
Synthesis of triarylborane-centered N-heterocyclic carbene cages with tunable photophysical properties†
Yi-Fan
Zhang
,
Ya-Wen
Zhang
,
Xin
Li
,
Li-Ying
Sun
* and
Ying-Feng
Han
 *
*
Key Laboratory of Synthetic and Natural Functional Molecule of the Ministry of Education, Xi'an Key Laboratory of Functional Supramolecular Structure and Materials, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, P. R. China. E-mail: lysun@nwu.edu.cn; yfhan@nwu.edu.cn
First published on 30th January 2023
Abstract
Triarylborane-based discrete metal-carbene supramolecular cages [M3(1)2](PF6)3 (M = Ag, Au) were synthesized and characterized. The new hexacarbene assemblies show a significant solvatochromic effect in solvents of different polarity. Furthermore, the reversible fluoride binding property of [Au3(1)2](PF6)3 was investigated by UV-vis absorption and fluorescence titrations. This work holds promise for future developments in the area of highly emissive and stimulus-responsive NHC-metal assemblies.
Triarylborane (TAB)-involving functional materials has received significant attention over the past few years owing to their potential utility in numerous applications, such as catalysis,1 organic optoelectronic materials,2 anion sensors,3 biological imaging4 and frustrated Lewis pairs (FLPs).1,5 Due to the inherent high reactivity of the boron center, its structural geometry and optical properties can be altered in the presence of suitable Lewis bases, such as fluoride ions.6 Compared with a few examples of triarylborane-containing metal–organic frameworks (MOFs) that were described,7 their applications in discrete organometallic supramolecular assemblies remain underexplored.8
During the past decade, continuous efforts have been made to construct supramolecular assemblies through coordination-driven self-assembly for different applications.9 In this regard, functional ligands are attractive as building blocks to bring potential applications to the final structures. Among them, discrete supramolecular assemblies held together by metal-carbene bonds have attracted considerable attention. A variety of N-heterocyclic carbene (NHC) donor types have been used to construct supramolecular metal-NHC assemblies.10,11 The obtained discrete NHC-based supramolecular systems have found wide applications in homogeneous catalysis,12 metal drugs,13 and materials science.14 Recent investigations on the exploration of metal-NHC-involving supramolecular assemblies have focused on their stimuli-responsive luminescence properties.15 Given the excellent luminescence and photoelectronic properties of TAB, we became interested in synthesizing a new tris-NHC ligand featuring the TAB scaffold, which may bestow the cage with unique optoelectronic properties.
We herein present a new example of a discrete AgI hexacarbene supramolecular cage [Ag3(1)2](PF6)3 containing Lewis acidic triarylborane-bridged NHC ligands. The assembly can be readily transformed to the corresponding AuI analogue [Au3(1)2](PF6)3. Both metal-carbene supramolecular cages [Ag3(1)2](PF6)3 and [Au3(1)2](PF6)3 show solvent-induced luminescence response properties. The position of the corresponding luminescence band was found to be red shifted with an increase in solvent polarity. Notably, the boron centers of the metal carbene assemblies are prone to complex formation with Lewis bases and altering absorption and emission properties.
The palladium-catalyzed Suzuki cross-coupling of 5-bromo-2-pyridinecarboxaldehyde with boronate ester compounds provided borane-containing pyridinecarbaldehydes 2 (Scheme S1, Fig. S1 and S2, ESI†). Then, the imine condensation of 2 with 2,4,6-trimethylaniline in the presence of an acid catalyst led to the formation of compound 3. Subsequently, the reaction of compound 3 with paraformaldehyde in the presence of 4 M HCl/1,4-dioxane solution, followed by anion exchange, yielded the desired triarylborane-bridged NHC precursor H3-1(PF6)3. The formation of NHC precursor H3-1(PF6)3 was confirmed by nuclear magnetic resonance (NMR) spectroscopy (1H, 13C{1H}, 2D NMR) and high resolution-electrospray ionization (HR-ESI) mass spectrometry (Fig. S3–S7, ESI†).
