
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInverting glucuronidation of hymecromone in situ by catalytic nanocompartments†
Maria
Korpidou
 a,
Viviana
Maffeis
ab,
Ionel Adrian
Dinu
ab,
Cora-Ann
Schoenenberger
ab,
Wolfgang P.
Meier
ab and
Cornelia G.
Palivan
*ab
a,
Viviana
Maffeis
ab,
Ionel Adrian
Dinu
ab,
Cora-Ann
Schoenenberger
ab,
Wolfgang P.
Meier
ab and
Cornelia G.
Palivan
*ab
aDepartment of Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4058, Basel, Switzerland. E-mail: cornelia.palivan@unibas.ch
bNCCR-Molecular Systems Engineering, Mattenstrasse 24a, BPR 1095, 4058, Basel, Switzerland
First published on 25th April 2022
Abstract
Glucuronidation is a metabolic pathway that inactivates many drugs including hymecromone. Adverse effects of glucuronide metabolites include a reduction of half-life circulation times and rapid elimination from the body. Herein, we developed synthetic catalytic nanocompartments able to cleave the glucuronide moiety from the metabolized form of hymecromone in order to convert it to the active drug. By shielding enzymes from their surroundings, catalytic nanocompartments favor prolonged activity and lower immunogenicity as key aspects to improve the therapeutic solution. The catalytic nanocompartments (CNCs) consist of self-assembled poly(dimethylsiloxane)-block-poly(2-methyl-2-oxazoline) diblock copolymer polymersomes encapsulating β-glucuronidase. Insertion of melittin in the synthetic membrane of these polymersomes provided pores for the diffusion of the hydrophilic hymecromone–glucuronide conjugate to the compartment inside where the encapsulated β-glucuronidase catalyzed its conversion to hymecromone. Our system successfully produced hymecromone from its glucuronide conjugate in both phosphate buffered solution and cell culture medium. CNCs were non-cytotoxic when incubated with HepG2 cells. After being taken up by cells, CNCs produced the drug in situ over 24 hours. Such catalytic platforms, which locally revert a drug metabolite into its active form, open new avenues in the design of therapeutics that aim at prolonging the residence time of a drug.
1. Introduction
Living organisms use metabolism for the biotransformation of endogenous and exogenous substances, such as drugs. During the metabolic processes, hydrophilic derivatives are created which lead to rapid excretion and elimination from the body.1 In most cases, drugs with shorter half-life tend to act quickly with their effects wearing off rapidly. As a result, more frequent administrations or higher doses are needed which can lead to abuse of these compounds and even addiction.2,3 Hence, the alteration of the pharmacodynamic and pharmacokinetic parameters of rapidly metabolized drugs is the focus of intense research. One state-of-the-art approach involves the entrapment or encapsulation of the active molecules in lipid or polymer-based nanocarriers that offer a more controlled and/or targeted drug delivery.4–6 These delivery systems were shown to enhance the biodistribution, pharmacokinetics, stability and solubility of the active compounds.7 However, low drug loading capacity, toxicity associated with high concentrations and environmental concerns are major limitations of these nanocarriers when it comes to mass production and market release.8An important step in overcoming these limitations is the development of nanocompartments confining enzymes able to convert prodrugs into drugs in order to achieve a spatially and/or temporally controlled drug production.9–12 These biosynthetic nanocompartments protect the encapsulated enzymes from their surroundings, leading to prolonged activity and less immunogenicity.13–15 When appropriately selected, polymer-based compartments offer several advantages over liposomes,16,17 such as enhanced mechanical and colloidal stability,18 tuneable permeability19 and stimuli responsiveness.20,21 Enzyme biopharmaceuticals have been encapsulated in different types of nanocompartments in order to improve their properties, including stability against degradation or reduced toxicity.22 For example, L-asparaginase (ASNase), used to treat leukaemias and lymphomas has been encapsulated in polyion complex vesicles (PICsomes)23 and in permeable, asymmetric polymersomes.24 When systemically injected into mice, ASNase PICsomes exhibited sustained conversion of L-asparagine in the blood stream due to their prolonged blood circulation compared with free ASNase.23 While in this study the enzyme itself was acting as the therapeutic, and the shielding effect from the encapsulation was pivotal, encapsulation of β-galactosidase in carbohydrate-b-poly(propylene glycol) derived vesicles (CAPsomes), which are intrinsically permeable to small molecular weight molecules, afforded the conversion of a co-administered prodrug into an active compound in addition to protecting the enzyme from degradation.11 Advances in nanocompartments designed for enzyme replacement therapy,23,25,26 cancer treatment11,12 and other applications10,27,28in vitro and/or in vivo have increased the interest and research in the field. Inverting metabolic inactivation of a drug in situ is a novel concept that has yet to be explored.
Hymecromone or 4-methylumbelliferone (4-MU) is an inhibitor of hyaluronan synthesis and primarily used as a drug in bile therapy.29 More recently, its effectiveness in treating other diseases such as cancer,30 type 1 diabetes31 and COVID-1932 is being explored. Hymecromone is rapidly metabolized to 4-methylumbelliferyl glucuronide (4-MUG) in the liver.33 Owing to its poor pharmacokinetics and quick elimination from the body (oral bioavailability <3%, t½ = 28 min) due to glucuronidation, the daily administered hymecromone dose may rise up to 2400 mg.30,34,35
In this study, we created catalytic nanocompartments based on polymersomes rendered permeable by melittin biopores that confine β-glucuronidase (GUS) in their cavity. These catalytic nanocompartments (CNCs) are designed to catalyse the conversion of a pro-drug or metabolite from its glucuronide form to the active compound in vitro and in cells (Fig. 1). We chose bacterial GUS for encapsulation as it cleaves the glucuronide moiety from a variety of molecules under a broad range of conditions (pH 5.5–7.8 and temperatures up to 60 °C),36–38 and explored the production of active 4-MU from the glucuronide conjugate 4-MUG which is the main 4-MU metabolite found in the body.39
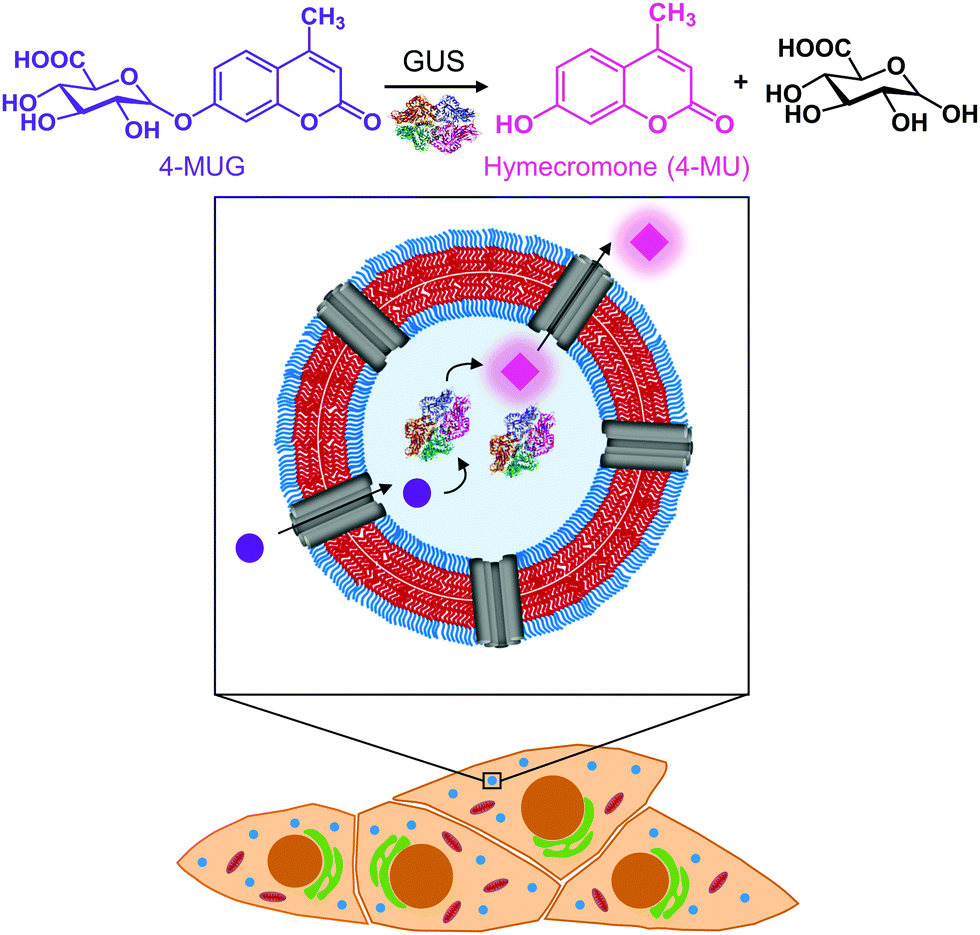 | ||
| Fig. 1 Schematic representation of the catalytic nanocompartment containing GUS and its enzymatic activity. Melittin pores in the PDMS25-b-PMOXA10 membrane provide a pathway to from the compartment interior where β-glucuronidase (GUS) catalyses the production of hymecromone (4-MU) from its glucuronide conjugate (4-MUG) within cells that have taken up CNCs. | ||
Polymersomes self-assembled from poly(dimethylsiloxane)-block-poly(2-methyl-2-oxazoline) amphiphilic block copolymers are recognized for their stealth properties, non-toxicity, and biocompatibility.16,40 Hence, we generated polymersomes by self-assembly of PDMS25-b-PMOXA10, a diblock copolymer where the short hydrophobic PDMS block facilitates melittin insertion resulting in the permeabilization of the polymersome membrane.41–43 A porous membrane was pivotal to our CNC design in order to provide a pathway for hydrophilic substrates and products to and from the nanocompartment cavity containing GUS.41,43,44 In the present study, we report the physicochemical characterization of GUS CNCs and their enzymatic efficiency in phosphate buffered solution (PBS) and cell culture medium. To explore the potential of CNCs for therapeutic applications, we examined cellular toxicity, uptake into cells and intracellular activity of GUS CNCs. In this regard, we used HepG2 cells, since they are derived from liver where hymecromone is mainly glucuronidated.33 Our catalytic nanocompartments are unique in that they represent a prototype for the locally confined transformation of a prodrug/metabolite to an active compound which makes them a stepping stone toward a whole new field of biomedical applications.
