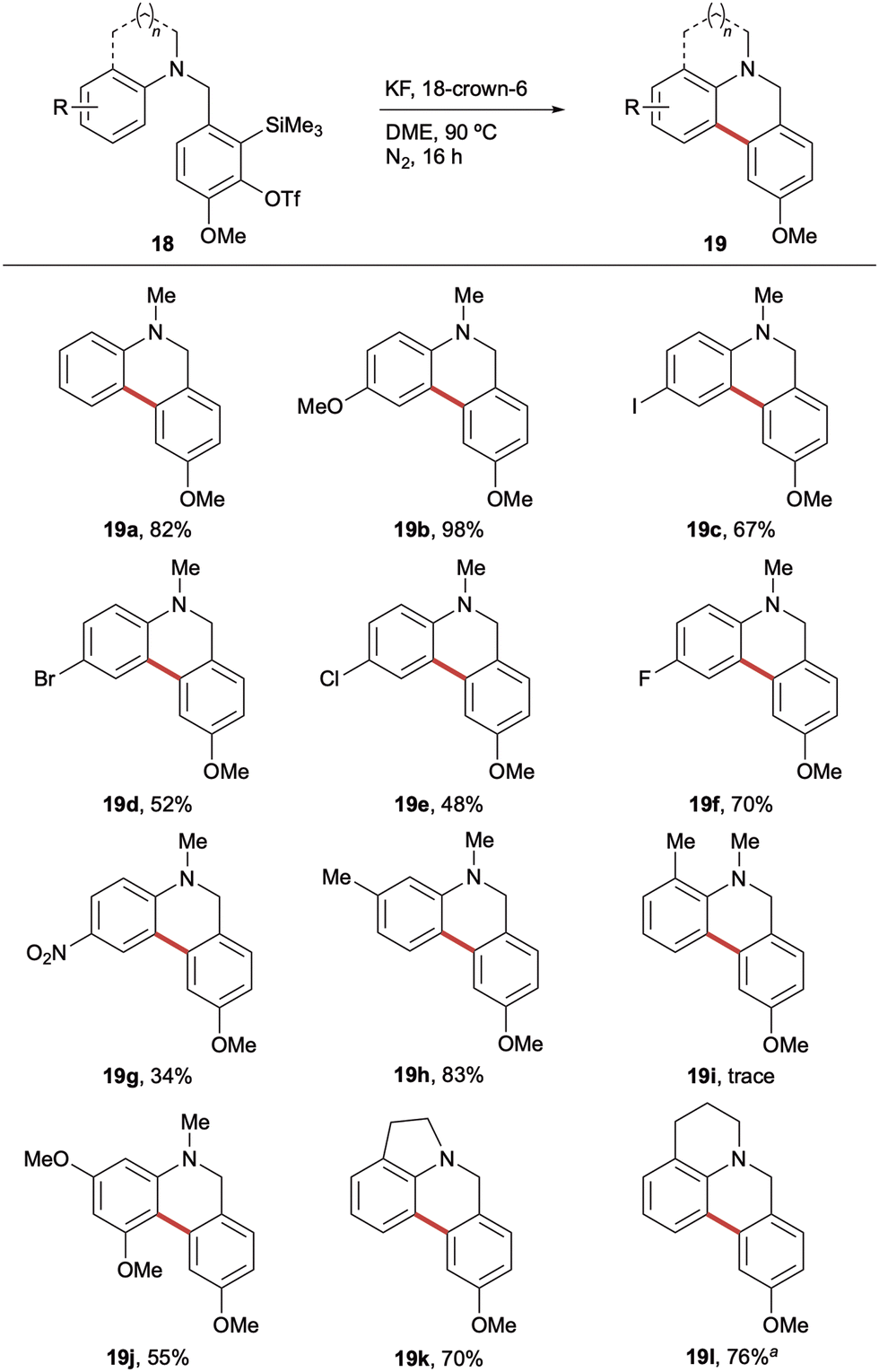 | ||
| Scheme 3 Synthesis of 5,6-dihydrophenanthridine derivatives 15. Reaction conditions: aryl amine (1.0 equiv.), KF (2.0 equiv.), 18-crown-6 (2.0 equiv.), DME (0.01 M), 90 °C, 16 h, N2 atmosphere. Yields of isolated products throughout. a1H NMR yield vs. dibromomethane internal standard, note: 19l partially oxidised upon purification to give isolated 1:1.2 mixture of 19l and corresponding phenanthridinone 20l. | ||
Having established the formation of dihydrophenanthridines from a range of electron rich and electron deficient aryl amines, we next investigated whether the product selectivity could be reversed to instead access phenanthridinones. During the initial optimisation studies, dihydrophenathridine 19a had been found to be susceptible to partial oxidation upon exposure to air, whilst 19l had part-oxidised during purification. This afforded mixtures of the dihydrophenanthridines and phenanthridinones; however a discrete second oxidative step was always required to effect complete conversion. With a view to accessing the phenanthridinone derivatives in a single step from the analogous aryl amine precursors 18, the reaction was performed in the presence of a range of oxidants, with most attempts affording mixtures of the two cyclisation products. However, it was extremely pleasing to observe that when 18a and 18l were exposed to the previously optimised cyclisation conditions, only in air rather than under an inert atmosphere, it resulted in a complete switch of selectivity to exclusively afford the corresponding phenanthridin-6(5H)-ones 20a and 20l in 62% and 79% yields respectively (Scheme 4).15