The reaction of TAB-bridged trisimidazo[1,5-a]pyridinium salt H3-1(PF6)3 with a slight excess of Ag2O at 55 °C for 14 h afforded the trinuclear AgI hexacarbene [Ag3(1)2](PF6)3 in an excellent yield of 96% (Scheme 1). The formation of hexacarbene metallocage [Ag3(1)2](PF6)3 was initially verified by multinuclear NMR spectroscopy and HR-ESI mass spectrometry (Fig. S8–S14, ESI†). The 1H NMR spectrum of complex [Ag3(1)2](PF6)3 featured no resonance for H1 protons of the imidazo[1,5-a]pyridinium group, which was previously observed for H3-1(PF6)3 at δ = 9.15 ppm in CD3CN (Fig. 1a and b). The peaks in the 1H NMR spectrum of [Ag3(1)2](PF6)3 showed pronounced shifts with respect to those of the NHC precursor H3-1(PF6)3. Specifically, the resonance of H11 protons of [Ag3(1)2](PF6)3 was downfield shifted, while the other protons of the imidazo[1,5-a]pyridinium group were upfield shifted as compared to the NHC precursor. Additionally, the 13C{1H} NMR spectrum of [Ag3(1)2](PF6)3 displayed the typical resonance of a carbene carbon atom at δ = 175.8 ppm. The formation of the complex [Ag3(1)2](PF6)3 was further confirmed by HR-ESI mass spectrometry, by the highest intensity peak at m/z = 793.9169 (calcd for [Ag3(1)2]3+ 793.9517) (Fig. 1c).
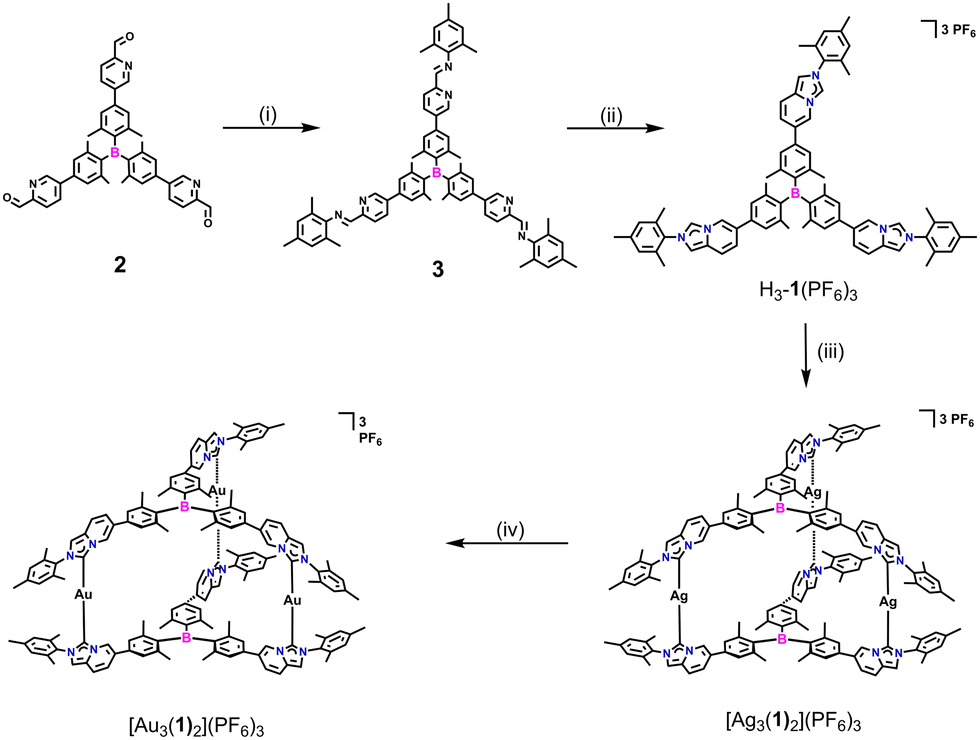 | ||
| Scheme 1 Synthesis of the triarylborane-bridged NHC precursor H3-1(PF6)3 and trinuclear hexacarbene complexes. (i) 2,4,6-Trimethylaniline, CH3COOH, CHCl3/EtOH, reflux; (ii) (a) paraformaldehyde, HCl, 25 °C; (b) NH4PF6, MeOH, 25 °C, yield 51%; (iii) Ag2O, CH3CN, 55 °C, yield 96%; (iv) AuCl(THT), CH3CN, 25 °C, yield 83%. | ||
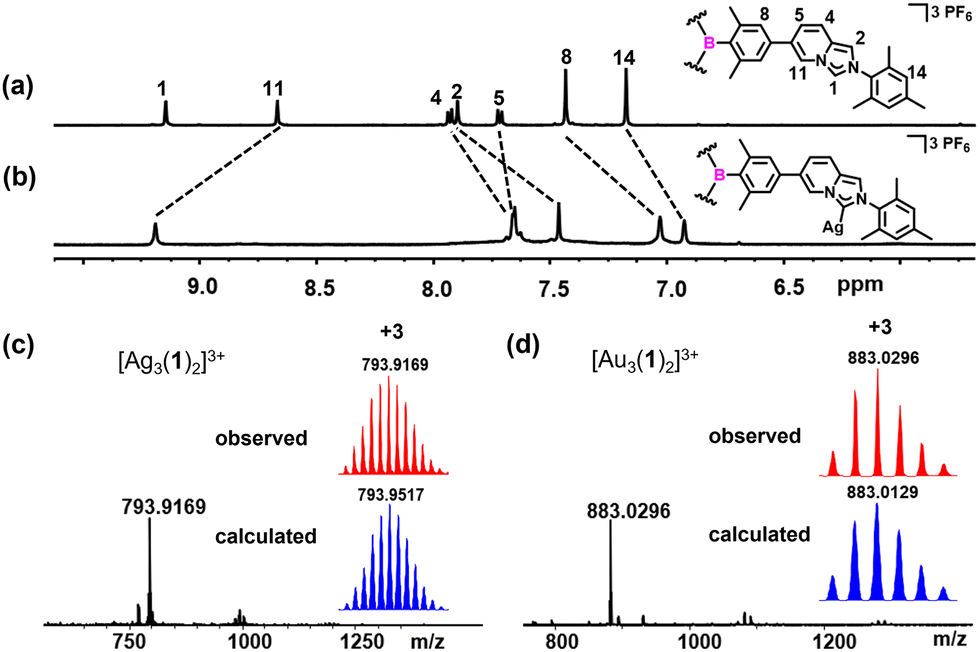 | ||
| Fig. 1 Sections of the 1H NMR spectra (600 MHz, CD3CN) of (a) H3-1(PF6)3 and (b) [Ag3(1)2](PF6)3. The HR-ESI mass spectra of (c) [Ag3(1)2](PF6)3 and (d) [Au3(1)2](PF6)3 (experimental in red, calculated in blue). | ||