2. Experimental
2.1 Materials
B-glucuronidase (GUS, E. coli Type VII-A), melittin (from honey bee venom), 4-methylumbelliferyl-β-D-glucuronide (4-MUG), proteinase K (from Tritirachium album), fluorescent dye Atto647, penicillin, streptomycin, L-glutamine, Sepharose® (4B, 45–165 μm beads diameter), and Whatman® Nucleopore™ Track-Etched membranes (100 nm) were purchased from Sigma-Aldrich (USA). Atto488 N-hydroxysuccinimide ester (Atto488 NHS-Ester) was purchased from ATTO-TEC (Germany). Enhanced Pierce bicinchonic acid (BCA) assay, and wheat germ agglutinin – Alexa Fluor™ 555 conjugate were purchased from Thermo Fisher Scientific (USA). Phosphate buffered saline (PBS) and fetal bovine serum (FBS) were purchased from BioConcept (Switzerland). Minimum essential medium and non-essential amino acids (NEAA) were purchased from Gibco Life Sciences (USA). CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) and Hoechst 33342 trihydrochloride trihydrate were purchased from Invitrogen (USA).2.2 Nanocompartment preparation
Synthesis and characterization of amphiphilic diblock copolymer poly(dimethylsiloxane)25-block-poly(2-methyl-2-oxazoline)10 (PDMS25-b-PMOXA10) were described previously.41 A membrane thickness of 12 ± 0.8 nm for self-assembled PDMS25-b-PMOXA10 polymersomes was determined from cryogenic transmission electron micrographs.42Polymersomes containing enzymes, henceforth termed catalytic nanocompartments (CNCs) and 'empty' polymersomes were prepared using the film rehydration method. In brief, a thin film of PDMS25-b-PMOXA10 (5 mg mL−1 polymer in EtOH) was formed by rotary evaporation of the solvent (100 rpm at 40 °C, 160 mbar for 45 min). For the preparation of permeabilized CNCs containing GUS (GUS–melCNCs), the polymer film was rehydrated in the dark in PBS (pH 7.2) containing β-glucuronidase (0.4 mg mL−1, 25 kU) and melittin (50 μM), by stirring overnight at room temperature (RT). Polymersomes without melittin pores and enzyme (PSs), melittin-permeabilized polymersomes (melPSs), and CNCs without melittin (GUS–CNCs) were obtained by rehydrating the polymer film with PBS, PBS and melittin, and PBS and β-glucuronidase, respectively. For Atto647-labeled polymersomes (Atto647-PSs), the film was rehydrated with PBS containing Atto647 (0.02 mg mL−1). Following self-assembly, nanocompartments were incubated with proteinase K (0.05 mg mL−1) for 2 hours at 37 °C to remove non-encapsulated protein, followed by extrusion (10 times) through a 100 nm Whatman Nuclepore polycarbonate membrane. Size exclusion chromatography (SEC) on a Sepharose® 4B (45–165 μm beads diameter) column equilibrated in PBS was performed for further purification. Nanocompartment suspensions were stored at 4 °C until further use.
2.3 Characterization of nanocompartments
Static light scattering (SLS) experiments were performed on a light scattering spectrometer (LS instruments, Switzerland), equipped with a He–Ne 21 mW laser (λ = 632.8 nm) at scattering angles from 30° to 135° at 25 °C. The samples were diluted to 0.03 mg mL−1 final concentration of polymer in order to reduce multiple scattering. The radius of gyration (Rg) was obtained from the SLS data using Guinier plots, while the hydrodynamic radius (Rh) was obtained from DLS.45
For fluorescence correlation spectroscopy (FCS), an inverted laser scanning confocal microscope (LSM 880, Carl Zeiss, Germany) with a water immersion objective (Zeiss C/Apochromat, M = 40, NA = 1.2) was used. An Argon laser (wavelength 488 nm) with appropriate filter (MBS 488) was used to excite Atto488. The pinhole size (34 μm, 1 AU) was adjusted before recording FCS curves of the free dye.
