
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive and core cross-linked micelles developed by NiCCo-PISA of helical poly(aryl isocyanide)s†
Sètuhn
Jimaja
 abc,
Spyridon
Varlas
b,
Jeffrey C.
Foster
b,
Daniel
Taton
c,
Andrew P.
Dove
*b and
Rachel K.
O'Reilly
*b
abc,
Spyridon
Varlas
b,
Jeffrey C.
Foster
b,
Daniel
Taton
c,
Andrew P.
Dove
*b and
Rachel K.
O'Reilly
*b
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
bSchool of Chemistry, University of Birmingham, Edgbaston B15 2TT, UK. E-mail: a.dove@bham.ac.uk; r.oreilly@bham.ac.uk
cLaboratoire de Chimie des Polymères Organiques, Université de Bordeaux/CNRS École Nationale Supérieure de Chimie, de Biologie & de Physique, 33607 Cedex Pessac, France
First published on 13th June 2022
Abstract
We report the synthesis of redox- and pH-sensitive block copolymer micelles that contain chiral cores composed of helical poly(aryl isocyanide)s. Pentafluorophenyl (PFP) ester-containing micelles synthesised via nickel-catalysed coordination polymerisation-induced self-assembly (NiCCo-PISA) of helical poly(aryl isocyanide) amphiphilic diblock copolymers are modified post-polymerisation with various diamines to introduce cross-links and/or achieve stimulus-sensitive nanostructures. The successful introduction of the diamines is confirmed by Fourier-transform infrared spectroscopy (FT-IR), while the stabilisation effect of the cross-linking is explored by dynamic light scattering (DLS). The retention of the helicity of the core-forming polymer block is verified by circular dichroism (CD) spectroscopy and the stimuli-responsiveness of the nanoparticles towards a reducing agent (L-glutathione, GSH) and pH is evaluated by following the change in the size of the nanoparticles by DLS. These stimuli-responsive nanoparticles could find use in applications such as drug delivery, nanosensors or biological imaging.
1. Introduction
Smart nanomaterials that undergo alteration when subjected to a particular stimulus have received significant levels of attention.1–4 The design and development of these stimuli-responsive materials are of interest for applications such as drug delivery,5–8 biological imaging,7,9,10 chiroptical materials11 and sensing.12–14 A wide variety of different triggers have been developed, including pH,12,15–20 temperature,19,21–25 light,26–31 magnetic field,32–34 CO2,35 glucose,36–38 β-cyclodextrin,39 ions,14,40,41 electric potential,42 and redox potential.16,43–46 Systems with a range of different responses can be achieved as a result of an alteration in the solubility of the nanoparticles (NPs), such as changes in size19,24,31 and morphology,47–49 light emission,11,13 and permeability.27,28,50,51 Additionally, reactive cross-linkers can be employed to impart responsiveness to the resulting three-dimensional polymeric network. Cross-linked nanoparticles are of interest for their improved stability, which allows the conservation of the nanoparticle morphology under changes to their solvation state and/or concentration.A potential application of stimuli-responsive self-assembled NPs is their triggered disassembly, usually intended for the design of drug delivery systems that necessitates a release of their cargo in a specific microenvironment, e.g. acidic or reducing conditions found in tumours. Cross-links that are cleaved under these conditions allow for a controlled stimulus-responsive disassembly. For example, cross-linkers containing disulfide bonds that are reactive towards thiols and reducing compounds,43,45,52,53 or acetals that can be cleaved under acidic conditions16,54,55 have been successfully employed.
Helical polymers that present a static structure display optical activity that leads them to play an important role in areas such as chiral recognition27,56,57 or catalysis.58–62 We hypothesised that the combination of helicity alongside stimuli-responsiveness could lead to materials of potential importance in the domain of asymmetric catalysis or drug-delivery. For instance, one could envision a chiral nanoreactor that would only react with one enantiomer of a racemic mixture, which would thus eliminate complex purification steps for on-demand delivery of compounds with high enantiopurity. Helical polymers, such as polyisocyanides, have been successfully employed as stimuli-responsive building blocks. In a noteworthy report, Wu and co-workers developed a dual-responsive nanomaterial that responded both to oxidation and pH changes able to release its cargo in a cell-like environment.63,64 Indeed, combining two triggers (oxidation and pH) increased the solubility of the core in water, causing the micelles to disassemble and release their cargo.65 However, the synthetic methodologies used to achieve stimuli-responsive NPs proved time- and resource-consuming and give NP dispersions with a relatively low content of polymer. An alternative method to readily prepare nanostructures is polymerisation-induced self-assembly (PISA), which is a versatile technique to achieve nano-objects at high solid concentrations using simple procedures.66–71 PISA is a one-pot self-assembly process that has been employed with various polymerisation techniques71–74 in a wide range of solvents.75–78 The adaptability of PISA and the ability to achieve a pure phase of the desired morphology in a predictable manner makes it ideal for development of new nanomaterials.79–85
Herein, we report the development of core-functionalised poly(aryl isocyanide) nanostructures synthesised by nickel-catalysed coordination polymerisation-induced self-assembly (NiCCo-PISA). Subsequent post-polymerisation modification (PPM) of pentafluorophenyl (PFP) activated ester units43,86 using a variety of primary diamines, enabled cross-linking of the core of the derived micelles. The latter cross-linking step provided stability and/or stimuli-responsive properties to the final nanostructures. The stimuli-responsiveness, and resultant disassembly, of the resulting cross-linked nanoparticles in a reducing environment or low pH was monitored by dynamic light-scattering (DLS). We think the combination of helicity and stimuli-responsiveness into nano-objects will open new avenues for delivery of chiral therapeutics. Moreover, the micelles as chiral platform developed by NiCCo-PISA were easily modified to introduce the wanted moiety, which paves the road to other applications such as enantioselective catalyst and circularly polarised light emitters.