Stirring an acetonitrile solution of [Ag3(1)2](PF6)3 with three equivalents of [AuCl(THT)] (THT = tetrahydrothiophene) for 24 h at ambient temperature gave exclusively the trinuclear AuI hexacarbene complex [Au3(1)2](PF6)3 in 83% yield. All the three AgI in [Ag3(1)2](PF6)3 can be substituted by AuI to give the homonuclear complex [Au3(1)2](PF6)3 without destroying the three-dimensional metallosupramolecular assembly. The conversion of the sliver complex to the gold one was characterized by 1H, 13C{1H}, 19F, 31P, and 2D NMR spectroscopy and HR-ESI mass spectrometry (Fig. S15–S20, ESI†). HR-ESI mass spectrometry of [Au3(1)2](PF6)3 showed the highest intensity peak at m/z = 883.0296 (calcd for [Au3(1)2]3+ 883.0129) (Fig. 1d).
In addition, single-crystal X-ray diffraction unambiguously confirmed the molecular structure of the complex [Ag3(1)2](PF6)3. Crystals suitable for X-ray diffraction were grown by the slow diffusion of diethyl ether into a saturated acetonitrile solution at room temperature. Complex [Ag3(1)2](PF6)3 crystallizes in the monoclinic C2/c space group and the central B atoms have an ideal trigonal-planar geometry with the sum of the C8-B-C5, C5-B-C9, and C8-B-C9 angles being 359.9° (Fig. 2a). As shown in Fig. 2b, the distance between the two boron atoms was measured to be 7.102(5) Å. Due to the steric repulsion between the six ortho-methyl groups around the boron atom, tricoordinate central boron atoms formed a propeller-like structure, which prevented π-stacking interaction between the two triarylborane units. Indeed, complex [Ag3(1)2](PF6)3 exists as a racemic mixture in solution and crystallizes as a racemic modification of the P and M enantiomers (Fig. S21, ESI†).16 Furthermore, weak intermolecular C–H⋯π interactions were found between the N-mesityl group and the imidazo[1,5-a]pyridine ring in the