For FCS measurements, 20 μL of the free fluorophore, fluorophore labelled-enzyme or polymersomes in PBS (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 dilution), were placed on a 0.15 mm thick glass coverslip mounted on the microscope stage. Fluorescence signals from free fluorophore, Atto488-labeled enzyme and polymersomes loaded with Atto488-labeled enzyme were measured in a real time (5 s with 30 repetitions) and autocorrelation function was obtained by a QuickFit 3.0 software calculator. The experimental autocorrelation curves for the free fluorophore were fitted according to eqn (1) with a one component diffusion model:
:2 dilution), were placed on a 0.15 mm thick glass coverslip mounted on the microscope stage. Fluorescence signals from free fluorophore, Atto488-labeled enzyme and polymersomes loaded with Atto488-labeled enzyme were measured in a real time (5 s with 30 repetitions) and autocorrelation function was obtained by a QuickFit 3.0 software calculator. The experimental autocorrelation curves for the free fluorophore were fitted according to eqn (1) with a one component diffusion model:
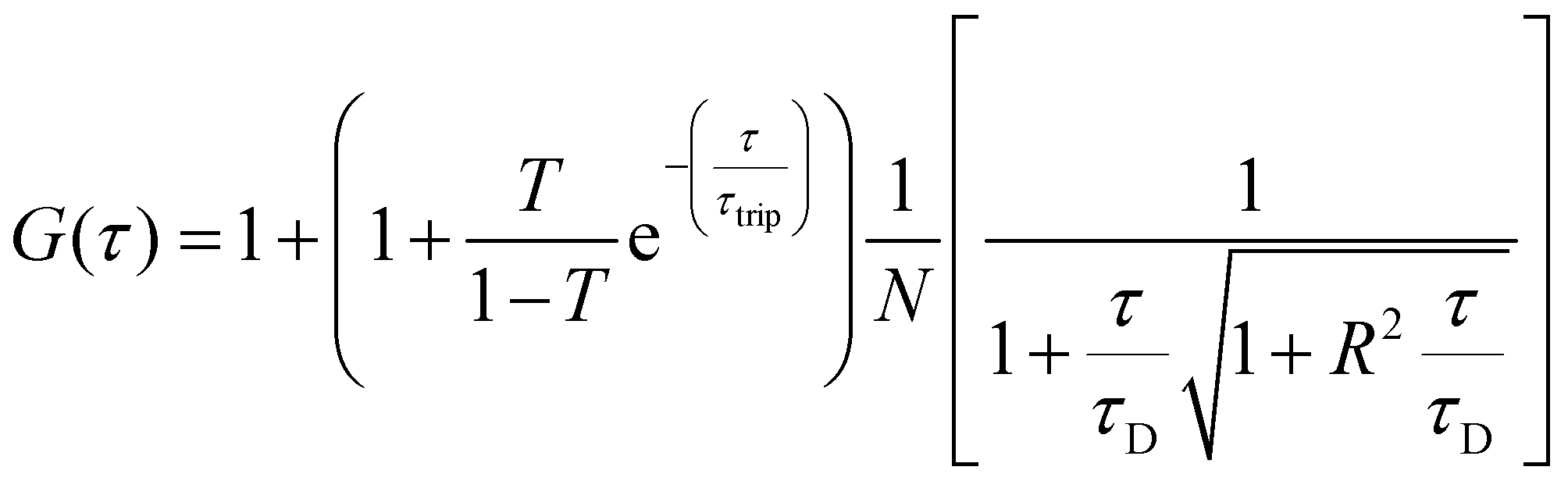 | (1) |
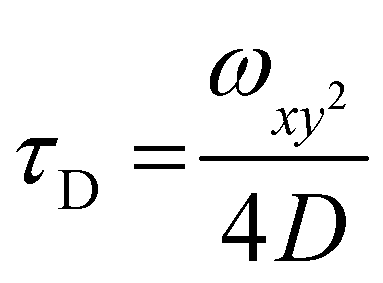 | (2) |
Two component diffusion model, presented in eqn (3) was used for fitting the experimental autocorrelation curves for the free labelled enzyme and the nanocompartment encapsulating labelled enzyme:
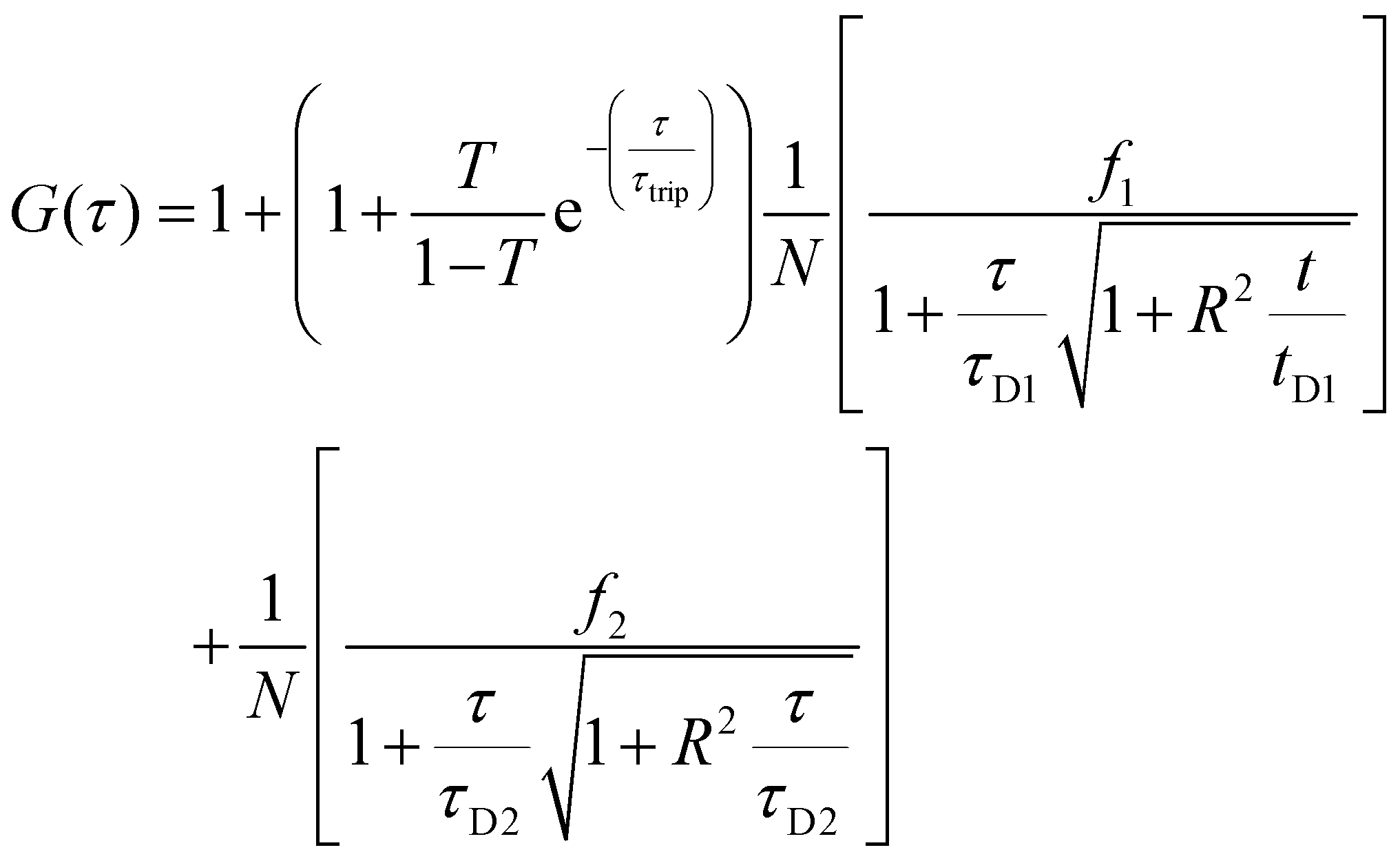 | (3) |
The number of dye molecules per enzyme (NPE) was calculated by eqn (4):
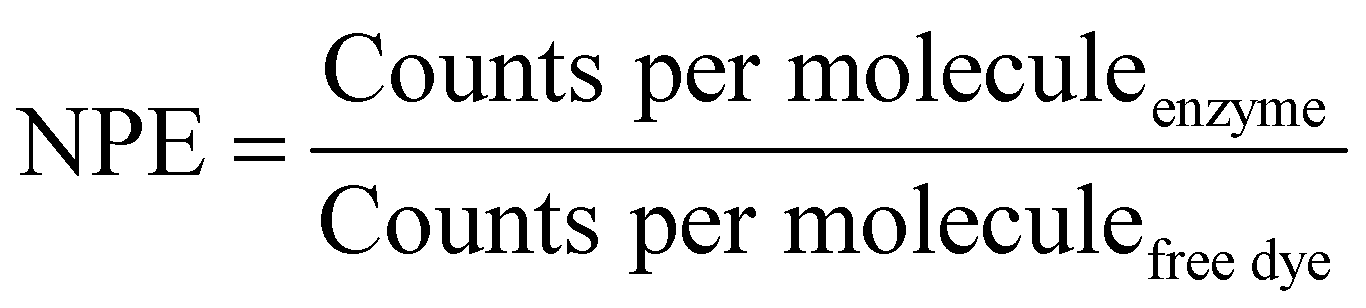 | (4) |
The number of enzymes per nanoreactor (NPN) was calculated by eqn (5):
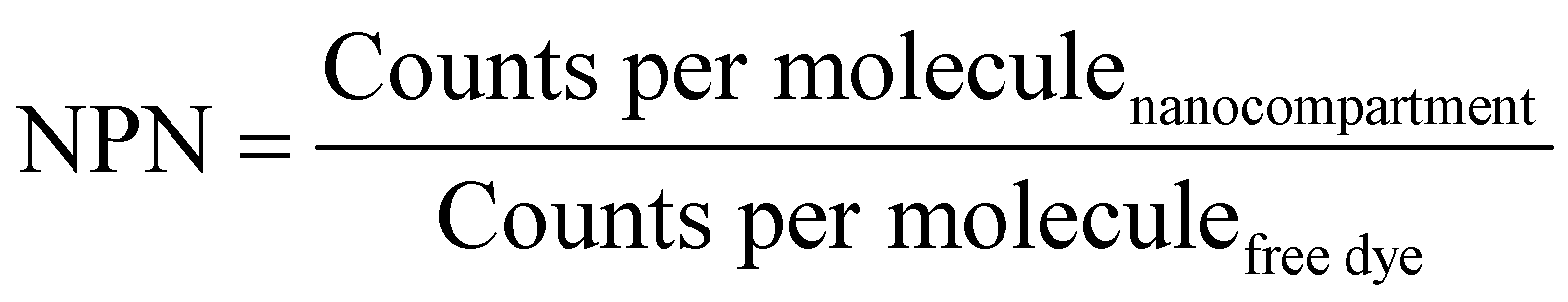 | (5) |
:1 (v/v) for 1 hour at 37 °C. The solution was filtered through 0.2 μm nylon membrane, 4 mm filter (Whatman™, General Electric, U.K.) and added at a 1:2 ratio to the BCA reagent. Samples and GUS standards were incubated for 2 hours at 37 °C, and the absorbance was measured at 562 nm using a SpectraMax id3 plate reader (Molecular Devices, USA).
2.4 Enzyme activity assays
Fluorescence assays were performed using 96-well, flat bottom black plates (Thermo Fisher Scientific). The increase of fluorescence was measured at λex (365 nm) and λem (445 nm) in a SpectraMax id3 microplate reader (Molecular Devices, USA). 4-Methylumbelliferyl-β-D-glucuronide (4-MUG, 5 μM) was added to CNCs (1.25 mg mL−1, [GUS] 24 U mL−1) or the respective amount of free enzyme in solution (24 U mL−1) in a final volume of 200 μL PBS or minimum essential medium containing 10% FBS ('MEM') per well. 4-MUG (5 μM) without CNCs or free enzyme was added in PBS or MEM as control. All assay conditions were performed in triplicate. Fluorescence emission was monitored for 90 min at 37 °C.For estimating drug production, the fluorescence intensity of 0, 5, 10, 20, 40 μM 4-MU either in PBS or MEM was measured and a calibration curve was prepared. Hymecromone production from 4-MUG by free and encapsulated GUS was calculated based on the calibration curve at time points when the reaction reached maximum fluorescence intensity (t = 10 min for free GUS in PBS, t = 90 min for GUS–melCNCs in PBS, t = 30 min for free GUS in MEM, t = 60 min for GUS–melCNCs in MEM).
For the evaluation of enzyme stability, free GUS, GUS–melCNCs and GUS–CNCs were stored in PBS for 2 months at 4 °C. After filtration through a 100 nm Whatman Nuclepore polycarbonate membrane, 5 μM 4-MUG were added and the increase in fluorescence was monitored for 90 min at 37 °C. The activity of aged samples was compared to that of freshly prepared samples at 18 min (time point of 4-MU maximum catalysed by free GUS) and at 60 min (time point of 4-MU maximum catalysed by GUS–CNCs).
Kinetic parameters for free enzyme and GUS–melCNCs were calculated using the Michaelis–Menten model:
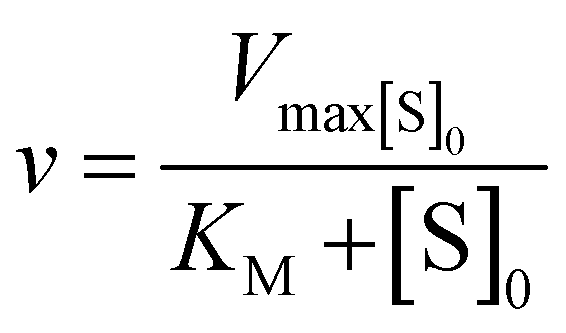
where v is the velocity of the enzyme, Vmax is the maximum velocity at saturating concentration, [S]0 is initial the concentration of the substrate S, KM is the Michaelis–Menten constant, kcat is the turnover number, and [E]0 is the concentration of catalytic sites, equivalent with the concentration of enzyme.