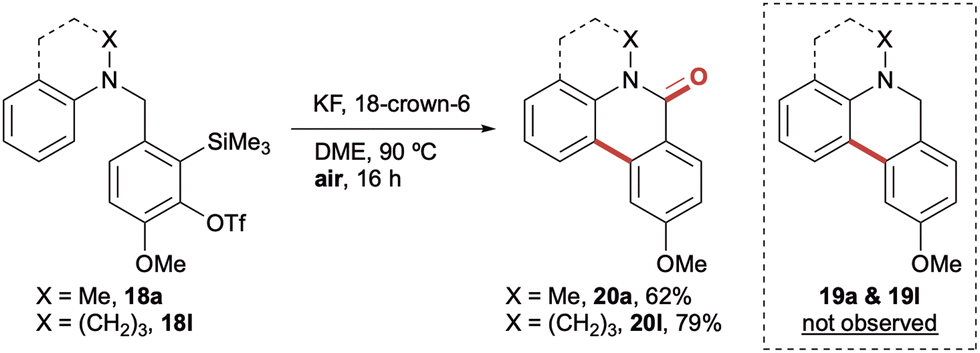 | ||
| Scheme 4 Cyclisation in air exclusively affords phenanthridinones 20a and 20l. Reaction conditions: aryl amine (1.0 equiv.), KF (2.0 equiv.), 18-crown-6 (2.0 equiv.), DME (0.01 M), 90 °C, 16 h, air. Yields of isolated products throughout. | ||
Finally, N-benzyl (18m) and N-allyl (18n) aniline precursors were also found to be amenable to cyclisation (Scheme 5). Given the increased potential for subsequent protecting group cleavage, the comparable yields obtained for the analogous N-benzyl (19m, 85%), N-allyl (19n, 69%) and N-Me (19a, 82%) dihydrophenanthridines were particularly encouraging for the synthetic utility of the method.
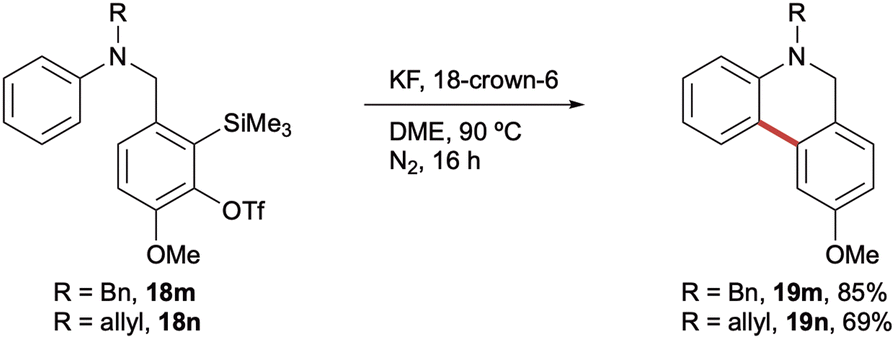 | ||
| Scheme 5 5,6-Dihydrophenanthridines bearing alternative N-protecting groups. Reaction conditions are as shown in Scheme 3. Yields of isolated products throughout. | ||
In conclusion, the cyclisation of aryl amines onto N-tethered arynes has been developed under mild reaction conditions using a novel silylaryl triflate precursor. This enabled the new biaryl linkage to be generated by SEAr using both electron rich and poor aryl amines and furnished a range of phenanthridine derivatives with handles for subsequent functionalisation. Selective access to dihydrophenanthridines or the corresponding phenanthridinones is dependent upon a simple switch between anaerobic and aerobic reaction environments.