molecular stacking diagram in which the separation between the point-centroids was 3.497(2) Å (Fig. S23, ESI†). Moreover, the bond length and the bond angle parameters of each component of the complex cation [Ag3(1)2]3+ were measured (Ag–CNHC bond lengths, 2.036(10)–2.082(10) Å, CNHC–Ag–CNHC bond angles, 176.4(5)°–178.2(4)°, and N–CNHC–N bond angles, 101.1(8)°–104.0(8)°). These parameters fall within the range previously reported for related AgI polycarbene assemblies.17
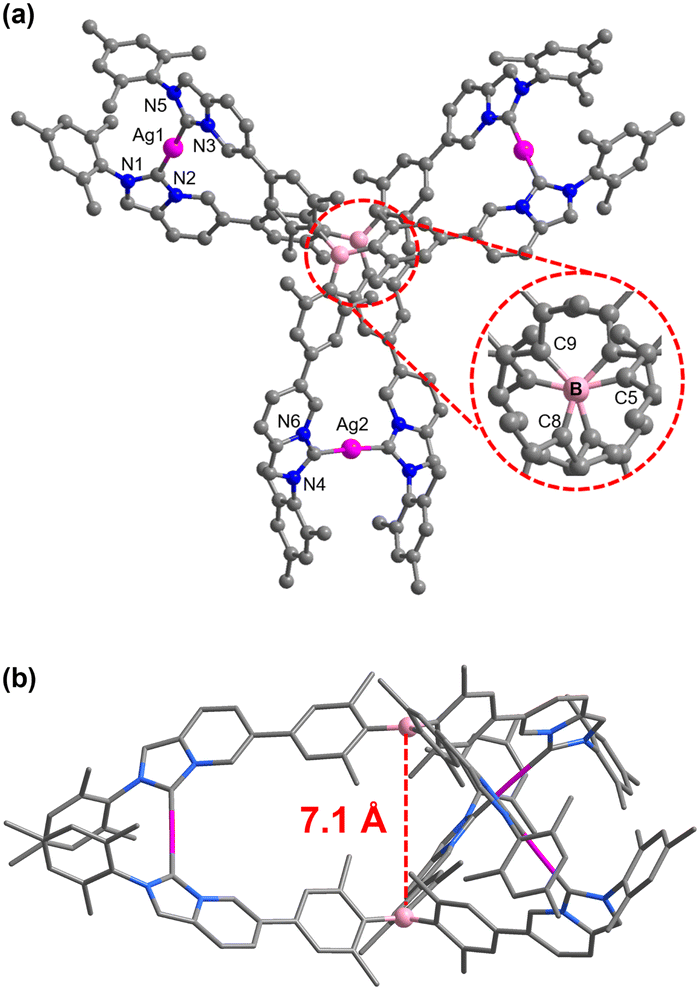 | ||
| Fig. 2 Molecular structure of the trication [Ag3(1)2]3+ in [Ag3(1)2](PF6)3: (a) top view, (b) side view. Hydrogen atoms and counteranions are omitted for clarity; color code: B, pink; Ag, violet; N, blue; C, gray. | ||
Since triarylboranes are promising fluorescent skeleton molecules,18 we further investigated the optical properties of the NHC precursor and its AgI and AuI complexes. The UV-vis absorption spectra of the NHC precursor and the trinuclear hexacarbene metal complexes showed similar types of absorption curves in the range of 220–400 nm (Fig. 3a). The lower energy band results from the imidazo[1,5-a]pyridinium groups to boron intramolecular charge transfer (ICT) and the higher energy band can be attributed to the boron aryl center π–π* transitions.19 Upon excitation at 330 nm, the NHC precursor H3-1(PF6)3 and trinuclear hexacarbene complexes showed different emission curves in acetonitrile solution, as λem = 418 nm for H3-1(PF6)3, 487 nm for [Ag3(1)2](PF6)3 and 478 nm for [Au3(1)2](PF6)3. A red-shift of the emission maximum was observed for the NHC precursor upon metalation (Fig. 3b). The absolute fluorescence quantum yields determined for the solutions of H3-1(PF6)3, [Ag3(1)2](PF6)3, and [Au3(1)2](PF6)3 were 21.76, 19.19, and 1.46%, respectively. All the photophysical data are summarized in Table S2 (ESI†).
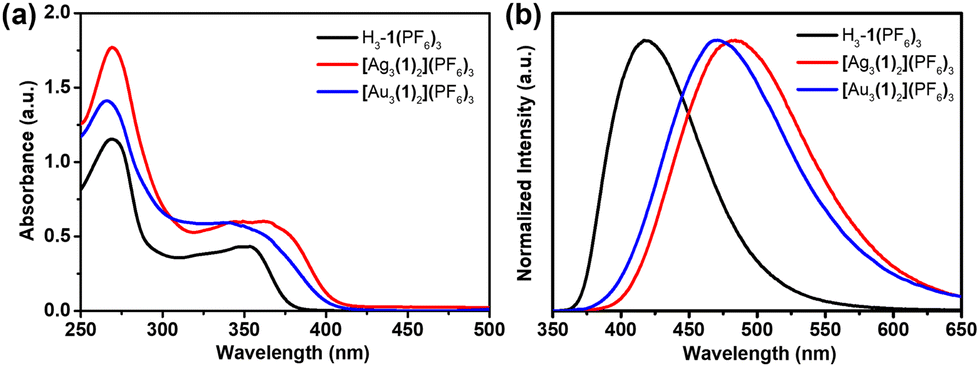 | ||
| Fig. 3 (a) The UV-vis absorption and (b) normalized emission spectra of H3-1(PF6)3, [Ag3(1)2](PF6)3, and [Au3(1)2](PF6)3 in acetonitrile solution (λex = 330 nm, c = 10−5 M, and T = 298 K). | ||
As illustrated in Fig. 4, the fluorescence emission spectra of the hexacarbene complexes in both polar (e.g., DMSO, DMF, and MeCN) and nonpolar solvents (e.g., DCE, DCM, and THF) were highly tunable over a broad spectrum from 450 nm to 550 nm. The emission bands were red-shifted with an increase in the solvent polarity, showing the characteristics of highly solvent-dependent emission, donor–acceptor charge transfer, and a highly polarized excited state (Fig. S24 and S25, ESI†).20 To gain an insight into the emission characteristics of the NHC precursor and the trinuclear hexacarbene complexes, DFT theoretical calculations were used to reveal their electronic structures and electron transition orbits (Fig. S26, ESI†). The HOMO and LUMO energy levels of H3-1(PF6)3 are −11.24 and −7.38 eV, respectively. The HOMO energy levels of the complexes [Ag3(1)2](PF6)3 and [Au3(1)2](PF6)3 are −9.86 eV, while the LUMO energy levels of the complexes [Ag3(1)2](PF6)3 and [Au3(1)2](PF6)3 are −6.39 and −6.43 eV, respectively. The HOMO is mainly localized in the imidazo[1,5-a]pyridine group, while the LUMO is in the triarylborane group. This finding further suggests that the low-energy emission is mainly due to the ICT transfer between triarylborane (π) and boron-bound phenyl rings (π*).
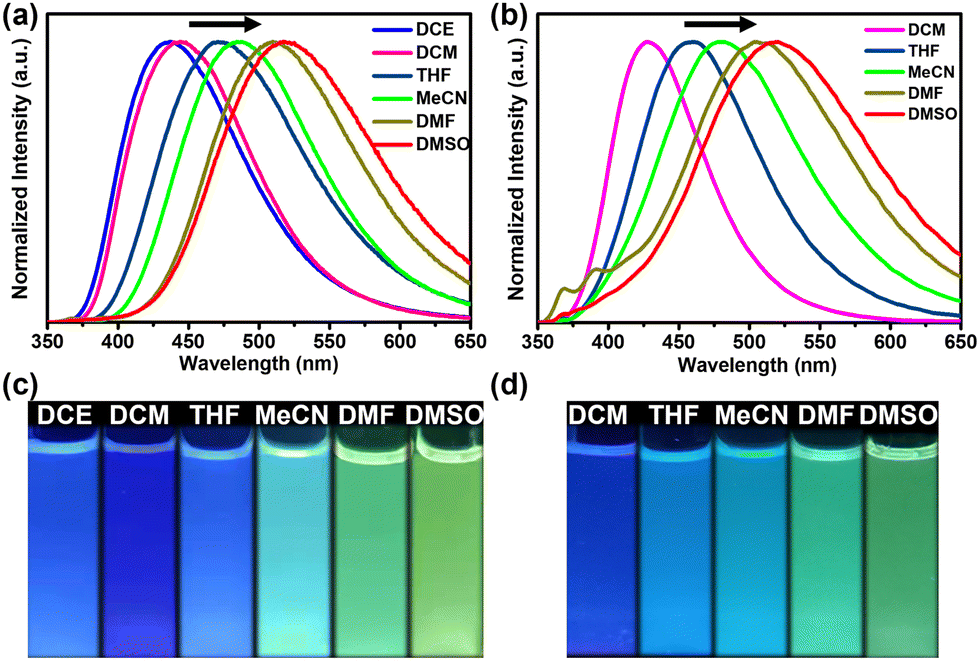 | ||
| Fig. 4 The normalized fluorescence emission spectra of (a) [Ag3(1)2](PF6)3 and (b) [Au3(1)2](PF6)3 in various solvents (λex = 330 nm, c = 10−5 M, and T = 298 K). Photographs of (c) [Ag3(1)2](PF6)3 and (d) [Au3(1)2](PF6)3 in various solvents under UV light. | ||
Considering that the interaction between the ligands and gold atoms further increases the stability of the complex, we use the trinuclear hexacarbene cage [Au3(1)2](PF6)3 in fluoride ion sensing that was monitored by absorption and emission studies. In the UV-vis absorption spectra, the complex [Au3(1)2](PF6)3 featured a broad low-energy absorption band centered at 360 nm, which was gradually quenched upon the addition of an increasing amount of fluoride ions, and the peak at 265 nm also decreased (Fig. 5a). The absorption spectral change can be assigned primarily to the binding of F− with the boron center.21 This corresponding change can be rationalized as a result of the interruption of the π-conjugation extending through the vacant p-orbital of the boron atom by forming the corresponding fluoroborate.22 In the fluorescence titration, the emission band of the complex [Au3(1)2](PF6)3 at 478 nm was quenched by the addition of fluoride ions (Fig. 5b). This finding indicated that the low energy absorption in [Au3(1)2](PF6)3 was mainly assignable to the triarylborane-centered π → pπ (B) charge transfer transition.23 According to previous reports,8 we guessed that the cage and fluoride ions were bound in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, and a nonlinear least-squares curve fitting procedure and the conclusion of the conducted Job's plot experiments provided further evidence (Fig. S27 and S28, ESI†).
:2, and a nonlinear least-squares curve fitting procedure and the conclusion of the conducted Job's plot experiments provided further evidence (Fig. S27 and S28, ESI†).
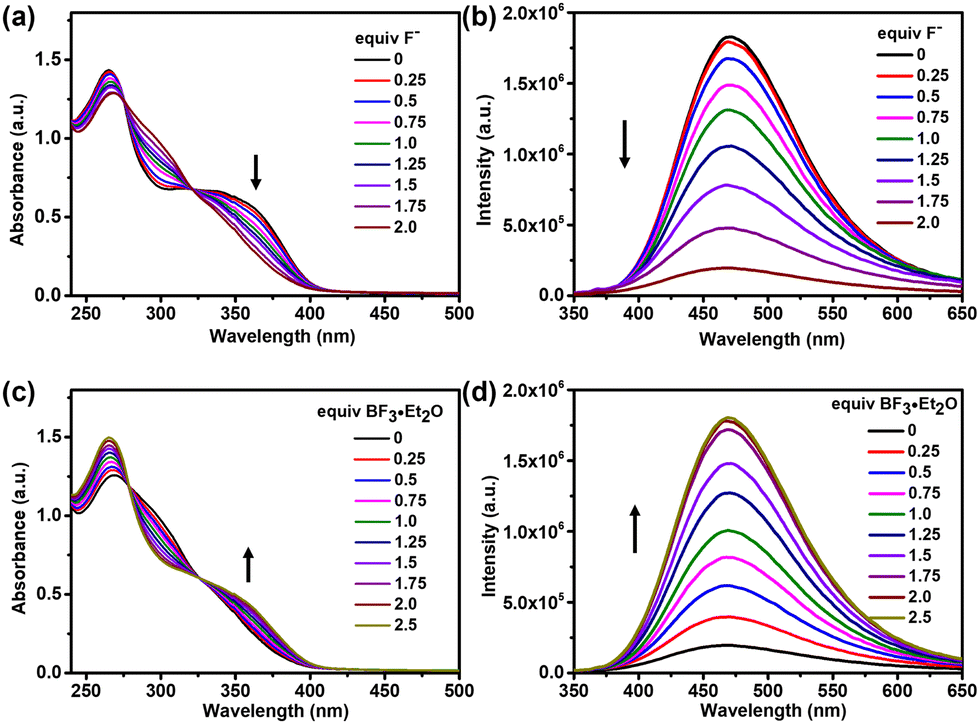 | ||
| Fig. 5 (a) The UV-vis absorption and (b) fluorescence emission spectra of [Au3(1)2](PF6)3 with TBAF (tetrabutylammonium fluoride). (c) The UV-vis absorption and (d) fluorescence emission spectra of [Au3(1)2](PF6)3 with BF3·Et2O in the presence of 2 equivalents of TBAF (CH3CN, λex = 330 nm, c = 10−5 M, and T = 298 K). | ||
Subsequently, the reversibility of fluoride binding in the complex [Au3(1)2](PF6)3 was studied using BF3·Et2O as an external Lewis acid. Both the UV-vis absorption and fluorescence emission spectra of [Au3(1)2](PF6)3 showed reversible processes (Fig. 5c and d), which demonstrated that the AuI hexacarbene cage was stable in the presence of excess fluoride ions.
In summary, we have successfully synthesized the trigonal prismatic AgI and AuI hexacarbene cages by a metal-controlled self-assembly approach from triarylborane-based tris-NHC ligands and silver or gold ions. The molecular structure of [Ag3(1)2](PF6)3 was confirmed by X-ray crystallography. Notably, the fluorescence spectra of the trinuclear hexacarbene complexes show some degree of dependence on the solvent polarity. In particular, the fluoride binding and removal properties of [Au3(1)2](PF6)3 were investigated by the UV-vis absorption and fluorescence titrations. The results showed that the triarylborane-based cage had a strong fluorine-binding ability. The results of this study not only provided insight into the design principles of metal coordination-driven self-assembly but also demonstrate the ON/OFF and OFF/ON fluorescence switching in the presence and absence of fluoride ions, opening a new route for the development of chemical sensors.
This work was supported by the National Natural Science Fund for Distinguished Young Scholars of China (No. 22025107), the National Youth Top-notch Talent Support Program of China and the National Natural Science Foundation of China (22101225), Natural Science Foundation of Shaanxi Province (2021JQ-432), Xi’an Key Laboratory of Functional Supramolecular Structure and Materials, and the FM&EM International Joint Laboratory of Northwest University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC.
- M. Elbing and G. C. Bazan, Angew. Chem., Int. Ed., 2008, 47, 834–838 CrossRef CAS PubMed.
- (a) C. A. Swamy and P. P. Thilagar, Inorg. Chem., 2014, 53, 2776–2786 CrossRef PubMed; (b) M.-S. Yuan, X. Du, Z. Liu, T. Li, W. Wang, E. V. Anslyn and J. Wang, Chem. – Eur. J., 2018, 24, 9211–9216 CrossRef CAS PubMed.
- S. M. Berger and T. B. Marder, Mater. Horiz., 2022, 9, 112–120 RSC.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- (a) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed; (b) Q. Xia, J. Zhang, X. Chen, C. Cheng, D. Chu, X. Tang, H. Li and Y. Cui, Coord. Chem. Rev., 2021, 435, 213783 CrossRef CAS.
- (a) Y. Liu, X. Xu, Q. Xia, G. Yuan, Q. He and Y. Cui, Chem. Commun., 2010, 46, 2608–2610 RSC; (b) S. Shyshkanov, T. N. Nguyen, F. M. Ebrahim, K. C. Stylianou and P. J. Dyson, Angew. Chem., Int. Ed., 2019, 58, 5371–5375 CrossRef CAS PubMed; (c) T. N. Nguyen, I. M. Harreschou, J.-H. Lee, K. C. Stylianou and D. W. Stephan, Chem. Commun., 2020, 56, 9600–9603 RSC; (d) B. A. Blight, R. Guillet-Nicolas, F. Kleitz, R.-Y. Wang and S. Wang, Inorg. Chem., 2013, 52, 1673–1675 CrossRef CAS PubMed; (e) F. M. Ebrahim, T. N. Nguyen, S. Shyshkanov, A. Gładysiak, P. Favre, A. Zacharia, G. Itskos, P. J. Dyson and K. C. Stylianou, J. Am. Chem. Soc., 2019, 141, 3052–3058 CrossRef CAS PubMed.
- (a) N. Mihara, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2019, 58, 12497–12501 CrossRef CAS PubMed; (b) J. Y. Ryu, J. M. Lee, N. V. Nghia, K. M. Lee, S. Lee, M. H. Lee, P. J. Stang and J. Lee, Inorg. Chem., 2018, 57, 11696–11703 CrossRef CAS PubMed.
- (a) Z. Wang, L. He, B. Liu, L.-P. Zhou, L.-X. Cai, S.-J. Hu, X.-Z. Li, Z. Li, T. Chen, X. Li and Q.-F. Sun, J. Am. Chem. Soc., 2020, 142, 16409–16419 CrossRef CAS PubMed; (b) P.-F. Cui, Y.-J. Lin, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2020, 142, 8532–8538 CrossRef CAS PubMed; (c) P.-F. Cui, X.-R. Liu, Y.-J. Lin, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2022, 144, 6558–6565 CrossRef CAS PubMed; (d) X. Gao, Z. Cui, Y.-R. Shen, D. Liu, Y.-J. Lin and G.-X. Jin, J. Am. Chem. Soc., 2021, 143, 17833–17842 CrossRef CAS PubMed.