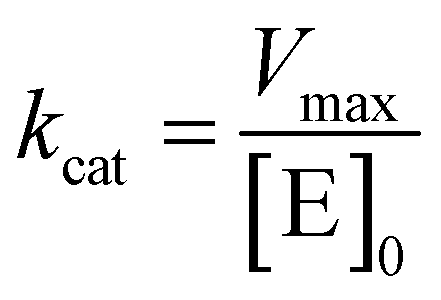
GUS–melCNCs or free GUS in solution ([GUS] 24 U mL−1) were incubated with increasing concentrations of 4-MUG in PBS (30, 90, 150, 300, 600, 750 μM) or MEM (80, 120, 200, 400, 800 μM). The increase in fluorescence associated with the production of hymecromone was monitored over 1 hour at 37 °C, and KM, Vmax, and kcat values were calculated.
2.5 Cell culture
HepG2 cells (hepatocellular carcinoma, human; ATCC® HB-8065™) were routinely cultured in minimum essential medium supplemented with 10% FBS, 100 U mL−1 penicillin, 100 U mL−1 streptomycin, 1% L-glutamine and 1% non-essential amino acids. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.000 cells (in 200 μL MEM) in each well of an ibidi 8-well chambered glass bottom coverslip (Vitaris, Switzerland). After 24 hours, the medium was replaced with fresh MEM containing either Atto647-labeled polymersomes (Atto647-PSs, 1.25 mg mL−1) or the respective amount of PBS as a control. After 24 h incubation, cells were gently washed with PBS (3×). Nuclei were stained by incubating cells with a 20000-fold dilution of Hoechst 33342 fluorescent dye (20 mins, 37 °C), followed by 3 washing steps with PBS. Cells were fixed with 4% paraformaldehyde (15 mins at RT), followed by rinsing with PBS (3×). Fixed cells were incubated with wheat germ agglutinin–Alexa Fluor™ 555 conjugate (200-fold dilution, 10 min at RT) for membrane staining. The cells were washed with PBS (3×) and imaged by confocal laser scanning microscopy (LSM 880, Carl Zeiss, Germany) using an oil immersion objective (Zeiss, 63× Plan-Apochromat, NA 1.4). Images were recorded using a 633 nm HeNe laser to visualize Atto647-PSs (detection range: 643–758 nm), a 561 nm DPSS 5561-10 laser for Alexa Fluor™ 555 (detection range: 570–615 nm), and a UV laser for Hoechst 33342 detection (detection range: 415–470 nm). The images were analyzed using the ZEN 3.2 software (blue edition) and Imaris software (Bitplane) for 3D reconstructions.
3. Results and discussion
3.1 Formation and characterization of CNCs
We selected the diblock copolymer PDMS25-b-PMOXA10 to generate catalytic nanocopmartments (CNCs) because its short block lengths result in the self-assembly of polymersomes whose thin membrane is conducive to the insertion of pore-forming membrane proteins.27,46 In addition, polymersomes assembled from block copolymers based on PDMS and PMOXA are predisposed for biomedical applications as many times they have been shown to be non-toxic and biocompatible.16,41 However, in the body, these block copolymers are non-degradable and excreted mainly via feces.47 The nanocompartments were formed by the film rehydration method where organic solvents that can adversely affect enzymes and peptides are avoided.41,48 To obtain membrane-permeabilized CNCs encapsulating bacterial β-glucuronidase (GUS–melCNCs), a thin PDMS25-b-PMOXA10 film was rehydrated with a mixture of enzyme and pore-forming melittin in phosphate buffered saline (PBS). In addition, polymersomes containing GUS but lacking melittin pores (GUS–CNCs) (Fig. S1, S3 and S4, ESI†) and polymersomes permeabilized with melittin but lacking enzymes (melPSs) were prepared as controls. All nanocompartments prepared in this study were incubated with proteinase K prior to their SEC purification in order to remove non-encapsulated enzyme and melittin where applicable.10The morphology of our polymersomes was characterized by a combination of light scattering, TEM and NTA.41 DLS measurements indicated an average diameter of 136 ± 37 nm for GUS–melCNCs and 137 ± 37 nm for GUS–CNCs (Fig. 2A, Fig. S1A, ESI†). The hydrodynamic radius (Rh) revealed by the DLS profile and the radius of gyration (Rg) obtained by SLS (Fig. S2 and S3, ESI†) are presented in Table 1. The Rg/Rh ratio or shape parameter, ρ, was around 1 (Table 1), which is typical for the morphology of hollow spheres.41,49 The vesicular nature of the polymersomes was further corroborated by TEM (Fig. 2B, Fig. S1B, ESI†). Polymersome concentration was assessed by NTA measurements which also confirmed the narrow size distribution of CNCs (Fig. S4, ESI†). Comparison of GUS–melCNCs with non-permeabilized GUS–CNCs by light scattering techniques, NTA and TEM indicated that melittin-induced membrane permeability had no effect on morphology or dispersity of the nanocompartments. Similarly, we and others have shown that morphology of polymersomes harbouring other enzymes was barely affected by enzyme encapsulation.27,41,50
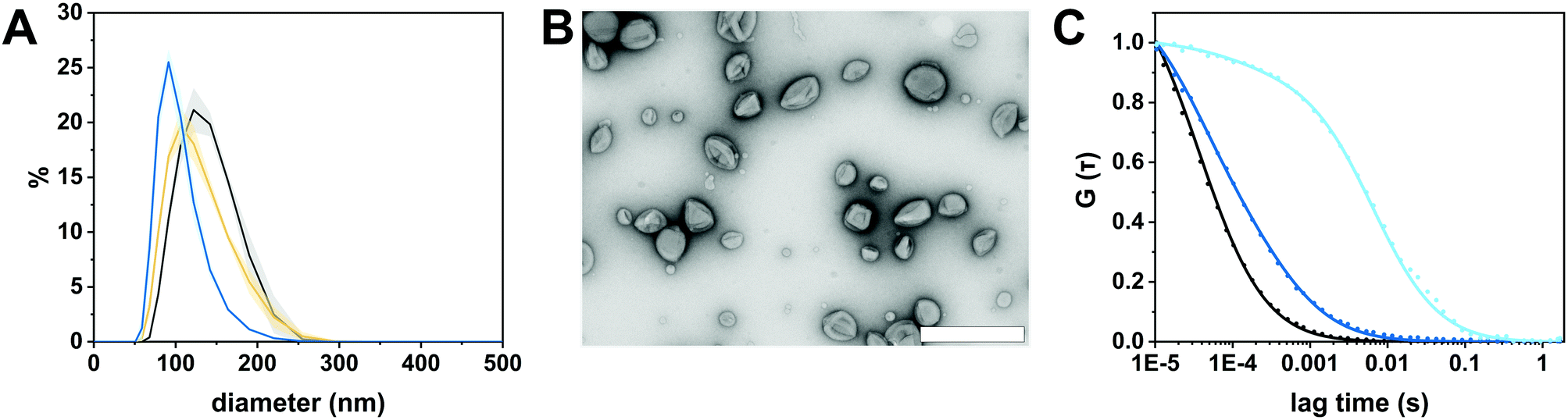 | ||
| Fig. 2 Characterization of GUS-melCNCs. (A) Size distribution of GUS-melCNCs measured by DLS (black: intensity, yellow: volume, blue: number; curves represent mean ± s.d. of 3 replications). (B) TEM micrograph of GUS-melCNCs showing the deflated structure typical for hollow spheres and the variation in their size. (Scale bar: 500 nm) (C) Normalized FCS autocorrelation curves (solid lines) for free Atto488 dye (black), Atto488-GUS (blue) and Atto488-GUS-melCNCs (cyan). Symbols represent raw data and solid lines represent fitted curves. | ||
| Radius of gyration (Rg) (nm) | Hydrodynamic radius (Rh) (nm) | ρ (Rg/Rh) | Concentration (polymersomes mL−1) | |
|---|---|---|---|---|
| GUS–melCNC | 69 ± 6 | 68 ± 7 | 1.01 ± 0.3 | (3.5 ± 0.3) × 1012 |
| GUS–CNC | 62 ± 6 | 66 ± 2 | 0.95 ± 0.1 | (3.3 ± 0.1) × 1012 |
The autocorrelation curves of standalone Atto488-GUS and CNCs encapsulating Atto488-labeled enzyme were compared to free dye (Fig. 2C and Fig. S1C, ESI†). A shift of the diffusion time (τD) calculated from the FCS autocorrelation curve of Atto488-GUS to increased values indicated the successful labelling of the enzyme (τD of free dye compared to τD of Atto488-GUS, Table S1, ESI†), τD of Atto488-GUS being directly correlated to its size.
A further increase of the diffusion time was observed for Atto488-GUS–melCNCs, indicating encapsulated enzyme (τD of Atto488-GUS–CNCs compared to τD of Atto488-GUS, Table S1, ESI†). By using Stockes–Einstein equation, we calculated the respective sizes of the nanocompartments derived from the FCS diffusion times. Their values were in agreement with the sizes obtained by LS and NTA, indicating that there was no tendency of the CNCs to aggregate (Table S1, ESI†). As the overall catalytic activity of CNCs largely depends on the amount of enzyme encapsulated,10,27,41,44 we evaluated the encapsulation of GUS by FCS. Based on the molecular brightness of the free fluorophore, Atto488-GUS and Atto488-GUS–melCNCs, we calculated that the enzyme was labelled on average with 1 dye molecule (see eqn (4) above), and 2 molecules of GUS (see eqn (5) above) were encapsulated per CNC. To estimate the number of melittin pores per GUS–melCNC, corresponding polymersomes were prepared that lack GUS but are melittin-permeabilized (melPSs). The total GUS–melCNC concentration (Fig. S4, ESI†) and the total amount of melittin present in the melPSs solution was calculated and correlated with the average number of melittin monomers constituting each pore (12),51 resulting in approximately 200 pores per GUS–melCNC. According to our previous study on melittin pores in PMOXA-b-PDMS-b-PMOXA membranes, we expect a pore diameter of at least 1 nm.46 Our data regarding size and distribution of GUS containing CNCs, enzyme encapsulation (Fig. S5, ESI†)41,52,53 and melittin pore formation43,46 are in good agreement with other studies addressing nanocompartments self-assembled by film rehydration of PDMS-b-PMOXA block copolymers.
3.2 Catalytic conversion of 4-MUG to hymecromone
The enzymatic activity of free and encapsulated enzyme in solution was evaluated by monitoring the conversion of 4-MUG (5 μM) to hymecromone (4-MU) at 445 nm under different conditions (Fig. 3).54 Comparison of GUS activity of free versus encapsulated enzyme in PBS is shown in Fig. 3A. Upon addition of 4-MUG to free enzyme (Fig. 3A, blue squares), fluorescence associated with the catalytic conversion to hymecromone rapidly increased and reached a plateau after about 10 min. In contrast, hymecromone production by GUS–melCNCs (Fig. 3A, cyan circles) constantly increased up to 80 min, and then appeared to plateau. A corresponding behavior was reported for other CNCs44 and is attributable to the diffusion time of substrate and product through the (melittin) pores of CNCs. Consistent with this notion, when GUS–CNCs without pores were incubated with 4-MUG, fluorescence at 445 nm remained minimal over time (Fig. 3A, yellow rhombi). This highlights the importance of melittin pores as in their absence, the polymer membrane does not allow for the passive diffusion of 4-MUG towards the confined enzymes. Furthermore, the absence of fluorescence confirms that the incubation of GUS–CNCs with proteinase K prior to their purification successfully eliminated unencapsulated enzymes attached to the outer surface of the membrane. Similarly, 4-MUG by itself showed no increase in fluorescence in PBS (Fig. 3A, black triangles) or cell culture medium with 10% fetal bovine serum (FBS; Fig. 3B, white triangles) which suggests that in the absence of the enzyme, the hydrolysis of the ether bond and therefore production of 4-MU, does not occur. This is in accordance with clinical data showing that only a small percentage of hymecromone glucuronidation is reversible by the β-glucuronidase present in the bacteria of the small intestine.55 The stability of the CNCs and their enzymatic activity were assessed with samples stored in PBS at 4 °C for 2 months (Fig. S6, ESI†). All our GUS–melCNCs retained their size and virtually full enzymatic activity.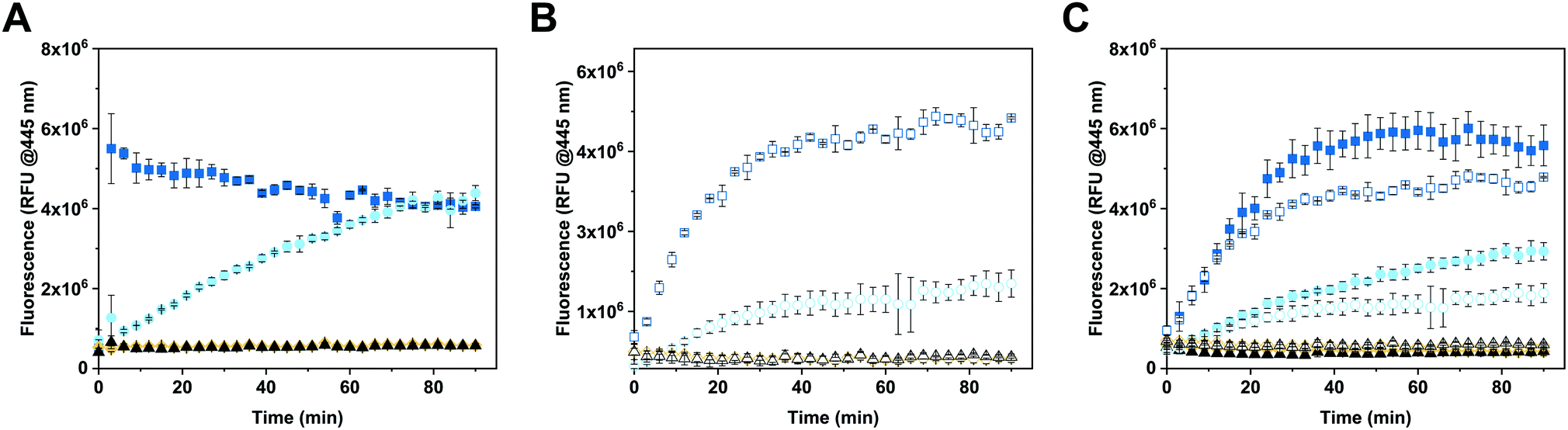 | ||
| Fig. 3 Enzymatic efficiency of CNCs at 37 °C. (A) 4-MUG (5 μM) conversion to 4-MU in PBS, (B) in cell culture medium supplemented with 10% fetal bovine serum (FBS), and (C) in PBS (filled symbols) and cell culture medium (empty symbols), each supplemented with 10% FBS. Squares represent free GUS (24 U mL−1), circles: GUS-melCNCs, rhombi: GUS-CNCs triangles: corresponding reaction mix without enzyme. Measurements represent triplicates, symbols might overlap. | ||
In order to more closely mimic the environmental conditions of living systems,56 the activity of free and encapsulated GUS was assessed in culture medium containing 10% FBS (Fig. 3B). Upon addition of 4-MUG to free GUS (Fig. 3B, white squares), fluorescence slowly increased over the first 30 min and then reached a plateau, suggesting molecular crowding related to the presence of 10% FBS decreased the efficiency of hymecromone production. In the case of GUS–melCNCs (Fig. 3B, white circles), the time course of hymecromone production was similar to that in PBS (Fig. 3A, cyan circles), suggesting that the confined enzyme was barely affected by protein-rich serum. The amount of hymecromone produced was estimated based on a 4-MU reference curve (Fig. S7, ESI†). In PBS, approximately 4.5 μM hymecromone were produced by the free enzyme and 3 μM by GUS–melCNCs. In complete cell culture medium, free enzyme was able to produce approximately 4.3 μM of hymecromone, while GUS–melCNCs produced 2.2 μM. The lower levels of hymecromone production by confined GUS are likely to be caused by the restriction of diffusion by the compartment membrane. Furthermore, we investigated the possibility that the drug was entrapped within the polymer membrane (Fig. S8, ESI†). Based on a calibration curve using different concentrations of hymecromone, we calculated that only 4% of the drug produced remains associated with the nanocompartment.
To explore whether the FBS in the cell culture medium accounted for the difference in hymecromone production between free and encapsulated GUS (Fig. 3B), we compared its production in PBS containing 10% FBS (Fig. 3C, filled symbols) to culture medium with 10% FBS (Fig. 3C, empty symbols). Indeed, the difference in hymecromone production between free and encapsulated enzyme was similar for PBS and culture medium. In fact, the FBS interfered with the kinetics of hymecromone production for the free enzyme but not for GUS–melCNCs. Because the GUS inside CNCs is protected by the membrane and only small molecules can diffuse through the pores, molecular crowding brought about by FBS mainly affected the activity of the free enzyme. Notably, in the highly complex environment of living cells, we expect other small molecules to affect the intracellular production of hymecromone by GUS–melCNCs.
3.3 Kinetic analysis of catalytic nanocompartments
The kinetic analysis of both free and encapsulated GUS (GUS–melCNCs) was assessed first in PBS, and then in complete cell culture medium (Table 2).56 The kinetic analysis of the reaction was modelled in a first approximation with the Michaelis–Menten kinetics since the substrate (4-MUG) was added in excess (from 100 to 4000-fold molar excess of [4-MUG] over [GUS]) (Fig. S9, ESI†).57 CNCs without melittin pores (GUS–CNCs) were not amenable to kinetic analysis since they did not exhibit any drug producing activity (Fig. 3, yellow rhombi).| Free GUS (PBS) | GUS–melCNCs (PBS) | Free GUS (MEM) | GUS–melCNCs (MEM) | |
|---|---|---|---|---|
| K M, Michaelis–Menten constant, Vmax, maximal enzyme velocity, kcat, turnover rate | ||||
| K M (μM) | (1.2 ± 0.4) × 103 | (3.8 ± 1.4) × 102 | (1.9 ± 0.2) × 102 | (3.4 ± 1) × 102 |
| V max (μmoles s−1) | (7 ± 1.7) × 10 | (4.2 ± 1) × 10 | (4.2 ± 0.4) × 10 | (3.3 ± 0.2) × 10 |
| k cat (1 s−1) | (2.7 ± 0.6) × 102 | (1.6 ± 0.4) × 102 | (1.6 ± 0.2) × 102 | (1.3 ± 0.1) × 102 |
The encapsulation of enzymes in polymersomes influences their kinetic parameters in several ways: the velocity of the reaction is decreased and the affinity for the substrate is increased.44,58 Accordingly, in PBS, KM and Vmax of GUS–melCNCs decreased 3 and 1.5 times, respectively, compared to free enzyme (Table 2). Lower KM values of GUS–melCNCs reflect a higher affinity for the substrate44,50,59 which is attributable to the increased probability of an interaction between enzyme and substrate within the confined reaction space. The turnover parameter, kcat, is decreased 1.5-fold for GUS–melCNCs. As kcat indicates the maximum number of 4-MUG molecules that are converted to hymecromone per enzyme per second, any reduction in GUS–melCNCs versus free GUS is associated with limited diffusion of 4-MUG and 4-MU by the polymer membrane.44
Comparison of the kinetic parameters of free GUS in PBS and MEM revealed a decrease in Vmax, KM and kcat in MEM, which could be expected considering the complexity of cell culture medium.60,61 It is conceivable that proteins, vitamins and nutrients present in cell culture media might interact with the substrate and/or enzyme, thereby decreasing the efficiency of catalysis. In contrast, the kinetic parameters of the encapsulated GUS in PBS and MEM are comparable. These results taken together suggest that the catalytic efficiency of our GUS–melCNCs is predominantly governed by the influx and efflux of the substrate and product molecules. Based on the enzymatic activity of GUS–melCNCs in cell culture medium we next assessed their activity and efficiency of producing hymecromone in living cells.
3.4 Hymecromone production in cells
In the body, glucuronidation of hymecromone occurs mainly in the liver. Therefore, we chose the liver-derived HepG2 cancer cell line for studying the effects of GUS CNCs in vitro.62 Our choice was further endorsed by recent findings showing an association of hymecromone with anticancer properties.63,64 Confocal laser scanning microscopy of HepG2 cells incubated with Atto647-encapsulating polymersomes (Atto647-PSs) at 1.25 mg mL−1 for 24 hours revealed the presence of Atto647-PSs throughout the cytoplasm (Fig. 4A1–2, Fig. S10A–C, ESI†). Control cells that had not been treated with Atto647-PSs did not show any fluorescence (Fig. 4B, Fig. S10D–F, ESI†).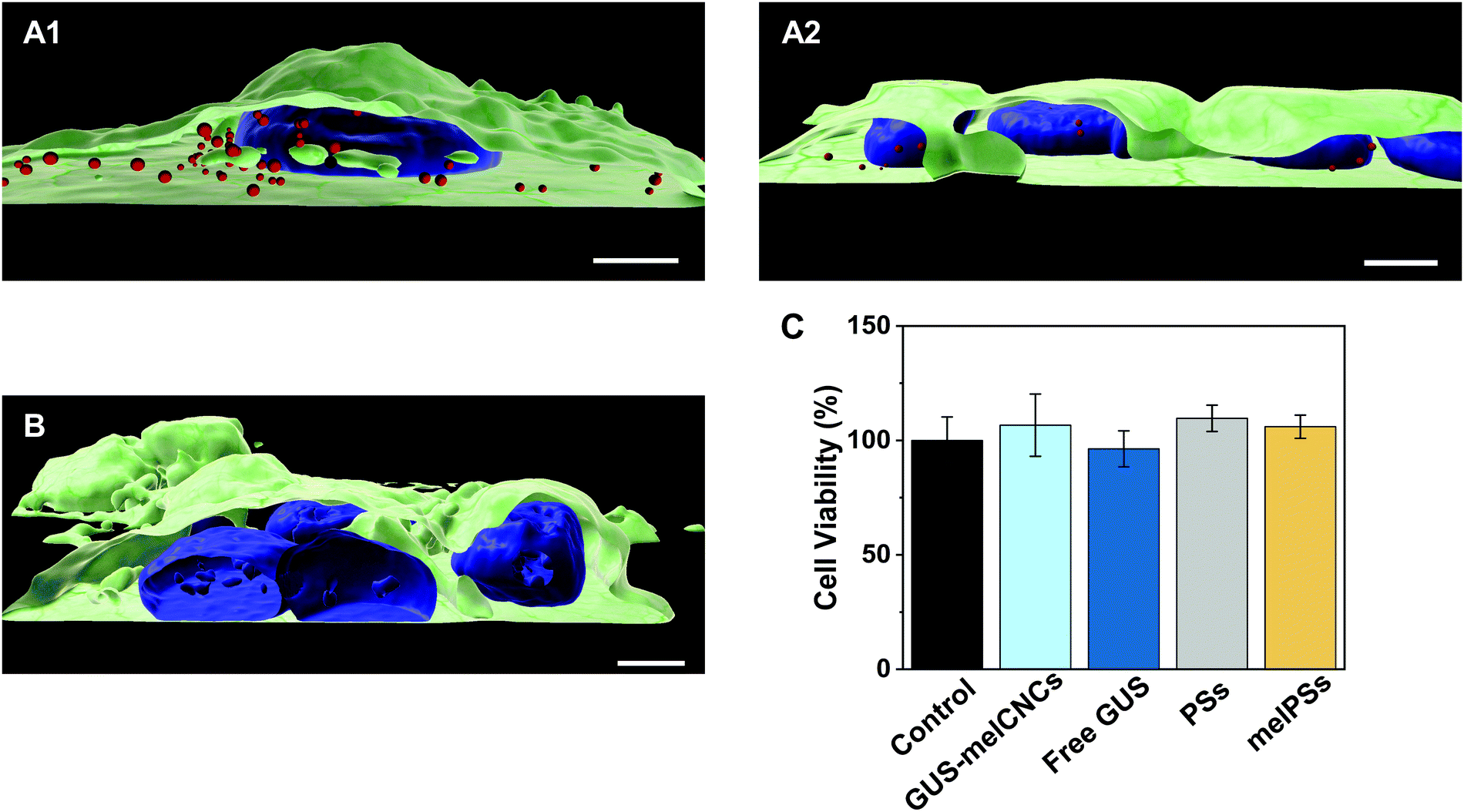 | ||
| Fig. 4 Uptake of Atto647-PSs by HepG2 cells. 3D reconstructions of multiple confocal sections of HepG2 cells incubated with Atto647-PSs (1.25 mg mL−1) for 24 hours (A12, red spheres). (B) HepG2 cells incubated with the respective amount of PBS. Nuclei (blue) were stained with Hoechst 33342 fluorescent dye, cellular membranes (green) with Atto555-WGA fluorescent dye. Scale bar, 5 μm. (C) Cell viability as percentage of HepG2 cells incubated with only PBS (control, black), GUS-melCNCs (cyan), free GUS (blue), PSs (grey) and melPSs (yellow). Graph shows mean ± s.d. of three independent experiments. | ||
We addressed the cytotoxicity of CNCs and free enzyme by carrying out MTS proliferation assays with HepG2 cells (Fig. 4C).65 None of the nanocompartments nor the free enzyme had a negative impact on the cell viability. Furthermore, although melittin is a bee venom, when inserted as pores in the CNC membrane at the concentrations indicated, it apparently had no adverse effect on cell proliferation.
To explore the ability of CNCs to produce hymecromone in living cells, we incubated HepG2 cells with either GUS–melCNCs, GUS–CNCs, free GUS or the respective amount of PBS for 24 hours. Subsequently, fresh culture medium containing 400 μM 4-MUG was added as, at this concentration, hymecromone was shown to inhibit hyaluronan synthesis in cancer cell lines,66–69 and cultures were returned to the 37 °C incubator. The change in fluorescence was recorded at 0, 2, 4, 8, 12 and 24 hours (Fig. 5). After 24 hours, cells that had taken up GUS–melCNCs, showed a fluorescence signal that corresponds to approximately 12 μM of hymecromone (see Fig. S7B and S11, ESI†). In fact, this amount might be an underestimation as some of the hymecromone produced in situ will be metabolized to 4-MUG in cells. Furthermore, although there is a possibility of intracellular components blocking individual melittin pores, considering their transient formation and their number per CNC, a potential blockage would only be a minor drawback. No increase in fluorescence was observed when untreated cells were incubated with 4-MUG (Fig. 5, black triangles), an indication that HepG2 cells by themselves are not able to produce such levels of hymecromone. However, we cannot exclude that small amounts of 4-MUG are converted to 4-MU by endogenous GUS (Fig. 5, Fig. S11, black triangles, ESI†).33 In addition, 4-MUG was not converted to 4-MU in cells that were treated with non-permeabilized GUS–CNCs (Fig. 5, yellow rhombi) or with polymersomes lacking GUS (PSs, melPSs, Fig. S12, ESI†).
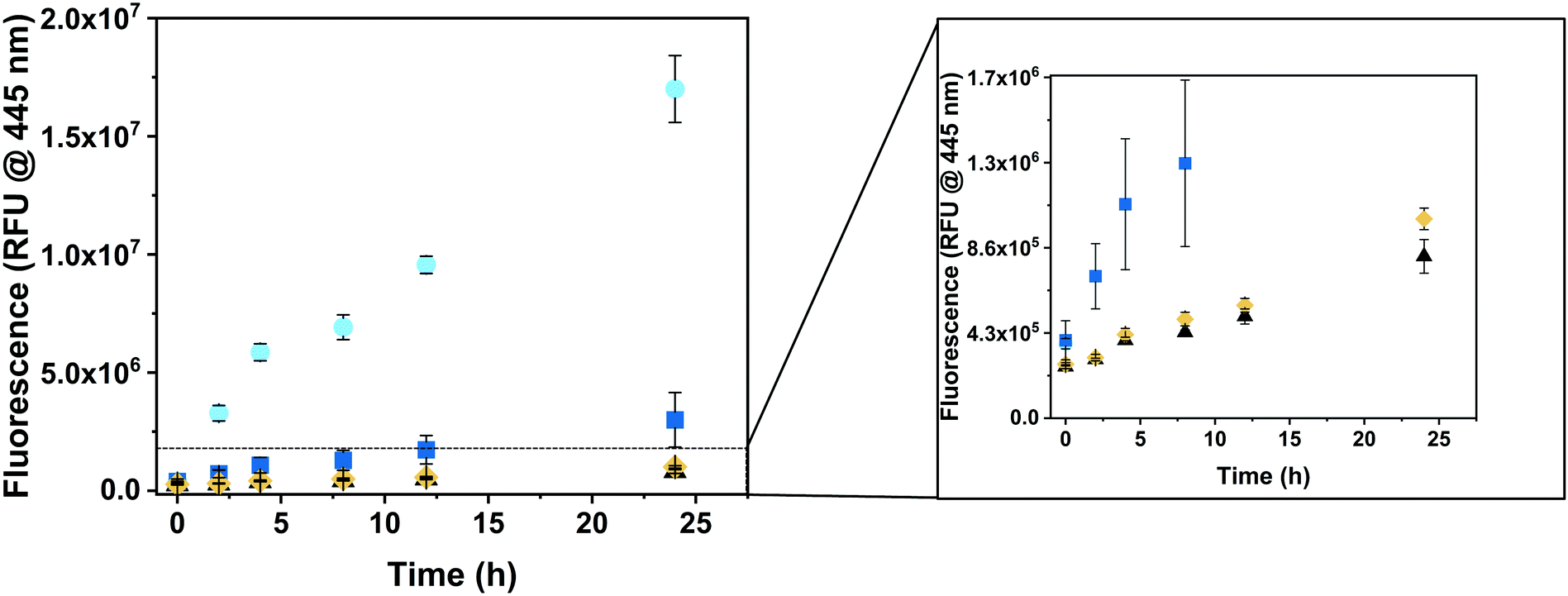 | ||
| Fig. 5 Intracellular production of hymecromone. HepG2 cells were incubated for 24h with CNCs for uptake, washed to remove extracellular CNCs, then exposed to a single dose of 400 μM 4-MUG, after which the increase in fluorescence was recorded at 0, 2, 4, 8, 12 and 24 h. GUS-melCNCs (cyan circles), free GUS (blue squares), GUS-CNCs (yellow rhombi), PBS (black triangles). Graph shows mean ± s.d. of three independent experiments. | ||
Free β-glucuronidase applied in clinical treatment was shown to be quickly degraded and to elicit an immune response in patients with Sly syndrome.70,71 One of the advantages of polymersomes is that they provide a protective shell for the encapsulated enzyme and thereby prolong its activity.13,14 Consistent with this notion, the increase in fluorescence was significantly higher in cells incubated with GUS–melCNCs compared to those incubated with free GUS (Fig. 5, blue squares), confirming that our nanocompartments shield GUS from proteolysis and degradation inside cells.
Our PDMS-b-PMOXA based CNCs are prone to accumulation in the liver47,72 where they encounter high levels of metabolized hymecromone.73 Undoubtedly, differences in pharmaco-kinetics will have to be taken into account in order to optimize GUS–melCNC efficiency in vivo. Beforehand, however, further studies are necessary to elucidate the bio-distribution of our PDMS-b-PMOXA based nanocompartments.
4. Conclusions
Here, we demonstrate the formation of catalytic nanocompartments containing β-glucuronidase that are able to produce hymecromone in HepG2 cells over 24 hours by inverting glucuronidation naturally occurring in the drug's metabolism. The locally confined catalysis of 4-MUG to 4-MU conversion inside cells led to 17-fold higher levels of the drug compared to 4-MUG converted by endogenous glucuronidase. To the best of our knowledge, this is the first report of an intracellular conversion of a metabolite into an active drug. We believe that our nanosystem opens new approaches for extending the lifetime of drugs by counteracting metabolism.Author contributions
M. K., V. M., I. A. D. and W. P. M. conceived the project. M. K. conducted the experiments. M. K. V. M. and CA.S analysed the data. W. P. M. and C. G. P. supervised the project. The manuscript was written through contributions of all authors. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is part of BIOMOLMACS and has received funding from the European Union's Horizon2020 research and innovation programme under the Marie Skłodowska–Curie grant agreement N° 859416. Additional financial support has been obtained from National Centre of Competence in Research – Molecular Systems Engineering (NCCR-MSE) and the University of Basel. The authors further thank Dr V. Mihali for TEM images, Dr M. Skowicki for the support in cell imaging, and Dr M. Kyropoulou for fruitful discussions. The authors also thank Y. Qutbuddin (Max Planck Institute of Biochemistry, Prof. Dr P. Schwille, Munich, Germany) for the support in the drug entrapment study. M.K. personally thanks EU ITN BIOMOLMACS for research funding and support.References
- L. A. Stanley, in Pharmacognosy: Fundamentals, Applications and Strategy, ed. S. Badal and R. Delgoda, Academic Press, London, 1st edn, 2017, pp. 527–545 Search PubMed.
- L. P. Longo and B. Johnson, Am. Fam. Physician, 2000, 61, 2121–2128 CAS.
- B. Sproule, B. Brands, S. Li and L. Catz-Biro, Can. Fam. Physician, 2009, 55, 68–69 Search PubMed.
- S. Cao, Z. Lv, S. Guo, G. Jiang and H. Liu, J. Drug Delivery Sci. Technol., 2021, 61, 102124 CrossRef CAS.
- C. Tan, J. Wang and B. Sun, Biotechnol. Adv., 2021, 48, 107727 CrossRef CAS PubMed.
- W. Gu, J. An, H. Meng, N. Yu, Y. Zhong, F. Meng, Y. Xu, J. J. L. M. Cornelissen, Z. Zhong, W. X. Gu, H. Meng, N. Yu, Y. N. Zhong, F. H. Meng, Z. Y. Zhong, J. J. L. M. Cornelissen, J. N. An and Y. Xu, Adv. Mater., 2019, 31, 1904742 CrossRef CAS PubMed.
- K. K. Upadhyay, A. K. Mishra, K. Chuttani, A. Kaul, C. Schatz, J. F. Le Meins, A. Misra and S. Lecommandoux, Nanomedicine, 2012, 8, 71–80 CrossRef CAS PubMed.
- V. Dhapte and V. Pokharkar, in Green Synthesis, Characterization and Applications of Nanoparticles, ed. A. Shukla and S. Iravani, Elsevier, London, 1st edn, 2019, pp. 321–345 Search PubMed.
- J. F. Mukerabigwi, Z. Ge and K. Kataoka, Chem. – Eur. J., 2018, 24, 15706–15724 CrossRef CAS PubMed.
- K. Langowska, C. G. Palivan and W. Meier, Chem. Commun., 2013, 49, 128–130 RSC.
- T. Nishimura, Y. Sasaki and K. Akiyoshi, Adv. Mater., 2017, 29, 1702406 CrossRef PubMed.
- W. Ke, J. Li, F. Mohammed, Y. Wang, K. Tou, X. Liu, P. Wen, H. Kinoh, Y. Anraku, H. Chen, K. Kataoka and Z. Ge, ACS Nano, 2019, 13, 2357–2369 CAS.
- R. A. J. F. Oerlemans, S. B. P. E. Timmermans and J. C. M. van Hest, ChemBioChem, 2021, 22, 2051–2078 CrossRef CAS PubMed.
- A. Belluati, I. Craciun, C. E. Meyer, S. Rigo and C. G. Palivan, Curr. Opin. Biotechnol, 2019, 60, 53–62 CrossRef CAS PubMed.
- X. Li, X. Zhao, R. Lv, L. Hao, F. Huo and X. Yao, Macromol. Biosci., 2021, 21, 2000424 CrossRef CAS PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- K. Kumar Upadhyay, J. F. Le Meins, A. Misra, P. Voisin, V. Bouchaud, E. Ibarboure, C. Schatz and S. Lecommandoux, Biomacromolecules, 2009, 10, 2802–2808 CrossRef PubMed.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 8203–8208 CrossRef CAS.
- J. Du and R. K. O’Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- H. Oliveira, E. Pérez-Andrés, J. Thevenot, O. Sandre, E. Berra and S. Lecommandoux, J. Controlled Release, 2013, 169, 165–170 CrossRef CAS PubMed.
- F. Bialas, D. Reichinger and C. F. W. Becker, Enzyme Microb. Technol., 2021, 150, 109864 CrossRef CAS PubMed.
- D. Sueyoshi, Y. Anraku, T. Komatsu, Y. Urano and K. Kataoka, Biomacromolecules, 2017, 18, 1189–1196 CrossRef CAS PubMed.
- C. Z. Bueno, A. C. Apolinário, A. Duro-Castano, A. Poma, A. Pessoa, C. O. Rangel-Yagui and G. Battaglia, ACS Macro Lett., 2020, 9, 1471–1477 CrossRef CAS.
- Y. Anraku, A. Kishimura, M. Kamiya, S. Tanaka, T. Nomoto, K. Toh, Y. Matsumoto, S. Fukushima, D. Sueyoshi, M. R. Kano, Y. Urano, N. Nishiyama and K. Kataoka, Angew. Chem., Int. Ed., 2016, 55, 560–565 CrossRef CAS PubMed.
- M. Spulber, P. Baumann, S. S. Saxer, U. Pieles, W. Meier and N. Bruns, Biomacromolecules, 2014, 15, 1469–1475 CrossRef CAS PubMed.
- C. E. Meyer, J. Liu, I. Craciun, D. Wu, H. Wang, M. Xie, M. Fussenegger and C. G. Palivan, Small, 2020, 16, 1906492 CrossRef CAS PubMed.
- B. S. Kim, M. Naito, H. Chaya, M. Hori, K. Hayashi, H. S. Min, Y. Yi, H. J. Kim, T. Nagata, Y. Anraku, A. Kishimura, K. Kataoka and K. Miyata, Biomacromolecules, 2020, 21, 4365–4376 CrossRef CAS PubMed.
- S. Takeda and M. Aburada, J. Pharmacobiodyn., 1981, 4, 724–734 CrossRef CAS PubMed.
- N. Nagy, I. Gurevich, H. F. Kuipers, S. M. Ruppert, P. L. Marshall, B. J. Xie, W. Sun, A. V. Malkovskiy, J. Rajadas, M. Grandoch, J. W. Fischer, A. R. Frymoyer, G. Kaber and P. L. Bollyky, J. Biol. Chem., 2019, 294, 7864–7877 CrossRef CAS PubMed.
- N. Nagy, G. Kaber, P. Y. Johnson, J. A. Gebe, A. Preisinger, B. A. Falk, V. G. Sunkari, M. D. Gooden, R. B. Vernon, M. Bogdani, H. F. Kuipers, A. J. Day, D. J. Campbell, T. N. Wight and P. L. Bollyky, J. Clin. Invest., 2015, 125, 3928–3940 CrossRef PubMed.
- W. Li, S. Yang, P. Xu, D. Zhang, Y. Tong, L. Chen, B. Jia, A. Li, C. Lian, D. Ru, B. Zhang, M. Liu, C. Chen, W. Fu, S. Yuan, C. Gu, L. Wang, W. Li, Y. Liang, Z. Yang, X. Ren, S. Wang, X. Zhang, Y. Song, Y. Xie, H. Lu, J. Xu, H. Wang and W. Yu, EBioMedicine, 2022, 76, 103861 CrossRef CAS PubMed.
- D. Turgeon, J. S. Carrier, E. Lévesque, D. W. Hum and A. Bélanger, Endocrinology, 2001, 142, 778–787 CrossRef CAS PubMed.
- H. F. Kuipers, N. Nagy, S. M. Ruppert, V. G. Sunkari, P. L. Marshall, J. A. Gebe, H. D. Ishak, S. G. Keswani, J. Bollyky, A. R. Frymoyer, T. N. Wight, L. Steinman and P. L. Bollyky, Clin. Exp. Immunol., 2016, 185, 372–381 CrossRef CAS PubMed.
- E. R. Garrett, J. Venitz, K. Eberst and J. J. Cerda, Biopharm. Drug Dispos., 1993, 14, 13–39 CrossRef CAS PubMed.
- X. Song, Z. Jiang, L. Li and H. Wu, Front. Chem. Sci. Eng., 2014, 8, 353–361 CrossRef CAS.
- S.-J. Yeom, G. H. Han, M. Kim, K. K. Kwon, Y. Fu, H. Kim, H. Lee, D.-H. Lee, H. Jung and S.-G. Lee, PLoS One, 2017, 12, e0170398 CrossRef PubMed.
- P. M. Kemp and K. D. Cliburn, Federal Aviation Administration Comparison of Species-Specific β-Glucuronidase Hydrolysis of Cannabinoid Metabolites in Human Urine, Oklahoma City, 2015.
- E. R. Garrett and J. Venitz, J. Pharm. Sci., 1994, 83, 115–116 CrossRef CAS PubMed.
- K. Kiene, S. H. Schenk, F. Porta, A. Ernst, D. Witzigmann, P. Grossen and J. Huwyler, Eur. J. Pharm. Biopharm., 2017, 119, 322–332 CrossRef CAS PubMed.
- C. E. Meyer, I. Craciun, C.-A. Schoenenberger, R. Wehr and C. G. Palivan, Nanoscale, 2021, 13, 66–70 RSC.
- R. Wehr, E. C. dos Santos, M. S. Muthwill, V. Chimisso, J. Gaitzsch and W. Meier, Polym. Chem., 2021, 37, 5377–5389 RSC.
- V. Maffeis, A. Belluati, I. Craciun, D. Wu, S. Novak, C. A. Schoenenberger and C. G. Palivan, Chem. Sci., 2021, 12, 12274–12285 RSC.
- A. Belluati, I. Craciun, J. Liu and C. G. Palivan, Biomacromolecules, 2018, 19, 4023–4033 CrossRef CAS PubMed.
- A. Guinier and C. B. Walker, in Small-angle scattering of X-rays, ed. Mayer M. G., John Wiley & Sons, Inc., New York, 1st edn, 1955 Search PubMed.
- A. Belluati, V. Mikhalevich, S. Y. Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, Biomacromolecules, 2019, 21, 701–715 CrossRef PubMed.
- K. Liu, X. Wang, X. Li-Blatter, M. Wolf and P. Hunziker, ACS Appl. Bio Mater., 2020, 3, 6919–6931 CrossRef CAS PubMed.
- P. Baumann, M. Spulber, O. Fischer, A. Car and W. Meier, Small, 2017, 13, 1603943 CrossRef PubMed.
- D. Daubian, J. Gaitzsch and W. Meier, Polym. Chem., 2020, 11, 1237–1248 RSC.
- Q. Chen, H. Schönherr and G. J. Vancso, Small, 2009, 5, 1436–1445 CrossRef CAS PubMed.
- J. H. Lin and A. Baumgaertner, Biophys. J., 2000, 78, 1714–1724 CrossRef CAS PubMed.
- A. Moquin, J. Ji, K. Neibert, F. M. Winnik and D. Maysinger, ACS Omega, 2018, 3, 13882–13893 CrossRef CAS PubMed.
- M. Mertz and K. Castiglione, Int. J. Mol. Sci., 2021, 22, 7134 CrossRef CAS PubMed.
- T. Martin, R.-V. Wöhner, S. Hummel, L. Willmitzer and W. B. Frommer, in Gus Protocols: Using the GUS Gene as a Reporter of Gene Expression, ed. S. R. Gallagher, Academic press, San Diego, 1st edn, 1992, pp. 23–43 Search PubMed.
- P. Dashnyam, R. Mudududdla, T. J. Hsieh, T. C. Lin, H. Y. Lin, P. Y. Chen, C. Y. Hsu and C. H. Lin, Sci. Rep., 2018, 8, 1–12 CAS.
- A. Zotter, F. Bäuerle, D. Dey, V. Kiss and G. Schreiber, J. Biol. Chem., 2017, 292, 15838–15848 CrossRef CAS PubMed.
- L. A. Segel and M. Slemrod, SIAM Rev., 1989, 31, 446–477 CrossRef.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O’Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- Q. Chen, G. R. Kristin, H. Schönherr and G. J. Vancso, ChemPhysChem, 2010, 11, 3534–3540 CrossRef CAS PubMed.
- K. van Eunen and B. M. Bakker, Perspect. Sci., 2014, 1, 126–130 CrossRef.
- R. Eisenthal, M. J. Danson and D. W. Hough, Trends Biotechnol., 2007, 25, 247–249 CrossRef CAS PubMed.
- R. M. Hoffmann, G. Schwarz, C. Pohl, D. J. Ziegenhagen and W. Kruis, Dtsch. Med. Wochenschr., 2005, 130, 1938–1943 CrossRef CAS PubMed.
- B. Kundu, P. Saha, K. Datta and S. C. Kundu, Biomaterials, 2013, 34, 9462–9474 CrossRef CAS PubMed.
- F. Piccioni, M. Malvicini, M. G. Garcia, A. Rodriguez, C. Atorrasagasti, N. Kippes, I. T. Piedra Buena, M. M. Rizzo, J. Bayo, J. Aquino, M. Viola, A. Passi, L. Alaniz and G. Mazzolini, Glycobiology, 2012, 22, 400–410 CrossRef CAS PubMed.
- T. M. Buttke, J. A. McCubrey and T. C. Owen, J. Immunol. Methods, 1993, 157, 233–240 CrossRef CAS PubMed.
- T. Nakamura, M. Funahashi, K. Takagaki, H. Munakata, K. Tanaka, Y. Saito and M. Endo, Biochem. Mol. Biol. Int., 1997, 43, 263–268 CAS.
- V. B. Lokeshwar, L. E. Lopez, D. Munoz, A. Chi, S. P. Shirodkar, S. D. Lokeshwar, D. O. Escudero, N. Dhir and N. Altman, Cancer Res., 2010, 70, 2613–2623 CrossRef CAS PubMed.
- I. Kakizaki, K. Kojima, K. Takagaki, M. Endo, R. Kannagi, M. Ito, Y. Maruo, H. Sato, T. Yasuda, S. Mita, K. Kimata and N. Itano, J. Biol. Chem., 2004, 279, 33281–33289 CrossRef CAS PubMed.
- I. Kakizaki, K. Takagaki, Y. Endo, D. Kudo, H. Ikeya, T. Miyoshi, B. A. Baggenstoss, V. L. Tlapak-Simmons, K. Kumari, A. Nakane, P. H. Weigel and M. Endo, Eur. J. Biochem., 2002, 269, 5066–5075 CrossRef CAS PubMed.
- P. S. Kishnani, AAPS Adv. Pharm. Sci. Ser., 2015, 19, 9–21 Search PubMed.
- B. H. Biela, L. A. Khawli, P. Hu and A. L. Epstein, Cancer Biother. Radiopharm., 2003, 18, 339–353 CrossRef CAS PubMed.
- C. De Vocht, A. Ranquin, R. Willaert, J. A. Van Ginderachter, T. Vanhaecke, V. Rogiers, W. Versées, P. Van Gelder and J. Steyaert, J. Controlled Release, 2009, 137, 246–254 CrossRef CAS PubMed.
- N. Nagy, H. F. Kuipers, A. R. Frymoyer, H. D. Ishak, J. B. Bollyky, T. N. Wight and P. L. Bollyky, Front. Immunol., 2015, 6, 123 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00243d |
| This journal is © The Royal Society of Chemistry 2022 |