We are grateful to the EPSRC (EP/M026221/1, CRJ), QMUL (studentships to FIMI & AYRW), CSC (studentship to WS) and DAAD RISE (GB-CH-3538, studentship to MU) for financial support. We thank the National Mass Spectrometry Facility at Swansea University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) F. Viladomat, J. Bastida, G. Tribo, C. Codina and M. Rubiralta, Phytochemistry, 1990, 29, 1307 CrossRef CAS; (b) R. B. Macgregor, R. M. Clegg and T. M. Jovin, Biochemistry, 1987, 26, 4008 CrossRef CAS PubMed; (c) Z. Jin, Nat. Prod. Rep., 2009, 26, 363 RSC.
- (a) V. Holl, D. Coelho, D. Weltin, P. Dufour and P. Bischoff, Anticancer Res., 2000, 20, 3233 CAS; (b) T. C. Johnstone, S. M. Alexander, W. Lin and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 116 CrossRef CAS PubMed; (c) G. T. Tan, M. J. Pezzuto and A. D. Kinghorn, J. Nat. Prod., 1991, 54, 143 CrossRef CAS PubMed; (d) G. Y. Zuo, F. Y. Meng, X. Y. Hao, Y. L. Zhang, G. C. Wang and G. L. Xu, J. Pharm. Pharm. Sci., 2008, 11, 90 CrossRef CAS PubMed; (e) X. J. Yang, F. Miao, Y. Yao, F. J. Cao, R. Yang, Y. N. Ma, B. Q. Qin and L. Zhou, Molecules, 2012, 17, 13026 CrossRef CAS PubMed; (f) M. Rivaud, A. Mendoza, M. Sauvain, A. Valentin and V. Jullian, Bioorg. Med. Chem., 2012, 20, 4856 CrossRef CAS PubMed.
- (a) N. Stevens, N. O’Connor, H. Vishwasrao, D. Samaroo, E. R. Kandel, D. L. Atkins, C. M. Drain and N. J. Turro, J. Am. Chem. Soc., 2008, 130, 7182 CrossRef CAS PubMed; (b) Y.-L. Chen, F.-H. Li and Z.-S. Bo, Macromolecules, 2010, 43, 1349 CrossRef CAS; (c) P. Pang, X. Miao, L. Ying, G. Kong, C. Che and W. Deng, J. Phys. Chem. C, 2020, 124, 5665 CrossRef CAS.
- Selected reviews: (a) F. Rafiee, Appl. Organomet. Chem., 2017, 31, 12 CrossRef; (b) N. Kshirsager, R. Sonawane, S. Pathan, G. Kamble and G. P. Singh, Lett. Org. Chem., 2021, 19, 434 CrossRef; (c) R. R. Aleti, A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Molecules, 2021, 26, 5560 CrossRef CAS PubMed.
- (a) B. A. Lorsbach and M. Kurth, J. Chem. Rev., 1999, 99, 1549 CrossRef CAS PubMed; (b) R. Alonso, P. J. Campos, B. Garcia and M. A. Rodriguez, Org. Lett., 2006, 8, 3521 CrossRef CAS PubMed; (c) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed; (d) F. Portela-Cubillo, J. S. Scott and J. C. Walton, J. Org. Chem., 2008, 73, 5558 CrossRef CAS PubMed; (e) C. Tang, Y. Yuan and N. Jiao, Org. Lett., 2015, 17, 2206 CrossRef CAS PubMed; (f) S. P. Marsden, A. E. McGonagle and D. McKeever-Abbas, Org. Lett., 2008, 10, 2589 CrossRef CAS PubMed; (g) M. Lysén, J. L. Kristensen, P. Vedsø and M. Begtrup, Org. Lett., 2002, 4, 257 CrossRef PubMed; (h) Q. Wang, X. Dong, T. Xiao and L. Zhou, Org. Lett., 2013, 15, 4846 CrossRef CAS PubMed; (i) H. A. McManus, M. J. Fleming and M. Lautens, Angew. Chem., Int. Ed., 2007, 46, 433 CrossRef PubMed.