2. Results and discussion
2.1 Cross-linking of NiCCo-PISA micelles
Building on the NiCCo-PISA-derived functionalisable micelles presented in our earlier works,86,87 the synthesis of stimulus-responsive nanostructures through a PPM methodology was envisioned. Namely, a functionalisable poly(PEG-ester aryl isocyanide)20-block-poly(menthyl-ester aryl isocyanide15-co-pentafluorophenyl-ester aryl isocyanide15), i.e. P(PAIC)20-b-P(MAIC15-co-FAIC15) or D50% (diblock polymer containing 50% of functionalisable core units), and its non-functionalisable counterpart, i.e. P(PAIC)20-b-P(MAIC30) or D0%, were synthesised via NiCCo-PISA in DMSO at 5 wt% solids content. These polyisocyanides were synthesised in earlier studies, which contain complete characterisation data for the polymers.86,87 Basic characterisation data, including molecular weight and dispersity, is summarized here (Table 1).| Sample | D DLS DMSOa (nm) | D DLS H2Oa (nm) | D DLS THFa (nm) | D TEM (nm) | Zeta pot. (mV) | CD360c (mdeg) |
|---|---|---|---|---|---|---|
| a Hydrodynamic diameter measured by DLS at a 173° angle. PD is in parenthesis. b Average diameter measured from dry-state TEM images from water suspension of NPs. c CD signal at λ = 360 nm in THF. d No assemblies in THF. | ||||||
| D0% | 20 (0.18) | 58 (0.23) | —d | 21 ± 7 | −15 ± 5 | 14 |
| D50% | 20 (0.25) | 140 (0.15) | —d | 18 ± 4 | +7 ± 7 | 8.7 |
| D50% + HDA | 20 (0.14) | 100 (0.20) | 28 (0.24) | 15 ± 3 | −1 ± 3 | 6.2 |
| D50% + CA | 19 (0.10) | 180 (0.29) | 26 (0.20) | 16 ± 3 | −13 ± 3 | 6.5 |
| D50% + AEE | 22 (0.15) | 55 (0.18) | 26 (0.16) | 15 ± 3 | −9 ± 9 | 8.9 |
Here, direct cross-linking of the D50% micelles in DMSO was attempted in order to impart stability and stimulus-responsiveness to the nanostructures. Three cross-linkers were investigated: 1,6-hexanediamine (HDA) as a non-responsive cross-linker; cystamine (CA) that contains a redox-sensitive disulfide moiety and 2-[1-(2-amino-ethoxy)-1-methyl-ethoxy]-ethylamine (AEE) that bears a pH-sensitive acetal linker (Scheme 1). Redox and pH stimuli were selected as these are potentially useful for tumour targeting.45,88–93 Conditions from a previous report were used for the cross-linking experiments.86
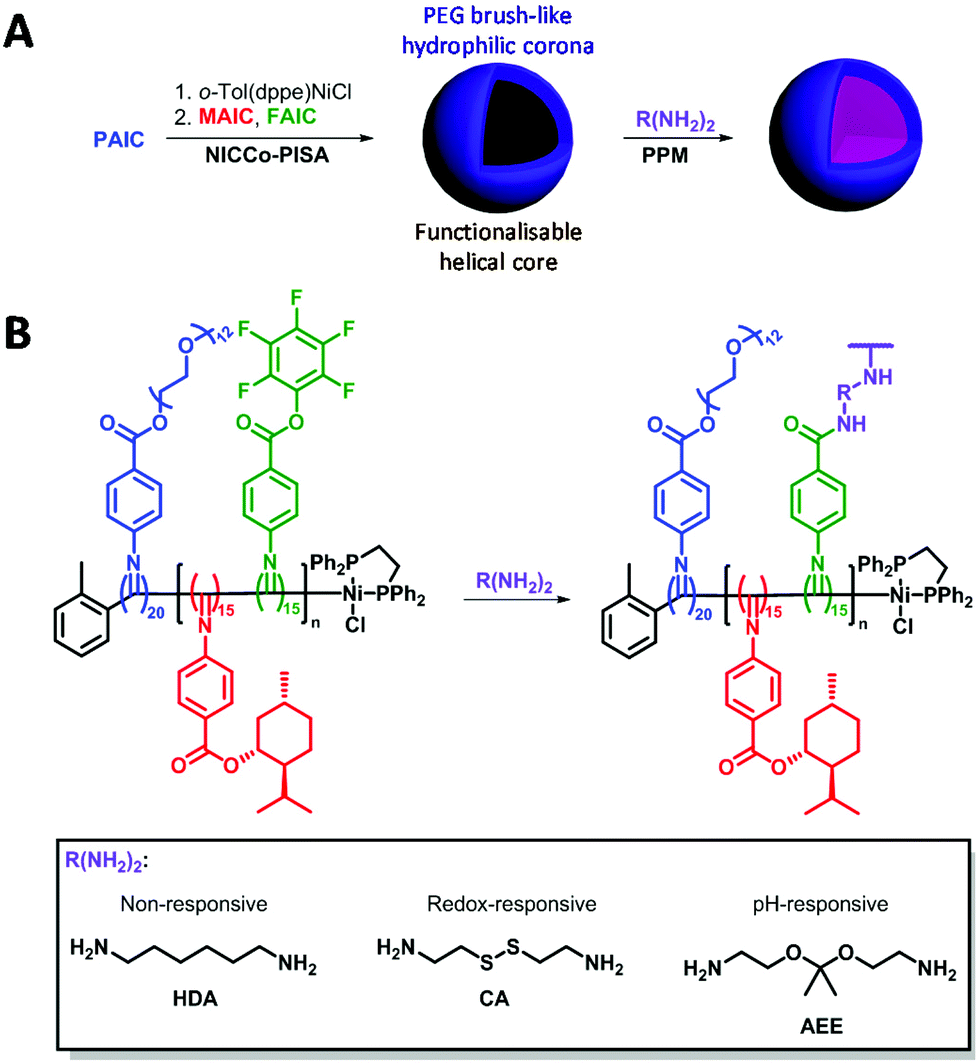 | ||
| Scheme 1 (A) Overview of the NiCCo-PISA of block copolymer micelles and post-polymerisation modification (PPM). (B) Detailed PPM procedure for the development of cross-linked and stimuli-responsive nanomaterials. | ||
The NiCCo-PISA suspension of non-functionalised D50% NPs was reacted with 0.6 equivalents of each of the diamine cross-linkers for three days in DMSO at 50 °C. These PPM reactions were repeated using non-functional particles D0% (i.e. P(PAIC)20-b-P(MAIC30)), as controls. The reaction mixtures were dialysed against water to remove any unreacted diamines and an aliquot was freeze-dried prior to analysis. Analysis of the purified copolymer samples by FT-IR (Fig. 1) was used to determine the completion of the core cross-linking reaction in each case. The disappearance of three vibration signals indicated the absence of PFP esters: 1520 cm−1 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic bond signal, 1240 cm−1 for the C–O ester bond and 1030 cm−1 for the C–F bond (Fig. 1C). Moreover, the signal from the CO ester at 1755 cm−1 was replaced by a weaker and broader signal at 1750 cm−1, which originated from the newly formed amide bond (CO stretching) (Fig. 1D). Finally, a new broad signal appeared at 3350 cm−1, which verified the formation of new amide N–H bonds (Fig. 1A and B). 19F NMR analysis of the cross-linked materials also showed the disappearance of the PFP group, which indicated that the reactions were highly efficient (Fig. S1†).
C aromatic bond signal, 1240 cm−1 for the C–O ester bond and 1030 cm−1 for the C–F bond (Fig. 1C). Moreover, the signal from the CO ester at 1755 cm−1 was replaced by a weaker and broader signal at 1750 cm−1, which originated from the newly formed amide bond (CO stretching) (Fig. 1D). Finally, a new broad signal appeared at 3350 cm−1, which verified the formation of new amide N–H bonds (Fig. 1A and B). 19F NMR analysis of the cross-linked materials also showed the disappearance of the PFP group, which indicated that the reactions were highly efficient (Fig. S1†).
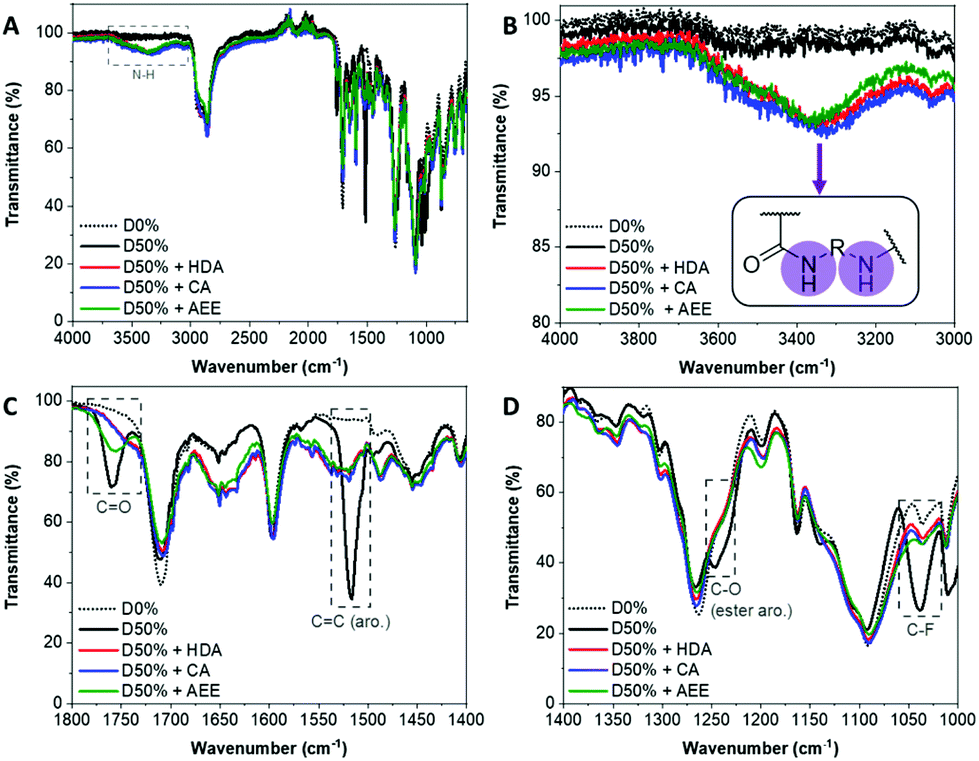 | ||
| Fig. 1 FT-IR spectra of the D0% (dotted black line) and D50% copolymer (black line) cross-linked with HDA (red line), CA (blue line) and AEE (green line). (A) Full spectrum. (B) Zoom in the 4000–3000 cm−1 region. (C) Zoom in the 1800–1400 cm−1 region. (D) Zoom in the 1400–1000 cm−1 region. | ||
DLS analysis of the nano-objects obtained after cross-linking indicated no major change in the NP size distribution, showing sizes around 20 nm and polydispersities (PDs) in line with the micelles prior to cross-linking (Fig. 2A–C, S3† and Table 1). These cross-linked nanostructures were subsequently transferred from DMSO to water by dialysis, which led to a change from transparent solutions in DMSO to opaque suspensions in aqueous media, for D0%, D50%, D50% + HDA and D50% + CA. Further analysis by DLS showed an increase in size of the particles from 20 nm in DMSO to 100–200 nm in water (Fig. 2D and S3†). In contrast, the size of D50% + AEE (55 nm) was found to be approximately the same to the originally determined value in DMSO. These larger apparent nano-object sizes could have been a consequence of the swelling of the NPs in water, their aggregation or a combination of these two effects. The possibility for the nano-objects to aggregate was investigated by determining the zeta-potential of the suspension of micelles in water, which was found to vary between +7 to −15 mV (Table 1). It thus appeared that the surface charges were too low to prevent aggregation via electrostatic repulsion, providing an explanation for the increase in size upon transferring the micelles to water.
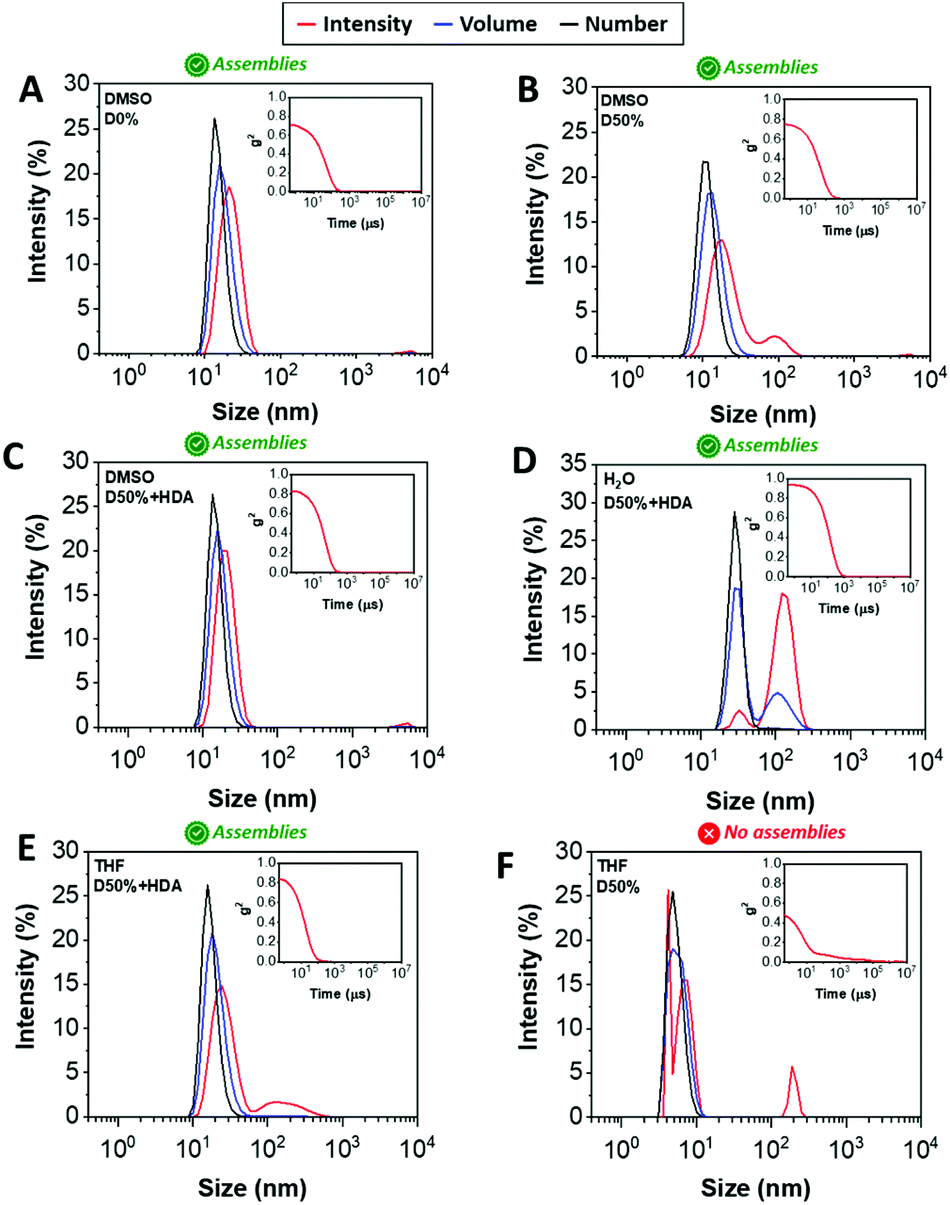 | ||
| Fig. 2 Size distributions of (A) D0%, (B) D50%, and (C) D50% + HDA in DMSO, (D) water, (E) THF, and (F) D50% in THF obtained by DLS. The intensity (red line), volume (blue line) and number (black line) distributions are displayed. The insets show the corresponding correlograms in each case. | ||
One way to verify whether polymeric NPs are successfully stabilised by cross-linking is to transfer them into a good solvent for both constituent blocks. Non-cross-linked nano-objects would be expected to disassemble under such conditions, which is typically reflected by a reduced scattering intensity by DLS. In contrast, efficient cross-linking should lead to NPs showing approximately the same size to those collected in selective solvent (i.e. DMSO). The dialysed micelles were thus freeze-dried and re-solvated in THF, which proved to be a good solvent for both blocks. Analysis of these THF solutions by DLS confirmed the presence of nano-objects of similar dimensions to the NPs suspended in DMSO (Fig. 2E and S3†), while the non-cross-linked nano-objects D0% displayed no self-assembly (Fig. 2F). These results demonstrated that efficient cross-linking took place using the different diamines, HDA, CA and AEE, providing stabilising effect to the cross-linked cores.
The morphology of the cross-linked nano-objects suspended in water was then investigated by transmission electron microscopy (TEM). Spherical micelles with similar sizes to those obtained in DMSO were observed, which supported the hypothesis that large sizes as measured by DLS originated from local flocculation (Fig. S8†). The TEM images showed the presence of a population of individual micelles and revealed that the morphology of the spherical NPs did not change. For the isolated particles, the average diameters measured from the dry-state TEM images (upon counting >250 particles in each case) were 15–21 nm, slightly smaller than the size measured by DLS in DMSO. This can be explained by shrinkage of the micelles caused upon drying during TEM grid preparation.
Retention of the helicity of the micellar cores was investigated by circular dichroism (CD) spectroscopy of the cross-linked micelles in THF, to ensure solvation of both blocks at 0.5 mg mL−1. This CD analysis after cross-linking indicated a slight decrease of the CD signal at λ = 360 nm (CD360) for D50% + HDA and D50% + CA, from 8.7 mdeg for the unreacted copolymer to 6.2 mdeg and 6.5 mdeg, respectively, for the cross-linked micelles (Fig. 3). However, helicity was mainly retained (>70% compared to D50%) after cross-linking. No loss in helicity was reported for D50% + AEE. This is promising for applications that would leverage the helical core of the nano-objects. This also establishes that the core's helices could withstand the reaction conditions employed during the PPM step.
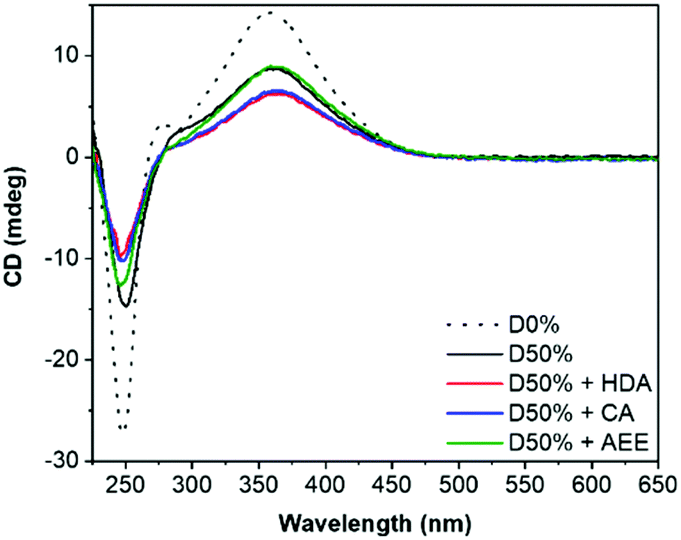 | ||
| Fig. 3 CD (THF, 0.5 mg mL−1) spectra of the unsubstituted D50% and its cross-linked counterparts. D0% is shown as reference. | ||
2.2 Redox- and pH-responsive NiCCo-PISA micelles
Core-cross-linked micelles D50% + CA and D50% + AEE were expected to exhibit responsiveness towards reducing environment and low pH, respectively (Fig. 4A and B). The stimuli-responsive behaviour of D50% + CA and D50% + AEE NPs was thus investigated using DLS analysis. CA cross-linked NPs (D50% + CA) in water at a concentration of 0.2 mg mL−1 were subjected to L-glutathione (GSH) reducing agent at a concentration of 10 mM. The disulfide bond was expected to be cleaved at this [GSH], which would result in the formation of more solvophilic thiol moieties inside the core and change the assembly of the nano-objects. 1H NMR spectroscopy analysis of the cross-linked structures yielded broad signals with poor resolution (Fig. S2†). Moreover, TEM analysis was also not employed since analogous polyisocyanide nanoparticles exhibit inherent aggregation effects, which would obfuscate any changes to particle size.86,87 Instead, the size distribution of the NPs was monitored by DLS over a period of 4 weeks, in the presence of reducing agent. The redox-responsive NPs showed a size increase over this time period (Fig. 4C). This observed change in size can be explained by the gradual increase in the core's hydrophilicity, which led to a swelling of the NPs. Moreover, the cross-link density in the micelle cores slowly decreased, resulting in “looser” particles that can either swell or aggregate, or both. The increase in size was found to be progressive over a period of several days, which proved to be a slower process as compared to previously reported systems.43,45 This could be linked to a number of elements including the hydrophobicity of the core, aggregation of NPs and solubility. First, the high hydrophobicity of the NPs core – that contained L-menthyl side-chains – could slow down the access of the hydrophilic reducing agent.94 Secondly, the aggregation behaviour of the NPs might further limit the diffusion which might have delayed the disassembly of the systems into unimers and could explain the continuous increase in size. Also, the switch from stabilising disulfide bridges to thiol functionalities increased the exchange rate of the unimers, favouring micelle aggregation or rearrangement, which in turn led to the size increase observed for D50% + CA, from 155 nm to 220 nm. Finally, it was likely that the thiol-functionalised copolymers display only a limited solubility in water. As cleavage of disulfides by GSH is reversible, the high concentration of thiols in D50% + CA micellar cores might have favoured the reformation of disulfide-type cross-links, which could explain the slow change in size. D0% treated with GSH showed no sign of swelling, aggregation or disassembly and no apparent change in size. This proved that the CA cross-linked micelles are effectively triggered by the presence of a reducing agent, changing the size distribution of the NPs.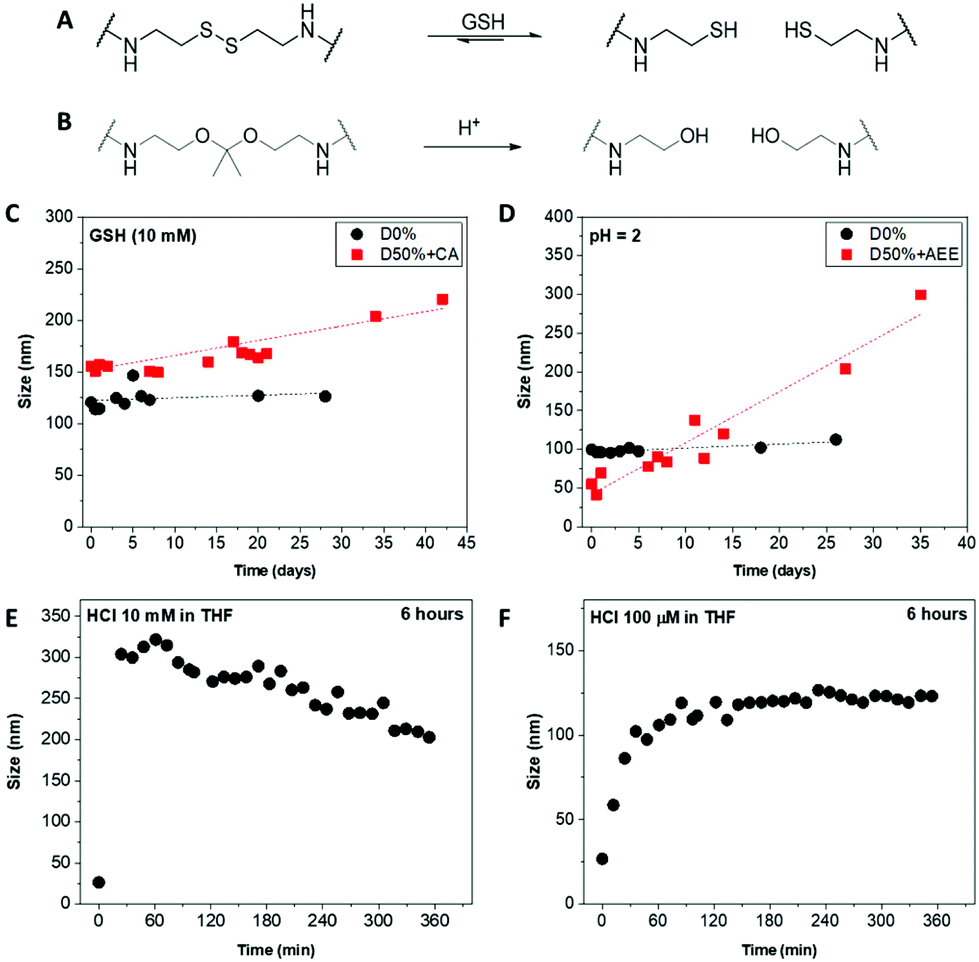 | ||
| Fig. 4 Scheme of (A) the GSH-triggered cleavage of the cystamine disulfide linkers and (B) the acid-triggered cleavage of the acetal linkers. Size evolution of (C) the CA-modified nanostructures upon treatment with GSH and the AEE-modified nanostructures at (D) pH = 2 in water, (E) 10 mM HCl and (F) 100 μM HCl in THF monitored by DLS. | ||
AEE cross-linked NPs (D50% + AEE) were treated under acidic conditions to assess their pH-responsive behaviour. The acetal linkers were thus cleaved into alcohols, which led to a response. The NPs were incubated in water at pH 2 for 4 weeks and their size evolution was monitored by DLS over this period (Fig. 4D). The D50% + AEE micelles exhibited a steady increase of their size from 55 nm at t0 to 205 nm after 4 weeks, demonstrating their responsiveness to an acid stimulus. On the other hand, only a slight change in size was observed for the unreactive non-cross-linked NPs, D0%, from 100 nm to 110 nm, confirming the responsivity conferred by the AEE moiety.
With the aim of circumventing the slow rate of response in water, the acid-triggered cleavage of D50% + AEE NPs was also performed in THF. The size change was hypothesised to be much faster as a consequence of the better solubility of the core in THF relatively to water. First, the D50% + AEE NPs suspension was reacted with 10 mM HCl in THF, which led to a fast size change of the NPs as evidenced by DLS (Fig. 4E). However, the response time frame, monitored every 10 minutes by DLS, was shorter than the time between the measurement points. There, the size of the nanoparticles even begins to decrease after the rapid initial increase. This observation alludes to potential disassembly of the nanostructures although complete disassembly was not observed under our experimental conditions. To slow down the process, a concentration of 100 μM of HCl in THF was employed resulting in expectedly slower size change (Fig. 4F). In 3 h, the size monitored by DLS increased from 20 nm to 120 nm where it reached a plateau as a result of the swelling of the micelles. This monitoring established that the NiCCo-PISA micelles were indeed pH-sensitive.
3. Conclusions
Nano-objects with cross-linked helical cores were readily achieved by NiCCo-PISA, followed by a straightforward PPM methodology involving the reaction between pentafluorophenyl ester-based units and various diamine molecules. The stability of the cross-linked micelles was confirmed by DLS measurements in THF, while the preservation of the helicity was confirmed by CD spectroscopy. NPs that were core cross-linked by cystamine exhibited redox-triggered response, which could be monitored by DLS in water, whereas NPs modified with the acetal-containing cross-linker were found to respond to changes in pH, as evidenced by DLS. Although complete disassembly of nanostructures was not observed in this study, we are actively investigating this process. Nevertheless, changes in core polarity could possibly still facilitate efficient delivery of payload from within the particles. Thus, this study expands the potential of NiCCo-PISA towards applications of the resulting smart nanomaterials in drug delivery and in sensors, or as nanoreactors where the chiral core could be leveraged for enantioselectivity.Author contributions
S. J. and J. C. F. conceived this study. S. J. performed the experiments and analysed the data. S. V. obtained the TEM images. D. T., A. P. D. and R. K. O. obtained the funding for this research project. S. J. wrote the original draft. J. C. F., S. V., D. T., A. P. D. and R. K. O. reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Union (SUSPOL-EJD 642671), ERC (grant number 615142), EPSRC, and the University of Birmingham.Notes and references
- M. Yoshida and J. Lahann, ACS Nano, 2008, 2, 1101–1107 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- X. Liu, Y. Yang and M. W. Urban, Macromol. Rapid Commun., 2017, 38, 1700030 CrossRef.
- S. Sur, A. Rathore, V. Dave, K. R. Reddy, R. S. Chouhan and V. Sadhu, Nano-Struct. Nano-Objects, 2019, 20, 100397 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- O. V. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef.
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 24, 3878–3886 CrossRef CAS PubMed.
- Q. Wan, K. Wang, C. He, M. Liu, G. Zeng, H. Huang, F. Deng, X. Zhang and Y. Wei, Polym. Chem., 2015, 6, 8214–8221 RSC.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- H. Li, H. Li, W. Wang, Y. Tao, S. Wang, Q. Yang, Y. Jiang, C. Zheng, W. Huang and R. Chen, Angew. Chem., Int. Ed., 2020, 59, 4756–4762 CrossRef CAS PubMed.
- S. Maji, B. Cesur, Z. Zhang, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2016, 7, 1705–1710 RSC.
- Q. Yan, J. Yuan, W. Yuan, M. Zhou, Y. Yin and C. Pan, Chem. Commun., 2008, 6188–6190 RSC.
- D.-Q. Feng, W. Zhu, G. Liu and W. Wang, RSC Adv., 2016, 6, 96729–96734 RSC.
- J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- M. C. Arno, R. J. Williams, P. Bexis, A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Biomaterials, 2018, 180, 184–192 CrossRef CAS PubMed.
- C. Lu and M. Urban, ACS Nano, 2015, 9, 3119–3124 CrossRef CAS.
- M. Motornov, J. Zhou, M. Pita, V. Gopishetty, I. Tokarev, E. Katz and S. Minko, Nano Lett., 2008, 8, 2993–2997 CrossRef CAS.
- W. Agut, A. Brûlet, C. Schatz, D. Taton and S. Lecommandoux, Langmuir, 2010, 26, 10546–10554 CrossRef CAS.
- N. Deirram, C. Zhang, S. S. Kermaniyan, A. P. R. Johnston and G. K. Such, Macromol. Rapid Commun., 2019, 40, 1800917 CrossRef PubMed.
- D. E. Bergbreiter, B. L. Case, Y.-S. Liu and J. W. Caraway, Macromolecules, 1998, 31, 6053–6062 CrossRef CAS.
- S. Kanaoka, N. Yagi, Y. Fukuyama, S. Aoshima, H. Tsunoyama, T. Tsukuda and H. Sakurai, J. Am. Chem. Soc., 2007, 129, 12060–12061 CrossRef CAS PubMed.
- H. A. Zayas, A. Lu, D. Valade, F. Amir, Z. Jia, R. K. O'Reilly and M. J. Monteiro, ACS Macro Lett., 2013, 2, 327–331 CrossRef CAS PubMed.
- S. M. Lee and Y. C. Bae, Macromolecules, 2014, 47, 8394–8403 CrossRef CAS.
- R. Keogh, L. D. Blackman, J. C. Foster, S. Varlas and R. K. O'Reilly, Macromol. Rapid Commun., 2020, 41, 1900599 CrossRef CAS PubMed.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- T. Yan, F. Li, S. Qi, J. Tian, R. Tian, J. Hou, Q. Luo, Z. Dong, J. Xu and J. Liu, Chem. Commun., 2020, 56, 149–152 RSC.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. Read de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS.
- S. Tsoi, J. Zhou, C. Spillmann, J. Naciri, T. Ikeda and B. Ratna, Macromol. Chem. Phys., 2013, 214, 734–741 CrossRef CAS.
- K. Kumar, C. Knie, D. Bléger, M. A. Peletier, H. Friedrich, S. Hecht, D. J. Broer, M. G. Debije and A. P. H. J. Schenning, Nat. Commun., 2016, 7, 11975 CrossRef CAS PubMed.
- T. Zhao, P. Wang, Q. Li, A. A. Al-Khalaf, W. N. Hozzein, F. Zhang, X. Li and D. Zhao, Angew. Chem., Int. Ed., 2018, 57, 2611–2615 CrossRef CAS PubMed.
- W. Agut, D. Taton, A. Brûlet, O. Sandre and S. Lecommandoux, Soft Matter, 2011, 7, 9744–9750 RSC.
- A. P. Majewski, A. Schallon, V. Jérôme, R. Freitag, A. H. E. Müller and H. Schmalz, Biomacromolecules, 2012, 13, 857–866 CrossRef CAS PubMed.
- J. Thévenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099–7116 RSC.
- L. Yu, Y. Zhang, X. Dai, Q. Xu, L. Zhang and J. Tan, Chem. Commun., 2019, 55, 11920–11923 RSC.
- J. Zhang, N. Ma, F. Tang, Q. Cui, F. He and L. Li, ACS Appl. Mater. Interfaces, 2012, 4, 1747–1751 CrossRef CAS PubMed.
- D. Wang, T. Liu, J. Yin and S. Liu, Macromolecules, 2011, 44, 2282–2290 CrossRef CAS.
- J. Zhang, J. Tanaka, P. Gurnani, P. Wilson, M. Hartlieb and S. Perrier, Polym. Chem., 2017, 8, 4079–4087 RSC.
- W. Deng, J. Chen, A. Kulkarni and D. H. Thompson, Soft Matter, 2012, 8, 5843–5846 RSC.
- L.-J. Chen, G.-Z. Zhao, B. Jiang, B. Sun, M. Wang, L. Xu, J. He, Z. Abliz, H. Tan, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 5993–6001 CrossRef CAS PubMed.
- M. J. Bergman, J. S. Pedersen, P. Schurtenberger and N. Boon, Soft Matter, 2020, 16, 2786–2794 RSC.
- S. H. Kim, J. H. Kim, H. J. Choi and J. Park, RSC Adv., 2015, 5, 72387–72393 RSC.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2019, 40, 1800460 CrossRef.
- J. Tanaka, G. Moriceau, A. Cook, A. Kerr, J. Zhang, R. Peltier, S. Perrier, T. P. Davis and P. Wilson, Macromolecules, 2019, 52, 992–1003 CrossRef.
- C. Maiti, S. Parida, S. Kayal, S. Maiti, M. Mandal and D. Dhara, ACS Appl. Mater. Interfaces, 2018, 10, 5318–5330 CrossRef CAS PubMed.
- Y. Gao and C.-M. Dong, Polym. Chem., 2017, 8, 1223–1232 RSC.
- Y. Wang, P. Xing, S. Li, M. Ma, M. Yang, Y. Zhang, B. Wang and A. Hao, Langmuir, 2016, 32, 10705–10711 CrossRef CAS PubMed.
- R. Deng, Y. Ning, E. R. Jones, V. J. Cunningham, N. J. W. Penfold and S. P. Armes, Polym. Chem., 2017, 8, 5374–5380 RSC.
- S. M. North and S. P. Armes, Polym. Chem., 2020, 11, 2147–2156 RSC.
- L. Thomi, P. Schaefer, K. Landfester and F. R. Wurm, Macromolecules, 2016, 49, 105–109 CrossRef CAS.
- X. Han, J. Zhang, C.-Y. Qiao, W.-M. Zhang, J. Yin and Z.-Q. Wu, Macromolecules, 2017, 50, 4114–4125 CrossRef CAS.
- A. N. Koo, H. J. Lee, S. E. Kim, J. H. Chang, C. Park, C. Kim, J. H. Park and S. C. Lee, Chem. Commun., 2008, 6570–6572 RSC.
- Y. Li, K. Xiao, J. Luo, W. Xiao, J. S. Lee, A. M. Gonik, J. Kato, T. A. Dong and K. S. Lam, Biomaterials, 2011, 32, 6633–6645 CrossRef CAS PubMed.
- H. He, Y. Bai, J. Wang, Q. Deng, L. Zhu, F. Meng, Z. Zhong and L. Yin, Biomacromolecules, 2015, 16, 1390–1400 CrossRef CAS.
- S. E. Paramonov, E. M. Bachelder, T. T. Beaudette, S. M. Standley, C. C. Lee, J. Dashe and J. M. J. Fréchet, Bioconjugate Chem., 2008, 19, 911–919 CrossRef CAS PubMed.
- T. Miyabe, H. Iida, A. Ohnishi and E. Yashima, Chem. Sci., 2012, 3, 863–867 RSC.
- Q. Wang, B.-F. Chu, J.-H. Chu, N. Liu and Z.-Q. Wu, ACS Macro Lett., 2018, 7, 127–131 CrossRef CAS PubMed.
- M. Reggelin, S. Doerr, M. Klussmann, M. Schultz and M. Holbach, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5461 CrossRef CAS PubMed.
- D. Bécart, V. Diemer, A. Salaün, M. Oiarbide, Y. R. Nelli, B. Kauffmann, L. Fischer, C. Palomo and G. Guichard, J. Am. Chem. Soc., 2017, 139, 12524–12532 CrossRef PubMed.
- T. Yamamoto, R. Murakami and M. Suginome, J. Am. Chem. Soc., 2017, 139, 2557–2560 CrossRef CAS PubMed.
- B. Zhao, J. R. Deng and J. P. Deng, ACS Macro Lett., 2017, 6, 6–10 CrossRef CAS PubMed.
- L. Zhou, B.-F. Chu, X.-Y. Xu, L. Xu, N. Liu and Z.-Q. Wu, ACS Macro Lett., 2017, 6, 824–829 CrossRef CAS.
- H. Yamashita, T. Misawa, M. Oba, M. Tanaka, M. Naito, M. Kurihara and Y. Demizu, Bioorg. Med. Chem., 2017, 25, 1846–1851 CrossRef CAS PubMed.
- W.-M. Zhang, J. Zhang, Z. Qiao, H.-Y. Liu, Z.-Q. Wu and J. Yin, Polym. Chem., 2018, 9, 4233–4242 RSC.
- Y. Chen, Z.-H. Zhang, X. Han, J. Yin and Z.-Q. Wu, Macromolecules, 2016, 49, 7718–7727 CrossRef CAS.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef.
- C. Grazon, P. Salas-Ambrosio, E. Ibarboure, A. Buol, E. Garanger, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Angew. Chem., Int. Ed., 2019, 59, 622–626 CrossRef PubMed.
- C. Liu, C. Y. Hong and C. Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- J. C. Foster, S. Varlas, L. D. Blackman, L. A. Arkinstall and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 10672–10676 CrossRef CAS.
- D. Cordella, F. Ouhib, A. Aqil, T. Defize, C. Jérôme, A. Serghei, E. Drockenmuller, K. Aissou, D. Taton and C. Detrembleur, ACS Macro Lett., 2017, 6, 121–126 CrossRef CAS PubMed.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- A. Hanisch, P. Yang, A. N. Kulak, L. A. Fielding, F. C. Meldrum and S. P. Armes, Macromolecules, 2016, 49, 192–204 CrossRef CAS.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 18964–18970 CrossRef CAS PubMed.
- S. Jimaja, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2021, 12, 105–112 RSC.
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226–232 CrossRef CAS PubMed.
- X. An, A. Zhu, H. Luo, H. Ke, H. Chen and Y. Zhao, ACS Nano, 2016, 10, 5947–5958 CrossRef CAS PubMed.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- M. Yin, Y. Bao, X. Gao, Y. Wu, Y. Sun, X. Zhao, H. Xu, Z. Zhang and S. Tan, J. Mater. Chem. B, 2017, 5, 2964–2978 RSC.
- M. Chen, J. Hu, L. Wang, Y. Li, C. Zhu, C. Chen, M. Shi, Z. Ju, X. Cao and Z. Zhang, Sci. Rep., 2020, 10, 14447 CrossRef CAS PubMed.
- G. Chen, Y. Wang, R. Xie and S. Gong, J. Controlled Release, 2017, 259, 105–114 CrossRef CAS PubMed.
- Y. Tian, R. Tian, L. Chen, R. Jin, Y. Feng, Y. Bai and X. Chen, Macromol. Rapid Commun., 2019, 40, 1800824 CrossRef PubMed.
- R. J. Carrazzone, J. C. Foster, Z. Li and J. B. Matson, Eur. Polym. J., 2020, 141, 110077 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00397j |
| This journal is © The Royal Society of Chemistry 2022 |