- (a) P. J. Altmann and A. Pöthig, Angew. Chem., Int. Ed., 2017, 56, 15733–15736 CrossRef CAS PubMed; (b) P. J. Altmann and A. Pöthig, J. Am. Chem. Soc., 2016, 138, 13171–13174 CrossRef CAS PubMed; (c) L.-L. Ma, Y. Li, X. Li, L. Zhang, L.-Y. Sun and Y.-F. Han, Angew. Chem., Int. Ed., 2022, 61, e202208376 CAS.
- (a) N. Sinha, L. Stegemann, T. T. Y. Tan, N. L. Doltsinis, C. A. Strassert and F. E. Hahn, Angew. Chem., Int. Ed., 2017, 56, 2785–2789 CrossRef CAS PubMed; (b) N. Sinha and F. E. Hahn, Acc. Chem. Res., 2017, 50, 2167–2184 CrossRef CAS PubMed; (c) M.-M. Gan, J.-Q. Liu, L. Zhang, Y.-Y. Wang, F. E. Hahn and Y.-F. Han, Chem. Rev., 2018, 118, 9587–9641 CrossRef CAS PubMed; (d) S. Ibáñez, M. Poyatos and E. Peris, Acc. Chem. Res., 2020, 53, 1401–1413 CrossRef PubMed; (e) Z.-E. Zhang, Y.-Y. An, B. Zheng, J.-P. Chang and Y.-F. Han, Sci. China: Chem., 2021, 64, 1177–1183 CrossRef CAS; (f) X.-X. Liu, Y. Li, X. Li, F. E. Hahn and Y.-F. Han, Sci. China: Chem., 2021, 64, 1709–1715 CrossRef CAS; (g) R. C. Nishad, S. Kumar and A. Rit, Angew. Chem., Int. Ed., 2022, 61, e202206788 CrossRef CAS PubMed.
- (a) E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed; (b) T. Liu, S. Bai, L. Zhang, F. E. Hahn and Y.-F. Han, Natl. Sci. Rev., 2022, 9, nwac067 CrossRef CAS PubMed.
- W. Liu and R. Gust, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS.
- (a) A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed; (b) R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS PubMed; (c) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- (a) Y. Li, T. Yang, N. Li, S. Bai, X. Li, L.-L. Ma, K. Wang, Y. Zhang and Y.-F. Han, CCS Chem., 2022, 4, 732–743 CrossRef CAS; (b) Y. Li, Y.-Y. An, J.-Z. Fan, X.-X. Liu, X. Li, F. E. Hahn, Y.-Y. Wang and Y.-F. Han, Angew. Chem., Int. Ed., 2020, 59, 10073–10080 CrossRef CAS PubMed.
- Y. Liu, X. Xu, F. Zheng and Y. Cui, Angew. Chem., Int. Ed., 2008, 47, 4538–4541 CrossRef CAS PubMed.
- (a) A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS PubMed; (b) Y.-F. Han, G.-X. Jin and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 9263–9266 CrossRef CAS PubMed.
- (a) K. Parab, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 12879–12885 CrossRef CAS PubMed; (b) J. Liu, S. Zhang, C. Zhang, J. Dong, C. Shen, J. Zhu, H. Xu, M. Fu, G. Yang and X. Zhang, Chem. Commun., 2017, 53, 11476–11479 RSC; (c) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (d) S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Organometallics, 2018, 37, 1900–1909 CrossRef CAS.
- (a) B. H. Choi, J. H. Lee, H. Hwang, K. M. Lee and M. H. Park, Organometallics, 2016, 35, 1771–1777 CrossRef CAS; (b) P. Sudhakar, K. K. Neena and P. Thilagar, J. Mater. Chem. C, 2017, 5, 6537–6546 RSC.
- S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Inorg. Chem., 2020, 59, 3142–3151 CrossRef CAS PubMed.
- (a) F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286–17289 CrossRef CAS PubMed; (b) S. W. Kwak, H. Kwon, J. H. Lee, H. Hwang, M. Kim, Y. Chung, Y. Kim, K. M. Lee and M. H. Park, Dalton Trans., 2018, 47, 5310–5317 RSC.
- (a) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CrossRef CAS; (b) Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- H. Lee, S. Jana, J. Kim, S. U. Lee and M. H. Lee, Inorg. Chem., 2020, 59, 1414–1423 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supporting figures and tables. CCDC 2167062. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06584c |
| This journal is © The Royal Society of Chemistry 2023 |