- (a) T. Kametani and K. Ogasawara, J. Chem. Soc. C, 1967, 2208 RSC; (b) S. V. Kessar, N. Parkash and G. S. Joshi, J. Chem. Soc., Perkin Trans. 1, 1973, 1158 RSC; (c) S. V. Kessar, M. Singh and P. Balakrishnan, Indian J. Chem., 1974, 12, 323 CAS; (d) J. P. Gillespie, L. G. Amoros and F. R. Stermitz, J. Org. Chem., 1974, 39, 3239 CrossRef CAS PubMed; (e) T. Nakanishi and M. Suzuki, Org. Lett., 1999, 1, 985 CrossRef CAS PubMed; (f) J. Pawlas and M. Begtrup, Org. Lett., 2002, 4, 2687 CrossRef CAS PubMed; (g) J. Barluenga, F. J. Fananas, R. Sanz and Y. Fernandez, Chem. – Eur. J., 2002, 8, 2034 CrossRef CAS PubMed; (h) R. Sanz, Y. Fernandez, M. P. Castroviejo, A. Perez and F. J. Fananas, Eur. J. Org. Chem., 2007, 62 CrossRef CAS; (i) R. S. Reddy, C. Lagishetti, S. Chen, I. N. Chaithanya Kiran and Y. He, Org. Lett., 2016, 18, 4586 CrossRef PubMed.
- Selected examples: (a) C. Lu, A. V. Dubrovskiy and R. C. Larock, J. Org. Chem., 2012, 77, 8648 CrossRef CAS PubMed; (b) Y. Yang, H. Huang, L. Wu and Y. Liang, Org. Biomol. Chem., 2014, 12, 5351 RSC; (c) X. Peng, W. Wang, C. Jiang, D. Sun, Z. Xu and C.-H. Tung, Org. Lett., 2014, 16, 5354 CrossRef CAS PubMed; (d) S. Pimparkar and M. Jeganmohan, Chem. Commun., 2014, 50, 12116 RSC; (e) M. Feng, B. Tang, N. Wang, H.-X. Xu and X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 14960 CrossRef CAS PubMed; (f) C. Zhu and T. R. Hoye, J. Am. Chem. Soc., 2022, 144, 7750 CrossRef CAS PubMed; (g) R. Fang, S. Liu, Q. Yan, Y. Wei, J. Wang, Y. Lan and J. Tan, Chem. Sci., 2023, 14, 4278 RSC.
- Selected reviews: (a) R. Sanz, Org. Prep. Proced. Int., 2008, 40, 215 CrossRef CAS; (b) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (c) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191 RSC; (d) R. W. Hoffmann and K. Suzuki, Angew. Chem., Int. Ed., 2013, 52, 2655 CrossRef CAS PubMed; (e) D. Pérez, D. Peña and E. Guitían, Eur. J. Org. Chem., 2013, 5981 CrossRef; (f) J.-A. Garcia-Lopez and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766 RSC; (g) O. J. Diamond and T. J. Marder, Org. Chem. Front., 2017, 4, 891 RSC; (h) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044 RSC; (i) J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892 CrossRef CAS PubMed.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211 CrossRef CAS.
- T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208 CrossRef CAS PubMed.
- (a) P. Trinchera, W. Sun, J. E. Smith, D. Palomas, R. Crespo-Otero and C. R. Jones, Org. Lett., 2017, 19, 4644 CrossRef CAS PubMed; (b) F. I. M. Idiris, C. E. Majesté, G. B. Craven and C. R. Jones, Chem. Sci., 2018, 9, 2873 RSC; (c) W. Sun, P. Trinchera, N. Kurdi, D. Palomas, R. Crespo-Otero, S. Afshinjavid, F. Javid and C. R. Jones, Synthesis, 2018, 4591 CAS; (d) Y. Yang, Y. Xu and C. R. Jones, Eur. J. Org. Chem., 2019, 5196 CrossRef CAS; (e) E. A. Neal, A. Y. R. Werling and C. R. Jones, Chem. Commun., 2021, 57, 1663 RSC; (f) Y. Xu, R. Xie, Q. Li, J. Feng, H. Luo, Q. Ye, Z. Guo, Y. Cao, M. Palma, G. Chai, M.-M. Titirici and C. R. Jones, Small, 2023, 2302795 CrossRef PubMed.
- Unable to isolate pure sample of by-product. 1H NMR spectroscopic and mass spectrometric analyses of reaction mixture supported tentative assignment: postulated attack of aniline nitrogen onto second molecule of aryne affords quaternary ammonium species that could undergo cleavage of labile N-benzyl bond.
- J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 15798 CrossRef CAS PubMed.
- H. Takikawa, A. Nishii, H. Takiguchi, H. Yagashita, M. Tanaka, K. Hirano, M. Uchiyama, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 12440 CrossRef CAS PubMed.
- H. Wang, Z. Wang, H. Huang, J. Tan and K. Xu, 18-Crown-6 ether was found to promote oxidation. No reduction in phenanthridinone yield when reaction conducted in the presence of TEMPO (1.3 equivalents) and no substrate-TEMPO adducts observed, Org. Lett., 2016, 18, 5680 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03027j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |